

CobB serves as a lysine de-2-hydroxyisobutyrylation enzyme to regulate metabolism and cell growth in prokaryotes.
Abstract
Lysine 2-hydroxyisobutyrylation (Khib) has recently been shown to be an evolutionarily conserved histone mark. Here, we report that CobB serves as a lysine de-2-hydroxyisobutyrylation enzyme that regulates glycolysis and cell growth in prokaryotes. We identified the specific binding of CobB to Khib using a novel self-assembled multivalent photocrosslinking peptide probe and demonstrated that CobB can catalyze lysine de-2-hydroxyisobutyrylation both in vivo and in vitro. R58 of CobB is a critical site for its de-2-hydroxyisobutyrylase activity. Using a quantitative proteomics approach, we identified 99 endogenous substrates that are targeted by CobB for de-2-hydroxyisobutyrylation. We further demonstrated that CobB can regulate the catalytic activities of enolase (ENO) by removing K343hib and K326ac of ENO simultaneously, which account for changes of bacterial growth. In brief, our study dissects a Khib-mediated molecular mechanism that is catalyzed by CobB for the regulation of the activity of metabolic enzymes as well as the cell growth of bacteria.
INTRODUCTION
Lysine 2-hydroxyisobutyrylation (Khib), a new histone mark, was first reported on histone in eukaryotic cells (1). Recent studies showed that Khib was a kind of conserved posttranslational modification (PTM) and widely existed in eukaryotes and prokaryotes from histone to nonhistone proteins (2, 3). In addition to its function in transcriptional regulation, Khib can also repress the catalytic activities of glycolytic enzymes in HCT116 cells (4). Our previous study has indicated that this PTM can change the enzymatic activity of metabolic enzymes in vitro, and carbon sources such as pyruvate, glucose, and 2-hydroxyisobutyrate (HIBA) influence the extent of 2-hydroxyisobutyrylation in Proteus mirabilis (3). Therefore, Khib is closely related to metabolic regulation. However, how Khib affects metabolism in vivo in prokaryotes remains unclear.
Lysine acylation has been thought to be one of the most extensive PTMs and plays a key role in diverse biological processes such as in gene transcription, energy metabolism, and signal transduction (5–10). These dynamic PTMs are usually regulated by the enzymes that write or erase the lysine acylation. For example, lysine acetyltransferases (KATs) and lysine deacetylases (KDACs) have been reported to catalyze acylation or deacylation reactions for different modifications (11–14). For 2-hydroxyisobutyrylation, H4K8hib of Saccharomyces cerevisiae was mediated by the histone KDAC Rpd3p and Hos3p. Esa1p in budding yeast and its homolog Tip60 in humans could add Khib to substrate proteins (2). In addition, histone deacetylase 2 (HDAC2) and HDAC3 were identified as the major enzymes that remove Khib in human cells (15). The latest research showed that EP300 functions as a lysine 2-hydroxyisobutyryltransferase (4). However, the regulatory enzymes in prokaryotes are still unknown, and the physiological effects of the regulation of Khib in prokaryotes are also unclear.
Mimic peptide–based affinity purification has been thought to be an effective approach for the discovery of PTM readers and KDACs. Recently, we developed a novel PTM probe based on a self-assembled multivalent photocrosslinking technique for the identification of PTM binders, which enable us to obtain a more comprehensive profiling of PTM binders compared with previous reports because of its unique advantages on affinity enrichment (16).
Here, we demonstrated that CobB serves as a lysine de-2-hydroxyisobutyrylation enzyme that regulates glycolysis and cell growth in bacteria. We first identified the specific affinity binding of CobB to Khib by integrating a novel self-assembled multivalent photoaffinity probe with mass spectrometric (MS) analysis. Next, we revealed that CobB can catalyze lysine de-2-hydroxyisobutyrylation both in vivo and in vitro, and R58 of CobB is essential for lysine de-2-hydroxyisobutyrylation by a series of biochemical experiments. Using a stable isotope labeling by amino acids in cell culture (SILAC)–based quantitative proteomics approach, we quantified relative differences in 2-hydroxyisobutyrylation site between wild-type (WT) Escherichia coli and the corresponding CobB knockout (KO) cells. Ninety-nine endogenous substrates that are targeted by CobB for de-2-hydroxyisobutyrylation were identified. Then, we found that overexpression of CobB significantly reduced Khib levels and increased the cell growth rate. Last, our results showed that CobB can regulate the catalytic activities of ENO by reducing K343hib and K326ac of ENO simultaneously, which may underlie the changes of cell growth. In brief, our study reveals a CobB-catalyzed, Khib-dependent molecular mechanism for the regulation of cellular glycolysis and bacterial growth.
RESULTS
Discovery of specific binders of Khib
Khib has unique features that differ from the widely studied lysine PTMs, such as acetylation (Kac) and succinylation (Ksucc) (Fig. 1A), due to its distinctive chemical structure, specific genomic distributions, and varied dynamics among diverse model systems (1, 3, 17, 18). To uncover the function of this PTM in prokaryotes, we screened its specific binding proteins by combining MS analysis and a self-assembled multivalent photocrosslinking peptide probe, which is a powerful tool developed by our group to capture the binders that recognize lysine PTMs (16). As shown in Fig. 1B, we prepared the H3K9hib peptide [ARTKQTAR-K(hib)-STGGKAC] as the experimental probe and the H3K9 peptide (ARTKQTARKSTGGKAC) as the control. The two probes were incubated with whole-protein lysates of P. mirabilis. The resulting enrichment was subjected to proteomics profiling; thus, CobB was identified as a distinctly specific binder with H3K9hib (Fig. 1C and table S1). CobB has been known to be sirtuin-2–like enzyme that functions as a deacetylase and a desuccinylase. Therefore, we rationally hypothesized that CobB may be a lysine de-2-hydroxyisobutyrylase.
Fig. 1. Capture of the binding proteins of Khib.
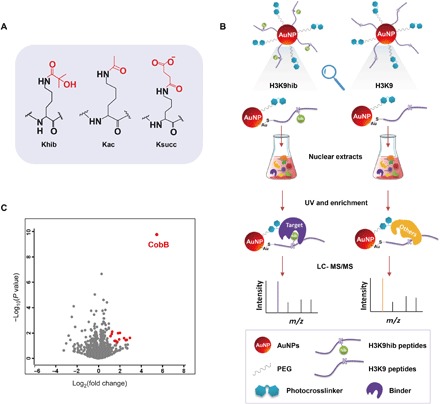
(A) Structures of acetylation, succinylation, and 2-hydroxyisobutyrylation. (B) Workflow of the capture and enrichment of binding proteins of Khib by self-assembled multivalent photoaffinity peptide probes. UV, ultraviolet; AuNP, gold nanoparticles. (C) Volcano plot of enrichment result. The identified proteins with an abundance increase of more than twofold (log2[S/C] > 1) and P value less than 0.05 were marked red.
CobB catalyzes lysine de-2-hydroxyisobutyrylation in vivo
To examine whether CobB is an active lysine de-2-hydroxyisobutyrylase in vivo, we cultured WT and CobB-overexpressing P. mirabilis and blotted whole-cell lysates with pan-antibodies against seven newly reported acyllysines (acetyllysine, succinyllysine, 2-hydroxyisobutyryllysine, malonyllysine, glutaryllysine, crotonoyllysine, and β-hydroxybutyryllysine). Compared with the WT, CobB overexpression caused an obvious decrease in Khib, Kac, and Ksucc, whereas other acylations were substantially unchanged (Fig. 2A). To further confirm the universality of this phenomenon in prokaryotes, we repeated the above experiments in E. coli BL21 [CobB overexpression with or without isopropyl-β-d-thiogalactopyranoside (IPTG) induction and WT] and found that, similar to P. mirabilis, only Khib, Kac, and Ksucc were significantly down-regulated (Fig. 2B). Further, using the protein lysate from WT E. coli MG1655 cells and CobB KO E. coli MG1655 cells cultured in Luria-Bertani (LB) medium, Western blotting analysis showed that CobB KO cells had no obvious change in acylation compared with WT cells (Fig. 2C). However, when the cells were cultured in M9 medium, the levels of Khib, Kac, and Ksucc increased in CobB KO E. coli (Fig. 2D). These results proved that CobB has significant impact on the global Khib like acetylation and succinylation in vivo.
