

Summary
A novel negative‐stranded (ns) RNA virus associated with a severe citrus disease reported more than 80 years ago has been identified. Transmission electron microscopy showed that this novel virus, tentatively named citrus concave gum‐associated virus, is flexuous and non‐enveloped. Notwithstanding, its two genomic RNAs share structural features with members of the genus Phlebovirus, which are enveloped arthropod‐transmitted viruses infecting mammals, and with a group of still unclassified phlebo‐like viruses mainly infecting arthropods. CCGaV genomic RNAs code for an RNA‐dependent RNA polymerase, a nucleocapsid protein and a putative movement protein showing structural and phylogenetic relationships with phlebo‐like viruses, phleboviruses and the unrelated ophioviruses, respectively, thus providing intriguing evidence of a modular genome evolution. Phylogenetic reconstructions identified an invertebrate‐restricted virus as the most likely ancestor of this virus, revealing that its adaptation to plants was independent from and possibly predated that of the other nsRNA plant viruses. These data are consistent with an evolutionary scenario in which trans‐kingdom adaptation occurred several times during the history of nsRNA viruses and followed different evolutionary pathways, in which genomic RNA segments were gained or lost. The need to create a new genus for this bipartite nsRNA virus and the impact of the rapid and specific detection methods developed here on citrus sanitation and certification are also discussed.
Keywords: citrus disease, concave gum, nsRNA virus, phylogeny, virus adaptation
Introduction
Negative‐stranded (ns) RNA viruses are amongst the most threatening pathogens of humans, animals and plants (Elliott and Brennan, 2014; Hubálek et al., 2014; Kormelink et al., 2011). With a single‐stranded RNA genome that needs to be transcribed by the viral RNA‐dependent RNA polymerase (RdRp) before translation, they are classified according to the number of genomic components, which may range from one to several segments. The taxonomy of nsRNA viruses has been revisited recently (https://talk.ictvonline.org/taxonomy), with those infecting plants being grouped into: (i) the family Rhabdoviridae (order Mononegavirales); (ii) the genus Ophiovirus (family Ophioviridae); and (iii) the genera Emaravirus (family Fimoviridae), Tospovirus (family Tospoviridae) and Tenuivirus (family Phenuiviridae), now grouped in the new order Bunyavirales (https://talk.ictvonline.org/taxonomy). This order, in addition to the plant‐infecting viruses, includes vertebrate‐infecting (genera Orthohantavirus, Orthonairovirus, Orthobunyavirus and Phlebovirus) and arthropod‐restricted (genera Goukovirus, Herbevirus, Orthojonvirus, Orthoferavirus and Orthophasmavirus) viruses. Except for members of the genus Tenuivirus (Falk and Tsai, 1998), the members of the order Bunyavirales are enveloped by a membrane in which viral glycoproteins are embedded (Walter and Barr, 2011). Most of these viruses are transmitted by arthropods, in which they also replicate, thus showing a typical dual host tropism.
The first arthropod‐restricted bunya‐like viruses, now classified in the genera Goukovirus and Herbevirus (https://talk.ictvonline.org/taxonomy), were reported a few years ago (Marklewitz et al., 2011, 2013). More recently, metagenomics approaches have allowed the discovery of many novel nsRNA viruses that apparently only infect invertebrates, among which several novel bunya‐related viruses were also included (Li et al., 2015; Shi et al., 2016; Tokarz et al., 2014). The great genetic diversity of these novel nsRNA viruses and the phylogenetic relationships with the other nsRNA genera indicate that extant vertebrate and plant nsRNA viruses originated from ancestor viruses infecting arthropods (Junglen, 2016; Marklewitz et al., 2011, Li et al., 2015).
Several novel nsRNA viruses have also been identified in nematodes (Bekal et al., 2011), other invertebrates (Shi et al., 2016) and plant‐infecting fungi (Kondo et al., 2013; Marzano et al., 2016), thus complicating the origin and evolution of nsRNA viruses (Dudas and Obbard, 2015). In addition, although genomic recombination and reassortment are considered to be major molecular drivers of nsRNA virus evolution (Han and Worobey, 2011; McDonald et al., 2016), the evolutionary routes followed by phylogenetically related viruses to adapt to their extant hosts (vertebrates or plants) are difficult to envisage. Striking examples include the plant‐infecting tospoviruses and tenuiviruses, and the vertebrate‐infecting phleboviruses, which are segmented nsRNA viruses with some of their genomic components being ambisense: although their origin from a common ancestor probably infecting an arthropod has been proposed (Falk and Tsai, 1998; Kormelink et al., 2011; Li et al., 2015; Marklewitz et al., 2015), the evolutionary history of their adaptation to plants or vertebrates remains unclear.
Here, we report a novel nsRNA virus from citrus with unique molecular and phylogenetic features that could contribute to broaden the evolutionary adaptation route followed by plant nsRNA viruses. This novel virus is associated with the graft‐transmissible citrus disease concave gum‐blind pocket (CG), first observed in the early 1930s (Fawcett, 1936), but the aetiology of which has remained unknown. The two genomic RNAs of this novel virus have molecular signatures previously reported in phleboviruses and code for three proteins showing phylogenetic relationships with phlebo‐like viruses, phleboviruses and ophioviruses, respectively, thus providing intriguing evidence of ancient reassortment and recombination events between ancestors of viruses nowadays infecting animal and/or plant hosts. The need to create a new genus for the classification of this bipartite nsRNA virus and implications in defence strategies to counteract its spread are also discussed.
Results
Identification of a novel nsRNA virus in CG‐affected plants
Citrus psorosis disease and other citrus diseases, including ringspot, impietratura, cristacortis and CG, were initially considered to be caused by the same infectious agent, mainly because of the similar leaf symptoms (fleck and oak‐leaf patterns) induced on graft‐inoculated indicator plants (Moreno et al., 2015). When Citrus psorosis virus (CPsV, genus Ophiovirus) was identified as the agent of citrus psorosis disease, the involvement of this virus in CG, impietratura and cristacortis disorders was excluded (da Graça et al., 1991), leaving open the question of the aetiology of these diseases. In 2015, trees showing deeply depressed trunk concavities frequently associated with leaf chlorotic flecking resembling typical symptoms of CG disease (Fig. 1a–c) were observed in a citrus varietal collection in southern Italy. CPsV infection was excluded by serological and molecular analyses (data not shown) and, as expected for CG‐affected trees (Roistacher, 1991), chlorotic flecking and oak‐leaf patterns were transiently detected on young leaves of sweet orange [Citrus sinensis (L.) Osbeck, cv. Madame vinous] and Dweet tangor (C. reticulata Blanco × C. sinensis) seedlings graft inoculated with bark from the symptomatic trees (Fig. 1d,f).
Figure 1.

Symptoms of citrus concave gum disease. (a, b) Deeply depressed concavities on trunks and (c) chlorotic flecking observed in sweet orange trees (symptoms observed on the CGW2 tree are shown in a and c). (d) Chlorotic flecks and (f) oak‐leaf pattern in seedlings (cv. Madam vinous) graft inoculated with bark patches from a diseased tree. (e) Symptomless leaf from a negative control (cv. Madam vinous).
One CG‐affected tree [CGW2, cv. Tarocco grafted on sour orange (C. aurantium L.) rootstock] from the citrus collection was selected for further investigation of the aetiology of the disease. In the absence of information on whether the potential viral agent accumulated preferentially in the leaves or bark, two independent cDNA libraries of small RNAs (sRNAs) of 16–30 nucleotides (nt) were generated from both sources of the CGW2 tree. An sRNA library from leaves of a symptomless and CPsV‐free tree (W4) of the same cultivar, grown in the same field, and testing negative to bioassays, was used as a negative control. The deep sequenced reads from these libraries (Table S1, see Supporting Information) were assembled in de novo contigs, the sequences of which were searched in databases. No contigs with sequences from previously reported viruses were found, whereas contigs with sequences identical to citrus exocortis, hop stunt and citrus dwarfing viroids were retrieved from the libraries of the symptomatic and non‐symptomatic trees (CGW2 and W4). In contrast, in the CGW2‐derived library, but not in that from the W4 tree, contigs encoding polypeptides sharing slight sequence identity with the RdRp or nucleocapsid protein of previously reported phlebo or phlebo‐like viruses were identified (Table S2, see Supporting Information). Two additional contigs coding for polypeptides with weak sequence identity to the movement protein (MP) of nsRNA viruses infecting plants were also found (Table S2). Although the significance of the blastx matches was generally low, all of these contigs were considered to be noteworthy because the corresponding sequences were not found when exhaustively searched in the citrus genome database, indicating that they did not come from host RNAs and suggesting that they could be partial RNA sequences of a novel nsRNA virus distantly related to those reported previously.
To further support this hypothesis, primer pairs to amplify sequences of contigs that blast‐matched with previously reported viral RdRp, nucleocapsid and MP proteins were designed (Table S3, see Supporting Information), and a multiplex reverse transcription‐polymerase chain reaction (mRT‐PCR) was developed to detect them simultaneously. The mRT‐PCR assays, followed by cloning and sequencing of the expected amplicons, showed the close association of the potential viral sequences to each other in CGW2 and in three other CG‐affected trees, and their absence in three symptomless citrus controls from the same field (Fig. S1, see Supporting Information). The findings supported the possible involvement of a single CG‐associated virus and excluded the possibility that the viral sequences were from an insect or another organism contaminating the CGW2 RNA preparations used for the generation of the sRNA libraries. On the basis of additional data from a field survey and bioassays (see below), the name citrus concave gum‐associated virus was tentatively assigned to this potential new virus.
CCGaV genome is probably composed of only two segments
By cloning and sequencing overlapping cDNAs, two RNAs (RNA1 and RNA2) were assembled, whose termini were determined by 5′ and 3′ RNA rapid amplification of cDNA ends (RACE). RNA1 and RNA2 are composed of 6681 and 2703 nt, respectively, sharing at their 5′ and 3′ terminal regions stretches (up to 26 nt) of almost identical sequences (Fig. 2a) that are also largely complementary to each other (Fig. 2b), a typical feature of members of the order Bunyavirales. Moreover, the nine 5′ and 3′ terminal nucleotides of both RNAs are almost identical to those reported previously at the respective RNA termini of phleboviruses (Elliott and Brennan, 2014) and tenuiviruses (Falk and Tsai, 1998) (Fig. 2c), suggesting a closer relationship of CCGaV with the latter viruses.
Figure 2.

Terminal sequences of citrus concave gum‐associated virus genomic RNAs. (a) Alignment of 5′ (left) and 3′ (right) termini of CCGaV RNAs. (b) Panhandle structures formed by the 5′ and 3′ termini of CCGaV RNAs. (c) Alignment of CCGaV RNA1 termini with those of other negative‐stranded RNA (nsRNA) viruses. Identical nucleotides are in grey. BUNV, Bunyamwera virus; DUGV, Dugbe virus; EMARaV, European mountain ash ringspot‐associated virus; GOUV, Gouleako virus; HTNV, Hantaan virus; RGSV, rice grassy stunt virus; RSV, rice stripe virus; RVFV, Rift Valley fever virus; SFTSV, severe fever with thrombocytopenia syndrome; TOSV, Toscana virus; TSWV, tomato spotted wilt virus; UUKV, Uukuniemi virus.
