

Summary
We conducted a comprehensive analysis of virulence in the fungal wheat pathogen Zymoseptoria tritici using quantitative trait locus (QTL) mapping. High‐throughput phenotyping based on automated image analysis allowed the measurement of pathogen virulence on a scale and with a precision that was not previously possible. Across two mapping populations encompassing more than 520 progeny, 540 710 pycnidia were counted and their sizes and grey values were measured. A significant correlation was found between pycnidia size and both spore size and number. Precise measurements of percentage leaf area covered by lesions provided a quantitative measure of host damage. Combining these large and accurate phenotypic datasets with a dense panel of restriction site‐associated DNA sequencing (RADseq) genetic markers enabled us to genetically dissect pathogen virulence into components related to host damage and those related to pathogen reproduction. We showed that different components of virulence can be under separate genetic control. Large‐ and small‐effect QTLs were identified for all traits, with some QTLs specific to mapping populations, cultivars and traits and other QTLs shared among traits within the same mapping population. We associated the presence of four accessory chromosomes with small, but significant, increases in several virulence traits, providing the first evidence for a meaningful function associated with accessory chromosomes in this organism. A large‐effect QTL involved in host specialization was identified on chromosome 7, leading to the identification of candidate genes having a large effect on virulence.
Keywords: image analysis, Mycosphaerella graminicola, QTL mapping, quantitative virulence, Zymoseptoria tritici
Introduction
Much plant pathology research has been oriented towards an understanding of the classic gene‐for‐gene (GFG) interaction (Flor, 1955) between the pathogen and its host, largely because early plant breeding programmes focused on the introgression of major, qualitative resistance genes (R genes) to control plant pathogens. Because most pathogens evolved quickly to defeat major R genes, and quantitative resistance appeared more durable, the genetic basis of quantitative resistance became an area of intensive investigation, leading eventually to the identification of host genes encoding quantitative resistance (Krattinger et al., 2009; Moore et al., 2015; St. Clair, 2010). The term ‘aggressiveness’ is typically used by plant pathologists to describe the quantitative degree of damage caused by a pathogen strain. In the broader literature, virulence is defined as the quantitative degree of damage caused by a pathogen to its host. We use the latter definition to be consistent with the wider field of life sciences. In contrast with quantitative resistance in the host, quantitative virulence in pathogens has been studied in much less detail and remains poorly understood (Lannou, 2011; Pariaud et al., 2009), although quantitative virulence appears to be common in plant pathogens (Pariaud et al., 2009).
Given the broad spectrum in observed virulence phenotypes, the genetic architecture of virulence may be mediated by both single genes of large effect that follow the GFG model (Flor, 1955) and by many genes that make smaller contributions to quantitative virulence. Quantitative traits often exhibit a continuous distribution in a population and are typically governed by an independent assortment of multiple alleles with small individual effects, with different combinations of alleles responsible for the range of observed phenotypes in recombining populations (Mackay, 2001). Numerous confounding factors, such as dominance, pleiotropy and additive allele effects, also contribute to the genetic architecture of a trait (Hansen, 2006). In order to disentangle individual loci contributing to overall virulence phenotypes, we utilized quantitative trait locus (QTL) mapping, an established technique for the determination of the genetic architecture of quantitative traits. QTL mapping has been successfully used to identify candidate genes underlying traits of interest in plants (Alonso‐Blanco and Méndez‐Vigo, 2014), animals (Solberg Woods, 2014) and humans (Almasy and Blangero, 2009). It has also been employed in fungi (Larraya et al., 2003), although its use in fungi remains rare compared with other eukaryotes (Foulongne‐Oriol, 2012).
Virulence in the wheat pathogen Zymoseptoria tritici (formerly Mycosphaerella graminicola), causal agent of Septoria tritici blotch (STB), is a predominantly quantitative trait (Stewart and McDonald 2014; Zhan et al., 2007). Zymoseptoria tritici is the most damaging wheat pathogen in Europe (Jørgensen et al., 2014; O'Driscoll et al., 2014) and is considered to be an important fungal pathogen worldwide (Dean et al., 2012). Under optimal conditions, yield losses can reach 50% (Eyal et al., 1987). Even with the use of resistant cultivars and regular fungicide treatments, yield losses of 5%–10% can be expected (Fones and Gurr, 2015). Ten to fourteen days after infection, chlorotic areas begin to appear that later become necrotic lesions. Pycnidia containing asexual pycnidiospores develop mostly within these necrotic lesions in the substomatal cavities. Pycnidiospores are exuded from a pycnidium in a gelatinous cirrhus during periods of high humidity and are spread throughout the plant canopy by rain splash. Numerous asexual infection cycles occurring during a growing season are the main cause of STB epidemics in the field.
Despite its agricultural importance, the mechanisms responsible for virulence in Z. tritici and their underlying genetics remain poorly understood. Nineteen genes affecting virulence in Z. tritici have been described and functionally characterized (Table S1, see Supporting Information). Most of these genes have developmental regulatory functions or affect inherent fitness without having a specific role in virulence. Host resistance is also quantitative, with 21 genes and 89 genomic regions implicated in resistance to Z. tritici (Brown et al., 2015). The quantitative nature of virulence in Z. tritici makes QTL mapping an ideal technique to elucidate the genetic determinants of quantitative virulence, as already validated using several other quantitative traits in Z. tritici (Lendenmann et al., 2014, 2015, 2016).
Epidemic development is strongly affected by the ability of a pathogen to reproduce and cause subsequent infections (Parlevliet, 1979); hence, the reproductive output of a pathogen is an important determinant of the total damage it can cause during an epidemic. Damage caused by plant pathogens is frequently assessed by the quantification of the area covered by disease lesions on the host. However, a pathogen's ability to induce lesions does not necessarily reflect its ability to reproduce. Indeed, several studies have shown that pathogen damage, as indicated by the plant area covered by disease lesions, can be independent of pathogen reproduction, as indicated by the number of spores produced during an infection (Bruns et al., 2014; Habgood, 1977; Halama et al., 1999; Pariaud et al., 2009; Twizeyimana et al., 2014), suggesting that these traits may be under separate genetic control. The contribution of each virulence component combines to produce quantitative disease phenotypes.
The success of a QTL mapping project depends on the availability of genetic markers and accurate phenotypic data. With the current availability of genomic tools that can generate very large numbers of genetic markers comparatively easily and cheaply, the limiting factor in most studies is accurate phenotyping (Furbank and Tester, 2011). Using image analysis to phenotype Z. tritici infection allows for the measurement of both leaf damage, based on the percentage of the leaf covered by necrotic lesions, and pathogen reproduction, based on the number and size of the pycnidia formed on the infected leaves (Stewart and McDonald 2014; Stewart et al., 2016).
The aim of this study was to investigate the genetic architecture of virulence traits in Z. tritici. We conducted a QTL mapping study using two mapping populations derived from parental isolates exhibiting varying degrees of virulence. Combining digital image analysis with a large number of restriction site‐associated DNA sequencing (RADseq) single nucleotide polymorphism (SNP) markers in a large number of offspring allowed us to genetically separate traits related to host damage and pathogen reproduction. We also identified novel candidate genes involved in host specialization and virulence.
Results
Phenotyping
A phenotyping method based on digital image analysis (Stewart and McDonald, 2014) was used to measure the percentage leaf area covered by lesions (PLACL), pycnidia density (number of pycnidia per square centimetre of leaf), pycnidia size and pycnidia melanization in two Z. tritici mapping populations (3D1 × 3D7 and 1A5 × 1E4) which have also been used to study melanization (Lendenmann et al., 2014), fungicide sensitivity (Lendenmann et al., 2015) and temperature sensitivity (Lendenmann et al., 2016). The pairs of parental isolates used to make the crosses were chosen based on the differing levels of virulence observed in previous work (Zhan et al., 2005). To investigate the effect of host on virulence, we used cultivars Runal (moderately susceptible to Z. tritici) and Titlis (moderately resistant) (Courvoisier et al., 2015). All phenotypes showed a continuous distribution in both crosses. Transgressive segregation was evident for all phenotypes, with some progeny exhibiting more extreme phenotypes than the parental isolates (Fig. 1). Progeny from cross 3D1 × 3D7 were significantly more virulent on Runal than Titlis, in terms of mean pycnidia size (F = 3.93, P = 0.048), pycnidia melanization (F = 22.7, P < 0.001), PLACL (F = 21.4, P < 0.001) and pycnidia density (F = 19.2, P < 0.001). In cross 1A5 × 1E4, PLACL (F = 22.7, P < 0.001) and pycnidia melanization (F = 17.7, P < 0.001) were significantly higher in Runal, whereas pycnidia size (F = 11.5, P < 0.001) and pycnidia density (F = 73.6, P < 0.001) were significantly higher in Titlis. Across both crosses, a total of 540 710 pycnidia were counted and their sizes and melanization were measured. In order to evaluate whether the counting and measurement of pycnidia provide an accurate estimate of spore number, the spore output was measured in a subset of 10 isolates with the smallest mean pycnidia size and 10 isolates with the largest mean pycnidia size. The mean number of spores per pycnidium was 2598. The mean spore size, as measured by length, was 17.6 μm. There were significant positive correlations between pycnidia size and spore size (r 2 = 0.111, P = 0.001) and between pycnidia size and the number of spores per pycnidium (r 2 = 0.112, P = 0.001). These findings indicate that pycnidia size is correlated with the reproductive output of Z. tritici.
Figure 1.
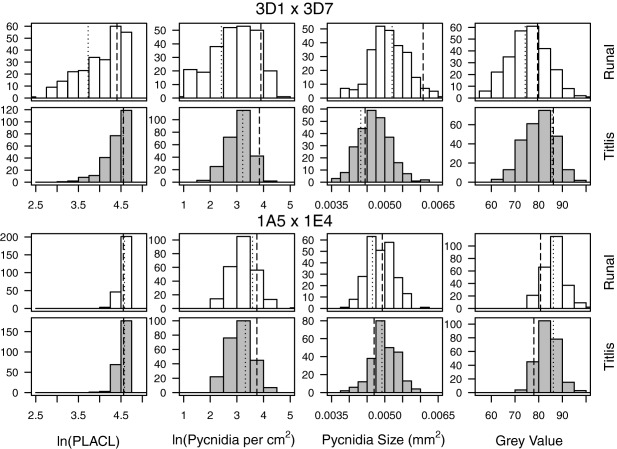
Frequency histograms of virulence phenotypes from the Zymoseptoria tritici mapping populations 3D1 × 3D7 (top panels) and 1A5 × 1E4 (bottom panels) on the wheat cultivars Runal (white bars) and Titlis (grey bars). Vertical lines represent phenotype values of the parental isolates: dotted line, 3D1 and 1A5; broken line, 3D7 and 1E4. PLACL, percentage leaf area covered by lesions.
QTL mapping
We used QTL mapping to elucidate the genetic architecture of the quantitative virulence phenotypes. QTLs were identified for all phenotypes. All QTLs in cross 3D1 × 3D7 were found only on Runal and mapped to a single large‐effect QTL on chromosome 7 (Fig. 2, left). In 1A5 × 1E4, the same QTL for pycnidia density was found on chromosome 5 on both cultivars. A second QTL for pycnidia density was found on chromosome 9 in Titlis and on chromosome 3 in Runal. PLACL in Titlis and pycnidia grey value in Runal both mapped to the same QTL on chromosome 5. Pycnidia size also mapped to the same QTL on chromosome 3 in cv. Runal (Fig. 2, right). No QTLs were shared between the two crosses. The 95% confidence interval for a QTL ranged from 91.3 kb for the chromosome 5 QTL in 1A5 × 1E4 to 717.7 kb for the chromosome 7 QTL for grey value on Runal in 3D1 × 3D7.