Fig. 2. CobB catalyzes lysine de-2-hydroxyisobutyrylation in vivo.
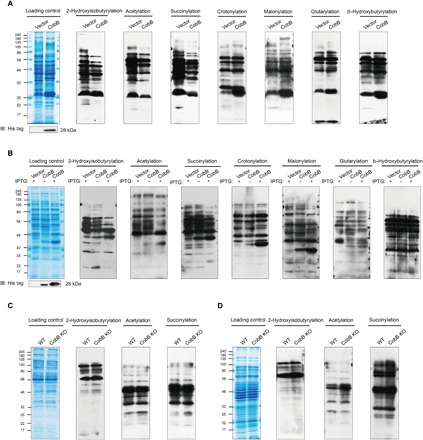
(A) Western blotting (IB) analysis of lysine acylation in protein lysates from CobB-overexpressing P. mirabilis cells. (B) Western blotting analysis of lysine acylation in protein lysates from CobB-overexpressing E. coli BL21 (DE3) cells. (C) Western blotting analysis of lysine acylation in protein lysates from npdA KO E. coli MG1655 cells cultured in LB medium. (D) Western blotting analysis of lysine acylation in protein lysates from npdA KO E. coli MG1655 cells cultured in M9 medium.
Arg58 of CobB is an important active site for de-2-hydroxyisobutyrylation
It was reported that Tyr92 and Arg95 of CobB in E. coli are important sites for desuccinylation (19). On the basis of sequence alignment, we found that Tyr92 and Arg95 of CobB in E. coli are conserved in P. mirabilis (Tyr55 and Arg58) (Fig. 3A). Therefore, we predict that Tyr55 and Arg58 of CobB in P. mirabilis are likely to be the key sites for its de-2-hydroxyisobutyrylase activity. To test this possibility, we expressed and purified WT and mutated CobB (Y55F and R58M) and then performed isothermal titration calorimetry (ITC) assays using the aforementioned gpmA Khib peptides (Fig. 3B). Compared with the binding affinity of the Khib peptide to WT CobB (2.03 μM), the affinity with the Khib peptide of Y55F CobB (2.34 μM) is basically unchanged, but the affinity of R58M CobB (15.92 μM) significantly decreased eight times. The ITC result indicated that R58, instead of Y55, of CobB is important for CobB to bind with Khib. Furthermore, we used a matrix-assisted laser desorption/ionization–time-of-flight (MALDI-TOF) MS assay to calculate the de-2-hydroxyisobutyrylation kinetic parameters (kcat, Km, and kcat/Km) of WT CobB and mutated CobB (Y55F and R58M). The kinetic measurements (Fig. 3C and fig. S1) confirmed that the de-2-hydroxyisobutyrylase activity of WT CobB (kcat = 0.225 ± 0.011 s−1, Km = 13.3 ± 1.6 μM, kcat/Km = 1.7 × 104 s−1 μM−1) was similar to that of Y55F CobB (kcat = 0.101 ± 0.006 s−1, Km = 12.2 ± 2.0 μM, kcat/Km = 8.3 × 103 s−1 μM−1), but when Arg58 was mutated to Met, the de-2-hydroxyisobutyrylase activity was not detected. This result further demonstrated that R58 is an important binding site for de-2-hydroxyisobutyrylation of CobB. Last, we overexpressed CobB and the R95M mutant in CobB KO E. coli with that transformed with T vector as a control. When the whole-cell lysates were blotted with pan-antibodies against 2-hydroxyisobutyrylation, we found that CobB overexpression caused a significant down-regulation of Khib compared with control, but the R95M-overexpressing mutant remained basically unchanged (Fig. 3D). These three groups of experiments demonstrated that R58 (conserved site R95 in E. coli CobB) is a key site influencing the de-2-hydroxyisobutyrylase activity of CobB in P. mirabilis.
Fig. 3. Characterization of de-2-hydroxyisobutyrylation of CobB.
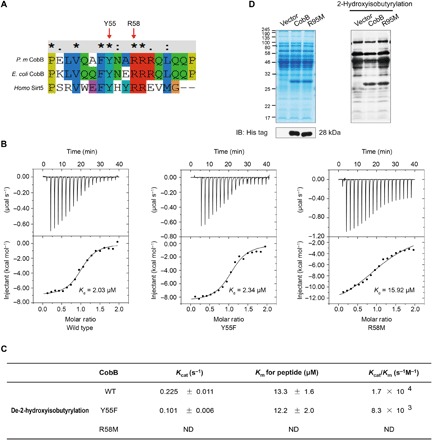
(A) Multiple sequence alignment of CobB from different species by Clustal×2. (B) ITC analysis of the affinity of recombinant CobB (WT) and mutated CobB (Y55F and R58M) with the 2-hydroxyisobutyrylated peptide [YGDEQVK(hib)QWR]. (C) De-2-hydroxyisobutyrylation kinetic parameters (kcat, Km, and kcat/Km) of WT and mutated CobB (Y55F and R58M). ND means “not determined” because of a lack of enzyme saturation with the highest substrate concentration used. (D) Western blotting analysis of Khib in protein lysates from npdA KO E. coli MG1655 cells overexpressed with pTrc99a, CobB, and CobB-mutated R95M and cultured in M9 medium.
CobB catalyzes lysine de-2-hydroxyisobutyrylation in vitro
To further confirm the enzymatic activities of CobB, we carried out a series of in vitro experiments. We first resolved whole-cell lysates of P. mirabilis in SDS–polyacrylamide gel electrophoresis (SDS-PAGE) gel and then transferred the proteins from the gel to the nitrocellulose (NC) membrane, and after incubating the recombinant CobB with the NC membrane, we performed Western blot assay. The results show that Khib, Kac, and Ksucc levels are decreased in many different protein bands, as shown in Fig. 4A. The findings further indicate that CobB can play as a de-2-hydroxyisobutyrylase, a deacetylase, and a desuccinylase targeting different proteins in vitro. For further proof, we carried out a lysine deacylation reaction using the synthetic peptide bearing Khib residues [YGDEQVK(hib)QWR, a 2-hydroxyisobutyrylated fragment of protein gpmA from P. mirabilis], Kac residues [YGDEQVK(ac)QWR, from gpmA of P. mirabilis], and Ksucc residues [YGDEQVK(su)QWR, from gpmA of P. mirabilis]; these PTM sites have been identified in our experiments (fig. S2). Liquid chromatography–MS (LC-MS) was used to monitor de-2-hydroxyisobutyrylation of the peptide by CobB and MS/MS spectrum to verify its veracity (Fig. 4B and fig. S3). Many deacylation modifications require NAD [nicotinamide adenine dinucleotide (oxidized form)] as a cofactor. We found that CobB efficiently catalyzed the hydrolysis of the 2-hydroxyisobutyryl peptide only in the presence of NAD+ (nicotinamide adenine dinucleotide); in contrast, the de-2-hydroxyisobutyrylated peptide was not detected by the reaction without NAD+, suggesting NAD-dependent de-2-hydroxyisobutyrylation mechanisms such as deacetylation and desuccinylation (Fig. 4, C and D, and fig. S3). The de-2-hydroxyisobutyrylation reaction can be inhibited by nicotinamide (NAM) (a class III HDAC inhibitor), but the sensitivity of CobB to NAM is different in removing different acyl groups. NAM had the greatest inhibitory effect on Khib and had the least effect on Ksucc. This result indicates that CobB can catalyze lysine de-2-hydroxyisobutyrylation in a Khib peptide in vitro and that NAD+ is the cofactor of this reaction, which can be inhibited efficiently by NAM. The de-2-hydroxyisobutyrylated peptide, which was treated with the R58M mutant, was not detectable; in contrast, deacetylated peptides completely replaced acetylated peptides and desuccinylated peptides partially replaced succinylated peptides. These results suggested that R58 of CobB in P. mirabilis is not a key site for its deacetylation activity but is essential for de-2-hydroxyisobutyrylation and also necessary for desuccinylation, albeit less critical. These in vitro experiments on protein and peptide levels demonstrated that CobB is a lysine de-2-hydroxyisobutyrylase.
