

Abstract
A subset of genetic variants found through screening of patients with hereditary breast and ovarian cancer syndrome (HBOC) and Lynch syndrome impact RNA splicing. Through target enrichment of the transcriptome, it is possible to perform deep‐sequencing and to identify the different and even rare mRNA isoforms. A targeted RNA‐seq approach was used to analyse the naturally‐occurring splicing events for a panel of 8 breast and/or ovarian cancer susceptibility genes (BRCA1, BRCA2, RAD51C, RAD51D, PTEN, STK11, CDH1, TP53), 3 Lynch syndrome genes (MLH1, MSH2, MSH6) and the fanconi anaemia SLX4 gene, in which monoallelic mutations were found in non‐BRCA families. For BRCA1, BRCA2, RAD51C and RAD51D the results were validated by capillary electrophoresis and were compared to a non‐targeted RNA‐seq approach. We also compared splicing events from lymphoblastoid cell‐lines with those from breast and ovarian fimbriae tissues. The potential of targeted RNA‐seq to detect pathogenic changes in RNA‐splicing was validated by the inclusion of samples with previously well characterized BRCA1/2 genetic variants. In our study, we update the catalogue of normal splicing events for BRCA1/2, provide an extensive catalogue of normal RAD51C and RAD51D alternative splicing, and list splicing events found for eight other genes. Additionally, we show that our approach allowed the identification of aberrant splicing events due to the presence of BRCA1/2 genetic variants and distinguished between complete and partial splicing events. In conclusion, targeted‐RNA‐seq can be very useful to classify variants based on their putative pathogenic impact on splicing.
Keywords: targeted RNA‐seq, alternative splicing, inherited breast/ovarian cancer syndrome, lynch syndrome, BRCA1/2
Short abstract
What's new?
Hereditary familial breast/ovarian cancer (HBOC) syndrome involves numerous pathogenic variants, including variants of uncertain clinical significance (VUS). A subset of VUS, however, is suspected to influence RNA splicing, leading to the expression of potentially pathological transcript isoforms. Here, using a targeted RNA‐seq approach, naturally occurring splice isoforms were described for BRCA1/2, RAD51C, RAD51D, and eight additional tumor‐suppressor genes that are associated with HBOC and Lynch syndrome. The targeted RNA‐seq approach also identified aberrant splicing events associated with the presence of BRCA1/2 genetic variants and successfully distinguished complete from incomplete splicing events, which is of major importance in determining pathogenicity.
Introduction
Pathogenic variants in the BRCA1/2 genes account for about 15–20% of the families with hereditary familial breast/ovarian cancer syndrome (HBOC). Recent studies have demonstrated that RAD51C and RAD51D should also be included in the genetic screening of ovarian cancer patients.1, 2, 3, 4, 5 Pathogenic variants in other known genes, such as PTEN, TP53, CDH1, STK11/LKB1, and NBS1 account for less than 10% of the non‐BRCA HBOC families.3, 6, 7, 8, 9 Monoallelic mutations in the Fanconi Anaemia SLX4 (or FANCP) gene were also found in non‐BRCA families, although at an extremely low percentage.10, 11 Cases of Lynch syndrome, account for 2–3% of all colorectal cancers and are associated with germline pathogenic variants in the MLH1, MSH2, MSH6 and PMS2 genes. Women are also at risk of endometrial and ovarian cancers.12
A large percentage of the sequence variants in the BRCA1 and BRCA2 genes that are detected by routine mutation screening are variants of uncertain (clinical) significance (VUS). A subset of VUS may affect splicing by disturbing the recognition of the donor and acceptor splice sites (DSS and ASS, respectively) or by disrupting intronic and exonic cis‐elements necessary for the regulation of splicing.13, 14 The effect of genetic variants in canonical DSS and ASS can be well predicted using in silico tools.15, 16, 17 For intronic or exonic splice enhancer/silencer elements the predictive power remains limited. To confirm or exclude an effect of variants on mRNA splicing experimental in vitro work is needed: usually, RT‐PCR followed by Sanger sequencing. These experiments can be laborious and time‐consuming. In addition, RT‐PCR experiments are usually limited to the region containing the sequence variant of interest and thereby do not assess a putative effect of this variant on the overall splicing architecture of the mRNA. Moreover, these RT‐PCR experiments often lead to the detection of non‐canonical mRNA isoforms present in both HBOC patients and healthy controls.18 Genetic variants may lead to significantly changed expression of these transcripts’ isoforms and, when there is loss of the reference transcript, may be pathogenic. Known exceptions to this situation are in‐frame deletion/insertion splicing events which lead to protein isoforms that retain tumour suppressor function, such as BRCA1Δ9,1019 or BRCA2Δ12.20 When designing splicing assays, it is important to take all isoforms into account to either: a) consider them when analysing expression levels of the reference transcript, or b) target them more specifically to measure isoform‐specific expression level changes. The ENIGMA consortium of investigators has recently published a comprehensive list of all naturally‐occurring BRCA1/2 isoforms found by RT‐PCR/capillary analysis.21, 22 Such an extensive analysis remains lacking for many other human genes.
Nowadays, with the aid of RNA‐seq, it is possible to analyse transcription events at an unprecedented depth.23, 24 Through target‐enrichment of a subset of the transcriptome, the different and even rare mRNA isoforms can be detected.25, 26, 27 Thousands of new isoforms and low abundant transcripts have been identified using this approach. Therefore, we sought to analyse in depth the naturally‐occurring splicing events for a panel of tumour suppressor genes that are associated with HBOC and Lynch syndrome. We initially validated the targeted RNA‐seq approach using previously published data for BRCA1 and BRCA2.21, 22 Then a detailed analysis of RAD51C and RAD51D transcription was performed to obtain an extensivelist of naturally‐occurring isoforms. These results were validated by capillary electrophoresis (CE) and compared to a (non‐targeted) RNA‐seq approach. Splicing events from lymphoblastoid cell‐lines were compared to those from breast, ovarian, and ovarian fimbria tissues. Furthermore, we also assessed the diagnostic potential of targeted RNA‐seq to detect pathogenic changes in RNA‐splicing by the inclusion of samples with known effects on BRCA1/2‐splicing.
Material and Methods
Bait design
We selected 12 genes, spanning a total region of 688,440 bp (Supporting Information Table S1). Double tiling SureSelect baits (Agilent Technologies) were custom designed by Agilent for the regions of interest using two approaches: a) covering the known transcripts (which allows selecting baits for annotated splicing events from Agilent own data) and b) covering the genomic region (including introns and 1 Kb upstream and downstream). Duplicate baits were removed. A list of all baits is available on request.
Cell cultures
We used lymphoblastoid cell‐lines (LCLs) from 2 BRCA1‐ and 2 BRCA2‐mutation carriers (BRCA1:c.5467+5G>C, BRCA1:[c.594‐2A+c.641A>G], BRCA2:c.8632+1G>A, BRCA2:c.9501+3A>T) previously generated by the Kathleen Cuningham Consortium for Research into Familial Breast Cancer (kConFab) as described elsewhere.18 Culture conditions and RNA isolation are described in the Supporting Information methods.
Library preparation
SureSelect RNA Target Enrichment for Illumina Paired‐End Multiplexed Sequencing kit (Agilent; protocol version 2.2.1) was used. Briefly, 200 ng of mRNA were chemically fragmented and double‐stranded cDNA was synthesized. After end‐repair and dA‐ligation to the 3′‐end of the cDNA fragments, paired‐end adaptors were ligated. cDNA of about 250 bp was isolated with two rounds of clean‐up with SPRI beads (AMPure XP, Agencourt) according to instructions. After amplifying the cDNA library for 11 cycles, the quality and quantity of each sample were determined with the 2100 Bioanalyzer (Agilent Technologies) and the Qubit 1.27 (Invitrogen), respectively. The prepared libraries were hybridized with the custom‐designed SureSelect Oligo Capture library during 24 h at 65 °C. An amplification step of 12 cycles was used to add index tags. The quantity and quality of the samples were assessed as described above. The index‐tagged sample libraries were pooled to an equimolar (4 nM) amount. 20pM were subject to cluster amplification and sequenced on a HiSeq2000 instrument (Illumina) using the TruSeq SBS kit‐HS (2x100 cycles) on a single lane.
Read alignment
The STAR aligner (Version 2.4.1d) was used to map read pairs to H. sapiens reference genome HS.GRCh37 (iGenomes).28 The only set parameter for index construction was ‐‐sjdbOverhang 92. Explicitly adjusted parameters used in STAR include ‐‐outFilterMultimapNmax 2, −‐outFilterMismatchNmax 20 and ‐‐chimSegmentMin 0. Duplicate read pairs removal was performed with Picard tools (https://github.com/broadinstitute/picard). Alignment of the raw reads to specific events is described in the Supporting Information methods. Start and end positions from STAR output refer to the first nucleotide in the intron (AG|gu) and last nucleotide of the intron (ag|G), respectively.
