

Summary
Leptosphaeria maculans, the causal agent of stem canker disease, colonizes oilseed rape (Brassica napus) in two stages: a short and early colonization stage corresponding to cotyledon or leaf colonization, and a late colonization stage during which the fungus colonizes systemically and symptomlessly the plant during several months before stem canker appears. To date, the determinants of the late colonization stage are poorly understood; L. maculans may either successfully escape plant defences, leading to stem canker development, or the plant may develop an ‘adult‐stage’ resistance reducing canker incidence. To obtain an insight into these determinants, we performed an RNA‐sequencing (RNA‐seq) pilot project comparing fungal gene expression in infected cotyledons and in symptomless or necrotic stems. Despite the low fraction of fungal material in infected stems, sufficient fungal transcripts were detected and a large number of fungal genes were expressed, thus validating the feasibility of the approach. Our analysis showed that all avirulence genes previously identified are under‐expressed during stem colonization compared with cotyledon colonization. A validation RNA‐seq experiment was then performed to investigate the expression of candidate effector genes during systemic colonization. Three hundred and seven ‘late’ effector candidates, under‐expressed in the early colonization stage and over‐expressed in the infected stems, were identified. Finally, our analysis revealed a link between the regulation of expression of effectors and their genomic location: the ‘late’ effector candidates, putatively involved in systemic colonization, are located in gene‐rich genomic regions, whereas the ‘early’ effector genes, over‐expressed in the early colonization stage, are located in gene‐poor regions of the genome.
Keywords: Brassica napus, effector waves, isochores, Leptosphaeria maculans, RNA sequencing, transcriptomics
Introduction
The understanding of plant pathogens and their infection cycle is critical to the efficient control of plant diseases. However, this is a great challenge as phytopathogens, especially fungal pathogens, may have complex life cycles, different host plants or different feeding strategies. The high diversity of fungal life cycles and of the interactions of fungi with their host plants, but also with their environment, necessitates a large transcriptional plasticity and the ability to set up in time different unrelated pathogenic programmes.
During infection, plant pathogens produce an arsenal of molecules to favour the infection process and to successfully colonize the host. Among these, effectors are able to prevent the recognition by the host plant, to interfere with plant defence pathways or to modify the host cell physiology (Lo Presti et al., 2015; Wang et al., 2015). Secreted proteins, often small (<300 amino acids) and cysteine‐rich, secondary metabolites or small RNAs, have been identified to act as effectors (de Jonge et al., 2011; Lo Presti et al., 2015). During an infection cycle, distinct expression patterns of effectors have been observed, depending on the infection stage and the infection structure. In Colletotrichum higginsianum, specific sets of effectors are expressed in appressoria prior to penetration, in the subsequent biotrophic phase and in the final necrotrophic stage (Kleemann et al., 2012). Waves of effectors at different infection stages and in different tissues have also been observed in other fungi, such as Puccinia striiformis, Melampsora larici‐populina, Zymoseptoria tritici and Magnaporthe oryzae (Cantu et al., 2013; Dong et al., 2015; Hacquard et al., 2012; Mirzadi Gohari et al., 2015). In the oomycete Phytophthora sojae, the two observed waves of effectors have antagonistic effects: the first wave, expressed even before infection, is involved in cell death suppression, whereas the second wave triggers cell death (Wang et al., 2011).
Leptosphaeria maculans is a fungal pathogen of Brassicaceae, responsible for the most important disease of oilseed rape (Brassica napus): phoma stem canker disease, also termed blackleg. This fungus is spread across Europe, America and Australia (Fitt et al., 2006), causing over US$900 million losses per year (Fitt et al., 2008). Leptosphaeria maculans has an especially complex life and pathogenic cycle which makes it a relevant model to study the setting up of developmental programmes. Its life cycle encompasses a saprophytic stage, two biotrophic stages, including an endophytic life cycle in tissues, and two necrotrophic stages. Surviving on stem residues during its saprophytic stage, the fungus produces sexual spores (ascospores). The resulting spores that land on the aerial organs of plants germinate and the hyphae penetrate leaves and cotyledons through stomata. Once in the leaves, the fungus has a short biotrophic stage of 5–15 days during which it colonizes the apoplast of plant tissues (Fitt et al., 2006). Then, the fungus switches to necrotrophy and causes leaf spots (Rouxel and Balesdent, 2005). Following this primary leaf infection, L. maculans progresses in the stem tissues from the leaves to the crown during a long endophytic systemic colonization (Hammond et al., 1985; Huang et al., 2014; Rouxel and Balesdent, 2005). The colonization is completely symptomless and may last up to 9 months in winter oilseed rape (West et al., 2001). Following this stage, L. maculans shifts to necrotrophy again, leading to the damaging stem canker responsible for plant lodging. To date, we have virtually no information on the L. maculans and B. napus determinants operating during this exceptionally long symptomless colonization of the stem.
The L. maculans genome is structured into equilibrated and low GC‐content blocks, called GC‐isochores (51.0% GC; average size, 70.4 kb) and AT‐isochores (33.9% GC; average size, 38.6 kb), respectively (Rouxel et al., 2011). GC‐isochores are rich in housekeeping genes and poor in transposable elements, whereas AT‐isochores have heterochromatin characteristics, being gene poor, enriched in transposable elements and showing a low rate of recombination. AT‐isochores were found to be enriched in genes encoding effector candidates, i.e. genes coding for small secreted proteins and with low expression in vitro and high expression in planta at 7 and 14 days post‐inoculation (dpi) in cotyledons (Rouxel et al., 2011; Soyer et al., 2014). The expression of these effector candidates is under a chromatin‐based control: the silencing of two genes involved in heterochromatin assembly and maintenance, LmHP1 and LmDIM5, leads to de‐repression in vitro of effectors located in AT‐isochores (Soyer et al., 2014). It has been hypothesized that, during leaf infection, AT‐isochores switch from a heterochromatin status to a euchromatin status, enabling a concerted expression of effector genes present in AT‐isochores.
The seven cloned avirulence genes of L. maculans (AvrLm1, AvrLm2, AvrLm3, AvrLm4‐7, AvrLmJ1, AvrLm6 and AvrLm11) (Balesdent et al., 2013; Fudal et al., 2007; Ghanbarnia et al., 2014; Gout et al., 2006; Parlange et al., 2009; Plissonneau et al., 2015; Van de Wouw et al., 2014) and the candidate effector gene LmCys2 (Fudal et al., 2007) are part of this set of genes strongly expressed in the first stages of plant infection and located in AT‐isochores. AvrLm1, AvrLm6 and AvrLm4‐7 show reduced expression at the stem necrosis stage (Fudal et al., 2007; Parlange et al., 2009). In addition to the effector candidate genes located in AT‐isochores, numerous effector candidates (529) are predicted to be located in GC‐isochores and are not differentially expressed in the early infection stage, questioning their effector function (Rouxel et al., 2011). These data also question which effectors, if any, are produced during the long endophytic life and stem canker development and what can be their regulation. In particular, we postulate that effectors are also necessary for the endophytic stage, allowing the fungus to remain unnoticed during many months within plant tissues.
With both L. maculans (Rouxel et al., 2011) and B. napus (Chalhoub et al., 2014) genomes being available, the objective of our study was to test this hypothesis by deciphering the fungal molecular determinants mobilized during systemic colonization and by identifying candidate effectors specific to this infection stage. For this purpose, we first performed an RNA sequencing (RNA‐seq) pilot project to characterize the expression profile of L. maculans genes during interaction with B. napus at the infection stages corresponding to endophytic stem colonization and the development of stem necrosis. Following the validation of this approach, we explored, through a validation RNA‐seq experiment and quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR), the genes over‐expressed during the systemic colonization stage, mainly focusing on effector candidates.
Results
A pilot RNA‐seq project demonstrates that fungal transcripts can be detected in symptomless infected stems
The first question to be raised was whether or not we would be able to detect sufficient fungal transcripts in infected stems to conduct an RNA‐seq analysis. For this purpose, we performed an RNA‐seq pilot project at two key steps of the infection cycle: the early and late colonization stages (Fig. 1). The early colonization stage corresponded to primary infection following inoculation of cotyledons with three isolates (NzT4, G06‐E107 and the reference isolate JN3). Samples, termed infected cotyledons (ICs), were collected at 7 dpi for RNA extraction. The late colonization stage corresponded to stems submitted to systemic colonization following petiole inoculation with JN3. Stem samples were collected at 24 dpi. Not knowing whether we would be able to detect fungal transcript in symptomless stem tissues, we extracted RNA from three different types of infected stem tissue: infected but symptomless stem tissue (S), necrotic stem tissue (N) and tissues located in advance of necrosis, below necrosis stem tissue (BN) (Fig. 1).
Figure 1.