As phenoviruses and tenuiviruses have a genome composed of at least three RNAs, we next investigated whether the CCGaV genome has only two components. Moreover, considering that additional genomic segments could code for protein(s) with no known homologues, RT‐PCR and in silico approaches independent of protein homologues, and similar to those used previously for an emaravirus (Zheng et al., 2017), were applied to identify RNAs containing the conserved CCGaV RNA termini. RT‐PCR with specific primers derived from these conserved termini (Table S3) failed to identify additional RNA components (data not shown), which were also not found when deeply searched in a novel cDNA library generated using a double‐stranded RNA (dsRNA)‐enriched preparation from CGW2 bark. Such a library was sequenced with Illumina HiSeq 2x125 technology that provided longer reads (up to 125 nt) than the sRNA libraries (Table S1). When reads of 55–125 nt were aligned to RNA1 and RNA2, both RNAs were completely covered (average coverage depth of 64x and 131x, respectively), generating consensus sequences fully consistent with those determined previously with other methods. Deeper analyses of the mapped reads revealed that the 5′ and 3′ termini of RNA2 were covered by 382 and 89 reads, and those of RNA1 by 2 and 16 reads, respectively, thus confirming that the actual termini of CCGaV genomic RNAs were indeed sequenced. However, reads with the 5′ or 3′ terminal stretches of 9 nt conserved in the CCGaV genomic RNAs, but differing in the rest of the sequence—a feature expected for additional segment(s) of the CCGaV genome—were not found.
Also fruitless were searches amongst the 26 577 total de novo contigs (length ≥ 140 nt) from the dsRNA library. Contigs covering RNA1 and RNA2 up to 95.3% and 74.2% of their length, respectively, were found, confirming that both viral genomic RNAs were largely represented in the library. However, exhaustive blastn and blastx searches did not allow the identification of additional sequences possibly related to viral RNAs. Furthermore, the sequences of those contigs that did not find blast matches were successfully mapped within the citrus genome, thus excluding the possibility that they could be derived from viral RNA(s) with low or no sequence identity to previously reported viruses. Interestingly, a specific search for contig(s) with sequence similarity to glycoproteins also failed. Altogether, these results support the view that CCGaV has a bipartite genome.
CCGaV RNA1 codes for an RdRp with typical signatures of nsRNA viruses
RNA1, the viral strand of which was detected by Northern blot hybridization in partially purified viral preparations (Fig. S2, see Supporting Information), contains a single open reading frame (ORF1), encoding a putative protein of 249.97 kDa (p250), and short 5′ and 3′ untranslated regions (UTRs) of 89 and 37 nt, respectively (Fig. 3a). A blastp search identified the RdRps of tentative phlebo‐ or phlebo‐like viruses infecting arthropods (ticks) as the proteins with the highest identity (E‐value: 2.0 e‐116) with p250, although restricted to a stretch of about 1700 amino acids in the central region of the latter (Table S4, see Supporting Information). Consistently, PFAM analyses showed, in the central region of p250 (positions 550–1241), the typical signatures of the RdRp of members of the order Bunyavirales (Pfam family: Bunya_RdRp; PF04196; E‐value: 5.2e‐47). Indeed, alignments of p250 with the RdRp of representative members of most genera in this taxon identified the five conserved motifs (Premotif A and motifs A–E) proposed to be part of the RdRp active site, with the highest similarity being shared with phleboviruses and tenuiviruses (Fig. 4a). Further analyses of the N‐terminal region of p250 (positions 1–128) disclosed that it contains the endonuclease domains [H68‐D80‐PD97–98‐(ExG109–111)‐K128] involved in cap‐snatching (Reguera et al., 2010), a translation strategy first reported and dissected in influenza virus A (family Orthomyxoviridae) (Plotch et al., 1981). Interestingly, the ExK domain found in bunyaviruses is replaced by the ExG domain in CCGaV p250, as in several orthomyxoviruses (Reguera et al., 2010). Altogether, these data indicate that p250 is the CCGaV RdRp.
Figure 3.
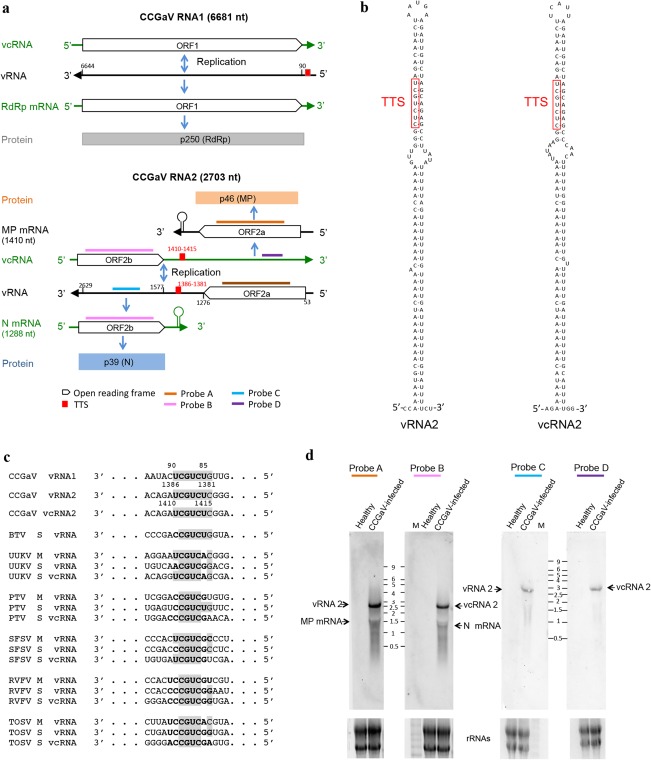
Citrus concave gum‐associated virus genome organization and expression strategy. (a) Schematic diagram. MP, movement protein; N, nucleocapsid; ORF, open reading frame; RdRp, RNA‐dependent RNA polymerase; vRNA, viral RNA; vcRNA, viral complementary RNA. (b) Stem loop and predicted transcription termination signal (TTS) motifs in the intergenic region (IR) of both RNA2 strands. (c) Alignment of TTS (in bold) identified in CCGaV RNAs with those of several phleboviruses and the phlebo‐like Bole tick virus (BTV) (TTS predicted in this study); nucleotides identical to CCGaV TTSs are in grey; PTV, Punta Toro virus; RVFV, Rift Valley fever virus; SFSV, sandfly fever Sicilian virus; TOSV, Toscana virus; UUKV, Uukuniemi virus. (d) Northern blot analysis of RNAs from citrus trees. M, Millennium RNA marker (Applied Biosystems/Ambion) with sizes (kb) indicated between the dashes. Equal loading assessed by ethidium bromide staining of rRNAs (bottom panels).
Figure 4.
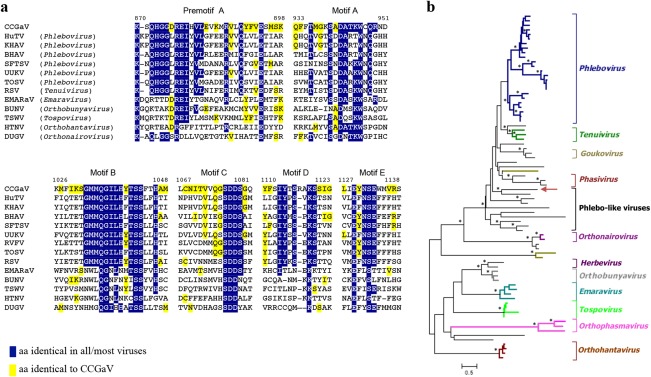
Relationships of citrus concave gum‐associated virus RNA‐dependent RNA polymerase (RdRp) with other negative‐stranded RNA (nsRNA) viruses. (a) Multiple alignment of RdRp conserved motifs. Positions in the CCGaV RdRp are reported. aa, amino acids; BHAV, Bhanja virus; BUNV, Bunyamwera virus; DUGV, Dugbe virus; EMARaV, European mountain ash ringspot‐associated virus; HTNV, Hantaan virus; HuTV, Huangpi tick virus 2; KHAV, Khasan virus; RSV, rice stripe virus; RVFV, Rift Valley fever virus; SFTSV, severe fever with thrombocytopenia syndrome; TOSV, Toscana virus; TSWV, tomato spotted wilt virus; UUKV, Uukuniemi virus. (b) Maximum‐likelihood (ML) phylogenetic analysis of polymerase proteins. The recognized genera and group of phlebo‐like viruses are shown. The black and ochre branches correspond to unclassified viruses infecting arthropods and fungi, respectively. Asterisks mark nodes with bootstrap values >90%. A detailed tree version is shown in Fig. S3 (see Supporting Information).
A maximum‐likelihood (ML) tree was inferred using the RdRp core sequence of CCGaV, of representative members of the order Bunyavirales and of a group of recently reported phlebo‐like viruses mostly composed of arthropod‐restricted viruses. In this phylogenetic tree, CCGaV was nested, together with phlebo‐like viruses, at a basal node of a supergroup including phleboviruses and tenuiviruses at more apical nodes (Figs 4b and S3, see Supporting Information). This and other clades in the ML phylogenetic tree are consistent with similar phylogenetic reconstructions inferred by others (Guterres et al., 2017; Li et al., 2015; Marklewitz et al., 2015). In addition, a similar tree topology was obtained by the Bayesian Markov Chain Monte Carlo (MCMC) analysis (see below), thus supporting the close phylogenetic relationships between the plant‐infecting CCGaV and some invertebrate‐specific nsRNA viruses.
CCGaV RNA2 is an ambisense RNA with an intergenic region (IR) containing transcription termination signals (TTSs) reported previously in phleboviruses
RNA2 is an ambisense RNA containing two ORFs, one in each polarity strand, and 5′ and 3′ UTRs of 52 and 74 nt, respectively (Fig. 3a). The two ORFs (ORF2a and ORF2b) are separated by an AU‐rich (76.3%) IR of 300 nt which, in both polarity strands, adopts structured conformations containing a long hairpin (Fig. 3b). Interestingly, the primary and secondary structures of the long hairpin are almost identical in the two RNA strands. As with other ambisense viruses of the genera Phlebovirus, Tenuivirus and Tospovirus, these structural elements could act as termination signals during genome transcription (Kormelink et al., 2011). In addition, the two hairpins contain, at the same relative position, a CUCUGCU sequence that is similar to the TTS first reported in both the negative‐sense M and ambisense S RNAs of several phleboviruses (Albariño et al., 2007), and also identified in the phlebo‐like Bole tick virus (BTV) (Fig. 3c).
