

Summary
The success of plant‐pathogenic fungi mostly relies on their arsenal of virulence factors which are expressed and delivered into the host tissue during colonization. The biotrophic fungal pathogen Ustilago hordei causes covered smut disease on both barley and oat. In this study, we combined cytological, genomics and molecular biological methods to achieve a better understanding of the molecular interactions in the U. hordei–barley pathosystem. Microscopic analysis revealed that U. hordei densely colonizes barley leaves on penetration, in particular the vascular system. Transcriptome analysis of U. hordei at different stages of host infection revealed differential expression of the transcript levels of 273 effector gene candidates. Furthermore, U. hordei transcriptionally activates core effector genes which may suppress even non‐host early defence responses. Based on expression profiles and novelty of sequences, knockout studies of 14 effector candidates were performed in U. hordei, which resulted in the identification of four virulence factors required for host colonization. Yeast two‐hybrid screening identified potential barley targets for two of the effectors. Overall, this study provides a first systematic analysis of the effector repertoire of U. hordei and identifies four effectors (Uvi1–Uvi4) as virulence factors for the infection of barley.
Keywords: barley, biotrophy, effectors, gene expression, Ustilago hordei
Introduction
Ustilago hordei is a facultative biotrophic fungal pathogen that causes covered smut disease on both barley and oat plants (Gaudet et al., 2010). Ustilago hordei is a member of the order Ustilaginales, which contains pathogens of several economically important crops, such as maize, wheat, oat, barley and sugar cane. Similar to other phytopathogenic smut fungi, the pathogenic development of U. hordei is coupled with sexual development. For successful infection, two haploid sporidia of opposite mating types fuse and form the infectious dikaryotic filament that subsequently penetrates the host seedling (Hu et al., 2002). On penetration, U. hordei proliferates both extracellularly and intracellularly in or just below the shoot meristem. The covered smut of barley proceeds in a mostly symptomless manner; however, at the late stage of infection, the proliferation of U. hordei in the spikelet of the barley inflorescence and the kernels of infected plants is manifested by masses of dark brown smut spores (Hu et al., 2002).
Secreted proteins are at the front line of host–microbe interactions. The secretome of pathogens varies considerably based on their pathogenic life style (Lowe and Howlett, 2012). Although necrotrophic plant pathogens typically secrete plant cell wall‐degrading enzymes and toxins to induce cell death and subsequently feed on host dead cells (Kubicek et al., 2014), the virulence of biotrophic pathogens is largely built on the secretion of effector proteins, which manipulate host physiology and immunity to promote disease establishment (Schuster et al., 2017). Effectors show diverse modes of action that are required at different stages of disease development; thus, their transcript levels fluctuate during colonization of the host (Kleemann et al., 2012; O'Connell et al., 2012). For example, Phytophthora sojae expresses the SNE1 effector to suppress the Necrosis‐ and Ethylene‐inducing Peptide 1‐Like Proteins (NLP1)‐mediated cell death activity during the biotrophic stage; by doing so it can fine‐tune the transition from the biotrophic to the necrotrophic stage (Kelley et al., 2010). Another example is the necrosis inducing protein 1 (NIP1) of Rhynchosporium commune, which is specifically induced on transition from the biotrophic to necrotrophic stage (Kirsten et al., 2012). Sequential expression of genes encoding putative effectors has also been observed for the obligate biotrophic rust fungus Melampsora larici‐populina (Duplessis et al., 2011). Ustilago maydis, a close relative of U. hordei, is considered as a model system for biotrophs. It has been reported that U. maydis deploys distinct sets of effector genes in a stage‐, host organ‐ and cell type‐specific manner (Lanver et al., 2018; Matei et al., 2018; Schilling et al., 2014; Skibbe et al., 2010). However, some effectors are constitutively expressed during all stages of colonization in order to modulate plant defence. Examples are the conserved core effector Pep1, which inhibits POX12 activity (Hemetsberger et al., 2012), and Pit2, which inhibits host plant papain‐like cysteine proteases (Mueller et al., 2013).
The genomics revolution of recent decades has increased our understanding of the genetic composition of a wide range of plant‐pathogenic fungi (Dean et al., 2005; Kämper et al., 2006; O'Connell et al., 2012; Schuster et al., 2017; de Wit et al., 2012). Several computational pipelines have been developed to identify the secretomes of phytopathogenic fungi, according to common hallmarks of fungal effectors, including the presence of a signal peptide for extracellular secretion, absence of transmembrane domains, molecular weight and cysteine content (Sperschneider et al., 2015, 2015; de Wit et al., 2012). Comparative genome analysis has revealed that, although closely related phytopathogenic fungi have core sets of effectors, a great number of fungal effectors are still lineage specific (Schuster et al., 2017). This makes the identification and characterization of effectors a challenging endeavour.
For the model system U. maydis, a rich toolbox for functional genetics is available (Kämper, 2004; Matei and Doehlemann, 2016; Spellig et al., 1996). Several of these tools have been successfully adapted for U. hordei in this and other studies, including targeted gene deletion. The availability of a complete genome sequence (Laurie et al., 2012) and the amenability to reverse genetics define the U. hordei–barley system as a good model to study the molecular basis of a fully biotrophic plant–pathogen interaction. In this study, we have analysed the effector repertoire of U. hordei by a transcriptome analysis at different stages of U. hordei infection. We functionally studied 14 effector candidates with regard to their contribution to U. hordei virulence. In addition, yeast two‐hybrid (Y2H) screens identified potential host targets of two effectors with significant virulence function.
Results
Infection of barley leaves by U. hordei
Ustilago hordei is a surface‐borne smut fungus that initially penetrates the coleoptile of barley seeds and grows just below the apical shoot meristem without causing any visible symptoms until formation of the inflorescence (Hu et al., 2002). Therefore, on a macroscopic level, successful U. hordei infection can only be evaluated at the very late stage of disease (2–3 months after inoculation). In order to set‐up a more rapid read‐out for U. hordei infection, we inoculated U. hordei into leaf whorls of 10‐day‐old barley seedlings, similar to the infection procedure that has been established for U. maydis on maize (Redkar et al., 2015). Fungal infection was monitored at 1, 2, 3, 4, 5 and 6 days post‐inoculation (dpi) via confocal microscopy, using calcofluor white for investigation at 1 dpi and WGA‐AF488/propidium iodide staining for the subsequent time points. The microscopic observations revealed that, at 20 h post‐inoculation (hpi), U. hordei can successfully form appressoria to penetrate barley leaves (Fig. 1A). At 2 dpi, the fungal hyphae grow extracellularly and intracellularly inside the leaf and move from one cell to another. At 3 dpi onwards, U. hordei increasingly colonizes the plant’s vascular system (Fig. 1A). Interestingly, we observed that, when U. hordei hyphae grow inside barley cells, they form structures, which structurally resemble haustoria (Fig. 1B). Unlike fungal clamps (Fig. 1B, first two photographs), these haustorium‐like structures (photographs 3 and 4) form multiple lobes and are found from 5 dpi onwards, but not at earlier time points.
Figure 1.
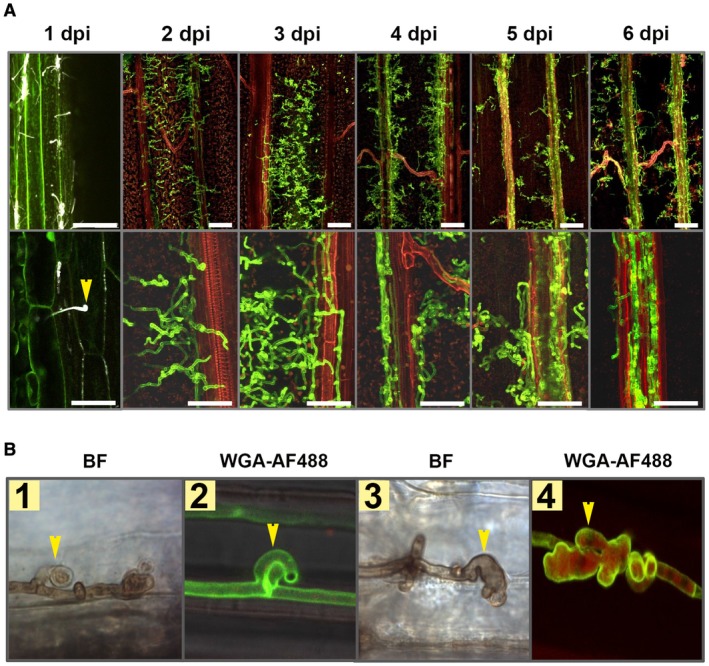
Ustilago hordei disease progression on barley leaves. (A) Wild‐type U. hordei (two mating types) was inoculated onto 10‐day‐old barley seedlings and disease progression was observed at 1, 2, 3, 4, 5 and 6 days post‐inoculation (dpi). Microscopic observations were performed with calcofluor white at 1 dpi and with WGA‐AF488/propidium iodide staining for the rest of the time points to visualize U. hordei colonization. Yellow arrow indicates U. hordei appressorium formation and host penetration. Scale bar at the top indicates 100 µm and at the bottom indicates 50 µm. Red represents plant cells and green represents fungal hyphae. (B) Ustilago hordei grows intracellularly in barley leaves and forms haustorium‐like structures during barley colonization. WGA‐AF488/propidium iodide staining and 3,3′‐diaminobenzidine (DAB) staining were performed to visualize U. hordei. Yellow arrows indicate clamps (photographs 1 and 2) and haustorium‐like structures (photographs 3 and 4). BF, bright field. The photographs were taken at 7 dpi. [Colour figure can be viewed at wileyonlinelibrary.com]
Transcriptome analysis of U. hordei during infection of barley leaves
To analyse the gene regulation of U. hordei at different stages of host infection, a whole‐genome transcriptome analysis was performed. The first time point for analysis was chosen at 20 hpi, when appressoria had formed and the fungus had started to penetrate epidermal host cells (Hof et al., 2014). In addition, samples were taken for RNA preparation at 40 hpi (early accommodation), 3 dpi (growth to vasculature) and 6 dpi (massive fungal proliferation). The transcriptome analysis was performed using a custom‐designed Agilent microarray, representing 7124 of the 7234 annotated U. hordei protein‐encoding open reading frames (for details, see Experimental procedures). Changes in U. hordei gene expression on the host plant were calculated relative to U. hordei yeast cells grown in axenic culture. Overall gene expression patterns at different transition stages during host colonization relative to axenic culture were visualized in heat maps (Fig. 2). Genes showing a two‐fold change in expression level were considered to be differentially expressed (log2‐fold change ≥ 1; false discovery rate (fdr) < 0.05; P < 0.005) (Fig. 2; Table S1‐All genes, see Supporting Information). The genome of U. hordei was analysed for genes that were differentially regulated in at least one of the tested time points. This analysis revealed that 5978 genes (83.9% of the total U. hordei genes) were differentially expressed in at least one of the tested infection time points (Fig. 2 ‐Genome; Table S1‐All genes). Although 2468 (34.6%) of the differentially expressed U. hordei genes were up‐regulated, 4078 (57.2%) were down‐regulated in at least one time point of barley colonization (Fig. 2 ‐Genome; Table S1‐All genes). During the very early time points of pathogenic development at 20 and 40 hpi, 29% (12.5% up‐ and 16.5% down‐regulated) and 65.2% (23.2% up‐ and 42% down‐regulated), respectively, of the total U. hordei genes underwent differential expression. The high numbers of differentially regulated genes at the very early stages of infection probably reflect the fundamental switch from saprophytic to biotrophic growth. In particular, at the 20‐hpi time point (penetration stage), the observed changes in gene expression may be partially linked to the developmental switch of U. hordei from yeast‐like growth to filamentous growth on the leaf surface. At 3 and 6 dpi of U. hordei colonization, 55.7% (19.5% up‐ and 36.2% down‐regulated) and 49.7% (16.8% up‐ and 32.9% down‐regulated), respectively, of the total U. hordei genes were differentially expressed. The highest differential expression ratio was observed at the 40‐hpi time point (Fig. 2; Table S1‐All genes). To predict the biological function of U. hordei differentially expressed genes, a Blast2Go (gene ontology, GO) analysis was performed (Table S1‐Blast2Go). In addition, a GO enrichment analysis was performed for all differentially expressed U. hordei genes for their molecular function (Fig. S1, see Supporting Information) (Zheng and Wang, 2008). In particular, two GO groups, including proteins with hydrolytic activity (GO: 0016798) and proteins with carbohydrate‐binding ability (GO: 0030246), were significantly enriched amongst the induced genes at all tested time points. The proteins of these GO groups were mainly sugar‐cleaving enzymes, such as xylanases, glucosidases, arabinofuranosidases, glucanases, cellulases and galactosidases (Table S1), which are involved in fungal or plant cell wall modification/degradation. These results are consistent with both the dimorphic and biotrophic life style of U. hordei. During the transition between saprobic yeast cell and filamentous infectious form, U. hordei remodels its cell wall and, in order to penetrate its host cell, also degrades plant cell walls. Amongst the negatively regulated genes in Fig. S1 (green boxes), four GO groups showed a significant enrichment, including enzymes with oxidoreductase activity (GO: 0016651, mainly NADH oxidoreductases), translation factors (GO: 0008135 and GO: 0003746), components of the ribosome (GO: 0003735) and transmembrane proteins of ion transport (GO: 0015077). In addition, 75 of 149 U. hordei transporters, including 11 sugar transporter, three ABC and 14 MFS multidrug transporters, were up‐regulated in at least one time point of colonization (Table S1‐All genes).
Figure 2.