Fig. 4. CobB catalyzes lysine de-2-hydroxyisobutyrylation in vitro.

(A) Western blotting showed that CobB can catalyze in vitro de-2-hydroxyisobutyrylation, deacetylation, and desuccinylation reactions in whole P. mirabilis cell proteins. (B to D) LC-MS detection of lysine de-2-hydroxyisobutyrylation, deacetylation, and desuccinylation activities of CobB in peptide substrates. Synthetic peptides [YGDEQVK(hib)QWR, YGDEQVK(ac)QWR, and YGDEQVK(su)QWR, from gpmA of P. mirabilis] were used as the substrates. The assays were carried out without CobB, with CobB, without NAD+, and with CobB in the presence of NAM (10 mM). A triangle and a circle indicate modified and unmodified peptides, respectively.
Comparative analysis of Khib, Kac, and Ksucc in P. mirabilis
The above studies have demonstrated CobB-catalyzed regulation of Khib in vivo and in vitro. To explore the biological significance of Khib regulation, we compared the relationship of Khib with Kac and Ksucc in P. mirabilis at the proteome level. For acetylation and succinylation, we performed a systematic analysis of Kac and Ksucc by combining the affinity enrichment and proteomics technique. For 2-hydroxyisobutyrylation, we obtained the data from our previous study (3). Last, we identified 2198 acetylated peptides that signified 2217 modified sites on 919 proteins and 1614 succinylated peptides that signified 1614 modified sites on 541 proteins (Fig. 5A and tables S2 and S3). Compared with Khib data showing 4724 2-hydroxyisobutyrylated sites on 1051 proteins, 475 lysine sites on 399 proteins overlapped with 2-hydroxyisobutyrylation, acetylation, and succinylation (Fig. 5, B and C). We also analyzed the position-specific amino acid frequency of the surrounding sequences (15 amino acids to both termini) of the 475 lysine residues using Motif-X. It can be seen from fig. S4A that leucine was drastically overrepresented in the −1 position. It indicates that lysine in this location was likely to co-occur with leucine in Khib, Kac and Ksucc in P. mirabilis. With Gene Ontology (GO) tool DAVID 6.7, functional annotation analysis showed that most of the 399 proteins are cytosolic proteins and have catalytic ability. Classification by pathway or biological process showed that most of the 399 proteins are related to metabolism (fig. S4B). The GO results suggested that three lysine modifications (Khib, Kac, and Ksucc) are all involved in the regulation of energy metabolism. Notably, further analysis of modified proteins in metabolic pathways revealed that almost all of the metabolic enzymes are modified (Khib, Kac, and Ksucc) in the tricarboxylic acid cycle and glycolysis (fig. S4C).
Fig. 5. Profiling of Kac, Ksucc, and Khib in P. mirabilis.
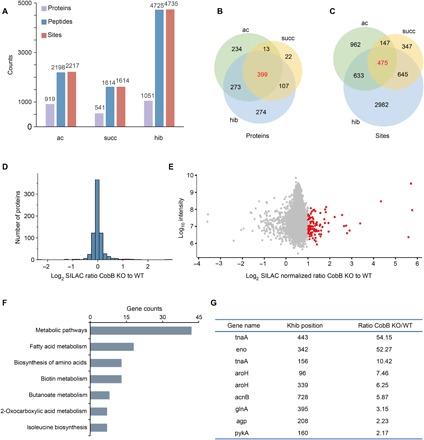
(A) Statistical analysis of the Kac, Ksucc, and Khib proteins, peptides, and sites. (B) Statistical analysis of the common acylated proteins. (C) Statistical analysis of the common acylated sites. (D) The histograms show experimentally determined relative protein abundance distributions for the samples used to analyze 2-hydroxyisobutyrylation. (E) The scatterplots show the ratio of Khib peptides in CobB KO versus WT E. coli cells in relation to average peptide intensities. (F) The bar graph shows pathways enriched in CobB-regulated Khib proteomes. (G) The list shows the carbon metabolic enzymes with a significant up-regulation of the Khib level under CobB KO conditions.
Identification of the endogenous substrate proteins for de-2-hydroxyisobutyrylation by CobB in E. coli
To better understand the endogenous substrates de-2-hydroxyisobutyrylated by CobB, we used a quantitative proteomics approach to characterize the effect of CobB gene deletion on the 2-hydroxyisobutyrylome in E. coli cells. Therefore, we quantified the relative abundance of Khib between WT and CobB KO E. coli. By combining SILAC, affinity enrichment, and two-dimensional LC with high-resolution MS, we identified 5799 Khib sites in 708 proteins (table S4). Normalizing the detected abundances to the expression of their corresponding proteins (Fig. 5D), we identified 99 significantly up-regulated Khib sites (more than twofold increase) in CobB KO E. coli (Fig. 5E). These up-regulated modifications may be potentially functional CobB-targeted substrates. To explore the biological significance of CobB regulation, we performed a pathway enrichment analysis by DAVID 6.7 on CobB-regulated Khib proteins. The result showed that many of the CobB-regulated Khib proteins were related with various metabolic processes (Fig. 5F), such as fatty acid metabolism, amino acid metabolism, and biotin metabolism. In addition, many carbon metabolic enzymes related to amino acid metabolism and glycolysis led to significant up-regulation (Fig. 5G). The analysis result suggested that the regulation of the Khib level by CobB may influence cellular metabolism. In this CobB-targeted Khib site of metabolic enzymes, we found that K342 on ENO in E. coli is a conserved site, with K343 on ENO in P. mirabilis. Our previous work has illustrated that it is a negative regulatory modification on ENO activity in P. mirabilis (3). Therefore, we focused on the modified site on ENO for further biological function.
CobB regulates ENO activity and influences cell growth by erasing Khib and Kac
On the basis of the conservation analysis (Fig. 6A), we found that K326 and K343 of ENO were conserved among species (K326 and K343 of ENO in P. mirabilis were conserved sites, with K325 and K342 in E. coli). To avoid confusion, here, we defined the functional sites as K326 and K343, indicating that K326, like K343, might also be important for an evolutionarily conserved function. In this study, we identified acetylation in K326 of ENO (Fig. 6B and fig. S4D), which predicted a regulatory role of acetylated K326 in ENO activity. To confirm this hypothesis, we locked acetylated K326 of ENO by Q and 2-hydroxyisobutyrylated K343 by T, and we cloned, overexpressed, and purified ENO (ENO-K326Q, ENO-K343T, and ENO-K326Q/K343T) of P. mirabilis as a His-tagged fusion protein. We measured the enzymatic activity of ENO and its mutants. The results showed that mutations of K326Q, K343T, and both K326Q and K343T caused a decrease in the enzymatic activity compared with WT ENO, and the lowest ENO activity is examined in the K326Q/K343T double mutant (Fig. 6C). It was indicated that Khib and Kac jointly regulate the enzymatic activity of ENO.
Fig. 6. CobB regulates ENO activity and influences cell growth by erasing Khib and Kac.
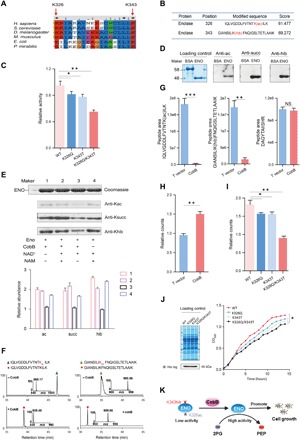
(A) Multiple sequence alignment of ENO from different species by Clustal×2. (B) Identification of K326ac and K343hib in ENO. (C) Detection of the activity of ENO WT and its mutants (K343T, K326Q, and K326Q/K343T) (data are means ± SEM from three independent assays; *P < 0.05, **P < 0.001). (D) Western blotting determined the Khib, Kac, and Ksucc levels of purified recombinant ENO. (E) Western blotting revealed that NAD-dependent CobB can catalyze de-2-hydroxyisobutyrylation/deacetylation/desuccinylation with ENO. ENO was incubated with CobB in the presence of NAD+ (0.5 mM) for 10 hours at 25°C. NAM (10 mM) was added to the reaction as a CobB inhibitor. (F) LC-MS detection of lysine de-2-hydroxyisobutyrylation and deacetylation activities of CobB with ENO peptides [GIANSILI-K(hib)FNQIGSLTETLAAIK, IQLVGDDLFVTNTK(Ac)ILK]. (G) Comparison of the quantities of K326ac and K343hib in T vector and CobB-overexpressing P. mirabilis (data are presented as means ± SEM, and P values were determined by two-tailed Student’s t test; NS, not significant; P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001). (H) Measurement of the PEP amounts of T vector and CobB-overexpressing P. mirabilis (data are presented as means ± SEM; P values were determined by two-tailed Student’s t test; **P < 0.01). (I) Measurement of the PEP amounts of ENO-overexpressing P. mirabilis that transformed ENO WT and mutation (K326Q, K343T, and K326Q/K343T) vectors (data are means ± SEM from three independent assays; *P < 0.05, **P < 0.001). (J) Measurement growth curve of ENO-overexpressing P. mirabilis that transformed ENO WT and mutation (K326Q, K343T, and K326Q/K343T) vectors (data are means ± SEM from three independent assays, *P < 0.05). OD600, optical density at 600 nm. (K) Graphic model as discussed in the text. CobB can promote bacterial growth by effective de-2-hydroxyisobutyrylation of K343 and deacetylation of K326 of ENO. 2PG, 2-phosphoglycerate.
To further test whether CobB can regulate Khib and Kac of ENO, using the unmodified bovine serum albumin (BSA) as control, we determined that the recombinant ENO purified from E. coli was 2-hydroxyisobutyrylated, acetylated, and succinylated, but unmodified BSA was not detected (Fig. 6D), and we incubated recombinant ENO with CobB in the presence or absence of NAD+ or NAM. As shown in Fig. 6E, only in the presence of NAD+, the three modifications (Khib, Kac, and Ksucc) of ENO decrease significantly, but there was no significant change in the modifications of ENO when NAD+ and NAM coexisted. Therefore, it was confirmed that CobB can regulate the three modifications of ENO in vitro. To confirm this conclusion, we further examined the modifications on the peptide level. We also carried out a lysine deacylation reaction using the synthetic peptide bearing K343hib residues [GIANSILIK(hib)FNQIGSLTETLAAIK, from ENO) and K326ac residues (IQLVGDDLFVTNTK(ac)ILK, from ENO]. LC-MS/MS was used to monitor de-2-hydroxyisobutyrylation and deacetylation of the peptides by CobB; the results showed that CobB efficiently catalyzed the hydrolysis of the K343hib and K326ac peptides of ENO in vitro (Fig. 6F).
Further, a parallel reaction monitoring (PRM) assay was performed on WT and CobB-overexpressing P. mirabilis to confirm that CobB has the ability to hydrolyze K343hib and K326ac of ENO in vivo, and the peptides of ENO (DAGYTAIISHR) were detected for normalization. We found that the K326ac and K343hib peptides [IQLVGDDLFVTNTK(ac)ILK and GIANSILIK(hib)FNQIGSLTETLAAIK] were significantly reduced in CobB-overexpressing P. mirabilis compared with WT. Meanwhile, the normalized peptides remained unchanged (Fig. 6G and table S5). Next, we detected that the count of phosphoenolpyruvate (PEP) was higher in CobB-overexpressing P. mirabilis than in WT (Fig. 6H). Therefore, it was confirmed that CobB can effectively de-2-hydroxyisobutyrylate K343 and deacetylate K326 of ENO in vivo. We also detected the counts of PEP of ENO and mutated ENO (K326Q, K343T, and K326Q/K343T)–overexpressing P. mirabilis and found that the amount of PEP detected in ENO-overexpressing P. mirabilis is more than that in mutated ENO-overexpressing P. mirabilis, and that in the ENO-K326Q/K343T–overexpressing P. mirabilis was the lowest (Fig. 6I). This result is consistent with our experimental results for determining ENO activity in vitro (Fig. 6C). Together, we conclude that CobB reducing K326ac and K343hib of ENO can up-regulate the activity of ENO.
Comparing the growth curves of P. mirabilis overexpressing different ENO variants, we found that the growth rate of the WT CobB-overexpressing strain was faster than that of the mutated R58M CobB-overexpressing P. mirabilis in M9 minimal medium with 0.2% glucose (fig. S4E). Moreover, we also found that the growth rate of ENO-overexpressing P. mirabilis was faster than that of mutated ENO-overexpressing P. mirabilis, and the growth rate of ENO-K326Q/K343T–overexpressing P. mirabilis was the lowest (Fig. 6J). It was indicated that the activity of ENO can influence cell growth by regulating glycolysis. Together, this study demonstrated that CobB can regulate ENO activity and influence cell growth by erasing K343hib and K326ac (Fig. 6K).
DISCUSSION
With the discovery of novel types of lysine PTMs, emerging data indicate that diverse modifications are associated with cellular physiology and disease pathogenesis in eukaryotes (20–24). Meanwhile, studies on new PTMs in prokaryotes are also flourishing, and many of them have revealed the importance of regulatory functions of PTMs in prokaryotes. For example, acetylated proteins are involved in cellular physiology in diverse prokaryotes, including central metabolism, mRNA splicing, protein synthesis and degradation, cell morphology, cell cycle, and bacterial virulence (25–32). Ksucc regulated the activity of acetyl-coenzyme A synthetase (Acs) in Mycobacterium tuberculosis (33). Lysine malonylation was involved in metabolism in E. coli and photosynthesis in Cyanobacteria (9, 34). Given the important biological significance of the above-listed lysine PTMs in prokaryotes, it is conceivable to anticipate that Khib also likely regulates the protein function and thereby modulates the physiological processes in prokaryotes. We previously found that the Khib level varies in the presence of distinct carbon sources and can regulate the activity of metabolic enzymes in P. mirabilis (3). Different from the properties of other PTMs such as Kac and Ksucc, Khib has a unique chemical structure (Fig. 1A), implying that this novel PTM may play a unique role in the regulation of protein structure and function. However, the study of the Khib function is hindered by a lack of knowledge of its regulatory enzymes. It is of significance to find the regulatory enzymes for this PTM in prokaryotes.
In this study, we identified the specific binding of CobB to Khib by taking advantage of the unique, sensitive probe that enables the detection of the PTM-mediated weak and transient protein-protein interaction (Fig. 1, B and C) (16). CobB has been known to perform as a deacetylase and a desuccinylase (19, 35–37), predicting its possible function as an eraser of 2-hydroxyisobutyrylation in bacteria. Here, four lines of evidence demonstrate that CobB can catalyze de-2-hydroxyisobutyrylation (Figs. 2 and 3). First, our in vivo experiments showed that overexpression of CobB in both P. mirabilis and E. coli causes a significant down-regulation of the Khib level on whole-cell lysates (Fig. 2, A and B). Consistently, the CobB KO system and rescue experiments also supported the same conclusion (Figs. 2C and 3D). We found 99 endogenous substrates targeted by CobB for de-2-hydroxyisobutyrylation using a quantitative proteomics approach. Second, using an in vitro assay, we demonstrated that the recombinant CobB can catalyze de-2-hydroxyisobutyrylation on both protein and peptide levels. We also revealed that the de-2-hydroxyisobutyrylation process is NAD+ dependent and can be inhibited by NAM, a known deacetylase inhibitor (36). However, the inhibition effect is different for Khib compared with Kac and Ksucc (Fig. 4). Third, we revealed the catalytic properties of CobB as a de-2-hydroxyisobutyrylase by determining the thermodynamic binding constant of CobB with substrates as well as the kinetic parameters of enzymes. Last, we found that R58 is the key residue for the de-2-hydroxyisobutyrylase and desuccinylase activity of CobB, but is dispensable for its deacetylase activity (Fig. 3), suggesting that distinct active pockets of CobB are used for its deacylation function. The different structure of these three substrates (Fig. 1A) may explain why the different active pocket of CobB is used for deacylations. Together, we found and proved that CobB is a de-2-hydroxyisobutyrylase by a series of in vivo and in vitro evidence.
We observed that CobB served as a lysine de-2-hydroxyisobutyrylation enzyme that removes K343hib of ENO. In addition, K343hib reduction in ENO modulates related metabolic pathways to regulate the cell growth of P. mirabilis. Different from a recent report that K281hib can increase the activity of ENO1 (homology with ENO in bacteria) in human cells (4), our data revealed that K343hib decreases the activity of ENO in P. mirabilis. These results indicated that Khib can play opposite roles in different residues of the same protein. Another interesting finding is that K326ac and K343hib of ENO can jointly reduce the activity of ENO and that this regulation has a synergized effect (Fig. 6), indicating that Kac and Khib can co-regulate metabolism in bacteria. Therefore, it is likely that various modifications co-regulate biological processes through competition or coordination between modifications. Moreover, we observed that the level of K343hib and K326ac is closely linked with ENO activity and bacterial growth. However, the extent of the role that K343hib and K326ac play in the regulation of ENO function and cell growth is currently unclear. In addition, the molecular basis for the regulation of ENO activity by K343hib and K326ac is also unknown. Future investigation in this regard will offer more insight into the mechanism of regulatory roles of these two PTMs in protein function and biological processes.
In summary, we found that CobB is a lysine de-2-hydroxyisobutyrylase involving glycolysis regulation in bacteria and revealed that R58 is a key residue for CobB to fulfill its de-2-hydroxyisobutyrylation activity. Our results also showed that CobB can regulate the catalytic activities of ENO by reducing K343hib and K326ac of ENO simultaneously, which further affects bacterial growth. This study explored the regulatory enzyme and function of Khib and, at the same time, provided the endogenous repertoire of potentially functional CobB-targeted substrates for de-2-hydroxyisobutyrylation. Therefore, we considered that Khib-mediated biological functions in bacteria were only the tip of the iceberg. Our work paved the way for future understanding of the diverse functions of Khib in regulating biological processes.
MATERIALS AND METHODS
Strains and reagents
E. coli MG1655 cells and CobB KO E. coli MG1655 cells were obtained from Tolo Biotech. E. coli BL21 (DE3) was from TIANGEN. P. mirabilis strain ATCC29906 was provided by A. Rozalski and A. Palusiak from the University of Lodz. All the anti-acyllysine antibodies were purchased from PTM Biolabs Inc. All the synthetic peptides were generated by SciLight Biotechnology, LLC. Other reagents are listed in table S6.
Affinity pull-down of Khib binders
Peptides (8 mmol) and HS-PEG-Bpa (4 mmol) were added to 200-μl solution of gold nanoparticles, and the mixture was shaken at room temperature for 12 hours. Then, the functionalized gold nanoparticles were washed three times with H2O and incubated with 0.5 mg of P. mirabilis whole-cell lysate for 12 hours at 4°C on a rotation wheel. Next, the mixture was irradiated at 365 nm on ice for 20 min using a UVP crosslinker (Analytik Jena). After centrifugation, the precipitates were washed twice with wash buffer 1 [4 M urea in phosphate-buffered saline (PBS)], followed by two washes with wash buffer 2 [50 mM tris-HCl, 200 mM NaCl, 2.5 mM KCl, 2.5 mM MgCl2, and 1 mM ZnCl2 (pH 7.8)] and one wash with wash buffer 3 (100 mM NH4HCO3). The precipitation was suspended in 50 μl of 100 mM NH4HCO3 and 1 μg of trypsin to digest overnight at 37°C. The eluted peptide was separated by high-speed centrifugation, and then 5 mM dithiothreitol (DTT) was added to react at 65°C heat block for 1 hour. After cooling down to room temperature, 15 mM iodoacetamide was added to react for 45 min, followed by another 30-min reaction after 30 mM cysteine was added. Peptides were desalted with the C18 material and further analyzed by high-performance LC (HPLC)–MS/MS.
Construction of CobB- and ENO-overexpressing P. mirabilis and E. coli strains
The method was described previously (3, 38). Briefly, plasmids T vector–CobB–His and T vector–ENO–His were constructed to generate CobB- and ENO-expressing strains, respectively, and pET28a-CobB, pET28a-ENO, pTRC99a-CobB-His, and pTRC99a-CobB-R95M-His were constructed for recombinant protein expression. We cloned CobB and ENO genes and selected Sph I and Sal I as restriction enzyme cutting sites to insert the genes into T vector, Bam HI and Xho I as restriction enzyme cutting sites to insert the genes into pET28a, and Bam HI and Xba I as restriction enzyme cutting sites to insert the genes into pTRC99a. The constructed vectors of T vector, T vector–CobB–His, and T vector–ENO–His were transformed into P. mirabilis for overexpressing strains and cultured in LB medium containing ampicillin (50 μg/ml) at 37°C with 220 rpm for a night. The constructed vectors of pET28a-CobB and pET28a-ENO were transformed into E. coli BL21 (DE3) and cultured in LB medium containing ampicillin (50 μg/ml) at 37°C in shaking flasks to an optical density at 600 nm of 0.6 to 0.8. Next, cells were induced with 0.05 and 0.15 mM IPTG at 37°C for 4 hours and then harvested for protein expression. The constructed vectors of pTRC99a, pTRC99a-CobB-His, and pTRC99a-CobB-R95M-His were transformed into CobB KO E. coli and cultured in M9 medium containing ampicillin (50 μg/ml) at 37°C in shaking flasks to an optical density at 600 nm of 0.6 to 0.8. Then, the cells were induced with 0.1 mM IPTG at 37°C for 4 hours and then harvested. The primers for polymerase chain reaction (PCR) are listed in table S6.
In vitro de-2-hydroxyisobutyrylation, deacetylation, and desuccinylation activity assay for CobB using whole-protein lysate
The assay was performed as described in our previous study (3). Briefly, P. mirabilis strain ATCC29906 was cultured overnight in LB medium at 37°C and then harvested and extracted during the exponential growth phase by centrifugation. Protein lysates (10 μg) were resolved by SDS-PAGE gel and transferred to the polyvinylidene difluoride membranes. The membranes were incubated with or without 50 μg of CobB in 2 ml of reaction buffer [50 mM tris-HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mM DTT, 1 mM NAD+, and 0.1% PEG-8000 (polyethylene glycol, molecular weight 8000) (pH 8.0)] at 37°C overnight. Then, the membranes were blocked and probed with anti-Khib antibody, anti-Kac antibody, and anti-Ksucc antibody, respectively. Coomassie blue staining was used as a loading control.
Nano-HPLC assay for determination of the deacylated peptides of CobB
2-Hydroxyisobutyrylated, acetylated, or succinylated peptides (50 μM) with 1 μM CobB were incubated in a reaction buffer [50 mM tris-HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mM DTT, and 1 mM NAD+ (pH 8.5)] for 2 hours at 37°C. The reaction was then quenched with 1 volume of 10% (v/v) trifluoroacetic acid and spun for 10 min at 18,000g (Beckman Coulter Microfuge) to separate the enzyme from the reaction. The sample was separated by a 50-min HPLC gradient [linear gradient from 2 to 35% HPLC buffer B (0.1% formic acid in acetonitrile) for 35 min and then to 90% buffer B for 15 min]. The supernatant was then analyzed using nano-HPLC–MS/MS. The HPLC elute was electrosprayed directly into an Orbitrap Q Exactive mass spectrometer (Thermo Fisher Scientific, Waltham, MA).
ITC measurements
The experiments were performed at 37°C on a MicroCal iTC200 titration calorimeter (Malvern Instruments). The reaction cell containing 200 μl of 50 μM proteins was titrated with 19 injections [0.5 μl (first) and 2 μl (all subsequent injections) of 500 μM peptides]. The binding isotherm was fit with Origin 7.0 software package (OriginLab, Northampton, MA) that uses a single set of independent sites to determine the thermodynamic binding constants and stoichiometry.
Determination of kcat and Km of CobB
WT and mutated (Y55F and R58M) CobB were incubated with peptides in the reaction buffer [50 mM tris-HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, 1 mM DTT, and 1 mM NAD+ (pH 8.5)] at 37°C for 2 min. The concentrations of the 2-hydroxysiobutyrylated peptide were 2, 5, 8, 10, 12, 16, 32, 40, 60, 142, and 285 μM. One volume of 10% (v/v) trifluoroacetic acid was added to quench the reaction. After cleaning with C18 ZipTips, the resulting peptides were analyzed with an Autoflex III TOF/TOF mass spectrometer (Bruker Daltonics). The measurements were carried out in a reflex positive-ion mode with delayed ion extraction. Before analysis, the instrument was externally calibrated with a mixture of peptide standards. 2,5-dihydroxybenzoic acid (DHB) was used as the matrix for the analysis of labeled/unlabeled peptides. The sample aliquots (1.0 μl) were placed onto a MALDI plate, and then 1.0 μl of the DHB matrix was added and dried at room temperature before MS analysis. An acceleration voltage of 20 kV was used. MS data were analyzed using FlexAnalysis software for spectral processing and peak detection.
Enrichment and identification of acetylated and succinylated peptide assay
For enrichment and identification of acetylation, we used the previously reported method similar to that for 2-hydroxyisobutyrylation (3). Briefly, after whole-cell proteins were digested, tryptic peptides were fractionated using high-pH reversed-phase HPLC, and then acetylated peptides were enriched using agarose-conjugated anti-acetyl lysine antibody. Each sample of enriched Kac peptides was analyzed by HPLC-MS/MS, and the final result was verified using MaxQuant software (v1.5.2.8). In contrast to acetylation assay, the samples used for succinylation were not fractionated by high-pH reversed-phase HPLC.
SILAC labeling of E. coli and HPLC-MS/MS analysis
WT E. coli and CobB KO E. coli strains were cultured in M9 minimal medium with 0.2% glucose and l-lysine (100 mg/ml) (CobB KO) or 13C6-lysine (WT), respectively. Then, cells were harvested during the mid-exponential phase of growth and lysate, and HPLC fraction and HPLC-MS/MS analysis were prepared for acylated peptide enrichment as described above.
Database search and data filter criteria
The database search and filter criteria were performed according to reported research (4). Briefly, the acquired MS/MS data were searched using MaxQuant (v.1.5.5.1) against the UniProt E. coli K12 protein database, with an overall false discovery rate for peptides of less than 1%. Peptide sequences were searched using trypsin specificity and by allowing a maximum of two missed cleavages. The minimal peptide length was set to seven. Carbamidomethylation on Cys was specified as fixed modification. 2-Hydroxyisobutyryl on lysine as well as oxidation of methionine and acetylation on the peptide N terminus were fixed as variable modifications. Mass tolerances for precursor ions were set at ±10 ppm for precursor ions and ±0.02 Da for MS/MS. 2-Hydroxyisobutyrylated peptides with a score of <40 and a localization probability of <0.75 were further excluded. After quantifying the protein expression levels of the sample, we normalized all the ratios of quantified Khib peptides by the ratios of the corresponding protein levels.
Site-directed mutagenesis and purification of CobB and ENO
Using our previously reported method (3), the mutated sites of genes were introduced into the T vector–ENO–His, pET28a-CobB, and pET28a-ENO by PCR. To purify the recombinant proteins, the pET28a-CobB mutated strains and pET28a-ENO mutated strains were cultured in LB medium containing canalicillin (50 μg/ml), and cells were induced with 0.05 mM (for CobB and mutations) or 0.15 mM (for ENO and mutations) IPTG at 37°C for 4 hours. The proteins were harvested from cultured cells and purified with HisPur Ni-NTA Resin. The primers for PCR are listed in table S6.
ENO activity assay
The activity of purified ENO, ENO-K343T, ENO-K326Q, and ENO-K343T/K326Q was measured as described (39). Briefly, the purified proteins were added in the reaction buffer [100 mM Hepes, 10 mM MgCl2, 7.7 mM KCl, and 8 mM 2-phosphoglycerate (2PG) (pH 7.0)], and samples without protein served as a blank. After preincubation at 37°C for 5 min, the reactions were measured at 240 nm on a spectrophotometer to detect the production of PEP.
In vitro assay to determine CobB de-2-hydroxyisobutyrylation, deacetylation, and desuccinylation activity on ENO
The purified recombinant CobB and ENO were incubated in a reaction buffer [50 mM tris-HCl, 137 mM NaCl, 2.7 mM KCl, 1 mM MgCl2, and 1 mM DTT (pH 8.5)] with or without 1 mM NAD+ and in the presence or absence of 10 mM NAM. The mixture was incubated at 25°C for 10 hours. The reaction products were analyzed by SDS-PAGE and Western blotting with anti–2-hydroxysiobutyrylation, anti-acetylation, and anti-succinylation.
PRM analysis
Quantitation using PRM was performed on an Orbitrap Q Exactive Plus mass spectrometer with an EASY-Spray source coupled to a Nano-LC system (EASY-nLC 1000, Thermo Fisher Scientific, Waltham, MA). Samples were loaded and separated by reversed-phase chromatography. Upon LC direct injection, peptides were resolved with a 125-min HPLC gradient [linear gradient from 2 to 35% HPLC buffer B (0.1% formic acid in acetonitrile) in 110 min and then to 90% buffer B in 15 min]. The eluate was examined by MS using Q Exactive, which was coupled to the Nano-LC online. After a full-scan event, the MS/MS scans in the PRM mode were triggered by an inclusion list. A full mass spectrum was detected in the Orbitrap at a resolution of 70,000 [automatic gain control (AGC) target was set to 3E6; the maximum injection time was 50 ms; and the mass/charge ratio (m/z) range was 400 to 1350], followed by 20 MS/MS scans on the Orbitrap at a resolution of 17,500 (AGC target was 1E5, and the maximum injection time was 100 ms) in a data-independent procedure. The mass window for precursor ion selection was 1.6 m/z. The isolation window for MS/MS was set at 2.0 m/z. The normalized collision energy (NCE) was 27% with higher-energy collisional dissociation (HCD). Three biological replicates were performed. The PRM data were analyzed using Skyline daily.
Measurement of the concentration of PEP
The concentrations of PEP were measured fluorometrically according to the kits’ manufacturer’s guidelines (MAK102, Sigma-Aldrich) (40). P. mirabilis was homogenized in 3 N HClO4 on ice in a 1:5 ratio (mg wet mass: μl of 3 N HClO4). The samples were neutralized with 3 M KHCO3 to achieve pH 7.4 and centrifuged (13,000g, 4°C, 5 min). Three milliliters of the resulting supernatants was incubated in 100 ml of master mix for 30 min and 1 hour for the measurement of the PEP concentrations. Last, the fluorescence intensity (excitation, 535 nm; emission, 587 nm) of the samples was measured. The concentrations of PEP were calculated by building the calibration curve for 0 to 0.5 nmol standards.
Drawing growth curve
We first screened out overexpressing strains expressing the same recombinant protein by His tag antibody. The overnight cultured, overexpressing strains were diluted 1:1000 in LB medium or M9 medium and incubated at 37°C with 96-well plates, and each strain was added to six wells. The absorbance at 600 nm at initiation was set as a blank and then measured every hour using a spectrophotometer. All the growth dynamic curves were drawn using Origin 8.0.
Supplementary Material
Acknowledgments
Funding: This work was supported by the National Natural Science Foundation of China (nos. 21874100 and 21275077), National Basic Research Program of China (no. 2016YFC0903000), Tianjin Municipal Science and Technology Commission (no. 14JCYBJC24000), and Talent Excellence Program from Tianjin Medical University. Author contributions: K.Z. supervised experiments. H.D. and K.Z. designed experiments, analyzed the data, and wrote the manuscript. H.D. carried out cell culture, enzymatic assay, metabolomics survey, and molecular biological analysis. H.D., G.Z., and C.C. carried out the proteomic survey. G.Z. and C.C. developed the probe for the identification of binding proteins. H.D., X.B., S.T., D.H., E.F., and K.Z. carried out data collection, analysis, and interpretation. All authors discussed the results and commented on the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The MS proteomics data have been deposited to the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) via the iProX partner repository with the dataset identifier PXD013377.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw6703/DC1
Fig. S1. Kinetic curves of enzymatic reaction.
Fig. S2. MS/MS spectrum of lysine-acylated peptides (YGDEQVKQWR) identified in P. mirabilis.
Fig. S3. MS/MS spectrum of 2-hydroxyisobutyrylated, acetylated, and succinylated peptides under different incubation conditions.
Fig. S4. Comparative analysis of Khib, Kac, and Ksucc in P. mirabilis.
Table S1. The quantification result of H3K9hib and H3K9 probe enrichment in P. mirabilis protein lysate.
Table S2. The identification result of Kac of P. mirabilis.
Table S3. The identification result of Ksucc of P. mirabilis.
Table S4. The quantification result of 2-hydroisobutyrylated proteomes with CobB KO E. coli and WT E. coli.
Table S5. The PRM result of comparison of the quantities of K326ac and K343hib in T vector and CobB-overexpressing P. mirabilis.
Table S6. The list of reagents, bacterial strains, and plasmids used in this paper.
REFERENCES AND NOTES
- 1.Dai L., Peng C., Montellier E., Lu Z., Chen Y., Ishii H., Debernardi A., Buchou T., Rousseaux S., Jin F., Sabari B. R., Deng Z., Allis C. D., Ren B., Khochbin S., Zhao Y., Lysine 2-hydroxyisobutyrylation is a widely distributed active histone mark. Nat. Chem. Biol. 10, 365–373 (2014). [DOI] [PubMed] [Google Scholar]
- 2.Huang J., Luo Z., Ying W., Cao Q., Huang H., Dong J., Wu Q., Zhao Y., Qian X., Dai J., 2-Hydroxyisobutyrylation on histone H4K8 is regulated by glucose homeostasis in Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. U.S.A. 114, 8782–8787 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dong H. Y., Guo Z., Feng W., Zhang T., Zhai G., Palusiak A., Rozalski A., Tian S., Bai X., Shen L., Chen P., Wang Q., Fan E., Cheng Z., Zhang K., Systematic identification of lysine 2-hydroxyisobutyrylated proteins in Proteus mirabilis. Mol. Cell. Proteomics 17, 482–494 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang H., Tang S., Ji M., Tang Z., Shimada M., Liu X., Qi S., Locasale J. W., Roeder R. G., Zhao Y., Li X., EP300-mediated lysine 2-hydroxyisobutyrylation regulates glycolysis. Mol. Cell 70, 663–678.e6 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montellier E., Rousseaux S., Zhao Y., Khochbin S., Histone crotonylation specifically marks the haploid male germ cell gene expression program. Bioessays 34, 187–193 (2012). [DOI] [PubMed] [Google Scholar]
- 6.Rardin M. J., He W., Nishida Y., Newman J. C., Carrico C., Danielson S. R., Guo A., Gut P., Sahu A. K., Li B., Uppala R., Fitch M., Riiff T., Zhu L., Zhou J., Mulhern D., Stevens R. D., Ilkayeva O. R., Newgard C. B., Jacobson M. P., Hellerstein M., Goetzman E. S., Gibson B. W., Verdin E., SIRT5 regulates the mitochondrial lysine succinylome and metabolic networks. Cell Metab. 18, 920–933 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Choudhary C., Weinert B. T., Nishida Y., Verdin E., Mann M., The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol. 15, 536–550 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Hirschey M. D., Zhao Y. M., Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol. Cell. Proteomics 14, 2308–2315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qian L. L., Nie L., Chen M., Liu P., Zhu J., Zhai L., Tao S.-c., Cheng Z., Zhao Y., Tan M., Global profiling of protein lysine malonylation in Escherichia coli reveals its role in energy metabolism. J. Proteome Res. 15, 2060–2071 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Sabari B. R., Zhang D., Allis C. D., Zhao Y., Metabolic regulation of gene expression through histone acylations. Nat. Rev. Mol. Cell Biol. 18, 90–101 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du J., Zhou Y., Su X., Yu J. J., Khan S., Jiang H., Kim J., Woo J., Kim J. H., Choi B. H., He B., Chen W., Zhang S., Cerione R. A., Auwerx J., Hao Q., Lin H., Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 334, 806–809 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sabari B. R., Tang Z., Huang H., Yong-Gonzalez V., Molina H., Kong H. E., Dai L., Shimada M., Cross J. R., Zhao Y., Roeder R. G., Allis C. D., Intracellular crotonyl-CoA stimulates transcription through p300-catalyzed histone crotonylation. Mol. Cell 58, 203–215 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tan M., Peng C., Anderson K. A., Chhoy P., Xie Z., Dai L., Park J., Chen Y., Huang H., Zhang Y., Ro J., Wagner G. R., Green M. F., Madsen A. S., Schmiesing J., Peterson B. S., Xu G., Ilkayeva O. R., Muehlbauer M. J., Braulke T., Mühlhausen C., Backos D. S., Olsen C. A., McGuire P. J., Pletcher S. D., Lombard D. B., Hirschey M. D., Zhao Y., Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab. 19, 605–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wei W., Liu X., Chen J., Gao S., Lu L., Zhang H., Ding G., Wang Z., Chen Z., Shi T., Li J., Yu J., Wong J., Class I histone deacetylases are major histone decrotonylases: Evidence for critical and broad function of histone crotonylation in transcription. Cell Res. 27, 898–915 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huang H., Luo Z., Qi S., Huang J., Dai L., Dai J., Zhao Y., Landscape of the regulatory elements for lysine 2-hydroxyisobutyrylation pathway. FASEB J. 31, 926.1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhai G. J., Dong H., Guo Z., Feng W., Jin J., Zhang T., Chen C., Chen P., Tian S., Bai X., Shi L., Fan E., Zhang Y., Zhang K., An efficient approach for selective enrichment of histone modification readers using self-assembled multivalent photoaffinity peptide probes. Anal. Chem. 90, 11385–11392 (2018). [DOI] [PubMed] [Google Scholar]
- 17.Yu Z., Ni J., Sheng W., Wang Z., Wu Y., Proteome-wide identification of lysine 2-hydroxyisobutyrylation reveals conserved and novel histone modifications in Physcomitrella patens. Sci. Rep. 7, 15553 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meng X., Xing S., Perez L. M., Peng X., Zhao Q., Redoña E. D., Wang C., Peng Z., Proteome-wide analysis of lysine 2-hydroxyisobutyrylation in developing rice (Oryza sativa) Seeds. Sci. Rep. 7, 17486 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Colak G., Xie Z., Zhu A. Y., Dai L., Lu Z., Zhang Y., Wan X., Chen Y., Cha Y. H., Lin H., Zhao Y., Tan M., Identification of lysine succinylation substrates and the succinylation regulatory enzyme CobB in Escherichia coli. Mol. Cell. Proteomics 12, 3509–3520 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen Y., Zhao W., Yang J. S., Cheng Z., Luo H., Lu Z., Tan M., Gu W., Zhao Y., Quantitative acetylome analysis reveals the roles of SIRT1 in regulating diverse substrates and cellular pathways. Mol. Cell. Proteomics 11, 1048–1062 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miao J., Lawrence M., Jeffers V., Zhao F., Parker D., Ge Y., Sullivan W. J. Jr., Cui L., Extensive lysine acetylation occurs in evolutionarily conserved metabolic pathways and parasite-specific functions during Plasmodium falciparum intraerythrocytic development. Mol. Microbiol. 89, 660–675 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rousseaux S., Khochbin S., Histone acylation beyond acetylation: Terra Incognita in chromatin biology. Cell J. 17, 1–6 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sadhukhan S., Liu X., Ryu D., Nelson O. D., Stupinski J. A., Li Z., Chen W., Zhang S., Weiss R. S., Locasale J. W., Auwerx J., Lin H., Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc. Natl. Acad. Sci. U.S.A. 113, 4320–4325 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen C., Xue J., Sun T., Guo H., Zhang L., Meng Y., Wang H., Succinyl-proteome profiling of a high taxol containing hybrid Taxus species (Taxus x media) revealed involvement of succinylation in multiple metabolic pathways. Sci. Rep. 6, 21764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jones J. D., O’Connor C. D., Protein acetylation in prokaryotes. Proteomics 11, 3012–3022 (2011). [DOI] [PubMed] [Google Scholar]
- 26.Zhang K., Zheng S., Yang J., Chen Y., Cheng Z., Comprehensive profiling of protein lysine acetylation in Escherichia coli. J. Proteome Res. 12, 844–851 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Xu J. Y., Xu Y., Xu Z., Zhai L.-H., Ye Y., Zhao Y., Chu X., Tan M., Ye B.-C., Protein acylation is a general regulatory mechanism in biosynthetic pathway of Acyl-CoA-derived natural products. Cell Chem. Biol. 25, 984–995.e6 (2018). [DOI] [PubMed] [Google Scholar]
- 28.Pisithkul T., Patel N. M., Amador-Noguez D., Post-translational modifications as key regulators of bacterial metabolic fluxes. Curr. Opin. Microbiol. 24, 29–37 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Zhou M., Xie L., Yang Z., Zhou J., Xie J., Lysine succinylation of Mycobacterium tuberculosis isocitrate lyase (ICL) fine-tunes the microbial resistance to antibiotics. J. Biomol. Struct. Dyn. 35, 1030–1041 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Gaviard C., Broutin I., Cosette P., Dé E., Jouenne T., Hardouin J., Lysine succinylation and acetylation in Pseudomonas aeruginosa. J. Proteome Res. 17, 2449–2459 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Ren J., Sang Y., Lu J., Yao Y.-F., Protein acetylation and its role in bacterial virulence. Trends Microbiol. 25, 768–779 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Liang W., Malhotra A., Deutscher M. P., Acetylation regulates the stability of a bacterial protein: Growth stage-dependent modification of RNase R. Mol. Cell 44, 160–166 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang M., Wang Y., Chen Y., Cheng Z., Gu J., Deng J., Bi L., Chen C., Mo R., Wang X., Ge F., Succinylome analysis reveals the involvement of lysine succinylation in metabolism in pathogenic Mycobacterium tuberculosis. Mol. Cell. Proteomics 14, 796–811 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ma Y., Yang M., Lin X., Liu X., Huang H., Ge F., Malonylome analysis reveals the involvement of lysine malonylation in metabolism and photosynthesis in cyanobacteria. J. Proteome Res. 16, 2030–2043 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Starai V. J., Celic I., Cole R. N., Boeke J. D., Escalante-Semerena J. C., Sir2-dependent activation of acetyl-CoA synthetase by deacetylation of active lysine. Science 298, 2390–2392 (2002). [DOI] [PubMed] [Google Scholar]
- 36.Gallego-Jara J., Conesa A. É., de Diego Puente T., Terol G. L., Díaz M. C., Characterization of CobB kinetics and inhibition by nicotinamide. PLOS ONE 12, e0189689 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao K., Chai X., Marmorstein R., Structure and substrate binding properties of cobB, a Sir2 homolog protein deacetylase from Escherichia coli. J. Mol. Biol. 337, 731–741 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Tsai Y.-L., Wang M.-C., Hsueh P.-R., Liu M.-C., Hu R.-M., Wu Y.-J., Liaw S.-J., Overexpression of an outer membrane protein associated with decreased susceptibility to carbapenems in Proteus mirabilis. PLOS ONE 10, e0120395 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feng Y., Pan X., Sun W., Wang C., Zhang H., Li X., Ma Y., Shao Z., Ge J., Zheng F., Gao G. F., Tang J., Streptococcus suis enolase functions as a protective antigen displayed on the bacterial cell surface. J. Infect. Dis. 200, 1583–1592 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Bartok O., Teesalu M., Ashwall-Fluss R., Pandey V., Hanan M., Rovenko B. M., Poukkula M., Havula E., Moussaieff A., Vodala S., Nahmias Y., Kadener S., Hietakangas V., The transcription factor Cabut coordinates energy metabolism and the circadian clock in response to sugar sensing. EMBO J. 34, 1538–1553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw6703/DC1
Fig. S1. Kinetic curves of enzymatic reaction.
Fig. S2. MS/MS spectrum of lysine-acylated peptides (YGDEQVKQWR) identified in P. mirabilis.
Fig. S3. MS/MS spectrum of 2-hydroxyisobutyrylated, acetylated, and succinylated peptides under different incubation conditions.
Fig. S4. Comparative analysis of Khib, Kac, and Ksucc in P. mirabilis.
Table S1. The quantification result of H3K9hib and H3K9 probe enrichment in P. mirabilis protein lysate.
Table S2. The identification result of Kac of P. mirabilis.
Table S3. The identification result of Ksucc of P. mirabilis.
Table S4. The quantification result of 2-hydroisobutyrylated proteomes with CobB KO E. coli and WT E. coli.
Table S5. The PRM result of comparison of the quantities of K326ac and K343hib in T vector and CobB-overexpressing P. mirabilis.
Table S6. The list of reagents, bacterial strains, and plasmids used in this paper.