Non‐targeted RNA‐seq library preparation and mapping
Described in the Supporting Information methods.
Nomenclature
The description of genetic variants follows the Human Genetic Variation Society (HGVS) approved guidelines,29 where c.1 (and r.1) is the A of the ATG translation initiation codon. Alternative splicing events are those incorporating splice junctions not present in the reference transcripts (BRCA1: NM_007294, lacking exon 4 as initially described,30 BRCA2: NM_000059, RAD51C: NM_058216, RAD51D: NM_002878). Splicing events in other genes were not annotated. The Supporting Information data provided shows the genomic positions (HS.GRCh37) of the splicing events detected. We described splicing events using the after symbols: Δ (skipping),▾(insertion), p (acceptor shift) and q (donor shift); see Supporting Information Figure S1. In case there is a new cassette exon we add a letter after the intron number, and we use A, B or C for the different cassette insertion events. For example, if, between exons 2 and 3 of the reference transcript, 2 cassette insertion events occur, these would be ▾2A and ▾2B. The letter designation was the same when events shared the acceptor splice site. A sub‐index (skipping) or a super‐index (insertion) indicates the number of nucleotides involved in the alternative event.
Capillary electrophoresis
Capillary electrophoresis (CE) was conducted for RAD51C and RAD51D as previously reported.21, 22 CE analyses were performed in cDNAs obtained from control Lymphoblastoid cell‐lines (LCLs) generated by kConFab, Tempus‐stabilized (Thermofisher) peripheral blood RNA from healthy control individuals, commercially available RNA from a non‐malignant breast tissue (Clontech 636,576), and commercially available RNA from a pool of non‐malignant ovarian tissues (Clontech 636,555). cDNA was amplified with various combinations of forward and FAM‐labeled reverse primers spanning the full sequence of the reference transcripts (sequences are available upon request) and products were visualized with CE. In some cases, splicing isoforms were verified by automated Sanger sequencing.
Quantitative evaluation of the targeted‐enrichment RNA‐seq
Samples with known splicing events in BRCA1 (c.5467+5G>C and c.[594‐2A>G; 641A>G]) and BRCA2 (c.8632+1G>A and c.9501+3A>T) were used for evaluation.18 The use of targeted RNA‐seq to detect pathogenic changes in splicing was assessed taking into account the after: 1) detection of increased expression of splicing events in one sample compared to the other samples; 2) distinction of partial splicing events (variant allele still expresses the reference transcript) and complete splicing events (no residual expression of the reference transcript from the variant allele). The in‐house developed QURNAS‐tool (unpublished data), available at https://hdl.handle.net/10441/LY8ZQ4. A brief description of the tool is described in Supporting Information methods. Analysis of the read counts for reference exon‐exon junctions, as described in the Supporting Information methods, was used to determine the expression of the reference transcript.
Results
In total, 425,665,943 reads were obtained for the 4 LCL samples. From these, 19% non‐duplicate read pairs were uniquely mapped on the reference genome and about 7% of these were mapped onto the genes of interest (Supporting Information Table S2).
Performance test
Initially, we established whether the read depth of the RNA‐seq experiment was sufficient to achieve our objective to obtain an extensivelist of splicing events for a given gene. Therefore, we compared the list of detected BRCA1/2 splicing events in our RNA‐seq data to previously published naturally occurring events (Supporting Information Tables S3 and S4).21, 22 Supporting Information Figures S2 and S3 depict the splice junctions that were identified in BRCA1 and BRCA2, as well as their relative expression. Compared to previously identified/reported BRCA1 splicing events,21, 22 we detected 63 out of 67 events (94%), missing 3 multicassette and 1 mixed biotype event (Supporting Information Table S3). For BRCA2, we were initially able to detect 34 out of 36 known splicing events (Supporting Information Table S4),21, 22 missing the identification of a cassette and one mixed biotype event. Overall our method allowed to detect more known events than a previous targeted RNA‐seq study.31 Nevertheless, we did not find 2 BRCA1 and 1 BRCA2 junctions described in that study. So, we aligned the raw reads to the already known events not found by the STAR aligner and visually inspected the outcome. The BRCA1 events described by ENIGMA were indeed not present in our samples, but new events from Davy et al., ins 2A (donor splice site) and Δ15q were identified in all 4 samples with 282 and 144 reads, respectively. Also, the three BRCA2 remaining splicing events were detected. BRCA2Δ6q89,7 contains only 2 nucleotides of exon 6, which likely caused problems for the STAR aligner. BRCA2Δ18 was found in 3 out of 4 samples (138 reads). Intron 17 contains a rare GC donor splice site,32 but we were able to detect the normal 17–18 exon‐exon junction, as well as the 18–19 junction, and other GC‐donor splice sites. The donor site of exon 20C (previously described as 20B31) was also detected with 40 reads. It is unclear why the STAR aligner did not detect these events in our data.
In addition to the previously described events, a high number of new events was detected. This created the need to set a threshold: splice junctions must be present in at least one sample with a minimum of 25 reads, independently of the number of samples in which they were observed. Using this criterion, over 20 new events were found for each gene (not described in Gencode, Ensembl, or published21, 22, 31), as described in Supporting Information Tables S3 and S4. Since CE was shown to be very sensitive for characterization and relative semi‐quantitative analysis of splicing events,21, 22 we reanalysed some of the unresolved CE peaks from the previous studies, taking into account the targeted RNA‐seq data. PCR products with sizes consistent with some of the newly identified events could indeed be identified. More specifically, we observed CE evidence of 6 BRCA1 and 5 BRCA2 events not previously described. Most events that were not confirmed by CE were large retention/insertion events, which give technical limitations for CE. Moreover, events in the 3′ and 5′ ends of the genes could not be tested with CE or other PCR‐based methods.
RAD51C and RAD51D splicing events
Once we established that our RNA‐seq experiment performed well for BRCA1/2 genes, we analysed the data for RAD51C and RAD51D genes, using the above‐mentioned threshold (at least one sample with a minimum of 25 reads). Splicing events and their relative expression are depicted in Supporting Information Figures S4 and S5. We detected 46 and 36 alternative splicing events (Tables 1 and 2) with expression levels ranging from 0.02–6% to 0.05–61% of the reference RAD51C and RAD51D exon‐exon junctions, respectively (see Supporting Information Methods for details on the estimation strategy). Of the alternative splicing events, 14 and 11 events detected in RAD51C and RAD51D, respectively, were not previously described in Ensembl, Gencode or Davy et al.31 It is noteworthy that in 3 of 4 samples a frameshift isoform of RAD51D lacking exon 3 was more abundant than the reference transcript (isoform 1) and the isoform containing a downstream alternative exon 3 (Supporting Information Fig. S5).
Table 1.
Events detected by STAR in RAD51C with RNA‐seq compared to the reference sequence NM_058216
| Genomic coordinates start and end1 | Event description | HGVS nomenclature | Biotype | Functional Annotation2 | Read counts | CE3 | Davy et al.3 | G/E4 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Targeted‐LCLs5 | LCLs | breast | fimbria | |||||||||
| 56,769,994 | 56,772,291 | Δ1q156 | r.‐11_145del | donor shift | Non‐coding | 1,172 | 28 | 12 | 6 | Y | Y | GE |
| 56,769,994 | 56,770,711 | Δ1q156+▾1A | r.‐11_145del+r.145_146ins145+563_145+862 | mixed | PTC‐NMD | 62 | 0 | 0 | 2 | Y | N | G |
| 56,770,047 | 56,772,291 | Δ1q103 | r.43_145del | donor shift | PTC‐NMD | 3,777 | 80 | 82 | 26 | Y | Y | GE |
| 56,770,047 | 56,770,711 | Δ1q103+▾1A | r.43_145del+r.145_146ins145+563_145+862 | mixed | PTC‐NMD | 125 | 4 | 1 | 0 | Y | Y | G |
| 56,770,047 | 56,772,294 | Δ1q103_Δ2p3 | r.43_148del | mixed | PTC‐NMD | 31 | 2 | 1 | 0 | Y | Y | |
| 56,770,047 | 56,774,053 | Δ1q103_Δ2 | r.43_404del | mixed | PTC‐NMD | 40 | 0 | 0 | 0 | N | N | |
| 56,770,321 | 56,772,291 | ▾1q171 | r.145_146ins145+1_145+171 | donor shift | PTC‐NMD | 272 | 7 | 1 | 0 | Y | Y | |
| 56,770,480 | 56,772,291 | ▾1q330 | r.145_146ins145+1_145+330 | donor shift | PTC‐NMD | 59 | 0 | 0 | 0 | N | N | G |
| 56,770,150 | 56,770,711 | ▾1Aa300 | r.145_146ins145+563_145+862 | cassette | PTC‐NMD | 401/571 | 23/20 | 6/3 | 0 | Y | Y | GE |
| 56,771,173 | 56,772,291 | ▾1Ab461 | r.145_146ins145+563_145+1,023 | cassette | PTC‐NMD | 401/723 | 23/21 | 6/10 | 0 | Y | Y | GE |
| 56,770,150 | 56,772,294 | Δ2p3 | r.146_148del | acceptor shift | No FS | 187 | 1 | 0 | 0 | Y | Y | |
| 56,770,150 | 56,774,053 | Δ2 | r.146_404del | cassette | PTC‐NMD | 788 | 0 | 2 | 0 | Y | Y | GE |
| 56,772,376 | 56,774,053 | Δ2q175 | r.230_404del | donor shift | PTC‐NMD | 19 | 0 | 0 | 0 | Y | N | |
| 56,772,529 | 56,774,053 | Δ2q22 | r.383_404del | donor shift | PTC‐NMD | 24 | 0 | 0 | 0 | Y | N | |
| 56,772,551 | 56,780,556 | Δ3 | r.405_571del | cassette | PTC‐NMD | 372 | 63 | 2 | 0 | Y | Y | GE |
| 56,772,551 | 56,787,219 | Δ3,4 | r.405_705del | multicassette | PTC‐NMD | 67 | 0 | 0 | 0 | N | N | |
| 56,772,578 | 56,774,053 | ▾2q27 | r.404_405ins404+1_404+27 | donor shift | PTC‐NMD | 553 | 9 | 0 | 0 | Y | Y | |
| 56,774,221 | 56,776,553 | ▾3A (alternative 3′ end) | r.571_572ins571+2334_571+3,395+r.572_1131del | Terminal modification | intronic STOP+polyA | 15/− | 0/− | 0/− | 0/− | ‐ | N | |
| 56,774,221 | 56,777,235 | ▾3B (alternative 3′ end) | r.571_572ins571+3016_571+3,394+r.572_1131del | Terminal modification | intronic STOP+polyA | 310/− | 18/− | 6/− | 0/− | ‐ | Y | GE |
| 56,774,221 | 56,780,637 | Δ4p81 | r.572_652del | acceptor shift | No FS | 178 | 5 | 1 | 0 | Y | Y | |
| 56,774,221 | 56,780,667 | Δ4p111 | r.572_682del | acceptor shift | No FS | 32 | 1 | 0 | 0 | N | N | |
| 56,774,221 | 56,787,219 | Δ4 | r.572_705del | cassette | PTC‐NMD | 444 | 10 | 0 | 0 | Y | Y | |
| 56,774,221 | 56,798,106 | Δ4,5 | r.572_837del | multicassette | PTC‐NMD | 17 | 0 | 0 | 0 | Y | N | |
| 56,774,221 | 56,783,238 | Δ4,▾4A145 | r.572_705del+r.705_706ins705+2549_705+2,693 | mixed | PTC‐NMD | 17 | 2 | 0 | 0 | Y | Y | |
| 56,780,691 | 56,783,238 | ▾4A145 | r.705_706ins705+2549_705+2,693 | cassette | PTC‐NMD | 4144/4016 | 233/180 | 8/0 | 12/0 | Y | Y | GE |
| 56,780,691 | 56,783,849 | ▾4B120 | r.705_706ins705+3160_706–3,251 | cassette | PTC‐NMD | 456/332 | 28/24 | 1/1 | 0/0 | Y | Y | GE |
| 56,780,691 | 56,785,151 | ▾4C48 | r.705_706ins706‐2068_706–2021 | cassette | No FS | 25/34 | 1/1 | 0/0 | 0/0 | Y | N | |
| 56,780,691 | 56,798,106 | Δ5 | r.706_837del | cassette | No FS | 38 | 0 | 0 | 0 | Y | Y | |
| 56,787,352 | 56,787,956 | ▾5A57 | r.837_838ins837+606_837+662 | cassette | PTC‐NMD | 161/54 | 22/10 | 0/0 | 0/0 | Y | N | |
| 56,787,352 | 56,791,366 | ▾5B75 | r.837_838ins837+4,016_837+4,089 | cassette | No FS | 36/4 | 0/0 | 0/0 | 0/0 | Y | N | |
| 56,787,352 | 56,797,876 | ▾5C100 | r.837_838ins838‐230_838–131 | cassette | PTC‐NMD | 881/327 | 29/24 | 5/1 | 0/2 | Y | Y | |
| 56,787,352 | 56,797,928 | ▾5D48 | r.837_838ins838‐178_838–131 | cassette | PTC‐NMD | 199/327 | 0/24 | 0/1 | 0/2 | Y | N | |
| 56,787,352 | 56,801,400 | Δ6 | r.838_904del | cassette | PTC‐NMD | 200 | 6 | 0 | 0 | Y | Y | GE |
| 56,787,352 | 56,809,844 | Δ6,7 | r.838_965del | multicassette | PTC‐NMD | 397 | 1 | 0 | 0 | Y | Y | |
| 56,787,352 | 56,809,841 | Δ6,7+▾8p3 | r.838_965del+r.965_966ins966‐3_966–1 | mixed | PTC‐NMD | 153 | 0 | 2 | 0 | Y | Y | |
| 56,787,352 | 56,811,478 | Δ6_8 | r.838_1026del | multicassette | No FS | 81 | 3 | 0 | 0 | Y | Y | |
| 56,798,174 | 56,801,378 | ▾7p22 | r.904_905ins905‐22_905–1 | acceptor shift | PTC‐NMD | 39 | 0 | 0 | 0 | Y | N | |
| 56,798,174 | 56,809,844 | Δ7 | r.905_965del | cassette | PTC‐NMD | 3,382 | 48 | 7 | 2 | Y | Y | GE |
| 56,798,174 | 56,809,841 | Δ7+▾8p3 | r.905_r.965del+r.965_966ins966‐3_966–1 | mixed | PTC‐NMD | 227 | 1 | 0 | 0 | Y | Y | G |
| 56,798,174 | 56,811,478 | Δ7,8 | r.905_1026del | multicassette | PTC‐NMD | 677 | 5 | 1 | 0 | Y | Y | GE |
| 56,801,462 | 56,803,052 | ▾7A72 | r.965_966ins965+1592_965+1,663 | cassette | PTC‐NMD | 46/39 | 23/13 | 0/0 | 0/0 | Y | N | |
| 56,801,462 | 56,807,546 | ▾7B122 | r.965_966ins966‐2298_966–2,177 | cassette | PTC‐NMD | 612/1091 | 18/23 | 3/2 | 2/0 | Y | Y | G |
| 56,807,669 | 56,809,841 | ▾7B122+▾8p3 | r.965_966ins966‐2298_966–2,177+r.965_966ins966‐3_966–1 | mixed | PTC‐NMD | 48 | 0 | 0 | 2 | Y | Y | |
| 56,801,462 | 56,809,841 | ▾8p3 | r.965_966ins966‐3_966–1 | acceptor shift | No FS | 3,381 | 84 | 14 | 10 | Y | Y | GE |
| 56,801,462 | 56,811,478 | Δ8 | r.966_1026 | cassette | PTC‐NMD | 49 | 3 | 0 | 0 | Y | Y | |
| 56,809,906 | 56,811,484 | Δ9p6 | r.1027_1032del | acceptor shift | No FS | 21 | 1 | 0 | 0 | Y | N | |
Combination of individual splicing events was inferred from CE‐data.
Genomic coordinates on chr 17, human genome built GRCh37.
PTC‐NMD, premature‐stop codon‐ nonsense mRNA‐mediated decay; FS, frameshift.
Y, event was found; N, event was not found. ‐: not tested.
Events described in Gencode or Ensemble are shown with a G or E, respectively. GE is used if an event is described in both databases.
Read counts are shown as the average or read counts in the 4 samples. 2 numbers are shown for inserted exons.
Table 2.
Events detected by STAR in RAD51D with RNA‐seq compared to reference NM_002878. Combination of individual splicing events was inferred from CE‐data
| Genomic coordinates start and end1 | Description | HGVS nomenclature | Biotype | Functional Annotation2 | Read counts | CE3 | Davy et al.3 | G/E4 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Targeted‐LCLs5 | LCLs | breast | fimbria | |||||||||
| 33,448,761 | 33,455,841 | Δ5′‐gen‐5’‐UTR | r.‐2,256 _‐2127del | terminal modification | unknown | 27 | 0 | 0 | 0 | ‐ | N | |
| 33,448,756 | 33,455,841 | Δ5’‐gen‐5’‐UTR | r.‐2,256 _‐2124del | terminal modification | unknown | 9 | 0 | 0 | 0 | ‐ | N | |
| 33,446,192 | 33,448,309 | Δ5’_1 | r.‐1678_82del | Terminal modification+ cassette | Non‐Coding | 25 | 0 | 0 | 0 | ‐ | N | GE |
| 33,434,467 | 33,448,309 | Δ5’_3 | r.‐1678_263del | Terminal modification+ multicassette | Non‐Coding | 12 | 0 | 0 | 0 | ‐ | N | G |
| 33,433,501 | 33,448,309 | Δ5’_5 | r.‐1678_480del | Terminal modification+ multicassette | Non‐Coding | 10 | 0 | 0 | 0 | ‐ | N | GE |
| 33,446,192 | 33,447,065 | Δ1q515_1 | r.‐433_82del | Terminal modification+ cassette | Non‐Coding | 23 | 0 | 2 | 0 | ‐ | N | GE |
| 33,446,192 | 33,446,718 | Δ1q168_1 | r.‐86_82del | Terminal modification+ cassette | Non‐Coding | 68 | 3 | 0 | 0 | ‐ | Y | GE |
| 33,434,467 | 33,446,550 | Δ2,3 | r.83_263del | multicassette | No Fs | 70 | 1 | 0 | 0 | Y | N | |
| 33,433,501 | 33,446,550 | Δ2_5 | r.83_480del | multicassette | PTC‐NMD | 41 | 1 | 0 | 0 | Y | N | |
| 33,434,467 | 33,446,129 | Δ3 | r.145_263del | cassette | PTC‐NMD | 11,531 | 166 | 14 | 2 | Y | Y | GE |
| 33,444,057 | 33,446,129 | Δ3+▾3A179 | r.145_263del+r.263_264ins263+1464_263+1,642 | cassette+cassette | No Fs | 704 | 8 | 4 | 0 | Y | Y | GE |
| 33,443,812 | 33,446,129 | Δ3+ ▾3B98 | r.145_263del+r.263_264ins263+1709_263+1806 | cassette+cassette | PTC‐NMD | 12 | 0 | 0 | 0 | Y | N | |
| 33,434,142 | 33,446,129 | Δ3,4 | r.145_345del | multicassette | No Fs | 191 | 4 | 1 | 0 | Y | Y | |
| 33,433,501 | 33,446,129 | Δ3_5 | r.145_480del | multicassette | No Fs | 7,372 | 54 | 46 | 10 | Y | Y | GE |
| 33,433,497 | 33,446,129 | Δ3_6p4 | r.145_484del | multicassette+ aceptor shift | PTC‐NMD | 23 | 1 | 0 | 0 | Y | N | |
| 33,430,564 | 33,446,129 | Δ3_6 | r.145_576del | multicassette | No Fs | 55 | 2 | 1 | 0 | Y | Y | |
| 33,444,057 | 33,445,519 | ▾3A179 | r.263_264ins263+1464_263+1,642 | cassette | PTC‐NMD | 788/1795 | 52/44 | 7/5 | 1/0 | Y | Y | GE |
| 33,443,812 | 33,445,519 | ▾3B98 | r.263_264ins263+1709_263+1806 | cassette | PTC‐NMD | 14/62 | 11/2 | 0/0 | 0/0 | N | Y | |
| 33,434,142 | 33,443,877 | ▾3A179+Δ4 | r.263_264ins263+1464_263+1,642+264_345del | mixed | PTC‐NMD | 20 | 0 | 0 | 0 | N | N | G |
| 33,433,501 | 33,443,877 | ▾3A179+Δ4,5 | r.263_264ins263+1464_263+1,642+r.264_480del | mixed | PTC‐NMD | 120 | 7 | 6 | 0 | Y | Y | GE |
| 33,434,142 | 33,445,519 | Δ4 | r.264_345del | cassette | PTC‐NMD | 87 | 5 | 1 | 0 | Y | N | GE |
| 33,433,501 | 33,445,519 | Δ4,5 | r.264_480del | multicassette | PTC‐NMD | 428 | 27 | 1 | 1 | Y | Y | GE |
| 33,434,142 | 33,434,401 | Δ4q17 | r.329_345del | donor shift | PTC‐NMD | 33 | 3 | 0 | 0 | Y | N | |
| 33,434,081 | 33,434,384 | Δ5p61 | r.346_406del | acceptor shift | PTC‐NMD | 104 | 11 | 1 | 0 | Y | Y | GE |
| 33,433,501 | 33,434,384 | Δ5 | r.346_480del | cassette | No Fs | 1,651 | 28 | 20 | 5 | Y | Y | GE |
| 33,433,497 | 33,434,006 | Δ6p4 | r.481_484del | acceptor shift | PTC‐NMD | 81 | 0 | 0 | 0 | Y | Y | |
| 33,433,310 | 33,433,404 | ▾6A163 | r.576_577ins576+96_576+258 | cassette | PTC‐NMD | 68/1278 | 0/54 | 2/2 | 0/0 | Y | N | |
| 33,433,269 | 33,433,404 | ▾6B122 | r.576_577ins576+137_576+258 | cassette | PTC‐NMD | 1220/1278 | 45/54 | 5/2 | 0/0 | Y | Y | GE |
| 33,433,202 | 33,433,404 | ▾6C55 | r.576_577ins576+204_576+258 | cassette | PTC‐NMD | 89/1278 | 1/54 | 0/2 | 0/0 | Y | N | |
| 33,428,385 | 33,433,404 | Δ7,8 | r.577_738del | multicassette | No Fs | 99 | 2 | 1 | 0 | Y | Y | |
| 33,428,385 | 33,429,523 | ▾8Aa121 | r.738_739ins738+629_738+749 | cassette | PTC‐NMD | 5/12 | 0/1 | 0/0 | 0/0 | N | N | |
| 33,428,385 | 33,429,523 | ▾8B117 | r.738_739ins738+633_738+749 | cassette | No Fs | 3/12 | 0/1 | 0/0 | 0/0 | Y | N | |
| 33,428,401 | 33,430,272 | ▾9p16 | r.738_739ins739‐16_739–1 | acceptor shift | PTC‐NMD | 31 | 0 | 0 | 0 | N | Y | |
| 33,428,049 | 33,428,219 | Δ10p7 | r.904_910del | acceptor shift | FS‐alternative stop | 108 | 1 | 0 | 0 | Y | Y | |
| 33,427,911 | 33,428,219 | Δ10p145 | r.904_*61del | acceptor shift | FS‐alternative stop | 89 | 0 | 4 | 0 | N | Y | |
| 33,353,581 | 33,428,219 | Δ10 | r.904_*3161del | Terminal modification | To be defined | 139 | 0 | 0 | 0 | ‐ | N | G |
Genomic coordinates on chr 17, human genome built GRCh37.
PTC‐NMD, premature‐stop codon‐ nonsense mRNA‐mediated decay; FS, frameshift.
Y, event was found; N, event was not found. ‐: not tested.
Events described in Gencode or Ensemble are shown with a G or E, respectively. GE is used if an event is described in both databases.
Read counts are shown as the average or read counts in the 4 samples. 2 numbers are shown for inserted exons.
CE was used both as a confirmation of the RNA‐seq results and to help solve intricate events. Analysis with CE enables, at least to some extent, the identification of co‐occurring events, which is not directly possible using solely RNA‐seq data for events that are not captured in one read. For example, alternative cassette exons which result from the combination of two splicing events can be imputed from the exact CE‐sizing data. Of the new splicing events, 23/27 RAD51C (88%, 1 event was not tested) and 13/20 RAD51D (76%, 3 events not tested) were confirmed by CE. Events not evaluated are located either at the 5′ and 3′ ends of the transcripts hindering an efficient, sensitive PCR‐reaction. Furthermore, CE and PCR followed by sequencing also allowed identification of combinations of multicassette exons that are not adjacent to each other in each gene: RAD51CΔ1q103+Δ3, RAD51DΔ3+▾3A179+Δ4,5 and RAD51DΔ3+▾3A179+Δ4_6 (Δ4_6 was found below the threshold). Possibly, other event combinations exist, but they were not extensively tested.
Some CE peaks were difficult to be associated with splicing events. Four inferred events initially only found by CE were tested by mapping the raw data to them. RAD51C▾5D33 was found to be present in all 4 samples with 199 reads on average. Others are either not present in our samples or we missed the prediction of the event.
We also compared our initial results with non‐targeted RNA‐seq data from an immortalized lymphocyte cell line, and normal breast and fimbria tissues (Tables 1, 2, 3). It is important to note that the average number of reads for the reference exon‐exon junctions of RAD51C and RAD51D varied among the different experiments. In the targeted RNA‐seq on LCLs we obtained an average of 18,868 reads [9389‐33,956], whereas it was 347 [118–484] for non‐targeted LCLs, 134 [44–226] for normal breast tissue and 10 [2‐16] for normal fimbria tissue. This, together with the fact that some events detected by targeted RNA‐seq were also found in breast or ovarian tissue by CE (data not shown), indicates that the lower number of splice isoforms found in the normal breast and fimbria tissue is not related to tissue‐specific transcription regulation, but due to lack of coverage in non‐targeted RNA‐seq experiments.
Table 3.
Number of individual splicing events detected by STAR for RAD51C and RAD51D per tissue type and sequencing approach
| Targeted RNA‐seq LCLs | Non‐targeted RNA‐seq | |||
|---|---|---|---|---|
| LCLs | normal breast | normal fimbria | ||
| RAD51C | ||||
| ≥ 25 reads1 | 55 | 39 | 23 | 9 |
| < 25 reads2 | 13 | 10 | 1 | 0 |
| Not in the targeted3 | 5 | 1 | 0 | |
| RAD51D | ||||
| ≥ 25 reads1 | 40 | 25 | 18 | 5 |
| < 25 reads2 | 13 | 6 | 2 | 0 |
| Not in the targeted3 | 0 | 1 | 0 | |
There is a difference between the number of events described here and those shown in Tables 1 and 2, because here we count all separate splicing events as listed in the STAR output, whereas in the previous tables part of the separate splicing events were combined, e.g., to describe a cassette insertion, as imputed from CE data.
The events in the targeted RNA‐seq are used as reference.
Due to the large amount of data, only events that are found by other method/tissue are taken into account.
Events that are completely absent in the targeted RNA‐seq data, but detected in non‐targeted RNA‐seq.
Interestingly, despite the lower coverage, RAD51CΔ8,9+▾10 and RAD51DΔ3,4+▾5p182 events were only observed in breast tissue by the non‐targeted approach (Table 3). Additional 5 RAD51C events (▾1A351; ▾1A461+▾2p28; ▾5A57+ ▾5C100; ▾5A57+▾5D48; ▾931) were only observed in non‐targeted RNA‐seq of LCLs. None of these 7 events was observed after specific alignment of the raw targeted RNA‐seq data for blood cells. These can be tissue‐specific isoforms and/or a reflection of interindividual variability (events that are not present in one or more individuals). In CE tests, which were performed for multiple samples (average of 8 [2‐32] samples), interindividual variability was observed for 54% of the splicing events. One particular event was only present in 16% of the samples. Interindividual variability was also observed among our 4 samples with targeted RNA‐seq, although this was Amostly observed for lower expressed events. Yet, only the RAD51D:r.‐2256_‐2124del was observed in one single sample. It is noteworthy that also among the splicing events in other genes, the events observed in a single sample are a minority, i.e. 2 for CDH1 and 1 for MLH1.
DSS and ASS that gave rise to the new events detected by targeted RNA‐seq in RAD51C and RAD51D were tested for in silico prediction (data not shown). Most events used a combination of previously known splice sites. Two new splice sites were predicted with scores >80% by different in silico tools present in Alamut Visual 2.8 (Interactive Biosoftware, Rouen, France). One junction used a non‐canonical GC donor splice site. In Alamut, only the Human Splicing Finder (HSF) tool generates scores for GC‐donor‐sites, although these sites are known to be as strong as the canonical GT splice donor site since they are also processed by the standard U2‐type spliceosome. For the non‐canonical splice site RAD51C:c.705+2693, HSF indicated an 83.2% chance of being a splice GC‐donor site. Overall, the good correlation with splice‐prediction scores indicates that the events observed, but not necessarily confirmed by CE, are true events rather than artefacts.
Our next step was to evaluate whether there could be in‐frame skipping events in the additional 8 genes tested that could potentially rescue the protein function. This type of information proved to be crucial to explain the non‐pathogenic effect of BRCA1Δ9,1019 and BRCA2Δ12.20 However, no high‐expressed in‐frame events (compared to reference junctions) were detected and practically all exons seem to be relevant for protein function based on protein domains (UniProtKB, InterPro and Nextprot databases). We cannot exclude that combinations of frameshift events could result in in‐frame transcripts, but the function might still be compromised. For a summary of the findings and the list of splicing events see the Supporting Information results and tables.
Quantitative analysis
We also sought to investigate whether targeted‐enriched RNA‐seq could be used in a clinical diagnostic setting, i.e. to find clinically relevant aberrations in splicing caused by genetic variants in individual samples. For this reason, samples with previously well‐characterized splicing events in either one of the BRCA1/2 genes were used. To identify putative pathogenic splicing events in RNA‐seq data, it is important to be able to: 1) detect de novo or increased expression of splicing events in one sample compared to other samples using QURNAS (unpublished data); 2) know if the expression of reference transcript is decreased, by inferring loss of the reference exon‐exon junctions. The latter will give an indication about partial or complete aberrant splicing events. In general, for tumour suppressor genes like BRCA1/2, complete splicing, which is characterized by the absence of reference transcript expression from the variant allele, is more likely to be pathogenic.34, 35 Table 4 and Figure 1 show our results. In brief, these are in agreement with previous results obtained with conventional RT‐PCR.
Table 4.
Splicing events occurring in BRCA1/2 due to genetic variants and respective number of reads for each sample and enrichment scores calculated by QURNAS
| Sample nr | Mutation (rs number) | Description | Reads sample 1 | Reads sample 2 | Reads sample 3 | Reads sample 4 | Enrichment score1 | Previously reported as pathogenic?2 [refs] |
|---|---|---|---|---|---|---|---|---|
| 1 | BRCA1:c.5467+5G>C | Δ23 | 8,798 | 92 | 89 | 50 | 25 | No 36 |
| (rs397509287) | Δ22,23 | 97 | 25 | 25 | 17 | 0.3 | Uncertain 37 | |
| Δ22 | 149 | 381 | 430 | 324 | 0.2 | Yes 38 | ||
| Δ21 | 120 | 65 | 111 | 41 | 0.5 | |||
| 2 | BRCA1:c.594‐2A>C+c.641A>G | Δ10 | 83 | 3,689 | 134 | 76 | 9 | No 19 |
| Δ9,10 | 5,349 | 13,973 | 8,300 | 3,927 | 2 | |||
| (rs80358033 + | Δ9 | 93 | 117 | 197 | 63 | 0.3 | ||
| rs55680408) | ins21bp3 | 72 | 183 | 112 | 79 | 0.4 | ||
| Δ9,10,11 | 19 | 90 | 41 | 33 | 0.3 | |||
| Δ10,11 | 28 | 77 | 5 | 9 | 0.2 | |||
| Δ11q4 | 2,192 | 3,931 | 3,749 | 1,568 | 1.0 | |||
| 3 | BRCA2:c.8632+1G>A | Δ20 | 14 | 22 | 3,072 | 20 | 5 | Yes 39 |
| Δ19 | 347 | 360 | 324 | 630 | 0.4 | |||
| (rs397507997) | Δ20, ins64bp5 | 2 | 0 | 747 | 0 | 1.4 | ||
| Δ19,20 | 0 | 0 | 434 | 7 | 1.3 | |||
| ret17bp4 | 0 | 0 | 253 | 0 | 1.2 | |||
| ret17bp,ins64bp5 | 0 | 0 | 52 | 0 | N.A. | |||
| Δ19,20,ins64bp5 | 0 | 0 | 57 | 0 | 0.17 | |||
| Δ20,ins93bp5 | 0 | 0 | 51 | 0 | 0.09 | |||
| ins64bp | 1,284 | 1,249 | 441 | 1,192 | 0.3 | |||
| 1941 | 2,417 | 1,563 | 1903 | N.A. | ||||
| ins93bp5 | 187 | 270 | 71 | 173 | 0.17 | |||
| 1941 | 2,417 | 1,563 | 1903 | N.A. | ||||
| 4 | BRCA2:c.9501+3A>T (rs61757642) | Δ25 | 0 | 7 | 2 | 1923 | 10 | No 36, 40, 41 |
The reads for the mutation‐carrier and high enrichment scores are highlighted in bold.
The enrichment score shown is for the carrier of the mutation described in the second column.
Yes—the variant was previously described as pathogenic; No‐ the variant was previously described as non‐pathogenic; Uncertain‐ the variant was classified as being a variant of uncertain clinical significance.
Event previously not detected in controls [Whiley et al, Clin Chem, 2014].
There are 3 other transcripts that include Δ11q (Δ9,11q; Δ9,10,11q; Δ10,11q), but RNA‐seq results do not allow to distinguish them, since they are a combination of splice events, i.e., Δ9, Δ10 or Δ9,10 with Δ11q.
Newly described event.
Figure 1.
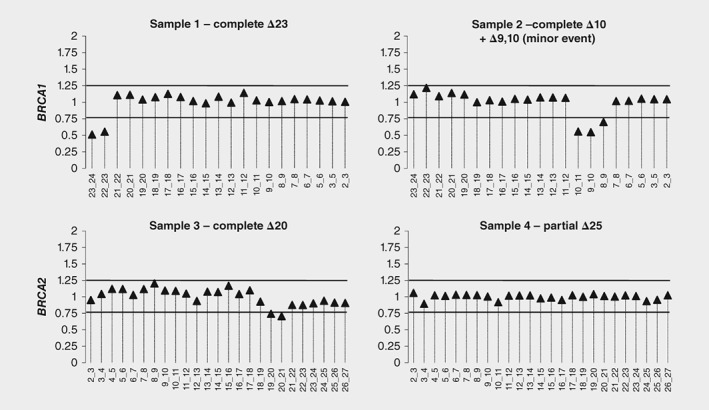
Normalized abundance of reference exon–exon junctions. Quantification of the reference exon‐exon junctions allows to determine loss of the reference transcript and, therefore, helps to distinguish between complete and partial splice events.
Sample 1, carrying variant BRCA1:c.5467+5G>C, showed a strong enrichment for out‐of‐frame exon 23 skipping (Table 4) and is accompanied by a decrease of the local reference exon‐exon junctions (Fig. 1), indicating loss of the reference transcript. The deletion of this exon, which codes for the second BRCT domain, leads to a premature stop codon within the last exon. This information suggests that c.5467+5G>C could be pathogenic like other variants leading to BRCA1Δ23.42, 43, 44 Initial multifactorial analysis had predicted this variant as likely not pathogenic,36 however this was based on few data and the most recent classification for this variant is that it is a class 3 (unclassified).37 A recent study, which used saturation genome editing to predict the functional effects of thousands of BRCA1 variants, reports this variant as having loss of function.38 There are no other studies that confirm complete loss of the reference transcript from the variant allele. So, future studies are required to improve the classification of this variant. Additional splicing events, previously described in this sample, did not seem to be enriched according to QURNAS. Yet, there was a slight increase in reads for BRCA1Δ22,23, accompanied by a decrease of the normal isoform BRCA1Δ22 (Table 4). QURNAS might be missing enrichment of BRCA1Δ22,23 because it is a minor event compared to BRCA1Δ23—97 reads and 8798 reads, respectively—and it was also found in the other samples (22 average reads).
Sample 2, carrying BRCA1:c.594‐2A>C in cis with c.641A>G, showed two enriched events: a strongly enriched event (enrichment score = 9) leading to out‐of‐frame exon 10 skipping and a weakly enriched event (enrichment score = 2) leading to in‐frame exons 9 and 10 skipping. The latter was present in all samples, already at a relatively high expression level. In fact, this is a major naturally‐occurring alternative splicing event as previously published.21 Another event using a cryptic acceptor site 21 bp upstream of exon 10, was previously described to be associated with the presence of the variant.18 However, its detection in all 4 samples of our study shows that it is a naturally‐occurring event with slightly increased expression in this carrier (Table 4). Analysis of the reference splice sites showed that 3 reference exon‐exon junctions involved in the alternative splicing are decreased (Fig. 1). This indicates that there is loss of the reference transcript, consistent with dPCR data from another study.19 It is noteworthy that, probably due to the high expression of the in‐frame BRCA1Δ9,10 transcript, which can produce functional protein, the BRCA1:[c.594‐2A>C;c.641A>G] allele is not pathogenic.19
Our results for sample 3, carrying BRCA2:c.8632+1G>A, confirmed that the major effect of this variant is out‐of‐frame exon 20 deletion (enrichment score = 5), as previously published.18, 39 Previously described minor events, combined deletion of exons 19 and 20, and deletion of exon 20 combined with an insertion of 64 bp of intron 20 (c.8633‐1327_8633–1264ins), were also slightly increased. In addition, two new events were identified in the presence of the variant. One leads to the activation of a cryptic splice site located 17 nt downstream of exon 20: c.8632_8633ins8632+1_8632+17. The expression of the naturally‐occurring event c.8632_8633ins8633‐1356_8633–1264 is decreased, whereas the combination of this event with skipping of exon 20 is increased (Table 4). Analysis of loss of reference splice sites showed that the exon‐exon junctions between exons 19/20 as well as 20/21 are decreased (Fig. 1), which indicates that there is loss of the reference transcript. Our results for c.8632+1G>A are in agreement with previous studies and confirm that it is pathogenic.39
The QURNAS’ results for sample 4, carrier of BRCA2:c.9501+3A>T, indicated that out‐of‐frame deletion of exon 25 was the most prominent splicing event caused by the variant, with an enrichment score of 10. The intron 23 retention, previously described as a minor event occurring in this carrier,18 could not be confirmed. The raw read counts for the exons 24/25 junction were 16,890 (ranging from 15,966 to 23,858 in the other samples) and 28,849 (ranging from 27,518 to 33,774 in the other samples) for exons 25/26 junction. In contrast, the read counts for the aberrant boundary between exons 24/26 were only 1923 reads. Subsequent analysis of the relative expression levels of the reference exon‐exon junctions confirmed that BRCA2Δ25 is incomplete, as previously demonstrated.40, 41 These results are compatible with the fact that c.9501+3A>T is not pathogenic.36
Discussion
Every gene undergoes alternative splicing, which is crucial in shaping transcriptome variation and proteome diversity. Changes in alternative splicing are also often associated with cancer. In order to recognize pathogenic splicing events, it is important to have a thorough understanding of the natural variation in splicing of expressed transcripts under healthy conditions. Therefore, the aim of the study was to identify naturally occurring alternative splicing in transcripts from 12 tumour suppressor genes. The first task was to evaluate whether the targeted RNA‐seq approach was able to identify splice junctions across the whole gene simultaneously at high‐sensitivity. To accomplish this, BRCA1/2 genes were used as controls since extensive analysis of the splice isoforms repertoire of these genes was previously conducted using PCR‐based techniques21, 22 and recently also by targeted RNA‐seq.31 Our results show that the approach used in our study was able to identify almost all previously described BRCA1/2 splicing events, i.e. 93% of the splicing events were detected with our standard analysis. Additional events, missed with STAR, were found after specific mapping. Five previously reported naturally occurring BRCA1 splicing events were not found in our four LCL samples, which seems to be due to the interindividual variability. Transcripts resulting from the combination of different splice events were also not always possible to detect. This is because the sequencing read‐length often does not allow to know which events co‐occur. This can be overcome with synthetic long‐read sequencing (10x Genomics technology, www.10xgenomics.com), single molecule sequencing using PacBio sequencer, or sequencing of long‐range PCR products using MinION nanopore sequencing, as previously reported for BRCA1.45 Nevertheless, it is noteworthy that the sequencing coverage used was high enough to detect additional new events occurring at low expression levels. Most genes had a sufficiently high expression (reference exon‐exon junctions over 10,000 reads), except CDH1 and SLX4. As most of the events at low expression levels are probably due to stochastic effect of the splicing machinery, resulting from random combinations of splice sites and usage of weak splice and are assumed to have no biological significance,46 we set a threshold for the events to be described. The list of new events would otherwise be too extensive.
Once it was established that the approach used had sufficient sensitivity to detect virtually all previously known and even new BRCA1/2 alternative splicing events, RAD51C and RAD51D data was analysed. Using the above‐mentioned read threshold (at least one sample with a minimum of 25x coverage), 24% and 30% of the detected events are described for the first time for RAD51C and RAD51D, respectively. The majority of these were confirmed by CE. In‐frame events are of particular interest, since they do not lead to NMD and may lead to (partially) functional proteins. Within the BRCA1/2 transcripts, examples of functional isoforms (having tumour suppressor activity) are BRCA1Δ9,1019 and BRCA2Δ12.20 In the absence of reference transcript and increased expression of these isoforms, there remains tumor‐suppressor function. In contrast, BRCA1Δ16,17, BRCA2Δ3 and BRCA2Δ17 are examples of pathogenic in‐frame deletions since these proteins lack important functional domains and tumor‐suppressor activity.33, 47, 48 For RAD51C and RAD51D, practically all exons code for functional domains (UniprotKB, InterPro and Nextprot databases). No in‐frame deletions that could lead to a functional protein were identified. Our findings for the additional eight genes analysed were similar.
In general, the frequency of alternative splicing depends on species complexity and cell type. It changes also during development and upon cellular differentiation, indicating that alternative splicing is an important cellular mechanism for the fine‐tuning of gene expression both temporally and spatially.49, 50 Therefore, RNA‐seq data collected from healthy breast and fimbria tissues was analysed and compared to blood with the aim of finding different splice patterns between the tissues. The number of isoforms found in breast and fimbria was smaller than that found in LCLs. However, since we did not perform targeted RNA‐seq in these tissue samples, it is not possible to make a good comparison. The mean coverage of the reference exon‐exon junctions is more than 50 times larger in the targeted sequencing compared to non‐targeted sequencing of LCLs. Compared to the data from breast and fimbria tissues, it is 214 and 1380 times higher, respectively. This coverage difference seen for the reference exon‐exon junctions limits our conclusions about the number and type of alternative isoforms in these tissues. Similarly, publicly available data on the GTEx portal (www.gtexportal.org; version 4.1, build # 201) shows very low read numbers over reference exon‐exon junctions and even lower for several known splicing events. Only sequencing at very high coverage, such as can be achieved with targeted RNA‐seq, will provide sufficient insight into the different isoforms in the breast and fimbria tissues.
The samples used in our study contain BRCA1/2 variants leading to well defined aberrant splicing events which were all detected in the targeted RNA‐seq data. Importantly, we could also correctly assess whether the events were complete or partial, which is crucial information to infer their pathogenicity. Therefore, targeted RNA‐seq can be used to map RNA splicing for a complete locus with one test and can detect potential pathogenic splicing events in a gene, provided that the gene of interest is expressed in the available tissue. This technique can make a major contribution in the classification of genetic variants as either neutral or pathogenic, based on their effect on splicing, reducing the burden of VUS in genetic counselling.
In summary, here we describe an updated overview of the normal splicing events of BRCA1/2, and provide for the first time an extensive catalogue of normal RAD51C and RAD51D alternative splicing. We also provide an overview of normal alternative splicing for eight additional tumour suppressor genes. In‐frame exon deletions that could potentially rescue protein function were not identified. The data can be further used in the design and interpretation of RNA‐experiments to assess the effect of variants with a putative effect on splicing based on RNA‐seq and conventional RT‐PCR. Without targeted enrichment of the genes of interest, we would have not been able to detect splicing events that occur in these genes to the extent and depth that was achieved. Furthermore, we validated our RNA‐seq protocol in combination with the in‐house developed QURNAS software for the identification of significant changes in splicing and developed a method to distinguish complete from partial loss of reference transcript. This is crucial information in finding aberrant splicing events caused by genetic variants and determining their clinical relevance.
Supporting information
Supplementary figure 1 – Annotation used for description of splicing events. Here we illustrate hypothetical events occurring in the region of exon 7 compared to a full‐length (FL) reference transcript that contains exon 7. Splicing events are shown with solid black lines, whereas alternative splicing events are represented with dashed grey lines and cassette insertion events are represented in grey boxes.
Supplementary Figure 2 – Schematic representation of the events found in BRCA1. Exon order provided from 5′ to 3′ as this gene is on the reverse strand. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the each read count to the maximum read count.
Supplementary Figure 3 – Schematic representation of the events found in BRCA2. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the i‐th occurrence to the maximum occurrence.
Supplementary Figure 4 – Schematic representation of the events found in RAD51C. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the i‐th occurrence to the maximum occurrence.
Supplementary Figure 5 – Schematic representation of the events found in RAD51D. Exon order provided from 5′ to 3′ as this gene is on the reverse strand. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the i‐th occurrence to the maximum occurrence.
Supplementary table S1‐ Selected genes and respective genomic coordinates and region size.
Supplementary Table S2: Alignment statistics
Supplementary Table S3 – Performance test: detection of naturally occurring splice events detected in BRCA1 and new events
Supplementary Table S4 ‐ Performance test: detection of naturally occurring splice events detected in BRCA2 and new events
Appendix S1: Splicing events all genes for submission
Appendix S2: Major splice events detected in additional tumour suppressor genes
Appendix S3: Supplementary methods
Acknowledgements
We wish to thank Heather Thorne, Eveline Niedermayr, all the kConFab research nurses and staff, the heads and staff of the Family Cancer Clinics, and the Clinical Follow Up Study (which has received funding from the NHMRC, the National Breast Cancer Foundation, Cancer Australia, and the National Institute of Health (USA)) for their contributions to this resource, and the many families who contribute to kConFab. kConFab is supported by a grant from the National Breast Cancer Foundation, and previously by the National Health and Medical Research Council (NHMRC), the Queensland Cancer Fund, the Cancer Councils of New South Wales, Victoria, Tasmania and South Australia, and the Cancer Foundation of Western Australia. The authors also would like to thank Kasper Derks for executing the QURNAS software. RNA‐seq data collected from healthy breast was provided by the Sweden Cancerome Analysis Network ‐ Breast (SCAN‐B) and fimbria tissues by Ingrid Hedenfalk.
References
- 1. Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 2010;42:410–4. [DOI] [PubMed] [Google Scholar]
- 2. Clague J, Wilhoite G, Adamson A, et al. RAD51C Germline mutations in breast and ovarian cancer cases from high‐risk families. PLoS One 2011;6:e25632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Loveday C, Turnbull C, Ramsay E, et al. Germline mutations in RAD51D confer susceptibility to ovarian cancer. Nat Genet 2011;43:879–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Song H, Dicks E, Ramus SJ, et al. Contribution of Germline mutations in the RAD51B, RAD51C, and RAD51D genes to ovarian cancer in the population. J Clin Oncol 2015;33:2901–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jønson L, Ahlborn LB, Steffensen AY, et al. Identification of six pathogenic RAD51C mutations via mutational screening of 1228 Danish individuals with increased risk of hereditary breast and/or ovarian cancer. Breast Cancer ResTreat 2016;155:215–22. [DOI] [PubMed] [Google Scholar]
- 6. van der Groep P, van der Wall E, van Diest P. Pathology of hereditary breast cancer. Cell Oncol 2011;34:71–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vargas A, Reis‐Filho J, Lakhani S. Phenotype‐genotype correlation in familial breast cancer. J Mammary Gland Biol Neoplasia 2011;16:27–40. [DOI] [PubMed] [Google Scholar]
- 8. Foulkes WD. Inherited susceptibility to common cancers. N Engl J Med 2008;359:2143–53. [DOI] [PubMed] [Google Scholar]
- 9. Bennett RL. Cancer genetics in the clinic: the chalenges and responsabilities of counselling and treating women at risk In: Welcsh P, ed. The role of genetics in breast and reproductive cancersed. New York: Springer, 2009. 3–20. [Google Scholar]
- 10. Shah S, Kim Y, Ostrovnaya I, et al. Assessment of SLX4 mutations in hereditary breast cancers. PLoS One 2013;8:e66961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bakker JL, van Mil SE, Crossan G, et al. Analysis of the novel Fanconi anemia gene SLX4/FANCP in familial breast cancer cases. Hum Mutat 2013;34:70–3. [DOI] [PubMed] [Google Scholar]
- 12. Stoffel EM, Mangu PB, Gruber SB, et al. Hereditary colorectal cancer syndromes: American Society of Clinical Oncology clinical practice guideline endorsement of the familial risk–colorectal cancer: European Society for Medical Oncology clinical practice guidelines. J Clin Oncol 2015;33:209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cartegni L, Chew SL, Krainer AR. Listening to silence and understanding nonsense: exonic mutations that affect splicing. Nat Rev Genet 2002;3:285–98. [DOI] [PubMed] [Google Scholar]
- 14. Brandão RD, van Roozendaal K, Tserpelis D, et al. Characterisation of unclassified variants in the BRCA1/2 genes with a putative effect on splicing. Breast Cancer Res Treat 2011;129:971–82. [DOI] [PubMed] [Google Scholar]
- 15. Vreeswijk M, Kraan J, Klift H, et al. Intronic variants in BRCA1 and BRCA2 that affect RNA splicing can be reliably selected by splice‐site prediction programs. Hum Mutat 2008;30:107–14. [DOI] [PubMed] [Google Scholar]
- 16. Houdayer C, Caux‐Moncoutier V, Krieger S, et al. Guidelines for splicing analysis in molecular diagnosis derived from a set of 327 combined in silico/in vitro studies on BRCA1 and BRCA2 variants. Hum Mutat 2012;33:1228–38. [DOI] [PubMed] [Google Scholar]
- 17. Théry JC, Krieger S, Gaildrat P, et al. Contribution of bioinformatics predictions and functional splicing assays to the interpretation of unclassified variants of the BRCA genes. Eur J Hum Genet 2011;19:1052–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Whiley PJ, de la Hoya M, Thomassen M, et al. Comparison of mRNA splicing assay protocols across multiple laboratories: recommendations for best practice in standardized clinical testing. Clin Chem 2014;60:341–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de la Hoya M, Soukarieh O, López‐Perolio I, et al. Combined genetic and splicing analysis of BRCA1 c.[594‐2A>C; 641A>G] highlights the relevance of naturally occurring in‐frame transcripts for developing disease gene variant classification algorithms. Hum Mol Genet 2016;25:2256–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li L, Biswas K, Habib LA, et al. Functional redundancy of exon 12 of BRCA2 revealed by a comprehensive analysis of the c.6853A>G (p.I2285V) variant. Hum Mutat 2009;30:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Colombo M, Blok MJ, Whiley P, et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: a report from the ENIGMA consortium. Hum Mol Genet 2014;23:3666–80. [DOI] [PubMed] [Google Scholar]
- 22. Fackenthal JD, Yoshimatsu T, Zhang B, et al. Naturally occurring BRCA2 alternative mRNA splicing events in clinically relevant samples. J Med Genet 2016;53:548–58. [DOI] [PubMed] [Google Scholar]
- 23. Ozsolak F, Milos PM. RNA sequencing: advances, challenges and opportunities. Nat Rev Genet 2011;12:87–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wang Z, Gerstein M, Snyder M. RNA‐Seq: a revolutionary tool for transcriptomics. Nat Rev Genet 2009;10:57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Levin JZ, Berger MF, Adiconis X, et al. Targeted next‐generation sequencing of a cancer transcriptome enhances detection of sequence variants and novel fusion transcripts. Genome Biol 2009;10:R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mercer TR, Gerhardt DJ, Dinger ME, et al. Targeted RNA sequencing reveals the deep complexity of the human transcriptome. Nat Biotech 2012;30:99–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Halvardson J, Zaghlool A, Feuk L. Exome RNA sequencing reveals rare and novel alternative transcripts. Nucleic Acids Res 2013;41:e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. JTd D, Antonarakis SE. Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion. Hum Mutat 2000;15:7–12. [DOI] [PubMed] [Google Scholar]
- 30. Miki Y, Swensen J, Shattuck‐Eidens D, et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994;266:66–71. [DOI] [PubMed] [Google Scholar]
- 31. Davy G, Rousselin A, Goardon N, et al. Detecting splicing patterns in genes involved in hereditary breast and ovarian cancer. Eur J Hum Genet 2017;25:1147–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Burset M, Seledtsov IA, Solovyev VV. Analysis of canonical and non‐canonical splice sites in mammalian genomes. Nucleic Acids Res 2000;28:4364–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Carvalho M, Pino MA, Karchin R, et al. Analysis of a set of missense, frameshift, and in‐frame deletion variants of BRCA1. Mutat Res 2009;660:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Claes K, Poppe B, Machackova E, et al. Differentiating pathogenic mutations from polymorphic alterations in the splice sites of BRCA1 and BRCA2 . Genes Chromosomes Cancer 2003;37:314–20. [DOI] [PubMed] [Google Scholar]
- 35. Spurdle AB, Couch FJ, Hogervorst FBL, et al. Prediction and assessment of splicing alterations: implications for clinical testing. Hum Mutat 2008;29:1304–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Whiley PJ, Guidugli L, Walker LC, et al. Splicing and multifactorial analysis of intronic BRCA1 and BRCA2 sequence variants identifies clinically significant splicing aberrations up to 12 nucleotides from the intron/exon boundary. Hum Mutat 2011;32:678–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vallée MP, Di Sera TL, Nix DA, et al. Adding in silico assessment of potential splice aberration to the integrated evaluation of BRCA gene unclassified variants. Hum Mutat 2016;37:627–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Findlay GM, Daza RM, Martin B, et al. Accurate classification of BRCA1 variants with saturation genome editing. Nature 2018;562:217–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tesoriero AA, Wong EM, Jenkins MA, et al. Molecular characterization and cancer risk associated with BRCA1 and BRCA2 splice site variants identified in multiple‐case breast cancer families. Hum Mutat 2005;26:495. [DOI] [PubMed] [Google Scholar]
- 40. Bonnet C, Krieger S, Vezain M, et al. Screening BRCA1 and BRCA2 unclassified variants for splicing mutations using reverse transcription PCR on patient RNA and an ex vivo assay based on a splicing reporter minigene. J Med Genet 2008;45:438–46. [DOI] [PubMed] [Google Scholar]
- 41. Acedo A, Hernández‐Moro C, Curiel‐García Á, et al. Functional classification of BRCA2 DNA variants by splicing assays in a large Minigene with 9 exons. Hum Mutat 2015;36:210–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Gaildrat P, Krieger S, Théry J‐C, et al. The BRCA1 c.5434C>G (p.Pro1812Ala) variant induces a deleterious exon 23 skipping by affecting exonic splicing regulatory elements. J Med Genet 2010;47:398–403. [DOI] [PubMed] [Google Scholar]
- 43. Rouleau E, Lefol C, Moncoutier V, et al. A missense variant within BRCA1 exon 23 causing exon skipping. Cancer Genet Cytogenet 2010;202:144–6. [DOI] [PubMed] [Google Scholar]
- 44. Steffensen AY, Dandanell M, Jønson L, et al. Functional characterization of BRCA1 gene variants by mini‐gene splicing assay. Eur J Hum Genet 2014;22:1362–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. de Jong LC, Cree S, Lattimore V, et al. Nanopore sequencing of full‐length BRCA1 mRNA transcripts reveals co‐occurrence of known exon skipping events. Breast Cancer Res 2017;19:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Melamud E, Moult J. Stochastic noise in splicing machinery. Nucleic Acids Res 2009;37:4873–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Farrugia DJ, Agarwal MK, Pankratz VS, et al. Functional assays for classification of BRCA2 variants of uncertain significance. Cancer Res 2008;68:3523–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wu K, Hinson SR, Ohashi A, et al. Functional evaluation and cancer risk assessment of BRCA2 unclassified variants. Cancer Res 2005;65:417–26. [PubMed] [Google Scholar]
- 49. Barbosa‐Morais NL, Irimia M, Pan Q, et al. The evolutionary landscape of alternative splicing in vertebrate species. Science 2012;338:1587–93. [DOI] [PubMed] [Google Scholar]
- 50. Merkin J, Russell C, Chen P, et al. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 2012;338:1593–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary figure 1 – Annotation used for description of splicing events. Here we illustrate hypothetical events occurring in the region of exon 7 compared to a full‐length (FL) reference transcript that contains exon 7. Splicing events are shown with solid black lines, whereas alternative splicing events are represented with dashed grey lines and cassette insertion events are represented in grey boxes.
Supplementary Figure 2 – Schematic representation of the events found in BRCA1. Exon order provided from 5′ to 3′ as this gene is on the reverse strand. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the each read count to the maximum read count.
Supplementary Figure 3 – Schematic representation of the events found in BRCA2. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the i‐th occurrence to the maximum occurrence.
Supplementary Figure 4 – Schematic representation of the events found in RAD51C. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the i‐th occurrence to the maximum occurrence.
Supplementary Figure 5 – Schematic representation of the events found in RAD51D. Exon order provided from 5′ to 3′ as this gene is on the reverse strand. On top we show the reference‐transcript boundaries, followed by events that have been previously described and, in the bottom, events that were not previously described and that were observed at least 25 times in at least one of the four samples.
Normalized counts (norm. counts) = occurrence_i/max(occurrence_i), i.e. is the ratio of the i‐th occurrence to the maximum occurrence.
Supplementary table S1‐ Selected genes and respective genomic coordinates and region size.
Supplementary Table S2: Alignment statistics
Supplementary Table S3 – Performance test: detection of naturally occurring splice events detected in BRCA1 and new events
Supplementary Table S4 ‐ Performance test: detection of naturally occurring splice events detected in BRCA2 and new events
Appendix S1: Splicing events all genes for submission
Appendix S2: Major splice events detected in additional tumour suppressor genes
Appendix S3: Supplementary methods