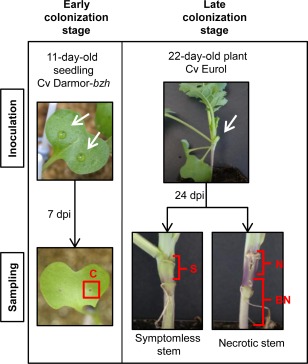
Samples used for the pilot RNA‐sequencing (RNA‐seq) project. Inoculations were performed on wounded cotyledons to analyse the transcriptome of the early colonization stage. The tissues of infected cotyledons (C) around the wounds were sampled at 7 days post‐inoculation (dpi). Samples corresponding to the late infection stage were obtained as follows: inoculations were performed on the wounded petiole of the second leaf and three different tissues were sampled: (i) 1 cm of stem starting from the inoculated petiole insertion for symptomless stem tissue (S); (ii) the necrotic stem tissue (N) for necrotic stems; and (iii) the tissue in advance of necrosis, the below necrosis stem tissue (BN), in which visible symptoms were not expressed. Controls consisted of tissue samples from plants inoculated with water. The white arrows indicate inoculation sites.
RNA‐seq produced between 64 and 189 million reads per library (Table 1), more than 83% of which mapped to the B. napus genome. Whatever the infection conditions and tissues, almost one‐half of the 101 040 predicted genes of B. napus were expressed (Table 1). In ICs, between 5% and 15% of the reads mapped to the L. maculans genome, with a similar number of genes (approximately 9000 genes) expressed by each of the three isolates. In necrotic stem tissues, 8.13% of the reads mapped to the L. maculans genome (Table 1). The proportion of reads corresponding to fungal genes was much lower in symptomless tissues of stems, with only 0.66% and 0.86% of reads mapping to L. maculans in S stems and BN stem tissues, respectively. Despite this low percentage of fungal transcripts, more than 5000 fungal genes were found to be expressed in these tissues (39.6% of the 12 613 predicted genes).
Table 1.
Mapping characteristics of RNA‐sequencing (RNA‐seq) reads to the reference genomes of Brassica napus and Leptosphaeria maculans in the pilot RNA‐seq project.
| Condition | Total no. of reads | Reads mapped on the B. napus genome (no./%) | Reads mapped on the L. maculans genome (no./%) | No. of B. napus expressed genes* | No. of L. maculans expressed genes* | ||
|---|---|---|---|---|---|---|---|
| Non‐inoculated cotyledons | 8.40E+07 | 8.25E+07 | (98%) | 6.72E+03 | (0.01%) | 54 825 | 9 |
| Non‐inoculated stems | 8.15E+07 | 7.97E+07 | (98%) | 8.51E+03 | (0.01%) | 58 655 | 20 |
| Inoculated cotyledons with NzT4 isolate | 8.05E+07 | 6.75E+07 | (84%) | 1.16E+07 | (14.37%) | 54 598 | 9175 |
| Inoculated cotyledons with G06‐E107 isolate | 1.07E+08 | 8.85E+07 | (83%) | 1.62E+07 | (15.18%) | 57 303 | 9395 |
| Inoculated cotyledons with JN3 isolate | 1.89E+08 | 1.76E+08 | (93%) | 1.03E+07 | (5.42%) | 59 849 | 9185 |
| Symptomless ST† | 9.08E+07 | 8.78E+07 | (97%) | 5.98E+05 | (0.66%) | 59 688 | 5875 |
| Below necrosis ST† | 6.42E+07 | 6.22E+07 | (97%) | 5.55E+05 | (0.86%) | 57 943 | 5611 |
| Necrotic ST† | 7.72E+07 | 6.93E+07 | (90%) | 6.28E+06 | (8.13%) | 57 483 | 9211 |
*Genes were considered to be expressed when more than 10 unique gene reads were mapped on a reference genome.
†ST, stem tissue inoculated with JN3 isolate at 24 days post‐inoculation.
The expression levels of fungal genes across the different colonization stages were visualized in the form of a heat map (Fig. 2). When focusing on genes showing differential patterns in the different infection conditions, two main categories of expression pattern were discriminated (Fig. 2). The first pool encompassed genes over‐expressed in cotyledons and repressed in all stem tissues. The second pool of genes contained genes with no or low expression in ICs, but with high expression in stem tissues. Most (60%) of the genes over‐expressed in stems were over‐expressed in all three different infected stem tissues (Fig. 2). Among the genes differentially expressed in the different conditions, we also searched for effector candidates, i.e. secreted proteins, and identified 66 ‘early’ (including the seven cloned avirulence genes, Fig. 2B) and 175 ‘late’ effector candidates produced in the early and late colonization stages, respectively.
Figure 2.
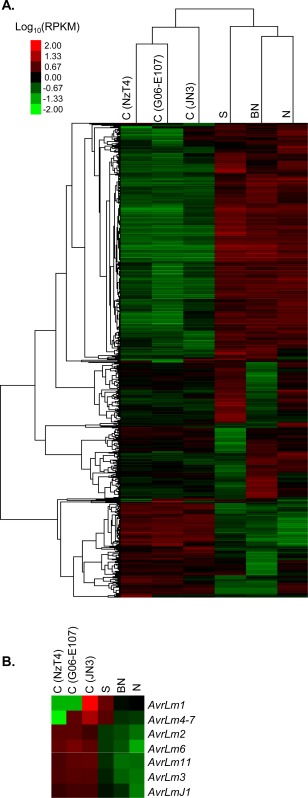
Heat map of Leptosphaeria maculans genes differentially expressed between early and late colonization stages in the pilot RNA‐sequencing (RNA‐seq) project. (A) Expression values were measured in infected cotyledons (IC, denoted as ‘C’) inoculated with three different L. maculans isolates (NzT4, G06‐E107 or JN3) and stems inoculated with JN3. Following stem inoculation, three types of tissue were collected: symptomless stem tissue (S), below necrosis stem tissue (BN) and necrotic stem tissue (N). (B) Focus on known avirulence genes of L. maculans. Note that AvrLm1 and AvrLm4‐7 are absent or inactivated in the genomes of the isolates NzT4 and G06‐E107. The differential expression patterns were based on the log10(RPKM) measured across all six conditions. The heat map was obtained with GeneCluster3.0 and visualized with TreeView. RPKM, reads per kilobase per million mapped reads.
The RNA‐seq approach was thus feasible and informative, even in the symptomless stem tissues. Furthermore, different effector candidates were expressed during cotyledon and stem colonization.
Different waves of effector expression take place in early and late colonization stages
To validate these data, we conducted an independent RNA‐seq project focused on the symptomless stem colonization. In this validation RNA‐seq project, three conditions were compared: symptomless ICs (at 7 dpi), early symptomless stem colonization (at 14 dpi) and an in vitro growth condition. Two independent biological replicates were sequenced per condition. For this project, we chose the closely related sister isolate of JN3, JN2 (also termed v23.1.2), showing a consistently higher level of aggressiveness on oilseed rape compared with JN3 (M. H. Balesdent, unpublished data). Most of the L. maculans predicted genes (approximately 10 800) were expressed in axenic culture (Table 2). The numbers of reads in inoculated cotyledons and stems were comparable with those of the pilot project. The percentages of reads mapping to the L. maculans genome in inoculated symptomless stems were also very low (0.52% and 0.31%), but more than 3000 fungal genes were expressed. This was slightly lower than the RNA‐seq pilot project, as the validation RNA‐seq analysis was performed at an earlier stem colonization stage (14 dpi) compared with the pilot project (24 dpi) to exclude genes involved in the development of stem necrosis.
Table 2.
Mapping characteristics of RNA‐sequencing (RNA‐seq) reads to the reference genomes of Brassica napus and Leptosphaeria maculans in the validation RNA‐seq experiment.
| Condition* | Replicate | Total no. of reads | Reads mapped on the B. napus genome (no./%) | Reads mapped on the L. maculans genome (no./%) | No. of B. napus expressed genes† | No. of L. maculans expressed genes† | ||
|---|---|---|---|---|---|---|---|---|
| Mycelium in vitro | 1 | 4.11E+07 | 4.29E+03 | (0.01%) | 4.00E+07 | (97.35%) | 4 | 10 775 |
| Mycelium in vitro | 2 | 4.57E+07 | 4.74E+03 | (0.01%) | 4.45E+07 | (97.36%) | 3 | 10 944 |
| Non‐inoculated cotyledons | 1 | 1.40E+08 | 1.27E+08 | (90.98%) | 1.52E+03 | (0.00%) | 52 288 | 0 |
| Non‐inoculated cotyledons | 2 | 1.77E+08 | 1.62E+08 | (91.31%) | 1.78E+04 | (0.01%) | 54 441 | 52 |
| Inoculated cotyledons | 1 | 1.56E+08 | 1.30E+08 | (83.36%) | 1.28E+07 | (8.16%) | 54 391 | 9315 |
| Inoculated cotyledons | 2 | 1.77E+08 | 1.38E+08 | (78.29%) | 2.41E+07 | (13.67%) | 54 591 | 9683 |
| Non‐inoculated stems | 1 | 1.88E+08 | 1.41E+08 | (75.23%) | 2.08E+03 | (0.00%) | 57 982 | 1 |
| Non‐inoculated stems | 2 | 1.38E+08 | 1.26E+08 | (91.68%) | 1.32E+03 | (0.00%) | 57 177 | 0 |
| Inoculated stems | 1 | 1.69E+08 | 1.55E+08 | (91.85%) | 8.72E+05 | (0.52%) | 58 997 | 5209 |
| Inoculated stems | 2 | 1.35E+08 | 1.23E+08 | (91.47%) | 4.21E+05 | (0.31%) | 58 259 | 3059 |
*Isolate JN2 was used in both in planta and in vitro conditions. In planta conditions correspond to cotyledons and stem tissue at 7 and 14 days post‐inoculation, respectively.