Figure 2.
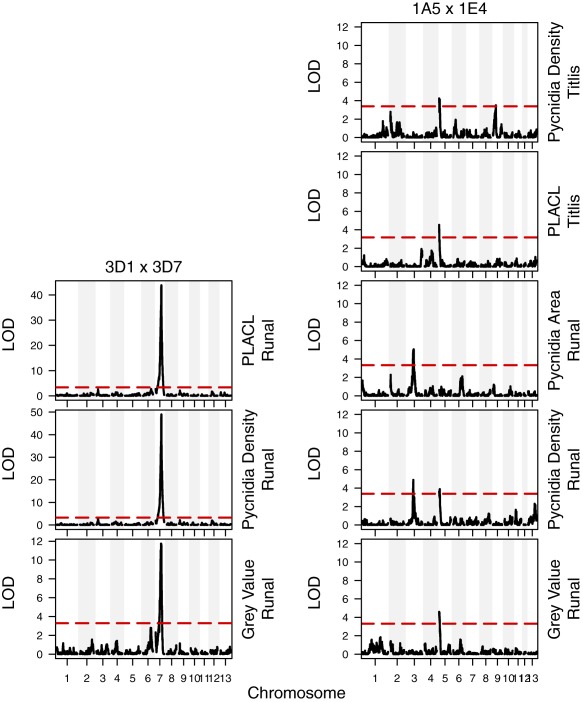
Logarithm of the odds (LOD) plots from quantitative trait locus (QTL) mapping of virulence traits in the Zymoseptoria tritici mapping populations 3D1 × 3D7 (left column) and 1A5 × 1E4 (right column) for the 13 core chromosomes (x‐axis). The broken horizontal line represents the 0.05 significance threshold calculated with 1000 permutations. PLACL, percentage leaf area covered by lesions.
The single, large‐effect QTL on chromosome 7 in cross 3D1 × 3D7 explained 54% of the phenotypic variance for PLACL, 57% of the variance for pycnidia density and 18% of the variance for pycnidia grey value. PLACL and pycnidia density shared the same confidence interval containing 35 genes. The wider confidence interval for the pycnidia grey value contained 227 genes (Table 1A). The allele from the 3D7 parent was responsible for higher PLACL and pycnidia density and decreased pycnidia melanization (Fig. 3).
Table 1.
Significant quantitative trait loci (QTLs) for in planta virulence traits from Zymoseptoria tritici mapping populations 3D1 × 3D7 (A) and 1A5 × 1E4 (B) on wheat cultivars Runal and Titlis.
| (A) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cultivar | Phenotype | Chr | LOD score | Variance explained (%) | LOD threshold | P | Confidence interval (Mb) | Number of genes | Mean phenotype (3D1 allele) | Mean phenotype (3D7 allele) |
| Runal | PLACL | 7 | 43.7 | 53.9 | 3.34 | <0.001 | 1.75–1.91 | 35 | 38.98 | 78.19 |
| Runal | Pycnidia/cm2 | 7 | 48.2 | 57.3 | 3.30 | <0.001 | 1.75–1.91 | 35 | 11.26 | 34.23 |
| Runal | Grey value* | 7 | 11.4 | 18.1 | 3.29 | <0.001 | 1.18–1.91 | 227 | 71.72 | 78.44 |
| (B) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Cultivar | Phenotype | Chr | LOD score | Variance explained (%) | LOD threshold | P | Confidence interval (Mb) | Number of genes | Mean phenotype (1A5 allele) | Mean phenotype (1E4 allele) |
| Runal | Pycnidia/cm2 | 3 | 4.9 | 8.6 | 3.38 | 0.003 | 1.27–1.87 | 209 | 24.79 | 32.13 |
| Runal | Pycnidia/cm2 | 5 | 3.8 | 7.0 | 3.38 | 0.018 | 1.36–2.27 | 280 | 31.02 | 26.60 |
| Runal | Pycnidia size (mm2 × 10−3) | 3 | 5.1 | 8.7 | 3.33 | 0.001 | 1.27–1.98 | 143 | 4.773 | 4.998 |
| Runal | Grey value* | 5 | 4.7 | 8.1 | 3.31 | 0.001 | 0.14–0.15 | 89 | 88.06 | 84.74 |
| Titlis | Pycnidia/cm2 | 5 | 4.15 | 7.2 | 3.38 | 0.006 | 1.36–2.27 | 280 | 28.03 | 22.62 |
| Titlis | Pycnidia/cm2 | 9 | 3.51 | 6.1 | 3.38 | 0.045 | 3.20–9.19 | 204 | 22.37 | 28.01 |
| Titlis | PLACL | 5 | 4.56 | 8.0 | 3.17 | 0.015 | 1.36–2.27 | 280 | 95.49 | 90.40 |
The number of genes within the QTL 95% confidence interval is based on the JGI reference annotation.
Chr, chromosome; LOD, logarithm of the odds; PLACL, percentage leaf area covered by lesions.
*The grey scale ranges from ‘0’ (black) to ‘255’ (white). Lower values indicate darker pycnidia.
Figure 3.
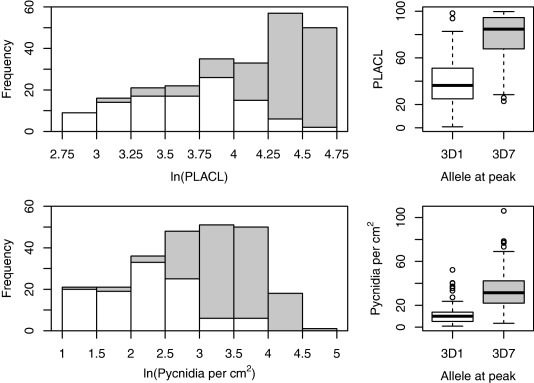
Phenotypic trait distribution of progeny that inherited either the more virulent allele from parent 3D7 (shown in grey) or the less virulent allele from parent 3D1 (shown in white) at the quantitative trait locus (QTL) located on chromosome 7. The percentage leaf area covered by lesions (PLACL) is shown in the top row and the pyncidia density is shown in the bottom row.
In cross 1A5 × 1E4, the variance explained by each QTL was smaller and ranged from 6.1% for pycnidia density on Titlis to 8.7% for pycnidia size on Runal. The number of genes within each confidence interval was generally higher than in cross 3D1 × 3D7, ranging from 89 for grey value in Runal to 280 in the QTL on chromosome 5 for pycnidia density in Runal and PLACL in Titlis (Table 1B). The 1A5 allele was responsible for the higher phenotype values in the QTL on chromosome 5, whereas the 1E4 allele was responsible for the higher phenotype values in the chromosome 3 and 9 QTLs.
Additive effects were observed for the two QTLs found for pycnidia density in 1A5 × 1E4. On Runal, isolates with the 1A5 allele at the chromosome 5 QTL peak and the 1E4 allele at the chromosome 3 QTL peak had significantly higher pycnidia density than isolates with the reverse alleles (F = 11.2, P < 0.001) (Table 2A). On Titlis, isolates with the 1E4 allele at the chromosome 9 QTL peak and the 1A5 allele at the chromosome 5 QTL peak had significantly higher phenotypes than isolates with the opposite allele combinations (F = 19.1, P < 0.001) (Table 2B). On both cultivars, isolates with the same parental allele at both QTL peaks were not different from each other and were intermediate to the isolates with a combination of alleles coming from different parents.
Table 2.
Additive allele effects for pycnidia density in cross 1A5 × 1E4 for cultivars Runal (A) and Titlis (B).
| (A) | ||||
|---|---|---|---|---|
| Cultivar | Chr 3 allele | Chr 5 allele | Mean phenotype | n |
| Runal | 1E4 | 1A5 | 39.28a | 43 |
| Runal | 1A5 | 1A5 | 26.10b | 32 |
| Runal | 1E4 | 1E4 | 26.96b | 44 |
| Runal | 1A5 | 1E4 | 23.09c | 46 |
| (B) | ||||
|---|---|---|---|---|
| Cultivar | Chr 5 allele | Chr 9 allele | Mean phenotype | n |
| Titlis | 1A5 | 1E4 | 33.38a | 37 |
| Titlis | 1A5 | 1A5 | 25.00b | 41 |
| Titlis | 1E4 | 1E4 | 25.15b | 54 |
| Titlis | 1E4 | 1A5 | 20.11c | 36 |
Chromosome (Chr) alleles represent the parental allele of the marker at the quantitative trait locus (QTL) peak. Phenotypes are the mean pycnidia density of all isolates with the corresponding allele combination. Superscript letters denote significant differences. n represents the number of isolates with each allele combination.
Effect of accessory chromosomes on virulence
Among the parental isolates used to make the crosses, all accessory chromosomes were present in 3D1, whereas 3D7 was missing chromosomes 14, 15, 18 and 21. In the 1A5 × 1E4 cross, chromosome 17 was absent in 1E4. QTLs could only be mapped on chromosomes present in both parental isolates. No QTLs were found on the accessory chromosomes in either cross.
The presence or absence of accessory chromosomes was established for each of the progeny in cross 3D1 × 3D7. Chromosome 14 was absent in 67 (26%) progeny, chromosome 15 in 68 (26%) progeny, chromosome 18 in 77 (30%) progeny and chromosome 21 in 73 (28%) progeny. All accessory chromosomes were present at a significantly higher frequency than the expected 1 : 1 Mendelian inheritance ratio (chromosome 14: χ 2 = 61.1, P < 0.001; chromosome 15: χ 2 = 59.1, P < 0.001; chromosome 18: χ 2 = 43.1, P < 0.001; chromosome 21: χ 2 = 49.9, P < 0.001). This is consistent with previous work which reported a skewed inheritance of accessory chromosomes in a subset of 48 isolates from the same population (Croll et al., 2013).
We studied the effect of chromosome presence–absence on all virulence traits for all accessory chromosomes in 3D1 × 3D7. On Runal, isolates with chromosome 21 had significantly larger pycnidia (F = 6.2, P = 0.014) than isolates without chromosome 21, but the effect size was small (η 2 = 0.023). On Titlis, isolates with chromosome 18 had significantly larger (F = 5.0, P = 0.027) and darker (F = 5.7, P = 0.018) pycnidia than isolates without chromosome 18. The effect sizes for pycnidia size (η 2 = 0.021) and pycnidia melanization (η 2 = 0.022) were small. A significant interaction between chromosomes 15 and 18 was also observed for pycnidia size (F = 7.0, P = 0.009), with isolates having both accessory chromosomes showing larger pycnidia, but the effect size of the interaction was small (η 2 = 0.028). No effects on PLACL or pycnidia density were found for any accessory chromosome.
Candidate genes for pathogen virulence within QTLs
In total, 918 candidate genes were identified in the 95% confidence intervals of all QTLs, including 227 from cross 3D1 × 3D7 and 691 from cross 1A5 × 1E4 (Table S1). No candidate genes were shared between the two crosses. In the literature to date, 19 genes have been implicated in virulence in Z. tritici and functionally characterized (Table S2, see Supporting Information). None of the genes previously shown to play a role in virulence were found within the QTLs.