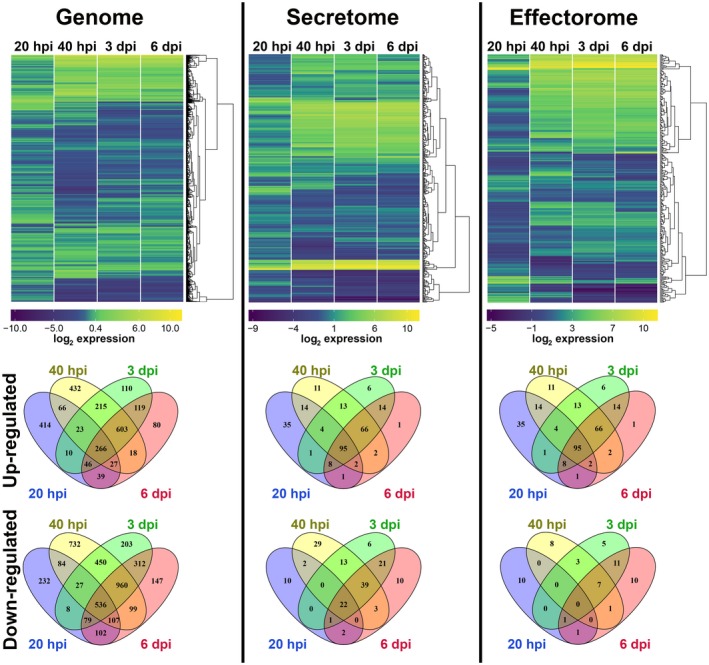
The hierarchical relation heat map for microarray analysis of Ustilago hordei in planta. The expression profiles of the whole genome, secretome and effectorome of U. hordei are depicted. Differentially expressed U. hordei genes, both up‐regulated (yellow) and down‐regulated (blue), in planta are shown as log2‐fold changes relative to in vitro axenic culture. Differentially expressed genes are plotted as Venn diagrams. [Colour figure can be viewed at wileyonlinelibrary.com]
Plant‐pathogenic fungi secrete different types of protein based on their growth environments. Although, in axenic culture, fungi mostly express and secrete catalytic enzymes to utilize carbon and nitrogen sources, in planta phytopathogenic fungi additionally secrete proteins (called effectors) that are known to be involved in fungal virulence with roles in the manipulation of host physiology. The types of secreted effector during host colonization determine the pathogenic life style of the fungus. For example, although necrotrophic fungi secrete more carbohydrate/plant cell wall‐degrading enzymes or toxins to kill their host cell, biotrophic fungi secrete proteins to avoid or suppress host defence responses to keep their host cell alive (Kämper et al., 2006; Lyu et al., 2015; Sprockett et al., 2011; de Wit et al., 2012). Thus, the cataloguing of the secreted proteins of fungi can provide hints about their life style. In this study, the catalogue of U. hordei secreted proteins was constructed using a computational pipeline (using SignalP, TargetP and TMHMM software to predict secreted proteins and exclude proteins with transmembrane domains) (Table S1‐Secretome). In total, 413 proteins (5.8%) were classified as secreted in U. hordei. BLAST homology searches against publicly available sequence databases at the National Center for Biotechnology Information (NCBI) revealed that 128 of these secreted proteins were novel, uncharacterized proteins, whereas 25 (19 Ustilaginaceae‐specific) were conserved, uncharacterized proteins (Table S1‐Secretome). The remaining secretome members had known or predicted functional domains, or had homology to proteins with characterized biological functions.
As there is no conserved sequence feature for the identification of effectors, fungal effector prediction relies on relatively broad criteria, such as secreted, small and cysteine‐rich proteins (de Jonge, 2012; Stergiopoulos and de Wit, 2009). However, several studies have shown the presence of effectors with a relatively large size and low/no cysteine content (Gout et al., 2006; Mueller et al., 2013; Ökmen et al., 2013). Because of this, more studies have started to include other criteria, such as in planta induction, in order to construct an effector catalogue (Guyon et al., 2014; Saunders et al., 2012; Sperschneider et al., 2014, 2014). So far, almost all described effectors are differentially up‐regulated in at least one time point of host colonization. Therefore, the effectorome of U. hordei was constructed solely based on the presence of a secretion signal and whether the transcript levels of the genes encoding the selected secreted proteins were different in at least one time point of barley colonization in comparison with growth in axenic culture. During barley infection, 273 U. hordei genes encoding secreted proteins were differentially up‐regulated in at least one time point of colonization (Table S1‐Effectorome). Thus, these 273 predicted secreted proteins (3.8% of the total gene number) were selected as effector candidates in the U. hordei–barley pathosystem, regardless of their size, cysteine content or absence/presence of known putative protein domains (Table S1‐Effectorome). Moreover, 130 U. hordei effector candidates were predicted to be apoplastic using ApoplastP (Sperschneider et al., 2018) (Table S1‐Apoplastome). The successful identification of all U. hordei orthologues of characterized U. maydis effectors, including UmPep1 (UMAG_01987; Hemetsberger et al., 2012), UmPit2 (UMAG_01375; Mueller et al., 2013), UmCmu1 (UMAG_05731; Djamei et al., 2011) and UmSee1 (UMAG_02239; Redkar et al., 2015), as well as the functionally characterized U. hordei effector Avr1 (UHOR_10022; Ali et al., 2014), in this U. hordei effectorome supports the reliability of this approach (Table S1‐Effectorome). Although 43.2% of the effectorome consisted of either hypothetical or uncharacterized proteins, 155 putative effector candidates showed conserved protein domains or similarity to characterized proteins (Table S1‐Effectorome). In total, the U. hordei secretome contained 55 carbohydrate‐degrading enzymes, 31 of which were up‐regulated in planta (Tables 1 and S1‐Secretome and Effectorome). Moreover, two proteins (UHOR_03469 and UHOR_07804) with a predicted carbohydrate‐binding module were also identified (Table 1). In addition, four proteins that were annotated as ribonuclease and one endo/exo nuclease were found to be secreted (Table 1). Furthermore, the effectorome of U. hordei was mined for proteins that had at least one predicted nuclear localization signal (NLS), resulting in 21 effector candidates with a predicted NLS signal (Table S2, see Supporting Information). The presence of a predicted NLS signal in these secreted proteins indicates that they may target the host cell nucleus.
Table 1.
Carbohydrate‐degrading enzymes and nucleases present in the Ustilago hordei effectorome.
| Carbohydrate‐degrading enzymes present in the U. hordei effectorome | |||
|---|---|---|---|
| Protein ID | UMAG_Orthologue | Description | Amino acid Length |
| UHOR_03500 | UMAG_02111 | α,β‐Hydrolase | 320 |
| UHOR_00039 | UMAG_10000 | Glycosyl hydrolase | 857 |
| UHOR_08862 | UMAG_10975 | Glycoside hydrolase | 354 |
| UHOR_07381 | UMAG_05704 | Glycoside hydrolase | 371 |
| UHOR_06098 | UMAG_04032 | Glycoside hydrolase | 803 |
| UHOR_00370 | UMAG_00235 | Glycoside hydrolase family 5 | 464 |
| UHOR_05270 | UMAG_03411 | Glycoside hydrolase family 10 | 342 |
| UHOR_00509 | UMAG_00330 | Glycoside hydrolase family 16 | 353 |
| UHOR_03204 | UMAG_05223 | Glycoside hydrolase family 30 | 661 |
| UHOR_07931 | UMAG_05528 | Glycosyl hydrolase 53 domain‐containing | 424 |
| UHOR_07793 | NO | Glycoside hydrolase family 93 | 383 |
| UHOR_08685 | UMAG_06120 | Glycoside hydrolase family 115 | 1128 |
| UHOR_02067 | UMAG_01377 | Extracellular cellulase allergen Asp F7 | 583 |
| UHOR_07788 | UMAG_12007 | Related to cellulase | 468 |
| UHOR_04296 | UMAG_05792 | Related to chitin deacetylase precursor | 490 |
| UHOR_01593 | UMAG_01022 | Barwin‐like endoglucanase | 160 |
| UHOR_03357 | UMAG_06332 | Related to Endoglucanase 1 precursor | 385 |
| UHOR_07693 | UMAG_04816 | Related to Endoglucanase 1 precursor | 323 |
| UHOR_04056 | UMAG_02523 | Related to Endoglucanase 1 precursor | 289 |
| UHOR_06823 | UMAG_04364 | Related to EXG1‐exo‐β‐1,3‐glucanase | 482 |
| UHOR_04077 | UMAG_04503 | Arabinanase levansucrase invertase | 466 |
| UHOR_02718 | UMAG_01829 | Related to α‐l‐arabinofuranosidase I precursor | 707 |
| UHOR_06745 | UMAG_04309 | Probable α‐l‐arabinofuranosidase precursor | 327 |
| UHOR_02584 | UMAG_01734 | Glucan 1,3‐β‐glucosidase | 331 |
| UHOR_00702 | UMAG_00446 | Probable β‐glucosidase | 819 |
| UHOR_08622 | UMAG_06075 | Related to β‐glucosidase | 839 |
| UHOR_05461 | UMAG_03551 | Related to glucose oxidase | 631 |
| UHOR_02907 | UMAG_01957 | Related to mannosyl‐oligosaccharide α‐1,2‐mannosidase | 528 |
| UHOR_03214 | UMAG_05229 | Related to β‐mannosidase precursor | 1059 |
| UHOR_06909 | UMAG_04422 | Related to endo‐1,4‐β‐xylanase b | 637 |
| UHOR_07547 | UMAG_10671 | Pectin lyase B precursor | 888 |
| Putative carbohydrate‐binding protein found in U. hordei effectorome | |||
| UHOR_07804 | UMAG_05439 | Chitin‐binding | 302 |
| UHOR_03469 | UMAG_11464 | Carbohydrate‐binding module family 50 | 374 |
| Putative nucleases found in U. hordei effectorome | |||
| UHOR_08127 | NO | Related to small nuclear ribonucleosome d3 | 225 |
| UHOR_04748 | UMAG_03023 | Related to ribonuclease Trv | 298 |
| UHOR_03174 | NO | Related to ribonuclease T1 | 119 |
| UHOR_02675 | UMAG_10881 | Probable ribonuclease T1 | 128 |
| UHOR_05247 | UMAG_03381 | Endonuclease exonuclease phosphatase family | 300 |
Expression of U. hordei effector candidate genes is adapted to the host plant during epidermal penetration
For the obligate biotroph Blumeria graminis f. sp. hordei (powdery mildew), similar gene expression patterns were found during the early stages of infection of either the host plant barley or the non‐host plant Arabidopsis thaliana (Hacquard et al., 2013). As our previous findings on organ‐ and cell type‐specific gene expression in the maize smut U. maydis showed high plasticity of the fungal transcriptome depending on the host tissue (Matei et al., 2018; Skibbe et al., 2010), we analysed U. hordei gene expression on penetration (20 hpi) of the non‐host plant (maize) relative to the host plant (barley). Comparison of the gene expression during host and non‐host penetration events revealed differential gene expression of U. hordei (Fig. 3). In total, 2066 and 2237 U. hordei genes were differentially expressed on barley and maize plants, respectively (Fig. 3). Among these genes, 249 and 317 U. hordei genes were specifically up‐regulated only on barley and maize, respectively, and 295 and 398 U. hordei genes were specifically down‐regulated only on barley and maize, respectively (Fig. 3). For the genes encoding predicted secreted proteins, 197 and 197 U. hordei genes were differentially expressed on barley and maize plants, respectively (Fig. 3). For the genes encoding effector candidates, 37 and 27 U. hordei genes were specifically up‐regulated on barley and maize plants, respectively; 14 and 24 U. hordei genes encoding effector candidates were specifically down‐regulated on barley and maize, respectively (Fig. 3). Moreover, 14 secreted proteins that were not differentially expressed in any tested time point of barley infection showed significant induction on maize (Table S1‐Secretome). Amongst these secreted proteins were two carboxypeptidases (UHOR_02798 and UHOR_05981), one protein that showed similarity to plant PR‐1 (pathogenesis‐related protein 1) (UHOR_01820), one hydrophobin 3 (UHOR_06926), one ribonuclease (UHOR_04170), one member of glycosyltransferase family 24 (UHOR_02240) and one protein that contained a glucan‐binding domain (UHOR_07386). However, U. hordei homologues for the previously identified effectors Pep1 (UHOR_02965), Pit2 (UHOR_02064) and Cmu1 (UHOR_07433), which all are required for the suppression of early defence responses, showed a remarkably similar expression pattern on host‐ and non‐host plants (Table S1‐Effectorome) (Djamei et al., 2011; Hemetsberger et al., 2012; Mueller et al., 2013).
Figure 3.
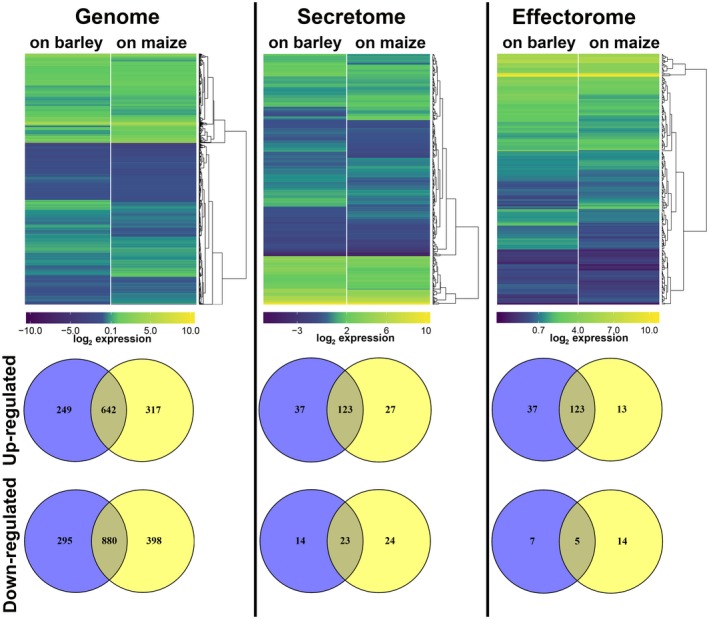
The hierarchical relation heat map for microarray analysis of Ustilago hordei wild‐type on host barley and non‐host maize plants. The expression profiles of the whole genome, secretome and effectorome of U. hordei are depicted. Differentially expressed U. hordei genes which are up‐regulated (yellow) and down‐regulated (blue) in planta are shown as log2‐fold changes relative to in vitro axenic culture. Differentially expressed genes are plotted as Venn diagrams. [Colour figure can be viewed at wileyonlinelibrary.com]
Identification of U. hordei effectors with virulence functions
To confirm the transcriptomic data, real‐time quantitative polymerase chain reaction (RT‐qPCR) was performed for 16 effector gene candidates, which were selected for further characterization (Table S1‐Effectors for KO). The RT‐qPCR results confirmed the transcriptional induction of the selected effector candidates (Figs 4, S2, see Supporting Information). The selection of candidate effector genes was performed according to their expression patterns during barley and maize colonization. The selection criteria were as follows: (a) effector genes only expressed at the very early time point of barley penetration (20 hpi); (b) effector genes only expressed at early time points (20 and 40 hpi); (c) effector genes more strongly expressed at very early time points (20 and 40 hpi) compared with late time points (3 and 6 dpi); (d) effector genes more strongly expressed at late time points (3 and 6 dpi) compared with early time points (20 and 40 hpi); (e) effector genes only up‐regulated on barley, but not on non‐host maize, or more highly expressed on barley compared with maize plants; and (f) effector genes with genome‐wide highest induced transcript levels (top 25 genes) (Table S1‐Effectors for KO). To test the eventual role of the selected 16 effector candidates for U. hordei virulence on barley, they were targeted for gene deletion via homologous recombination. The effector candidate genes were replaced by a hygromycin resistance cassette in the two compatible U. hordei mating strains (4857‐4 Mat‐1 and 4857‐5 Mat‐2). The targeted gene deletion in the mutants was confirmed via PCR using genomic DNA (Fig. S3A–F, see Supporting Information). In addition, RT‐qPCR analysis was performed to confirm the absence of transcripts resulting from the deleted genes during the infection of barley (Fig. S3B,F). The U. hordei deletion mutants could be generated for only 14 of the selected genes, whereas, for two genes (UHOR_07510 and UHOR_14478), no gene deletion could be confirmed. For all 14 genes, at least two independent mutants of U. hordei were tested in plant infections. All mutant strains were tested for growth in liquid culture and filament formation assays on charcoal plates compared with U. hordei wild‐type (Fig. S4A,B, see Supporting Information). To quantify host colonization of the mutant strains relative to U. hordei wild‐type, fungal biomass was quantified via qPCR at 6 dpi using a standardized amount of genomic DNA isolated from infected barley leaves (Fig. S5, see Supporting Information and Experimental procedures). The U. hordei mutant for the pep1 effector gene (∆uhpep1; UHOR_02965), which has been described previously to be essential for barley infection (Hof et al., 2014), was used as an additional control in all experiments. This approach revealed a significant reduction in infection for four effector candidate genes, including UHOR_02413, UHOR_02700, UHOR_03756 and UHOR_05101 (Fig. 5), whereas deletion of the other 10 candidates did not result in significantly reduced virulence (Fig. S5). Interestingly, a traditional disease scoring of floral symptoms which appeared 3–4 months after infection (for details, see Experimental procedures) showed drastically reduced symptoms caused by mutants for UHOR_02413, UHOR_02700, UHOR_03756 and UHOR_05101 (Fig. S4C). This observation suggests that the U. hordei effectors required for the colonization of seedling leaves are also important for the systemic infection of barley that ultimately results in floral symptoms. The four effectors that were identified as virulence factors were renamed as Ustilago hordei virulence factors 1–4 (Uvi1– Uvi4).
