

Abstract
Presynaptic kainate receptors regulate synaptic transmission in several brain areas but are not known to have this action at immature mossy fiber (MF) terminals, which during the first week of postnatal life release GABA, which exerts into targeted cells a depolarizing and excitatory action. Here, we report that, during the first week of postnatal life, endogenous activation of GluK1 receptors by glutamate present in the extracellular space severely depresses MF-mediated GABAergic currents [GABAA-mediated postsynaptic currents (GPSCs)]. Activation of GluK1 receptors was prevented by treating the slices with enzymatic glutamate scavengers that enhanced the clearance of glutamate from the extracellular space. The depressant effect of GluK1 on MF-GPSCs was mediated by a metabotropic process sensitive to pertussis toxin. In the presence of U73122 (1-[6-[[(17b)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione), a selective inhibitor of phospholipase C, along the transduction pathway downstream to G-protein, GluK1 activation increased the probability of GABA release, thus unveiling the ionotropic action of this receptor. In line with this type of action, we found that GluK1 enhanced MF excitability by directly depolarizing MF terminals via calcium-permeable cation channels. Furthermore, GluK1 dynamically regulated the direction of spike time-dependent plasticity occurring by pairing MF stimulation with postsynaptic spiking and switched spike time-dependent potentiation into depression. The GluK1-induced depression of MF-GPSCs would prevent excessive activation of the CA3 associative network by the excitatory action of GABA and the emergence of seizures in the immature brain.
Introduction
Kainate receptors (KARs) are highly expressed in the CNS, where they are involved in different physiological functions (Jane et al., 2009). Five different receptor subtypes have been cloned, GluK1-5, which coassemble in various combinations to form functional receptors with distinct pharmacological and physiological properties (Collingridge et al., 2009). Although at some excitatory connections KARs carry at least in part current charges of synaptic responses (Castillo et al., 1997; Vignes and Collingridge, 1997; Mulle et al., 1998; Cossart et al., 2002), at presynaptic sites they exert a powerful control of transmitter release (Lerma, 2003). In addition, evidence has been provided that, in several CNS regions, KARs control cell excitability through a metabotropic type of action, which involves the activation of G-protein and intracellular signal cascades (Rodríguez-Moreno and Lerma, 1998; Rozas et al., 2003; Melyan et al., 2004; Lerma, 2006; Rodríguez-Moreno and Sihra, 2007). The hippocampus is endowed with KARs with preponderant presynaptic localization (Represa et al., 1987; Ben-Ari and Cossart, 2000). In particular, activation of KARs localized on glutamatergic mossy fibers (MFs), the axons of dentate gyrus granule cells, enhances glutamate release particularly during frequency-dependent facilitation, a form of short-term plasticity characteristic of MF–CA3 synapses (Contractor et al., 2001; Lauri et al., 2001a; Schmitz et al., 2001) (but see Kwon and Castillo, 2008). This effect has been attributed to kainate-induced depolarization of presynaptic boutons or axon terminals (Lauri et al., 2001b; Kamiya et al., 2002; Nicoll and Schmitz, 2005) and the release of calcium from local stores (Scott et al., 2008). Interestingly, KARs are present also on presynaptic GABAergic terminals, where they exert either a facilitatory (Mulle et al., 2000; Cossart et al., 2001) or a depressant (Clarke et al., 1997; Rodríguez-Moreno et al., 1997; Rodríguez-Moreno and Lerma, 1998; Maingret et al., 2005) effect on GABA release. Activation of these receptors relies on the source of glutamate, on the spatiotemporal feature of glutamate release in the extracellular space, and their affinity for glutamate.
Several lines of evidence suggest a role of KARs in neuronal development. Signaling via presynaptic GluK1 containing KARs has been shown to be critical for regulating the number of functional glutamatergic synapses (Vesikansa et al., 2007) and the balance between GABAergic and glutamatergic transmission, which control correlated network activity (Lauri et al., 2005). Interestingly, during the first week of postnatal life, the main neurotransmitter released from MF terminals is GABA (Safiulina et al., 2006), which exerts a depolarizing and excitatory action on targeted cells (Sivakumaran et al., 2009). MF-evoked glutamatergic currents comprising a kainate component start appearing after postnatal day 6 (P6) (Marchal and Mulle, 2004).
Here, we addressed whether, immediately after birth, MF-induced GABAA-mediated postsynaptic currents (GPSCs) in CA3 principal cells are controlled by presynaptic KARs. We report that, at this developmental stage, MF-GPSCs are downregulated by G-protein-coupled GluK1 receptors, which are endogenously activated by glutamate present in the extracellular medium. We further demonstrated that, at these synapses, GluK1 receptors dynamically regulate the direction of spike time-dependent plasticity (STDP).
Materials and Methods
Slice preparation.
Experiments were performed on hippocampal slices from P2 to P5 Wistar rats as previously described (Gasparini et al., 2000). All experiments were performed in accordance with the European Community Council Directive of 24 November 1986 (86/609EEC) and were approved by local authority veterinary service. Briefly, animals were decapitated after being anesthetized with an intraperitoneal injection of urethane (2 g/kg). The brain was quickly removed from the skull and placed in ice-cold artificial CSF (ACSF) containing the following (in mm): 130 NaCl, 3.5 KCl, 1.2 NaH2PO4, 27 NaHCO3, 1.3 MgCl2, 2 CaCl2, 25 glucose, saturated with 95% O2 and 5% CO2, pH 7.3–7.4. Transverse hippocampal slices (400 μm thick) were cut with a vibratome and stored at room temperature (20–24°C) in a holding bath containing the same solution as above. After a recovery period of at least 1 h, an individual slice was transferred to the recording chamber, in which it was continuously superfused with oxygenated ACSF at a rate of 2–3 ml/min at 33–35°C.
Electrophysiological recordings.
Electrophysiological experiments were performed from CA3 pyramidal cells using the whole-cell configuration of the patch-clamp technique in current- or voltage-clamp mode. Neurons were visualized using an upright microscope (Olympus BX51WI) equipped with differential interference contrast optics and infrared video camera. Patch electrodes were pulled from borosilicate glass capillaries (Hilgenberg). They had a resistance of 4–6 MΩ when filled with an intracellular solution containing the following (in mm): 140 KCl, 1 MgCl2, 10 HEPES, 4 MgATP, 0.5 EGTA, pH 7.3. In some cases, guanosine 5′-[β-thio]diphosphate (GDPβS) (Sigma-Aldrich) at the concentration of 0.3 mm was included into the patch pipette to block postsynaptic G-protein-coupled receptors. Recordings were made with a patch-clamp amplifier (Axopatch 200A; Molecular Devices). The access resistance was repetitively monitored every 5 min with a voltage step and was <20 MΩ, and the results were discarded if it changed >15–20%.
Mossy fiber GABAA-mediated postsynaptic currents (GPSCs) were evoked at 0.05 Hz from a holding potential of −70 mV. We used minimal stimulation of the granule cells in the dentate gyrus to activate only one or few presynaptic fibers. According to the technique described by Jonas et al. (1993) and Allen and Stevens (1994), the stimulation intensity was decreased until only a single axon was activated. This was achieved when the mean amplitude of the postsynaptic currents and failure probability remained constant over a range of stimulus intensities near threshold for detecting a response (see Fig. 1A,B) (Safiulina et al., 2006). An abrupt increase in the mean peak amplitude of synaptic currents was observed when the stimulus intensity was further increased. This all-or-none behavior led us to assume that only a single fiber was stimulated. In addition, the latency and the shape of individual synaptic responses remained constant for repeated stimuli. The monosynaptic nature of synaptic currents was supported by the unimodal and narrow latencies and rise time distributions, which remained constant when the extracellular Ca2+/Mg2+ concentration ratio was reduced from 2:1.3 to 1:3 (Safiulina et al., 2006).
Figure 1.

AMPA receptors do not contribute to unitary postsynaptic currents evoked in CA3 principal cells by minimal stimulation of granule cells in the dentate gyrus. A, Unitary synaptic currents evoked in a CA3 pyramidal cell at P4 with different stimulation intensities before (Control) and during application of GYKI 52466 (30 μm). Each trace is the average of 15–20 responses (including failures). B, Peak amplitudes of synaptic currents represented in A are plotted as a function of stimulus intensities. The bars are SEM, and the dashed lines are the averaged amplitude of GPSCs. Latency (C) and rise time (D) distributions of individual currents evoked in a CA3 pyramidal cell by minimal stimulation of granule cells in the dentate gyrus in the absence (Control) and in the presence of GYKI 52466. Note that, in the presence of GYKI 52466, the unimodal distribution of latencies and rise times of individual responses did not change.