Northern blot assays support a role of IR in the expression strategy of CCGaV RNA2. When total nucleic acid preparations from CCGaV‐infected leaves were separately tested with digoxigenin (DIG)‐labelled, strand‐specific riboprobes targeting the respective coding region (Fig. 3a), the full‐length viral RNA2 (vRNA2) or viral complementary (vcRNA2) and the respective mRNAs (expected sizes of about 1300 and 1500 nt, respectively) were detected (Fig. 3d). In contrast, only the full‐length vRNA2 and vcRNA2 were detected when the same preparations were examined with two probes targeting non‐coding regions downstream of the IR in each polarity strand, thus showing that transcription of the two viral mRNAs is not extended beyond the IR.
CCGaV RNA2 codes for a putative MP and a nucleocapsid protein
ORF2a encodes a putative protein of 407 amino acids (45.83 kDa, p46, Fig. 3a). SMART searches for conserved protein domains were negative and only a stretch of 170 amino acids shared 45% identity (76 amino acids) with the MP of CPsV (Robles Luna et al., 2013). Although the significance of such a similarity was low (Table S4), multiple alignment with promals3d showed that p46 and MPs of the 30K superfamily share the same core domain formed by an α‐helix followed by seven β‐strands and a nearly invariant aspartate (D) (Melcher, 2000; Mushegian and Elena, 2015) (Fig. S4, see Supporting Information). Interestingly, the MPs of several ophioviruses, including CPsV, have been shown recently to belong to the 30K superfamily (Borniego et al., 2016; Mushegian and Elena, 2015). In addition, ML phylogenetic analyses with the MP of representative nsRNA viruses infecting plants showed that CCGaV p46 is clustered together with the MPs of ophioviruses (Fig. S5, see Supporting Information), thus suggesting the involvement of p46 in viral trafficking. Further experimental evidence is needed to confirm such a functional role for p46.
The complementary strand of RNA2 contains ORF2b (Fig. 3a), which encodes a putative protein of 350 amino acids (39.44 kDa, p39). blastp search found significant matches with segments of about 200 amino acids of the nucleocapsid of several phleboviruses, although the identity was relatively low (25%–27%; E‐value: 3.0 e‐10 to 6.0 e‐4) (Table S4, see Supplementary Information). Accordingly, PFAM search identified a stretch of 218 amino acids (from amino acid 90 to 307) in p39 closely related to a conserved domain in the nucleocapsids of tenuiviruses and phleboviruses (Tenui_N; PF05733; E‐value: 4.2e‐14). However, ML phylogenetic analysis using p39 and the nucleocapsid proteins of representative viruses in the family Phenuiviridae clustered CCGaV with phleboviruses, in a different cluster with respect to the plant‐infecting tenuiviruses (Fig. S6, see Supporting Information). In addition, Phyre2 analysis identified, with 100% confidence, the nucleocapsid of the Rift Valley fever virus and other phleboviruses as the top template to model the tertiary structure of about 62% of the full p39 (216 residues, from amino acid 106 to 342), further supporting the close structural relationships between p39 and the nucleocapsids of phleboviruses.
CCGaV is a non‐enveloped virus with filamentous particles containing nucleocapsid protein
The actual expression and structural role of p39 were confirmed by electron microscopy and serology. Filamentous, flexuous, virus‐like particles, 200–300 nm long and 6 nm wide, were observed by transmission electron microscopy (TEM) in preparations from sap (Fig. 5a) and partially purified sap (Fig. 5b) from CCGaV‐infected citrus leaves, but not from non‐infected leaves. Similar particles were also observed when sap preparations from affected trees were briefly fixed with glutaraldehyde before staining, thus supporting the absence of a viral envelope.
Figure 5.
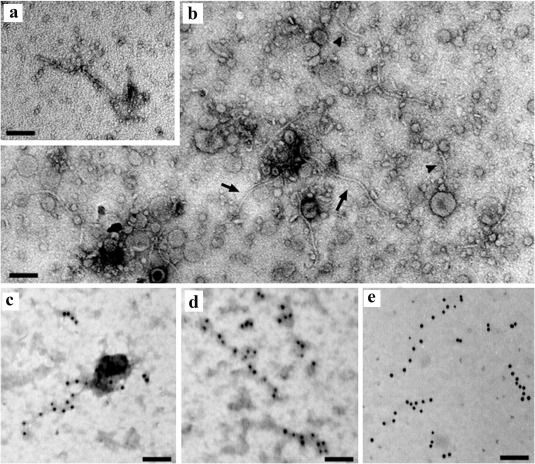
Electron micrograph of citrus concave gum‐associated virus (CCGaV) particles. (a) Negative staining of a particle from the sap. (b) Partially purified dip preparation with elongated flexuous particles of 200–300 nm (arrows) and shorter fragments thereof (arrowheads). (c–e) Immunolabelled gold particles, stained (c, d) and unstained (e). Bar, 100 nm.
These particles were decorated by gold immunolabelling using a polyclonal antiserum raised against a synthetic peptide from the N‐terminal region of p39 (Fig. 5c–e), thus confirming the expression and structural role of this protein as the nucleocapsid of flexuous and filamentous virions.
CCGaV most probably evolved from an invertebrate‐infecting ancestor
In an attempt to determine whether the plant‐infecting CCGaV actually originated from an invertebrate‐restricted phlebo‐like virus, as suggested by ML analyses and several structural features of its RNAs and proteins, an ancestral Bayesian MCMC phylogenetic reconstruction of the ancestral host was performed using the viral RdRps of representative segmented nsRNA viruses and including in the analysis viruses infecting fungi, plants, vertebrates and/or invertebrates.
The inferred MCMC tree (Fig. 6) showed significant posterior probability values at most nodes and a topology congruent with the ancestral reconstruction of Bunyavirales and other nsRNA viruses proposed recently (Li et al., 2015; Marklewitz et al., 2015). The tree shows that all nsRNA virus groups, including those infecting plants, most probably originated from a common invertebrate‐infecting ancestor. Interestingly, the plant‐infecting CCGaV also probably originated from an invertebrate‐infecting ancestor virus, as indicated by the high host posterior probability of its most proximal node (92.8%). Moreover, the basal position of this node suggests that adaptation of CCGaV to plants predated adaptation of phleboviruses and tenuiviruses to vertebrates and plants, respectively. The other plant‐infecting viruses (emaraviruses and tospoviruses) also appear to have evolved from invertebrate‐infecting viruses, but they are phylogenetically more distant from CCGaV (Fig. 6).
Figure 6.
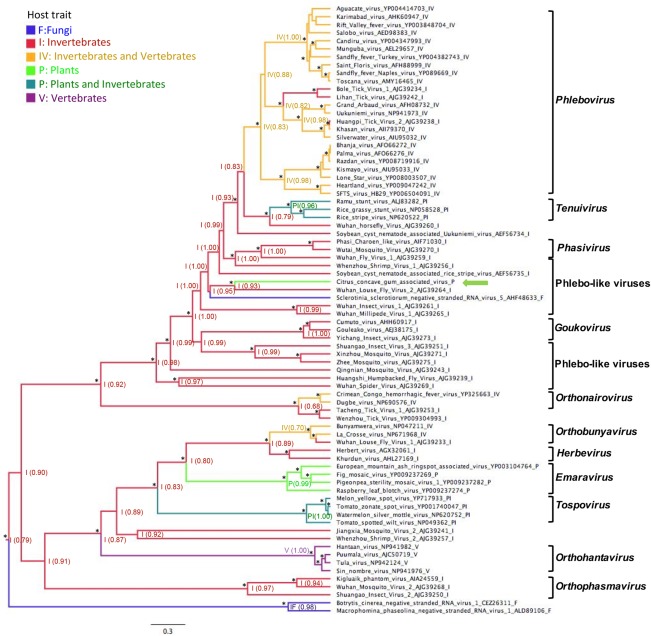
Ancestral state reconstruction of host trait based on the RNA‐dependent RNA polymerase (RdRp). The most probable host state is reported at each node with the posterior probability shown in parentheses. Asterisks denote a Bayesian posterior probability of >90%. Branches are colour coded according to the host state and are proportional to the genetic distances. The scale bar indicates substitutions per amino acid site. Square brackets delimit clusters of viruses belonging to the same genus, the name of which is on the right. citrus concave gum‐associated virus is indicated by a green arrow.
RNA1 and RNA2 are closely associated with each other and with CG disease, and are graft transmissible to indicator plants
A total of 86 citrus trees of the species sweet orange, clementine (C. clementina Hort. Ex Tan.) and mandarin (C. reticulata Blanco), from the Campania region (southern Italy), were tested for CCGaV infection. CCGaV RNA1 and RNA2, always associated with each other, were detected by mRT‐PCR in most (83.7%) trees showing typical CG symptoms on the trunk (Table 1). In contrast, only one of 43 non‐symptomatic samples tested positive. In line with these findings, CCGaV was graft transmitted from sweet orange trees expressing CG symptoms (isolates CGW1 and CGW2) to seedlings of four different citrus indicator species [Madame vinous sweet orange, grapefruit (C. paradisi Macf.), rough lemon (C. jambhiri Lush) and Dweet tangor (C. reticulata Blanco × C. sinensis)] (Table S5, see Supporting Information). Symptoms of fleck and oak‐leaf pattern (Fig. 1d,f) were sporadically and transiently observed in young leaves of the inoculated Madame vinous seedlings. Altogether, these data provide firm evidence for the association of CCGaV with CG disease in sweet orange, mandarin and clementine.
Table 1.
Preliminary survey of citrus concave gum‐associated virus (CCGaV) in several citrus species.
| Symptomatic plants | Asymptomatic plants | |||
|---|---|---|---|---|
| Species | Tested | CCGaV positive | Tested | CCGaV positive |
| Citrus sinensis | 19 | 16 | 22 | 1 |
| C. clementina | 14 | 13 | 8 | 0 |
| C. reticulata | 10 | 7 | 13 | 0 |
| Total | 43 | 36 (83.7%) | 43 | 1 (2.3%) |
Discussion
The present investigation of the aetiology of concave gum, a severe disease of citrus, allowed the identification of a novel nsRNA virus (CCGaV) closely related to nsRNA viruses infecting vertebrates or invertebrates. The two genomic CCGaV RNAs show conserved self‐complementary 3′ and 5′ termini sharing significant sequence identity with those of the genomic RNAs of several phlebo and phlebo‐like viruses. Moreover, Northern blot hybridization showed that the AU‐rich IR of the bicistronic ambisense CCGaV RNA2 probably regulates transcription termination of both the viral and viral complementary mRNAs, a feature shared by the IR of several ambisense genomic components of phleboviruses, tospoviruses and tenuiviruses (Kormelink et al., 2011). However, the presence of a sequence stretch (CUCUGCU) in both the viral and viral complementary strands of the CCGaV IR, resembling the TTS motif reported previously in the vertebrate‐infecting phleboviruses, supports a closer relationship of CCGaV with the latter taxon. Although this motif was reported in non‐structured IRs (Albariño et al., 2007), the IR of CCGaV RNA2 forms a long hairpin in both polarity strands, with the CUCUGCU stretch on the top of this structural element. This situation is common to the S RNA of the Punta Toro phlebovirus (Emery and Bishop, 1987) which also contains similar TTSs in a hairpin structure in both polarity strands. Interestingly, a GUCUGCU motif also exists in the 5′ terminal sequence of the monocistronic CCGaV RNA1, thus suggesting that it could also act as a TTS of the viral RdRp mRNA.