Figure 4.
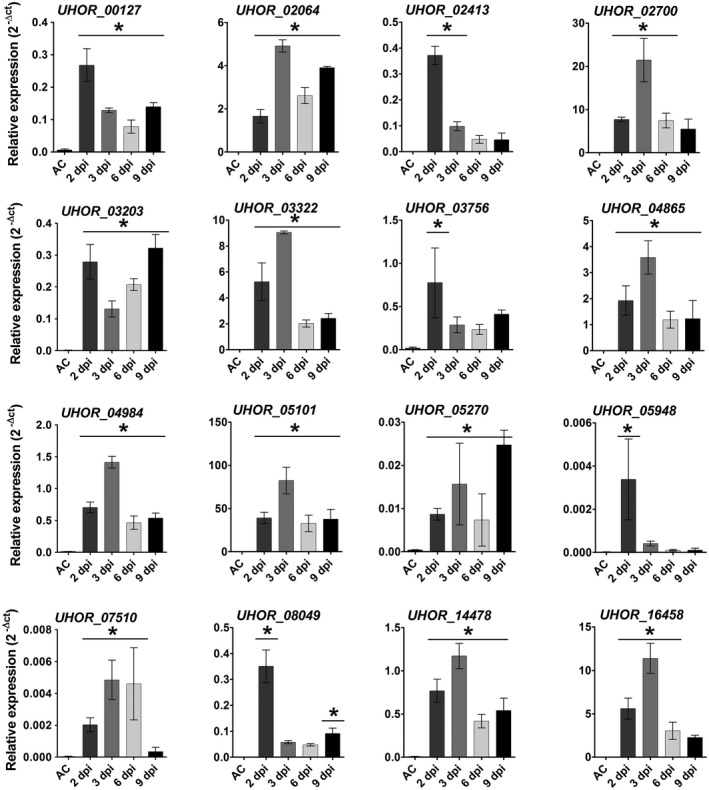
Expression profile of Ustilago hordei effector candidates in axenic culture (AC) and in planta. Quantitative real‐time polymerase chain reaction (qRT‐PCR) was performed to assess the expression profiles of the 16 U. hordei effector candidates. Expression levels were normalized using the U. hordei peptidylprolyl isomerase (Ppi) gene. A one‐way analysis of variance (ANOVA) was performed. Significant differences compared with axenic culture are shown with an asterisk (*P<0.05). Error bars represent the standard deviation of three biological replicates.
Figure 5.
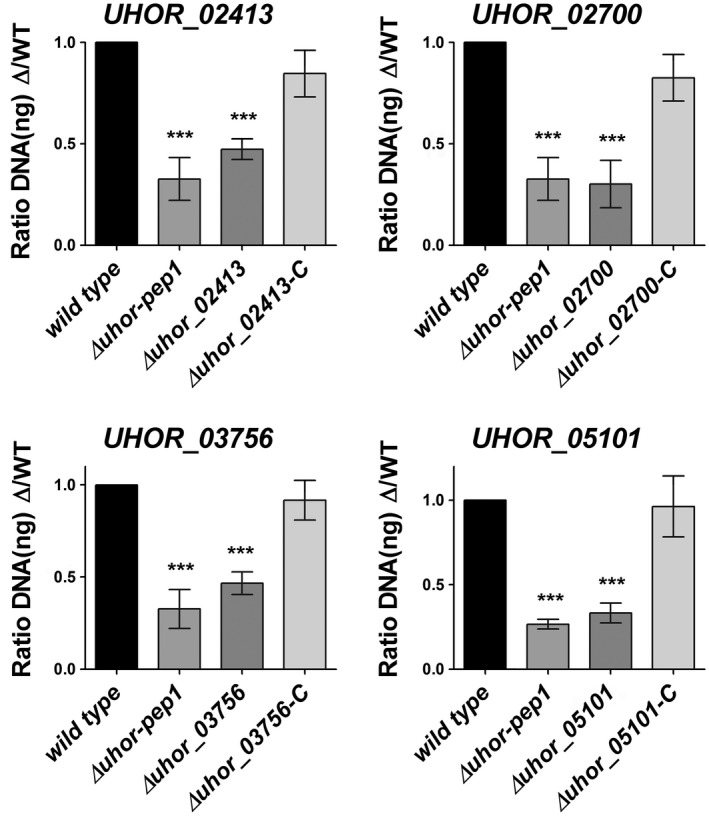
Gene deletion and complementation of Ustilago hordei effector genes. The virulence of the U. hordei wild‐type (WT), four effector mutants and four complementation strains was assessed by the quantification of fungal biomass at 6 days post‐inoculation (dpi). The U. hordei Pep1 deletion mutant was used as a positive control for virulence reduction. The infected barley leaves at 6 dpi were harvested and analysed via quantitative polymerase chain reaction (qPCR) using genomic DNA and the Ppi1 genes from U. hordei as a standard. The fungal biomass was deduced from a standard curve. A one‐way analysis of variance (ANOVA) was performed and followed by a Bonferroni test. Significant differences are indicated with asterisks (***P < 0.001). Error bars represent the standard deviation of at least three biological repeats.
To confirm that the observed phenotype was specifically caused by deletion of the targeted effector gene candidates, genetic complementation was performed for the four effector genes whose deletion resulted in virulence reduction. In all cases, the complemented mutants showed wild‐type virulence (Fig. 5). As UHOR_02413 (Uvi1), UHOR_02700 (Uvi2), UHOR_03756 (Uvi3) and UHOR_05101 (Uvi4) are significant virulence factors for the U. hordei–barley pathosystem, further functional characterization was focused on these four effectors.
Functional analysis of the four U. hordei virulence effectors
On pathogen invasion, host plants induce several layers of defence responses [pathogen‐associated molecular pattern (PAMP)‐triggered immunity (PTI) and effector‐triggered immunity (ETI)] to restrict further pathogen growth. Successful pathogens must evade or suppress these defence barriers. To assess whether any of the four characterized effectors (Uvi1–Uvi4) had the ability to suppress programmed cell death (PCD), two established assays were deployed. First, a BAX1‐mediated cell death suppression assay (Hückelhoven, 2004; Watanabe and Lam, 2009) was performed. Both BAX1 and each of the selected effector candidates (Uvi1, Uvi2, Uvi3 and Uvi4) were co‐expressed in yeast cells. The expression of these four U. hordei effectors in yeast cells was confirmed via western blot analysis (Fig. S6A, see Supporting Information). Suppression of cell death was monitored on galactose‐containing medium to induce the expression of BAX1 and U. hordei effectors. The known inhibitor of BAX1‐mediated cell death, BCL‐XL, was used as a positive control. As expected, BCL‐XL suppressed BAX1‐mediated cell death; however, none of the selected effectors resulted in the suppression of BAX1‐mediated cell death in this assay (Fig. S7A, see Supporting Information). As the mechanisms of cell death induction in yeast differ significantly from the situation in plants, these four effectors were also tested for INF1‐mediated cell death suppression in Nicotiana benthamiana via an Agrobacterium‐mediated transient transformation assay. Although the expression of C‐terminally tagged Uvi1, Uvi2 and Uvi3 in N. benthamiana leaves was confirmed via western blot analysis, the Uvi4 protein could not be detected (Fig. S6C). However, when C‐terminally eGFP‐tagged Uvi4 was expressed in tobacco, a green fluorescent protein (GFP) signal was visible under the GFP channel, suggesting that the C‐terminal haemagglutinin (HA) tag could be removed in tobacco (Fig. S6D). INF1 is an extracellular elicitin from Phytophthora infestans which acts as a PAMP to induce the hypersensitive response (HR) in N. benthamiana (Kamoun et al., 1997). The P. infestans effector Avr3aKI and a mutated version, Avr3a∆Y147, were used as positive and negative controls, respectively (Bos et al., 2010). However, although the co‐expression of INF1 with Avr3a and Avr3a∆Y147 confirmed the previously published suppression of INF1‐mediated cell death by Avr3aKI, none of the tested effectors showed INF1‐mediated cell death suppression activity in this assay (Fig. S7B). In summary, this indicates that, at least in the tested conditions, none of the four virulence factors suppresses plant cell death at either the PAMP‐induced level (INF1) or via mitochondrial PCD (BAX1).
An alternative strategy to gain insights into effector function is the search for interacting host proteins. The effectors Uvi1, Uvi2, Uvi3 and Uvi4, which contribute significantly to U. hordei virulence, were chosen for an interaction screen via the well‐established Y2H assay. To this end, a custom cDNA library was generated from U. hordei‐infected barley leaves (for details, see Experimental procedures). Prior to the library screens, correct expression of the four effectors in yeast was tested via western blotting. Although Uvi1, Uvi2 and Uvi3 were successfully expressed in yeast cells, no signal for Uvi4 could be detected from yeast cells (Fig. S6B). Therefore, Uvi4 was excluded from the Y2H experiment. For Uvi1, screening of the barley–U. hordei cDNA library revealed three potential interaction partners: the barley heavy metal‐associated (HvHMA) protein (id: BAK00981.1), a barley domain unknown function protein (HvDuf866) (id: BAJ98443.1) and a lipid transfer protein (HvLTP) (id: CAA65680.1). For all three proteins, SignalP analysis predicted an N‐terminal signal peptide for extracellular secretion. The coding regions of the three genes were cloned from barley for verification of the screening result in one‐to‐one Y2H assays. Although this confirmed the interaction of Uvi1 with HvHMA and HvDuf866 (Fig. 6 ), HvLTP showed autoactivation and was therefore considered to be a false positive candidate. For Uvi2, two potential interaction partners were identified: one thaumatin‐like protein 4 (HvTLP4, id: AF355455_1) and a NAC transcription factor (id: AM500855.1). However, in one‐to‐one interaction assays, only the interaction of HvTLP4 with Uvi2 was confirmed using the full‐length version of the respective barley genes (Fig. 6). For Uvi3, an SNF1‐related protein kinase (SnRK1) was identified as a possible interactor partner. However, this interaction could not be confirmed in the one‐to‐one interaction assay using the full‐length version of the respective barley gene.
Figure 6.
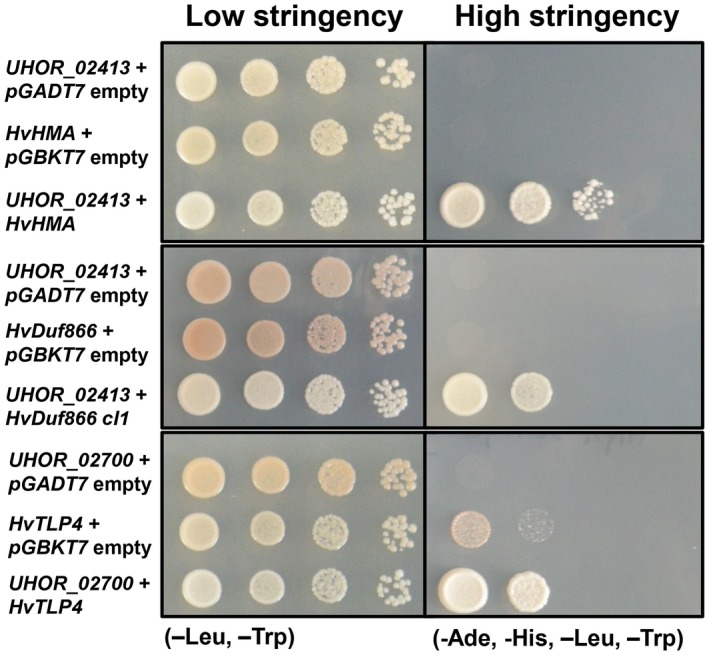
In vivo interaction of Ustilago hordei effectors with Hordeum vulgare target proteins via a yeast two‐hybrid (Y2H) system. Interaction of pGBKT7‐UHOR_02413 and pGADT7‐HvHMA, pGBKT7‐UHOR_02413 and pGADT7‐HvDUF866, and pGBKT7‐UHOR_02700 and pGADT7‐HvTLP4 was confirmed via Y2H assay. The empty vectors were used as negative controls for the Saccharomyces cerevisiae AH109 strain. Different dilutions (100–10–3) of each sample were dropped on low‐ [–leucine (‐Leu) and –tryptophan (‐Trp)] and high‐ [‐Leu, ‐Trp, –adenine (‐Ade) and –histidine (‐His)] stringency plates. The photographs were taken after 3–4 days of incubation at 28 ºC. Growth on high‐stringency plates indicates interaction.