†Genes were considered to be expressed when more than 10 unique gene reads were mapped on a reference genome.
The heat map (Fig. 3) showed more complex patterns of gene expression because of the inclusion of the axenic growth condition, with a high reproducibility in gene expression patterns between the two biological replicates of each tested condition. Four main patterns were observed: the first encompassed genes specifically over‐expressed during the early colonization stages, i.e. over‐expressed in cotyledons compared with stem and in vitro conditions (pattern B in Fig. 3). This group of 725 differentially expressed genes (P < 0.05) comprised all known avirulence genes. In the pilot RNA‐seq project, known avirulence genes were not only under‐expressed in symptomless stems, but also in necrotic stem tissues and BN tissues (Fig. 2B), thus confirming that L. maculans avirulence genes are over‐expressed only at the onset of cotyledon colonization, but under‐expressed during systemic colonization of the stem and during the stem necrosis stage. The second pattern encompassed genes over‐expressed during the late colonization stages, i.e. over‐expressed in stems compared with cotyledon and in vitro conditions (pattern C in Fig. 3); 1050 genes were found in this group (P < 0.05). Despite the use of different isolates in the two projects, the expression levels of the ‘early’ and ‘late’ genes observed in this validation RNA‐seq project correlated at 79.5% with those observed in the pilot RNA‐seq project (P < 2.2E‐16) (Fig. 4). In addition to these two patterns, we also identified genes specifically under‐expressed in stems (pattern A in Fig. 3) or cotyledons (pattern D in Fig. 3) compared with axenic growth. Being especially focused on the identification of effectors involved in late oilseed rape colonization, the genes whose expression is repressed during plant infection were not investigated further here. The top 50 most expressed genes in early and late colonization stages are listed in Table S2 (see Supporting Information).
Figure 3.
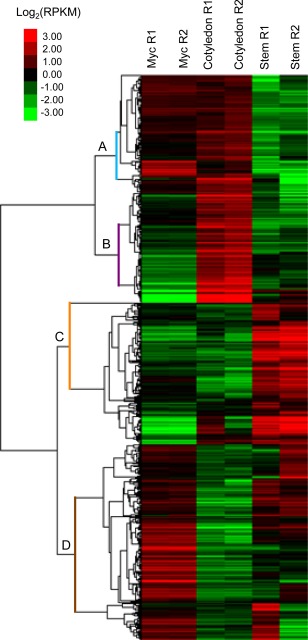
Heat map of Leptosphaeria maculans genes differentially expressed (P < 0.05) in vitro (Myc) and during early (Cotyledon) and late (Stem) colonization in the second (validation) RNA‐sequencing (RNA‐seq) experiment. The differential expression patterns were based on the log2(RPKM) measured across all six conditions. The heat map was obtained with GeneCluster3.0 and visualized with TreeView. Two independent biological replicates (R1 and R2) are displayed for each condition. RPKM, reads per kilobase per million mapped reads.
Figure 4.
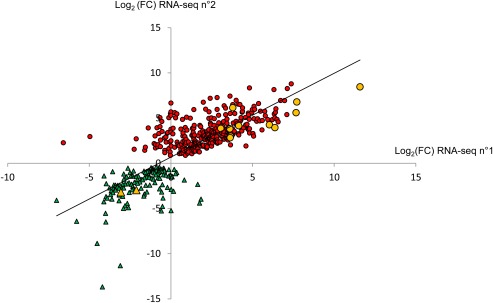
Comparison of expression values of ‘early’ and ‘late’ genes identified in the pilot RNA‐sequencing (RNA‐seq n°1) and in the validation RNA‐sequencing (RNA‐seq n°2) project. The fold change (FC) corresponds to the ratio between the expression value in symptomless stem tissues and that in infected cotyledons in each of the RNA‐seq experiments. The circles and triangles represent the genes over‐expressed in stems and under‐expressed in cotyledons, and the genes over‐expressed in cotyledons and under‐expressed in stems, respectively. The yellow symbols correspond to genes selected for quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) validation.
Effector candidates were then searched for in these pools of ‘early’ and ‘late’ genes. Compared with previously published analyses (Rouxel et al., 2011), the criteria for the selection of effector gene candidates were slightly modified. Here, the effector genes were selected based on their ability to be secreted, i.e. with a predicted signal peptide, without glycosylphosphatidylinositol (GPI) anchor or additional transmembrane domain, but no additional filter according to the protein size or cysteine content was applied. According to these criteria, the genome of L. maculans contained 1741 genes (13.8%) predicted to be secreted. In the set of 725 genes over‐expressed in cotyledons and under‐expressed in stem tissues, we identified 139 early effector candidates (19%). Of the 1050 genes putatively involved in the late colonization stages, 307 secreted proteins (29%) were considered as late effector candidates. The transcriptome was thus strongly enriched in genes encoding for secreted proteins during both the early and late colonization stages of oilseed rape (P < 1.28E‐04). Among the early and late effector candidates identified in the RNA‐seq pilot project, 79% and 65%, respectively, were common to this validation RNA‐seq experiment.
Characterization of the early and late colonization stages
Using Blast2GO, we searched for gene ontologies (GO) characteristic of the early and late colonization stages. Only 23% of ‘early’ genes and 10% of ‘late’ genes had a predicted GO with a significant enrichment (P < 0.05) compared with the whole genome (Fig. 5). Comparison of the two stages, however, indicated the set‐up of distinct strategies during the early and late colonization stages. In early colonization, we observed significant enrichments in translation processes and trafficking towards the membrane (Fig. 5A). Twenty‐one of the top 50 genes over‐expressed in early colonization were associated with translation processes (Table S2). Twenty‐five hypothetical or predicted genes were among the top 50 genes, as well as five avirulence genes, AvrLm3, AvrLm4‐7, AvrLmJ1, AvrLm6 and AvrLm11. Similar to early expressed genes, around 50% of the top 50 most highly over‐expressed genes in late colonization were classified as hypothetical or predicted genes. However, the GO enrichment analysis showed a significant enrichment in oxidoreductase and hydrolase activities during late colonization (Fig. 5B). Secreted proteins involved in the extracellular region were also significantly enriched during late colonization. Twenty‐nine of the top 50 genes were associated with these GO categories (Table S2).
Figure 5.
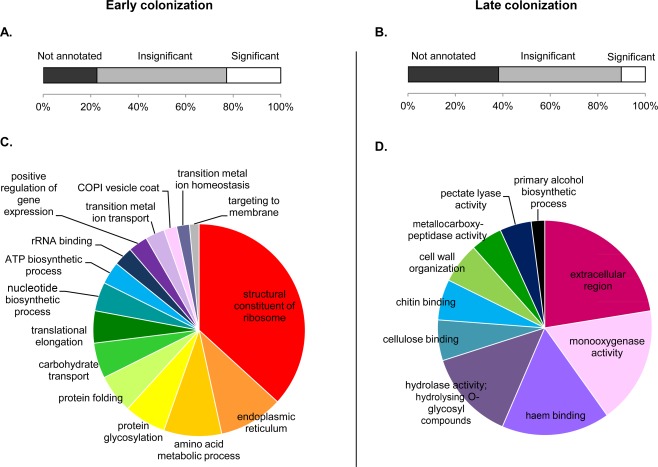
Gene ontology (GO) enrichment analysis of the Leptosphaeria maculans genes over‐expressed in early (cotyledon) (A, C) and late (stem) (B, D) colonization stages compared with the whole genome. Genes were separated into three classes (A, B): genes with no annotated GO (in black), those with insignificant enrichment GO compared with the whole genome (in grey) and those with significant enrichment (in white). Only significantly enriched GO categories are represented [false discovery rate (FDR) < 0.05] (C, D). This analysis was performed with Blast2GO.
Despite the absence of filtering based on protein size and cysteine content, the secreted proteins expressed during early or late colonization stages shared common characteristics with previously described early effectors expressed during cotyledon colonization. (i) Their size was significantly smaller than those of all predicted proteins of L. maculans: the mean sizes for early and late effector candidates were 372 amino acids (P = 5.5E‐06) and 384 amino acids (P = 3.1E‐10), respectively, vs. 479 amino acids for all predicted proteins of L. maculans. (ii) They were also richer in cysteines: 2.4% (P = 3.9E‐07) and 2.0% (P = 3.3E‐03) for early and late candidates, respectively, vs. 1.7% in the whole proteome. (iii) There was no predicted function for 61% and 50% of the early and late effector candidates, respectively; 19% early and 17% late candidates displayed no significant homology (E‐value < 1E‐10) with other species. In the top 50 genes over‐expressed during the early colonization stage, 56% of the genes without predicted function showed no significant homology with other organisms, whereas, in the late colonization stage, only 29% of the genes without predicted function showed no significant homology with other organisms (Table S2).
qRT‐PCR validation of late effector expression patterns
The RNA‐seq experiments enabled us to identify candidate effector genes specifically over‐expressed during stem colonization (Fig. 4). We validated the expression profiles for 11 selected late effectors by qRT‐PCR. These genes were chosen from the most highly expressed genes during stem colonization (Fig. 4, Table S2). Two genes encoding for avirulence effectors involved in early colonization (AvrLm3 and AvrLm4‐7) and two genes whose expression varied little in the RNA‐seq experiment (ribosomal protein S30A and H+/ATPase) were also included as controls. The expression of ribosomal protein S30A and H+/ATPase genes did not differ by more than factors of 7 and 2, respectively, across the different infected tissues (Fig. 6). The low level of expression in cotyledons and the over‐expression in infected stems at 24 dpi was confirmed for all the 11 late effector candidates tested. Conversely, the known avirulence genes, AvrLm3 and AvrLm4‐7, were found to be highly expressed in cotyledons and expressed at a low level in stems (Fig. 6). RNA‐seq data were thus validated by qRT‐PCR experiments for the 15 genes investigated.