When the probability of synaptic failures in response to a first stimulus was near 1 (failures were estimated by visual discrimination), we applied a second pulse at 50 ms interval. If a response to a second stimulus appeared in 15–30 consecutive trials (silent to the first stimulus), we considered this synapse “presynaptically” silent.
MF inputs were identified on the basis of their sensitivity to group III metabotropic glutamate receptor (mGluR) agonist l(+)-2-amino-4-phosphonobutyric acid (l-AP4) (10 μm) (Gutiérrez et al., 2003; Kasyanov et al., 2004; Safiulina et al., 2006), their strong paired-pulse facilitation, and short-term frequency-dependent facilitation (Safiulina et al., 2006). GPSCs were blocked by bicuculline or picrotoxin. In contrast to MF inputs, GABAergic inputs from interneurons were insensitive to l-AP4 (Walker et al., 2001; Safiulina et al., 2006). To block G-protein-mediated signals, slices were incubated overnight in a medium containing pertussis toxin (PTx) (5 μg/ml).
In some experiments, antidromic action potentials were recorded from visually identified granule cells in dentate gyrus. In this case, the intracellular solution contained K-gluconate (150 mm) instead of KCl. Extracellular stimuli (at 0.3 Hz; duration, 150 s) were delivered via a stimulation electrode positioned in stratum lucidum ∼200 μm away from the granule cell layer.
STDP was induced in current-clamp mode by pairing MF stimulation with postsynaptic spikes. MF-GPSCs were recorded first in voltage-clamp mode for 5–10 min to obtain a stable baseline. Then, STDP was induced by pairing presynaptic stimulation of granule cells in the dentate gyrus with postsynaptic spiking. This sequence (MF stimulation–postsynaptic spike) was repeated 10 times at 0.1 Hz. In the case of positive pairing (pre vs post), a delay of 15 ms was introduced between presynaptic stimulation and postsynaptic spiking, whereas, in the case of negative pairing (post vs pre), the delay between postsynaptic spiking and presynaptic stimulation was 50 ms. Changes in synaptic efficacy were monitored by recording synaptic currents for additional 20–30 min after pairing.
The drugs used were as follows: 6,7-dinitroquinoxaline-2,3-dione (DNQX), l-AP4, picrotoxin (PTX), 4-(8-methyl-9H-1,3-dioxolo[4,5-h][2,3]benzodiazepin-5-yl)-benzenamine hydrochloride (GYKI 52466), (S)-1-(2-amino-2-carboxyethyl)-3-(2-carboxybenzyl)pyrimidine-2,4-dione (UBP 302), (RS)-2-amino-3-(3-hydroxy-5-tert-butylisoxazol-4-yl)propanoic acid (ATPA), d-(−)-2-amino-5-phosphonopentaoic acid (d-AP5), (2S)-2-amino-2-[(1S,2S)-2-carboxycycloprop-1-yl]-3-(xanth-9-yl) propanoic acid (LY341495), philanthotoxin (PhTx), PTx, 1-[6-[[(17b)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino]hexyl]-1H-pyrrole-2,5-dione (U73122) (all purchased from Tocris Bioscience); glutamate scavenger system [glutamic-pyruvic transaminase (GPT) plus pyruvate], 4-[[4-formyl-5-hydroxy-6-methyl-3-[(phosphonooxy)methyl]-2-pyridinyl] azo]-1,3-benzenedisulfonic acid tetrasodium salt (PPADS), dihydro-β-erythroidine (DHβE), atropine, GDPβS, baclofen, and 3-[[(3,4-dichlorophenyl) methyl]amino]propyl]diethoxymethyl)phosphinic acid (CGP 52432) were purchased from Sigma-Aldrich.
All drugs were dissolved in water, ethanol, or dimethylsulfoxide (DMSO), as required. UBP 302 was dissolved in 1 eq NaOH. DNQX, GYKI 52466, and LY341495 were dissolved in DMSO. The final concentration of DMSO in the bathing solution was 0.1%. At this concentration, DMSO alone did not modify the shape or the kinetics of synaptic currents. Drugs were applied in the bath via a three-way tap system, by changing the superfusion solution to one differing only in its content of drug(s). The ratio of flow rate to bath volume ensured complete exchange within 1–2 min.
Data acquisition and analysis.
Data were acquired and digitized with an analog-to-digital converter (Digidata 1200; Molecular Devices) and stored on a computer hard disk. Acquisition and analysis of evoked responses were performed with Clampfit 9 (Molecular Devices). Data were sampled at 20 kHz and filtered with a cutoff frequency of 1 kHz. Mean GPSCs amplitude was obtained by averaging successes and failures. The paired-pulse ratio (PPR) was calculated as the mean amplitude of the synaptic response evoked by the second stimulus over that evoked by the first one. The coefficient of variation was calculated as the ratio between the SD of synaptic currents amplitude and the mean. Unless otherwise stated, data are presented as mean ± SEM. Quantitative comparisons were based on Student's paired or unpaired t test, as required, and values of p < 0.05 were considered to be significant.
Results
Tonic activation of presynaptic GluK1 receptors downregulates GABA release from MF terminals
Whole-cell patch-clamp recordings were performed from CA3 principal cells in hippocampal slices obtained from P2–P5 rats. In agreement with previous studies (Gutiérrez et al., 2003; Kasyanov et al., 2004; Safiulina et al., 2006; Safiulina and Cherubini, 2009; Sivakumaran et al., 2009), during the first week of postnatal life, minimal stimulation of granule cells in the dentate gyrus elicited low-probability GPSCs, which were completely blocked by bicuculline (10 μm) or picrotoxin (100 μm). Depolarizing the cell to positive potentials (+40 mV) produced bicuculline-sensitive outward currents with latency, onset, and deactivation kinetics similar to those obtained at −70 mV, further indicating that they were mediated by GABAA receptors (data not shown) (Safiulina et al., 2006). Consistent with previous observations (Safiulina et al., 2006; Safiulina and Cherubini, 2009), bath application of GYKI 52466 (30 μm), which at this concentration selectively blocks AMPA receptors, did not modify the amplitude, the shape, the latency, and rise time of individual responses, indicating that AMPA receptors do not contribute to synaptic currents (Fig. 1A–D). On average, on five cells, the amplitude of MF-evoked GPSCs was 40 ± 2 and 39 ± 1 pA (p = 0.8), the rise time was 1.8 ± 0.2 and 1.9 ± 0.3 ms (p = 0.4), and latency was 2.8 ± 0.2 and 2.8 ± 0.3 ms (p = 0.8), before and during GYKI 52466 application, respectively. Interestingly, the addition of DNQX (50 μm) (which blocks both AMPA and kainate receptors) to GYKI 52466 significantly enhanced the amplitude of single fiber-evoked GPSCs, an effect that was associated with an increase in the number of successes (Fig. 2A). The time course of DNQX effects on the amplitude of GPSCs (successes plus failures) obtained in eight cells is represented in Figure 2B. As summarized in supplemental Figure S1, A and B (available at www.jneurosci.org as supplemental material), the peak amplitude of GPSCs (successes plus failures) was 30 ± 4 and 53 ± 10 pA before and after DNQX, respectively (p = 0.01), whereas the success rate changed from 0.34 ± 0.05 to 0.57 ± 0.08 (p = 0.006). These effects were associated with a significant reduction of the PPR (from 1.33 ± 0.23 to 0.67 ± 0.11; p = 0.001) (supplemental Fig. S1C, available at www.jneurosci.org as supplemental material) and a significant increase in the inverse squared value of the coefficient of variation (CV−2) of responses amplitude (from 0.73 ± 0.15 to 2.28 ± 0.44; p = 0.03) (supplemental Fig. S1D, available at www.jneurosci.org as supplemental material). Although changes in success rate may be indicative of presynaptic or postsynaptic changes, PPR and CV−2 are primarily used to evaluate changes in probability of transmitter release (Zucker and Regehr, 2002). These effects occurred in the absence of any change in input resistance or in the holding current of the recorded neurons. As expected for MF-mediated responses, GPSCs were significantly reduced in amplitude by group III mGluR agonist l-AP4 (10 μm) and completely blocked by picrotoxin (100 μm) (Fig. 2B) (Gutiérrez et al., 2003; Kasyanov et al., 2004; Safiulina et al., 2006). Synaptic currents originating from GABAergic interneurons could be readily distinguished from MF-GPSCs since unlike the latter, which exhibited paired-pulse facilitation, they exhibited paired-pulse depression and were insensitive to l-AP4 (the peak amplitude of synaptic currents was 39 ± 8 and 36 ± 9 pA before and during l-AP4, respectively; p = 0.1; n = 7) (Walker et al., 2001; Safiulina et al., 2006) (data not shown).