In summary, we have identified two possible barley interaction partners, i.e. HvHMA and HvDuf866, for Uvi1, whereas HvTLP4 is the only possible interactor partner for Uvi2.
Discussion
Ustilago hordei is a seed‐borne pathogen that initially penetrates barley coleoptiles; the fungus grows just below the apical shoot meristem without demonstrating any visible symptoms until the occurrence of sori filled with dark teliospores in flowers (Hu et al., 2002). Therefore, macroscopic evaluation of disease symptoms is only possible at the very late stages of the disease (2–3 months after inoculation) (Hu et al., 2002). In this study, we have developed a more rapid readout assay to evaluate the disease symptoms of U. hordei during barley infection. Two compatible U. hordei strains were needle inoculated into leaf whorls of 12‐day‐old barley seedlings, similar to the inoculation procedure established for the maize smut U. maydis (Redkar et al., 2015). Microscopic observation of U. hordei on barley inoculation showed that the infectious filaments penetrate the leaf epidermis via appressorium formation and, subsequently, the fungus proliferates both extracellularly and intracellularly. At 3 dpi, the fungal filaments grow towards vascular tissue and, at 6 dpi, U. hordei massively proliferates around and inside the host vascular tissue. Biomass quantification of U. hordei on barley via qPCR was stable at 6 dpi, which represents a dramatic reduction in experimental workflow compared with the traditional disease scoring of floral symptoms (which takes around 3–4 months). Interestingly, U. hordei develops intracellular structures, which show morphological similarity to haustoria. These special feeding structures are typically formed by biotrophic fungi or oomycetes to utilize nutrients from their hosts (Slusarenko and Schlaich, 2003; Yi and Valent, 2013). Although it has been reported that the smut fungus Ustacystis waldsteiniae can form haustoria in its host plant Waldsteinia geoides (Bauer et al., 1995), to our knowledge, no previous studies have reported the formation of haustoria‐like structures in U. hordei.
To facilitate host infection, biotrophic pathogens deploy large repertoires of effectors, which often show specific transcriptional induction during host colonization. Our transcriptome analysis showed the significant induction of 273 U. hordei effector genes, which is comparable with previous findings for the maize smut U. maydis (261 effector genes) (Skibbe et al., 2010). Some effectors (95 genes) showed constitutively high expression during all the tested stages of U. hordei infection. This group contains homologues of the known U. maydis effectors Pep1 and Pit2, which are both essential for fungal virulence (Hemetsberger et al., 2015, 2015; Mueller et al., 2013). However, the majority of U. hordei effector genes (178 genes) showed transient transcriptional induction, suggesting a stage‐specific activity of these putative virulence factors. Sequential expression of putative effector genes has been observed for other plant pathogens, including the rust fungus Melampsora larici‐populina and the hemibiotrophic anthracnose pathogen Colletotrichum higginsianum (Duplessis et al., 2011; Kleemann et al., 2012; O'Connell et al., 2012). Kleemann et al. (2012) proposed that C. higginsianum expresses effectors that are required for the suppression of host immune responses to maintain host cell viability during the early biotrophic stage; however, on switching from the biotrophic stage to the necrotrophic stage, the expression of NLP1 is increased to induce host cell death (Kleemann et al., 2012). Although U. hordei is strictly biotrophic, it may express different effector waves that are infection stage specific, thus eventually resembling organ‐ or cell type‐specific interactions, as recently described for the U. maydis–maize interaction (Matei et al., 2018; Schilling et al., 2014; Skibbe et al., 2010).
Predictions of fungal effectors mostly rely on whether the proteins contain an extracellular signal peptide, have a relatively large size and low/no cysteine content (de Jonge, 2012; Sperschneider et al., 2016; Stergiopoulos and de Wit, 2009). However, recent studies have revealed that not all effectors fit into these criteria (Gout et al., 2006; Mueller et al., 2013; Ökmen et al., 2013). As almost all characterized effectors are differentially up‐regulated in at least one time point of host colonization, more and more studies have prioritized in planta‐expressed secreted proteins as effector candidates (Guyon et al., 2014; Saunders et al., 2012; Sperschneider et al., 2014). In this study, we defined the U. hordei effectorome solely based on the presence of a secretion signal and significant up‐regulation during host colonization compared with axenic growth. The presence of all functionally characterized U. maydis effector orthologues, including UmPep1 (UMAG_01987) (Hemetsberger et al., 2012), UmPit2 (UMAG_01375) (Mueller et al., 2013), UmCmu1 (UMAG_05731) (Djamei et al., 2011) and UmSee1 (UMAG_02239) (Redkar et al., 2015), in this effector catalogue indicates the reliability of the constructed U. hordei effectorome.
Previous comparative genomic analyses have shown that the numbers of plant cell wall‐degrading enzymes in necrotrophic or hemibiotrophic fungi are significantly greater than in biotrophic fungi (Lyu et al., 2015; Sprockett et al., 2011; de Wit et al., 2012). Lyu et al. (2015 reported that, in the necrotrophic phytopathogenic fungus Sclerotinia sclerotiorum, the expression of 73 of a total number of 186 genes encoding for plant cell wall‐degrading‐associated enzymes was significantly up‐regulated during infection. The combination of microarray and Blast2Go analyses revealed the presence of 31 putative carbohydrate‐degrading enzymes that were significantly up‐regulated on host penetration. This result also confirms that biotrophs have a smaller repertoire of carbohydrate‐degrading enzymes than more aggressive plant pathogens. The effector repertoire of U. hordei contains cellulases, xylanases, β‐glucosidase, pectinases, α‐l‐arabinofuranosidases and exo‐ and endo‐glucanases, which can hydrolyse the main components of either plant or fungal cell walls. Recently, several studies have shown that fungal‐derived cellulases, endo‐1,4‐β‐xylanases and α‐l‐arabinofuranosidases are required for full virulence of Magnaporthe oryzae during rice colonization (Nguyen et al., 2011; Van Vu et al., 2012; Wu et al., 2016). Ustilago hordei may secrete these glycosyl hydrolase proteins into the host apoplast to degrade the host cell wall in order to facilitate host penetration or cell‐to‐cell passage by loosening the plant cell wall, or to possibly release nutrients from the host cell wall to support its own growth. We have also identified one chitin‐binding protein and one LysM (CBM family 50) protein in the effectorome of U. hordei. These chitin‐binding proteins and other cell wall‐degrading enzymes could be used to suppress or avoid host immunity by sequestering fungal‐ or plant‐derived microbe‐associated molecular pattern (MAMP) or damage‐associated molecular pattern (DAMP) molecules, respectively (de Jonge et al., 2010; Marshall et al., 2011; Mentlak et al., 2012).
In addition to carbohydrate‐degrading enzymes, U. hordei also differentially expresses several proteases, including carboxypeptidases, metalloproteases, aspartyl and serine proteases. Although there have been many studies on the function of secreted fungal protease inhibitors that inhibit host serine or cysteine proteases to suppress host defence responses (Lozano‐Torres et al., 2012; Mueller et al., 2013; Rooney et al., 2005; Tian et al., 2004, 2004), the roles of secreted fungal proteases and their substrate specificities in host–microbe interactions are still elusive. A knowledge of the substrate specificities of these enzymes will provide new insights into our understanding of their contribution to virulence.
Although many studies have been performed to understand the mechanism of non‐host resistance in host plants, only a few studies have investigated how plant pathogens behave on non‐host plants. Recently, Hacquard et al. (2013 reported that the obligate biotroph B. graminis f. sp. hordei (powdery mildew) shows similar gene expression patterns on two very divergent hosts, barley and Arabidopsis (on the immunocompromised pen2/pad4/sag101 triple mutant), at very early time points on penetration (at 6–24 hpi). In contrast, we have shown that U. hordei transcriptionally activates different sets of genes during host (barley) and non‐host (maize) penetration at 20 hpi. In total, U. hordei expresses 160 and 150 genes encoding secreted proteins on host and non‐host plants, respectively. Moreover, 37 and 27 of these genes are specifically expressed only on barley and maize plants, respectively. These results indicate that U. hordei transcriptionally activates its secretome even during penetration attempts on a non‐host plant in order to suppress the plant immune system. Furthermore, U. hordei keeps some effectors ‘up its sleeve’ for non‐host penetration events, which are not expressed on the host plant. Consistent with these results, it has been reported that the generalist root endophyte Serendipita indica also transcriptionally activates different sets of effectors during the colonization of different host plants in a host‐dependent manner (Lahrmann et al., 2013). By expressing its virulence factors, which are involved in the suppression of the host plant immune system, on non‐host plants, pathogens may obtain an evolutionary potential for possible host‐jump events. To check whether U. hordei also attempts to suppress early non‐host defence responses, the expression levels of the characterized core effectors of smuts were checked on both barley and maize plants (Schuster et al., 2017). Surprisingly, the U. hordei genes encoding homologues of Pep1, Pit2 and Cmu1 proteins, which are required for suppression of early defence responses, including reactive oxygen species (ROS) accumulation (Hemetsberger et al., 2012), host cysteine protease activity (Mueller et al., 2013) and salicylic acid accumulation (Djamei et al., 2011), respectively, in the U. maydis–maize system, show remarkably similar expression patterns on both barley and maize plants. These data suggest that U. hordei attempts to penetrate and suppress very early non‐host defence responses on maize. Consistent with these results, Hof et al. (2014) have reported that U. maydis can perform pep1‐mediated suppression of barley ROS accumulation on non‐host barley plants at 20 hpi.
In order to obtain further insights into the roles of highly expressed effector candidates during barley infection, 14 U. hordei effector candidates were selected for gene deletion. The selection was performed on the basis of their expression level and pattern at different time points on host or non‐host plants. Biomass quantification of the 14 U. hordei mutants on infected barley leaves revealed four effector genes (Uvi1, Uvi2, Uvi3 and Uvi4) that were required for full virulence. BLAST analysis showed that, although Uvi2 and Uvi4 are uncharacterized proteins, Uvi1 shows some similarity to expansins, proteins involved in plant cell wall loosening (Cosgrove, 2000). Therefore, Uvi1 may be involved in barley cell wall loosening during cell‐to‐cell passage. The Uvi3 protein has six predicted Ish1 (induced in stationary phase) domains. Proteins containing this domain are putative stress‐responsive nuclear envelope proteins that are localized in the nuclear envelope and plasma membrane (Taricani et al., 2002). It has been reported that the Ish1 protein from Saccharomyces pombe interacts with the nuclear Bis1 protein, which is widely conserved in a wide range of organisms, including humans, Drosophila and Arabidopsis. Bis1 could be involved in RNA processing and protein degradation (Taricani et al., 2002). Uvi3 may also interact with a homologue of the Bis1 protein in barley to manipulate host RNA processing or protein degradation.
The Y2H library screen revealed barley TLP4, a member of the PR‐5 class of proteins with a predicted extracellular secretion signal (Liu et al., 2010), as a potential host interaction partner of Uvi2. It has been shown that TLPs play a role in antifungal activities in several plant‐pathogenic fungi, including Trichoderma viride, Botrytis cinerea, Fusarium oxysporum, Physalospora piricola and Mycosphaerella arachidicola (Chu and Ng, 2003; Ho et al., 2007; Singh et al., 2013). Consistently, heterologous expression of TLP in tobacco plants induced enhanced resistance against Rhizoctonia solani and higher levels of PR gene expression (Chu and Ng, 2003). These results suggest that barley TLP4 may also have antifungal activity to inhibit U. hordei colonization. If this is the case, by interacting with TLP4, Uvi2 may inhibit its antifungal activity to support U. hordei growth. The Y2H screen for Uvi1 gave two potential unrelated interactor candidates with a predicted extracellular secretion signal. One of the candidates had a heavy metal‐associated domain (HMA domain) and the other had a Duf866 domain. According to the integrated decoy hypothesis, during the arms race between hosts and their pathogens, some plants have integrated protein domains targeted by effector proteins into their receptor proteins to lure effectors and, subsequently, to induce the immune system (Kroj et al., 2016; Maqbool et al., 2015). Recently, it has been reported that a rice resistance protein has an integrated HMA domain, indicating that the HMA‐containing host protein could be targeted by pathogen effectors (Kroj et al., 2016; Maqbool et al., 2015). Although cell death suppression assays, including BAX1‐mediated and Inf1‐mediated cell death suppression assays, were performed for further characterization of these four effectors, we could not identify any effector that could suppress either of these cell death mechanisms.
In this study, we systematically investigated the putative effector candidates of U. hordei during barley colonization. We identified 273 secreted effector candidates which were differentially up‐regulated in planta. The results revealed that, in order to establish colonization, U. hordei transcriptionally activates consecutive effector waves to manipulate host metabolism at different infection stages. Moreover, we have also shown that U. hordei up‐regulates core effectors to suppress early non‐host defence responses, including ROS accumulation, PLCP activation and salicylic acid production. This indicates that U. hordei attempts to infect non‐host plants with full intention, but is blocked by further non‐host immune responses. Deletion studies for highly expressed effector candidates revealed four effectors that contribute significantly to U. hordei virulence on barley, and potential interacting host proteins were identified for two of these virulence factors.
Experimental Procedures
Fungal and plant materials
Ustilago hordei (4857‐4 Mat‐1 and 4857‐5 Mat‐2) cultures were incubated in YEPSlight (0.4% yeast extract, 0.4% peptone and 2% saccharose) liquid medium at 22 °C with shaking at 200 rpm. The susceptible barley cultivar (Golden Promise) was grown in a glasshouse at 70% relative humidity, 22 °C during the day and night, a light/dark regime of 15 h/9 h and 100 W/m2 supplemental light when the sunlight influx intensity was less than 150 W/m2.
Nicotiana benthamiana plants were grown in a growth chamber at 22 °C and 70% relative humidity with a photoperiod of 12 h.
Nucleic acid methods
Total RNA was isolated from axenic culture and U. hordei‐infected barley plants (at 20 and 40 hpi, and 3 and 6 dpi). The third leaf of infected barley plants was collected by cutting 1 cm below the injection needle site and approximately 3 cm of the leaf was taken for RNA extraction. For the 20‐hpi time point, melted latex was used to collect penetrated fungal materials. Samples were then frozen in liquid nitrogen and ground using a mortar and pestle under constant liquid nitrogen cooling conditions. Total RNA was isolated from the fine powder using the TRIzol® extraction method (Invitrogen, Karlsruhe, Germany) according to the manufacturer’s instructions. Total RNA was then cleaned with a Turbo DNA‐Free™ Kit (Ambion/Applied Biosystems, Darmstadt, Germany), according to the manufacturer’s instructions, to remove any DNA contamination.
The synthesis of cDNA was performed with 1 µg of total RNA using the First Strand cDNA Synthesis Kit (ThermoFisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s instructions. RT‐qPCR analysis was performed using SYBR® Green Supermix (BioRad, Munich, Germany) according to the manufacturer’s instructions. RT‐qPCR oligonucleotide pairs were designed with Primer3 Plus (Table S3, see Supporting Information) (Untergasser et al., 2007). The efficiency and specificity of the oligonucleotide pairs were determined with a dilution series of genomic DNA before use. The reaction was performed in a Bio‐Rad iCycler system using the following conditions: 2 min at 95 °C, followed by 45 cycles of 30 s at 95 °C, 30 s at 61 °C and 30 s at 72 °C. The expression levels of U. hordei effector genes were calculated relative to the U. hordei peptidylprolyl isomerase gene (UhPpi, UHOR_05685). The RT‐qPCR data were analysed using the 2–ΔΔCt method (Livak and Schmittgen, 2001). Error bars in all figures showing RT‐qPCR data represent the standard deviation of the raw data from at least three biological replicates. The primers used for RT‐qPCR are listed in Table S3.
Fungal biomass quantification was performed using qPCR analysis as described previously, but employing genomic DNA isolated from U. hordei wild‐type‐ and mutant‐infected barley leaves. DNA isolation from infected barley leaves was performed using a MasterPure™ Complete DNA&RNA Purification Kit (Epicentre®, Illumina®, Madison, Wisconsin, USA) according to the manufacturer’s instructions. The U. hordei UhPpi gene (UHOR_05685) was used as a reference gene. A standard curve was constructed using serial dilutions of U. hordei genomic DNA (100, 10, 1, 0.1, 0.01 and 0.001 ng/µL) employing UhPpi as a reference gene. Logarithms (base 10) of DNA concentrations were plotted against the crossing point of Ct values. The results are the average of three biological repeats.
All PCRs were performed in 25‐µL reaction mixtures using Phusion© DNA polymerase (Thermo Scientific, Bonn, Germany), following the manufacturer’s instructions, with 100 ng of genomic DNA or cDNA as template. All primers introduced into the reaction are listed in Table S3. The amplified DNA was then used for the cloning process. All PCRs were run in a PTC‐200 (Peletier Thermal Cycler, MJ Research, Quebec, Canada) PCR machine. Nucleic acids (i.e. deriving from PCRs or restriction digests) were purified from 1% TAE agarose gels with a ‘Wizard SV Gel and Purification System Kit’ (Promega, Fitchburg, Wisconsin, USA) according to the manufacturer’s instructions.
Plasmid DNA isolation from bacterial cells was performed using a QIAprep Mini Plasmid Prep Kit (QIAGEN, Venlo, Netherlands), according to the manufacturer’s information, after the principle of alkaline lysis.
Cloning and fungal transformation
For knockout and complementation plasmid constructions, standard molecular biology methods were used according to the molecular cloning laboratory manual of Sambrook et al. (1989). Ustilago hordei gDNA was used for the amplification of PCR products for the construction of deletion and complementation constructs, and cDNA was employed for the amplification of genes for expression constructs, using appropriate primer pairs (Table S3). After restriction of the PCR fragments with appropriate restriction enzymes, they were ligated into a vector that was digested with the same restriction enzymes using T4‐DNA ligase (New England Biolabs, Frankfurt‐am‐Main, Germany) according to the manufacturer’s instructions. The sequences of all constructs were confirmed via sequencing at the GATC‐Biotech Company (Cologne, Germany). Gateway cloning was performed with a pENTR™/D‐TOPO™ Cloning Kit (ThermoFisher Scientific, Waltham, Massachusetts, USA) to obtain pDONR vectors according to the manufacturer’s instructions, and Gateway™ LR Clonase™ Enzyme mix was used to construct pK2GW7 and pEZY45 destination constructs according to the manufacturer’s instructions. All vector constructs, primer pairs and restriction sites are indicated in Table S3. For all heterologous gene expression assays, UHOR_0241324‐226aa (Gene Bank: CCF53559.1), UHOR_0270022‐224aa(Gene Bank: CCF53255.1), UHOR_0375628‐503aa(Gene Bank: CCF51752.1) and UHOR_0510124‐703aa(Gene Bank: CCF51983.1) were cloned without the signal peptide sequences.
Escherichia coli transformation was performed via heat shock assay according to standard molecular biology methods (Sambrook et al., 1989). Transformation of Saccharomyces cerevisiae ESM356.1 was performed as described in the yeast protocol handbook (Clontech, Mountain View, CA, USA). Gene replacement and transformation of U. hordei protoplasts were conducted according to Kämper (2004). For both gene deletion and complementation studies, a minimum of two colonies was selected for phenotypic observation. Only when all independent mutants showed the same virulence reduction phenotype was the observed phenotype considered to be true. Complementation of deletion mutants was performed in the native locus of the gene using a nourseothricin (Nat) resistance cassette (final concentration of 25 µg/mL).
Virulence assay
For virulence assay, both wild‐type and mutant strains of U. hordei were grown in YEPSlight liquid medium at 22 °C with 200 rpm shaking to obtain an optical density at 600 nm (OD600) of 0.6–0.8. The cells were then centrifuged at 2744 × g for 10 min at room temperature and resuspended in sterile water supplemented with 0.1% Tween‐20 to obtain OD600 of 3.0. Then, the U. hordei suspension was injected into the centre of the leaf whorl of 10–12‐day‐old barley seedlings (three‐leaf stage) with a syringe with a needle. All infection assays were performed in three biological replicates with at least 16 plants. The quantification of U. hordei fungal biomass in infected barley plants was performed at 6 dpi using qPCR with a standardized amount of genomic DNA (200 ng/µL) as the template. Before genomic DNA isolation, infected barley leaves were cleaned (wiped) with 1% Tween‐20 water to remove U. hordei sporidia from the leaf surface. Genomic DNA was isolated from barley leaves infected by the wild‐type fungus and mutants, diluted to a final concentration of 200 ng/µL. A standard curve was constructed using serial dilutions of U. hordei genomic DNA (100, 10, 1, 0.1 and 0.01 ng/µL), employing UHOR_05685 (UHOR_Ppi) as a reference gene. The base 10 logarithms of DNA concentrations were plotted against the crossing point of Ct values obtained from qPCR. The ∆uhpep1 mutant of U. hordei was used as a positive control for biomass reduction. For classical U. hordei infection assay, two haploid cultures of opposite mating types were mixed (1 : 1, v/v) to obtain a final concentration of OD600 = 1.0. Barley seeds were dehulled and surface sterilized by incubation with 70% ethanol for 3 min, followed by incubation with 1% bleach for 10 min, and rinsed four times with sterile water. Subsequently, the surface‐sterilized barley seeds were treated with U. hordei cell suspension and a vacuum of 413 kPa was applied twice for 15 min. After removal of excessive fungal inoculum, the barley seeds were incubated for 6 h at room temperature before sowing in soil. Approximately 2–3 months after inoculation, disease symptoms were scored at barley heading.
Bioinformatics analysis
Conventionally secreted U. hordei proteins were predicted using SignalP (Nielsen et al., 1997) to identify the N‐terminal signal peptide and, subsequently, the TMHMM software program (Krogh et al., 2001) was used to exclude proteins with one or more transmembrane domains. The proteins whose transmembrane domains (sequence length of less than 30 amino acids) overlapped with their signal peptides were not excluded from the list. The protein sequences were then analysed with TargetP (Emanuelsson et al., 2000) to predict the extracellular localization.
The Blast2GO program was used to predict multiple associated GO categories for each protein with default parameter settings and given the BLAST results against the non‐redundant database of NCBI as input (GO term database was last updated on 25 April 2017) (Conesa et al., 2005). In addition, each protein sequence was scanned for the presence of protein domains using InterproScan v4.4, including domain profiles from Pfam.
Microarray analysis
Samples for microarray analyses were processed using the One Color Quick Amp Labeling Kit, One Color Spike‐In Kit, Gene Expression Hybridization Kit, Gene Expression Wash Buffer Kit (Agilent, Santa Clara, California, USA) and RNeasy Mini Kit (Qiagen, Venlo, Netherlands) according to the manufacturers’ instructions. One‐hundred nanograms of total RNA were used as input for reverse transcription. Microarray analyses were performed on 8 × 15k microarrays (Agilent) and scanned using the following parameters: profile, AgilentHD_GX_1Color; slide ID, AutoDetect; channels, G; scan region, Agilent HD (61 mm × 21.6 mm); resolution, 2 µm; Tiff, 20 bit; RPMT, 100%; GPMT, 100%; XDS, no XDR. The image was processed in Agilent Feature Extraction, and data analysis was performed using the Partek® Genomics Suite Software (Partek Inc., St. Louis, Missouri, USA). Changes in U. hordei gene expression in planta (at 20 and 40 hpi, and 3 and 6 dpi) were calculated relative to U. hordei yeast‐like cells grown in axenic culture. Expression data were submitted to Gene Expression Omnibus (https://www.ncbi.nlm.nih.gov/geo/) (Accession Number: GSE117325).
Y2H assay
The U. hordei–barley Y2H library was constructed using a Make Your Own ‘Mate&Plate™’ Library System (Clontech Laboratories, Takara Bio Company, Shimogyo‐ku, Kyoto, Japan) according to the manufacturer’s instructions. To construct the Y2H library, total RNA was isolated from 20‐hpi, 40‐hpi, 3‐dpi and 6‐dpi U. hordei‐infected barley plants.
To identify the host targets of UHOR_0241324‐226aa, UHOR_0270022‐243aa and UHOR_0375628‐503aa effectors, the corresponding genes were cloned without their signal peptide sequences into pGBKT7 to be in frame with the Gal4 DNA‐binding domain and a Myc tag. Using the ‘Mate&Plate™’ Library System, the U. hordei–barley Y2H library was screened with these three effectors according to the manufacturer’s instructions.
To confirm the results of the Y2H screen, all potential interaction partners were cloned into pGADT7 to be in frame with the Gal4 activation domain and an HA tag. pGBKT7‐UHOR_02413, pGBKT7‐UHOR_02700 and pGBKT7‐UHOR_03756, and their potential interaction partners in pGADT7, were co‐transformed into the S. cerevisiae AH109 strain. As a negative control, co‐transformation was also performed with empty pGADT7 and pGBKT7. Saccharomyces cerevisiae transformation was performed according to Yeast Transformation System II protocol. Drop assays for Y2H interaction were performed according to Mueller et al. (2013). From each dilution, 5 µL were dropped onto low‐stringency [–leucine (‐Leu) and –tryptophan (‐Trp)] and high‐stringency [‐Leu, ‐Trp, –adenine (‐Ade) and –histidine (‐His)] SD plates, and the plates were incubated at 28 °C for 5 days. Photographs were taken after 5 days. Growth on high‐stringency plates indicates candidates for protein–protein interaction.
Cell death suppression assays
To assess whether any of these selected effector candidates possess activity to suppress PCD, both BAX1‐mediated cell death and INF1‐mediated cell death suppression assays were performed according to Kawai et al. (1999) and Bos et al. (2010), respectively.
For the BAX1‐mediated cell death suppression assay, the corresponding effector genes were cloned without their signal peptide sequences into the pEZY45 Gateway expression vector to be in frame with the B42 transcription activator domain and an HA tag. BAX1‐mediated cell death suppression assays were performed according to Kawai et al. (1999). The ESM356.1 yeast strain was co‐transformed with an effector candidate and BAX1 under the control of a galactose inducible promoter. Drop assay was performed with 5 µL of different dilutions of cultures on SD solid medium (0.67% w/v yeast nitrogen base, 0.06% v/v dropout solution without uracil and tryptophan, and 2% agar, pH 5.8) supplemented with 2% glucose or 2% galactose and 1% raffinose to induce the expression of the effector candidate and BAX1 genes. Yeast cells were incubated for 4 days at 28 °C to monitor BAX1‐mediated cell death induction. The BAX1‐mediated cell death inhibitor, BCL‐XL, and an empty vector were used as positive and negative controls, respectively, for cell death suppression.
For the INF1‐mediated cell death suppression assay, all selected effector candidates were cloned into the pK2GW7 Gateway binary expression vector for Agrobacterium‐mediated transient transformation assays (ATTAs). The INF1‐mediated cell death suppression assay was performed according to Bos et al. (2010). Phytophthora infestans cell death inducing INF1 (pGR106‐FLAG) and pK2GW7‐UHOR_02413, pK2GW7‐UHOR_02700, pK2GW7‐UHOR_03756 and pK2GW7‐UHOR_05101 were co‐infiltrated into 5–6‐week‐old N. benthamiana leaves via ATTA. Phytophthora infestans Avr3aKI (pGR106‐FLAG) and Avr3aΔY (pGR106‐FLAG) were used as positive and negative controls for INF1‐mediated cell death suppression, respectively (Bos et al., 2010). INF1‐mediated cell death suppression was observed at 4 dpi. The pGR106‐INF1, pGR106‐Avr3aKI and pGR106‐Avr3aΔY constructs were kindly provided by the group of Sophien Kamoun (The Sainsbury Laboratory, Norwich Research Park, Norwich, UK).
Extraction of proteins from yeast cells for western blot
A single colony of the respective yeast strain was inoculated in 3 mL of liquid SD medium without tryptophan. The cell suspension was incubated at 28 °C with shaking at 200 rpm to obtain OD600 = 0.8. The cell suspension was transferred to a 2‐mL reaction tube and centrifuged at 9600 ×g for 3 min. The supernatant was discarded and the pellet was resuspended in 100 µL of 2 × sodium dodecylsulfate (SDS) loading dye. Glass beads (0.3 g) were added to the suspension and the sample was boiled at 98 °C for 5 min. Subsequently, the sample was shaken for 10 min at 1800 rpm using a Vibrax‐VXR shaker (IKA, Staufen, Germany). The sample was again boiled at 98 °C for 5 min. Finally, 25 µL of the sample were used for western blot analysis. Western blot was performed according to Mueller et al. (2013).