When RdRp and nucleocapsid proteins are considered, CCGaV also appears to share close structural motifs with phleboviruses and invertebrate‐infecting phlebo‐like viruses. Accordingly, the ML and MCMC phylogenetic trees inferred using RdRp amino acid sequences highlighted that phlebo‐like viruses are those with the closest phylogenetic links with CCGaV.
Although the data reported above strongly support evolutionary relationships of CCGaV with phleboviruses, other traits discriminate it from these and other genera in the order Bunyavirales. Contrary to the latter viruses, which have a tripartite genome, CCGaV has a genome most probably formed by only two RNAs, as no additional genomic component, potentially encoding glycoprotein, was found by RT‐PCR or by in silico search strategies independent of protein homologues. The CCGaV bipartite nature resembles the genome of several phlebo‐like viruses infecting invertebrates that are composed only of L and S segments (Li et al., 2015; Shi et al., 2016; Tokarz et al., 2014).
From a structural point of view, the flexuous and non‐enveloped CCGaV virions also diverge from the roundish and enveloped virions of phleboviruses. In this context, CCGaV appears similar to tenuiviruses, which are also non‐enveloped nsRNA viruses (Falk and Tsai, 1998), although the genome composition (four to six segments) is largely different. The non‐enveloped virions of CCGaV also support the absence of additional genomic component(s) encoding glycoprotein(s). Lack of glycoprotein(s) has also been reported in other phlebo‐like viruses infecting arthropods and recurrently observed in the invertebrate virosphere, especially in nsRNA viruses (Li et al., 2015; Shi et al., 2016). Interestingly, the ability to code for glycoproteins has been preserved in the genome of all nsRNA plant viruses that are also hosted or vectored by arthropods (nucleorhabdoviruses, cytorhabdoviruses, emaraviruses, tospoviruses and tenuiviruses). In contrast, the ophioviruses and varicosaviruses, which are plant restricted or transmitted by fungi (Lot et al., 2002), do not code for any glycoprotein. In this respect, it is noteworthy that the available information indicates that CCGaV is most probably not transmitted by an animal vector. Indeed, on the one hand, we have shown that CCGaV is closely associated with concave gum disease, but, on the other, vector‐mediated transmission of the CG infectious agent has not been shown so far (Roistacher, 1991). Altogether, these differential features of CCGaV with respect to the other nsRNA viruses suggest that the former could be the type member of a novel nsRNA genus.
The recent identification of a huge number of arthropod‐restricted nsRNA viruses and their phylogenetic relationships with viruses infecting divergent host taxa support the proposal that all the extant nsRNA viruses, including those with dual tropism (invertebrates/vertebrates or invertebrates/plants) and those restricted to vertebrates, plants or fungi, derived from ancestors exclusively infecting arthropods (Guterres et al., 2017; Junglen, 2016; Li et al., 2015). The Bayesian phylogeny‐based reconstruction of the ancestral host using the RdRp (Fig. 6) is in line with this hypothesis and shows that CCGaV most probably also had an invertebrate‐infecting ancestor. Moreover, the CCGaV tree branch is nested directly with that of an invertebrate‐restricted phlebo‐like virus and far from the other plant‐infecting viruses. Therefore, similar to the vertebrate‐infecting viruses (Junglen, 2016), plant nsRNA viruses are grouped in paraphyletic clades, suggesting that the adaptation of invertebrate‐infecting viruses to plants evolved independently several times during the evolutionary history of nsRNA viruses. It has been proposed that the switch from mono to dual tropism could have been promoted by recombination and/or reassortment of a pre‐existing ‘housekeeping‐replication module’ of genes with an ‘interactive module’ containing the genes needed for the adaptation to a new host taxon (Dolja and Koonin, 2011). In plant‐infecting viruses, such an ‘interactive module’ might correspond to the MP and RNA silencing suppressor genes, allowing systemic invasion of the host plant and impairment of its antiviral defence mechanism based on RNA silencing, respectively (Dolja and Koonin, 2011; Hedil and Kormelink, 2016). A similar adaptation pathway has been proposed for the phleboviruses and orthobunyaviruses infecting vertebrates, with the difference that, in these cases, the interactive module acquired by the invertebrate‐restricted ancestor was needed to impair the vertebrate antiviral defences (Marklewitz et al., 2011).
Interestingly, in the ML tree inferred with the MPs (Fig. S5), CCGaV clusters with members of the family Ophioviridae, thus pointing to a different origin of the gene coding for this protein with respect to the RdRp and nucleocapsid genes, which, instead, are closely related to those of phenuiviruses. These data show that CCGaV displays the typical modular genome evolution proposed for most eukaryotic viruses (Koonin et al., 2015), with the acquisition of the MP gene being the key step in the adaptation of the invertebrate‐infecting ancestor of CCGaV to plants. Such an ancestor most probably gained the MP through lateral gene transfer, which is an event recurrently observed during the evolution of nsRNA viruses infecting invertebrates (Shi et al., 2016). However, in contrast with the multisegmented tenuiviruses and emaraviruses, the CCGaV ancestor did not acquire additional genomic RNAs during its evolution and adaptation to its plant host. The CCGaV bipartite genome instead suggests a loss of a genomic segment with respect to the ancestor. This evolutionary pathway, unique among the known nsRNA viruses infecting plants, recalls again that proposed for the recently identified bipartite phlebo‐like viruses restricted to invertebrates (Li et al., 2015; Shi et al., 2016; Tokarz et al., 2014). Moreover, in contrast with the tri‐segmented tospoviruses, CCGaV did not incorporate the MP gene in a genomic RNA also encoding the glycoprotein. Instead, it integrated the MP gene in the same RNA coding for the nucleocapsid protein, showing a unique genome organization amongst plant‐infecting nsRNA viruses. Whether the putative MP also has suppressor activity of RNA silencing remains to be investigated. In this respect, it is noteworthy that the MP of several plant viruses also shows RNA silencing suppressor activity (Csorba et al., 2015; Robles Luna et al., 2017).
Altogether, the molecular, biological and phylogenetic features of CCGaV indicate that it could be the representative member of a novel virus lineage that probably followed adaptation pathways to its plant host independent of those reported previously for other plant‐infecting nsRNA viruses. Thus, the discovery of CCGaV further expands the huge diversity of nsRNA viruses recently highlighted in invertebrates (Li et al., 2015; Shi et al., 2016; Tokarz et al., 2014).
Finally, within the framework of this study, an RT‐PCR method for the efficient and specific detection of CCGaV was developed. Moreover, the polyclonal antiserum against the nucleocapsid protein used in TEM can be employed for reliable serological detection of this virus. Therefore, new tools are available for field surveys, for the improvement of sanitation and certification programmes of propagated citrus plants and for the investigation of the relationships, if any, of CCGaV with cristacortis and impietratura, which are the two other citrus diseases still awaiting identification of their aetiological agent.
Experimental Procedures
RNA isolation and deep sequencing of cDNA libraries
Symptomless and CG‐affected citrus trees (W4 and CGW2, respectively), used for the generation of cDNA libraries sequenced by next‐generation sequencing (NGS), were grown in the same field in a varietal collection in Eboli, Salerno province (Italy). They tested negative to CPsV infection when assayed by enzyme‐linked immunosorbent assay (ELISA) and RT‐PCR performed according to Martín et al. (2004). Citrus trees in the same field and in other areas of the Campania region (Italy) were assayed within the framework of the survey.
Total nucleic acids were extracted with phenol–chloroform from leaves or bark from green stems, recovered by ethanol precipitation and processed as reported previously (Di Serio et al., 2010) to generate cDNA libraries of sRNAs (16–30 nt), which were sequenced (run 1 × 50) on an Illumina Genome HiScan Analyzer by Fasteris custom service (Fasteris, Geneva, Switzerland). Nucleic acid preparations enriched in dsRNAs were obtained by extracting bark with buffer‐saturated phenol and partitioning the nucleic acids by chromatography on non‐ionic cellulose CF‐11 (Whatman, Maidstone, UK) with STE [50 mm Tris‐HCl, pH 7.2, 100 mm NaCl, 1 mm ethylenediaminetetraacetic acid (EDTA)] containing 16% ethanol (Morris and Dodds, 1979). The dsRNA‐enriched fraction was recovered by ethanol precipitation and digested with TURBO DNase (Ambion, Foster City, CA, USA). After removing ribosomal RNAs (rRNAs) by the Ribo‐Zero Plant leaf kit (Illumina, San Diego, CA, USA), an RNA‐seq cDNA library was generated according to standard Illumina procedures, and pair‐end sequencing (2 × 125) was performed on an Illumina Genome Analyzer HiSeq 2500 (Fasteris, Switzerland).
Sequencing of the complete viral genome
Raw sequenced reads were filtered for quality, trimmed and de novo assembled into larger contigs using Velvet Software 1.2.08 (Zerbino and Birney, 2008) with k‐mer 15–17 and 39 for the sRNA and dsRNA libraries, respectively. The contigs were screened for homologous viral sequences by blastx on the National Center for Technology Information (NCBI) databases. To exclude non‐viral sequences, contigs with identity e‐values of >0.001 were further searched in the entire NCBI database and in the citrus genome database (https://phytozome.jgi.doe.gov/pz/portal.html#!info?alias=Org_Csinensis). Specific primers (Table S3) were designed into putative viral contigs to generate overlapping cDNA by RT‐PCR. RNA preparation, RT‐PCR solutions and conditions were similar to those reported previously (Navarro et al., 2017), except that the polymerase extension step ranged from 1 to 3 min, depending on the size of the expected PCR product. The termini of the viral genomic RNAs were determined by RACE. Briefly, in the case of 5′ RACE, total RNA or dsRNA‐enriched preparations were reverse transcribed using a specific primer (Table S3) and, after the addition of a poly(dG) tail, the tailed cDNAs were PCR amplified with appropriate primers (Table S3). In contrast, to perform 3′ RACE, the Escherichia coli Poly(A) polymerase (New England Biolabs, Hitchin, UK) was used to add a polyA tail to the RNA. Polyadenylated RNAs were reverse transcribed using the primer Poly(dT) tail 3′RACE (Table S3) and PCR amplified with suitable primers (Table S3). Amplicons were gel purified, cloned into pGEMT‐Easy vector (Promega, Madison, Wisconsin, USA) and sequenced by Sanger Sequencing Custom Service (Macrogen, Amsterdam, the Netherlands).