Figure 2.

Endogenous activation of presynaptic kainate receptors downregulates MF-GPSCs. A, Superimposed individual traces of MF-GPSCs evoked in the presence of GYKI 52466 (30 μm) and GYKI 52466 plus DNQX (50 μm). Note that, in this and in the following experiments, GYKI 52466 was always present in the bathing solution in control conditions. Below, Averaged traces (successes plus failures). Note that DNQX enhanced the amplitude of the first response and reduced the number of synaptic failures. B, Summary plot showing the mean amplitude of GPSCs obtained in eight cells in the presence of DNQX, l-AP4, and PTX. Vertical bars are SEM. The horizontal dashed line refers to the mean amplitude value measured before DNQX. C, Superimposed individual traces (above) and average traces (below) of MF-GPSCs evoked in the presence of GYKI 52466 (30 μm) and GYKI 52466 plus UBP 302 (10 μm). D, As in B, but in the presence of UBP 302, l-AP4, and PTX (n = 19).
To exclude the possibility of an indirect effect via kainate-induced modulation of other receptors known to depress transmitter release such as GABAB receptors (Safiulina and Cherubini, 2009), nicotine and muscarinic acetylcholine receptors (Maggi et al., 2004), purinergic P2Y receptors (Zhang et al., 2003; Safiulina et al., 2005), and mGluRs (Scanziani et al., 1997), in a set of experiments (n = 18) DNQX was applied in the presence of CGP 52432 (1 μm), DHβE (50 μm), atropine (1 μm), PPADS (50 μm), and LY341495 (100 μm), selective antagonists for these receptors. Also in this case, DNQX induced a significant increase in GPSCs amplitude similar to that obtained in the absence of the blockers, suggesting a direct effect of DNQX on kainate receptors localized on MF terminals (supplemental Fig. S2, available at www.jneurosci.org as supplemental material).
To identify which kainate receptor subtype was involved in the observed effects, we used UBP 302, which is a selective GluK1 kainate receptor antagonist (More et al., 2004; Jane et al., 2009). The rationale behind was that, during postnatal development, GluK1 receptors are highly expressed in the hippocampus (Bettler et al., 1990; Bahn et al., 1994; Ritter et al., 2002) and previous work from immature CA3 pyramidal cells demonstrated the involvement of GluK1 in modulation of spontaneous, network-driven giant depolarizing potentials (GDPs) (Lauri et al., 2005), which are highly expressed in the CA3 area (Ben-Ari et al., 2007). Like DNQX, UBP 302 (10 μm) caused a significant increase in amplitude of GPSCs (Fig. 2C,D). As summarized in supplemental Figure S3A (available at www.jneurosci.org as supplemental material), the peak amplitude of GPSCs was 48 ± 8 and 84 ± 15 pA before and after UBP 302, respectively (n = 19; p = 0.001). UBP 302 caused also a significant increase in success rate (from 0.4 ± 0.04 to 0.7 ± 0.03; p = 0.001) (supplemental Fig. S3B, available at www.jneurosci.org as supplemental material), a significant decrease in PPR (from 1.4 ± 0.2 to 0.6 ± 0.1; n = 16; p = 0.001) (supplemental Fig. S3C, available at www.jneurosci.org as supplemental material), and a significant increase in CV−2 (from 0.97 ± 0.2 to 3.3 ± 0.9; p = 0.001; n = 19) (supplemental Fig. S3D, available at www.jneurosci.org as supplemental material). In six cases, stimulation of granule cells in the dentate gyrus failed to produce any synaptic response to the first stimulus (over at least 30 consecutive trials). However, occasional responses to the second stimulus suggested that these synapses were “presynaptically” silent (Gasparini et al., 2000; Kasyanov et al., 2004; Safiulina and Cherubini, 2009; Sivakumaran et al., 2009). Application of UBP 302 to “presynaptically” silent cells induced the appearance of synaptic responses to the first stimulus (supplemental Fig. S4, available at www.jneurosci.org as supplemental material). In these cases, the PPR could not be measured because calculated ratios are infinitely large when the mean amplitude of the first response is close to zero. Compatible with a lack of quantal release, in these cases also the CV−2 was close to zero.
In contrast to UBP 302, pharmacological activation of GluK1 receptors with ATPA (1 μm), a selective GluK1 agonist (Clarke et al., 1997), reversibly decreased the amplitude of MF-GPSCs (from 54 ± 9 to 22 ± 4 pA; n = 11; p = 0.001) (Fig. 3A–C), an effect that was accompanied with a decrease in the number of successes (0.6 ± 0.1 to 0.35 ± 0.1; p = 0.001) (Fig. 3D), an increase in PPR (0.8 ± 0.1 to 1.3 ± 0.2; n = 7; p = 0.008) (Fig. 3E), and a decrease in the CV−2 (1.5 ± 1.3 to 1.1 ± 0.2; n = 11; p = 0.01) (Fig. 3F). It is worth noting that for ATPA experiments we used cells with high release probability and therefore exhibiting a low PPR (ranging from 0.4 to 1.3). Together, these data strongly indicate that presynaptic GluK1 receptors depress GABA release from immature MF terminals.
Figure 3.

Bath application of the selective GluK1 agonist ATPA mimics the effects of endogenous glutamate on kainate receptors. A, Averaged traces of GPSCs evoked in a CA3 pyramidal cell by stimulation of granule cells in the dentate gyrus, in control condition and after addition of ATPA (1 μm). B, Summary plot for 11 cells. C–F, Amplitude (C), successes (D), PPR (E), and inversed square of CV (F) measured in individual cells before and after application of ATPA. **p < 0.01; ***p < 0.001.
Presynaptic GluK1 receptors are endogenously activated by glutamate present in the extracellular space
How could presynaptic GluK1 receptors be activated if, early in postnatal development, the main neurotransmitter released by MF is GABA? One possibility is that these receptors are tonically activated by endogenous glutamate present in the extracellular space. To test this hypothesis, in another set of experiments, we used an enzymatic glutamate scavenger system (GPT plus pyruvate) (Overstreet et al., 1997; Min et al., 1998) on the assumption that enhancing the clearance of glutamate from the extracellular space prevents the activation of presynaptic kainate receptors. GPT catalyzes the conversion of glutamate and pyruvate to α-ketoglutarate and alanine. To exclude any effect via mGluRs (Scanziani et al., 1997), we routinely added in the bathing solution the broad spectrum mGluR1–8 antagonist LY341495 (100 μm) (Fitzjohn et al., 1998). The scavenger mimicked in all respects the facilitating effects of UBP 302 on GPSCs (the peak amplitude of GPSCs was 44 ± 8 and 73 ± 10 pA before and after the scavenger, respectively; n = 7; p = 0.003) (Fig. 4A,B). The scavenger caused also a significant increase in success rate (from 0.5 ± 0.1 to 0.8 ± 0.1; p = 0.01) (Fig. 4C), a significant decrease in PPR (from 1.5 ± 0.2 to 0.7 ± 0.1; n = 7; p = 0.008) (Fig. 4D), and a significant increase in CV−2 (from 1.02 ± 0.2 to 3.4 ± 0.94; n = 7; p = 0.01) (Fig. 4E), further suggesting a presynaptic site of action. In addition, the facilitating effect of the scavenger on GPSCs was fully occluded by subsequent application of UBP (the peak amplitude of GPSCs was 73 ± 10 and 70 ± 12 pA in the presence of the scavenger and the scavenger plus UBP 302, respectively; n = 7) (Fig. 4B). Application of the scavenger after UBP 302 was also ineffective (n = 5; p = 0.3) (data not shown). These results indicate that, early in postnatal life, depression of GABA release from MF terminals occurs via activation of GluK1 receptors by endogenous glutamate present in the extracellular space.
Figure 4.

Ambient glutamate activates presynaptic GluK1 kainate receptors on mossy fibers terminals. A, Averaged traces of GPSCs evoked by stimulation of granule cells in the dentate gyrus before and after addition of the glutamate scavenger (GPT plus pyruvate) and the scavenger plus UBP 302. The broad-spectrum mGluR1-8 antagonist LY341495 (100 μm) was present throughout the experiments. B, Pooled data from seven cells to show that the glutamate scavenger increased the mean peak amplitude of GPSCs and fully occluded the effects of UBP 302. Error bars indicate SEM. C–E, Successes (C), PPR (D), and inversed square of CV (E) measured in individual cells before and after application of the glutamate scavenger. **p < 0.01.