Figure 6.
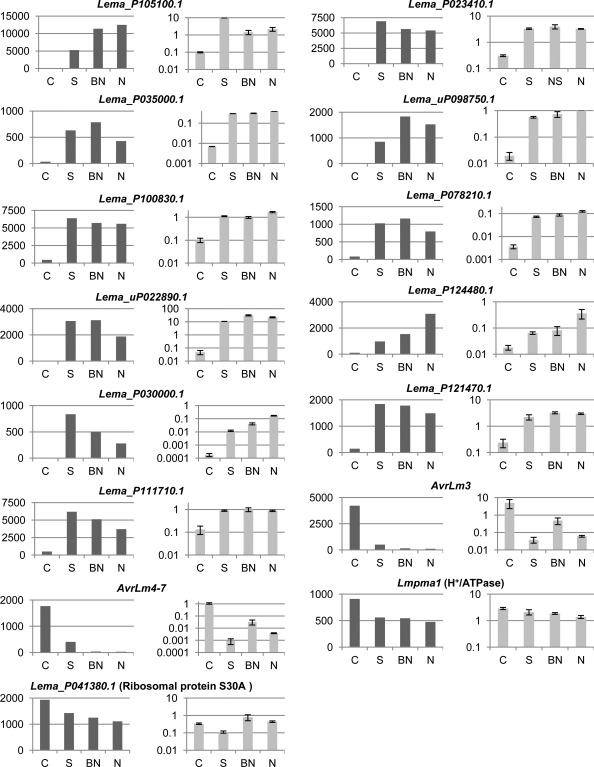
Validation of the RNA‐sequencing (RNA‐seq) experiment by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) for 15 genes, including 11 late effector candidates. The expression level in RNA‐seq data is represented by the RPKM (reads per kilobase per million mapped reads) value indicated in dark grey. In qRT‐PCR experiments, the mean expression normalized with actin is represented in light grey. The expression levels were measured in infected cotyledons (IC, denoted as ‘C’), in infected symptomless stem (S), in below necrosis stem tissue (BN) and in necrotic tissue (N) of infected stem. Error bars represent the standard error for two biological and three technical replicates.
Late effector and early effector candidates do not share the same genomic location and regulation of expression
We analysed the genomic location of the 307 late effector candidate genes compared with the 139 early effector candidate genes (Table 3). As described previously (Rouxel et al., 2011), AT‐isochores were significantly enriched in early effector candidate genes (χ2 test, P = 9.6E‐11) (Table 3). In contrast, nearly all late effector candidate genes were located in GC‐isochores and there was no enrichment of late effector candidates in AT‐isochores. In contrast, only three late effector candidate genes were found in AT‐isochores, and they were not the most highly expressed compared with other late effector candidates [mean RPKM (reads per kilobase per million mapped reads) in infected stems <200]. In addition, late and early candidate effector genes displayed the same GC content as their genomic environment: late effectors located in GC‐isochores were GC‐equilibrated (>51% GC), whereas the early effectors located in AT‐isochores were impoverished in GC (<45% GC) (Rouxel et al., 2011).
Table 3.
Genomic location of Leptosphaeria maculans genes over‐expressed at early (cotyledon infection) or late (stem infection) colonization stages.
| Secreted proteins* | Non‐secreted proteins | |||||||
|---|---|---|---|---|---|---|---|---|
| Total number | GC‐isochores† (%) | Borders‡ (%) | AT‐isochores§ (%) | Total number | GC‐isochores† (%) | Borders‡ (%) | AT‐isochores§ (%) | |
| All predicted genes | 1741 | 90.1 | 6.4 | 3.6 | 10 944 | 95.9 | 3.3 | 0.8 |
| Genes over‐expressed during early colonization | 139 | 77.0 | 7.9 | 15.1 | 586 | 94.7 | 3.6 | 1.7 |
| Genes over‐expressed during late colonization | 307 | 92.8 | 6.2 | 1.0 | 743 | 96.2 | 3.4 | 0.4 |
*Proteins with no predicted transmembrane signal, no glycosylphosphatidylinositol (GPI) anchor and with a secretion signal were considered as secreted proteins.
†GC isochores: genomic blocks with equilibrated GC content (Rouxel et al., 2011).
‡Borders: transition regions (859 ± 385 bp) between AT‐ and GC‐isochores.
§AT‐isochores: genomic blocks with low GC content.
To further explore the impact of the different genomic locations of early and late effector genes, we investigated the impact of chromatin structure modification on the expression of effector candidates. AT‐isochores are heterochromatin‐like regions, whereas GC‐isochores are postulated to mostly correspond to euchromatin. Leptosphaeria maculans transformants in which LmHP1 and LmDIM5 expression was silenced have been generated previously, and gene expression in the transformants compared with that in the wild‐type was analysed with oligo‐arrays (Soyer et al., 2014). From this data, we evaluated the impact of the silencing of LmHP1 and LmDIM5 on the expression of early and late effector candidates (Table 4). In axenic culture, the silencing of LmHP1 and LmDIM5 had no impact on the expression of most of the late effector candidates (Table 4). In contrast, 38% and 19% of the early effector genes localized in the AT‐isochore were de‐repressed in siLmHP1 and siLmDIM5, respectively.
Table 4.
Percentage of Leptosphaeria maculans effector candidates up‐regulated or down‐regulated in vitro in a silenced‐LmHP1 or silenced‐LmDIM5 background, according to their genomic location.
| Effector category | Genomic location | Total no. of genes | Over‐expressed in siLmHP1‡ (%) | Repressed in siLmHP1‡ (%) | Over‐expressed in siLmDIM5‡ (%) | Repressed in siLmDIM5‡ (%) |
|---|---|---|---|---|---|---|
| Early effector candidates* | GC‐isochores§ | 107 | 4 | 2 | 3 | 6 |
| Borders¶ | 11 | 18 | 9 | 9 | 27 | |
| AT‐isochores** | 21 | 38 | 5 | 14 | 5 | |
| Late effector candidates† | GC‐isochores§ | 285 | 2 | 5 | 4 | 7 |
| Borders¶ | 19 | 5 | 21 | 0 | 32 | |
| AT‐isochores** | 3 | 0 | 0 | 0 | 0 |
*Early effectors refer to effector genes showing a peak of expression during cotyledon infection (7 days post inoculation) and no or low expression during stem colonization and in vitro.
†Late effectors refer to effector genes showing no or low expression during cotyledon infection and in vitro, and peaks of expression during stem colonization.
‡The expression level is measured in in vitro conditions and compared with that of the wild‐type isolate. Both LmHP1 and LmDIM5 silencing have been shown previously to impact on the heterochromatic structure of AT‐isochores in L. maculans (Soyer et al., 2014).
§GC‐isochores, genomic blocks with equilibrated GC content (Rouxel et al., 2011).
¶Border, transition regions (859 ± 385 bp) between AT‐ and GC‐isochores.
**AT‐isochores, genomic blocks with low GC content (Rouxel et al., 2011).
Discussion
Transcriptomic analyses of microorganism–plant interactions are usually performed on infected leaves (Garnica et al., 2013; Hacquard et al., 2012, 2013; de Jonge et al., 2012; Kawahara et al., 2012; Kleemann et al., 2012; Mirzadi Gohari et al., 2015), root knobs (Haegeman et al., 2013) or nodules (Boscari et al., 2013). In such tissues, transcripts of the microorganism are usually sufficient for detection and analysis. However, in the infected stem and, in particular, in the symptomless stem of oilseed rape plants, the host biomass is much more important than that of L. maculans and we were unsure whether or not it would be possible to detect significant levels of fungal transcripts. In other pathosystems, the difficulty of obtaining sufficient pathogen reads has been overcome through the use of different strategies. For example, transcriptomic analyses of Candida albicans, a pathogen of mammals, were achieved with specific amplification of fungal reads after whole RNA extraction (Thewes et al., 2007), or additional extraction of fungal RNAs from infected hosts (Amorim‐Vaz et al., 2015). During Arabidopsis thaliana infection by Dickeya dadantii, transcriptomic analyses required the purification of bacterial cells from infected leaf tissues (Chapelle et al., 2015). In the pilot RNA‐seq experiment reported here, we managed to detect the expression of up to 47% of L. maculans predicted genes during stages of tissue colonization, even when only 0.66% of reads corresponded to the fungus; no specific enrichment strategy had to be applied to detect more fungal transcripts. The validation RNA‐seq experiment performed at an even earlier stage of systemic colonization (14 dpi) also confirmed that this approach is achievable and reliable in infected plant tissues, such as stems, even without visible symptoms. Last, the sensitivity and accuracy of RNA‐seq analysis were confirmed by qRT‐PCR analysis for 15 selected genes. This opens up the possibility for a better understanding of the determinants of systemic colonization of oilseed rape by L. maculans and of the molecular dialogue taking place between the plant and the fungus during the lengthy colonization of plant tissues.