To search for additional viral genomic segments, primers were designed in the conserved terminal ends of CCGaV RNA1 and RNA2 (Table S3) and employed in RT‐PCR amplification reactions using RNA preparations from CCGaV‐infected and non‐infected trees. An in silico strategy of sequential searching was also adopted to identify putative additional viral RNA segments without homology with previously known viral sequences in databases. To this end, reads of 55–125 nt from the dsRNA‐enriched library were exhaustively screened for the identification of those containing at their ends the same nine nucleotides conserved at the 5′ and 3′ termini of RNA1 and RNA2. Alignments of deep‐sequenced reads to CCGaV genomic RNAs were performed using Bowtie (Langmead et al., 2009) implemented in the MacVector Assembler platform (15.1.5, MacVector, Inc. Apex, North Carolina, USA).
Sequence analysis and phylogenetic trees
The mfold webserver (Zucker, 2003) was used to predict RNA secondary structures. Potential ORFs and conserved protein domains were identified using ORF Finder at the NCBI (http://www.ncbi.nlm.nih.gov/gorf/gorf.html) and PFAM (http://pfam.xfam.org/) databases (Finn et al., 2014), respectively. The Phyre2 web portal (Kelley et al., 2015) was used for modelling, prediction and analyses of CCGaV proteins. When indicated, multiple alignments of protein sequences and/or structures were performed using promals3d (http://prodata.swmed.edu/promals/promals.php; Pei et al., 2008).
The RdRp phylogenetic analysis was performed using the core amino acid sequence in the proteins encoded by 76 nsRNA viruses, including CCGaV and representative members of the genera Phlebovirus, Orthonairovirus, Orthobunyavirus, Tospovirus, Orthohantavirus, Emaravirus, Tenuivirus, Phasmavirus, Goukovirus and Herbevirus, and several unclassified viral species related to these genera that have been identified recently in arthropods and fungi. Multiple alignments were performed with the Expresso structural alignment algorithm available at the TCoffee webserver (http://tcoffee.crg.cat/apps/tcoffee) (Armougom et al., 2006). After removing poorly aligned regions with TrimAl (Capella‐Gutiérrez et al., 2009), an alignment with a total of 279 positions was generated. The RdRp‐based phylogenetic tree inferred using the ML method was based on the best‐fit LG amino acid substitution model (Le and Gascuel, 2008), with a gamma distribution of rate variation with four categories and a subtree pruning and regrafting (SPR) topology searching algorithm. The statistical support of the clades was measured by a heuristic search with 100 bootstraps The analysis was implemented in mega7 (Kumar et al., 2016). The same alignment was used to infer the MCMC phylogenetic tree in the Beast 2.4.4 software package (Bouckaert et al., 2014). The ProtTest 3.4 (Abascal et al., 2005) best‐fit amino acid substitution model was LG with gamma distributed rate variation across sites. A strict clock model and a constant population‐size coalescent prior were used. The convergence and the effective sample size of parameter estimates were examined with Tracer v1.6 (http://beast.bio.ed.ac.uk/Tracer). The maximum clade credibility (MCC) tree was generated by Tree Annotator v2.4.4 (http://beast.bio.ed.ac.uk/treeannotator) from 10 000 trees after removing a burn‐in of 10% and including a description of ancestral host associations. Nodal support was summarized as posterior probabilities.
The ML phylogenetic trees of the MP and nucleocapsid proteins were inferred in the CLC Main Workbench 7.7.3 package (protein best‐fit substitution model JTT) and mega7 (protein best‐fit substitution model LG + G), respectively, using a multiple sequence alignment generated by COBALT (https://www.ncbi.nlm.nih.gov/tools/cobalt/re_cobalt.cgi), from which poorly aligned regions were removed with TrimAl (Capella‐Gutiérrez et al., 2009).
Northern blot assays
Total nucleic acid preparations from bark (3 µg) or partially purified viral preparations were fractionated by electrophoresis on denaturing formaldehyde‐agarose gel, transferred by capillarity to nylon membranes (Hybond‐N, Amersham, Little Chalfont, UK) and immobilized by UV irradiation. The membranes were hybridized with DIG‐labelled riboprobes, specific for the viral strand of CCGaV RNA1 (probe: from nt 3503 to 3987) or different regions of viral and viral complementary strands of CCGaV RNA2 (probe A: from nt 86 to 1252; probe B: from nt 1796 to 2488; probe C: from nt 1796 to 2068; probe D: from nt 581 to 734). Pre‐hybridization and hybridization were performed as described previously (Hajizadeh et al., 2012). The membranes were analysed with a Chemidoc Touch Imaging system (Bio‐Rad, Hercules, CA, USA). rRNAs stained with ethidium bromide were used as equal loading controls.
Field survey and bioassays
A field survey was performed in 2016 using a multiplex RT‐PCR assay developed to detect simultaneously both CCGaV RNA1 and RNA2. Three primer pairs (Bunya‐cit.1F/Bunya‐cit.4R; CG.15/CG.20; CG.18/CG.19) (Table S3) were designed to generate PCR amplicons of 485, 292 and 154 nt from regions of the CCGaV genome coding for the RdRP (RNA1), nucleocapsid and MP proteins (RNA2), respectively. Total nucleic acids (100 ng) were extracted and reverse transcribed as reported previously (Navarro et al., 2017). PCR amplification was performed using 2 µL of the cDNA reaction and 1.25 units of GoTaq polymerase (Promega) in a reaction volume of 25 µL, containing a final concentration of 0.2 µm of each primer. The cycling conditions were as follows: initial denaturation at 94 ºC for 3 min, followed by 32 cycles at 94 ºC for 30 s, 55 ºC for 30 s, 72 ºC for 30 s, and a final extension step at 72 ºC for 7 min. The reaction products were analysed by electrophoresis on 2% agarose gels buffered in TAE (0.04 m Tris–acetate, 1 mm EDTA, pH 8) and visualized by UV light after ethidium bromide staining.
Bioassays were performed using, as source of inoculum, two trees affected (CGW1 and CGW2 trees) and one tree non‐affected (W4) by CG disease. These trees were grown in the same field located in Eboli (Salerno province, Italy). Seedlings (height, 25–30 cm) of sweet orange cv. Madam vinous, grapefruit, rough lemon and dweet tangor were graft inoculated by chip budding and maintained under controlled conditions in a glasshouse. The grafted plants were inspected weekly or at shorter intervals for 6 months.
TEM
TEM observations were performed using sap and partially purified preparations of virus particles. Sap was extracted from leaves with 0.05 m phosphate buffer (pH 7.2) and, when indicated, the extract was briefly fixed with 1% glutaraldehyde before staining. Partially purified virus particles were obtained by the clarification and precipitation steps of the protocol reported by Alioto et al. (1999). TEM samples were prepared as follows. A carbon‐coated copper/rhodium grid (400 mesh; TAAB Laboratories Equipment Ltd., Aldermaston, Berkshire, UK) was floated for 2 min on a small drop (50 µL) of sample (sap, sap briefly fixed in 1% glutaraldehyde or partially purified preparation) and rinsed with 200 µL of double‐distilled water. Negative staining was performed with 200 µL of 2% w/v uranyl acetate solution (TAAB Laboratories Equipment Ltd.). After draining off the excess staining solution, the specimen was transferred for examination to a Philips (Amsterdam, the Netherlands) Morgagni 282D transmission electron microscope, operating at 60 kV. Immunogold labelling was carried out as described by Louro and Lesemann (1984) with some modifications. Carbonated grids were floated for a few minutes on a drop of sample, rinsed in 0.05 m phosphate‐buffered saline (PBS) containing 0.01% Tween 20 and 0.01% Triton (PBSTT) for 20 min, and incubated for 2 h at room temperature on a drop (dilution 1 : 1 with PBSTT) of rabbit polyclonal antibody (GenScript, Piscataway, New Jersey, USA) against the N‐terminal region of the p39 protein encoded by the CCGaV ORF2b (PKFATRDGDDGQEPC). After floating on a drop of PBSTT for 10 min, the grids were exposed overnight at 4 °C to a preparation of colloidal gold of 10 nm in diameter conjugated with anti‐rabbit antibodies (Sigma, St. Louis, Missouri, USA) diluted 1 : 50 with PBSTT. Grids were rinsed with PBS and glass‐distilled water prior to viewing under the electron microscope. The negative staining procedure was performed in the same manner as the dip method. The size of the virus particles was the mean of 15 independent measurements.
Accession numbers and virus names
CCGaV RNA1 and RNA2 have the GenBank accession numbers KX960111 and KX960112, respectively. Virus names are abbreviated as follows: BTV, Bole tick virus 1, KM817731; BUNV, Bunyamwera virus, NC_001925; DUGV, Dugbe virus, NC_004159; EMARaV, European mountain ash ringspot‐associated virus, NC_013105; GOUV, Gouleako virus, HQ541738; HTNV, Hantaan virus, NC_005222; PTV, Punta Toro virus, NC_027201, KR912210; RGSV, rice grassy stunt virus, NC_002323; RSV, rice stripe virus, NC_003755; RVFV, Rift Valley fever virus, NC_014397, NC_014396, NC_014395; SFTSV, severe fever with thrombocytopenia syndrome, NC_018136; SFSV, sandfly fever Sicilian virus, NC_015412, NC_015411, EF201822; TOSV, Toscana virus, NC_006319, NC_006320, NC_006318; TSWV, tomato spotted wilt virus, NC_002052; UUKV, Uukuniemi virus, NC_005214, NC_005220, NC_005221.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Detection of the novel viral RNAs in citrus plants by multiplex reverse transcription‐polymerase chain reaction (mRT‐PCR). Lanes 1, 2 and 4, citrus plants not showing symptoms of concave gum (CG) disease; lane 3, citrus tree with typical symptoms of CG and used for the generation of cDNA libraries sequenced by Illumina technology; lanes 5–7, citrus plants showing typical symptoms of CG; M, 100‐bp DNA ladder. The positions of expected amplicons of RNA1 (485 bp) and RNA2 (292 and 154 bp) are indicated on the right by full and broken arrows, respectively.
{kind=link}
Fig. S2 Detection of citrus concave gum‐associated virus (CCGaV) RNA1 by Northern blot hybridization with a digoxigenin (DIG)‐labelled riboprobe specific for the viral strand. Lane 1, nucleic acids extracted from a partially purified viral preparation from a concave gum (CG)‐affected tree (CGW2); lane 2, nucleic acid preparation from a non‐symptomatic citrus tree (W4) obtained as for lane 1; lane M, Millennium RNA marker (Applied Biosystems/Ambion) with sizes (kb) indicated on the right.