In‐depth investigation of the chromosome 7 QTL
The QTL for PLACL and pycnidia density in cross 3D1 × 3D7 on Runal had a high LOD score, a low number of candidate genes within the 95% confidence interval and explained approximately one‐half of the observed phenotypic variance. Existing gene models were manually checked for accuracy, sequence polymorphisms between the two parental isolates were analysed and gene expression during the infection cycle was investigated (Table S3, see Supporting Information). Population genomic approaches were used to investigate the genomic features of the region in a natural population.
Re‐annotation of the genes in the chromosome 7 QTL
Within the 95% confidence interval, only 10 of the 35 gene models from the reference annotation were convincingly supported by RNA sequencing (RNAseq) reads. The annotations of 20 genes were replaced with those of Grandaubert et al. (2015). Four genes not present in any previous annotation were discovered and supported by our RNAseq data (J. Palma‐Guerrero et al., 2017). The annotations of four additional genes were altered slightly from those of Grandaubert et al. (2015) or the JGI. After re‐annotation, 38 genes were found in the 95% confidence interval (Tables 3 and S4, see Supporting Information). Among the newly annotated genes, gene ontology (GO) terms could be assigned to 17 genes, whereas the remaining 21 genes had no known function. Six genes within the QTL encode proteins with a predicted signal peptide, but lack a transmembrane domain and are therefore predicted to be secreted. Among these six genes, two meet the criteria for being small secreted effector‐like proteins (<300 amino acids, secreted and cysteine rich).
Table 3.
Re‐annotated candidate genes located in a quantitative trait locus (QTL) for pycnidia density and percentage leaf area covered by lesions (PLACL) on chromosome 7 in Zymoseptoria tritici.
| Gene name | Description | Protein length | Signal peptide | Cysteine (%) (3D7) | Transmembrane (TM) domain |
|---|---|---|---|---|---|
| Zt_QTL7_1 a | ‐ | 504 | N | 2.4 | N |
| Zt_QTL7_2 a | ‐ | 228 | Y | 10.7 | N |
| Zt_QTL7_3 a | ‐ | 357 | N | 7.1 | N |
| Zt_QTL7_4 b | Hypothetical protein | 471 | N | 2.6 | Y |
| Zt_QTL7_5 a | ‐ | 198 | Y | 9.2 | N |
| Zt_QTL7_6 b | AMP‐binding enzyme family | 921 | N | 2.0 | Y |
| Zt_QTL7_7 b | Serine carboxypeptidase | 966 | N | 2.2 | N |
| Zt_QTL7_8 b | Hypothetical protein | 879 | N | 3.8 | N |
| Mycgr3T100839 c | Fe‐containing alcohol dehydrogenase | 1251 | N | 1.9 | N |
| Mycgr3T100840 c | Vacuolar membrane pq loop repeat | 972 | N | 1.5 | Y |
| Mycgr3T105313 c | Trichothecene efflux pump | 1788 | N | 1.3 | Y |
| Mycgr3T44611 c | Radical S‐adenosyl methionine domain‐containing 2 | 1074 | N | 3.1 | Y |
| Mycgr3T44946 c | Hypothetical protein | 666 | N | 0.5 | N |
| Mycgr3T45542 c | Lipoate ligase B | 720 | N | 2.5 | N |
| Mycgr3T74280 c | Prolyl 4‐hydroxylase | 1503 | N | 1.6 | N |
| Mycgr3T94634 c | Hypothetical protein | 525 | N | 2.3 | N |
| Mycgr3T94648 c | Hypothetical protein | 1143 | N | 0.3 | N |
| Mycgr3T94659 c | Hypothetical protein | 972 | N | 1.5 | N |
| Zt09_model_7_00553 d | Ferric‐chelate reductase (Fre2) | 2085 | N | 1.9 | Y |
| Zt09_model_7_00555 d | Hypothetical protein | 255 | N | 2.4 | N |
| Zt09_model_7_00556 d | Hypothetical protein | 1038 | N | 1.7 | N |
| Zt09_model_7_00557 d | Hypothetical protein | 378 | N | 0.0 | N |
| Zt09_model_7_00558 d | Major allergen alt a1, partial | 540 | Y | 2.8 | N |
| Zt09_model_7_00559 d | Hypothetical protein | 789 | N | 1.5 | Y |
| Zt09_model_7_00561 d | Hypothetical protein | 1302 | N | 2.1 | N |
| Zt09_model_7_00562 d | Hypothetical protein | 3663 | Y | 3.4 | N |
| Zt09_model_7_00563 d | Hypothetical protein | 1851 | N | 0.2 | N |
| Zt09_model_7_00564 d | Hypothetical protein | 1761 | N | 0.9 | Y |
| Zt09_model_7_00567 d | Hypothetical protein | 651 | N | 0.5 | N |
| Zt09_model_7_00569 d | Acid phosphatase | 456 | N | 2.3 | Y |
| Zt09_model_7_00571 d | Adenylate kinase | 696 | N | 0.0 | N |
| Zt09_model_7_00573 d | ‐ | 420 | N | 3.6 | N |
| Zt09_model_7_00574 d | Glycoside hydrolase | 933 | N | 3.2 | N |
| Zt09_model_7_00575 d | Glycoside hydrolase family 36 | 2238 | Y | 0.4 | N |
| Zt09_model_7_00577 d | Ca2+‐modulated non‐selective cation channel polycystin | 1917 | Y | 1.1 | N |
| Zt09_model_7_00578 d | Urease accessory | 780 | N | 1.2 | N |
| Zt09_model_7_00579 d | CFEM domain‐containing | 1230 | Y | 4.6 | Y |
| Zt09_model_7_00581 d | Hypothetical protein | 294 | N | 3.1 | N |
AMP, adenosine monophosphate; ‐, Unknown function.
Superscripts denote the source of the gene annotation: anew genes not present in previous annotations; bannotations modified from existing annotations; cannotation from the JGI reference annotation; dannotation from Grandaubert et al. (2015). Descriptions from blast2go, signal peptide predictions from signalP and TM domains from TMHMM. Protein length in amino acids and percentage of cysteines in the parental isolate 3D7.
Identification of high‐priority candidate genes within the chromosome 7 QTL
Sequence variation within gene coding regions of the chromosome 7 QTL was investigated in the parental genomes. Eighty‐two sequence variants were identified, comprising 51 synonymous and 31 non‐synonymous mutations, as well as four insertions/deletions (indels). Seventeen genes were found to have at least one mutation affecting the amino acid sequence. Nineteen genes showed differential expression in planta between the two parental isolates for at least one time point during the infection process. Several high‐priority candidate genes were identified based on a combination of attributes, including putative function, sequence variation and/or expression levels during infection. Candidate gene Zt_QTL7_5 is not present in any existing annotation and blast searches showed no similarities to other proteins. With a length of 65 amino acids, six cysteine residues and a signal peptide domain, it fulfils the criteria of a typical small secreted effector protein. Its peak expression is between 12 and 14 days post‐infection (dpi), during the transition from symptomless growth to the onset of chlorosis (Fig. 4). It is the second most expressed gene in the QTL confidence interval, but shows significantly lower expression in 3D7 (the more virulent parent) at 14 dpi.
Figure 4.
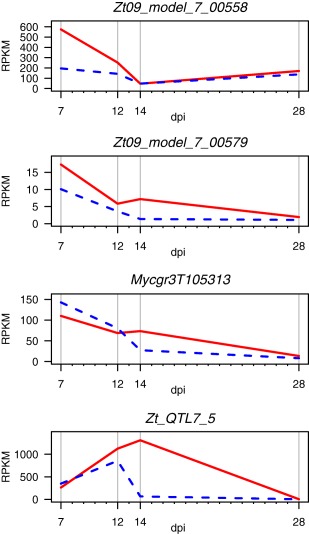
Gene expression profiles of Zymoseptoria tritici candidate genes at 7, 12, 14 and 28 days post‐infection (dpi). Solid red lines represent parental isolate 3D1 and broken blue lines represent parental isolate 3D7. RPKM, reads per kilobase of transcript per million mapped reads.
Candidate gene 00558 is also a small secreted protein (SSP) containing an Alternaria alternata allergen domain. This domain is unique to the Dothideomycete and Sordariomycete classes of fungi (Chruszcz et al., 2012) and can induce major allergic reactions in the human respiratory system (Bush and Portnoy, 2001). This gene shows peak expression during the symptomless phase and lower expression in 3D7 (Fig. 4). Candidate gene 105313 is a fungal‐specific transporter from the Major Facilitator Superfamily (MFS). It contains a trichothecene efflux domain that has been shown to play a role in toxin secretion in Fusarium (Alexander et al., 1999). The parental alleles differed by two non‐synonymous SNPs. Candidate gene 00579 is a fungal‐specific membrane protein containing seven transmembrane domains, a CFEM domain and a signal peptide with a peak expression at 7 dpi (Fig. 4).
Population genomics of genes in the chromosome 7 QTL
As QTL mapping is highly cross specific, genetic variation in the chromosome 7 QTL was investigated in a natural population of 28 re‐sequenced Z. tritici isolates from Switzerland. The 163‐kb confidence interval contained an alternation of gene‐rich and gene‐poor regions. A large gene‐poor region (found between c. 1.79 and 1.84 Mb in the reference genome assembly) contained only two genes (Fig. 5E). This gene‐poor region contained all transposable elements (TEs) found within the confidence interval (Fig. 5D). Three classes of TEs were found in this region, including Copia, Gypsy and Tad1 elements. The gene‐rich regions contained a higher density of SNPs than the average on the chromosome, reaching up to 120 SNPs per kilobase in some regions (Fig. 5C). The genotyping rate was high in the gene‐rich region. The gene‐poor and TE‐rich regions contained few SNPs and had a low genotyping rate (Fig. 5C), and generally had a lower GC content than the gene‐rich regions (Fig. 5B).
Figure 5.
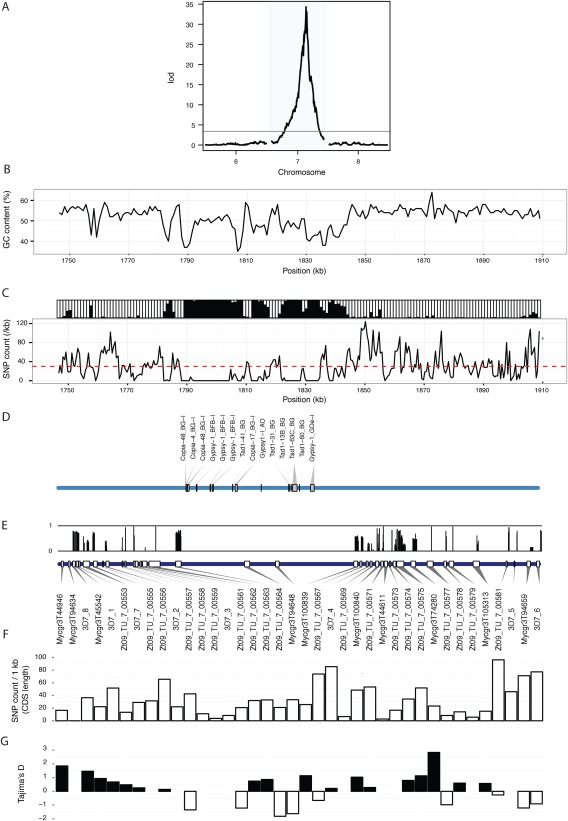
(A) Population genomic characterization of the quantitative trait locus (QTL) located on chromosome 7 (region 1746 to 1909 kb) in the reference isolate IPO323 and a population of 28 Zymoseptoria tritici isolates drawn from a single field in Switzerland. (B) GC content in the IPO323 reference genome. (C) Single nucleotide polymorphism (SNP) density (bottom) and genotyping rate (top) in the 28 field isolates. Black rectangles represent the frequency of isolates with a segmental deletion. The horizontal line represents the mean SNP density across chromosome 7 in the 28 isolates. (D) Transposable element locations in the IPO323 reference genome. (E) Bottom: location of coding sequences in the IPO323 reference genome. Top: frequency of the allele carried by 3D7 in the 28 field isolates for the SNPs shared by the 3D1 and 3D7 parents in the gene coding sequences. (F) SNP density per kilobase in the coding sequences of the genes. Only SNPs with a genotyping rate of ≥90% were included. (G) Tajima's D statistic per gene coding sequence. Tajima's D values for genes with less than 10 SNPs are not shown.
Genes within the confidence interval were highly variable for the number of SNPs within the natural population (Fig. 5F). Seven genes contained fewer than 10 SNPs per kilobase, whereas nine genes contained more than 50 SNPs per kilobase. On average, the genes within the confidence interval contained 33 SNPs per kilobase. Tajima's D statistic for 27 genes having 10 or more SNPs showed an alternation of clusters of genes with positive D values and genes with negative D values (Fig. 5G). Genes with negative D values are more likely to be under purifying selection, whereas genes with positive D values are likely to be under balancing selection. This suggests variability in the selection processes acting on these genes, including a possibility of hitchhiking effects.
Discussion
Here, we report the most comprehensive QTL mapping analysis of fungal virulence to date, utilizing different mapping populations and different host cultivars, as well as measuring multiple traits associated with virulence. We were able to genetically dissect pathogen virulence into separate components, showing that different components of virulence can be under separate genetic control. QTLs were identified for all traits, including QTLs that were specific to mapping populations, cultivars and traits, as well as QTLs that were shared among traits within the same mapping population. We identified genome regions associated with single large effects, as well as regions of smaller additive effects.
The genetic architecture of virulence in Z. tritici is complex
Our results point to a complex genetic architecture involving multiple factors that combine to produce quantitative virulence. Strong evidence was found for transgressive segregation, with many progeny showing more extreme phenotypes than the parental isolates in both crosses. QTLs were specific to each cross and, in some cases, specific to a phenotype or cultivar. In all but one case, multiple traits mapped to the same QTL, an observation also made for other traits in Z. tritici (Lendenmann et al., 2014, 2015, 2016).
Both quantitative (Zhan et al., 2005) and large‐effect interactions consistent with the GFG hypothesis have been reported in Z. tritici (Brading et al., 2002; Kema et al., 2000). GFG interactions are frequently reported as conferring compatible/incompatible interactions (Flor, 1955). The large effect size of the 3D1 × 3D7 chromosome 7 QTL on one of the cultivars shows similarity to a GFG interaction between a major resistance gene in Runal and a corresponding avirulence effector gene in the 3D1 strain of the pathogen. In our case, the interaction explains approximately 50% of the overall variance for the virulence traits, which also show a continuous distribution in the mapping population (Fig. 1). With such a strong effect, a bimodal phenotype distribution may be present. When the progeny for 3D1 × 3D7 were split into categories according to the parental genotype found at the chromosome 7 QTL peak, two distinct distributions were found to contribute to the overall phenotype distribution (Fig. 3). These observations suggest that numerous additional genes of small effect may also be involved, in addition to the genotype‐by‐environment (G × E) interactions typically observed in quantitative phenotypes.
In contrast with the single large‐effect QTL in 3D1 × 3D7, numerous QTLs distributed across several chromosomes were found for the different phenotypes in cross 1A5 × 1E4. These QTLs had relatively small effects on phenotype and contained a large number of candidate genes. This suggests that the genetic architecture of virulence in 1A5 × 1E4 is more complex than that of 3D1 × 3D7, and demonstrates the contribution of both large and small effects on virulence.
Additive effects of QTLs on virulence
The multiple factor hypothesis (Morgan et al., 1915) based on the additive effects of many genes is the key principle underlying quantitative traits. Recombination generates progeny with different combinations of genes from the parents and can contribute to novel virulence phenotypes (Joseph et al., 2011; Kanvil et al., 2015). Strong additive effects were observed in cross 1A5 × 1E4 for the QTL for pycnidia density. For both cultivars, alternative alleles from the two parents combined to produce a higher phenotype. Isolates with the same parental allele at both QTLs showed lower virulence than isolates with the different parental alleles at the two QTLs. Transgressive segregation was observed for all phenotypes in both crosses. These findings illustrate the power of recombination to generate progeny phenotypes that are significantly different from their parents, an observation shared with other plant‐pathogenic fungi (Sommerhalder et al., 2010; Stefansson et al., 2013), and show how a range of quantitative virulence phenotypes can be generated through sexual reproduction in natural field populations.
The contribution of accessory chromosomes to virulence
By using the novel approach of associating virulence phenotypes with the presence or absence of specific accessory chromosomes, we discovered a significant correlation for some phenotypes, with higher virulence observed in offspring carrying certain accessory chromosomes. Accessory chromosomes have been shown to play an important role in virulence in other fungal pathogens (Akagi et al., 2009; Ma et al., 2010; Miao et al., 1991), but, until now, their role in Z. tritici was elusive (Stukenbrock et al., 2010). The increase in virulence was small, around 2%–3%, but significant. If this correlation also exists under natural field conditions, this increase in virulence and the associated increase in reproduction would represent a significant fitness advantage that could explain why accessory chromosomes are retained in natural field populations despite the fact that they are readily lost during meiosis and mitosis (Croll et al., 2013; Wittenberg et al., 2009).
Cultivar specificity
Cultivar specificity is well documented in Z. tritici (Ahmed et al., 1995, Cowger et al., 2000, Zhan et al., 2002) and is commonly reported as qualitative compatible/incompatible interactions (e.g. Brading et al., 2002) or as generally quantitative with isolates being more virulent or less virulent on a particular host (e.g. Zhan et al., 2005). In cross 3D1 × 3D7, all phenotypes were higher on the more susceptible cultivar as expected. However, in cross 1A5 × 1E4, PLACL was higher in Runal, whereas pycnidia size, density and melanization were higher in Titlis, the more resistant cultivar. In addition, in 1A5 × 1E4, the same chromosome 5 QTL was found for pycnidia density in both cultivars. However, a second QTL was found on chromosome 3 in Runal and on chromosome 9 in Titlis, suggesting that these QTL loci play a role in quantitative specialization to these two cultivars. This is in contrast with the findings of Mirzadi Gohari et al. (2015), who found a QTL at a similar position on chromosome 5 that was implicated in host specificity in a different set of cultivars. The results from the accessory chromosome presence–absence analysis also point towards cultivar specificity, with different accessory chromosomes affecting the same phenotype on different cultivars. Although 21 genes and 89 genomic regions have been associated with resistance to STB in wheat (Brown et al., 2015), the genetic basis of resistance in cultivars Runal and Titlis is currently unknown.
Contribution of pycnidia number and size to epidemic potential
The basic reproductive number (R 0) is the theoretical number of infections arising from a single infection event and can be useful in predicting the potential development of plant disease epidemics (van den Bosch et al., 2008). Pycnidia represent the asexual reproductive output of a Z. tritici strain, with larger pycnidia correlated with a greater number of conidia in earlier work (Gough, 1978). It has been postulated previously that the number and size of pycnidia could be important measures of virulence in Z. tritici (Stewart and McDonald 2014; Suffert et al., 2013). Our results show a significant correlation indicating that larger pycnidia bear both larger and more numerous spores.
The number and size of pycnidia influence R 0 and therefore provide a prediction of the epidemic potential associated with a pathogen strain under field conditions. In contrast, measurement of the leaf area covered by lesions (PLACL) gives an indication of the amount of damage caused by a pathogen strain on the host plant. The ability to directly measure both the epidemic potential of an isolate and the amount of damage it causes to its host represents a powerful improvement in the overall measurement of pathogen virulence that can also be used to improve measurements of resistance in the host (Stewart et al., 2016). Furthermore, the identification of a QTL for pycnidia size shows that this important trait can be under separate genetic control. A separate experiment oriented around the measurement of differences in host resistance and conducted using naturally infected plants under field conditions showed that host genotype could affect pycnidia formation independent of the damage caused by leaf lesions (Stewart et al., 2016). Theoretically, a pathogen strain which causes a high fraction of leaf area to be covered by lesions, but has a low R 0 (i.e. producing fewer pycnidia per infected leaf) may appear more damaging at a small spatial scale, but an isolate which has a higher R 0 (i.e. producing more pycnidia per infected leaf) has a greater potential to cause damage over larger spatial scales. From an epidemiological perspective, direct measurement of pathogen reproduction on a given host could inform disease management decisions oriented towards the inhibition of pathogen reproduction (e.g. through the search for resistance genes that lower pathogen reproduction, in addition to resistance genes that lower host damage), as well as providing a novel way of selecting for quantitative host resistance. The results from cross 1A5 × 1E4 illustrate the potential usefulness of this approach with different levels of pycnidia and PLACL found on the two different cultivars. One limitation of the current work is that phenotype data were collected at a single time point. As phenotypes, such as pycnidia size, could be associated with developmental stage, future work should consider adding a temporal dimension to the phenotype measurements.
Candidate genes affecting pathogen virulence
By combining several approaches, we were able to narrow down the candidate gene list for the chromosome 7 QTL affecting PLACL, pycnidia density and melanization, but it remains possible that different genes located within this QTL interact to affect all three traits. It is also possible that one gene in this QTL is primarily responsible for only one phenotype, which influences the other two without any genetic or regulatory connection, a situation referred to as spurious, vertical, relational or reactive pleiotropy (Paaby and Rockman, 2013). For example, it is plausible that a gene encoding higher PLACL would lead to greater pycnidia density that could, in turn, affect pycnidia size. The results from cross 1A5 × 1E4 offer support for the latter hypothesis, with multiple traits mapping to the same QTL on chromosome 5, although independent QTLs were found on different chromosomes for the same traits. In previous work, genes up‐regulated in the biotrophic phase of the infection cycle showed reduced pycnidia production in knockout mutants (Poppe et al., 2015), providing evidence that pycnidia development is somewhat dependent on prior processes.
Among the candidate genes within the chromosome 7 QTL, that which we consider to be most likely to explain the observed virulence is Zt_QTL7_5, a previously undescribed SSP that is highly expressed during the switch to necrotrophic growth. SSPs are common virulence factors in fungi that facilitate infection or elicit a response in the host (Lo Presti et al., 2015). SSPs often conform to the GFG paradigm by interacting with a major R gene in the host. A GFG interaction would explain the large effect of the chromosome 7 QTL on only one host. Interestingly, no significant sequence differences exist for this gene between the parental isolates, but the expression levels are significantly lower in 3D7, the more virulent parent. This is consistent with the theory of effector‐triggered immunity (ETI), in which the recognition of a pathogen effector triggers a defence response in the host. Another SSP, candidate gene 00558, contains a conserved domain known to elicit an allergic response in humans (Bush and Portnoy, 2001). It has additionally been implicated in virulence in Alternaria brasicicola on Arabidopsis (Cramer and Lawrence, 2004) and inhibits plant antimicrobial proteins (Gómez‐Casado et al., 2014). Given the existing knowledge of this protein, it appears that it could play a role in Z. tritici virulence. An MFS transporter implicated in toxin secretion was also located in the QTL. MFS transporters contribute to virulence by the secretion of fungal toxins and secondary metabolites, as well as the efflux of plant‐derived antimicrobial compounds (Coleman and Mylonakis, 2009). Candidate gene 00579 contains a CFEM domain that is unique to fungi and is found at significantly higher frequency in pathogenic than non‐pathogenic fungi (Zhang et al., 2015). Putative functions of the CFEM domain include cell surface receptors, signal transduction or adhesion of molecules in plant–pathogen interactions (Kulkarni et al., 2003). None of the genes previously described as having a role in virulence in Z. tritici were identified in this study, highlighting the usefulness of forward genetic approaches, such as QTL mapping, for the identification of novel virulence genes. Several of the newly identified candidate genes were not present in the reference genome annotation. Recent re‐annotations of the Z. tritici genome have generated significantly different sets of gene models (Grandaubert et al., 2015; Rothamstead Research: http://fungi.ensembl.org/Zymoseptoria_tritici). The existence of independent and conflicting genome annotations can impede the identification of candidate genes (Gibriel et al., 2016). A more accurate consensus annotation would greatly benefit future work on this pathogen.
Population genomics of the chromosome 7 QTL
Analysis of the large‐effect QTL on chromosome 7 in a Swiss field population revealed several interesting genomic features. The region was highly variable, with genes exhibiting variable numbers of SNPs, as well as evidence for both purifying and balancing selection. Genes involved in virulence are often under positive selection in plant pathogens (Lo Presti et al., 2015; Stukenbrock and McDonald, 2009), including in Z. tritici (Poppe et al., 2015). An island of TEs was found within a gene‐poor region of the QTL. The Z. tritici genome contains 16.7% repetitive elements (Dhillon et al., 2014). Effector genes show a tendency to cluster in gene‐poor regions of repetitive DNA that are TE‐rich in numerous filamentous fungal plant pathogens (Dong et al., 2015). TEs can influence the expression of effector genes by insertion into promoter regions (Ali et al., 2014) or through epigenetic gene silencing (Shaaban et al., 2010). We hypothesize that these processes may explain the differences in expression observed in Zt_QTL7_5.
Conclusions
This study highlights the complex nature of virulence in the wheat–Z. tritici pathosystem, illustrating that many factors contribute to the quantitative phenotypes. We show that virulence comprises different traits, some affecting host damage and others affecting pathogen reproduction, which can be under independent genetic control. In light of this, researchers and breeders should reconsider the best way to measure virulence in this pathosystem. We propose that more attention should be focused on resistance that reduces pathogen reproduction in order to decrease R 0 during epidemics.
Experimental Procedures
Two Z. tritici mapping populations composed of approximately 350 progeny from each of two crosses of Z. tritici strains sampled on the same day from the same Swiss wheat field (described in Lendenmann et al., 2014) were phenotyped in a glasshouse‐based seedling assay, as described in Stewart and McDonald (2014). The Swiss winter wheat cultivars ‘Runal’ and ‘Titlis’ (DSP Ltd., Delley, Switzerland) were inoculated using each offspring from both crosses, together with the parental isolates. Two plants of each cultivar were inoculated with each Z. tritici isolate to give two technical replicates. Each technical replicate was placed in a separate glasshouse compartment. This process was repeated three times over three consecutive weeks to generate three biological replicates, resulting in six replicates in total for each isolate–cultivar combination.
At 23 dpi, the second leaf from each plant was excised, photographed and phenotyped using automated image analysis, as described previously (Stewart and McDonald, 2014). The method was modified slightly to include a measure of pycnidia melanization. RGB images were converted to eight‐bit grey scale and the mean grey value for the pixels making up each pycnidium was calculated. The grey scale runs from ‘0’ (black) to ‘255’ (white). Grey values can be used as a proxy for the degree of melanization (Lendenmann et al., 2014). PLACL, pycnidia density (pycnidia/cm2 leaf area), pycnidia size (mm2) and pycnidia melanization were used as phenotypes for QTL mapping (see below). Any chlorotic, necrotic or otherwise unhealthy non‐green leaf tissue was treated as a lesion.
For the 3D1 × 3D7 progeny, the mean pycnidia size was calculated for each isolate on Runal. The 10 isolates showing the largest mean pycnidia size and the 10 showing the smallest mean pycnidia size were selected for further analysis. The leaves from the six replicates for each isolate were retrieved from storage. Each leaf was cut into approximately 30‐mm‐long sections. All sections from each leaf were placed into a 1.2‐mL collection microtube (Qiagen, Hilden, Germany) together with a 5 mm × 25 mm strip of filter paper; 100 μL of sterile water were added to each tube to moisten the filter paper. The tubes were capped and placed at 25 °C for 24 h to provide the humid environment needed to exude the cirrhi containing spores from pycnidia; 800 μL of water containing 0.001% TWEEN 20 were added to each tube. The tubes were vortexed for 20 s to suspend the released spores; 100 μL of the spore solution were placed into a black glass‐bottomed 96‐well plate (μClear, Greiner Bio‐One, Kremsmunster, Austria). Spores were imaged with an Olympus IX 81 (Shinjuku, Tokyo, Japan) inverted microscope coupled with a Hamamatsu ORCA‐ER camera (Hamamatsu City, Shizuoka, Japan) using transmission illumination. Four images were made from each well at 20 times magnification with a 10% overlap between images. Images from each well were stitched together using ImageJ (Rasband, 1997−2015). Each spore was counted and its length was measured using the line selection tool and the measure command in ImageJ (Fig. S1, see Supporting Information). The mean spore length was used as a proxy for spore size. The total number of spores per leaf was calculated (Fig. S1) and divided by the total number of pycnidia per leaf to derive the mean number of spores per pycnidium.
All analyses were performed in base R (R Core Team, 2012) unless specified otherwise. To normalize environmental variation between technical replicates and biological replicates and to account for G × E interactions, phenotypes were mean centred (Schielzeth, 2010). For each phenotype, the mean value for each glasshouse chamber and time point was calculated. This value was subtracted from all individual values from the corresponding glasshouse chamber and time point, resulting in a mean value of approximately zero for each glasshouse chamber time point. The mean of these transformed values was calculated for each progeny and used for subsequent analyses. PLACL and pycnidia density values were log transformed prior to mean centring. Mean centred values were used for analysis, but untransformed values are reported in the text and figures for clarity.
Phenotypic differences between wheat cultivars were calculated using analysis of variance (ANOVA). Effects on phenotypes of accessory chromosome presence or absence were calculated using ANOVA with the presence or absence of each chromosome as factors. Non‐significant factors were removed from the models. Effect sizes (η 2) were calculated using the lsr package (Navarro, 2015). Fisher's least‐significant difference (LSD) test was used to calculate the differences between isolates with different parental alleles at QTL peaks using the Agricolae package (de Mendiburu, 2014). Correlations between pycnidia size and spore size, and between pycnidia size and number of spores per pycnidia, were made with Pearson's correlation coefficient.
Progeny from both crosses were genotyped using RADseq based on a method adapted from Etter et al. (2011), as described previously (Lendenmann et al., 2014). The genetic map of Lendenmann et al. (2014) was used to perform QTL mapping. QTL mapping was performed using the r/QTL package (Arends et al., 2010) following the methods described in Lendenmann et al. (2014). Briefly, simple interval mapping (SIM) was used to identify QTLs, with 1000 genome‐wide permutations used to calculate significant logarithm of the odds (LOD) thresholds. Further analyses were performed on QTLs with significance levels of P < 0.05. The 95% confidence interval of each QTL was calculated using Bayesian credible intervals.
To establish the presence or absence of accessory chromosomes in 3D1 × 3D7, sequence reads from RADseq genotyping were aligned to the IPO323 reference genome (Goodwin et al., 2011). For each isolate, the mean read depth was calculated for each chromosome and divided by the mean sequence depth over all chromosomes. Thus, chromosomes with a read depth similar to the genome average have a value of ∼1 and were deemed to be present, whereas chromosomes with low or no read depth have a value of ∼0 and were deemed to be absent. Deviations from Mendelian inheritance among the accessory chromosomes exhibiting presence–absence polymorphisms in the parents were tested using a chi‐squared (χ 2) test.
Parent genome sequences (Croll et al., 2013) were aligned to the IPO323 reference genome. Sequence variants were identified using the unified genotyper within GATK (McKenna et al., 2010) and effects of variants found within genes were predicted using snpEFF (Cingolani et al., 2012a) and snpSIFT (Cingolani et al., 2012b), following the methods outlined in Lendenmann et al. (2014). The parental allele of the SNP marker closest to the QTL peak on chromosome 7 was used to identify the genotype of the QTL peak in each offspring.
The genes within the 95% confidence interval containing a QTL with large effect on chromosome 7 were manually re‐annotated using existing annotations from the IPO323 reference annotation (Goodwin et al., 2011) and Grandaubert et al. (2015), together with RNAseq data from IPO323 (Rudd et al., 2015) and from the 3D7 parental isolate (Palma‐Guerrero et al., 2017). Genes were annotated using blast2go (Conesa et al., 2005). Signal peptides were identified using signalP 4.1 (Petersen et al., 2011) and transmembrane domains were identified with TMHMM 2.0 (Krogh et al., 2001). The re‐annotated genes were used for all subsequent analyses of this region.
The RNAseq reads at 7, 12, 14 and 28 dpi from J. Palma‐Guerrero et al. (2017) were used to calculate expression levels for each gene within the chromosome 7 QTL. We used HTSeq v0.6.1 (Anders et al., 2015) to calculate gene counts for every sample with the ‘union’ model based on the new gene annotations. The Bioconductor edgeR v3.2.3 package (Robinson and Oshlack, 2010; Robinson and Smyth, 2008; Robinson et al., 2010) was used to normalize the gene counts across samples. RPKM (reads per kilobase of transcript per million mapped reads) values were generated using edgeR with the function ‘rpkm’, based on a vector containing the length of each re‐annotated gene.
The population genomics of the genes in the 95% confidence interval of the chromosome 7 QTL were characterized using 28 re‐sequenced Z. tritici isolates sampled from a single field in Switzerland. Sequences were aligned to the reference genome using Bowtie 2 version 2.2.3 (Langmead and Salzberg, 2012). SNPs among the isolates were identified using GATK tools (DePristo et al., 2011). GC content was calculated in the reference genome using 1000‐bp overlapping windows, with 100‐bp overlaps. The locations of transposable elements within the reference genome were identified using RepeatMasker (Smit et al., 2013−2015). The repeat library used for annotation was version 20150807 downloaded from Repbase in September 2015 (Jurka et al., 2005). The SNP genotyping rate in the 28 re‐sequenced isolates was calculated in windows of 1000 bp. The SNP density was calculated in windows of 500 bp for SNPs that were genotyped in >50% of the re‐sequenced isolates. The SNP density per coding sequence was calculated by dividing the number of SNPs with a genotyping rate >90% by the total coding sequence length. Tajima's D statistic (Tajima, 1989) was calculated per gene using Popgenome in R (Pfeifer et al., 2014). Only SNPs with a genotyping rate >90% were included. Tajima's D values for genes with less than 10 SNPs were not calculated.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Table S1 Detailed list of all genes found within the quantitative trait locus (QTL) 95% confidence intervals for all virulence traits in the Zymoseptoria tritici mapping populations 3D1 × 3D7 and 1A5 × 1E4.