Without being exhaustive, as it only focused on two discrete stages of plant colonization, our analysis identified one main general feature of the L. maculans pathogenic cycle: early effectors (some of which act as avirulence determinants) are expressed in the early stage of cotyledon infection, whereas different candidate effectors, with structural features similar to those of early effectors, are specifically produced during stem colonization and show kinetics of expression, genomic location and mode of regulation distinct from that of early effectors. We thus identified at least two main waves of effector expression in early colonization and during systemic colonization with novel putative effectors for functional studies. In addition to these two waves, the RNA‐seq pilot project also hinted at the existence of late effector candidates specifically expressed in necrotic stems or in advance of necrotic stem tissues. Of the 1739 predicted secreted proteins of the L. maculans genome, some may also be involved in particular infection stages not studied here, such as the progression of the fungus in the petiole. We also identified genes specifically under‐expressed during either cotyledon or stem colonization compared with axenic growth. This indicates that there is now a need for more extensive RNA‐seq analysis of the pathogen life cycle to obtain a more detailed knowledge of the genes mobilized throughout the L. maculans life cycle and their regulation.
One of the particularities of the L. maculans life cycle is the length of its systemic colonization during which no symptoms are visible and which can last for several months. In other hemibiotrophic or biotrophic fungal pathogens of annual crops, such as Z. tritici, Cladosporium fulvum, Colletotrichum spp. or M. oryzae, the asymptomatic biotrophic phase does not exceed 2 weeks (McDonald et al., 2015; Münch et al., 2008; Wilson and Talbot, 2009; de Wit et al., 2012). In other examples, Moniliophthora roreri and Moniliophthora perniciosa, the biotrophic stage can last 3–6 weeks after infection, during which the fungi manage to avoid plant defences (Meinhardt et al., 2014). The systemic colonization of L. maculans is one of the longest observed in fungal pathogens of annual crops, and resembles the lifestyle of some endophytes, such as Epichloë species. The RNA‐seq approach allowed us to identify 307 candidate effectors specific to the late colonization stages, expressed during stem infection. These effectors, like early effectors (Rouxel et al., 2011), are small cysteine‐rich secreted proteins, although no filter based on size or cysteine content was applied to identify them. Their expression pattern is clearly different from that of early effectors. The first waves of effector expression during the early colonization stages have been described by Haddadi et al. (2015) and Lowe et al. (2014). Our data are consistent with these findings, as approximately 70% of the genes or effectors found to be over‐expressed in cotyledons in these studies were also found here to be over‐expressed during cotyledon colonization (Fig. S1, see Supporting Information). The remaining 30% of genes not found in common could be expressed at one of the other stages of cotyledon colonization studied by Haddadi et al. (2015) or be isolate specific: for example, isolate IBCN18 used by Lowe et al. (2014) differs from isolate JN2 by the presence/absence of at least six of the currently known avirulence genes (Balesdent et al., 2002, 2005).
Comparing the few genes with GO annotation, we identified distinct GO enrichments between cotyledon and stem infection. In the early colonization stages, L. maculans had a high transcriptional activity related to trafficking towards membranes, indicating strong interactions with its host. Increased expression of genes involved in membrane trafficking at very early stages of plant infection have been described in other models, such as M. oryzae (Oh et al., 2008). We can speculate that, after germination on cotyledons, the fungus redirects transcriptional reprogramming towards the expression of the secreted proteins necessary for the first steps of pathogenicity. Lowe et al. (2014) also identified three genes encoding cytochrome P450 among the 20 most up‐regulated genes during early cotyledon colonization, which were not found in our study. During systemic colonization, we observed GO enrichments corresponding to plant cell wall degradation or modification. As also observed by Lowe et al. (2014), the enrichment in the expression of CAZy genes is not found in the early colonization step and could be specific to the systemic colonization stage. It is likely to allow the fungus to take up nutrients or to facilitate progression into the stem. We also observed GO enrichment corresponding to secreted proteins and chitin binding, which probably contribute to the evasion of plant defence responses (de Jonge et al., 2011). The oxido‐reduction GO group was also enriched during systemic colonization and may contribute to avoid the accumulation of reactive oxygen species produced by the plant, as has been suggested during symptomless leaf colonization of wheat by Z. tritici (Yang et al., 2015).
The duration of B. napus colonization by L. maculans, from cotyledon colonization to the appearance of necrosis in the stem, suggests a tight regulation of effector expression. One main result of our RNA‐seq analysis is that effector genes involved in early cotyledon colonization and in symptomless stem colonization do not share the same genomic compartment. The AT‐isochores act as a niche for L. maculans early effector genes expressed in cotyledons at 7 and 14 dpi (Haddadi et al., 2015, Rouxel et al., 2011; this study), whereas late effectors expressed during symptomless stem colonization are mainly located in GC‐isochores. Soyer et al. (2014) hypothesized that the environmental shift induced on leaf infection causes, through an as yet unknown signalling mechanism, a modification in the epigenetic status of AT‐isochores towards euchromatin status, thus enabling the concerted expression of the early effector genes located in AT‐isochores. Contrary to its effect on early effector gene expression, the silencing of HP1 or DIM5, two genes involved in heterochromatin maintenance, has no effect on the de‐repression of late effector candidate genes in vitro, which is consistent with their location in GC‐isochores. The specific location of late effectors in GC‐isochores is thus also consistent with their absence of contribution to the primary wave of effector gene expression. However, it also suggests (an)other regulatory system(s) to control the expression of late effectors, several weeks or months after primary infection.
The avirulence genes AvrLm1, AvrLm6 and AvrLm4‐7 are over‐expressed in the early infection stage and under‐expressed during the expression of stem necrosis (Fudal et al., 2007; Parlange et al., 2009) and during systemic colonization (this study). The same expression pattern is found here for all other cloned avirulence genes. With the identification of new effector genes probably involved in systemic colonization, we hypothesize that major resistance genes may exist and may be involved in the recognition of the pathogen and subsequent effector‐triggered immunity (ETI) in the late colonization stages, without interfering with the first steps of fungal colonization and with leaf spot development. The next hypothesis to address will be as follows: that adult resistance may not be based only on the quantitative effect of several plant genes, but may also involve resistance genes operating according to the gene‐for‐gene model at a later stage of infection. If they exist, they are putative targets of interest for a better understanding of adult‐stage resistance, the identification of novel resistance genes and the initiation of novel breeding strategies based on marker development or cloning of these new R genes.
Experimental procedures
Fungal and plant materials
RNA‐seq analyses of infected plants were performed using mostly the reference cultivar Darmor‐bzh (Chalhoub et al., 2014). In addition, for the RNA‐seq pilot project, the cultivar Eurol was used for late colonization conditions.
In the RNA‐seq pilot project, three different strains were used: G06‐E107, NzT4 and JN3. In the validation RNA‐seq experiment, the isolate JN2 was used. The isolate G06‐E107 is a field isolate obtained from leaf lesions in 2006 on cv. Exagone (Daverdin et al., 2012). The isolate NzT4 is a field isolate originating from New Zealand and obtained from a B. napus rapifera cultivar (Balesdent et al., 2005). The sequenced isolate JN3 (v23.1.3) (Rouxel et al., 2011) and the isolate JN2 (v23.1.2) are sister isolates from in vitro crosses between European field isolates (Balesdent et al., 2001). All four isolates are virulent towards Darmor‐bzh and Eurol. Fungal cultures were maintained, and sporulating cultures and conidia production were obtained, as described previously (Ansan‐Melayah et al., 1995). For transcriptomic analysis on axenic cultures of JN2, minimal medium MMII was inoculated with 1 mL of a conidial suspension at 108 spores/mL. It was grown for 3 days at 28°C with shaking at 100 rpm.
Plant inoculations
Cotyledons of 10‐day‐old seedlings of Darmor‐bzh were puncture inoculated with 10 µL of inoculum (107 pycnidiospores/mL), as described by Balesdent et al. (2001). Inoculations were performed with isolates G06‐E107, NzT4, JN3 or JN2. Seven days after inoculation, at a time of intense tissue colonization without expression of macroscopic symptoms, a 0.25‐cm2 area centred on the puncture was cut, frozen in liquid nitrogen and stored at −80°C until RNA extraction.
For the analysis of stem colonization, plants were inoculated at the three‐leaf developmental stage, 22 days after sowing. The petiole of the second leaf was cut at 1 cm from the leaf insertion (pilot RNA‐seq experiment) or just under the leaf blade (validation RNA‐seq experiment), and 10 µL of inoculum (107 pycnidiospores/mL) were placed over the wound. Inoculated plants were incubated in the dark for 48 h at room temperature under saturated humidity, and then transferred to a growth chamber at 16°C (night) and 24°C (day) with a 12‐h photoperiod.