{kind=link}
Fig. S3 Phylogenetic analysis based on the RNA‐dependent RNA polymerase (RdRp) conserved domain. Citrus concave gum‐associated virus (CCGaV) and representative members of the order Bunyavirales and several unclassified negative‐stranded RNA (nsRNA) viruses are considered. The maximum‐likelihood (ML) tree was inferred using mega7 software. The names of the viruses, the accession numbers and the respective host organisms (invertebrate, I; vertebrate, V; invertebrate and vertebrate, IV; plant, P; plant and invertebrate, PI; fungus, F) are shown at the branch tip. The number at each node is the result of bootstrap analysis (100 replicates). Tree branches are proportional to the genetic distances between the sequences; the scale bar indicates substitutions per amino acid site. The names of recognized genera and of a novel lineage (phlebo‐like viruses) related to phleboviruses are reported on the right.
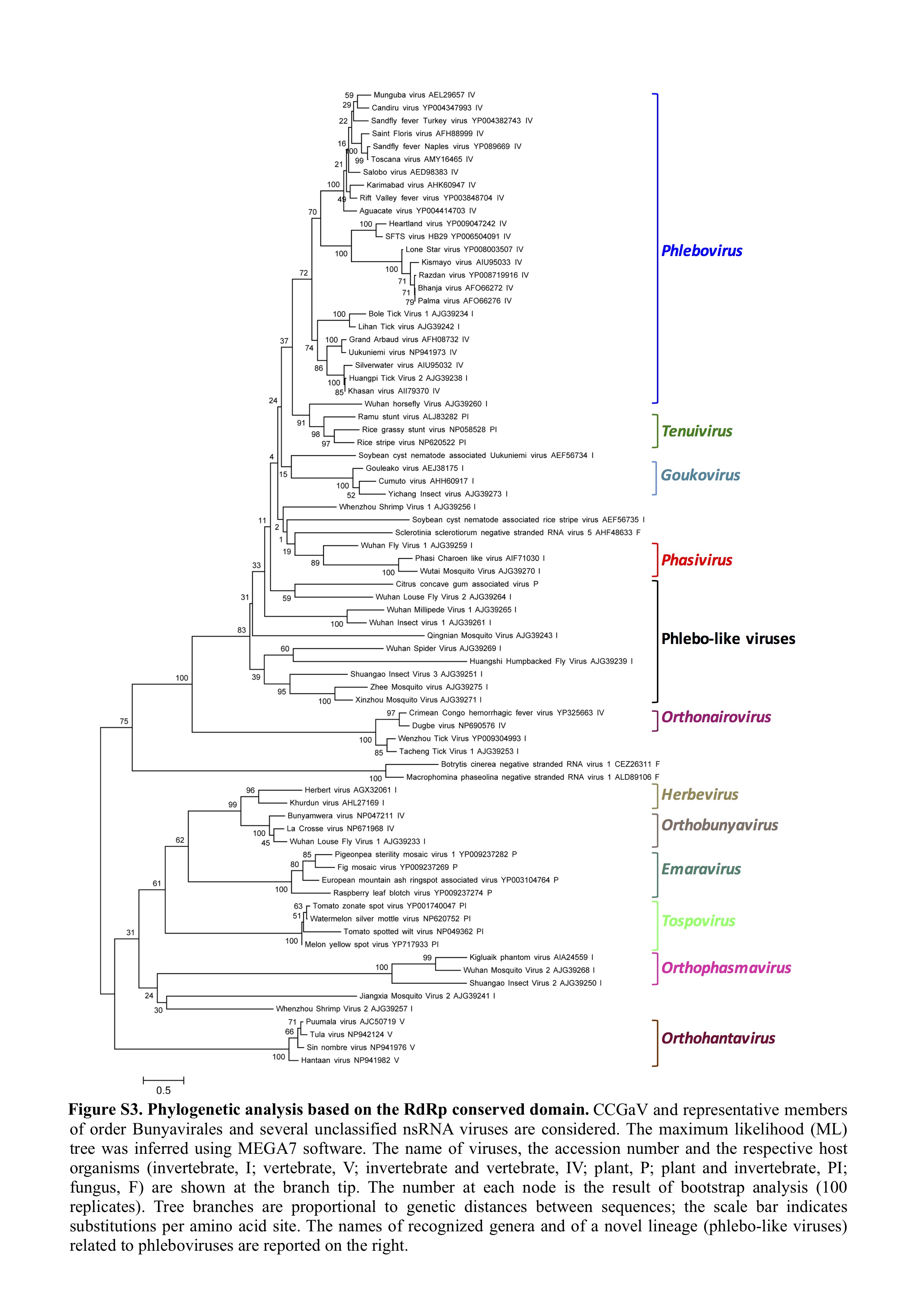{kind=link}
Fig. S4 Multiple sequence alignment of movement proteins from citrus concave gum‐associated virus (CCGaV) and plant viruses of the 30K superfamily. Sequences are from representative members of the virus genera in the 30K superfamily. CCGaV is shown in bold. The virus name, GenBank identifier, virus genus and distance in amino acids from the N‐terminus of the protein are shown on the left‐hand side of each sequence. Consensus secondary structure elements, predicted by PSIRED within the promals3d program, are reported at the top, with the strands and helices indicated by e and h, respectively. Amino acids that fold in helices and strands are in red and blue, respectively. The nearly invariant aspartic acid residue (the D motif) is shown in bold.
Fig. S5 Maximum‐likelihood phylogenetic tree inferred with the putative movement protein (MP) of citrus concave gum‐associated virus (CCGaV) and the MP of representative plant‐infecting viruses of the genera Emaravirus, Ophiovirus, Tenuivirus, Tospovirus, Nucleorhabdovirus and Varicosavirus. The number at each node is the result of bootstrap analysis (1000 replicates). Tree branches are proportional to the genetic distances between sequences and the scale bar at the bottom indicates substitutions per amino acid site. Square brackets delimit clusters of viruses belonging to the same genus, the name of which is on the right. The CCGaV branch is highlighted in red.
{kind=link}
Fig. S6 Maximum‐likelihood phylogenetic tree inferred with the p39 protein of citrus concave gum‐associated virus (CCGaV) and the nucleocapsid proteins of representative viruses of the four genera Phlebovirus, Goukovirus, Phasivirus and Tenuivirus of the family Phenuiviridae and some unclassified phlebo‐like viruses. The number at each node is the result of bootstrap analysis (100 replicates). Tree branches are proportional to the genetic distances between sequences and the scale bar at the bottom indicates substitutions per amino acid site. Square brackets delimit clusters of viruses belonging to the same genus, the name of which is on the right. The CCGaV branch is highlighted in red.
{kind=link}
Table S1 Number of reads obtained by deep sequencing of cDNA libraries of small RNAs (sRNAs) and double‐stranded RNA (dsRNA) from citrus.
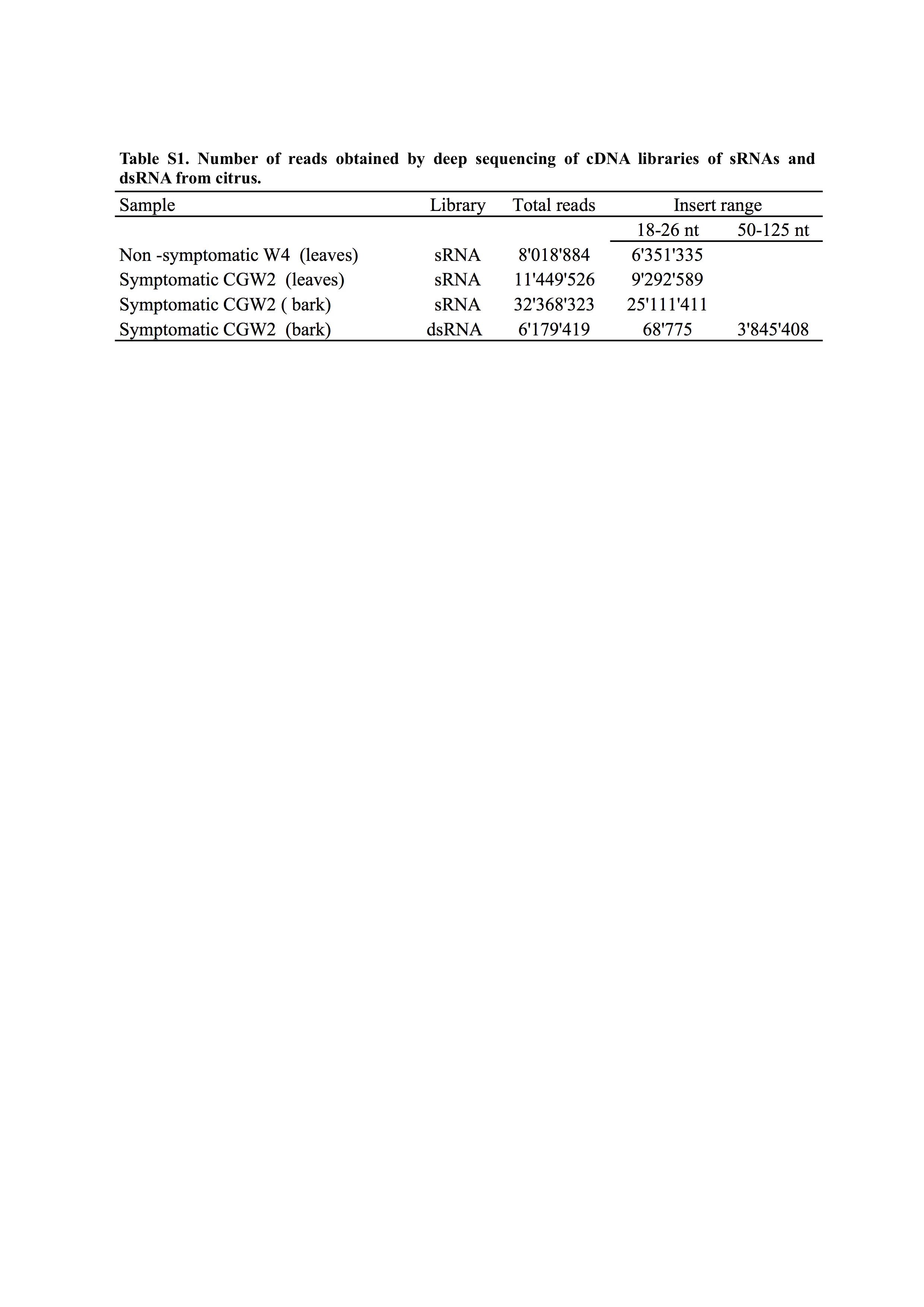{kind=link}
Table S2 blastx results of searches with contigs obtained by assembling small RNAs (sRNAs) from leaves and bark of a citrus tree showing concave gum symptoms.
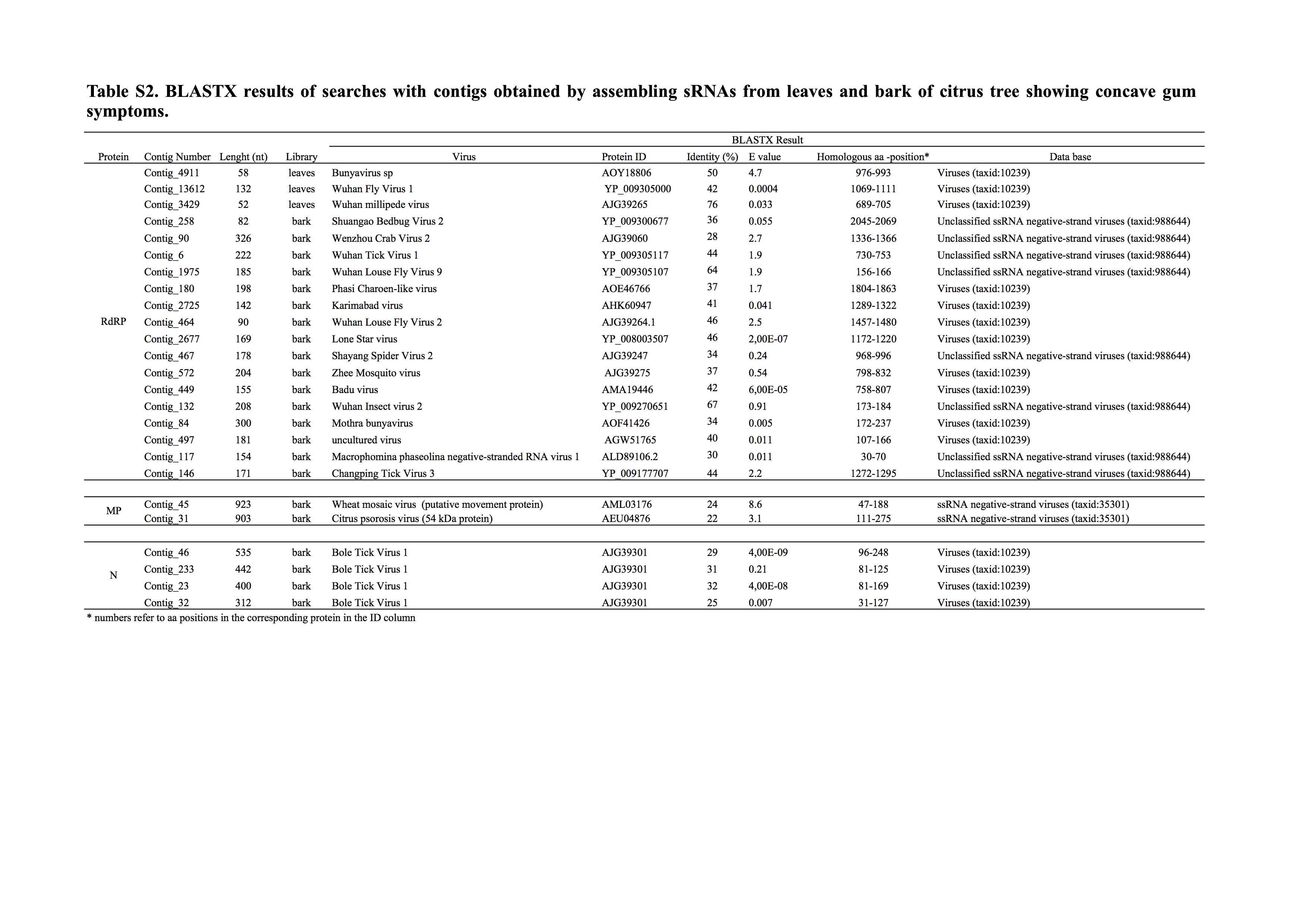{kind=link}
Table S3 List of primers used in this study.
{kind=link}
Table S4 Best blastp matches with citrus concave gum‐associated virus (CCGaV) proteins.
{kind=link}
Table S5 Results of bioassays.
{kind=link}
Acknowledgements
We wish to express our sincere thanks to Professors Maria Laura Cestaro and Patrizia Porretta, the Directors of the Istituto Tecnico Agrario Statale ‘G. Fortunato’ and Istituto Tecnico Agrario Statale ‘E. De Cillis’, respectively, for providing access to the citrus collections, to Valerio Verrina for technical assistance during the field survey, and to Professors Ricardo Flores and Giovanni Paolo Martelli for critical reading of the manuscript and for suggestions. This work was partially supported by the projects ‘URCOFI’ (funded by Regione Campania, DGR 388/2010, 23th April 2010) and ‘INNOCI’ (funded by Regione Puglia, DGR n. 903/2012, 15th May 2012).
Contributor Information
Daniela Alioto, Email: alioto@unina.it.
Francesco Di Serio, Email: francesco.diserio@ipsp.cnr.it.
References
- Abascal, F. , Zardoya, R. and Posada, D. (2005) ProtTest: selection of best‐fit models of protein evolution. Bioinformatics, 21, 2104–2105. [DOI] [PubMed] [Google Scholar]
- Albariño, C.G. , Bird, B.H. and Nichol, S.T. (2007) A shared transcription termination signal on negative and ambisense RNA genome segments of Rift Valley fever, sandfly fever Sicilian, and Toscana viruses. J. Virol. 81, 5246–5256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alioto, D. , Gangemi, M. , Deaglio, S. , Sposato, P. , Noris, E. , Luisoni, E. and Milne, R.G. (1999) Improved detection of citrus psorosis virus using polyclonal and monoclonal antibodies. Plant Pathol. 48, 735–741. [Google Scholar]
- Armougom, F. , Moretti, S. , Poirot, O. , Audic, S. , Dumas, P. , Schaeli, B. , Keduas, V. and Notredame, C. (2006) Expresso: automatic incorporation of structural information in multiple sequence alignments using 3D‐Coffee. Nucleic Acids Res. 34, W604–W608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekal, S. , Domier, L.L. , Niblack, T.L. and Lambert, K.N. (2011) Discovery and initial analysis of novel viral genomes in the soybean cyst nematode. J. Gen. Virol. 92, 1870–1879. [DOI] [PubMed] [Google Scholar]
- Borniego, M.B. , Karlin, D. , Peña, E.J. , Robles Luna, G. and García, M.L. (2016) Bioinformatic and mutational analysis of ophiovirus movement proteins, belonging to the 30K superfamily. Virology, 498, 172–180. [DOI] [PubMed] [Google Scholar]
- Bouckaert, R. , Heled, J. , Kühnert, D. , Vaughan, T. , Wu, C. , Xie, D. , Suchard, M.A. , Rambaut, A. and Drummond, A.J. (2014) BEAST 2: a software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 10, e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capella‐Gutiérrez, S. , Silla‐Martínez, J.M. and Gabaldón, T. (2009) TrimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics, 25, 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csorba, T. , Kontra, L. and Burgyán, J. (2015) Viral silencing suppressors: tools forged to fine‐tune host–pathogen coexistence. Virology, 479–480, 85–103. [DOI] [PubMed] [Google Scholar]
- da Graça, J.V. , Lee, R.F. , Moreno, P. , Civerolo, E.L. and Derrick, K.S. (1991) Comparison of isolates of citrus ringspot, psorosis, and other virus‐like agents of citrus. Plant Dis. 75, 613–616. [Google Scholar]
- Di Serio, F. , Martínez de Alba, A.E. , Navarro, B. , Gisel, A. and Flores, R. (2010) RNA‐dependent RNA polymerase 6 delays accumulation and precludes meristem invasion of a nuclear‐replicating viroid. J. Virol. 84, 2477–2489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolja, V.V. and Koonin, E.V. (2011) Common origins and host‐dependent diversity of plant and animal viromes. Curr. Opin. Virol. 1, 322–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudas, G. and Obbard, D.J. (2015) Are arthropods at the heart of virus evolution? eLife, 4, e06837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elliott, R.M. and Brennan, B. (2014) Emerging phleboviruses. Curr. Opin. Virol. 5, 50–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery, V.C. and Bishop, D.H. (1987) Characterization of Punta Toro S mRNA species and identification of an inverted complementary sequence in the intergenic region of Punta Toro phlebovirus ambisense S RNA that is involved in mRNA transcription termination. Virology, 156, 1–11. [DOI] [PubMed] [Google Scholar]
- Falk, B.W. and Tsai, J.H. (1998) Biology and molecular biology of viruses in the genus Tenuivirus . Annu. Rev. Phytopathol. 36, 139–163. [DOI] [PubMed] [Google Scholar]
- Fawcett, H.S. (1936) Citrus Diseases and Their Control. New York: McGraw‐Hill. [Google Scholar]
- Finn, R.D. , Bateman, A. , Clements, J. , Coggill, P. , Eberhardt, R.Y. , Eddy, S.R. , Heger, A. , Hetherington, K. , Holm, L. , Mistry, J. , Sonnhammer, E.L. , Tate, J. and Punta, M. (2014) Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guterres, A. , de Oliveira, R.C. , Fernandes, J. , de Lemos, E.R. and Schrago, C.G. (2017) New bunya‐like viruses: highlighting their relations. Infect. Genet. Evol. 49, 164–173. [DOI] [PubMed] [Google Scholar]
- Hajizadeh, M. , Navarro, B. , Bashir, N.S. , Torchetti, E.M. and Di Serio, F. (2012) Development and validation of a multiplex RT‐PCR method for the simultaneous detection of five grapevine viroids. J. Virol. Methods, 179, 62–69. [DOI] [PubMed] [Google Scholar]
- Han, G.Z. and Worobey, M. (2011) Homologous recombination in negative sense RNA viruses. Viruses, 3, 1358–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedil, M. and Kormelink, R. (2016) Viral RNA silencing suppression: the enigma of bunyavirus NSs proteins. Viruses, 8, 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubálek, Z. , Rudolf, I. and Nowotny, N. (2014) Arboviruses pathogenic for domestic and wild animals. Adv. Virus. Res. 89, 201–275. [DOI] [PubMed] [Google Scholar]
- Junglen, S. (2016) Evolutionary origin of pathogenic arthropod‐borne viruses—a case study in the family Bunyaviridae. Curr. Opin. Insect Sci. 16, 81–86. [DOI] [PubMed] [Google Scholar]
- Kelley, L.A. , Mezulis, S. , Yates, C.M. , Wass, M.N. and Sternberg, M.J. (2015) The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 10, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo, H. , Chiba, S. , Toyoda, K. and Suzuki, N. (2013) Evidence for negative‐strand RNA virus infection in fungi. Virology, 435, 201–209. [DOI] [PubMed] [Google Scholar]
- Koonin, E.V. , Dolja, V.V. and Krupovic, M. (2015) Origins and evolution of viruses of eukaryotes: the ultimate modularity. Virology, 479, 2–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kormelink, R. , Garcia, M.L. , Goodin, M. , Sasaya, T. and Haenni, A.L. (2011) Negative‐strand RNA viruses: the plant‐infecting counterparts. Virus Res. 162, 184–202. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. and Tamura, K. (2016) MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. , Trapnell, C. , Pop, M. and Salzberg, S.L. (2009) Ultrafast and memory‐efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le, S.Q. and Gascuel, O. (2008) An improved general amino acid replacement matrix. Mol. Biol. Evol. 25, 1307–1320. [DOI] [PubMed] [Google Scholar]
- Li, C.X. , Shi, M. , Tian, J.H. , Lin, X.D. , Kang, Y.J. , Chen, L.J. , Qin, X.C. , Xu, J. , Holmes, E.C. and Zhang, Y.Z. (2015) Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative‐sense RNA viruses. eLife, 4, e05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lot, H. , Campbell, R.N. , Souche, S. , Milne, R.G. and Roggero, P. (2002) Transmission by Olpidium brassicae of Mirafiori lettuce virus and lettuce big‐vein virus, and their roles in lettuce big‐vein etiology. Phytopathology, 92, 288–293. [DOI] [PubMed] [Google Scholar]
- Louro, D. and Lesemann, D.E. (1984) Use of protein A‐gold complex for specific labelling of antibodies bound to plant viruses. I. Viral antigens in suspensions. J. Virol. Methods, 9, 107–122. [DOI] [PubMed] [Google Scholar]
- Marklewitz, M. , Handrick, S. , Grasse, W. , Kurth, A. , Lukashev, A. , Drosten, C. , Ellerbrok, H. , Leendertz, F.H. , Pauli, G. and Junglen, S. (2011) Gouleako virus isolated from West African mosquitoes constitutes a proposed novel genus in the family Bunyaviridae . J. Virol. 85, 9227–9234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklewitz, M. , Zirkel, F. , Rwego, I.B. , Heidemann, H. , Trippner, P. , Kurth, A. , Kallies, R. , Briese, T. , Lipkin, W.I. , Drosten, C. , Gillespie, T.R. and Junglen, S. (2013) Discovery of a unique novel clade of mosquito‐associated bunyaviruses. J. Virol. 87, 12 850–12 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marklewitz, M. , Zirkel, F. , Kurth, A. , Drosten, C. and Junglen, S. (2015) Evolutionary and phenotypic analysis of live virus isolates suggests arthropod origin of a pathogenic RNA virus family. Proc. Natl. Acad. Sci. USA, 112, 7536–7541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martín, S. , Alioto, D. , Milne, R.G. , Garnsey, S.M. , García, M.L. , Grau, O. , Guerri, J. and Moreno, P. (2004) Detection of Citrus psorosis virus by ELISA, molecular hybridization, RT‐PCR and immunosorbent electron microscopy and its association with citrus psorosis disease. Eur. J. Plant Pathol. 110, 747–757. [Google Scholar]
- Marzano, S.Y. , Nelson, B.D. , Ajayi‐Oyetunde, O. , Bradley, C.A. , Hughes, T.J. , Hartman, G.L. , Eastburn, D.M. and Domier, L.L. (2016) Identification of diverse mycoviruses through metatranscriptomics characterization of the viromes of five major fungal plant pathogens. J. Virol. 90, 6846–6863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, S. , Nelson, M.I. , Turner, P.E. and Patton, J.T. (2016) Reassortment in segmented RNA viruses: mechanisms and outcomes. Nat. Rev. Microbiol. 14, 448–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher, U. (2000) The ‘30K’ superfamily of viral movement proteins. J. Gen. Virol. 81, 257–266. [DOI] [PubMed] [Google Scholar]
- Moreno, P. , Guerri, J. and García, M.L. (2015) The psorosis disease of citrus: a pale light at the end of the tunnel. J. Citrus Pathol. 2, 1–18. [Google Scholar]
- Morris, T.J. and Dodds, J.A. (1979) Isolation and analysis of double‐stranded RNA from virus‐infected plant and fungal tissue. Phytopathology, 69, 854–858. [Google Scholar]
- Mushegian, A.R. and Elena, S.F. (2015) Evolution of plant virus movement proteins from the 30K superfamily and of their homologs integrated in plant genomes. Virology, 476, 304–315. [DOI] [PubMed] [Google Scholar]
- Navarro, B. , Loconsole, G. , Giampetruzzi, A. , Aboughanem‐Sabanadzovic, N. , Ragozzino, A. , Ragozzino, E. and Di Serio, F. (2017) Identification and characterization of privet leaf blotch‐associated virus, a novel idaeovirus. Mol. Plant Pathol. 18, 925–936. doi: 10.1111/mpp.12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei, J. , Kim, B.H. and Grishin, N.V. (2008) PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotch, S.J. , Bouloy, M. , Ulmanen, I. and Krug, R.M. (1981) A unique cap(m7GpppXm)‐dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell, 23, 847–858. [DOI] [PubMed] [Google Scholar]
- Reguera, J. , Weber, F. and Cusack, S. (2010) Bunyaviridae RNA polymerases (L‐protein) have an N‐terminal, influenza‐like endonuclease domain, essential for viral cap‐dependent transcription. PLoS Pathog. 6, e1001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robles Luna, G. , Peña, E.J. , Borniego, M.B. , Heinlein, M. and Garcia, M.L. (2013) Ophioviruses CPsV and MiLBVV movement protein is encoded in RNA 2 and interacts with the coat protein. Virology, 441, 152–161. [DOI] [PubMed] [Google Scholar]
- Robles Luna, G. , Reyes, C.A. , Peña, E.J. , Ocolotobiche, E. , Baeza, C. , Borniego, M.B. , Kormelink, R. and García, M.L. (2017) Identification and characterization of two RNA silencing suppressors encoded by ophioviruses. Virus Res. 235, 96–105. [DOI] [PubMed] [Google Scholar]
- Roistacher, C.N. (1991) Graft‐Transmissible Diseases of Citrus: Handbook for Detection and Diagnosis. Rome: FAO. [Google Scholar]
- Shi, M. , Lin, X.D. , Tian, J.H. , Chen, L.J. , Chen, X. , Li, C.X. , Qin, X.C. , Li, J. , Cao, J.P. , Eden, J.S. , Buchmann, J. , Wang, W. , Xu, J. , Holmes, E.C. and Zhang, Y.Z. (2016) Redefining the invertebrate RNA virosphere. Nature, 540, 539–543. [DOI] [PubMed] [Google Scholar]
- Tokarz, R. , Williams, S.H. , Sameroff, S. , Sanchez‐Leon, M. , Jain, K. and Lipkin, W.I. (2014) Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J. Virol. 88, 11 480–11 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter, C.T. and Barr, J.N. (2011) Recent advances in the molecular and cellular biology of bunyaviruses. J. Gen. Virol. 92, 2467–2484. [DOI] [PubMed] [Google Scholar]
- Zerbino, D.R. and Birney, E. (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, Y. , Navarro, B. , Wang, G.Y. , Yang, Z. , Xu, W. , Zhu, C. , Wang, L. , Di Serio, F. and Hong, N. (2017) Actinidia chlorotic ringspot‐associated virus: a novel emaravirus infecting kiwifruit plants. Mol. Plant Pathol. 18, 569–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker, M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Detection of the novel viral RNAs in citrus plants by multiplex reverse transcription‐polymerase chain reaction (mRT‐PCR). Lanes 1, 2 and 4, citrus plants not showing symptoms of concave gum (CG) disease; lane 3, citrus tree with typical symptoms of CG and used for the generation of cDNA libraries sequenced by Illumina technology; lanes 5–7, citrus plants showing typical symptoms of CG; M, 100‐bp DNA ladder. The positions of expected amplicons of RNA1 (485 bp) and RNA2 (292 and 154 bp) are indicated on the right by full and broken arrows, respectively.
Fig. S2 Detection of citrus concave gum‐associated virus (CCGaV) RNA1 by Northern blot hybridization with a digoxigenin (DIG)‐labelled riboprobe specific for the viral strand. Lane 1, nucleic acids extracted from a partially purified viral preparation from a concave gum (CG)‐affected tree (CGW2); lane 2, nucleic acid preparation from a non‐symptomatic citrus tree (W4) obtained as for lane 1; lane M, Millennium RNA marker (Applied Biosystems/Ambion) with sizes (kb) indicated on the right.
Fig. S3 Phylogenetic analysis based on the RNA‐dependent RNA polymerase (RdRp) conserved domain. Citrus concave gum‐associated virus (CCGaV) and representative members of the order Bunyavirales and several unclassified negative‐stranded RNA (nsRNA) viruses are considered. The maximum‐likelihood (ML) tree was inferred using mega7 software. The names of the viruses, the accession numbers and the respective host organisms (invertebrate, I; vertebrate, V; invertebrate and vertebrate, IV; plant, P; plant and invertebrate, PI; fungus, F) are shown at the branch tip. The number at each node is the result of bootstrap analysis (100 replicates). Tree branches are proportional to the genetic distances between the sequences; the scale bar indicates substitutions per amino acid site. The names of recognized genera and of a novel lineage (phlebo‐like viruses) related to phleboviruses are reported on the right.
Fig. S4 Multiple sequence alignment of movement proteins from citrus concave gum‐associated virus (CCGaV) and plant viruses of the 30K superfamily. Sequences are from representative members of the virus genera in the 30K superfamily. CCGaV is shown in bold. The virus name, GenBank identifier, virus genus and distance in amino acids from the N‐terminus of the protein are shown on the left‐hand side of each sequence. Consensus secondary structure elements, predicted by PSIRED within the promals3d program, are reported at the top, with the strands and helices indicated by e and h, respectively. Amino acids that fold in helices and strands are in red and blue, respectively. The nearly invariant aspartic acid residue (the D motif) is shown in bold.
Fig. S5 Maximum‐likelihood phylogenetic tree inferred with the putative movement protein (MP) of citrus concave gum‐associated virus (CCGaV) and the MP of representative plant‐infecting viruses of the genera Emaravirus, Ophiovirus, Tenuivirus, Tospovirus, Nucleorhabdovirus and Varicosavirus. The number at each node is the result of bootstrap analysis (1000 replicates). Tree branches are proportional to the genetic distances between sequences and the scale bar at the bottom indicates substitutions per amino acid site. Square brackets delimit clusters of viruses belonging to the same genus, the name of which is on the right. The CCGaV branch is highlighted in red.
Fig. S6 Maximum‐likelihood phylogenetic tree inferred with the p39 protein of citrus concave gum‐associated virus (CCGaV) and the nucleocapsid proteins of representative viruses of the four genera Phlebovirus, Goukovirus, Phasivirus and Tenuivirus of the family Phenuiviridae and some unclassified phlebo‐like viruses. The number at each node is the result of bootstrap analysis (100 replicates). Tree branches are proportional to the genetic distances between sequences and the scale bar at the bottom indicates substitutions per amino acid site. Square brackets delimit clusters of viruses belonging to the same genus, the name of which is on the right. The CCGaV branch is highlighted in red.
Table S1 Number of reads obtained by deep sequencing of cDNA libraries of small RNAs (sRNAs) and double‐stranded RNA (dsRNA) from citrus.
Table S2 blastx results of searches with contigs obtained by assembling small RNAs (sRNAs) from leaves and bark of a citrus tree showing concave gum symptoms.
Table S3 List of primers used in this study.
Table S4 Best blastp matches with citrus concave gum‐associated virus (CCGaV) proteins.
Table S5 Results of bioassays.