The depressant effect of kainate on MF GPSCs is mediated by G-coupled receptors
At inhibitory synapses, kainate-induced depression of GABA release has been shown to involve the presynaptic activation of a PTx-sensitive Gi/o-protein-coupled kainate receptor (Lerma, 2006; Rodríguez-Moreno and Sihra, 2007). G-protein in turn would activate phospholipase C (PLC) leading to the release of calcium from intracellular stores. The concomitant activation of protein kinase C (PKC) would inhibit voltage-dependent N-type calcium channels known to control transmitter release (Castillo et al., 1994; Dunlap et al., 1995; Li et al., 2007). To elucidate whether the effects of kainate on GPSCs involved a G-protein, hippocampal slices were incubated overnight with a solution containing PTx (5 μg/ml). In these conditions, bath application of the GABAB receptor agonist baclofen (from a holding potential of −60 mV) failed to evoked any outward current (data not shown) (Lauri et al., 2005). UBP 302 applied to nine neurons previously exposed to PTx either did not modify the amplitude of GPSCs (the peak amplitude of GPSCs was 52 ± 11 and 58 ± 13 pA in the absence or in the presence of UBP 302, respectively; n = 5; p = 0.1) or depressed it (from 68 ± 8 to 42 ± 3 pA; n = 4; p = 0.02). Pooled data from all cells tested are represented in Figure 5A. Similarly, the amplitude of GPSC was unaltered when the scavenger was applied to slices incubated in PTx (on average, in six cells, the peak amplitude of GPSCs was 61 ± 14 and 59 ± 16 pA in the absence and in the presence of the scavenger, respectively; p = 0.6) (Fig. 5B, closed circles). Incubation with PTx affects G-protein-coupled receptors present on both presynaptic and postsynaptic membranes. To see whether the depression of MF-GPSCs involved G-coupled receptors localized on the presynaptic site, additional experiments (n = 5) were performed using GDPβS (0.3 mm) into the patch pipette. This treatment completely blocked postsynaptic GABAB receptors. Thus, in the presence of GDPβS, the GABAB receptor agonist baclofen (20 μm) failed to induce outward currents (baclofen evoked-currents were 53 ± 5 and 5 ± 2 pA in the absence or in the presence of GDPβS, respectively; n = 6; p = 0.001). However, as shown in the example of Figure 5C, baclofen, via presynaptic GABAB receptors, depressed spontaneous ongoing synaptic currents. Bath application of UBP 302 to neurons recorded with GDPβS was still able to enhance GPSCs amplitude (the peak amplitude of GPSCs varied from 29 ± 4 to 43 ± 5 pA; n = 5; p = 0.003) (Fig. 5D,E), indicating that the depression of MF-GPSCs is mediated by G-protein-coupled kainate receptors, present on MF terminals.
Figure 5.

The depressant effect of kainate on GPSCs involves a G-protein-mediated mechanism. A, Summary plot showing the effects of UBP 302 on the peak amplitude of GPSCs in controls (open symbols) and after overnight treatment with PTx (closed symbols). The insets above the graph represent average traces obtained in the presence of GYKI 52466 and GYKI 52466 plus UBP 302. B, As in A, but in the presence of the glutamate scavenger showing that also the effect of the scavenger is dependent on G-protein. C, Application of baclofen (20 μm; bar) induced an outward currents associated with a reduction of spontaneous synaptic events (top trace). Application of baclofen to a cell recorded with a patch pipette containing GDPβS failed to produce an outward current but was still able to depress the ongoing synaptic activity (bottom trace). D, E, GDPβS into the intrapipette solution did not affect UBP 302-induced GPSCs facilitation. D, Representative traces. E, Summary plot from five cells. Error bars indicate SEM.
As already mentioned, the most common transduction signaling pathway stimulated by G-protein involves PLC activation (Rodríguez-Moreno and Lerma, 1998; Rozas et al., 2003). Therefore, in the following experiments, we tried to disrupt the intracellular cascade downstream to G-protein activation with U73122 (10 μm), a selective PLC blocker. Surprisingly, we found that, unlike PTx, application of UBP 302 in the presence of U73122 caused a significant depression of MF-GPSCs, probably unmasking the ionotropic action of GluK1. The amplitude of MF-GPSCs was 50 ± 9 and 29 ± 11 pA in the absence or in the presence of UBP 302, respectively (n = 6; p = 0.01) (Fig. 6A,B). In contrast to UBP 302, application of the GluK1 agonist ATPA (1 μm) in the presence of U73122 induced a significant increase in amplitude of GPSCs (from 40 ± 5 to 61 ± 3 pA; n = 7; p = 0.01) (Fig. 6C,D). Similarly to UBP 302, the enzymatic glutamate scavenger system applied in the presence of U73122 caused a depressant effect on GPSCs, which was probably dependent on the relief of a tonic action of glutamate on GluK1 receptors (from 70 ± 3 to 30 ± 2 pA; n = 11; p < 0.001) (Fig. 6E,F). The lack of UBP 302-induced depression of GPSCs amplitude observed in some neurons exposed to PTx could be explained by the fact that incubating the slices with PTx for several hours may interfere with other processes not directly linked to KAR activation.
Figure 6.

Blocking PLC with U73122 reveals the ionotropic action of GluK1 on MF-GPSCs. A, Averaged traces of MF-GPSCs evoked in a CA3 principal cell in the presence of U73122 (10 μm; Control) and U73122 plus UBP 302 (10 μm). B, Summary plot showing the mean amplitude of GPSCs obtained in six cells in the presence of U73122 before and during application of UBP 302 (bar). The amplitude of synaptic responses is normalized to those obtained before UBP 302 (dashed line). In the presence of UBP 302, the amplitude of the synaptic responses was significantly different from controls (p = 0.01). C–F, As in A and B, but in the presence of ATPA (1 μm) (C, D) and the glutamate scavenger (GPT plus pyruvate) (E, F). Note that both the ATPA-induced potentiation and the scavenger-induced depression of GPSCs amplitude were significantly different from controls (p = 0.01 and p < 0.001, respectively). Error bars indicate SEM.
Activation of presynaptic kainate receptors enhances MF excitability
In juvenile animals, activation of presynaptic kainate receptors has been found to directly depolarize via cation channels glutamatergic MF (Kamiya and Ozawa, 2000) or GABAergic terminals (Semyanov and Kullmann, 2001), thus lowering the threshold for antidromic action potential generation. To test whether activation of GluK1 receptors localized on immature MF terminals exhibits similar characteristics, we recorded antidromic spikes from single granule cells (held at −70 mV) in response to stimulation of MFs via a stimulation electrode positioned in stratum lucidum. When we used stimuli able to evoke antidromic spikes in >50% of trials, application of UBP 302 (10 μm) reversibly decreased the probability of successes from 0.6 ± 0.05 to 0.15 ± 0.02 (n = 7; p = 0.001) (Fig. 7A,B). In contrast, ATPA reversibly increased the probability of successes when antidromic spikes were evoked by weak stimuli (<50% of successes). The success rate was 0.38 ± 0.04 and 0.72 ± 0.04 in the absence or in the presence of ATPA, respectively (n = 7; p = 0.001) (Fig. 7C,D). Since GluK1 receptors are known to be highly permeable to calcium (Lauri et al., 2003; Scott et al., 2008), in another set of experiments we applied PhTx (3 μm), which blocks calcium-permeable AMPA/kainate receptor (Fletcher and Lodge, 1996). As UBP 302, PhTx significantly reduced the number of successes from 0.59 ± 0.04 to 0.15 ± 0.05 (n = 7; p = 0.001) (Fig. 7E,F). In the presence of PhTx, we increased the stimulation intensity to obtain again >50% of successes, and then we applied UBP 302. In this case, the drug failed to produce any effect. The success rate was 0.58 ± 0.09 and 0.61 ± 0.09 before and during UBP 302, respectively (n = 7; p = 0.08) (Fig. 7G,H). In line with an ionotropic type of action, exposing the slices to PTx did not alter UBP 302-induced reduction in the success rate of antidromic spikes (0.62 ± 0.08 and 0.16 ± 0.06; n = 5; p = 0.002) (data not shown). These data indicate GluK1 increases MF excitability through the activation of calcium-permeable cationic channels and depolarization of MF terminals.
Figure 7.
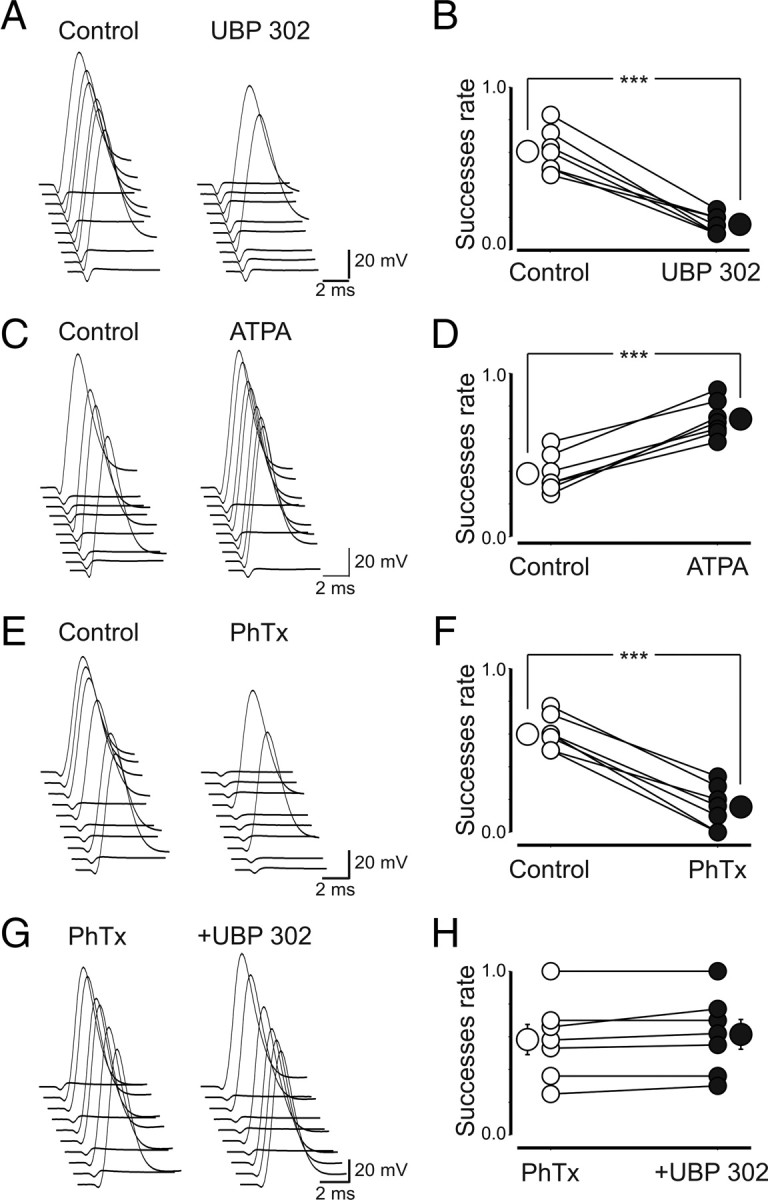
GluK1 receptors sensitive to philantotoxin control MF excitability. A, Consecutive traces showing antidromic spikes recorded in granule cells on stimulation of MF in stratum lucidum before and during application of UBP 302 (note that the stimulus strength was set to obtain >50% of successes). B, Summary plot of UBP 302 effects on success rate (n = 7). C, D, In cells with <50% of successes, ATPA enhanced MF excitability and the success rate (n = 7). E, F, PhTx (3 μm) mimicked the effects of UBP 302 (n = 7). G, H, In the presence of philantotoxin, UBP was not effective (n = 7). ***p < 0.001. Error bars indicate SEM.
GluK1Rs control the direction of spike time-dependent plasticity
In a previous study, we demonstrated that, depending on the relative timing, pairing afferent stimulation with postsynaptic spiking induced bidirectional changes in synaptic efficacy (Sivakumaran et al., 2009). In particular, a persistent increase in synaptic strength was observed when MF stimulation preceded postsynaptic spiking. To avoid possible contamination with glutamatergic events, experiments were routinely performed in the presence of d-AP5 and DNQX to block NMDA and AMPA/kainate receptors, respectively. Therefore, to see whether GluK1 receptors control spike time-dependent long-term potentiation (LTP), in the present experiments, the pairing procedure was repeated only in the presence of GYKI 52466, to prevent the activation of AMPA receptors. Pairing consisted in correlating (in current-clamp mode) MF stimulation with postsynaptic spikes (10 times at 0.1 Hz) (see Materials and Methods) using, as in previous work, a delay of 15 ms (corresponding to the peak of the synaptic responses) (Fig. 8A,B). As shown in the representative example of Figure 8C, pairing (arrow) induced a reduction in amplitude of GPSCs, which persisted for at least 20 min. On average, the peak amplitude of GPSCs (successes plus failures) was 42 ± 6 and 19 ± 2 pA before and 20 min after pairing, respectively (p = 0.003; n = 11) (Fig. 8C). This effect was associated with a significant decrease in success rate (from 0.46 ± 0.06 to 0.25 ± 0.06; p = 0.005), in the inversed square of the coefficient of variation of responses amplitude (from 1.67 ± 0.49 to 0.96 ± 0.29; p = 0.02), and a significant increase in PPR (from 0.86 ± 0.27 to 1.97 ± 0.47; n = 8; p = 0.01). To see whether spike time-dependent depression was mediated via an ionotropic or a metabotropic type of action, similar experiments were repeated in the presence of the selective PLC blocker U73122 (10 μm). In this condition, the pairing procedure failed to produce any effect (the peak amplitude of GPSCs was 60 ± 3 and 58 ± 2 pA before and after pairing, respectively; n = 7; p = 0.5) (Fig. 8D), suggesting the involvement of a G-protein-coupled KAR in STDP. When UBP 302 was added to GYKI 52466, the pairing procedure produced a persistent potentiation of synaptic responses that was in all respects similar to that described previously (Sivakumaran et al., 2009) (Fig. 8E). In summary, the amplitude of GPSCs varied from 84 ± 15 to 124 ± 18 pA (p = 0.001). This effect was associated with an increase in success rate (from 0.5 ± 0.05 to 0.8 ± 0.06; p = 0.001), in CV−2 (from 1.19 ± 0.2 to 4.1 ± 1; p = 0.01), and a decrease in PPR (from 1.5 ± 0.2 to 0.8 ± 0.2; n = 9; p = 0.004). In additional experiments, negative pairing (postsynaptic spiking preceding MF stimulation with a delay of 50 ms) induced (in the presence of GYKI 52466) long-term depression similar in all respect to that induced in the presence of DNQX (Sivakumaran et al., 2009), suggesting that KARs are not involved in this form of synaptic plasticity (the peak amplitude of GPSCs was 116 ± 22 and 53 ± 10 pA before and 20 min after pairing, respectively; n = 7; p = 0.004) (data not shown). These results indicate that presynaptic GluK1 control STD-LTP at immature MF–CA3 synapses.
Figure 8.

At immature MF–CA3 synapses, presynaptic kainate receptors control the direction of STDP. A, Schematic representation of the experimental design. B, The stimulation of granule cells in the dentate gyrus (pre) preceded the postsynaptic spike (post) by 15 ms (Δt). C, Summary plot of the mean peak amplitude of GPSCs recorded in the presence of GYKI 52466 before and after pairing (arrow at time 0; n = 11). The dashed line represents the mean amplitude of GPSCs before pairing. The insets represent averaged GPSCs obtained from a single neuron before and after pairing. Note that pairing induced synaptic depression. D, Summary plot of MF-GPSCs amplitude obtained in the presence U73122 versus time (n = 7). Note that blocking PLC with U73122 failed to produce any effect on GPSCs amplitude. E, As in C, but in the presence of UBP 302 (n = 9). In this case, pairing induced synaptic potentiation. Error bars indicate SEM.
Discussion
The present experiments from the immature hippocampus have revealed the presence of functional presynaptic kainate receptors on GABAergic MF terminals. Endogenous activation of these receptors by glutamate present in the extracellular medium reduces the probability of GABA release contributing in some cases to synapses silencing. Moreover, GluK1 receptors present on MF terminals dynamically regulate the direction of STDP being able to switch spike time-dependent potentiation into depression.
In previous studies, we provided evidence that, during the first week of postnatal life, the main neurotransmitter released from MF terminals is GABA (Kasyanov et al., 2004; Safiulina et al., 2006; Safiulina and Cherubini, 2009; Sivakumaran et al., 2009). MF-mediated glutamatergic responses start appearing during the second week of postnatal life (Amaral and Dent, 1981). In particular, kainate-mediated EPSCs have been detected only after P6, in coincidence with the appearance of large-amplitude AMPA responses and with the onset of low-frequency facilitation (Marchal and Mulle, 2004). This period tightly correlates with that in which GABAA-mediated synaptic responses shift from the depolarizing to the hyperpolarizing direction (Cherubini et al., 1991; Ben-Ari et al., 2007). In the present experiments, GPSCs originated from MF since they exhibited strong paired-pulse facilitation and were sensitive to group III mGluR agonist l-AP4 (Gutiérrez, 2005; Safiulina et al., 2006). In this respect, they could be easily distinguished from those originating from GABAergic interneurons, which were insensitive to l-AP4 and exhibited paired-pulse depression (Walker et al., 2001; Safiulina et al., 2006). MF-mediated synaptic responses did not carry AMPA-mediated components since GYKI 52466, at the concentration used to block AMPA receptors, failed to modify the amplitude, rise time, or latency of synaptic currents. However, in the presence of GYKI 52466, synaptic currents were reversibly enhanced by DNQX or UBP 302, indicating that presynaptic KARs downregulate GABA release from MF terminals. In this respect, our results are similar to those obtained by Maingret et al. (2005) on GABAA-mediated postsynaptic currents evoked in neonatal CA1 pyramidal neurons by electrical stimulation of GABAergic axons. In the immature hippocampus, presynaptic GluK1 receptors have been well documented (Bahn et al., 1994; Bettler et al., 1990; Ritter et al., 2002), and a recent study has provided evidence that at CA3–CA1 synapses functional GluK1 receptors downregulate glutamate release (Lauri et al., 2006). Moreover, in the CA3 hippocampal region, activation of GluK1 receptors reversibly blocks spontaneous network-driven bursts such as GDPs (Lauri et al., 2005). In our case, the depression of MF-GPSCs by KARs was not mediated indirectly via other signaling molecules known to inhibit GABA release, since DNQX was still able to enhance the amplitude of GPSCs when applied in the presence of various receptor antagonists including those for GABAB, nicotinic, muscarinic, P2Y, and mGlu.
Several lines of evidence suggest that GluK1 receptors localized on MF terminals reduce the probability of GABA release: (1) the increase in amplitude of MF-GPSCs by DNQX or UBP 302 was associated with an enhanced success rate and a reduction in paired-pulse ratio, considered an index of presynaptic release probability (Zucker and Regehr, 2002). (2) The GluK1 agonist ATPA induced a powerful depression of MF-evoked synaptic responses, an effect associated with a significant increase in transmitter failures and in the paired-pulse ratio. (3) Decreasing the concentration of “ambient” glutamate with the scavenger prevented the activation of GluK1 receptors and mimicked the effects of KAR antagonists. Previous studies from different brain structures have demonstrated that physiological modifications in glutamate concentration may cause a switch in KAR function from facilitation to inhibition (Jiang et al., 2001; Delaney and Jahr, 2002; Braga et al., 2003; Youn and Randic, 2004; Lerma, 2006). It is therefore likely that, early in postnatal development, a high level of glutamate in the extracellular space, maintained by a less efficient glutamate transport mechanism (Danbolt, 2001; Diamond, 2005) and a poorly developed diffusional barrier (Jansson et al., 2000), facilitates the activation of high-affinity KARs with consequent reduction in the probability of transmitter release.
KARs have been shown to exert both an ionotropic and a metabotropic type of action (for review, see Lerma, 2006). In particular, the depression of transmitter release seems to occur via G-protein-coupled KARs (Rodríguez-Moreno and Lerma, 1998; Cunha et al., 2000; Frerking et al., 2001; Lauri et al., 2005, 2006). Also in our case, the depressant effect of kainate on GABA release was likely dependent on a metabotropic type of mechanism since the potentiating effects of both UBP 302 and the glutamate scavenger on GPSCs were prevented by PTx. The signaling pathway likely involved the release of calcium from intracellular stores, the activation of phospholipase C and PKC with consequent inhibition of voltage-dependent calcium channels (Rodríguez-Moreno and Lerma, 1998; Rozas et al., 2003). Interestingly, blocking PLC with U73122, downstream to G-protein activation, unveiled a potentiating effect of GluK1 on MF GPSCs. This was probably dependent on the ionotropic type of action of this receptor and was similar to that observed in some cells exposed to PTx. This type of action was clearly responsible for GluK1-induced increase in MF excitability, in which calcium flux through calcium-permeable KARs depolarized presynaptic terminals and lowered the threshold for antidromic spikes (Kamiya and Ozawa, 2000; Schmitz et al., 2001; Semyanov and Kullmann, 2001; Maingret et al., 2005). How can an increased MF excitability be reconciled with a depression of GABA release? Although, according to Kamiya and Ozawa (2000), a downregulation of transmitter release may occur via inactivation of Na+/Ca2+ channels or electrical shunting, this seems unlikely in view of the recent finding of KA-induced facilitation of action potential evoked calcium entry in MF boutons via a calcium store-dependent mechanism (Scott et al., 2008). In addition, it is worth noting that, unlike adults, immature MFs terminate in very small spherical expansions (Amaral and Dent, 1981) and do not exhibit use-dependent synaptic facilitation yet (Marchal and Mulle, 2004). It is unclear whether the dual signaling pathways (ionotropic and metabotropic), which depend on the common ionotropic GluK1 subunit, are independent or functionally coupled. In a previous study from dorsal root ganglion cells, it has been demonstrated that KA through GluK1 receptors induces a G-protein-dependent rise in [Ca2+]i, favoring its release from the internal stores (Rozas et al., 2003). As a matter of speculation, we favor the hypothesis that calcium entering through calcium-permeable KARs may directly or indirectly interfere with G-protein-mediated signaling leading to a dominant inhibitory action on MF-GPSCs. The interplay between these two different pathways has been recently shown to account for the PKC-dependent autoregulation of membrane KARs (Rivera et al., 2007). However, much work is needed to elucidate this issue.
Early in postnatal development, G-protein-dependent mechanisms linked to KA activation have been well established particularly at glutamatergic synapses (Lauri et al., 2005, 2006). These receptors have a very high affinity for glutamate, a condition required for being endogenously activated by glutamate present in the extracellular medium. With maturation, presynaptic KARs in an activity-dependent manner would be gradually lost and replaced by low-affinity ones, not longer able to be activated by ambient glutamate (Lauri et al., 2006). In the present experiments, tonic activation of presynaptic KARs by endogenous glutamate accounted for the persistent depression of MF-GPSCs observed after pairing presynaptic MF stimulation with postsynaptic spiking as demonstrated by the possibility to switch spike time-dependent depression into potentiation with UBP 302. Spike time-dependent depression, observed in the absence of AMPA/kainate receptor antagonists, involved the activation of a G-protein-coupled KAR since it was prevented by the selective PLC blocker U73122. In contrast, spike time-dependent potentiation of MF-GPSCs, observed in the presence of UBP 302, was shown to be dependent on intracellular calcium rise and release of BDNF (Sivakumaran et al., 2009). Although the precise mechanisms underlying these phenomena are still unclear, we cannot exclude the possibility that KA-induced synaptic depression may rely on a distinct calcium signal, which in turn may activate a different molecular pathway, as suggested by the calcium hypothesis (Caporale and Dan, 2008).
In conclusion, it is conceivable that, at immature MF–CA3 synapses, KA-induced depression of GABAergic transmission by ambient glutamate limits the excessive activation of the auto-associative CA3 network by the excitatory action of GABA, thus preventing the onset of seizures. These properties are likely to be critical for information processing and for the proper development of the adult hippocampal circuitry.
Footnotes
This work was supported by a grant from Ministero Istruzione Universitá e Ricerca.
References
- Allen C, Stevens CF. An evaluation of causes for unreliability of synaptic transmission. Proc Natl Acad Sci U S A. 1994;91:10380–10383. doi: 10.1073/pnas.91.22.10380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amaral DG, Dent JA. Development of the mossy fibers of the dentate gyrus: I. A light and electron microscopic study of the mossy fibers and their expansions. J Comp Neurol. 1981;195:51–86. doi: 10.1002/cne.901950106. [DOI] [PubMed] [Google Scholar]
- Bahn S, Volk B, Wisden W. Kainate receptor gene expression in the developing rat brain. J Neurosci. 1994;14:5525–5547. doi: 10.1523/JNEUROSCI.14-09-05525.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Cossart R. Kainate, a double agent that generates seizures: two decades of progress. Trends Neurosci. 2000;23:580–587. doi: 10.1016/s0166-2236(00)01659-3. [DOI] [PubMed] [Google Scholar]
- Ben-Ari Y, Gaiarsa JL, Tyzio R, Khazipov R. GABA: a pioneer transmitter that excites immature neurons and generates primitive oscillations. Physiol Rev. 2007;87:1215–1284. doi: 10.1152/physrev.00017.2006. [DOI] [PubMed] [Google Scholar]
- Bettler B, Boulter J, Hermans-Borgmeyer I, O'Shea-Greenfield A, Deneris ES, Moll C, Borgmeyer U, Hollmann M, Heinemann S. Cloning of a novel glutamate receptor subunit, GluR5: expression in the nervous system during development. Neuron. 1990;5:583–595. doi: 10.1016/0896-6273(90)90213-y. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Xie J, Li H. Bidirectional modulation of GABA release by presynaptic glutamate receptor 5 kainate receptors in the basolateral amygdala. J Neurosci. 2003;23:442–452. doi: 10.1523/JNEUROSCI.23-02-00442.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporale N, Dan Y. Spike timing-dependent plasticity: a hebbian learning rule. Annu Rev Neurosci. 2008;31:25–46. doi: 10.1146/annurev.neuro.31.060407.125639. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Weisskopf MG, Nicoll RA. The role of Ca2+ channels in hippocampal mossy fiber synaptic transmission and long-term potentiation. Neuron. 1994;12:261–269. doi: 10.1016/0896-6273(94)90269-0. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Cherubini E, Gaiarsa JL, Ben-Ari Y. GABA: an excitatory transmitter in early postnatal life. Trends Neurosci. 1991;14:515–519. doi: 10.1016/0166-2236(91)90003-d. [DOI] [PubMed] [Google Scholar]
- Clarke VR, Ballyk BA, Hoo KH, Mandelzys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Olsen RW, Peters J, Spedding M. A nomenclature for ligand-gated ion channels. Neuropharmacology. 2009;56:2–5. doi: 10.1016/j.neuropharm.2008.06.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Contractor A, Swanson G, Heinemann SF. Kainate receptors are involved in short- and long-term plasticity at mossy fiber synapses in the hippocampus. Neuron. 2001;29:209–216. doi: 10.1016/s0896-6273(01)00191-x. [DOI] [PubMed] [Google Scholar]
- Cossart R, Tyzio R, Dinocourt C, Esclapez M, Hirsch JC, Ben-Ari Y, Bernard C. Presynaptic kainate receptors that enhance the release of GABA on CA1 hippocampal interneurons. Neuron. 2001;29:497–508. doi: 10.1016/s0896-6273(01)00221-5. [DOI] [PubMed] [Google Scholar]
- Cossart R, Epsztein J, Tyzio R, Becq H, Hirsch J, Ben-Ari Y, Crépel V. Quantal release of glutamate generates pure kainate and mixed AMPA/kainate EPSCs in hippocampal neurons. Neuron. 2002;35:147–159. doi: 10.1016/s0896-6273(02)00753-5. [DOI] [PubMed] [Google Scholar]
- Cunha RA, Malva JO, Ribeiro JA. Pertussis toxin prevents presynaptic inhibition by kainate receptors of rat hippocampal [3H]GABA release. FEBS Lett. 2000;469:159–162. doi: 10.1016/s0014-5793(00)01272-2. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Delaney AJ, Jahr CE. Kainate receptors differentially regulate release at two parallel fiber synapses. Neuron. 2002;36:475–482. doi: 10.1016/s0896-6273(02)01008-5. [DOI] [PubMed] [Google Scholar]
- Diamond JS. Deriving the glutamate clearance time course from transporter currents in CA1 hippocampal astrocytes: transmitter uptake gets faster during development. J Neurosci. 2005;25:2906–2916. doi: 10.1523/JNEUROSCI.5125-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K, Luebke JI, Turner TJ. Exocytotic Ca2+ channels in mammalian central neurons. Trends Neurosci. 1995;18:89–98. [PubMed] [Google Scholar]
- Fitzjohn SM, Bortolotto ZA, Palmer MJ, Doherty AJ, Ornstein PL, Schoepp DD, Kingston AE, Lodge D, Collingridge GL. The potent mGlu receptor antagonist LY341495 identifies roles for both cloned and novel mGlu receptors in hippocampal synaptic plasticity. Neuropharmacology. 1998;37:1445–1458. doi: 10.1016/s0028-3908(98)00145-2. [DOI] [PubMed] [Google Scholar]
- Fletcher EJ, Lodge D. New developments in the molecular pharmacology of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate and kainate receptors. Pharmacol Ther. 1996;70:65–89. doi: 10.1016/0163-7258(96)00014-9. [DOI] [PubMed] [Google Scholar]
- Frerking M, Schmitz D, Zhou Q, Johansen J, Nicoll RA. Kainate receptors depress excitatory synaptic transmission at CA3→CA1 synapses in the hippocampus via a direct presynaptic action. J Neurosci. 2001;21:2958–2966. doi: 10.1523/JNEUROSCI.21-09-02958.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparini S, Saviane C, Voronin LL, Cherubini E. Silent synapses in the developing hippocampus: lack of functional AMPA receptors or low probability of glutamate release? Proc Natl Acad Sci U S A. 2000;97:9741–9746. doi: 10.1073/pnas.170032297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutiérrez R. The dual glutamatergic-GABAergic phenotype of hippocampal granule cells. Trends Neurosci. 2005;28:297–303. doi: 10.1016/j.tins.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Gutiérrez R, Romo-Parra H, Maqueda J, Vivar C, Ramìrez M, Morales MA, Lamas M. Plasticity of the GABAergic phenotype of the “glutamatergic” granule cells of the rat dentate gyrus. J Neurosci. 2003;23:5594–5598. doi: 10.1523/JNEUROSCI.23-13-05594.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jane DE, Lodge D, Collingridge GL. Kainate receptors: pharmacology, function and therapeutic potential. Neuropharmacology. 2009;56:90–113. doi: 10.1016/j.neuropharm.2008.08.023. [DOI] [PubMed] [Google Scholar]
- Jansson A, Lippoldt A, Mazel T, Bartfai T, Ogren SO, Syková E, Agnati LF, Fuxe K. Long distance signalling in volume transmission. Focus on clearance mechanisms. Prog Brain Res. 2000;125:399–413. doi: 10.1016/S0079-6123(00)25028-0. [DOI] [PubMed] [Google Scholar]
- Jiang L, Xu J, Nedergaard M, Kang J. A kainate receptor increases the efficacy of GABAergic synapses. Neuron. 2001;30:503–513. doi: 10.1016/s0896-6273(01)00298-7. [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fibre synapse on CA3 pyramidal cells of rat hippocampus. J Physiol. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S. Kainate receptor-mediated presynaptic inhibition at the mouse hippocampal mossy fibre synapse. J Physiol. 2000;523:653–665. doi: 10.1111/j.1469-7793.2000.t01-1-00653.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya H, Ozawa S, Manabe T. Kainate receptor-dependent short-term plasticity of presynaptic Ca2+ influx at the hippocampal mossy fiber synapses. J Neurosci. 2002;22:9237–9243. doi: 10.1523/JNEUROSCI.22-21-09237.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasyanov AM, Safiulina VF, Voronin LL, Cherubini E. GABA-mediated giant depolarizing potentials as coincidence detectors for enhancing synaptic efficacy in the developing hippocampus. Proc Natl Acad Sci U S A. 2004;101:3967–3972. doi: 10.1073/pnas.0305974101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon HB, Castillo PE. Role of glutamate autoreceptors at hippocampal mossy fiber synapses. Neuron. 2008;60:1082–1094. doi: 10.1016/j.neuron.2008.10.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Bortolotto ZA, Bleakman D, Ornstein PL, Lodge D, Isaac JT, Collingridge GL. A critical role of a facilitatory presynaptic kainate receptor in mossy fiber LTP. Neuron. 2001a;32:697–709. doi: 10.1016/s0896-6273(01)00511-6. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Delany C, Clarke VRJ, Bortolotto ZA, Ornstein PL, Isaac JTR, Collingridge GL. Synaptic activation of a presynaptic kainate receptor facilitates AMPA receptor-mediated synaptic transmission at hippocampal mossy fibre synapses. Neuropharmacology. 2001b;41:907–915. doi: 10.1016/s0028-3908(01)00152-6. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Bortolotto ZA, Nistico R, Bleakman D, Ornstein PL, Lodge D, Isaac JT, Collingridge GL. A role for Ca2+ stores in kainate receptor-dependent synaptic facilitation and LTP at mossy fiber synapses in the hippocampus. Neuron. 2003;39:327–341. doi: 10.1016/s0896-6273(03)00369-6. [DOI] [PubMed] [Google Scholar]
- Lauri SE, Segerstråle M, Vesikansa A, Maingret F, Mulle C, Collingridge GL, Isaac JT, Taira T. Endogenous activation of kainate receptors regulates glutamate release and network activity in the developing hippocampus. J Neurosci. 2005;25:4473–4484. doi: 10.1523/JNEUROSCI.4050-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauri SE, Vesikansa A, Segerstråle M, Collingridge GL, Isaac JT, Taira T. Functional maturation of CA1 synapses involves activity-dependent loss of tonic kainate receptor-mediated inhibition of glutamate release. Neuron. 2006;50:415–429. doi: 10.1016/j.neuron.2006.03.020. [DOI] [PubMed] [Google Scholar]
- Lerma J. Roles and rules of kainate receptors in synaptic transmission. Nat Rev Neurosci. 2003;4:481–495. doi: 10.1038/nrn1118. [DOI] [PubMed] [Google Scholar]
- Lerma J. Kainate receptor physiology. Curr Opin Pharmacol. 2006;6:89–97. doi: 10.1016/j.coph.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Li L, Bischofberger J, Jonas P. Differential gating and recruitment of P/Q-, N-, and R-type Ca2+ channels in hippocampal mossy fiber boutons. J Neurosci. 2007;7:13420–13429. doi: 10.1523/JNEUROSCI.1709-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Sola E, Minneci F, Le Magueresse C, Changeux JP, Cherubini E. Persistent decrease in synaptic efficacy induced by nicotine at Schaffer collateral-CA1 synapses in the immature rat hippocampus. J Physiol. 2004;559:863–874. doi: 10.1113/jphysiol.2004.067041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maingret F, Lauri SE, Taira T, Isaac JT. Profound regulation of neonatal CA1 rat hippocampal GABAergic transmission by functionally distinct kainate receptor populations. J Physiol. 2005;567:131–142. doi: 10.1113/jphysiol.2005.089474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchal C, Mulle C. Postnatal maturation of mossy fibre excitatory transmission in mouse CA3 pyramidal cells: a potential role for kainate receptors. J Physiol. 2004;561:27–37. doi: 10.1113/jphysiol.2004.069922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melyan Z, Lancaster B, Wheal HV. Metabotropic regulation of intrinsic excitability by synaptic activation of kainate receptors. J Neurosci. 2004;24:4530–4534. doi: 10.1523/JNEUROSCI.5356-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min MY, Rusakov DA, Kullmann DM. Activation of AMPA, kainate, and metabotropic receptors at hippocampal mossy fiber synapses: role of glutamate diffusion. Neuron. 1998;21:561–570. doi: 10.1016/s0896-6273(00)80566-8. [DOI] [PubMed] [Google Scholar]
- More JC, Nistico R, Dolman NP, Clarke VR, Alt AJ, Ogden AM, Buelens FP, Troop HM, Kelland EE, Pilato F, Bleakman D, Bortolotto ZA, Collingridge GL, Jane DE. Characterisation of UBP296: a novel, potent and selective kainate receptor antagonist. Neuropharmacology. 2004;47:46–64. doi: 10.1016/j.neuropharm.2004.03.005. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Pérez-Otaño I, Dickinson-Anson H, Castillo PE, Bureau I, Maron C, Gage FH, Mann JR, Bettler B, Heinemann SF. Altered synaptic physiology and reduced susceptibility to kainate-induced seizures in GluR6-deficient mice. Nature. 1998;392:601–615. doi: 10.1038/33408. [DOI] [PubMed] [Google Scholar]
- Mulle C, Sailer A, Swanson GT, Brana C, O'Gorman S, Bettler B, Heinemann SF. Subunit composition of kainate receptors in hippocampal interneurons. Neuron. 2000;28:475–484. doi: 10.1016/s0896-6273(00)00126-4. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Schmitz D. Synaptic plasticity at hippocampal mossy fibre synapses. Nat Rev Neurosci. 2005;6:863–876. doi: 10.1038/nrn1786. [DOI] [PubMed] [Google Scholar]
- Overstreet LS, Pasternak JF, Colley PA, Slater NT, Trommer BL. Metabotropic glutamate receptor mediated long-term depression in developing hippocampus. Neuropharmacology. 1997;36:831–844. doi: 10.1016/s0028-3908(97)00031-2. [DOI] [PubMed] [Google Scholar]
- Represa A, Tremblay E, Ben-Ari Y. Kainate binding sites in the hippocampal mossy fibers: localization and plasticity. Neuroscience. 1987;20:739–748. doi: 10.1016/0306-4522(87)90237-5. [DOI] [PubMed] [Google Scholar]
- Ritter LM, Vazquez DM, Meador-Woodruff JH. Ontogeny of ionotropic glutamate receptor subunit expression in the rat hippocampus. Brain Res Dev Brain Res. 2002;139:227–236. doi: 10.1016/s0165-3806(02)00572-2. [DOI] [PubMed] [Google Scholar]
- Rivera R, Rozas JL, Lerma J. PKC-dependent autoregulation of membrane kainate receptors. EMBO J. 2007;26:4359–4367. doi: 10.1038/sj.emboj.7601865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Lerma J. Kainate receptor modulation of GABA release involves a metabotropic function. Neuron. 1998;20:1211–1218. doi: 10.1016/s0896-6273(00)80501-2. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Sihra TS. Kainate receptors with a metabotropic modus operandi. Trends Neurosci. 2007;30:630–637. doi: 10.1016/j.tins.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Rodríguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- Rozas JL, Paternain AV, Lerma J. Noncanonical signaling by ionotropic kainate receptors. Neuron. 2003;39:543–553. doi: 10.1016/s0896-6273(03)00436-7. [DOI] [PubMed] [Google Scholar]
- Safiulina VF, Cherubini E. At immature mossy fibers-CA3 connections, activation of presynaptic GABAB receptors by endogenously released GABA contributes to synapses silencing. Front Cell Neurosci. 2009;3:1. doi: 10.3389/neuro.03.001.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina VF, Kasyanov AM, Sokolova E, Cherubini E, Giniatullin R. ATP contributes to the generation of network-driven giant depolarizing potentials in the neonatal rat hippocampus. J Physiol. 2005;565:981–992. doi: 10.1113/jphysiol.2005.085621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safiulina VF, Fattorini G, Conti F, Cherubini E. GABAergic signaling at mossy fiber synapses in neonatal rat hippocampus. J Neurosci. 2006;26:597–608. doi: 10.1523/JNEUROSCI.4493-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Salin PA, Vogt KE, Malenka RC, Nicoll RA. Use-dependent increases in glutamate concentration activate presynaptic metabotropic glutamate receptors. Nature. 1997;385:630–634. doi: 10.1038/385630a0. [DOI] [PubMed] [Google Scholar]
- Schmitz D, Mellor J, Frerking M, Nicoll RA. Presynaptic kainate receptors at hippocampal mossy fiber synapses. Proc Natl Acad Sci U S A. 2001;98:11003–11008. doi: 10.1073/pnas.191351498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott R, Lalic T, Kullmann DM, Capogna M, Rusakov DA. Target-cell specificity of kainate autoreceptor and Ca2+-store-dependent short-term plasticity at hippocampal mossy fiber synapses. J Neurosci. 2008;28:13139–13149. doi: 10.1523/JNEUROSCI.2932-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semyanov A, Kullmann DM. Kainate receptor-dependent axonal depolarization and action potential initiation in interneurons. Nat Neurosci. 2001;4:718–723. doi: 10.1038/89506. [DOI] [PubMed] [Google Scholar]
- Sivakumaran S, Mohajerani MH, Cherubini E. At immature mossy-fiber–CA3 synapses, correlated presynaptic and postsynaptic activity persistently enhances GABA release and network excitability via BDNF and cAMP-dependent PKA. J Neurosci. 2009;29:2637–2647. doi: 10.1523/JNEUROSCI.5019-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesikansa A, Sallert M, Taira T, Lauri SE. Activation of kainate receptors controls the number of functional glutamatergic synapses in the area CA1 of rat hippocampus. J Physiol. 2007;583:145–157. doi: 10.1113/jphysiol.2007.133975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Walker MC, Ruiz A, Kullmann DM. Monosynaptic GABAergic signaling from dentate to CA3 with a pharmacological and physiological profile typical of mossy fiber synapses. Neuron. 2001;29:703–715. doi: 10.1016/s0896-6273(01)00245-8. [DOI] [PubMed] [Google Scholar]
- Youn DH, Randic M. Modulation of excitatory synaptic transmission in the spinal substantia gelatinosa of mice deficient in the kainate receptor GluR5 and/or GluR6 subunit. J Physiol. 2004;555:683–698. doi: 10.1113/jphysiol.2003.057570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang JM, Wang HK, Ye CQ, Ge W, Chen Y, Jiang ZL, Wu CP, Poo MM, Duan S. ATP released by astrocytes mediates glutamatergic activity-dependent heterosynaptic suppression. Neuron. 2003;40:971–982. doi: 10.1016/s0896-6273(03)00717-7. [DOI] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]