In the pilot RNA‐seq project, 24 days after inoculation, plants were divided into two groups, depending on whether or not symptoms (stem necrosis) were visible. For stems without visible symptoms, 1 cm of the stem was sampled at the inoculated petiole insertion (Fig. 1). For stems with necrotic symptoms, the necrotic tissues and tissues in advance of necrosis, i.e. between the necrosis and stem base, were sampled separately. In the validation RNA‐seq experiment, 14 days after inoculation, 1 cm of the stem was sampled at the inoculated petiole insertion, before symptoms were visible. Eight different stem slices were pooled before extraction. Two independent replicates (corresponding to two distinct inoculation assays with different batches of spore suspension) were performed, extracted and sequenced separately. In both projects, plants inoculated with water were used as a negative control.
RNA manipulation and qRT‐PCR
Total RNA extraction and qRT‐PCR were performed as described by Fudal et al. (2007).
For the qRT‐PCR experiments, all RNA samples were adjusted to 3 μg of RNA and single‐strand cDNA was generated using oligo‐dT primed reverse transcription with PowerScript Reverse Transcriptase (Clontech, Palo Alto, CA, USA) according to the manufacturer's protocol. Water and uninfected plants were used as negative controls. For each condition, three technical replicates from two biological repeats were performed. The qRT‐PCR primers used are indicated in Table S3 (see Supporting Information). Ct values were analysed as described by Muller et al. (2002). Actin was used as a constitutive reference gene and EF1‐α expression relative to actin was used as a control.
RNA sequencing
In the pilot RNA‐seq project, RNAs from two biological replicates with five plants per replicate were pooled for each condition. The cDNA libraries and 50‐bp single read sequencing were performed by the Beijing Genomics Institute (BGI, Shenzen, China) using Illumina HiSeq2000.
In the validation RNA‐seq experiment, RNAs from two independent biological replicates were prepared for each tested condition (early symptomless cotyledon infection, symptomless stem infection or in vitro growth). cDNA libraries and 100‐bp paired‐read sequencing were performed by Genoscope (Evry, France) using Illumina HiSeq2500.
RNA analysis
RNA‐seq analyses were performed with the CLC Genomics Workbench v7.5.1 (Qiagen, Venlo, the Netherlands). Reads were mapped on the reference genomes of B. napus (Chalhoub et al., 2014) and L. maculans (JN3) (Rouxel et al., 2011).
Gene expression was measured using RPKM values (Mortazavi et al., 2008), which were used to cluster the fungal genes based on their expression level across the different infection stages. The data were visualized in the form of a heat map. Gene expression was normalized and differentially expressed genes were detected (P < 0.05) using DESeq2 (Love et al., 2014).
Gene ontology predictions were performed with Blast2GO v3.0.9. Annotation enrichment analyses were performed with Fisher's exact test, using a false discovery rate (FDR) < 0.05 as a filter.
Genes were clustered based on their expression level with Gene Cluster 3.0 by average linkage (de Hoon et al., 2004). Only genes with differential expression (log10(RPKM + 1) ≥ 1 in at least one condition, and Max[log10(RPKM + 1)] – Min[log10(RPKM + 1)] ≥ 1 across the different conditions were clustered. Clusters were visualized with Java Treeview (Saldanha, 2004).
The presence of a secretion signal was predicted with SignalP 4.1 and TargetP 1.1 (Emanuelsson et al., 2007). Transmembrane helix predictions in proteins were performed with TMHMM 2.0. GPI anchor predictions were performed with PredGPI (Pierleoni et al., 2008) and with big‐PI Predictor. Statistical analyses were performed with R (R Core Team, 2015).
Accession number of RNA‐seq data
GSE81756 (in Gene Expression Omnibus database).
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Comparison of Leptosphaeria maculans genes identified as over‐expressed during cotyledon colonization in different studies (Haddadi et al., 2015; Lowe et al., 2014) with the ‘early’ gene pool identified here. The early gene pool corresponds to genes up‐regulated in cotyledons [at 7 days post‐inoculation (dpi)] compared with stem colonization and in vitro conditions. Lowe et al. (2014) identified the 100 genes most highly up‐regulated in cotyledons at 7 dpi compared with in vitro conditions. Haddadi et al. (2015) identified 80 small secreted protein (SSP) encoding genes differentially expressed during cotyledon colonization at 2, 4, 6 and 8 dpi compared with in vitro conditions.
Table S1 List of Leptosphaeria maculans genes showing more than 10 reads mapped in non‐infected controls and excluded from subsequent analyses in the RNA‐sequencing pilot project.
Table S2 Top 50 most expressed genes in early and late colonization stages, identified in the validation RNA‐sequencing project.
Table S3 Primers used for validation of RNA‐sequencing data by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR).
Acknowledgements
The authors wish to thank Bertrand Auclair (INRA BIOGER) for plant management and Régine Delourme (INRA, UMR IGEPP) for Brassica napus Darmor‐bzh seeds. J.G. was funded by a joint grant from the Santé des Plantes et Environnement (SPE) INRA department and the Institute Terres Inovia (formely CETIOM). Cotyledon RNA‐seq experiments were funded by grants from the ONEMA‐CTPS (‘ICOSCOP’ project) and the INRA‐SMaCH Metaprogram ‘K‐Masstec’. The validation RNA‐seq experiment was funded by France Génomique ('LEPTOLIFE' project).
References
- Amorim‐Vaz, S. , Tran, V.D.T. , Pradervand, S. , Pagni, M. , Coste, A.T. and Sanglard, D. (2015) RNA enrichment method for quantitative transcriptional analysis of pathogens in vivo applied to the fungus Candida albicans . mBio, 6, e00942–e00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansan‐Melayah, D. , Balesdent, M.H. , Buée, M. and Rouxel, T. (1995) Genetic characterization of AvrLm1, the first avirulence gene of Leptosphaeria maculans . Phytopathology, 85, 1525–1529. [Google Scholar]
- Balesdent, M.H. , Attard, A. , Ansan‐Melayah, D. , Delourme, R. , Renard, M. and Rouxel, T. (2001) Genetic control and host range of avirulence toward Brassica napus cultivars Quinta and Jet Neuf in Leptosphaeria maculans . Phytopathology, 91, 70–76. [DOI] [PubMed] [Google Scholar]
- Balesdent, M.H. , Attard, A. , Kühn, M.L. and Rouxel, T. (2002) New avirulence genes in the phytopathogenic fungus Leptosphaeria maculans . Phytopathology, 92, 1122–1133. [DOI] [PubMed] [Google Scholar]
- Balesdent, M.H. , Barbetti, M.J. , Li, H. , Sivasithamparam, K. , Gout, L. and Rouxel, T. (2005) Analysis of Leptosphaeria maculans race structure in a worldwide collection of isolates. Phytopathology, 95, 1061–1071. [DOI] [PubMed] [Google Scholar]
- Balesdent, M.H. , Fudal, I. , Ollivier, B. , Bally, P. , Grandaubert, J. , Eber, F. , Chèvre, A.M. , Leflon, M. and Rouxel, T. (2013) The dispensable chromosome of Leptosphaeria maculans shelters an effector gene conferring avirulence towards Brassica rapa . New Phytol. 198, 887–898. [DOI] [PubMed] [Google Scholar]
- Boscari, A. , del Giudice, J. , Ferrarini, A. , Venturini, L. , Zaffini, A.L. , Delledonne, M. and Puppo, A. (2013) Expression dynamics of the Medicago truncatula transcriptome during the symbiotic interaction with Sinorhizobium meliloti: which role for nitric oxide? Plant Physiol. 161, 425–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantu, D. , Segovia, V. , MacLean, D. , Bayles, R. , Chen, X. , Kamoun, S. , Dubcovsky, J. , Saunders, D.G. and Uauy, C. (2013) Genome analyses of the wheat yellow (stripe) rust pathogen Puccinia striiformis f. sp. tritici reveal polymorphic and haustorial expressed secreted proteins as candidate effectors. BMC Genomics, 14, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. , Parkin, I.A.P. , Tang, H. , Wang, X. , Chiquet, J. , Belcram, H. , Tong, C. , Samans, B. , Corréa, M. , Silva, C.D. , Just, J. , Falentin, C. , Koh, C.S. , Clainche, I.L. , Bernard, M. , Bento, P. , Noel, B. , Labadie, K. , Alberti, A. , Charles, M. , Arnaud, D. , Guo, H. , Daviaud, C. , Alamery, S. , Jabbari, K. , Zhao, M. , Edger, P.P. , Chelaifa, H. , Tack, D. , Lassalle, G. , Mestiri, I. , Schnel, N. , Paslier, M.C.L. , Fan, G. , Renault, V. , Bayer, P.E. , Golicz, A.A. , Manoli, S. , Lee, T.H. , Thi, V.H.D. , Chalabi, S. , Hu, Q. , Fan, C. , Tollenaere, R. , Lu, Y. , Battail, C. , Shen, J. , Sidebottom, C.H.D. , Wang, X. , Canaguier, A. , Chauveau, A. , Bérard, A. , Deniot, G. , Guan, M. , Liu, Z. , Sun, F. , Lim, Y.P. , Lyons, E. , Town, C.D. , Bancroft, I. , Wang, X. , Meng, J. , Ma, J. , Pires, J.C. , King, G.J. , Brunel, D. , Delourme, R. , Renard, M. , Aury, J.M. , Adams, K.L. , Batley, J. , Snowdon, R.J. , Tost, J. , Edwards, D. , Zhou, Y. , Hua, W. , Sharpe, A.G. , Paterson, A.H. , Guan, C. and Wincker, P. (2014) Early allopolyploid evolution in the post‐Neolithic Brassica napus oilseed genome. Science, 345, 950–953. [DOI] [PubMed] [Google Scholar]