Staining of leaves with WGA‐AF488 and propidium iodide
WGA‐AF488 (Molecular Probes, Karlsruhe, Germany) and propidium iodide (Sigma‐Aldrich, St. Louis, Missouri, USA) staining were performed to monitor and track the fungal hyphae during host colonization. WGA‐AF488 stains fungal cell walls and propidium iodide stains plant cell walls. After U. hordei‐inoculated barley leaves had been bleached with pure ethanol, the samples were subsequently boiled for 2–3 h in 10% KOH at 85 °C. After boiling, the samples were neutralized with five washing steps using phosphate‐buffered saline (PBS) (pH 7.4). Subsequently the staining solution (1 µg/mL propidium iodide, 10 µg/mL WGA‐AF488; 0.02% Tween‐20 in PBS, pH 7.4) was vacuum infiltrated into the samples three to four times (5 min each) at 25 kPa using a desiccator. The infiltrated leaves were stored in PBS at 4 °C in the dark until microscopic analysis. WGA‐AF 488 used excitation at 488 nm and detection at 500–540 nm.
Supporting information
Fig. S1 Gene ontology enrichment analysis of differentially expressed Ustilago hordei genes by using GOEAST software. Red boxes represent GOs of gene groups that were significantly induced at all four time points. Green boxes represent GOs of gene groups that were significantly repressed at all times. More intense color indicates the higher statistical significance for the particular molecular function. p1 and p2 represent p‐values of the induced and repressed genes, respectively. A detailed list of the GO assignments is presented in Supplemental Table S1.
Fig. S2 Comparison of expression pattern of microarray vs. qRTPCR. Quantitative real‐time PCR (qRT‐PCR) was performed with 16 U. hordei effectors to confirm the microarray data. Expression levels of each gene used in RT‐qPCR were normalized by using the U. hordei Ppi gene. Relative gene expressions have been shown as log2‐fold changes relative to expression in axenic culture. Error bars represent standard deviation of three biological replicates.
Fig. S3 Confirmation of Ustilago hordei knock‐out strains via PCR. A) Schematic representation of U. hordei wild type (wt) and knock‐out (KO) gene locus before and after homologous recombination. The U. hordei UHOR_02413, UHOR_02700, UHOR_03756 and UHOR_05101 genes were replaced by hygromycin cassette. Since the length of inserted hygromycin cassette is bigger than replaced UHOR_02413, UHOR_02700 and UHOR_03756 genes, primer pairs that bind to flanking region of left border (LB) and right border (RB) of each gene were used to confirm knock‐out (Table S3). PCRs were performed with gDNAs isolated from both mating type of (Mate1:M1; Mate2:M2) wild type (wt) and knock‐out (KO) strains. Gene replacement of wt UHOR_02413 (B), UHOR_02700 (C) and UHOR_03756 (D) gene locus with hygromycin cassette was confirmed via PCR by using locus specific primer set (Table S3). RT‐qPCR was also performed for each gene to show the loss of expression in KO background compared to wt (B‐C). E) Since the length of inserted hygromycin cassette and UHOR_05101 genes was similar, F1‐R1 primer pair was used to confirm deletion of UHOR_05101 gene and F2‐R2 primer pair was used to confirm the locus integration (Table S3). F) While wt strains show PCR products with F1‐R1 primer pair, KO strains do not. On the other hand, while KO strains show PCR products with F2‐R2 primer pair, wt strains do not. RT‐qPCR was also performed to show loss of UHOR_05101 gene expression in KO background compared to wt. Error bars in RT‐qPCR results represent standard deviation of three biological replicates. Expression levels were normalized using U. hordei Ppi gene.
Fig. S4 A) Ustilago hordei filamentation test on charcoal plate. To opposite mating types of both wild type and effector deletion strains were grown in YEPS liquid medium till OD:0.8‐1.0. Subsequently, two mating type were mixed (1:1, v/v) to get OD:1.0 and 5 μl of the mixture was dropped on charcoal plate. Single mating type cultures were used as negative controls. Pictures were taken after 3 days incubation at room temperature. B) Growth rate of the U. hordei wild type and mutant strains in liquid YEPSlight. The OD600 was measured at the indicated time points. There was no significant difference between the U. hordei wild type and Δuhor_02413, Δuhor_02700, Δuhor_03756 and Δuhor_05101 mutant strains. C) Classical Ustilago hordei‐barley infection assay. Dehulled and surface sterilized barley seeds were inoculated with the U. hordei wild type and Δuhor_02413, Δuhor_02700, Δuhor_03756 and Δuhor_05101 mutant strains. Approximately 2‐3 months after inoculation, disease symptoms were scored at barley heading. This experiment was only performed one time.
Fig. S5 Biomass quantification of 14 Ustilago hordei effector mutants on barley. The virulence of the Ustilago hordei wild type and 14 effector mutants was performed by the quantification of fungal biomass at 6 dpi on barley leaf. The U. hordei Pep1 deletion mutant was used as a positive control for virulence reduction. The infected barley leaves at 6 day post‐inoculation (dpi) were harvested and analyzed via quantitative PCR by using genomic DNA and using the U. hordei Ppi1 genes as a standard. The fungal biomass was deduced from a standard curve. A oneway ANOVA was performed and followed by a Bonferroni test. Samples that showed significant differences compared to wild type are marked with asterisks (*, P< 0.001). Error bars represent the standard deviation of at least three biological repeats.
Fig. S6 Heterologous production of Ustilago hordei Uvi1‐4 in yeast cell and Nicotiana benthamiana. A) Western Blot (WB) analysis was performed with total proteins extract of ESM356 yeast strains transformed with pEZY45‐UHOR_02413‐HA, pEZY45‐UHOR_02700‐HA, pEZY45‐UHOR_03756‐HA and pEZY45‐ UHOR_05101‐HA to confirm the expression of these effectors. Empty pEZY15 transformed yeast cell was used as negative control. α‐HA antibody was used for WB. Expected protein sizes were indicated below figure. B) Western Blot (WB) analysis was performed with total proteins extract of AH109 yeast strains transformed with pGBKT7‐Myc‐UHOR_02413‐HA, pGBKT7‐Myc‐UHOR_02700‐HA and pGBKT7‐Myc‐UHOR_03756‐HA to confirm the expression of these effectors. Empty pGBKT7‐BDMyc transformed yeast cell was used as negative control. α‐HA antibody was used for WB. Expected protein sizes were indicated below figure. C) Western Blot (WB) analysis was performed to confirm expression of pK2GW7‐UHOR_02413‐HA, pK2GW7‐ UHOR_02700‐HA and pK2GW7‐UHOR_03756‐HA in Nicotiana benthamiana leaves transformed with Agrobacterium tumefaciens. αvHA antibody was used for WB. Expected protein sizes were indicated below figure. Red arrows indicate respective protein bands. D) Heterologous expression of pK2GW7‐GUSGFP, pK2GW7‐UHOR_02413‐GFP, pK2GW7‐UHOR_02700‐GFP, pK2GW7‐UHOR_03756‐GFP and pK2GW7‐UHOR_05101‐GFP in Nicotiana benthamiana leaves transformed with A. tumefaciens.
Fig. S7 Cell‐death suppression activity of four Ustilago hordei effectors. A) BAX1‐mediated cell death suppression assay. Cell death inducing pEZY45‐BAX1 and pEZY45‐UHOR_02413, pEZY45‐UHOR_02700 and pEZY45‐UHOR_03756 and pEZY45‐UHOR_05101 were co‐expressed under the control of a galactose inducible promoter in the ESM356.1 yeast strain. Empty pEZY45 and pEZY45‐Bcl‐XL were used as negative and positive controls for BAX1‐mediated cell death suppression, respectively. Pictures were taken after 4 days of incubation. The figure is representative of three biological replicates. B) INF1‐mediated cell death suppression assay. Phytophthora infestans cell death inducing INF1 (in pGR106‐FLAG) and pK2GW7‐UHOR_05101, pK2GW7‐UHOR_02413, pK2GW7‐UHOR_02700 and pK2GW7‐ UHOR_03756 were co‐infiltrated into 5‐6 week‐‐old Nicotiana benthamiana leaves via Agrobacterium‐mediated transformation. P. infestans Avr3AKI (pGR106‐FLAG) and Avr3aΔY (pGR106‐FLAG) was used as positive and negative control for INF1‐mediated cell death suppression, respectively. Pictures were taken at 4 dpi. The figure is representative of three biological replicates.
Table S1 Microarray analysis of the whole genome of Ustilago hordei. Blast2Go analysis of All genes, Secretome, Effectorome and Effectors for knock‐out (KO) microarray analyses are depicted in different excel sheets. Genes showing a two‐fold change in expression level were considered as differentially expressed (log2 fold change ≥1).
Table S2 Proteins with predicted NLS signal found in the Ustilago hordei effectorome.
Table S3 Constructs and oligonucleotides used in this study.
Acknowledgements
This work was supported by the Cluster of Excellence on Plant Science (CEPLAS). We thank Professor Regine Kahmann (Max Planck Institute for Microbiology, Marburg, Germany) for generous support of this work and Professor Karl‐Heinz Kogel (University Gießen, Germany) for providing access to the microarray facility. All members of AG Doehlemann are acknowledged for peeling off U. hordei infection structures from hundreds of barley seedling leaves using liquid‐latex. We also thank the group of Sophien Kamoun (The Sainsbury Laboratory, Norwich Research Park, Norwich, UK) for providing the constructs for INF1‐mediated cell death assay.
References
- Ali, S. , Laurie, J.D. , Linning, R. , Cervantes‐Chávez, J.A. , Gaudet, D. and Bakkeren, G. (2014) An immunity‐triggering effector from the barley smut fungus Ustilago hordei resides in an Ustilaginaceae‐specific cluster bearing signs of transposable element‐assisted evolution. PLoS Pathog. 10, e1004223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, R. , Oberwinkler, F. and Mendgen, K. (1995) Cellular interaction of the smut fungus Ustacystis waldsteiniae . Can. J. Bot. 73, 867–883. [Google Scholar]
- Bos, J.I.B. , Armstrong, M.R. , Gilroy, E.M. , Boevink, P.C. , Hein, I. , Taylor, R.M. , Zhendong, T. , Engelhardt, S. , Vetukuri, R.R. , Harrower, B. , Dixelius, C. , Bryan, G. , Sadanandom, A. , Whisson, S.C. , Kamoun, S. and Birch, P.R.J. (2010) Phytophthora infestans effector Avr3a is essential for virulence and manipulates plant immunity by stabilizing host E3 ligase CMPG1. Proc. Natl. Acad. Sci. USA, 107, 9909–9914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu, K.T. and Ng, T.B. (2003) Isolation of a large thaumatin‐like antifungal protein from seeds of the Kweilin chestnut Castanopsis chinensis . Biochem. Biophys. Res. Commun. 301, 364–370. [DOI] [PubMed] [Google Scholar]
- Conesa, A. , Gotz, S. , Garcia‐Gomez, J.M. , Terol, J. , Talon, M. and Robles, M. (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics, 21, 3674–3676. [DOI] [PubMed] [Google Scholar]
- Cosgrove, D.J. (2000) Loosening of plant cell walls by expansins. Nature, 407, 321–326. [DOI] [PubMed] [Google Scholar]
- Dean, R.A. , Talbot, N.J. , Ebbole, D.J. , Farman, M.L. , Mitchell, T.K. , Orbach, M.J. , Thon, M. , Kulkarni, R. , Xu, J.‐R. , Pan, H. , Read, N.D. , Lee, Y.‐H. , Carbone, I. , Brown, D. , Oh, Y.Y. , Donofrio, N. , Jeong, J.S. , Soanes, D.M. , Djonovic, S. , Kolomiets, E. , Rehmeyer, C. , Li, W. , Harding, M. , Kim, S. , Lebrun, M.‐H. , Bohnert, H. , Coughlan, S. , Butler, J. , Calvo, S. , Ma, L.‐J. , Nicol, R. , Purcell, S. , Nusbaum, C. , Galagan, J.E. and Birren, B.W. (2005) The genome sequence of the rice blast fungus Magnaporthe grisea . Nature, 434, 980–986. [DOI] [PubMed] [Google Scholar]
- Djamei, A. , Schipper, K. , Rabe, F. , Ghosh, A. , Vincon, V. , Kahnt, J. , Osorio, S. , Tohge, T. , Fernie, A.R. , Feussner, I. , Feussner, K. , Meinicke, P. , Stierhof, Y.‐D. , Schwarz, H. , Macek, B. , Mann, M. and Kahmann, R. (2011) Metabolic priming by a secreted fungal effector. Nature, 478, 395–398. [DOI] [PubMed] [Google Scholar]
- Duplessis, S. , Hacquard, S. , Delaruelle, C. , Tisserant, E. , Frey, P. , Martin, F. and Kohler, A. (2011) Melampsora larici‐populina transcript profiling during germination and timecourse infection of poplar leaves reveals dynamic expression patterns associated with virulence and biotrophy. Mol. Plant–Microbe Interact. 24, 808–818. [DOI] [PubMed] [Google Scholar]
- Emanuelsson, O. , Nielsen, H. , Brunak, S. and von Heijne, G. (2000) Predicting subcellular localization of proteins based on their N‐terminal amino acid sequence. J. Mol. Biol. 300, 1005–1016. [DOI] [PubMed] [Google Scholar]
- Gaudet, D.A. , Wang, Y. , Penniket, C. , Lu, Z.X. , Bakkeren, G. and Laroche, A. (2010) Morphological and molecular analyses of host and nonhost interactions involving barley and wheat and the covered smut pathogen Ustilago hordei . Mol. Plant–Microbe Interact. 23, 1619–1634. [DOI] [PubMed] [Google Scholar]
- Gout, L. , Fudal, I. , Kuhn, M.L. , Blaise, F. , Eckert, M. , Cattolico, L. , Balesdent, M.‐H. and Rouxel, T. (2006) Lost in the middle of nowhere: the AvrLm1 avirulence gene of the Dothideomycete Leptosphaeria maculans . Mol. Microbiol. 60, 67–80. [DOI] [PubMed] [Google Scholar]
- Guyon, K. , Balague, C. , Roby, D. and Raffaele, S. (2014) Secretome analysis reveals effector candidates associated with broad host range necrotrophy in the fungal plant pathogen Sclerotinia sclerotiorum . BMC Genomics, 15, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacquard, S. , Kracher, B. , Maekawa, T. , Vernaldi, S. , Schulze‐Lefert, P. and Ver Loren van Themaat, E. (2013) Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proc. Natl. Acad. Sci. 110, E2219–E2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemetsberger, C. , Herrberger, C. , Zechmann, B. , Hillmer, M. and Doehlemann, G. (2012) The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathog. 8, e1002684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemetsberger, C. , Mueller, A.N. , Matei, A. , Herrberger, C. , Hensel, G. , Kumlehn, J. , Mishra, B. , Sharma, R. , Thines, M. , Hückelhoven, R. and Doehlemann, G. (2015) The fungal core effector Pep1 is conserved across smuts of dicots and monocots. New Phytol. 206, 1116–1126. [DOI] [PubMed] [Google Scholar]
- Ho, V.S. , Wong, J.H. and Ng, T.B. (2007) A thaumatin‐like antifungal protein from the emperor banana. Peptides, 28, 760–766. [DOI] [PubMed] [Google Scholar]
- Hof, A. , Zechmann, B. , Schwammbach, D. , Hückelhoven, R. and Doehlemann, G. (2014) Alternative cell death mechanisms determine epidermal resistance in incompatible barley–Ustilago interactions. Mol. Plant–Microbe Interact. 27, 403–414. [DOI] [PubMed] [Google Scholar]
- Hu, G.G. , Linning, R. and Bakkeren, G. (2002) Sporidial mating and infection process of the smut fungus, Ustilago hordei, in susceptible barley. Can. J. Bot.‐Rev. Can. Bot. 80, 1103–1114. [Google Scholar]
- Hückelhoven, R. (2004) BAX Inhibitor‐1, an ancient cell death suppressor in animals and plants with prokaryotic relatives. Apoptosis, 9, 299–307. [DOI] [PubMed] [Google Scholar]
- deJonge, R. (2012) In silico identification and characterization of effector catalogs In: Plant Fungal Pathogens: Methods and Protocols (Bolton M.D. and Thomma B.P.H.J., eds), pp. 415–425. Totowa, NJ: Humana Press. [Google Scholar]
- de Jonge, R. , van Esse, H.P. , Kombrink, A. , Shinya, T. , Desaki, Y. , Bours, R. , van der Krol, S. , Shibuya, N. , Joosten, M.H.A.J. and Thomma, B.P.H.J. (2010) Conserved fungal LysM effector Ecp6 prevents chitin‐triggered immunity in plants. Science, 329, 953–955. [DOI] [PubMed] [Google Scholar]
- Kamoun, S. , van West, P. , de Jong, A.J. , de Groot, K.E. , Vleeshouwers, V.G. and Govers, F. (1997) A gene encoding a protein elicitor of Phytophthora infestans is down‐regulated during infection of potato. Mol. Plant–Microbe Interact. 10, 13–20. [DOI] [PubMed] [Google Scholar]
- Kamoun, S. , van West, P. , Vleeshouwers, V.G.A.A. , de Groot, K.E. and Govers, F. (1998) Resistance of Nicotiana benthamiana to Phytophthora infestans is mediated by the recognition of the elicitor protein INF1. Plant Cell, 10, 1413–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kämper, J. (2004) A PCR‐based system for highly efficient generation of gene replacement mutants in Ustilago maydis . Mol. Genet. Genomics, 271, 103–110. [DOI] [PubMed] [Google Scholar]
- Kämper, J. , Kahmann, R. , Bölker, M. , Ma, L.‐J. , Brefort, T. , Saville, B.J. , Banuett, F. , Kronstad, J.W. , Gold, S.E. , Müller, O. , Perlin, M.H. , Wösten, H.A.B. , de Vries, R. , Ruiz‐Herrera, J. , Reynaga‐Peña, C.G. , Snetselaar, K. , McCann, M. , Pérez‐Martín, J. , Feldbrügge, M. , Basse, C.W. , Steinberg, G. , Ibeas, J.I. , Holloman, W. , Guzman, P. , Farman, M. , Stajich, J.E. , Sentandreu, R. , González‐Prieto, J.M. , Kennell, J.C. , Molina, L. , Schirawski, J. , Mendoza‐Mendoza, A. , Greilinger, D. , Münch, K. , Rössel, N. , Scherer, M. , Vraneš, M. , Ladendorf, O. , Vincon, V. , Fuchs, U. , Sandrock, B. , Meng, S. , Ho, E.C.H. , Cahill, M.J. , Boyce, K.J. , Klose, J. , Klosterman, S.J. , Deelstra, H.J. , Ortiz‐Castellanos, L. , Li, W. , Sanchez‐Alonso, P. , Schreier, P.H. , Häuser‐Hahn, I. , Vaupel, M. , Koopmann, E. , Friedrich, G. , Voss, H. , Schlüter, T. , Margolis, J. , Platt, D. , Swimmer, C. , Gnirke, A. , Chen, F. , Vysotskaia, V. , Mannhaupt, G. , Güldener, U. , Münsterkötter, M. , Haase, D. , Oesterheld, M. , Mewes, H.‐W. , Mauceli, E.W. , DeCaprio, D. , Wade, C.M. , Butler, J. , Young, S. , Jaffe, D.B. , Calvo, S. , Nusbaum, C. , Galagan, J. and Birren, B.W. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature, 444, 97–101. [DOI] [PubMed] [Google Scholar]
- Kawai, M. , Pan, L. , Reed, J.C. and Uchimiya, H. (1999) Evolutionally conserved plant homologue of the Bax inhibitor‐1 (BI‐1) gene capable of suppressing Bax‐induced cell death in yeast(1). FEBS Lett. 464, 143–147. [DOI] [PubMed] [Google Scholar]
- Kelley, B.S. , Lee, S.J. , Damasceno, C.M. , Chakravarthy, S. , Kim, B.D. , Martin, G.B. and Rose, Jocelyn K.C. (2010) A secreted effector protein (SNE1) from Phytophthora infestans is a broadly acting suppressor of programmed cell death. Plant J. 62, 357–366. [DOI] [PubMed] [Google Scholar]
- Kirsten, S. , Navarro‐Quezada, A. , Penselin, D. , Wenzel, C. , Matern, A. , Leitner, A. , Baum, T., Seiffert, U. and Knogge, W. (2012) Necrosis‐inducing proteins of Rhynchosporium commune, effectors in quantitative disease resistance. Mol. Plant–Microbe Interact. 25, 1314–1325. [DOI] [PubMed] [Google Scholar]
- Kleemann, J. , Rincon‐Rivera, L.J. , Takahara, H. , Neumann, U. , van Themaat, E.V.L. , van der Does, H.C. , Hacquard, S., Stüber, K., Will, I., Schmalenbach, W. and Schmelzer, E. (2012) Sequential delivery of host‐induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum . PLoS Pathog. 8, e1002643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krogh, A. , Larsson, B. , von Heijne, G. and Sonnhammer, E.L. (2001) Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. [DOI] [PubMed] [Google Scholar]
- Kroj, T. , Chanclud, E. , Michel‐Romiti, C. , Grand, X. and Morel, J.B. (2016) Integration of decoy domains derived from protein targets of pathogen effectors into plant immune receptors is widespread. New Phytol. 210, 618–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicek, C.P. , Starr, T.L. and Glass, N.L. (2014) Plant cell wall‐degrading enzymes and their secretion in plant‐pathogenic fungi. Annu. Rev. Phytopathol. 52, 427–451. [DOI] [PubMed] [Google Scholar]
- Lahrmann, U. , Ding, Y. , Banhara, A. , Rath, M. , Hajirezaei, M.R. , Dohlemann, S. , von Wirén, N. , Parniske, M. and Zuccaro, A. (2013) Host‐related metabolic cues affect colonization strategies of a root endophyte. Proc. Natl. Acad. Sci. USA, 110, 13 965–13 970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanver, D. , Müller, A.N. , Happel, P. , Schweizer, G. , Haas, F.B. , Franitza, M. , Pellegrin, C. , Reissmann, S. , Altmüller, J. , Rensing, S.A. and Kahmann, R. (2018) The biotrophic development of Ustilago maydis studied by RNAseq analysis. Plant Cell, doi: 10.1105/tpc.17.00764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurie, J.D. , Ali, S. , Linning, R. , Mannhaupt, G. , Wong, P. , Gueldener, U. , Munsterkotter, M. , Moore, R. , Kahmann, R. , Bakkeren, G. and Schirawski, J. (2012) Genome comparison of barley and maize smut fungi reveals targeted loss of RNA silencing components and species‐specific presence of transposable elements. Plant Cell, 24, 1733–1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J.J. , Sturrock, R. and Ekramoddoullah, A.K. (2010) The superfamily of thaumatin‐like proteins: its origin, evolution, and expression towards biological function. Plant Cell Rep. 29, 419–436. [DOI] [PubMed] [Google Scholar]
- Livak, K.J. and Schmittgen, T.D. (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2‐ΔΔCT method. Methods, 25, 402–408. [DOI] [PubMed] [Google Scholar]
- Lowe, R.G.T. and Howlett, B.J. (2012) Indifferent, affectionate, or deceitful: lifestyles and secretomes of fungi. PLoS Pathog. 8,e1002515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozano‐Torres, J.L. , Wilbers, R.H. , Gawronski, P. , Boshoven, J.C. , Finkers‐Tomczak, A. , Cordewener, J.H. , America, A.H. , Overmars, H.A. , Van 't Klooster, J.W. , Baranowski, L. , Sobczak, M. , Ilyas, M. , van der Hoorn, R.A.l. , Schots, A. , de Wit, P.J.G.M. , Bakker, J. , Goverse, A. and Smant, G. (2012) Dual disease resistance mediated by the immune receptor Cf‐2 in tomato requires a common virulence target of a fungus and a nematode. Proc. Natl. Acad. Sci. USA, 109, 10 119–10 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu, X. , Shen, C. , Fu, Y. , Xie, J. , Jiang, D. , Li, G. and Cheng, J. (2015) Comparative genomic and transcriptional analyses of the carbohydrate‐active enzymes and secretomes of phytopathogenic fungi reveal their significant roles during infection and development. Sci. Rep. 5, 15 565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maqbool, A. , Saitoh, H. , Franceschetti, M. , Stevenson, C.E.M. , Uemura, A. , Kanzaki, H. , Kamoun, S. , Terauchi, R. and Banfield, M.J. (2015) Structural basis of pathogen recognition by an integrated HMA domain in a plant NLR immune receptor. eLife, 4, e08709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall, R. , Kombrink, A. , Motteram, J. , Loza‐Reyes, E. , Lucas, J. , Hammond‐Kosack, K.E. , Thomma, B. and Rudd, J. (2011) Analysis of two in planta expressed lysm effector homologs from the fungus Mycosphaerella graminicola reveals novel functional properties and varying contributions to virulence on wheat. Plant Physiol. 156, 756–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matei, A. and Doehlemann, G. (2016) Cell biology of corn smut disease–Ustilago maydis as a model for biotrophic interactions. Curr. Opin. Microbiol. 34, 60–66. [DOI] [PubMed] [Google Scholar]
- Matei, A. , Ernst, C. , Gunl, M. , Thiele, B. , Altmuller, J. , Walbot, V. , Usadel, B. and Doehlemann, G. (2018) How to make a tumour: cell type specific dissection of Ustilago maydis‐induced tumour development in maize leaves. New Phytol. 217(4), 1681–1695. [DOI] [PubMed] [Google Scholar]
- Mentlak, T.A. , Kombrink, A. , Shinya, T. , Ryder, L.S. , Otomo, I. , Saitoh, H. , Terauchi, R. , Nishizawa, Y. , Shibuya, N. , Thomma, B. and Talbot, N.J. (2012) Effector‐mediated suppression of chitin‐triggered immunity by Magnaporthe oryzae is necessary for rice blast disease. Plant Cell, 24, 322–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, A.N. , Ziemann, S. , Treitschke, S. , Assmann, D. and Doehlemann, G. (2013) Compatibility in the Ustilago maydis–maize interaction requires inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog. 9, e1003177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, Q.B. , Itoh, K. , Van Vu, B. , Tosa, Y. and Nakayashiki, H. (2011) Simultaneous silencing of endo‐beta‐1,4 xylanase genes reveals their roles in the virulence of Magnaporthe oryzae . Mol. Microbiol. 81, 1008–1019. [DOI] [PubMed] [Google Scholar]
- Nielsen, H. , Engelbrecht, J. , Brunak, S. and von Heijne, G. (1997) Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 10, 1–6. [DOI] [PubMed] [Google Scholar]
- O'Connell, R.J. , Thon, M.R. , Hacquard, S. , Amyotte, S.G. , Kleemann, J. , Torres, M.F. , Damm, U. , Buiate, E.A. , Epstein, L. , Alkan, N. , Altmüller, J. , Alvarado‐Balderrama, L. , Bauser, C.A. , Becker, C. , Birren, B.W. , Chen, Z. , Choi, J. , Crouch, J.A. , Duvick, J.P. , Farman, M.A. , Gan, P. , Heiman, D. , Henrissat, B. , Howard, R.J. , Kabbage, M. , Koch, C. , Kracher, B. , Kubo, Y. , Law, A.D. , Lebrun, M.‐H. , Lee, Y.‐H. , Miyara, I. , Moore, N. , Neumann, U. , Nordström, K. , Panaccione, D.G. , Panstruga, R. , Place, M. , Proctor, R.H. , Prusky, D. , Rech, G. , Reinhardt, R. , Rollins, J.A. , Rounsley, S. , Schardl, C.L. , Schwartz, D.C. , Shenoy, N. , Shirasu, K. , Sikhakolli, U.R. , Stüber, K. , Sukno, S.A. , Sweigard, J.A. , Takano, Y. , Takahara, H. , Trail, F. , van der Does, H.C. , Voll, L.M. , Will, I. , Young, S. , Zeng, Q. , Zhang, J. , Zhou, S. , Dickman, M.B. , Schulze‐Lefert, P. , Ver Loren van Themaat, E. , Ma, L.‐J. and Vaillancourt, L.J. (2012) Lifestyle transitions in plant pathogenic Colletotrichum fungi deciphered by genome and transcriptome analyses. Nat. Genet. 44, 1060–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ökmen, B. , Etalo, D.W. , Joosten, M.H. , Bouwmeester, H.J. , de Vos, R.C. , Collemare, J. , and de Wit, P.J. (2013) Detoxification of alpha‐tomatine by Cladosporium fulvum is required for full virulence on tomato. New Phytol. 198, 1203–1214. [DOI] [PubMed] [Google Scholar]
- Redkar, A. , Hoser, R. , Schilling, L. , Zechmann, B. , Krzymowska, M. , Walbot, V. and Doehlemann, G. (2015) A secreted effector protein of Ustilago maydis guides maize leaf cells to form tumors. Plant Cell, 27, 1332–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rooney, H.C. , van't Klooster, J.W., van der Hoorn, R.A., Joosten, M.H., Jones, J.D. and de Wit, P.J. (2005) Cladosporium Avr2 inhibits tomato Rcr3 protease required for Cf‐2‐dependent disease resistance. Science, 308, 1783–1786. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. , Fritsch, E.F. and Maniatis, T. (1989) Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory. [Google Scholar]
- Saunders, D.G. , Win, J. , Cano, L.M. , Szabo, L.J. , Kamoun, S. and Raffaele, S. (2012) Using hierarchical clustering of secreted protein families to classify and rank candidate effectors of rust fungi. PloS ONE, 7, e29847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling, L. , Matei, A. , Redkar, A. , Walbot, V. and Doehlemann, G. (2014) Virulence of the maize smut Ustilago maydis is shaped by organ‐specific effectors. Mol. Plant Pathol. 15, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster, M. , Schweizer, G. and Kahmann, R. (2017) Comparative analyses of secreted proteins in plant pathogenic smut fungi and related basidiomycetes. Fungal Genet. Biol. Available at 10.1016/j.fgb.2016.12.003. [DOI] [PubMed] [Google Scholar]
- Singh, N.K. , Kumar, K.R. , Kumar, D. , Shukla, P. and Kirti, P.B. (2013) Characterization of a pathogen induced thaumatin‐like protein gene AdTLP from Arachis diogoi, a wild peanut. PloS ONE, 8, e83963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbe, D.S. , Doehlemann, G. , Fernandes, J. and Walbot, V. (2010) Maize tumors caused by Ustilago maydis require organ‐specific genes in host and pathogen. Science, 328, 89–92. [DOI] [PubMed] [Google Scholar]
- Slusarenko, A.J. and Schlaich, N.L. (2003) Downy mildew of Arabidopsis thaliana caused by Hyaloperonospora parasitica (formerly Peronospora parasitica). Mol. Plant Pathol. 4, 159–170. [DOI] [PubMed] [Google Scholar]
- Spellig, T. , Bottin, A. and Kahmann, R. (1996) Green fluorescent protein (GFP) as a new vital marker in the phytopathogenic fungus Ustilago maydis . Mol. Gen. Genet. 252, 503–509. [DOI] [PubMed] [Google Scholar]
- Sperschneider, J. , Dodds, P.N. , Gardiner, D.M. , Manners, J.M. , Singh, K.B. and Taylor, J.M. (2015) Advances and challenges in computational prediction of effectors from plant pathogenic fungi. PLoS Pathog. 11, e1004806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperschneider, J. , Dodds, P.N. , Singh, K.B. and Taylor, J.M. (2018) ApoplastP: prediction of effectors and plant proteins in the apoplast using machine learning. New Phytol. 217, 1764–1778. [DOI] [PubMed] [Google Scholar]
- Sperschneider, J. , Gardiner, D.M. , Dodds, P.N. , Tini, F. , Covarelli, L. , Singh, K.B. , Manners, J.M. and Taylor, J.M. (2016) EffectorP: predicting fungal effector proteins from secretomes using machine learning. New Phytol. 210, 743–761. [DOI] [PubMed] [Google Scholar]
- Sperschneider, J. , Ying, H. , Dodds, P.N. , Gardiner, D.M. , Upadhyaya, N.M. , Singh, K.B. , Manners, J.M. and Taylor, J.M. (2014) Diversifying selection in the wheat stem rust fungus acts predominantly on pathogen‐associated gene families and reveals candidate effectors. Front. Plant Sci. 5, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprockett, D.D. , Piontkivska, H. and Blackwood, C.B. (2011) Evolutionary analysis of glycosyl hydrolase family 28 (GH28) suggests lineage‐specific expansions in necrotrophic fungal pathogens. Gene, 479, 29–36. [DOI] [PubMed] [Google Scholar]
- Stergiopoulos, I. and de Wit, P.J. (2009) Fungal effector proteins. Ann. Rev. Phytopathol. 47, 233–263. [DOI] [PubMed] [Google Scholar]
- Taricani, L. , Tejada, M.L. and Young, P.G. (2002) The fission yeast ES2 homologue, Bis1, interacts with the Ish1 stress‐responsive nuclear envelope protein. J. Biol. Chem. 277, 10 562–10 572. [DOI] [PubMed] [Google Scholar]
- Tian, M. , Benedetti, B. and Kamoun, S. (2005) A second Kazal‐like protease inhibitor from Phytophthora infestans inhibits and interacts with the apoplastic pathogenesis‐related protease P69B of tomato. Plant Physiol. 138, 1785–1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, M. , Huitema, E. , da Cunha, L. , Torto‐Alalibo, T. and Kamoun, S. (2004) A Kazal‐like extracellular serine protease inhibitor from Phytophthora infestans targets the tomato pathogenesis‐related protease P69B. J. Biol. Chem. 279, 26 370–26 377. [DOI] [PubMed] [Google Scholar]
- Untergasser, A. , Nijveen, H. , Rao, X. , Bisseling, T. , Geurts, R. and Leunissen, J.A.M. (2007) Primer3Plus, an enhanced web interface to Primer3. Nucleic Acids Res. 35, 71–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Vu, B. , Itoh, K. , Nguyen, Q.B. , Tosa, Y. and Nakayashiki, H. (2012) Cellulases belonging to glycoside hydrolase families 6 and 7 contribute to the virulence of Magnaporthe oryzae . Mol. Plant–Microbe Interact. 25, 1135–1141. [DOI] [PubMed] [Google Scholar]
- Watanabe, N. and Lam, E. (2009) Bax Inhibitor‐1, a conserved cell death suppressor, is a key molecular switch downstream from a variety of biotic and abiotic stress signals in plants. Int. J. Mol. Sci. 10, 3149–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Wit, P.J. , van der Burgt, A. , Ökmen, B. , Stergiopoulos, I. , Abd‐Elsalam, K.A. , Aerts, A.L. , Bahkali, A.H., Beenen, H.G., Chettri, P., Cox, M.P. and Datema, E. (2012) The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 8, e1003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, J. , Wang, Y. , Park, S.‐Y. , Kim, S.G. , Yoo, J.S. , Park, S. , Gupta, R. , Kang, K. Y. and Kim, S.T. (2016) Secreted alpha‐N‐arabinofuranosidase b protein is required for the full virulence of Magnaporthe oryzae and triggers host defences. PloS One, 11, e0165149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi, M. and Valent, B. (2013) Communication between filamentous pathogens and plants at the biotrophic interface. Annu. Rev. Phytopathol. 51, 587–611. [DOI] [PubMed] [Google Scholar]
- Zheng, Q. and Wang, X.J. (2008) GOEAST: a web‐based software toolkit for Gene Ontology enrichment analysis. Nucleic Acids Res. 36, W358–W363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Gene ontology enrichment analysis of differentially expressed Ustilago hordei genes by using GOEAST software. Red boxes represent GOs of gene groups that were significantly induced at all four time points. Green boxes represent GOs of gene groups that were significantly repressed at all times. More intense color indicates the higher statistical significance for the particular molecular function. p1 and p2 represent p‐values of the induced and repressed genes, respectively. A detailed list of the GO assignments is presented in Supplemental Table S1.
Fig. S2 Comparison of expression pattern of microarray vs. qRTPCR. Quantitative real‐time PCR (qRT‐PCR) was performed with 16 U. hordei effectors to confirm the microarray data. Expression levels of each gene used in RT‐qPCR were normalized by using the U. hordei Ppi gene. Relative gene expressions have been shown as log2‐fold changes relative to expression in axenic culture. Error bars represent standard deviation of three biological replicates.
Fig. S3 Confirmation of Ustilago hordei knock‐out strains via PCR. A) Schematic representation of U. hordei wild type (wt) and knock‐out (KO) gene locus before and after homologous recombination. The U. hordei UHOR_02413, UHOR_02700, UHOR_03756 and UHOR_05101 genes were replaced by hygromycin cassette. Since the length of inserted hygromycin cassette is bigger than replaced UHOR_02413, UHOR_02700 and UHOR_03756 genes, primer pairs that bind to flanking region of left border (LB) and right border (RB) of each gene were used to confirm knock‐out (Table S3). PCRs were performed with gDNAs isolated from both mating type of (Mate1:M1; Mate2:M2) wild type (wt) and knock‐out (KO) strains. Gene replacement of wt UHOR_02413 (B), UHOR_02700 (C) and UHOR_03756 (D) gene locus with hygromycin cassette was confirmed via PCR by using locus specific primer set (Table S3). RT‐qPCR was also performed for each gene to show the loss of expression in KO background compared to wt (B‐C). E) Since the length of inserted hygromycin cassette and UHOR_05101 genes was similar, F1‐R1 primer pair was used to confirm deletion of UHOR_05101 gene and F2‐R2 primer pair was used to confirm the locus integration (Table S3). F) While wt strains show PCR products with F1‐R1 primer pair, KO strains do not. On the other hand, while KO strains show PCR products with F2‐R2 primer pair, wt strains do not. RT‐qPCR was also performed to show loss of UHOR_05101 gene expression in KO background compared to wt. Error bars in RT‐qPCR results represent standard deviation of three biological replicates. Expression levels were normalized using U. hordei Ppi gene.
Fig. S4 A) Ustilago hordei filamentation test on charcoal plate. To opposite mating types of both wild type and effector deletion strains were grown in YEPS liquid medium till OD:0.8‐1.0. Subsequently, two mating type were mixed (1:1, v/v) to get OD:1.0 and 5 μl of the mixture was dropped on charcoal plate. Single mating type cultures were used as negative controls. Pictures were taken after 3 days incubation at room temperature. B) Growth rate of the U. hordei wild type and mutant strains in liquid YEPSlight. The OD600 was measured at the indicated time points. There was no significant difference between the U. hordei wild type and Δuhor_02413, Δuhor_02700, Δuhor_03756 and Δuhor_05101 mutant strains. C) Classical Ustilago hordei‐barley infection assay. Dehulled and surface sterilized barley seeds were inoculated with the U. hordei wild type and Δuhor_02413, Δuhor_02700, Δuhor_03756 and Δuhor_05101 mutant strains. Approximately 2‐3 months after inoculation, disease symptoms were scored at barley heading. This experiment was only performed one time.
Fig. S5 Biomass quantification of 14 Ustilago hordei effector mutants on barley. The virulence of the Ustilago hordei wild type and 14 effector mutants was performed by the quantification of fungal biomass at 6 dpi on barley leaf. The U. hordei Pep1 deletion mutant was used as a positive control for virulence reduction. The infected barley leaves at 6 day post‐inoculation (dpi) were harvested and analyzed via quantitative PCR by using genomic DNA and using the U. hordei Ppi1 genes as a standard. The fungal biomass was deduced from a standard curve. A oneway ANOVA was performed and followed by a Bonferroni test. Samples that showed significant differences compared to wild type are marked with asterisks (*, P< 0.001). Error bars represent the standard deviation of at least three biological repeats.
Fig. S6 Heterologous production of Ustilago hordei Uvi1‐4 in yeast cell and Nicotiana benthamiana. A) Western Blot (WB) analysis was performed with total proteins extract of ESM356 yeast strains transformed with pEZY45‐UHOR_02413‐HA, pEZY45‐UHOR_02700‐HA, pEZY45‐UHOR_03756‐HA and pEZY45‐ UHOR_05101‐HA to confirm the expression of these effectors. Empty pEZY15 transformed yeast cell was used as negative control. α‐HA antibody was used for WB. Expected protein sizes were indicated below figure. B) Western Blot (WB) analysis was performed with total proteins extract of AH109 yeast strains transformed with pGBKT7‐Myc‐UHOR_02413‐HA, pGBKT7‐Myc‐UHOR_02700‐HA and pGBKT7‐Myc‐UHOR_03756‐HA to confirm the expression of these effectors. Empty pGBKT7‐BDMyc transformed yeast cell was used as negative control. α‐HA antibody was used for WB. Expected protein sizes were indicated below figure. C) Western Blot (WB) analysis was performed to confirm expression of pK2GW7‐UHOR_02413‐HA, pK2GW7‐ UHOR_02700‐HA and pK2GW7‐UHOR_03756‐HA in Nicotiana benthamiana leaves transformed with Agrobacterium tumefaciens. αvHA antibody was used for WB. Expected protein sizes were indicated below figure. Red arrows indicate respective protein bands. D) Heterologous expression of pK2GW7‐GUSGFP, pK2GW7‐UHOR_02413‐GFP, pK2GW7‐UHOR_02700‐GFP, pK2GW7‐UHOR_03756‐GFP and pK2GW7‐UHOR_05101‐GFP in Nicotiana benthamiana leaves transformed with A. tumefaciens.
Fig. S7 Cell‐death suppression activity of four Ustilago hordei effectors. A) BAX1‐mediated cell death suppression assay. Cell death inducing pEZY45‐BAX1 and pEZY45‐UHOR_02413, pEZY45‐UHOR_02700 and pEZY45‐UHOR_03756 and pEZY45‐UHOR_05101 were co‐expressed under the control of a galactose inducible promoter in the ESM356.1 yeast strain. Empty pEZY45 and pEZY45‐Bcl‐XL were used as negative and positive controls for BAX1‐mediated cell death suppression, respectively. Pictures were taken after 4 days of incubation. The figure is representative of three biological replicates. B) INF1‐mediated cell death suppression assay. Phytophthora infestans cell death inducing INF1 (in pGR106‐FLAG) and pK2GW7‐UHOR_05101, pK2GW7‐UHOR_02413, pK2GW7‐UHOR_02700 and pK2GW7‐ UHOR_03756 were co‐infiltrated into 5‐6 week‐‐old Nicotiana benthamiana leaves via Agrobacterium‐mediated transformation. P. infestans Avr3AKI (pGR106‐FLAG) and Avr3aΔY (pGR106‐FLAG) was used as positive and negative control for INF1‐mediated cell death suppression, respectively. Pictures were taken at 4 dpi. The figure is representative of three biological replicates.
Table S1 Microarray analysis of the whole genome of Ustilago hordei. Blast2Go analysis of All genes, Secretome, Effectorome and Effectors for knock‐out (KO) microarray analyses are depicted in different excel sheets. Genes showing a two‐fold change in expression level were considered as differentially expressed (log2 fold change ≥1).
Table S2 Proteins with predicted NLS signal found in the Ustilago hordei effectorome.
Table S3 Constructs and oligonucleotides used in this study.