Table S2 Zymoseptoria tritici genes functionally characterized to date which show altered virulence phenotypes. Adapted from Orton et al. (2011).
Table S3 Expression values for genes within the chromosome 7 quantitative trait locus (QTL) for pycnidia density in Zymoseptoria tritici.
Table S4 Details of 38 re‐annotated genes within a quantitative trait locus (QTL) on chromosome 7 for pycnidia density in Zymoseptoria tritici in .gff format.
Fig. S1 Typical microscope images of Zymoseptoria tritici pycnidiospores showing many longer, overlapping spores (top left) and shorter, less dense spores (top right). Yellow lines represent spores counted and measured via a semi‐automated ImageJ macro (bottom panels).
Acknowledgements
The research was supported by a grant from the Swiss National Science Foundation (31003A_134755). RADseq libraries were prepared at the Genetic Diversity Center Zürich (GDC) and sequenced at the Quantitative Genomics Facility at the Department of Biosystems Science and Engineering (D‐BSSE) at the scientific central facilities of ETH Zürich. Microscopy was carried out at the Scientific Center for Optical and Electron Microscopy (ScopeM) of ETH Zürich. Technical assistance was provided by Renato Guidon, Thomas Kreienbu¨hl and Leandra Knecht. Cross 3D1 × 3D7 was made by M. Lendenmann. Cross 1A5 × 1E4 was made by Marcello Zala.
References
- Ahmed, H.U. , Mundt, C.C. and Coakley, S.M. (1995) Host–pathogen relationship of geographically diverse isolates of Septoria tritici and wheat cultivars. Plant Pathol. 44, 838–847. [Google Scholar]
- Akagi, Y. , Akamatsu, H. , Otani, H. and Kodama, M. (2009) Horizontal chromosome transfer, a mechanism for the evolution and differentiation of a plant‐pathogenic fungus. Eukaryot. Cell, 8, 1732–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander, J.N. , McCormick, P.S. and Hohn, M.T. (1999) TRI12, a trichothecene efflux pump from Fusarium sporotrichioides: gene isolation and expression in yeast. Mol. Genet. Genomics, 261, 977–984. [DOI] [PubMed] [Google Scholar]
- Ali, S. , Laurie, J.D. , Linning, R. , Cervantes‐Chávez, J.A. , Gaudet, D. and Bakkeren, G. (2014) An immunity‐triggering effector from the barley smut fungus Ustilago hordei resides in an Ustilaginaceae‐specific cluster bearing signs of transposable element‐assisted evolution. PLoS Pathog. 10, e1004223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almasy, L. and Blangero, J. (2009) Human QTL linkage mapping. Genetica, 136, 333–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso‐Blanco, C. and Méndez‐Vigo, B. (2014) Genetic architecture of naturally occurring quantitative traits in plants: an updated synthesis. Curr. Opin. Plant Biol. 18, 37–43. [DOI] [PubMed] [Google Scholar]
- Anders, S. , Pyl, P.T. and Huber, W. (2015) HTSeq—a Python framework to work with high‐throughput sequencing data. Bioinformatics, 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arends, D. , Prins, P. , Jansen, R.C. and Broman, K.W. (2010) R/qtl: high‐throughput multiple QTL mapping. Bioinformatics, 26, 2990–2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Bosch, F. , McRoberts, N. , van den Berg, F. and Madden, L.V. (2008) The basic reproduction number of plant pathogens: matrix approaches to complex dynamics. Phytopathology, 98, 239–249. [DOI] [PubMed] [Google Scholar]
- Brading, P.A. , Verstappen, E.C.P. , Kema, G.H.J. and Brown, J.K.M. (2002) A gene‐for‐gene relationship between wheat and Mycosphaerella graminicola, the Septoria tritici blotch pathogen. Phytopathology, 92, 439–445. [DOI] [PubMed] [Google Scholar]
- Brown, J.K.M. , Chartrain, L. , Lasserre‐Zuber, P. and Saintenac, C. (2015) Genetics of resistance to Zymoseptoria tritici and applications to wheat breeding. Fungal Genet. Biol. 79, 33–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruns, E. , Carson, M.L. and May, G. (2014) The jack of all trades is master of none: a pathogen's ability to infect a great number of host genotypes comes at a cost of delayed reproduction. Evolution, 68, 2453–2466. [DOI] [PubMed] [Google Scholar]
- Bush, R.K. and Portnoy, J.M. (2001) The role and abatement of fungal allergens in allergic diseases. J. Allergy Clin. Immunol. 107, S430–S440. [DOI] [PubMed] [Google Scholar]
- Chruszcz, M. , Chapman, M.D. , Osinski, T. , Solberg, R. , Demas, M. , Porebski, P.J. , Majorek, K.A. , Pomés, A. and Minor, W. (2012) Alternaria alternata allergen, Alt a 1 – a unique, β‐barrel protein dimer exclusively found in fungi. J. Allergy Clin. Immunol. 130, 241–247. e249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Platts, A. , Wang, L.L. , Coon, M. , Nguyen, T. , Wang, L. , Land, S.J. , Lu, X. and Ruden, D.M. (2012a) A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w(1118); iso‐2; iso‐3. Fly, 6, 80–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cingolani, P. , Patel, V.M. , Coon, M. , Nguyen, T. , Land, S.J. , Ruden, D.M. and Lu, X. (2012b) Using Drosophila melanogaster as a model for genotoxic chemical mutational studies with a new program, SnpSift. Front. Genet. 3, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman, J.J. and Mylonakis, E. (2009) Efflux in fungi: la piece de resistance. PLoS Pathog. 5, e1000486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conesa, A. , Götz, S. , García‐Gómez, J.M. , Terol, J. , Talón, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Courvoisier, N. , Häner, L.L. , Bertossa, M. , Thévoz, E. , Anders, M. , Stoll, P. , Weisflog, P. , Dugon, J. and Grünig, K. (2015) Liste Recommandée des Variétés de Céréales pour la Récolte 2016 Recherche Agronomique Suisse Available at: http://www.swissgranum.ch/88-2-Listes-recommandes.html. Accessed 21 December 2016.
- Cousin, A. , Mehrabi, R. , Guilleroux, M. , Dufresne, M. , Van Der Lee, T. , Waalwijk, C. , Langin, T. and Kema, G.H.J. (2006) The MAP kinase‐encoding gene MgFus3 of the non‐appressorium phytopathogen Mycosphaerella graminicola is required for penetration and in vitro pycnidia formation. Mol. Plant Pathol. 7, 269–278. [DOI] [PubMed] [Google Scholar]
- Cowger, C. , Hoffer, M.E. and Mundt, C.C. (2000) Specific adaptation by Mycosphaerella graminicola to a resistant wheat cultivar. Plant Pathol. 49, 445–451. [Google Scholar]
- Cramer, R.A. and Lawrence, C.B. (2004) Identification of Alternaria brassicicola genes expressed in planta during pathogenesis of Arabidopsis thaliana . Fungal Genet. Biol. 41, 115–128. [DOI] [PubMed] [Google Scholar]
- Croll, D. , Zala, M. and McDonald, B.A. (2013) Breakage‐fusion‐bridge cycles and large insertions contribute to the rapid evolution of accessory chromosomes in a fungal pathogen. PLoS Genet. 9, e1003567. doi: 10.1371/journal.pgen.1003567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derbyshire, M.C. , Michaelson, L. , Parker, J. , Kelly, S. , Thacker, U. , Powers, S.J. , Bailey, A. , Hammond‐Kosack, K. , Courbot, M. and Rudd, J. (2015) Analysis of cytochrome b(5) reductase‐mediated metabolism in the phytopathogenic fungus Zymoseptoria tritici reveals novel functionalities implicated in virulence. Fungal Genet. Biol. 82, 69–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean, R. , Van Kan, J.A.L. , Pretorius, Z.A. , Hammond‐Kosack, K.E. , Di Pietro, A. , Spanu, P.D. , Rudd, J.J. , Dickman, M. , Kahmann, R. , Ellis, J. and Foster, G.D. (2012) The top 10 fungal pathogens in molecular plant pathology. Mol. Plant Pathol. 13, 413–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M.A. , Banks, E. , Poplin, R. , Garimella, K.V. , Maguire, J.R. , Hartl, C. , Philippakis, A.A. , del Angel, G. , Rivas, M.A. , Hanna, M. , McKenna, A. , Fennell, T.J. , Kernytsky, A.M. , Sivachenko, A.Y. , Cibulskis, K. , Gabriel, S.B. , Altshuler, D. and Daly, M.J. (2011) A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nat Genet. 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhillon, B. , Gill, N. , Hamelin, R.C. and Goodwin, S.B. (2014) The landscape of transposable elements in the finished genome of the fungal wheat pathogen Mycosphaerella graminicola . BMC Genomics, 15, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, S. , Raffaele, S. and Kamoun, S. (2015) The two‐speed genomes of filamentous pathogens: waltz with plants. Curr. Opin. Genet. Dev. 35, 57–65. [DOI] [PubMed] [Google Scholar]
- Etter, P.D. , Bassham, S. , Hohenlohe, P.A. , Johnson, E.A. and Cresko, W.A. (2011) SNP discovery and genotyping for evolutionary genetics using RAD sequencing In Molecular Methods for Evolutionary Genetics (Orgogozo V. and Rockman V. M., eds), pp. 157–178. Totowa, NJ: Humana Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyal, Z. , Scharen, A.L. , Prescott, J.M. and van Ginkel, M. (1987) The Septoria Diseases of Wheat. Mexico, DF: CYMMIT. [Google Scholar]
- Flor, H.H. (1955) Host‐parasite interaction in flax rust—its genetics and other implications. Phytopathology, 45, 680–685. [Google Scholar]
- Fones, H. and Gurr, S. (2015) The impact of Septoria tritici blotch disease on wheat: an EU perspective. Fungal Genet. Biol. 79, 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foulongne‐Oriol, M. (2012) Genetic linkage mapping in fungi: current state, applications, and future trends. Appl. Microbiol. Biotechnol. 95, 891–904. [DOI] [PubMed] [Google Scholar]
- Furbank, R.T. and Tester, M. (2011) Phenomics ‐ technologies to relieve the phenotyping bottleneck. Trends Plant Sci. 16, 635–644. [DOI] [PubMed] [Google Scholar]
- Gibriel, H.A.Y. , Thomma, B.P.H.J. and Seidl, M.F. (2016) The age of effectors: genome‐based discovery and applications. Phytopathology, 106, 1206–1212. [DOI] [PubMed] [Google Scholar]
- Gómez‐Casado, C. , Murua‐García, A. , Garrido‐Arandia, M. , González‐Melendi, P. , Sánchez‐Monge, R. , Barber, D. , Pacios, L.F. and Díaz‐Perales, A. (2014) Alt a 1 from Alternaria interacts with PR5 thaumatin‐like proteins. FEBS Lett. 588, 1501–1508. [DOI] [PubMed] [Google Scholar]
- Goodwin, S.B. , M'barek, S. , Dhillon, B. , Wittenberg, A.H. , Crane, C.F. , Hane, J.K. , Foster, A.J. , Van der Lee, T.A. , Grimwood, J. , Aerts, A. , Antoniw, J. , Bailey, A. , Bluhm, B. , Bowler, J. , Bristow, J. , van der Burgt, A. , Canto‐Canche, B. , Churchill, A.C. , Conde‐Ferraez, L. , Cools, H.J. , Coutinho, P.M. , Csukai, M. , Dehal, P. , De Wit, P. , Donzelli, B. , van de Geest, H.C. , van Ham, R.C. , Hammond‐Kosack, K.E. , Henrissat, B. , Kilian, A. , Kobayashi, A.K. , Koopmann, E. , Kourmpetis, Y. , Kuzniar, A. , Lindquist, E. , Lombard, V. , Maliepaard, C. , Martins, N. , Mehrabi, R. , Nap, J.P. , Ponomarenko, A. , Rudd, J.J. , Salamov, A. , Schmutz, J. , Schouten, H.J. , Shapiro, H. , Stergiopoulos, I. , Torriani, S.F. , Tu, H. , de Vries, R.P. , Waalwijk, C. , Ware, S.B. , Wiebenga, A. , Zwiers, L.H. , Oliver, R.P. , Grigoriev, I.V. and Kema, G.H. (2011) Finished genome of the fungal wheat pathogen Mycosphaerella graminicola reveals dispensome structure, chromosome plasticity, and stealth pathogenesis. PLoS Genet. 7, e1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough, F.J. (1978) Effect of wheat host cultivars on pycnidiospore production by Septoria tritici . Phytopathology, 68, 1343–1345. [Google Scholar]
- Grandaubert, J. , Bhattacharyya, A. and Stukenbrock, E.H. (2015) RNA‐seq based gene annotation and comparative genomics of four fungal grass pathogens in the genus Zymoseptoria identify novel orphan genes and species‐specific invasions of transposable elements. G3, 5, 1323–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habgood, R.M. (1977) Resistance of barley cultivars to Rhynchosporium secalis . Trans. Br. Mycol. Soc. 69, 281–286. [Google Scholar]
- Halama, P. , Skajennikoff, M. and Dehorter, B. (1999) Tetrad analysis of mating type, mutations, esterase and aggressiveness in Phaeosphaeria nodorum . Mycol. Res. 103, 43–49. [Google Scholar]
- Hansen, T.F. (2006) The evolution of genetic architecture. Annu. Rev. Ecol. Evol. Syst. 37, 123–157. [Google Scholar]
- Jørgensen, L.N. , Hovmøller, M.S. , Hansen, J.G. , Lassen, P. , Clark, B. , Bayles, R. , Rodemann, B. , Flath, K. , Jahn, M. , Goral, T. , Jerzy Czembor, J. , Cheyron, P. , Maumene, C. , De Pope, C. , Ban, R. , Nielsen, G.C. and Berg, G. (2014) IPM strategies and their dilemmas including an introduction to www.eurowheat.org. J. Integr. Agric. 13, 265–281. [Google Scholar]
- Joseph, B. , Schwarz, R.F. , Linke, B. , Blom, J. , Becker, A. , Claus, H. , Goesmann, A. , Frosch, M. , Ller, T. , Vogel, U. and Schoen, C. (2011) Virulence evolution of the human pathogen Neisseria meningitidis by recombination in the core and accessory genome. PLoS One, 6, e18441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. and Walichiewicz, J. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Kanvil, S. , Collins, C.M. , Powell, G. and Turnbull, C.G.N. (2015) Cryptic virulence and avirulence alleles revealed by controlled sexual recombination in pea aphids. Genetics, 199, 581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kema, G.H.J. , Verstappen, E.C.P. and Waalwijk, C. (2000) Avirulence in the wheat Septoria tritici leaf blotch fungus Mycosphaerella graminicola is controlled by a single locus. Mol. Plant–Microbe Interact. 13, 1375–1379. [DOI] [PubMed] [Google Scholar]
- Kramer, B. , Thines, E. and Foster, A.J. (2009) MAP kinase signaling pathway components and targets conserved between the distantly related plant pathogenic fungi Mycosphaerella graminicola and Magnaporthe grisea . Fungal Genet. Biol. 46, 667–681. [DOI] [PubMed] [Google Scholar]
- Krattinger, S.G. , Lagudah, E.S. , Spielmeyer, W. , Singh, R.P. , Huerta‐Espino, J. , McFadden, H. , Bossolini, E. , Selter, L.L. and Keller, B. (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science, 323, 1360–1363. [DOI] [PubMed] [Google Scholar]
- Krogh, A. , Larsson, B. , von Heijne, G. and Sonnhammer, E.L.L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J Mol. Biol. 305, 567–580. [DOI] [PubMed] [Google Scholar]
- Kulkarni, R.D. , Kelkar, H.S. and Dean, R.A. (2003) An eight‐cysteine‐containing CFEM domain unique to a group of fungal membrane proteins. Trends Biochem. Sci. 28, 118–121. [DOI] [PubMed] [Google Scholar]
- Langmead, B. and Salzberg, S. (2012) Fast gapped‐read alignment with Bowtie 2. Nat. Methods, 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lannou, C. (2011) Variation and selection of quantitative traits in plant pathogens. Annu. Rev. Phytopathol. 50, 319–338. [DOI] [PubMed] [Google Scholar]
- Larraya, L.M. , Alfonso, M. , Pisabarro, A.G. and Ramirez, L. (2003) Mapping of genomic regions (quantitative trait loci) controlling production and quality in industrial cultures of the edible basidiomycete Pleurotus ostreatus . Appl. Environ. Microbiol. 69, 3617–3625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendenmann, M.H. , Croll, D. , Stewart, E.L. and McDonald, B.A. (2014) Quantitative trait locus mapping of melanization in the plant pathogenic fungus Zymoseptoria tritici . G3, 4, 2519–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lendenmann, M.H. , Croll, D. and McDonald, B.A. (2015) QTL mapping of fungicide sensitivity reveals novel genes and pleiotropy with melanization in the pathogen Zymoseptoria tritici . Fungal Genet. Biol. 80, 53–67. [DOI] [PubMed] [Google Scholar]
- Lendenmann, M.H. , Croll, D. , Palma‐Guerrero, J. , Stewart, E.L. and McDonald, B.A. (2016) QTL mapping of temperature sensitivity reveals candidate genes for thermal adaptation and growth morphology in the plant pathogenic fungus Zymoseptoria tritici . Heredity, 116, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Presti, L. , Lanver, D. , Schweizer, G. , Tanaka, S. , Liang, L. , Tollot, M. , Zuccaro, A. , Reissmann, S. and Kahmann, R. (2015) Fungal effectors and plant susceptibility. Annu. Rev. Plant. Biol. 66, 513–545. [DOI] [PubMed] [Google Scholar]
- Ma, L.J. , van der Does, H.C. , Borkovich, K.A. , Coleman, J.J. , Daboussi, M.J. , Di Pietro, A. , Dufresne, M. , Freitag, M. , Grabherr, M. , Henrissat, B. , Houterman, P.M. , Kang, S. , Shim, W.B. , Woloshuk, C. , Xie, X. , Xu, J.R. , Antoniw, J. , Baker, S.E. , Bluhm, B.H. , Breakspear, A. , Brown, D.W. , Butchko, R.A.E. , Chapman, S. , Coulson, R. , Coutinho, P.M. , Danchin, E.G.J. , Diener, A. , Gale, L.R. , Gardiner, D.M. , Goff, S. , Hammond‐Kosack, K.E. , Hilburn, K. , Hua‐Van, A. , Jonkers, W. , Kazan, K. , Kodira, C.D. , Koehrsen, M. , Kumar, L. , Lee, Y.H. , Li, L. , Manners, J.M. , Miranda‐Saavedra, D. , Mukherjee, M. , Park, G. , Park, J. , Park, S.Y. , Proctor, R.H. , Regev, A. , Ruiz‐Roldan, M.C. , Sain, D. , Sakthikumar, S. , Sykes, S. , Schwartz, D.C. , Turgeon, B.G. , Wapinski, I. , Yoder, O. , Young, S. , Zeng, Q. , Zhou, S. , Galagan, J. , Cuomo, C.A. , Kistler, H.C. and Rep, M. (2010) Comparative genomics reveals mobile pathogenicity chromosomes in Fusarium. Nature, 464, 367–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, T.F.C. (2001) The genetic architecture of quantitative traits. Annu. Rev. Genet. 35, 303–339. [DOI] [PubMed] [Google Scholar]
- Marshall, R. , Kombrink, A. , Motteram, J. , Loza‐Reyes, E. , Lucas, J. , Hammond‐Kosack, K.E. , Thomma, B.P.H.J. and Rudd, J.J. (2011) Analysis of two in planta expressed LysM effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiology, 156, 756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. and DePristo, M.A. (2010) The Genome Analysis Toolkit: A MapReduce framework for analyzing next‐generation DNA sequencing data. Genome Res. 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore, J.W. , Herrera‐Foessel, S. , Lan, C. , Schnippenkoetter, W. , Ayliffe, M. , Huerta‐Espino, J. , Lillemo, M. , Viccars, L. , Milne, R. , Periyannan, S. , Kong, X. , Spielmeyer, W. , Talbot, M. , Bariana, H. , Patrick, J.W. , Dodds, P. , Singh, R. and Lagudah, E. (2015) A recently evolved hexose transporter variant confers resistance to multiple pathogens in wheat. Nat. Genetics, 47, 1494–1498. [DOI] [PubMed] [Google Scholar]
- de Mendiburu, F. (2014) Agricolae: statistical procedures for agricultural research. R Package Version 1.1‐8. http://CRAN.R-project.org/package=agricolae. Accessed 21 December 2016. [Google Scholar]
- Mehrabi, R. and Kema, G.H.J. (2006) Protein kinase A subunits of the ascomycete pathogen Mycosphaerella graminicola regulate asexual fructification, filamentation, melanization and osmosensing. Mol. Plant Pathol. 7, 565–577. [DOI] [PubMed] [Google Scholar]
- Mehrabi, R. , van der Lee, T. , Waalwijk, C. and Kema, G.H.J. (2006a) MgSlt2, a cellular integrity MAP kinase gene of the fungal wheat pathogen Mycosphaerella graminicola, is dispensable for penetration but essential for invasive growth. Mol . Plant–Microbe Interact. 19, 389–398. [DOI] [PubMed] [Google Scholar]
- Mehrabi, R. , Awiers, L.H. , de Waard, M.A. and Kema, G.H.J. (2006b) MgHog1 regulates dimorphism and pathogenicity in the fungal wheat pathogen Mycosphaerella graminicola . Mol. Plant–Microbe Interact. 19, 1262–1269. [DOI] [PubMed] [Google Scholar]
- Mehrabi, R. , Ben M'barek, S. , van der Lee, T.A.J. , Waalwijk, C. , de Wit, P. and Kema, G.H.J. (2009) G alpha and G beta proteins regulate the cyclic AMP pathway that is required for development and pathogenicity of the phytopathogen Mycosphaerella graminicola . Eukaryot. Cell, 8, 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao, V.P. , Covert, S.F. and VanEtten, H.D. (1991) A fungal gene for antibiotic resistance on a dispensable (“B”) chromosome. Science, 254, 1773–1776. [DOI] [PubMed] [Google Scholar]
- Mirzadi Gohari, A. , Mehrabi, R. , Robert, O. , Ince, I.A. , Boeren, S. , Schuster, M. , Steinberg, G. , de Wit, P.J. and Kema, G.H. (2014) Molecular characterization and functional analyses of ZtWor1, a transcriptional regulator of the fungal wheat pathogen Zymoseptoria tritici . Mol. Plant Pathol. 15, 394–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadi Gohari, A. , Ware, S.B. , Wittenberg, A.H.J. , Mehrabi, R. , Ben M'barek, S. , Verstappen, E.C.P. , van der Lee, T.A.J. , Robert, O. , Schouten, H.J. , de Wit, P.P.J.G.M. and Kema, G.H.J. (2015) Effector discovery in the fungal wheat pathogen Zymoseptoria tritici . Mol Plant Pathol. 16, 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan, T.H. , Sturtevant, A.H. , Muller, H.J. and Bridges, C.B. (1915) The Mechanism of Mendelian Heredity. New York: Henry Holt and Company. [Google Scholar]
- Navarro, D. (2015) lsr: Companion to “Learning Statistics with R”. https://CRAN.R-project.org/package=lsr. Accessed 21 December 2016.
- O'Driscoll, A. , Kildea, S. , Doohan, F. , Spink, J. and Mullins, E. (2014) The wheat–Septoria conflict: a new front opening up? Trends Plant Sci. 19, 602–610. [DOI] [PubMed] [Google Scholar]
- Orton, E.S. , Deller, S. and Brown, J.K.M. (2011) Mycosphaerella graminicola: from genomics to disease control. Mol. Plant Pathol. 12, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paaby, A.B. and Rockman, M.V. (2013) The many faces of pleiotropy. Trends Genet. 29, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma‐Guerrero, J. , Ma, X. , Torriani, S.F.F. , Zala, M. , Francisco, C.S. , Hartmann, F.E. , Croll, D. and McDonald, B.A. (2017) Comparative transcriptome analyses in Zymoseptoria tritici reveal significant differences in gene expression among strains during plant infection. Mol. Plant–Microbe Interact. doi: 10.1111/mpp.12333. [DOI] [PubMed] [Google Scholar]
- Pariaud, B. , Robert, C. , Goyeau, H. and Lannou, C. (2009) Aggressiveness components and adaptation to a host cultivar in wheat leaf rust. Phytopathology, 99, 869–878. [DOI] [PubMed] [Google Scholar]
- Parlevliet, J.E. (1979) Components of resistance that reduce the rate of epidemic development. Annu. Rev. Phytopathol. 17, 203–222. [Google Scholar]
- Petersen, T.N. , Brunak, S. , von Heijne, G. and Nielsen, H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods, 8, 785–786. [DOI] [PubMed] [Google Scholar]
- Pfeifer, B. , Wittelsbürger, U. , Ramos‐Onsins, S.E. and Lercher, M.J. (2014) PopGenome: an efficient Swiss army knife for population genomic analyses in R. Mol. Biol. Evol. 31, 1929–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppe, S. , Dorsheimer, L. , Happel, P. and Stukenbrock, E.H. (2015) Rapidly evolving genes are key players in host specialization and virulence of the fungal wheat pathogen Zymoseptoria tritici (Mycosphaerella graminicola). PLoS Pathog. 11, e1005055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team . (2012) R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing; http://www.R-project.org. Accessed 21 December 2016. [Google Scholar]
- Rasband, W.S. (1997. –2015) ImageJ. Bethesda, MD: US National Institutes of Health; http://imagej.nih.gov/ij/. Accessed 21 December 2016. [Google Scholar]
- Robinson, M.D. and Oshlack, A. (2010) A scaling normalization method for differential expression analysis of RNA‐seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson, M.D. and Smyth, G.K. (2008) Small‐sample estimation of negative binomial dispersion, with applications to SAGE data. Biostatistics, 9, 321–332. [DOI] [PubMed] [Google Scholar]
- Robinson, M.D. , McCarthy, D.J. and Smyth, G.K. (2010) edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd, J. , Kanyuka, K. , Hassani‐Pak, K. , Derbyshire, M. , Andongabo, A. , Devonshire, J. , Lysenko, A. , Saqi, M. , Desai, N. , Powers, S. , Hooper, J. , Ambroso, L. , Bharti, A. , Farmer, A. , Hammond‐Kosack, K. , Dietrich, R. and Courbot, M. (2015) Transcriptome and metabolite profiling the infection cycle of Zymoseptoria tritici on wheat (Triticum aestivum) reveals a biphasic interaction with plant immunity involving differential pathogen chromosomal contributions, and a variation on the hemibiotrophic lifestyle definition. Plant Physiol. 167, 1158–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schielzeth, H. (2010) Simple means to improve the interpretability of regression coefficients. Methods Ecol. Evol. 1, 103–113. [Google Scholar]
- Shaaban, M. , Palmer, J.M. , El‐Naggar, W.A. , El‐Sokkary, M.A. , Habib, E.L.S.E. and Keller, N.P. (2010) Involvement of transposon‐like elements in penicillin gene cluster regulation. Fungal Genet. Biol. 47, 423–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smit, A.F.A. , Hubley, R. and Green, P. (2013. −2015) RepeatMasker Open‐4.0. http://www.repeatmasker.org. Accessed 21 December 2016.
- Solberg Woods, L.C. (2014) QTL mapping in outbred populations: successes and challenges. Physiol. Genomics, 46, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerhalder, R.J. , McDonald, B.A. , Mascher, F. and Zhan, J. (2010) Sexual recombinants make a significant contribution to epidemics caused by the wheat pathogen Phaeosphaeria nodorum . Phytopathology, 100, 855–862. [DOI] [PubMed] [Google Scholar]
- St. Clair, D.A. (2010) Quantitative disease resistance and quantitative resistance loci in breeding. Annu. Rev. Phytopathol. 48, 247–268. [DOI] [PubMed] [Google Scholar]
- Stefansson, T.S. , McDonald, B.A. and Willi, Y. (2013) Local adaptation and evolutionary potential along a temperature gradient in the fungal pathogen Rhynchosporium commune . Evol. Appl. 6, 524–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stergiopoulos, I. , Zwiers, L. and De Waard, M.A. (2003) The ABC transporter MgAtr4 is a virulence factor of Mycosphaerella graminicola that affects colonization of substomatal cavities in wheat leaves. Mol. Plant–Microbe Interact. 16, 689–698. [DOI] [PubMed] [Google Scholar]
- Stewart, E.L. and McDonald, B.A. (2014) Measuring quantitative virulence in the wheat pathogen Zymoseptoria tritici using high‐throughput automated image analysis. Phytopathology, 104, 985–992. [DOI] [PubMed] [Google Scholar]
- Stewart, E.L. , Hagerty, C.H. , Mikaberidze, A. , Mundt, C.C. , Zhong, Z. and McDonald, B.A. (2016) An improved method for measuring quantitative resistance to the wheat pathogen Zymoseptoria tritici using high throughput automated image analysis. Phytopathology, 106, 782–788. doi: 10.1094/PHYTO-01-16-0018-R [DOI] [PubMed] [Google Scholar]
- Stukenbrock, E.H. and McDonald, B.A. (2009) Population genetics of fungal and oomycete effectors involved in gene‐for‐gene interactions. Mol. Plant–Microbe Interact. 22, 371–380. [DOI] [PubMed] [Google Scholar]
- Stukenbrock, E.H. , Jorgensen, F.G. , Zala, M. , Hansen, T.T. , McDonald, B.A. and Schierup, M.H. (2010) Whole‐genome and chromosome evolution associated with host adaptation and speciation of the wheat pathogen Mycosphaerella graminicola . PLoS Genet. 6, e1001189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suffert, F. , Sache, I. and Lannou, C. (2013) Assessment of quantitative traits of aggressiveness in Mycosphaerella graminicola on adult wheat plants. Plant Pathol. 62, 1330–1341. [Google Scholar]
- Tajima, F. (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twizeyimana, M. , Ojiambo, P.S. , Bandyopadhyay, R. and Hartman, G.L. (2014) Use of quantitative traits to assess aggressiveness of Phakopsora pachyrhizi isolates from Nigeria and the United States. Plant Dis. 98, 1261–1266. [DOI] [PubMed] [Google Scholar]
- Wittenberg, A.H.J. , van der Lee, T.A.J. , Ben M'barek, S. , Ware, S.B. , Goodwin, S.B. , Kilian, A. , Visser, R.G.F. , Kema, G.H.J. and Schouten, H.J. (2009) Meiosis drives extraordinary genome plasticity in the haploid fungal plant pathogen Mycosphaerella graminicola . PLoS One, 4, e5863. doi: 10.1371/journal.pone.0005863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan, J. , Mundt, C.C. , Hoffer, M.E. and McDonald, B.A. (2002) Local adaptation and effect of host genotype on the rate of pathogen evolution: an experimental test in a plant pathosystem. Evolut. Biol. 15, 634–647. [Google Scholar]
- Zhan, J. , Linde, C.C. , Jurgens, T. , Merz, U. , Steinebrunner, F. and McDonald, B.A. (2005) Variation for neutral markers is correlated with variation for quantitative traits in the plant pathogenic fungus Mycosphaerella graminicola . Mol. Ecol. 14, 2683–2693. [DOI] [PubMed] [Google Scholar]
- Zhan, J. , Torriani, S.F.F. and McDonald, B.A. (2007) Significant difference in pathogenicity between MAT1‐1 and MAT1‐2 isolates in the wheat pathogen Mycosphaerella graminicola . Fungal Genet. Biol. 44, 339–346. [DOI] [PubMed] [Google Scholar]
- Zhang, Z.N. , Wu, Q.Y. , Zhang, G.Z. , Zhu, Y.Y. , Murphy, R.W. , Liu, Z. and Zou, C.G. (2015) Systematic analyses reveal uniqueness and origin of the CFEM domain in fungi. Sci. Rep. 5, 13 032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Table S1 Detailed list of all genes found within the quantitative trait locus (QTL) 95% confidence intervals for all virulence traits in the Zymoseptoria tritici mapping populations 3D1 × 3D7 and 1A5 × 1E4.
Table S2 Zymoseptoria tritici genes functionally characterized to date which show altered virulence phenotypes. Adapted from Orton et al. (2011).
Table S3 Expression values for genes within the chromosome 7 quantitative trait locus (QTL) for pycnidia density in Zymoseptoria tritici.
Table S4 Details of 38 re‐annotated genes within a quantitative trait locus (QTL) on chromosome 7 for pycnidia density in Zymoseptoria tritici in .gff format.
Fig. S1 Typical microscope images of Zymoseptoria tritici pycnidiospores showing many longer, overlapping spores (top left) and shorter, less dense spores (top right). Yellow lines represent spores counted and measured via a semi‐automated ImageJ macro (bottom panels).