- Chapelle, E. , Alunni, B. , Malfatti, P. , Solier, L. , Pédron, J. , Kraepiel, Y. and Van Gijsegem, F. (2015) A straightforward and reliable method for bacterial in planta transcriptomics: application to the Dickeya dadantii / Arabidopsis thaliana pathosystem. Plant J. 82, 352–362. [DOI] [PubMed] [Google Scholar]
- Daverdin, G. , Rouxel, T. , Gout, L. , Aubertot, J.N. , Fudal, I. , Meyer, M. , Parlange, F. , Carpezat, J. and Balesdent, M.H. (2012) Genome structure and reproductive behaviour influence the evolutionary potential of a fungal phytopathogen. PLoS Pathog. 8, e1003020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong, Y. , Li, Y. , Zhao, M. , Jing, M. , Liu, X. , Liu, M. , Guo, X. , Zhang, X. , Chen, Y. , Liu, Y. , Liu, Y. , Ye, W. , Zhang, H. , Wang, Y. , Zheng, X. , Wang, P. and Zhang, Z. (2015) Global genome and transcriptome analyses of Magnaporthe oryzae epidemic isolate 98‐06 uncover novel effectors and pathogenicity‐related genes, revealing gene gain and lose dynamics in genome evolution. PLoS Pathog. 11, e1004801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emanuelsson, O. , Brunak, S. , Heijne, G. and von Nielsen, H. (2007) Locating proteins in the cell using TargetP, SignalP and related tools. Nat. Protoc. 2, 953–971. [DOI] [PubMed] [Google Scholar]
- Fitt, B.D.L. , Brun, H. , Barbetti, M.J. and Rimmer, S.R. (2006) World‐wide importance of phoma stem canker (Leptosphaeria maculans and L. biglobosa) on oilseed rape (Brassica napus). Eur. J. Plant Pathol. 114, 3–15. [Google Scholar]
- Fitt, B.D.L. , Hu, B.C. , Li, Z.Q. , Liu, S.Y. , Lange, R.M. , Kharbanda, P.D. , Butterworth, M.H. and White, R.P. (2008) Strategies to prevent spread of Leptosphaeria maculans (phoma stem canker) onto oilseed rape crops in China; costs and benefits. Plant Pathol. 57, 652–664. [Google Scholar]
- Fudal, I. , Ross, S. , Gout, L. , Blaise, F. , Kuhn, M.L. , Eckert, M.R. , Cattolico, L. , Bernard‐Samain, S. , Balesdent, M.H. and Rouxel, T. (2007) Heterochromatin‐like regions as ecological niches for avirulence genes in the Leptosphaeria maculans genome: map‐based cloning of AvrLm6 . Mol. Plant–Microbe Interact. 20, 459–470. [DOI] [PubMed] [Google Scholar]
- Garnica, D.P. , Upadhyaya, N.M. , Dodds, P.N. and Rathjen, J.P. (2013) Strategies for wheat stripe rust pathogenicity identified by transcriptome sequencing. PLoS One, 8, e67150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghanbarnia, K. , Fudal, I. , Larkan, N.J. , Links, M.G. , Balesdent, M.H. , Profotova, B. , Fernando, W.G.D. , Rouxel, T. and Borhan, M.H. (2014) Rapid identification of the Leptosphaeria maculans avirulence gene AvrLm2, using an intraspecific comparative genomics approach. Mol. Plant Pathol. 16, 699–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gout, L. , Fudal, I. , Kuhn, M.L. , Blaise, F. , Eckert, M. , Cattolico, L. , Balesdent, M.H. and Rouxel, T. (2006) Lost in the middle of nowhere: the AvrLm1 avirulence gene of the Dothideomycete Leptosphaeria maculans . Mol. Microbiol. 60, 67–80. [DOI] [PubMed] [Google Scholar]
- Hacquard, S. , Joly, D.L. , Lin, Y.C. , Tisserant, E. , Feau, N. , Delaruelle, C. , Legué, V. , Kohler, A. , Tanguay, P. , Petre, B. , Frey, P. , Van de Peer, Y. , Rouzé, P. , Martin, F. , Hamelin, R.C. and Duplessis, S. (2012) A comprehensive analysis of genes encoding small secreted proteins identifies candidate effectors in Melampsora larici‐populina (Poplar Leaf Rust). Mol. Plant–Microbe Interact. 25, 279–293. [DOI] [PubMed] [Google Scholar]
- Hacquard, S. , Kracher, B. , Maekawa, T. , Vernaldi, S. , Schulze‐Lefert, P. and van Themaat, E.V.L. (2013) Mosaic genome structure of the barley powdery mildew pathogen and conservation of transcriptional programs in divergent hosts. Proc . Natl. Acad. Sci. USA, 110, E2219–E2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddadi, P. , Ma, L. , Wang, H. and Borhan, M.H. (2015) Genome‐wide transcriptome analyses provide insights into the lifestyle transition and effector repertoire of Leptosphaeria maculans during colonization of B. napus seedlings. Mol. Plant Pathol. DOI: 10.1111/mpp.12356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haegeman, A. , Bauters, L. , Kyndt, T. , Rahman, M.M. and Gheysen, G. (2013) Identification of candidate effector genes in the transcriptome of the rice root knot nematode Meloidogyne graminicola . Mol. Plant Pathol. 14, 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammond, K.E. , Lewis, B.G. and Musa, T.M. (1985) A systemic pathway in the infection of oilseed rape plants by Leptosphaeria maculans . Plant Pathol. 34, 557–565. [Google Scholar]
- de Hoon, M.J.L. , Imoto, S. , Nolan, J. and Miyano, S. (2004) Open source clustering software. Bioinformatics, 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Huang, Y.J. , Qi, A. , King, G.J. and Fitt, B.D.L. (2014) Assessing quantitative resistance against Leptosphaeria maculans (phoma stem canker) in Brassica napus (oilseed rape) in young plants. PLoS One, 9, e84924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara, Y. , Oono, Y. , Kanamori, H. , Matsumoto, T. , Itoh, T. and Minami, E. (2012) Simultaneous RNA‐seq analysis of a mixed transcriptome of rice and blast fungus interaction. PLoS One, 7, e49423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleemann, J. , Rincon‐Rivera, L.J. , Takahara, H. , Neumann, U. , van Themaat, E.V.L. , van der Does, H.C. , Hacquard, S. , Stüber, K. , Will, I. , Schmalenbach, W. , Schmelzer, E. and O'connell, R.J. (2012) Sequential delivery of host‐induced virulence effectors by appressoria and intracellular hyphae of the phytopathogen Colletotrichum higginsianum . PLoS Pathog. 8, e1002643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jonge, R. , Bolton, M.D. and Thomma, B.P. (2011) How filamentous pathogens co‐opt plants: the ins and outs of fungal effectors. Curr. Opin. Plant Biol. 14, 400–406. [DOI] [PubMed] [Google Scholar]
- de Jonge, R. , Esse, H.P. , , van Maruthachalam, K. , Bolton, M.D. , Santhanam, P. , Saber, M.K. , Zhang, Z. , Usami, T. , Lievens, B. , Subbarao, K.V. and Thomma, B.P.H.J. (2012) Tomato immune receptor Ve1 recognizes effector of multiple fungal pathogens uncovered by genome and RNA sequencing. Proc . Natl. Acad. Sci. USA, 109, 5110–5115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo Presti, L. , Lanver, D. , Schweizer, G. , Tanaka, S. , Liang, L. , Tollot, M. , Zuccaro, A. , Reissmann, S. and Kahmann, R. (2015) Fungal effectors and plant susceptibility. Annu. Rev. Plant Biol. 66, 513–545. [DOI] [PubMed] [Google Scholar]
