

The discovery of antibiotics revolutionized modern medicine and enabled us to cure previously deadly bacterial infections. However, a progressive increase in antibiotic resistance rates is a major and global threat for our health care system. Colistin represents one of our last-resort antibiotics that is still active against most Gram-negative bacterial pathogens, but increasing resistance is reported worldwide, in particular due to the plasmid-encoded protein MCR-1 present in pathogens such as Escherichia coli and Klebsiella pneumoniae. Here, we showed that colistin resistance in A. baumannii, a top-priority pathogen causing deadly nosocomial infections, is mediated through different avenues that result in increased activity of homologous phosphoethanolamine (PetN) transferases. Considering that MCR-1 is also a PetN transferase, our findings indicate that PetN transferases might be the Achilles heel of superbugs and that direct targeting of them may have the potential to preserve the activity of polymyxin antibiotics.
KEYWORDS: Acinetobacter baumannii, antibiotic resistance, colistin, eptA, ethanolamine transferase, mcr-1, pmrA
ABSTRACT
Nosocomial infections with Acinetobacter baumannii are a global problem in intensive care units with high mortality rates. Increasing resistance to first- and second-line antibiotics has forced the use of colistin as last-resort treatment, and increasing development of colistin resistance in A. baumannii has been reported. We evaluated the transcriptional regulator PmrA as potential drug target to restore colistin efficacy in A. baumannii. Deletion of pmrA restored colistin susceptibility in 10 of the 12 extensively drug-resistant A. baumannii clinical isolates studied, indicating the importance of PmrA in the drug resistance phenotype. However, two strains remained highly resistant, indicating that PmrA-mediated overexpression of the phosphoethanolamine (PetN) transferase PmrC is not the exclusive colistin resistance mechanism in A. baumannii. A detailed genetic characterization revealed a new colistin resistance mechanism mediated by genetic integration of the insertion element ISAbaI upstream of the PmrC homolog EptA (93% identity), leading to its overexpression. We found that eptA was ubiquitously present in clinical strains belonging to the international clone 2, and ISAbaI integration upstream of eptA was required to mediate the colistin-resistant phenotype. In addition, we found a duplicated ISAbaI-eptA cassette in one isolate, indicating that this colistin resistance determinant may be embedded in a mobile genetic element. Our data disprove PmrA as a drug target for adjuvant therapy but highlight the importance of PetN transferase-mediated colistin resistance in clinical strains. We suggest that direct targeting of the homologous PetN transferases PmrC/EptA may have the potential to overcome colistin resistance in A. baumannii.
INTRODUCTION
Antimicrobial resistance is a serious threat to global health systems, resulting in the loss of treatment options to fight a growing number of bacterial infections (1). Considering the paucity of newly developed antibiotics in the last decades, old antibiotics such as polymyxins have been increasingly used to treat infections caused by multidrug-resistant (MDR) Gram-negative pathogens (2–4). Nowadays, the polymyxin antibiotics polymyxin E (colistin) and polymyxin B represent the last resort for the treatment of serious Gram-negative infections, such as infections caused by carbapenem-resistant Enterobacteriaceae, MDR Pseudomonas aeruginosa, and MDR Acinetobacter baumannii (5, 6). Unfortunately, the increasing use of polymyxins to treat serious infections caused by these pathogens leads to a spread of resistance to these last-line drugs (7). There is a high unmet medical need for new drugs effective against Gram-negative bacteria to treat infections caused by these pathogens (8). Besides this, an alternative strategy resides in the recovery of colistin efficacy by blocking bacterial colistin resistance mechanisms. Antibiotic adjuvant therapies consist in the combination of a potent antibiotic with a nonantibiotic agent interfering with specific antibiotic resistance or virulence mechanisms. This strategy may provide a new tool to fight infections caused by drug-resistant pathogens by restoring or boosting the efficacy of an approved antibiotic (9).
Colistin resistance is conferred by lipopolysaccharide (LPS) modifications at the outer cell envelope. Reduction of the negative charge on LPS results in a reduced affinity of colistin to LPS (10). The two main LPS modifications conferring colistin resistance are the addition of 4-amino-4-deoxy-l-arabinose (AraN) and phosphoethanolamine (PetN) to the lipid A (11). The expression of LPS-modifying enzymes is regulated by the concerted action of several two-component systems (TCSs). In Enterobacteriaceae, PhoPQ and PmrAB TCSs regulate the expression of colistin resistance mechanisms, whereas in P. aeruginosa the PhoPQ, PmrAB, ParRS, ColRS, and CprRS TCSs seem to be involved (11). Plasmid-mediated colistin resistance has been recently reported in Enterobacteriaceae due to the PetN transferase MCR-1. The presence of MCR-1 on a plasmid leads to its rapid geographical and interspecies spread (12, 13). Nevertheless, mcr-1 seems to be restricted to Enterobacteriaceae species and has never been detected in A. baumannii. In A. baumannii, colistin resistance is mediated by PetN addition to the lipid A, and this resistance mechanism is regulated by the PmrAB TCS. In contrast to other pathogens, the AraN lipid A modification pathway is not present in A. baumannii (11), rendering A. baumannii a suitable pathogen to develop an adjuvant therapy approach to rejuvenate colistin efficacy by blocking the PmrAB TCS.
Colistin resistance in A. baumannii clinical isolates is associated with alterations in the pmrCAB operon. The pmrC gene codes for a PetN transferase, and pmrA and pmrB code for the TCS (14). It has been shown that mutations in the PmrAB TCS induce the overexpression of pmrC, leading to the modification of lipid A with PetN and colistin resistance (14–18). Because PmrA is the transcriptional regulator that triggers PmrC overexpression, inhibition of PmrA with a small molecule may potentially block PmrC overexpression and therefore switch off colistin resistance in A. baumannii (19). This study was designed to evaluate the clinical relevance of PmrA as a drug target to restore colistin efficacy in A. baumannii. We demonstrate that in the absence of PmrA-mediated expression of PmrC, transposition of an insertion sequence (IS) element leads to overexpression of the alternative highly similar PetN transferase EptA, which also confers colistin resistance in A. baumannii clinical isolates. Our results show that in all studied clinical isolates, overexpression of at least one PetN transferase (PmrC or various EptA variants) was responsible for colistin resistance, indicating that PetN transferases may be a suitable drug target to overcome colistin resistance in A. baumannii.
RESULTS
PmrA is not essential for colistin resistance in A. baumannii clinical isolates.
We deleted pmrA from the genome of a panel of 12 colistin-resistant A. baumannii strains to evaluate the transcriptional regulator PmrA as a potential drug target to rejuvenate colistin efficacy in A. baumannii. The strains in the panel consisted of recently isolated colistin-resistant clinical strains collected from diverse geographical origins. They belong to three distinct and highly successful clonal lineages, the international clone 1 (ST1), international clone 2 (ST2), and ST25 clonal lineages (Table 1) (20–22). All strains were classified as extensively drug resistant according to the criteria of Magiorakos et al. (23). These data underscore the clinical relevance and the diversity of the strain panel. The colistin-susceptible A. baumannii ATCC 17978 strain was included as a reference strain. In all strains, pmrA was deleted by applying a previously described method that allows efficient scarless genome engineering even in extensively resistant A. baumannii clinical isolates (24).
TABLE 1.
Characterization of the A. baumannii clinical isolate panel used in this studya
| Strain designation | Strain isolation |
MLST | MIC (μg/ml) of drug: |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Country | Yr | GENT | MERO | CIP | TZP | CTX | SXT | SAM | TET | ||
| ATCC 17978 | France | 1951 | 77 | 2 | 0.5 | 1 | 8/4 | 16 | >8/152 | 4/2 | 2 |
| BV94 | USA | 2011 | 2 | >128 | 32 | 256 | >256/4 | >256 | >8/152 | 16/8 | 32 |
| BV95 | Colombia | 2010 | 25 | 1 | 64 | 128 | 256/4 | 32 | >8/152 | 16/8 | >256 |
| BV172 | Israel | 2012 | 2 | >128 | 64 | 32 | 256/4 | >256 | >8/152 | 64/32 | >256 |
| BV173 | Greece | 2012 | 2 | >128 | >64 | 128 | >256/4 | >256 | >8/152 | 128/64 | >256 |
| BV174 | USA | 2012 | 2 | 8 | 64 | 256 | 256/4 | 256 | >8/152 | 32/16 | 32 |
| BV175 | Turkey | 2012 | 2 | 128 | 32 | 256 | >256/4 | 256 | >8/152 | 32/16 | 256 |
| BV185 | Mexico | 2013 | 2 | >128 | >64 | 128 | >256/4 | >256 | >8/152 | 64/32 | 256 |
| BV186 | USA | 2013 | 2 | 16 | 64 | 256 | >256/4 | >256 | >8/152 | 32/16 | 8 |
| BV187 | USA | 2013 | 2 | 32 | 64 | 256 | >256/4 | >256 | >8/152 | 16/8 | 8 |
| BV189 | Spain | 2013 | 2 | 128 | 32 | 128 | >256/4 | 256 | >8/152 | 32/16 | 16 |
| BV190 | Greece | 2012 | 1 | >128 | 64 | 64 | >256/4 | >256 | >8/152 | 64/32 | 256 |
| BV191 | China | 2013 | 2 | >128 | >64 | 256 | >256/4 | >256 | >8/152 | 128/64 | >256 |
| ATCC 25922 (quality control) | 1 | <0.06 | <0.25 | 4/4 | <0.25 | 0.125/2.34 | 4/2 | 2 | |||
Abbreviations: CIP, ciprofloxacin; CTX, cefotaxime; GENT, gentamicin; MERO, meropenem; MLST, multilocus sequence type; SAM, ampicillin-sulbactam; SXT, trimethoprim-sulfamethoxazole; TET, tetracycline; TZP, piperacillin-tazobactam. Classification of antibiotic resistance was done according to breakpoints published by the Clinical and Laboratory Standards Institute: susceptible (italics), intermediate (underlined), and resistant (bold) (34).
The colistin sensitivity of the parental clinical isolates and their corresponding pmrA knockout mutants (ΔpmrA) was determined by broth microdilution method. pmrA deletion reduced MICs 64- to 1,024-fold in 10 out of 12 initially colistin-resistant clinical isolates (83%), thus restoring susceptibility to colistin (MIC, ≤2 μg/ml) (Table 2). To our surprise, however, two strains (BV94 and BV189) retained colistin resistance even in the absence of pmrA.
TABLE 2.
Effect of loss of PmrA on colistin susceptibility and PmrB mutations in the strain panel
| Strain designation | Colistin MIC (μg/ml)a
|
PmrB mutations (amino acid substitutions) | |
|---|---|---|---|
| Wild type | ΔpmrA | ||
| ATCC 17978 | 0.25 | 0.25 | Reference |
| BV94 | 64 | 32 | Wild type |
| BV95 | 32 | 0.5 | L274W |
| BV172 | 256 | 1 | Q43L and L267F |
| BV173 | 128 | 1 | A138T and A226V |
| BV174 | 64 | 1 | Q277R |
| BV175 | 256 | 0.5 | L267W |
| BV185 | 256 | 0.25 | P233S |
| BV186 | 16 | 0.25 | Q277R |
| BV187 | 16 | 0.25 | Q277R |
| BV189 | 64 | 64 | Wild type |
| BV190 | 256 | 0.5 | A138T and A226V |
| BV191 | 256 | 0.25 | A138T and P233S |
Susceptibility breakpoint, ≤2 μg/ml. Susceptible, italics; resistant, bold.
We investigated the differences between strains that became susceptible after pmrA deletion and those that remained resistant by analyzing the sequence variations of the PmrAB TCS in the strain panel. The PmrA and PmrB sequences of the A. baumannii AYE, ACICU, and NIPH 146 strains were used as references for ST1, ST2, and ST25 clonal lineages, respectively. Nonsynonymous mutations were found only in the PmrB sensor kinase (Table 2). Interestingly, the two strains with an unaltered PmrB sequence were those that remained colistin resistant after pmrA deletion (BV94 and BV189). Our data suggest that colistin resistance in these two strains is not conferred by PmrA-mediated PmrC overexpression. We confirmed this hypothesis by quantifying the expression of pmrC using quantitative reverse transcription-PCR (qRT-PCR) (Fig. 1). The control strain ATCC 17978 and the two refractory strains BV94 and BV189 showed only marginal levels of pmrC expression. In contrast, the 10 other strains showed pmrC overexpression, and this overexpression was abolished in the ΔpmrA mutant.
FIG 1.
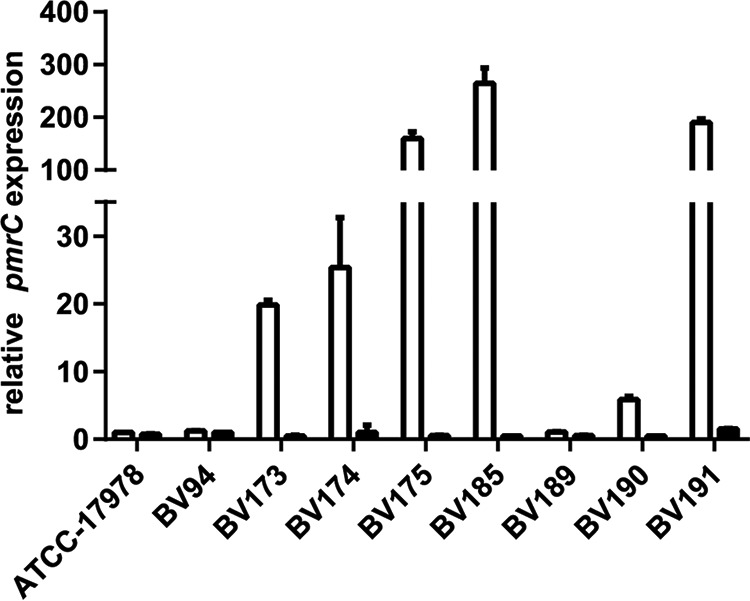
Quantification of pmrC expression levels in colistin-resistant A. baumannii clinical isolates and their ΔpmrA mutants. Expression levels of pmrC were quantified by qRT-PCR in colistin-resistant A. baumannii isolates (white bars) and their ΔpmrA mutants (black bars). The expression levels were normalized to the pmrC expression in the ATCC 17978 reference strain.
Taken together, colistin resistance in A. baumannii is predominantly conferred by mutations in the PmrB TCS sensor kinase that lead to overexpression of PmrC, as shown in 10 out of 12 clinical strains. However, some isolates (2 out of 12 strains in our panel) may use an alternative colistin resistance mechanism independent of PmrA-mediated PmrC overexpression to resist the antibacterial activity of colistin.
EptA, a PmrC homolog, is present in the A. baumannii strains of international clone 2.
Lesho et al. described the presence of the alternative PetN transferase EptA in A. baumannii (17). EptA and PmrC are homologous proteins with 93% amino acid identity, suggesting similar enzymatic activities. However, the role of EptA in A. baumannii colistin resistance is still unclear (17). To investigate the prevalence of eptA in A. baumannii, we took advantage of sequence differences between pmrC and eptA at the N- and C-terminal ends of the open reading frames and designed oligonucleotides (oVT152/oVT153) that can discriminate eptA from pmrC (Fig. 2A; see also Table S1 in the supplemental material). Using these eptA-specific primers, we detected eptA in all our international clone 2 strains but not in international clone 1 strains (Table 3 and Fig. S1). This finding was further confirmed by screening 12 additional isolates from the BioVersys strain collection (data not shown).
FIG 2.
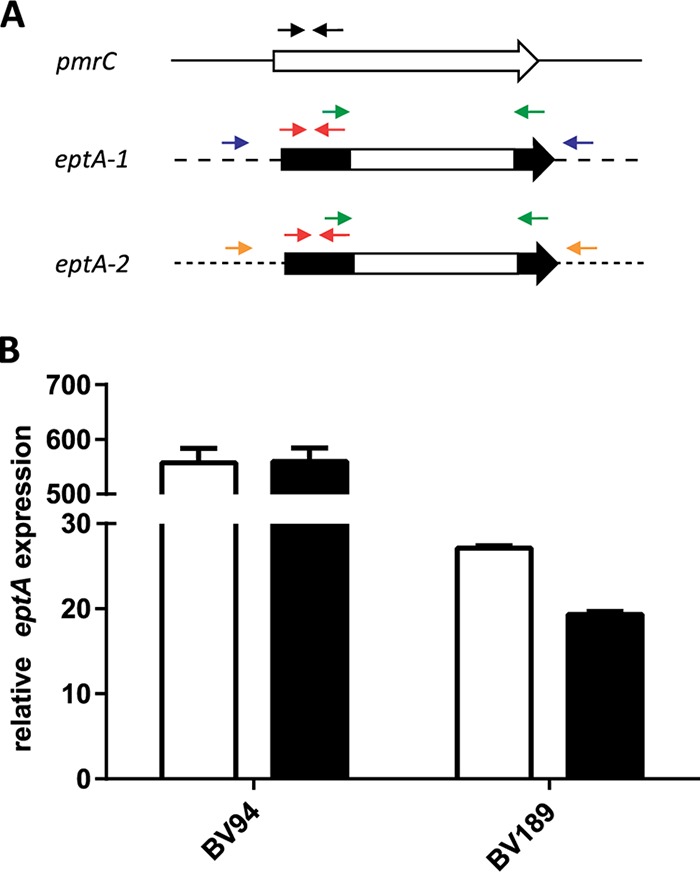
Discrimination and quantification of pmrC and eptA. (A) Schematic representation of differences in the pmrC, eptA-1, and eptA-2 coding sequence. Primers marked by black (oVT162/oVT163) and red (oVT164/oVT165) arrows were used to detect pmrC and eptA in qRT-PCR experiments, respectively. The primers marked by green arrows (oVT152/oVT153) were used to genotype the eptA isoforms. Primers marked by blue (oVT198/oVT199) and orange (oVT201/oVT202) arrows were used to discriminate eptA-1 and eptA-2, respectively. (B) Expression levels of eptA were quantified by qRT-PCR in colistin-resistant A. baumannii isolates BV94 and BV189 (white bars) and their ΔpmrA mutants (black bars). The expression levels were normalized to the pmrC expression in the ATCC 17978 reference strain.
TABLE 3.
Distribution of the eptA variants in the strain panel
| Strain designation | MLST | eptA variant(s) |
|---|---|---|
| ATCC 17978 | 77 | |
| BV94 | 2 | eptA-1, ISAbaI-eptA-2, ISAbaI-eptA-3 |
| BV95 | 25 | |
| BV172 | 2 | eptA-1 |
| BV173 | 2 | eptA-1 |
| BV174 | 2 | eptA-1, eptA-2 |
| BV175 | 2 | eptA-1 |
| BV185 | 2 | eptA-1 |
| BV186 | 2 | eptA-1, eptA-2 |
| BV187 | 2 | eptA-1, eptA-2 |
| BV189 | 2 | ISAbaI-eptA-1 |
| BV190 | 1 | |
| BV191 | 2 | eptA-1 |
Agarose gel of eptA-1 and eptA-2 genotyping in the strain panel. The genotyping of eptA-1 (upper gel) and eptA-2 (lower gel) was performed by PCR using primers oVT198/oVT199 and oVT201/oVT202, respectively. Lane 1, 2-log ladder (New England Biolabs); lane 2, ATCC 17978; lane 3, BV94; lane 4, BV95; lane 5, BV172; lane 6, BV173; lane 7, BV174; lane 8, BV175; lane 9, BV185; lane 10, BV186; lane 11, BV187; lane 12, BV189; lane 13, BV190; lane 14, BV191; lane 15, 2-log ladder. Compared to the expected sizes of 1,862 bp and 1,827 bp, the PCR products for BV94 eptA-2 and BV189 eptA-1 genotyping, respectively, are approximately 1 kb larger, corresponding to the presence of ISAbaI. Download FIG S1, PDF file, 0.3 MB (361.9KB, pdf) .
Copyright © 2019 Trebosc et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotides used in this study. Download Table S1, PDF file, 0.2 MB (189.2KB, pdf) .
Copyright © 2019 Trebosc et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
The integrated insertion element ISAbaI causes eptA overexpression in BV94 and BV189.
Two isoforms of eptA, eptA-1 and eptA-2 (GenBank accession numbers KC700024 and KC700023, respectively) have been described at different locations in the genome of various A. baumannii strains (17). Taking advantage of the different flanking regions, we designed primers able to discriminate eptA-1 from eptA-2 (oVT198/oVT199 and oVT201/oVT202, respectively) (Fig. 2A and Table S1). By genotyping the strain panel, we demonstrated that all strains that belong to the international clone 2 contained eptA-1 and four of them contained an additional copy of eptA-2 (Table 3 and Fig. S1). Interestingly, the PCR products obtained for eptA-2 in BV94 and eptA-1 in BV189 were approximately 1 kb larger than the expected fragment size. Sequencing of the PCR products identified the insertion element ISAbaI upstream of eptA-2 and eptA-1 in BV94 and BV189, respectively. The ISAbaI orientation enabled its strong promoter (Pout) to drive eptA overexpression as previously described for other antibiotic resistance determinants (Fig. 3) (25, 26).
FIG 3.

Representation of the different ISAbaI-eptA genomic regions present in BV94 and BV189. The nucleic acid sequence of the ISAbaI inverted repeats right and left (IRR and IRL, respectively) and Pout promoter are shown until the eptA start codon. The 9-bp target site duplications (TSD) up- and downstream of ISAbaI are not present for eptA-3, which is consistent with an ISAbaI-eptA-2 duplication. The junction between ABK1_3144 and ISAbaI-eptA-3 has been sequenced, while the sequence downstream of ABK1_2603 could not be resolved. The ABK1 gene annotation is shown according to the genomic sequence of A. baumannii strain 1656-2 (GenBank accession number NC_017162).
Using probes specific for eptA or pmrC, we quantified their respective expression levels in our strains. eptA was 550- and 25-fold higher expressed in BV94 and BV189, respectively, than the homologous isoform pmrC in the control strain ATCC 17978 (which does not contain eptA) (Fig. 2B). This eptA overexpression was not altered in the ΔpmrA mutant strains, suggesting that eptA expression in both strains is independent of the PmrAB TCS. These data suggest that an ISAbaI-driven eptA overexpression may represent an alternative and PmrAB-independent colistin resistance mechanism in A. baumannii clinical strains.
ISAbaI-driven eptA overexpression confers colistin resistance in A. baumannii clinical isolates.
To validate the hypothesis that ISAbaI-driven eptA overexpression confers colistin resistance in A. baumannii clinical isolates, we deleted eptA-1 in the clinical isolates BV189 and BV94 and determined MIC values. Indeed, BV189 (which carries ISAbaI upstream of eptA-1) became colistin susceptible upon eptA-1 deletion, indicating an essential role of ISAbaI-eptA-1 in conferring colistin resistance in this strain (Table 4). In contrast, BV94, carrying both eptA-1 and eptA-2 isoforms but carrying an ISAbaI insertion only upstream of eptA-2, remained colistin resistant after deletion of eptA-1, suggesting the key role of ISAbaI insertion for colistin resistance in A. baumannii. To further confirm the importance of ISAbaI, we constructed the double mutant BV94ΔeptA-1/ΔeptA-2 and evaluated its colistin susceptibility. A 4-fold MIC reduction was observed in the BV94ΔeptA-1/ΔeptA-2 mutant compared to BV94 and BV94ΔeptA-1, indicating that eptA-2 with an upstream ISAbaI is involved in the colistin resistance mechanism of BV94. However, we were surprised to see that BV94ΔeptA-1/ΔeptA-2 remained resistant to colistin with a MIC of 16 μg/ml, indicating that there must be yet another colistin resistance mechanism present in this isolate.
TABLE 4.
Recovery of colistin susceptibility after deletion of different eptA isoforms
| Strain | Colistin MIC (μg/ml)a
|
|||
|---|---|---|---|---|
| Wild type | ΔeptA-1 | ΔeptA-1/ΔeptA-2 | ΔeptA-1/ΔeptA-2/ΔeptA-3 | |
| BV189 | 128 | 0.5 | ||
| BV94 | 64 | 64 | 16 | 1 |
Susceptibility breakpoint, 2 μg/ml. Susceptible, italics; resistant, bold.
Three different eptA variants can confer colistin resistance in A. baumannii.
We genotyped the BV94ΔeptA-1/ΔeptA-2 double mutant and confirmed the successful deletion of eptA-1 and eptA-2. However, we detected the presence of at least one additional eptA copy (eptA-3) in the double mutant (Fig. S2). We performed a fusion primer and nested integrated PCR experiment (FPNI-PCR) to amplify the genomic flanking regions of the additional eptA-3 variant (27). Sequencing revealed the ISAbaI insertion element and the gene ABK1_2603 present upstream and downstream of eptA-3, respectively (Fig. 3). This eptA-3 gene context in BV94 was identical to the eptA-2 gene context present in the A. baumannii strain 1656-2 (GenBank accession number NC_017162). However, further upstream there were marked differences. ISAbaI-eptA-3 in BV94 was adjacent to the gene ABK1_3144, while ISAbaI-eptA-2 in 1656-2 was adjacent to a different gene (Fig. 3). We could not determine the downstream flanking region of ISAbaI-eptA3 ABK1_2603 in multiple attempts. Nevertheless, the 9-bp target site duplications (TSD) created by ISAbaI transposition could not be identified directly outside the ISAbaI upstream eptA-3 (25). In contrast, TSD were present next to the right and left inverted repeats of the ISAbaI upstream eptA-1 and eptA-2, which is consistent with a single transposition event. These observations indicate that ISAbaI did not insert upstream eptA-3 in a single transposition event, and therefore, ISAbaI-eptA-3 in BV94 might be a result of an ISAbaI-eptA-2 cassette gene duplication, implying that the ISAbaI-eptA colistin resistance determinant is contained in a mobile genetic element.
Agarose gel of eptA genotyping in the clinical isolate BV94 and its eptA knockout mutants. The genotyping of eptA isoforms was performed by PCR using primers oVT152/oVT153. Lane 1, 2-log ladder (New England Biolabs); lane 2, BV94; lane 3, BV94ΔeptA-1; lane 4, BV94ΔeptA-1/ΔeptA-2; lane 5, BV94ΔeptA-1/ΔeptA-2/ΔeptA-3. The triple mutant BV94ΔeptA-1/ΔeptA-2/ΔeptA-3 did not carry any other eptA isoform. Download FIG S2, PDF file, 0.3 MB (339.4KB, pdf) .
Copyright © 2019 Trebosc et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
We finally deleted the DNA fragment between ABK1_3144 and ABK1_3143 containing eptA-3 to confirm that ISAbaI-eptA-3 was responsible for the high residual colistin resistance in BV94ΔeptA-1/ΔeptA-2. In addition, we performed PCR-based eptA genotyping on the resulting triple mutant BV94ΔeptA-1/ΔeptA-2/ΔeptA-3 to exclude the presence of yet another eptA copy (Fig. S2). The loss of all 3 eptA isoforms in BV94ΔeptA-1/ΔeptA-2/ΔeptA-3 rendered this triple mutant susceptible to colistin, indicating that colistin resistance in BV94 was entirely conferred by the overexpression of EptA-isoforms (Table 4).
Targeting PetN transferases may overcome colistin resistance in A. baumannii.
We have shown that colistin resistance was mediated in 10 out of 12 analyzed clinical strains by PmrA-mediated overexpression of PmrC. In the remaining two strains, ISAbaI-driven EptA expression conferred colistin resistance. Taken together, in all tested clinical isolates colistin resistance was mediated by the overexpression of PetN transferases, suggesting that inhibition of these homologous enzymes with small molecules may have the potential to overcome colistin resistance in A. baumannii. Chin and colleagues recently suggested that the acetyl-galactosamine (GalNAc) deacetylase NaxD plays a role in colistin resistance in A. baumannii (28). In this report, the expression of NaxD, which was regulated by the PmrAB TCS, mediated galactosamine (GalN) addition to lipid A, conferring colistin resistance in A. baumannii. We performed additional experiments to exclude the possibility that the colistin resensitization observed in our clinical isolates after deletion of pmrA was based on a modulation of naxD expression and not pmrC expression. We first confirmed the PmrAB-controlled naxD expression based on qRT-PCR data for BV191 and BV191ΔpmrA. BV191 has a mutated PmrB that likely triggers PmrA-mediated pmrC overexpression (Table 2). Similarly, naxD expression was 15-fold higher in the colistin-resistant strain BV191 than the susceptible strain ATCC 17978 (Fig. 4). In BV191ΔpmrA, lacking the response regulator PmrA, pmrC and naxD overexpression was abolished, confirming that both genes were regulated by the PmrAB TCS. Notably, pmrC overexpression was 20-fold higher than naxD overexpression, suggesting a minor contribution of NaxD compared to PmrC in colistin resistance. To confirm the major role of PmrC in PmrA-mediated colistin resistance and to exclude that another PmrA-regulated gene, such as naxD, is involved in colistin resistance, we directly deleted the effector pmrC from the genome of BV191. The loss of PmrC rendered BV191 susceptible to colistin (MIC of 0.5 μg/ml) and resulted in a similar phenotype as in BV191ΔpmrA (Table 2). In contrast, naxD was still 15-fold overexpressed in BV191ΔpmrC (Fig. 4). This result suggests that overexpression of naxD is not sufficient to confer colistin resistance in BV191 and indicates that PmrC is the main effector of PmrA-mediated colistin-resistant A. baumannii strains, such as BV191.
FIG 4.
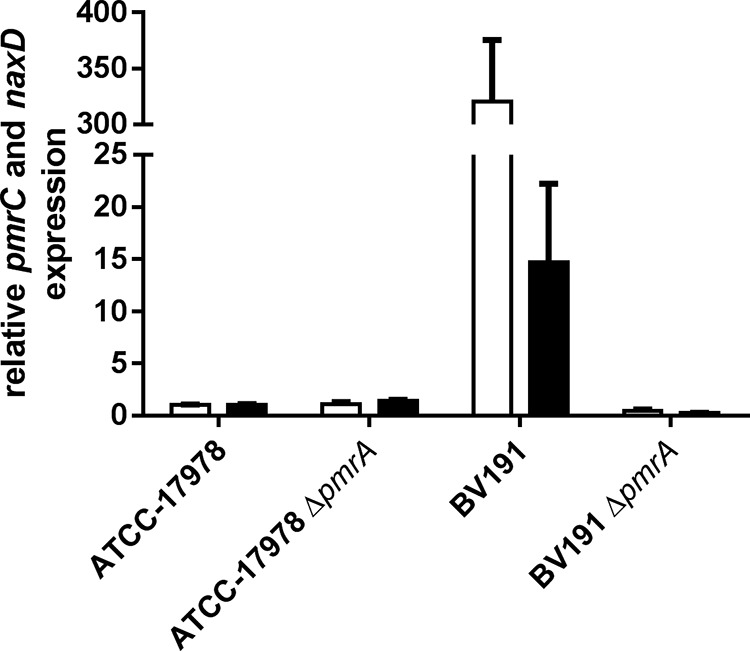
Quantification of pmrC and naxD expression levels in A. baumannii BV191 and its ΔpmrA mutants. The expression of pmrC (white bars) and naxD (black bars) was quantified by qRT-PCR and normalized to the gene expression level in the reference strain ATCC 17978.
DISCUSSION
Bacteria have evolved multiple ways to escape the hazardous action of antibiotics. In nosocomial infections, the individual strain history of antibiotic exposures during patient treatment may result in the development and accumulation of different resistance mechanisms in different strains of the same species. Therefore, it is important to study resistance mechanisms on multiple strains. Moreover, it is crucial to study these mechanisms on strains that developed resistance during patient treatment due to the discrepancy that may be observed between in vitro- and in vivo-developed mechanisms. For instance, A. baumannii polymyxin resistance is commonly mediated by LPS loss when A. baumannii is exposed to the drug in vitro, but this mechanism is not viable in vivo due to the strong fitness cost that it engenders (16, 29).
In this study, we dissected the mechanisms conferring colistin resistance in 12 clinically relevant A. baumannii strains. To our knowledge, this is the first time that colistin resistance is genetically characterized in a panel of A. baumannii clinical strains that developed resistance during patient treatment and not strains that artificially acquired resistance by in vitro selection/passaging. This gap in knowledge originates from the difficulties in manipulating the genome of A. baumannii colistin-resistant clinical strains. Indeed, as exemplified in our strain panel, such strains are generally resistant to all other antibiotics because colistin is used as a last option in the treatment of A. baumannii infections, only when other antibiotics fail. To break the barrier of antibiotic resistance in these strains, we applied a genome editing method based on a nonantibiotic resistance marker, which is efficient regardless of the resistance profile of the strain (24).
We demonstrated two different ways to overexpress PetN transferases that cause colistin resistance in A. baumannii clinical isolates (Fig. 5). The predominant colistin resistance mechanism found in 83% of the studied clinical isolates was mediated by pmrC overexpression. The overexpression of pmrC in these strains was entirely caused by mutations in the sensor kinase PmrB, although previous studies also found mutations in the response regulator PmrA (14, 15, 17). We found 7 different PmrB variants among the 10 PmrC-mediated colistin-resistant strains, indicating the diversity of mutations that lead to PmrC overexpression. Except fo r the A226V and P233S mutations, the identified PmrB mutations were not yet reported in A. baumannii (11, 15, 16).
FIG 5.

Schematic representation of A. baumannii colistin resistance mechanisms. The two pathways leading to phosphoethanolamine (PetN) transferase overexpression and colistin resistance are represented. The major A. baumannii PetN transferase overexpression pathway results from pmrC expression, which is activated by the transcriptional regulator PmrA previously phosphorylated (activated) by a mutated variant of the sensor kinase PmrB (PmrB*). Alternatively, A. baumannii PetN transferase overexpression can result from the integration of the ISAbaI insertion element upstream of an eptA isoform. PetN transferase enzymes decorate the outer membrane lipid A with PetN, thereby lowering the negative charge and preventing colistin binding. Potential PmrA inhibitors would only block the pmrC pathway (dark blue cross), while PetN transferase inhibitors would block lipid A modification (red cross) and restore colistin efficacy against A. baumannii.
Interestingly, we found two clinical isolates in which colistin resistance was conferred by a genomic insertion of ISAbaI, resulting in a strong overexpression of the pmrC homolog eptA. eptA-1 and eptA-2 genes have been previously identified in A. baumannii; however, their distribution, expression regulation, and role in colistin resistance were not assessed (17). Our study revealed that A. baumannii strains of the international clone 2, which represent the most problematic strains in hospitals, carry at least one eptA variant. In contrast, international clone 1 strains did not carry eptA. Our data further show that eptA expression is not regulated by the PmrAB TCS, but instead, integration of ISAbaI upstream of any eptA isoform is required to confer the resistance phenotype, presumably by ISAbaI-driven eptA overexpression. Consequently, detection of an eptA gene alone is not sufficient to classify A. baumannii strains as colistin resistant.
The analysis of PmrA as a potential drug target confirmed the importance of this protein in mediating colistin resistance in A. baumannii. However, the high prevalence of eptA and the ability of ISAbaI to integrate upstream of eptA and drive its expression independently of the PmrAB TCS disproved PmrA as a direct drug target for resensitization of A. baumannii to colistin (Fig. 5). An adjuvant therapy consisting of a PmrA inhibitor in combination with colistin would most likely select for ISAbaI-driven EptA-overexpressing colistin-resistant strains. As demonstrated by the two clinical isolates BV94 and BV189, such strains are already present in hospitals. One of the strains also contained a duplicated ISAbaI-eptA cassette, suggesting that this functional cassette mediating colistin resistance was present on a mobile element. The presence of a mobile colistin-resistance-mediating cassette increases the probability of intra- and interspecies transfer of the resistance pathways by the integration into plasmids. This phenomenon was recently illustrated with plasmid-carried PetN transferase mcr-1, which was initially found in China but rapidly has spread globally and in different species (12, 13). Nevertheless, mcr-1 seems to be limited to Enterobacteriaceae species and has never yet been detected in A. baumannii.
One of the major colistin resistance pathways in Enterobacteriaceae and P. aeruginosa is the addition of AraN to lipid A (11). Although we describe here two different ways to overexpress PetN transferases, our results suggest that colistin resistance in clinical A. baumannii isolates is exclusively conferred by PetN addition to lipid A. A recent study suggested that a GalN-based modification of lipid A may be involved in colistin resistance in A. baumannii (28). In contrast, our results suggest that alteration of the lipid A structure by addition of PetN plays the major role in colistin resistance in A. baumannii. It has also been described that loss of LPS may confer colistin resistance in A. baumannii (30). However, most of the LPS-deficient colistin-resistant mutants were obtained in vitro after colistin evolution, and it has been shown that these mutants are hypersusceptible to other antibiotic classes and are avirulent (16, 29). Emergence of LPS-deficient colistin-resistant mutants in patients is therefore unlikely.
In conclusion, the overexpression of homologous PetN transferases caused colistin resistance in all studied clinical isolates, but in some cases this occurred independently of PmrAB. The crystal structure of Neisseria meningitidis PetN transferase has been recently reported, and this enzyme has been proposed as a drug target for antivirulence and antiresistance drug development to treat Neisseria gonorrhoeae and N. meningitidis infections (31, 32). Our data suggest that a direct inhibitor of homologous PetN transferases PmrC and EptA may have the potential to overcome colistin resistance in A. baumannii clinical strains (Fig. 5).
MATERIALS AND METHODS
Bacterial strains, MIC, MLST, and oligonucleotides.
The A. baumannii reference strain ATCC 17978 and 12 extensively drug-resistant A. baumannii clinical isolates from the BioVersys proprietary strain collection were used in this study. The microdilution method was used to determine MICs according to the CLSI guidelines (33). Multiple locus sequence type (MLST) was determined according to the Pasteur scheme using specific primers (source: http://pubmlst.org/abaumannii/) (20). Oligonucleotides used in this study are listed in Table S1 in the supplemental material.
Genomic deletions of pmrA, eptA-1, eptA-2, eptA-3, and pmrC in A. baumannii clinical isolates.
Scarless deletions of pmrA, pmrC, and the eptA isoforms were performed using a two-step recombination method previously described (24).
DNA fragments corresponding to 700-bp up- and downstream genomic regions of the genes to be deleted were amplified by PCR and cloned in the multiple cloning site of the knockout platform pVT77. Oligonucleotides oVT49/oVT50 and oVT51/oVT52 were used to amplify the up- and downstream regions, respectively, of pmrA. The resulting DNA fragments were ligated and introduced into pVT77 previously digested by EcoRI and BamHI. Similarly, oligonucleotides oVT235/oVT236 and oVT237/oVT238 were used to amplify the flanking regions of eptA-1, and oligonucleotides oVT305/oVT306 and oVT307/oVT242 were used to amplify the flanking regions of eptA-2. The resulting DNA fragments for eptA-1 and eptA-2 were introduced into pVT77 previously digested by XhoI and XbaI using NEBuilder HiFi DNA assembly (New England Biolabs). For eptA-3 deletion, the genomic regions flanking the duplicated cassette were amplified using oVT390/oVT391 and oVT392/oVT393. The resulting DNA fragments were cloned into pVT77 previously digested with EcoRI and XbaI using NEBuilder HiFi DNA assembly. Last, the flanking regions of pmrC were amplified using oVT324/oVT325 and oVT326/oVT327, and the resulting DNA fragments were cloned into pVT77 previously digested with KpnI and PstI using NEBuilder HiFi DNA assembly.
The cloned knockout plasmids were transformed in E. coli conjugative strain MFDpir to proceed with the construction of markerless deletion in A. baumannii, as previously described (24). Briefly, after conjugation, genomic plasmid integration was selected on LB agar plates containing 100 μg/ml sodium tellurite. Clones were screened for up- or downstream integration by PCR using primer oVT8, which anneals on the plasmid, and oVT91, oVT243, oVT311, or oVT328, which anneals upstream of pmrA, eptA-1, eptA-2, or pmrC, respectively. For eptA-3, clones were screened using primers oVT8/oVT396 and oVT174/oVT397 for up- and downstream integration, respectively. Clones containing up- and downstream plasmid integrations were transferred on LB agar plates containing 1 mM isopropyl-β-d-1-thiogalactopyranoside and 200 μg/ml 3′-azido-3′-deoxythymidine to select for plasmid removal from the genome. Clones were screened for gene deletion and plasmid removal by PCR using primers oVT91/oVT92, oVT243/oVT244, oVT246/oVT311, oVT396/oVT397, and oVT328/oVT14 for pmrA, eptA-1, eptA-2, eptA-3, and pmrC, respectively. The genomic gene deletions were finally confirmed by DNA sequencing (Microsynth AG, Balgach, Switzerland).
Genotyping of pmrA, pmrB, and eptA.
A genomic DNA sequence including pmrA and pmrB was PCR amplified from all the strains of the panel using oVT91 and oCK292, and the PCR products were sent for sequencing (Microsynth AG, Balgach, Switzerland). The genotyping of eptA isoforms was performed by PCR using eptA-specific primers oVT152 and oVT153, which anneal on all eptA isoforms but not on pmrC (Fig. 2A). PCR using primers oVT198/oVT199 and oVT201/oVT202, which anneal on the flanking sides of eptA-1 and eptA-2, respectively, were used to discriminate between eptA isoforms (Fig. 2A).
qRT-PCR.
Quantitative reverse transcription-PCR was performed as previously described (24). The specific expression of the PetN transferases encoded by pmrC and eptA was evaluated using oVT162/oVT163 and oVT164/oVT165 primers, respectively (Fig. 2A). The expression of naxD was evaluated using oVT314/oVT315 primers. Expression levels were normalized to that of the housekeeping gene rpoD using the comparative threshold cycle (ΔΔCT) method. The expression of rpoD was evaluated using rpoD-qRT-F/rpoD-qRT-R primers.
FPNI-PCR.
Fusion primer and nested integrated PCR was performed as previously described (27). This method relies on a three-step PCR using arbitrary degenerated oligonucleotides fused to known adaptors and three sequence-specific oligonucleotides, which consist in our case of eptA-specific oligonucleotides. FPNI-PCR experiments were performed on the BV94ΔeptA-1/ΔeptA-2 mutant with two sets of three eptA-specific oligonucleotides, oligonucleotides oVT343, oVT344, and oVT345 and oligonucleotides oVT340, oVT341, and oVT342, to identify the sequence up- and downstream of the new eptA copy, respectively. The degenerated primers and the known adaptor primers were directly taken from the previously described method (27). Briefly, the first round of PCRs was performed using the degenerated primers and oVT343 for upstream identification and oVT340 for downstream identification. The second round of PCRs was performed with the first adaptor primer FSP1 and oVT344 for upstream identification and oVT341 for downstream identification. The last round of PCRs was performed with the second adaptor primer FSP2 and oVT345 for upstream identification and oVT342 for downstream identification. The brightest and most distinct PCR products obtained were sent for sequencing (Microsynth AG, Balgach, Switzerland).
ACKNOWLEDGMENTS
This project has received funding from the European Union’s Horizon 2020 research and innovation program for Vincent Trebosc and Valentina Lucchini under the Marie Skłodowska-Curie grant agreement numbers 607694 and 721484, respectively.
Footnotes
Citation Trebosc V, Gartenmann S, Tötzl M, Lucchini V, Schellhorn B, Pieren M, Lociuro S, Gitzinger M, Tigges M, Bumann D, Kemmer C. 2019. Dissecting colistin resistance mechanisms in extensively drug-resistant Acinetobacter baumannii clinical isolates. mBio 10:e01083-19. https://doi.org/10.1128/mBio.01083-19.
REFERENCES
- 1.Thabit AK, Crandon JL, Nicolau DP. 2015. Antimicrobial resistance: impact on clinical and economic outcomes and the need for new antimicrobials. Expert Opin Pharmacother 16:159–177. doi: 10.1517/14656566.2015.993381. [DOI] [PubMed] [Google Scholar]
- 2.Bassetti M, Merelli M, Temperoni C, Astilean A. 2013. New antibiotics for bad bugs: where are we? Ann Clin Microbiol Antimicrob 12:22. doi: 10.1186/1476-0711-12-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boucher HW, Talbot GH, Benjamin DK, Bradley J, Guidos RJ, Jones RN, Murray BE, Bonomo RA, Gilbert D, Infectious Diseases Society of America. 2013. 10 × ’20 progress—development of new drugs active against gram-negative bacilli: an update from the Infectious Diseases Society of America. Clin Infect Dis 56:1685–1694. doi: 10.1093/cid/cit152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li J, Nation RL, Turnidge JD, Milne RW, Coulthard K, Rayner CR, Paterson DL. 2006. Colistin: the re-emerging antibiotic for multidrug-resistant Gram-negative bacterial infections. Lancet Infect Dis 6:589–601. doi: 10.1016/S1473-3099(06)70580-1. [DOI] [PubMed] [Google Scholar]
- 5.Zavascki AP, Goldani LZ, Li J, Nation RL. 2007. Polymyxin B for the treatment of multidrug-resistant pathogens: a critical review. J Antimicrob Chemother 60:1206–1215. doi: 10.1093/jac/dkm357. [DOI] [PubMed] [Google Scholar]
- 6.Peleg AY, Hooper DC. 2010. Hospital-acquired infections due to Gram-negative bacteria. N Engl J Med 362:1804–1813. doi: 10.1056/NEJMra0904124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gales AC, Jones RN, Sader HS. 2011. Contemporary activity of colistin and polymyxin B against a worldwide collection of Gram-negative pathogens: results from the SENTRY Antimicrobial Surveillance Program (2006–09). J Antimicrob Chemother 66:2070–2074. doi: 10.1093/jac/dkr239. [DOI] [PubMed] [Google Scholar]
- 8.Pogue JM, Cohen DA, Marchaim D. 2015. Editorial commentary: polymyxin-resistant Acinetobacter baumannii: urgent action needed. Clin Infect Dis 60:1304–1307. doi: 10.1093/cid/civ044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pieren M, Tigges M. 2012. Adjuvant strategies for potentiation of antibiotics to overcome antimicrobial resistance. Curr Opin Pharmacol 12:551–555. doi: 10.1016/j.coph.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 10.Needham BD, Trent MS. 2013. Fortifying the barrier: the impact of lipid A remodelling on bacterial pathogenesis. Nat Rev Microbiol 11:467–481. doi: 10.1038/nrmicro3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olaitan AO, Morand S, Rolain J-M. 2014. Mechanisms of polymyxin resistance: acquired and intrinsic resistance in bacteria. Front Microbiol 5:643. doi: 10.3389/fmicb.2014.00643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu Y-Y, Wang Y, Walsh TR, Yi L-X, Zhang R, Spencer J, Doi Y, Tian G, Dong B, Huang X, Yu L-F, Gu D, Ren H, Chen X, Lv L, He D, Zhou H, Liang Z, Liu J-H, Shen J. 2016. Emergence of plasmid-mediated colistin resistance mechanism MCR-1 in animals and human beings in China: a microbiological and molecular biological study. Lancet Infect Dis 16:161–168. doi: 10.1016/S1473-3099(15)00424-7. [DOI] [PubMed] [Google Scholar]
- 13.Rolain J-M, Kempf M, Leangapichart T, Chabou S, Olaitan AO, Page SL, Morand S, Raoult D. 2016. Plasmid-mediated mcr-1 gene in colistin-resistant clinical isolates of Klebsiella pneumoniae in France and Laos. Antimicrob Agents Chemother 60:6994–6995. doi: 10.1128/AAC.00960-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Adams MD, Nickel GC, Bajaksouzian S, Lavender H, Murthy AR, Jacobs MR, Bonomo RA. 2009. Resistance to colistin in Acinetobacter baumannii associated with mutations in the PmrAB two-component system. Antimicrob Agents Chemother 53:3628–3634. doi: 10.1128/AAC.00284-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arroyo LA, Herrera CM, Fernandez L, Hankins JV, Trent MS, Hancock R. 2011. The pmrCAB operon mediates polymyxin resistance in Acinetobacter baumannii ATCC 17978 and clinical isolates through phosphoethanolamine modification of lipid A. Antimicrob Agents Chemother 55:3743–3751. doi: 10.1128/AAC.00256-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beceiro A, Moreno A, Fernández N, Vallejo JA, Aranda J, Adler B, Harper M, Boyce JD, Bou G. 2014. Biological cost of different mechanisms of colistin resistance and their impact on virulence in Acinetobacter baumannii. Antimicrob Agents Chemother 58:518–526. doi: 10.1128/AAC.01597-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lesho E, Yoon E-J, McGann P, Snesrud E, Kwak Y, Milillo M, Onmus-Leone F, Preston L, Clair KS, Nikolich M, Viscount H, Wortmann G, Zapor M, Grillot-Courvalin C, Courvalin P, Clifford R, Waterman PE. 2013. Emergence of colistin-resistance in extremely drug-resistant Acinetobacter baumannii containing a novel pmrCAB operon during colistin therapy of wound infections. J Infect Dis 208:1142–1151. doi: 10.1093/infdis/jit293. [DOI] [PubMed] [Google Scholar]
- 18.Park YK, Choi JY, Shin D, Ko KS. 2011. Correlation between overexpression and amino acid substitution of the PmrAB locus and colistin resistance in Acinetobacter baumannii. Int J Antimicrob Agents 37:525–530. doi: 10.1016/j.ijantimicag.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 19.Harris TL, Worthington RJ, Hittle LE, Zurawski DV, Ernst RK, Melander C. 2014. Small molecule downregulation of PmrAB reverses lipid A modification and breaks colistin resistance. ACS Chem Biol 9:122–127. doi: 10.1021/cb400490k. [DOI] [PubMed] [Google Scholar]
- 20.Diancourt L, Passet V, Nemec A, Dijkshoorn L, Brisse S. 2010. The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One 5:e10034. doi: 10.1371/journal.pone.0010034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sahl JW, Del Franco M, Pournaras S, Colman RE, Karah N, Dijkshoorn L, Zarrilli R. 2015. Phylogenetic and genomic diversity in isolates from the globally distributed Acinetobacter baumannii ST25 lineage. Sci Rep 5:15188. doi: 10.1038/srep15188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zarrilli R, Pournaras S, Giannouli M, Tsakris A. 2013. Global evolution of multidrug-resistant Acinetobacter baumannii clonal lineages. Int J Antimicrob Agents 41:11–19. doi: 10.1016/j.ijantimicag.2012.09.008. [DOI] [PubMed] [Google Scholar]
- 23.Magiorakos A-P, Srinivasan A, Carey RB, Carmeli Y, Falagas ME, Giske CG, Harbarth S, Hindler JF, Kahlmeter G, Olsson-Liljequist B, Paterson DL, Rice LB, Stelling J, Struelens MJ, Vatopoulos A, Weber JT, Monnet DL. 2012. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin Microbiol Infect 18:268–281. doi: 10.1111/j.1469-0691.2011.03570.x. [DOI] [PubMed] [Google Scholar]
- 24.Trebosc V, Gartenmann S, Royet K, Manfredi P, Tötzl M, Schellhorn B, Pieren M, Tigges M, Lociuro S, Sennhenn PC, Gitzinger M, Bumann D, Kemmer C. 2016. A novel genome-editing platform for drug-resistant Acinetobacter baumannii reveals an AdeR-unrelated tigecycline resistance mechanism. Antimicrob Agents Chemother 60:7263–7271. doi: 10.1128/AAC.01275-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mugnier PD, Poirel L, Nordmann P. 2009. Functional analysis of insertion sequence ISAba1, responsible for genomic plasticity of Acinetobacter baumannii. J Bacteriol 191:2414–2418. doi: 10.1128/JB.01258-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun J-R, Perng C-L, Chan M-C, Morita Y, Lin J-C, Su C-M, Wang W-Y, Chang T-Y, Chiueh T-S. 2012. A truncated AdeS kinase protein generated by ISAba1 Insertion correlates with tigecycline resistance in Acinetobacter baumannii. PLoS One 7:e49534. doi: 10.1371/journal.pone.0049534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Z, Ye S, Li J, Zheng B, Bao M, Ning G. 2011. Fusion primer and nested integrated PCR (FPNI-PCR): a new high-efficiency strategy for rapid chromosome walking or flanking sequence cloning. BMC Biotechnol 11:109. doi: 10.1186/1472-6750-11-109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chin C-Y, Gregg KA, Napier BA, Ernst RK, Weiss DS. 2015. A PmrB-regulated deacetylase required for lipid A modification and polymyxin resistance in Acinetobacter baumannii. Antimicrob Agents Chemother 59:7911–7914. doi: 10.1128/AAC.00515-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.García-Quintanilla M, Carretero-Ledesma M, Moreno-Martínez P, Martín-Peña R, Pachón J, McConnell MJ. 2015. Lipopolysaccharide loss produces partial colistin dependence and collateral sensitivity to azithromycin, rifampicin and vancomycin in Acinetobacter baumannii. Int J Antimicrob Agents [DOI] [PubMed] [Google Scholar]
- 30.Moffatt JH, Harper M, Adler B, Nation RL, Li J, Boyce JD. 2011. Insertion sequence ISAba11 is involved in colistin resistance and loss of lipopolysaccharide in Acinetobacter baumannii. Antimicrob Agents Chemother 55:3022–3024. doi: 10.1128/AAC.01732-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anandan A, Evans GL, Condic-Jurkic K, O’Mara ML, John CM, Phillips NJ, Jarvis GA, Wills SS, Stubbs KA, Moraes I, Kahler CM, Vrielink A. 2017. Structure of a lipid A phosphoethanolamine transferase suggests how conformational changes govern substrate binding. Proc Natl Acad Sci U S A 114:2218–2223. doi: 10.1073/pnas.1612927114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kahler CM, Nawrocki KL, Anandan A, Vrielink A, Shafer WM. 2018. Structure-function relationships of the neisserial EptA Enzyme responsible for phosphoethanolamine decoration of lipid A: rationale for drug targeting. Front Microbiol 9:1922. doi: 10.3389/fmicb.2018.01922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clinical and Laboratory Standards Institute. 2012. M07-A9. Methods for dilution antimicrobial susceptibility tests for bacteria that grow aerobically; approved standard—ninth edition. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
- 34.Clinical and Laboratory Standards Institute. 2014. M100-S24: performance standards for antimicrobial susceptibility testing; twenty-fourth informational supplement. Clinical and Laboratory Standards Institute, Wayne, PA. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Agarose gel of eptA-1 and eptA-2 genotyping in the strain panel. The genotyping of eptA-1 (upper gel) and eptA-2 (lower gel) was performed by PCR using primers oVT198/oVT199 and oVT201/oVT202, respectively. Lane 1, 2-log ladder (New England Biolabs); lane 2, ATCC 17978; lane 3, BV94; lane 4, BV95; lane 5, BV172; lane 6, BV173; lane 7, BV174; lane 8, BV175; lane 9, BV185; lane 10, BV186; lane 11, BV187; lane 12, BV189; lane 13, BV190; lane 14, BV191; lane 15, 2-log ladder. Compared to the expected sizes of 1,862 bp and 1,827 bp, the PCR products for BV94 eptA-2 and BV189 eptA-1 genotyping, respectively, are approximately 1 kb larger, corresponding to the presence of ISAbaI. Download FIG S1, PDF file, 0.3 MB (361.9KB, pdf) .
Copyright © 2019 Trebosc et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Oligonucleotides used in this study. Download Table S1, PDF file, 0.2 MB (189.2KB, pdf) .
Copyright © 2019 Trebosc et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.
Agarose gel of eptA genotyping in the clinical isolate BV94 and its eptA knockout mutants. The genotyping of eptA isoforms was performed by PCR using primers oVT152/oVT153. Lane 1, 2-log ladder (New England Biolabs); lane 2, BV94; lane 3, BV94ΔeptA-1; lane 4, BV94ΔeptA-1/ΔeptA-2; lane 5, BV94ΔeptA-1/ΔeptA-2/ΔeptA-3. The triple mutant BV94ΔeptA-1/ΔeptA-2/ΔeptA-3 did not carry any other eptA isoform. Download FIG S2, PDF file, 0.3 MB (339.4KB, pdf) .
Copyright © 2019 Trebosc et al.
This content is distributed under the terms of the Creative Commons Attribution 4.0 International license.