- Love, M.I. , Huber, W. and Anders, S. (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe, R.G.T. , Cassin, A. , Grandaubert, J. , Clark, B.L. , Van de Wouw, A.P. , Rouxel, T. and Howlett, B.J. (2014) Genomes and transcriptomes of partners in plant–fungal‐interactions between canola (Brassica napus) and two Leptosphaeria species. PloS One, 9, e103098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald, M.C. , McDonald, B.A. and Solomon, P.S. (2015) Recent advances in the Zymoseptoria tritici–wheat interaction: insights from pathogenomics. Front. Plant Sci. 6, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinhardt, L.W. , Costa, G.G.L. , Thomazella, D.P. , Teixeira, P.J.P. , Carazzolle, M.F. , Schuster, S.C. , Carlson, J.E. , Guiltinan, M.J. , Mieczkowski, P. , Farmer, A. , Ramaraj, T. , Crozier, J. , Davis, R.E. , Shao, J. , Melnick, R.L. , Pereira, G.A. and Bailey, B.A. (2014) Genome and secretome analysis of the hemibiotrophic fungal pathogen, Moniliophthora roreri, which causes frosty pod rot disease of cacao: mechanisms of the biotrophic and necrotrophic phases. BMC Genomics, 15, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirzadi Gohari, A. , Ware, S.B. , Wittenberg, A.H.J. , Mehrabi, R. , Ben M'barek, S. , Verstappen, E.C.P. , van der Lee, T.A.J. , Robert, O. , Schouten, H.J. , de Wit, P.P.J.G.M. and Kema, G.H.J. (2015) Effector discovery in the fungal wheat pathogen Zymoseptoria tritici . Mol. Plant Pathol. 16, 931–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortazavi, A. , Williams, B.A. , McCue, K. , Schaeffer, L. and Wold, B. (2008) Mapping and quantifying mammalian transcriptomes by RNA‐Seq. Nat. Methods, 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Muller, P.Y. , Janovjak, H. , Miserez, A.R. and Dobbie, Z. (2002) Processing of gene expression data generated by quantitative real‐time RT‐PCR. BioTechniques, 32, 1372–1374. 1376, 1378–1379. [PubMed] [Google Scholar]
- Münch, S. , Lingner, U. , Floss, D.S. , Ludwig, N. , Sauer, N. and Deising, H.B. (2008) The hemibiotrophic lifestyle of Colletotrichum species. J. Plant Physiol. 165, 41–51. [DOI] [PubMed] [Google Scholar]
- Oh, Y. , Donofrio, N. , Pan, H. , Coughlan, S. , Brown, D.E. , Meng, S. , Mitchell, T. and Dean, R.A. (2008) Transcriptome analysis reveals new insight into appressorium formation and function in the rice blast fungus Magnaporthe oryzae . Genome Biol. 9, R85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parlange, F. , Daverdin, G. , Fudal, I. , Kuhn, M.L. , Balesdent, M.H. , Blaise, F. , Grezes‐Besset, B. and Rouxel, T. (2009) Leptosphaeria maculans avirulence gene AvrLm4‐7 confers a dual recognition specificity by the Rlm4 and Rlm7 resistance genes of oilseed rape, and circumvents Rlm4‐mediated recognition through a single amino acid change. Mol. Microbiol. 71, 851–863. [DOI] [PubMed] [Google Scholar]
- Pierleoni, A. , Martelli, P.L. and Casadio, R. (2008) PredGPI: a GPI‐anchor predictor. BMC Bioinformatics, 9, 392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plissonneau, C. , Daverdin, G. , Ollivier, B. , Blaise, F. , Degrave, A. , Fudal, I. , Rouxel, T. and Balesdent, M.H. (2015) A game of hide and seek between avirulence genes AvrLm4‐7 and AvrLm3 in Leptosphaeria maculans . New Phytol. 209, 1613–1624. doi: 10.1111/nph.13736 [DOI] [PubMed] [Google Scholar]
- R Core Team (2015) R: A language and environment for statistical computing Vienna: R Foundation for Statistical Computing. http://www.R-project.org/.
- Rouxel, T. and Balesdent, M.H. (2005) The stem canker (blackleg) fungus, Leptosphaeria maculans, enters the genomic era. Mol. Plant Pathol. 6, 225–241. [DOI] [PubMed] [Google Scholar]
- Rouxel, T. , Grandaubert, J. , Hane, J.K. , Hoede, C. , van de Wouw, A.P. , Couloux, A. , Dominguez, V. , Anthouard, V. , Bally, P. , Bourras, S. , Cozijnsen, A.J. , Ciuffetti, L.M. , Degrave, A. , Dilmaghani, A. , Duret, L. , Fudal, I. , Goodwin, S.B. , Gout, L. , Glaser, N. , Linglin, J. , Kema, G.H.J. , Lapalu, N. , Lawrence, C.B. , May, K. , Meyer, M. , Ollivier, B. , Poulain, J. , Schoch, C.L. , Simon, A. , Spatafora, J.W. , Stachowiak, A. , Turgeon, B.G. , Tyler, B.M. , Vincent, D. , Weissenbach, J. , Amselem, J. , Quesneville, H. , Oliver, R.P. , Wincker, P. , Balesdent, M.H. and Howlett, B.J. (2011) Effector diversification within compartments of the Leptosphaeria maculans genome affected by repeat‐induced point mutations. Nat. Commun. 2, 202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldanha, A.J. (2004) Java Treeview—extensible visualization of microarray data. Bioinformatics, 20, 3246–3248. [DOI] [PubMed] [Google Scholar]
- Soyer, J.L. , El Ghalid, M. , Glaser, N. , Ollivier, B. , Linglin, J. , Grandaubert, J. , Balesdent, M.H. , Connolly, L.R. , Freitag, M. , Rouxel, T. and Fudal, I. (2014) Epigenetic control of effector gene expression in the plant pathogenic fungus Leptosphaeria maculans . PLoS Genet. 10, e1004227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thewes, S. , Kretschmar, M. , Park, H. , Schaller, M. , Filler, S.G. and Hube, B. (2007) In vivo and ex vivo comparative transcriptional profiling of invasive and non‐invasive Candida albicans isolates identifies genes associated with tissue invasion. Mol. Microbiol. 63, 1606–1628. [DOI] [PubMed] [Google Scholar]
- Van de Wouw, A.P. , Lowe, R.G.T. , Elliott, C.E. , Dubois, D.J. and Howlett, B.J. (2014) An avirulence gene, AvrLmJ1, from the blackleg fungus, Leptosphaeria maculans, confers avirulence to Brassica juncea cultivars. Mol. Plant Pathol. 15, 523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Weiberg, A. and Jin, H. (2015) Pathogen small RNAs: a new class of effectors for pathogen attacks. Mol. Plant Pathol. 16, 219–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Q. , Han, C. , Ferreira, A.O. , Yu, X. , Ye, W. , Tripathy, S. , Kale, S.D. , Gu, B. , Sheng, Y. , Sui, Y. , Wang, X. , Zhang, Z. , Cheng, B. , Dong, S. , Shan, W. , Zheng, X. , Dou, D. , Tyler, B.M. and Wang, Y. (2011) Transcriptional programming and functional interactions within the Phytophthora sojae RXLR effector repertoire. Plant Cell, 23, 2064–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West, J.S. , Kharbanda, P.D. , Barbetti, M.J. and Fitt, B.D.L. (2001) Epidemiology and management of Leptosphaeria maculans (phoma stem canker) on oilseed rape in Australia, Canada and Europe. Plant Pathol. 50, 10–27. [Google Scholar]
- Wilson, R.A. and Talbot, N.J. (2009) Under pressure: investigating the biology of plant infection by Magnaporthe oryzae . Nat. Rev. Microbiol. 7, 185–195. [DOI] [PubMed] [Google Scholar]
- de Wit, P.J.G.M. , van der Burgt, A. , Ökmen, B. , Stergiopoulos, I. , Abd‐Elsalam, K.A. , Aerts, A.L. , Bahkali, A.H. , Beenen, H.G. , Chettri, P. , Cox, M.P. , Datema, E. , de Vries, R.P. , Dhillon, B. , Ganley, A.R. , Griffiths, S.A. , Guo, Y. , Hamelin, R.C. , Henrissat, B. , Kabir, M.S. , Jashni, M.K. , Kema, G. , Klaubauf, S. , Lapidus, A. , Levasseur, A. , Lindquist, E. , Mehrabi, R. , Ohm, R.A. , Owen, T.J. , Salamov, A. , Schwelm, A. , Schijlen, E. , Sun, H. , van den Burg, H.A. , van Ham, R.C.H.J. , Zhang, S. , Goodwin, S.B. , Grigoriev, I.V. , Collemare, J. and Bradshaw, R.E. (2012) The genomes of the fungal plant pathogens Cladosporium fulvum and Dothistroma septosporum reveal adaptation to different hosts and lifestyles but also signatures of common ancestry. PLoS Genet. 8, e1003088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, F. , Li, W. , Derbyshire, M. , Larsen, M.R. , Rudd, J.J. and Palmisano, G. (2015) Unraveling incompatibility between wheat and the fungal pathogen Zymoseptoria tritici through apoplastic proteomics. BMC Genomics, 16, 362. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 Comparison of Leptosphaeria maculans genes identified as over‐expressed during cotyledon colonization in different studies (Haddadi et al., 2015; Lowe et al., 2014) with the ‘early’ gene pool identified here. The early gene pool corresponds to genes up‐regulated in cotyledons [at 7 days post‐inoculation (dpi)] compared with stem colonization and in vitro conditions. Lowe et al. (2014) identified the 100 genes most highly up‐regulated in cotyledons at 7 dpi compared with in vitro conditions. Haddadi et al. (2015) identified 80 small secreted protein (SSP) encoding genes differentially expressed during cotyledon colonization at 2, 4, 6 and 8 dpi compared with in vitro conditions.
Table S1 List of Leptosphaeria maculans genes showing more than 10 reads mapped in non‐infected controls and excluded from subsequent analyses in the RNA‐sequencing pilot project.
Table S2 Top 50 most expressed genes in early and late colonization stages, identified in the validation RNA‐sequencing project.
Table S3 Primers used for validation of RNA‐sequencing data by quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR).