

Summary
The ability of biotrophic fungi to metabolically adapt to the host environment is a critical factor in fungal diseases of crop plants. In this study, we analysed the transcriptome of maize tumours induced by Ustilago maydis to identify key features underlying metabolic shifts during disease. Among other metabolic changes, this analysis highlighted modifications during infection in the transcriptional regulation of carbohydrate allocation and starch metabolism. We confirmed the relevance of these changes by establishing that symptom development was altered in an id1 (indeterminate1) mutant that showed increased accumulation of sucrose as well as being defective in the vegetative to reproductive transition. We further established the relevance of specific metabolic functions related to carbohydrate allocation by assaying disease in su1 (sugary1) mutant plants with altered starch metabolism and in plants treated with glucose, sucrose and silver nitrate during infection. We propose that specific regulatory and metabolic changes influence the balance between susceptibility and resistance by altering carbon allocation to promote fungal growth or to influence plant defence. Taken together, these studies reveal key aspects of metabolism that are critical for biotrophic adaptation during the maize–U. maydis interaction.
Keywords: maize, RNA‐Seq, smut, starch, transcription factor, Zea mays
Introduction
Maize (Zea mays) is a staple crop that sustains the human population, either directly as a source of starch and lipids, or indirectly as feed for livestock. Furthermore, maize is a major source of industrial products, including starch and high‐fructose corn syrup and, more recently, biofuels (Henry, 2009). Maize is threatened by a number of pathogens that cause losses to biomass and seed production. Biotrophic pathogens depend on a living host for successful nutrient acquisition, and possess sophisticated mechanisms to infect the host, suppress plant defence and access nutrients. The fungus Ustilago maydis is a biotrophic basidiomycete pathogen capable of infecting all above‐ground parts of maize and teosinte. Infection is initiated by the fusion of two haploid yeast‐like cells of opposite mating type to form an infectious, filamentous dikaryon (Brefort et al., 2009; Kämper et al., 2006; Koeck et al., 2011; Kronstad and Leong, 1990; Mueller et al., 2008). The dikaryon forms an appressorium to penetrate the plant surface and the fungus grows into and between plant cells. After infection, the first obvious symptom is the formation of anthocyanins, leading to red coloration of plant tissues. Large tumours filled with fungal biomass are formed during the later stages of colonization, and these can lead to stunted plant growth. Fungal development in tumour tissue gives rise to teliospores that are eventually released to disperse the pathogen. Ustilago maydis is known to secrete an array of effectors and to adapt its infection strategy in an organ‐specific manner (Djamei et al., 2011; Hemetsberger et al., 2012; Kämper et al., 2006; van der Linde et al., 2012; Mueller et al., 2013, 2008; Redkar et al., 2015; Schilling et al., 2014; Skibbe et al., 2010; Tanaka et al., 2014).
Several studies have investigated maize gene expression during infection by U. maydis (Basse, 2005; Doehlemann et al., 2008; Gao et al., 2013; Horst et al., 2010a, 2010b; Skibbe et al., 2010; Voll et al., 2011). An early differential display approach revealed changes in the differentiation state of host tissue during infection, as well as altered expression of genes for secondary metabolism and defence (Basse, 2005). A subsequent analysis by Doehlemann et al. (2008) examined the transcriptome during disease progression at 12 h post‐inoculation (hpi), 24 hpi, 2 days post‐inoculation (dpi), 4 dpi and 8 dpi using an Affymetrix maize genome array containing 13 339 genes. Increasing numbers of differentially regulated genes were observed as infection progressed. The early time point of 12 hpi revealed differential expression for genes involved in the stress response, redox regulation and defence. Subsequent times revealed expression changes for additional functions for secondary metabolism, cell wall metabolism, protein metabolism, primary carbon metabolism, transcription and RNA processing, and transport. Notable processes influenced by U. maydis infection included a down‐regulation of the defence and cell death responses, as well as up‐regulation of hormone signalling functions (i.e. jasmonic acid and auxin biosynthesis/response). In addition, changes in the expression of genes for secondary metabolite synthesis were observed, including the induction of genes in the shikimate pathway for the synthesis of phenylalanine and tyrosine, the substrates for phenylalanine ammonia lyase (PAL). PAL expression was also induced in tumour tissue and this enzyme produces hydroxycinnamic acid derivatives leading to lignin and flavonoids, including anthocyanins, which are known to accumulate in infected tissue at early stages. For primary metabolism, it was observed that glycolysis, the tricarboxylic acid (TCA) cycle and lipid metabolism were induced in tumours and that the expression of genes for photosynthesis were reduced.
Skibbe et al. (2010) also performed transcriptome studies of infected adult tissue and identified a requirement for active cell proliferation for tumour formation, as well as transcriptional changes that reflected organ‐specific gene expression. The fungus appears to reprogram vegetative and reproductive development during tumour induction. Experimental infection of maize mutants with defects in hormone signalling revealed that gibberellin signalling is not needed for tumour formation on seedlings, but is needed for tumours on adult tissues. Disruption of gibberellin regulation in the knotted1 (kn1) mutant also resulted in normal tumours on seedlings and more abundant and larger tumours on adult leaves. A mutant defective in the auxin response also allowed normal tumour formation on vegetative tissue, but not on floral tissue. A detailed study by Gao et al. (2013) of anther infection also concluded that U. maydis employs specific functions to cause disease in different host organs. Interestingly, this study also identified transcription factors (TFs) as a prominent gene ontology (GO) term for maize genes that were up‐regulated during infection.
Finally, Voll et al. (2011) examined metabolome and transcriptome data to compare changes during infections by three biotrophic fungal pathogens: U. maydis, Blumeria graminis f. sp. hordei and Colletotrichum graminicola (a hemibiotroph). Notably, early stages of infection by all three pathogens involved strong regulation of the TCA cycle, nucleotide energy status and amino acid metabolism. In addition, general features of early interactions included the biosynthesis of asparagine and glutamine, together with defence‐associated branched chain and aromatic amino acids, a reduction in the Calvin cycle and/or starch metabolism with increased glycolysis and TCA cycle activity, and elevated photorespiration with reduced sucrose biosynthesis. These changes may be part of a common cereal response (Voll et al., 2011).
In general, previous transcriptome studies of U. maydis infection were limited by the sets of genes on the microarrays (i.e. 15%–30% of the transcripts for the maize genome). Furthermore, an engineered solo‐pathogenic strain was mainly used for these studies, and this strain shows reduced virulence compared with infection with compatible haploid strains (Leuthner et al., 2005). In this study, we employed RNA‐sequencing (RNA‐Seq) on seedling tissue infected with compatible haploid strains to expand the available data on the global changes in maize gene expression during tumour formation. Our goal was to test the functional significance of the transcriptional changes in the context of U. maydis adaptation to the changing metabolic landscape of the host during disease. We used information on transcriptional changes during tumour formation to generate testable hypotheses regarding the importance of altered transcriptional regulation and functions for carbohydrate allocation. We linked these functions with the success of U. maydis during disease by inoculating maize mutants impaired in these functions, as well as plants treated with sugars and silver nitrate. These studies provide insights into the role of metabolic changes related to carbohydrate availability in the plant–fungus interaction. We also examined the influence of chloroplast‐associated metabolic functions in an accompanying paper (Kretschmer et al. 2016).
Results
Tumour induction by U. maydis results in extensive remodelling of the maize transcriptome
We analysed differential gene expression during U. maydis induction of tumours by performing RNA‐Seq on uninfected and infected seedling samples at 10 days post‐inoculation (dpi). This time point was selected on the basis of the published time course analyses of the transcriptome using microarrays. For example, Doehlemann et al. (2008) performed a detailed time course analysis of infected versus non‐infected tissue with sampling at 0.5, 1, 2, 4 and 8 days. The number of differentially expressed genes continued to increase throughout the time course [see fig. 8 in Doehlemann et al. (2008)], and we therefore focused on a later time point when tumours were well developed under our growth conditions. At this relatively late stage, we expected to see extensive differences in gene expression that reflected the formation of physiologically and structurally novel tumour tissue relative to uninfected tissue. High‐quality reads were obtained for all libraries as summarized in the Experimental Procedures, as well as in Fig. S1A and Text S1 (see Supporting Information). The reads that mapped to the U. maydis genome were removed from the analysis and the remaining reads were aligned to the maize reference genome of the inbred line B73 (version 2), and expression values were calculated. No expression was detected for 45 973 of the known 108 894 maize transcripts, and transcripts for 5653 and 4150 genes were specific to the libraries from infected and uninfected tissue, respectively. In addition, 4086 genes were significantly up‐regulated and 5237 genes were significantly down‐regulated during tumour formation (P ≤ 0.05; Table S1, see Supporting Information). It should be noted that we employed the Z. mays variety Golden Bantam for infection and identified 6978 transcripts that mapped to the B73 reference genome, but not to any annotated genes; 311 of these transcripts were only expressed during infection, whereas 315 were only expressed in uninfected tissue (Table S2, see Supporting Information). We also validated the changes in transcript levels by quantitative polymerase chain reaction (qPCR) analysis on independent RNA samples prepared from seedling tumours (Fig. 1; Table S3, see Supporting Information). The trends in differential expression for eight genes selected to represent a range of fold changes were confirmed by qPCR, although the fold changes for ra2 (ramosa2, lateral organ boundaries protein) and bad1 (branch angle defective1, encoding a TF) were lower than detected by RNA‐Seq (Table S3).
Figure 8.
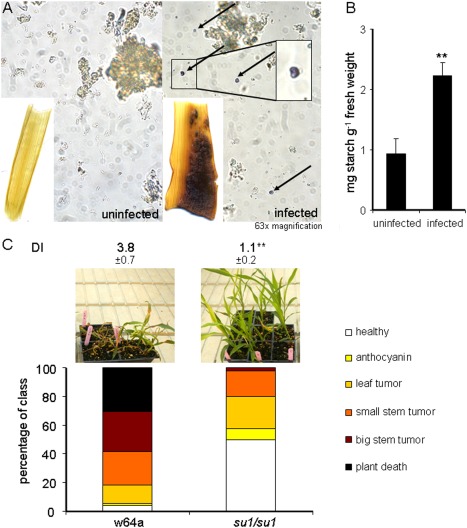
Carbohydrate metabolism of the host is changed during infection and is implicated in disease progression. (A) Infected and uninfected stem tissues at 10 days post‐inoculation (dpi) were stained for starch, and starch kernels were visually identified in infected and uninfected stem tissue. (B) Starch content of uninfected and infected stem tissue was determined as reducing sugars. (C) Disease symptom progression and disease index (DI) ratings for the su1/su1 mutant line and the maize wild‐type (wt; Su1/Su1) line at 14 dpi infected with Ustilago maydis. Standard deviations are shown. **Significant difference at P < 0.01. The plant symptoms were scored as described in Fig. 5.
Figure 1.
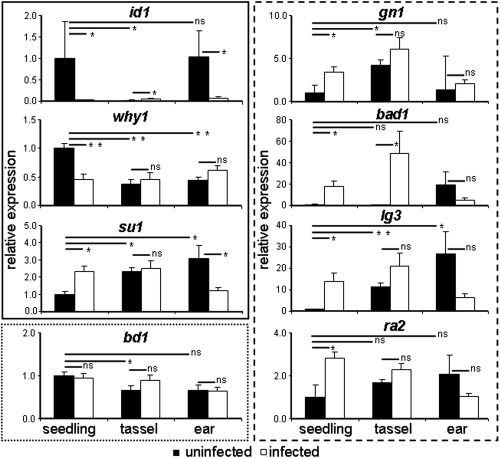
Zea mays gene expression in seedlings, tassels and ears in tumours induced by Ustilago maydis. The id1, why1 and su1 genes (in the box outlined with a solid line) showed constant gene expression in tumours, but with developmental expression changes. The bd1 gene (in the box outlined with the dotted line) showed unchanged expression independent of the developmental stage of the plant or infection status. The gn1, ra2, lg3 and bad1 genes (in the box outlined with the broken line) showed differential gene expression dependent on developmental state and infection state. The data were derived from three independent replicates. Standard error is shown. *P ≤ 0.05; **P ≤ 0.01; ns, not significant.
We extended the qPCR analysis to evaluate the impact of tumour formation on transcript levels in tumour tissue from tassels and ears. A previous report by Skibbe et al. (2010) documented organ‐specific gene expression during U. maydis infection. Our analysis identified three patterns of gene expression for the eight genes selected for quantitative reverse transcription‐polymerase chain reaction (qRT‐PCR) validation. The first pattern included three genes whose transcript levels were influenced by infection, but not in all tissues (Fig. 1). For example, id1 (indeterminate1, encoding a TF) was down‐regulated during tumour formation in seedlings (33.3‐fold) and ears (14.3‐fold), but slightly up‐regulated during tassel infection, although the latter expression level was quite low and may not have had biological significance. Similarly, why1 (whirly1, encoding a chloroplast single‐stranded nucleic acid‐binding protein) transcript levels were lower only in seedling tumours, and su1 (sugary1, encoding an isoamylase‐type starch‐debranching enzyme) transcripts were higher in seedling tumours and lower in ear tumours. The second pattern was represented by one gene, bd1, for which transcript levels were not influenced during infection in any tissue (Fig. 1). The third pattern included the four genes ra2, lg3 (liguleless3, encoding a TF), gn1 (gnarley1, encoding a TF) and bad1, whose transcript levels were influenced by tumour formation in all three tissues (Fig. 1). Taken together, the RNA‐Seq data, together with validation by qPCR, provide a robust view of the transcriptional response of maize during tumour formation.
Classification of differentially expressed genes highlights changes in metabolic functions, transposable elements (TEs) and TFs
We initially performed GO term enrichment analysis for the significantly up‐ or down‐regulated genes identified with the reference B73 genome (Table S4, see Supporting Information). GO term connections/networks of the enriched terms based on their P values were also visualized with REVIGO (Supek et al., 2011) (Figs 2 and 3). Enriched biological function GO terms for the down‐regulated genes included metabolism and biosynthesis (e.g. amino acid, organic acids and lipid metabolism), secondary metabolism (e.g. phenylpropanoid and lignin metabolism and photosynthesis), translation, RNA modification, DNA repair, cell stress and cellular homeostasis, including defence reactions (Fig. 2; Table S4). GO terms for up‐regulated genes included carbohydrate metabolism, cell wall modifications, lipid metabolism and cell cycle functions (Figs 3 and S2, see Supporting Information). Signalling functions, especially protein phosphorylation, were also enriched, as were functions for the response to biotic/abiotic stimuli related to cell death and the export of toxins, which may be linked to plant defence. Finally, terms for the export of amino acids and glutamine metabolism were over‐represented during infection. In general, the patterns of the observed transcript changes were in agreement with previous studies using microarrays (Doehlemann et al., 2008).
Figure 2.
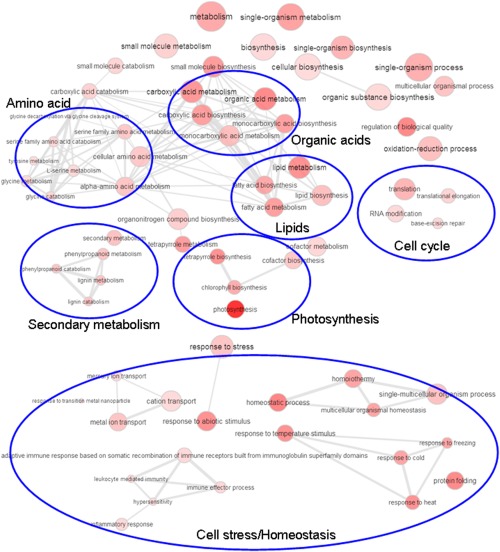
Enriched biological gene ontology (GO) term networks of Zea mays infected with Ustilago maydis (10 days post‐inoculation) for down‐regulated genes. More general GO terms are indicated by larger circles and the intensity of the red colour indicates the significance level of GO term enrichment based on the P value (see Table S4) for the GO term.
Figure 3.
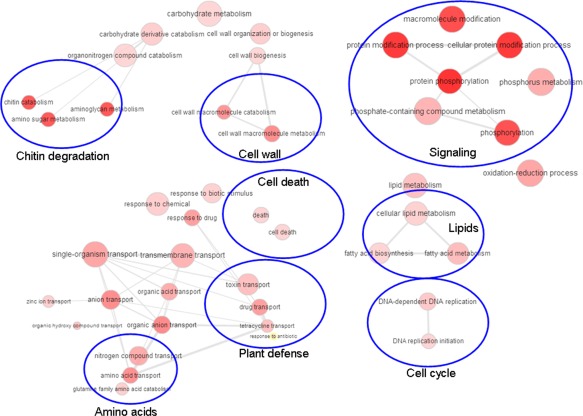
Enriched biological gene ontology (GO) term networks of Zea mays infected with Ustilago maydis (10 days post‐inoculation) for up‐regulated genes. More general GO terms are indicated by larger circles and the intensity of the red colour indicates the significance level of GO term enrichment based on the P value (see Table S4) for the GO term.
We also carried out a focused analysis of the approximately 1100 most differentially regulated genes with annotations in the B73 genome database and by performing blastp searches to identify additional potential functions. Approximately 50% of the transcripts showed no similarities to proteins with known functions, and the remaining transcripts were classified in the categories shown in Fig. 4A,B and Table S1. This analysis revealed changes in transcripts encoding TFs, signalling functions and plant defence proteins linked with secondary metabolism (e.g. phytoalexin production and terpene synthase expression). Further expression changes during tumour formation were observed for cell death and cell wall genes, as well as functions for general metabolism, nitrogen metabolism and transport. As noted above, metabolic functions with differential expression were related to carbohydrate, lipid and amino acid metabolism and transport. Changes in the expression of genes encoding hormone production and sensitivity were also observed. Interestingly, there were several functions only represented by down‐regulated genes during infection. The most prominent were genes encoding photosynthesis‐ and cell cycle‐related function, together with some central metabolic functions in the Calvin cycle, glycolysis and gluconeogenesis.
Figure 4.
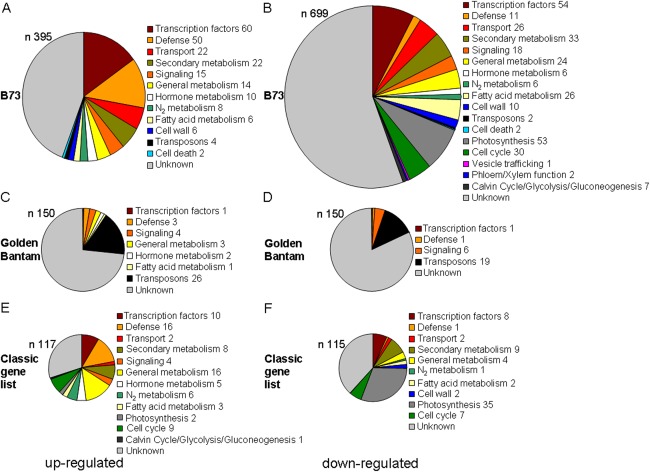
Categorization of the most up‐ and down‐regulated host genes in Ustilago maydis‐induced tumours at 10 days post‐inoculation (dpi). (A, C, E) Up‐regulated genes. (B, D, F) Down‐regulated genes. (A, B) Known transcripts from http://www.maizegdb.org; (C, D) transcripts from Golden Bantam; (E, F) genes from the classical gene list. n represents the number of genes in each chart. Numbers after the different categories indicate the number of genes in this category. The colour coding between the different charts signifies the same classification.
We also examined the 6978 transcripts for Z. mays variety Golden Bantam that mapped to the genome locations of the B73 reference genome shown in Table S2, but not to annotated genes, and employed blastx searches to identify potential functions for the top 150 up‐ and down‐regulated sequences among this group. Approximately 75% showed no sequence similarity to proteins with known function (Fig. 4C,D; Table S2). For those with similarities to known proteins, it was striking that 26 transcripts encoded by TEs were elevated during infection, whereas 19 others showed reduced expression. A re‐examination of the expression of the known TE transcripts for B73 also identified four TEs that were up‐regulated and two that were down‐regulated during infection. In general, these results suggest that elevated stress during tumour formation may interfere with genomic defence.
Our global analysis also included an examination of the expression of the approximately 575 classical genes/proteins which have been well studied by the maize community (Schnable and Freeling, 2011). These functions are interesting because of their economic implications for yield and quality. Amongst the strongly up‐regulated transcripts, the most interesting encoded functions for plant growth‐regulating factors, plant defence and sugar/starch metabolism (Fig. 4E,F; Tables S5 and S6, see Supporting Information). Other genes encoded both up‐ and down‐regulated transcripts for secondary and fatty acid metabolism that indicate shifts in specific pathways. Notably, down‐regulated transcripts encoded functions related to photosynthesis, the cytoskeleton and the cell wall. In general, the observed changes in the expression of genes in the classical gene list reflected the changes seen in the GO term analysis as noted above. Overall, our classification of differentially expressed transcripts and previous studies (Doehlemann et al., 2008; Horst et al., 2008, 2010a,b) revealed complex metabolic and cellular changes during tumour formation.
We classified these transcripts within the context of the major metabolic pathways defined in the genome analysis for maize (accessed April 2015), with a particular focus on carbohydrate metabolism, as described below (Monaco et al., 2013) (Fig. S2, Tables S1 and S5).
Finally, our analysis revealed that one notable group of differentially expressed genes encoded TFs. Specifically, we identified 114 TFs among the top regulated transcripts and classified them according to their developmental expression profiles (Tables 1 and S1). We observed that the majority of TFs with regulation during infection were expressed in two categories: leaf and coleoptile, and reproductive organs. Those expressed in the leaf and coleoptile were 21.7% and 50.0% up‐ and down‐regulated, respectively, whereas the TFs that were normally up‐regulated in reproductive organs (i.e. ears, tassels, silks, kernels) were 46.6% and 27.3% up‐ and down‐regulated during infection, respectively (Table 1). Lower percentages of TFs were identified in the categories of factors that had similar expression in all maize tissues or were preferentially expressed in roots. In general, these data suggest a reprogramming of TF expression, such that more prominent down‐regulation occurs for vegetative/photosynthetic leaf functions and up‐regulation occurs for functions expressed in reproductive/photosynthetic inactive tissue. Consistent with a shift in the expression of vegetative and reproductive functions during infection, a more detailed evaluation of the 114 TFs plus the classical gene list revealed that the expression of many genes known to regulate meristem size, organization, initiation and determinacy was changed in tumours (Table 2) (Bortiri and Hake, 2007; Eveland et al., 2014; Pautler et al., 2013; Tanaka et al., 2013). These included the four genes td1 (thick tassel dwarf1, encoding a CLAVATA1 leucine‐rich repeat receptor‐like kinase), wus1 (wuschel1, encoding a TF), br2 (brachytic2, encoding an ABC transporter) and id1, which were significantly down‐regulated in tumours, and 11 that were up‐regulated (P < 0.05).
Table 1.
Classification of Zea mays transcription factors regulated during Ustilago maydis infection in relation to the highest expression during normal maize development.
| Highest expression during maize development | Up‐regulated during infection (%; n = 60) | Down‐regulated during infection (%; n = 54) |
|---|---|---|
| Roots | 11.7 | 11.1 |
| Leaves | 21.7 | 50.0 |
| Germinating seeds | 3.3 | 0.0 |
|
Reproductive organs (ear, tassel, silks, kernel, husks) |
43.3 | 27.3 |
| Similar in all tissues | 18.3 | 9.3 |
| Unknown | 1.7 | 1.9 |
Table 2.
Expression of genes for meristem regulation in maize stem tumours induced by Ustilago maydis.
| Gene | Name | Long name | Fold change | P value |
|---|---|---|---|---|
| GRMZM2G139073 | si1 | silky1 | 167.61 | 0.00 |
| AC233943.1_FG002 | ra2 | ramosa2 | 42.96 (2.3 qPCR) | 0.01 |
| GRMZM2G005624 | gt1 | grassy tillers1 | 19.90 | 0.01 |
| GRMZM2G052890 | zag1 | zea agamous homolog1 | 18.18 | 0.00 |
| GRMZM2G014729 | ra3 | ramosa3 | 5.62 | 0.01 |
| GRMZM2G700665 | rap2 * | rap2.7 | 3.88 | 0.00 |
| GRMZM2G176175 | sid1 | sister of indeterminate spikelet1 | 2.21 | 0.05 |
| GRMZM2G104843 | ts1 | tassel seed1 | 1.94 | 0.04 |
| GRMZM2G098643 | pin1 | PIN‐formed protein1 | 1.65 | 0.03 |
| GRMZM2G040762 | fzt1 | fuzzy tassel1 | 1.65 | 0.04 |
| GRMZM2G315375 | br2 | brachytic2 | 0.59 | 0.00 |
| GRMZM2G300133 | td1 | thick tassel dwarf1 | 0.57 | 0.05 |
| GRMZM2G010929 | wus1 † | wuschel1 | 0.18 | 0.00 |
| GRMZM2G011357 | id1 | indeterminate growth1 | 0.05 | 0.00 |
Fold change: infected divided by uninfected, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively. Significant expression changes according to P value.
Note that one gene, zea floricaula/leafy1 (zfl1: GRMZM2G098813), showed expression only in infected tissue and thus the fold change could not be determined.
*Salvi et al. (2007).
†Salvo et al. (2014).
The ID1 transcriptional regulator of the vegetative to reproductive transition and carbohydrate allocation influences symptom development
To investigate the relevance of differential expression observed from our RNA‐Seq analysis, we focused on changes in functions related to carbohydrate metabolism. In particular, we were intrigued by the known role of the id1 gene in regulating both the transition to flowering and carbohydrate allocation in maize (Coneva et al., 2012). Specifically, mutations in id1 delay the vegetative to reproductive transition and prolong vegetative growth in ears and tassels (Wong and Colasanti, 2007). The gene is also expressed only in young leaves and is conserved in different plant species (Colasanti et al., 1998, 2006; Coneva et al., 2012). Importantly for our focus on the metabolic changes during disease, the transcriptome analyses of Coneva et al. (2007, 2012) revealed an influence of the id1 mutation on GO terms for the biological ‘carbohydrate metabolic process’, and specific regulation of genes for sucrose and starch metabolism was identified.
To directly test the role of ID1 in susceptibility to U. maydis, we obtained seeds segregating for the id1 mutation from a cross in the B73 background. We infected seedlings with U. maydis, and the plants were scored for disease symptoms after 2 weeks (Fig. 5). We also compared the wild‐type (wt) versus mutant genotype for id1 in each plant by PCR. The homozygous wt plants were highly susceptible to infection, with a disease index (DI) of 4.5, whereas the heterozygous plants and the homozygous mutants of id1 were less susceptible with DIs of 3.7 and 3.4, respectively (Fig. 5). The difference in DIs between the wt and heterozygous plants was not statistically significant (P = 0.084) compared with that of the wt and homozygous plants (P = 0.049). However, the disease symptoms for the heterozygous plants appeared to be intermediate in severity compared with those of the wt and homozygous plants. This was particularly true for the percentage of dead plants for each infection (wt, 60.0%; heterozygous plants, 41.7%; homozygous plants, 17.2%). We also observed a striking difference in the pigmentation of severely infected plants (i.e. those with stem tumours) (Fig. 5). The wt and heterozygous plants had dark red pigment formation in the stem and basal portions of the leaves (95.8 ± 7.2% and 100 ± 0.0% pigmentation, respectively), whereas only 8.5 ± 7.6% of the homozygous mutant plants had traces of a brownish red pigment. We speculate that this pigment is anthocyanin, because this product of secondary metabolism is routinely observed on U. maydis infection (Basse and Steinberg, 2004). In general, these results indicate that haploid insufficiency may affect some traits (e.g. susceptibility), but not others (e.g. anthocyanin), in the heterozygous line. Taken together, these observations prompted us to hypothesize that changes in carbohydrate metabolism in id1/id1 plants may contribute to altered susceptibility, and we investigated this possibility as described below.
Figure 5.
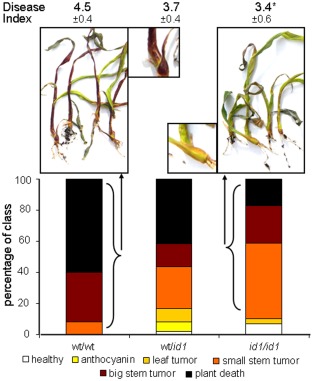
The transcription factor ID1 is important for the infection success of Ustilago maydis. The id1 homozygous (id1/id1) and heterozygous (Id1/id1) mutant lines and the corresponding wild‐type (wt) (Id1/Id1) line were infected with U. maydis. The infection progression was scored at 14 days post‐inoculation (dpi) and the data of three biological replicates are shown. Red pigment formation was observed in severely infected wt plants, which showed stem tumours or were dead (inset shows a close up view of the plant from the left photograph), compared with green homozygous id1/id1 mutant plants of the same infection class (inset shows a close up view of the plant from the right photograph). Standard deviations are shown and asterisk indicates a significant difference at P < 0.05 for the disease index (DI). DI was calculated for each biological replicate based on the following scoring scheme for symptoms: 0, healthy plants/no symptoms; 1, anthocyanin formation; 2, leaf tumours; 3, small stem tumours; 4, big stem tumours; 5, plant death. The overall percentages of plants of the three repeats in each category are indicated in the bar graphs and correlate with DI.
Carbohydrate metabolism plays an important role during tumour formation
In agreement with the discovery that ID1 influences the expression of genes for carbohydrate metabolism, Coneva et al. (2012) also performed a metabolite analysis which revealed higher sucrose and starch levels in id1/id1 mutant plants. This observation prompted us to test whether sucrose or glucose availability influenced disease outcome by injecting infected seedlings daily with solutions of glucose, sucrose or water over 14 days. We found that injections of sucrose and, more notably, of glucose into the infection site stimulated the virulence of U. maydis and led to a higher DI compared with the water control (Fig. 6A). These results suggest that an increased supply of carbon at the infection site improves pathogen growth, although there may also be a detrimental impact on host defence given the link between sucrose and plant immunity (Fig. 6B; Bolouri‐Moghaddam and Van den Ende, 2013; Roitsch et al., 2003).
Figure 6.
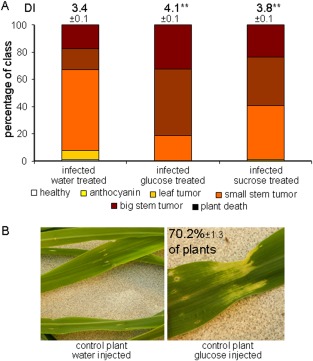
Sucrose and glucose supplementation increases the virulence of Ustilago maydis. Seedlings (14 days old) were infected with U. maydis and daily injections with water, sucrose or glucose were applied after the infection was established (see Experimental Procedures for details). (A) The plants were scored for disease symptoms (DI, disease index) after 14 days of infection and treatment as described in Fig. 5. (B) The glucose‐treated control plants (70.2%) showed signs of chlorosis near the injection sites, as shown in the photograph on the right. **Significant difference at P < 0.01. Standard deviations are shown.
We also investigated the role of sucrose metabolism in disease by treating plants with silver, because this metal is known to interfere with the ethylene‐dependent regulation of sugar signalling and anthocycanin production in Arabidopsis, potato, carnation and maize (Cournac et al., 1991; Hoeberichts et al., 2007; Jeong et al., 2010; Kwon et al., 2011; Rengel and Kordan, 1987). For example, Jeong et al. (2010) characterized a connection between sucrose, light, ethylene and anthocyanin production in Arabidopsis. In this case, ethylene was found to inhibit the expression of the regulators of anthocynanin production, which are induced by light and sucrose. Silver ions are also well‐known inhibitors of bacterial, fungal and plant invertases and some sucrose synthases in vitro (Duan et al., 1993; Ishimoto and Nakamura, 1997; Liu et al., 2006; Nishizawa et al., 1980; Pressey and Avants, 1980; Sung and Su, 1977; Warchol et al., 2002). For example, silver nitrate (4 µm) inhibits wheat invertase activity (Krishnan et al., 1985). We found that the addition of silver nitrate to the soil in increasing concentrations led to reduced plant growth and increased anthocyanin production in the basal portion of the plant stem, as reported previously for maize and Arabidopsis (Figs 7A,B and S3, see Supporting Information). Maize plants that were treated with silver and infected with U. maydis showed reduced disease symptoms and delayed infection progression in a concentration‐dependent manner (Figs 7C,D and S3). Specifically, 1 g and 2.5 g of silver nitrate per tray of plants reduced the DI by 1.1 and 1.3 units, respectively. The DI was reduced by 2 units at the higher concentration of 5 g (see Experimental Procedures for detailed information about silver concentrations in the soil). The reduced virulence correlated with increased plant survival (Figs 7 and S3). We also found that watering plants every 2–3 days with 50 mm silver nitrate during the infection period caused a slight reduction in virulence (<0.5 DI; data not shown). In general, these findings are consistent with the idea that disease progression is influenced by carbon availability and ethylene/sucrose signalling in infected tissue.
Figure 7.
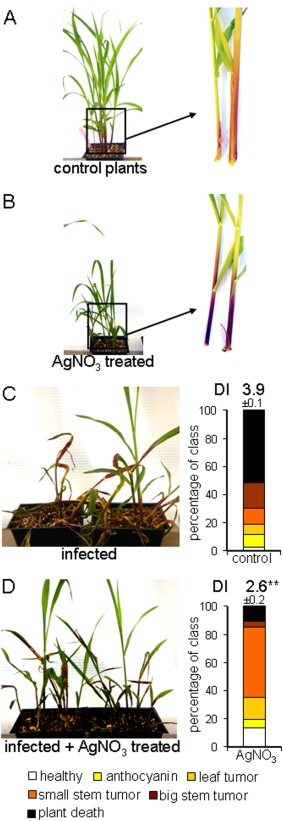
Silver treatment induces anthocyanin production and reduces disease progression in maize. (A) Untreated control plants showed unimpaired growth with little anthocyanin formation in the stem area. (B) Plants treated with 0.412 g AgNO3 per litre of soil showed reduced growth and increased anthocyanin production in the stems and basal parts of the leaves. Note that the plants were photographed at the same magnification in (A) and (B). (C) Disease symptoms and disease rating (DI, disease index) of untreated, but infected, control plants after 14 days. (D) Disease symptoms and disease rating of infected and AgNO3 (0.412 g/L soil)‐treated plants after 14 days. Plants were infected as 7‐day‐old seedlings with a mating‐compatible culture of wt strains at 5 × 106 cells/mL. AgNO3 was added to the soil at the beginning of the experiment. The experiment was repeated three times and with a range of additional silver concentrations (Fig. S3). Standard deviations are shown. **Statistically significant differences at P < 0.01. The plant symptoms were scored as described in Fig. 5.
We next examined starch metabolism during tumour formation because this aspect of U. maydis disease has not been thoroughly investigated and the published results for leaf tumours are somewhat contradictory (Doehlemann et al., 2008; Horst et al., 2008). In addition, Coneva et al. (2012) found that ID1 influenced the ratio of starch to sucrose, and this change in metabolism could potentially contribute to the reduced susceptibility of the id1/id1 mutant. We initially quantified starch content by staining tumour and uninfected stem tissue with Lugol's solution (Text S1). As shown in Fig. 8A, only tumour tissue showed strong staining. To verify that the staining was caused by starch, we quantified starch grains in tissue extracts and found 3.0 ± 1.5 per field of view in infected tissue versus 0.11 ± 0.33 in uninfected tissue (Fig. 8A). Furthermore, the amount of starch determined as reducing sugars per gram of plant material was 2.4 times higher in infected tissue relative to uninfected stems (Fig. 8B). We next obtained a maize mutant defective in starch metabolism and tested the susceptibility to U. maydis. Specifically, we used the sugary1 mutant su1 which shows altered starch production and is important for sweetcorn production (James et al., 1995). We noted that su1 transcripts were up‐regulated only 1.7‐fold during infection (2.1‐fold by qPCR), but that transcripts for other starch‐producing enzymes were also up‐regulated, consistent with an impact of tumour formation on starch metabolism. The other regulated genes included ae1 (amylose extender1, encoding a starch branching enzyme; 14.0‐fold), wx1 (waxy1, encoding a starch synthase; 8.0‐fold), du1 (dull endosperm1, encoding a starch synthase; 2.8‐fold) and sbe1 (starch branching enzyme1, encoding a starch branching enzyme; 2.5‐fold) (Table S1; Fig. S2). We compared disease development in the su1 mutant versus the corresponding wt W64A line and found dramatically increased resistance in the mutant (Fig. 8C). Tumours were rarely observed on mutant plants in three replicates of the assay and plant death was never observed (Fig. 8C). Together, these experiments are consistent with altered sucrose and starch metabolism during infection and an impact of these changes on susceptibility.
It is possible that U. maydis enzymatic activity influences starch production during infection directly via effectors or secreted enzymes. For example, some fungi possess secreted amylases to degrade external starch, and a search of the genome indicated that U. maydis had two candidate amylases (α‐amylase, um02300; γ‐amylase, um04064). The expression of the α‐amylase was unchanged during infection, whereas the γ‐amylase was up‐regulated 117‐fold (M. Kretschmer and J. W. Kronstad, unpublished results). Although neither protein is predicted to be secreted, we tested whether a haploid wt strain and the solo‐pathogenic strain SG200 could grow with 1% starch in minimal medium (MM) as a sole carbon source. Minimal growth was observed for either strain after 5 days of incubation and, importantly, the addition of 1% α‐amylase to the cultures with starch stimulated fungal growth (Fig. S4, see Supporting Information). Taken together, these results reveal that U. maydis infection influences carbohydrate metabolism/signalling and starch accumulation during tumour formation, but that the fungus is unable to use starch as a sole carbon source, at least in vitro.
Discussion
Transcriptome profiling of host–pathogen interactions generates testable hypotheses to determine whether the observed changes reflect adaptations that benefit the pathogen or the host. RNA‐Seq analysis provided a comprehensive view of the transcriptional changes in maize stem tumours, and we observed a differential expression of functions that was in general agreement with previous studies (Basse, 2005; Doehlemann et al., 2008; Gao et al., 2013; Horst et al., 2008, 2010a,b; Skibbe et al., 2010; Voll et al., 2011). In addition, we identified a novel impact of disease on meristem regulators, established a greater appreciation of the influence of infection on TFs, as noted by Gao et al. (2013), and identified the regulation of transcript levels for TEs. TE expression has been linked previously to the activation of plant defence (Grandbastien et al., 1997). For example, a TE in oats is highly expressed on wounding, exposure to UV light, jasmonic acid and salicylic acid, and infection with Puccinia coronata (Kimura et al., 2001). As discussed below, we also generated testable hypotheses regarding the significance of transcript changes in virulence assays with maize mutants (id1 and su1) and plants treated with sugars and silver nitrate. These approaches confirmed the influence of metabolic changes on fungal proliferation and symptom development, and distinguished between changes that benefit the pathogen versus the host. An overview of the metabolic functions in maize impacted by the interaction with U. maydis is presented in Fig. S5 (see Supporting Information).
Ustilago maydis is known to infect actively growing vegetative and reproductive tissue on all aerial parts of maize, and to target meristematic tissue with dividing cells for tumour formation (Gao et al., 2013; Walbot and Skibbe, 2010; Wenzler and Meins, 1987). Meristematic tissues and U. maydis‐induced tumours represent sink organs for the import of carbon and nitrogen from source tissues (Horst et al., 2010a, 2010b). Our analysis suggests that U. maydis disturbs functions in meristem maintenance and determinacy, leading to uncontrolled proliferation. We tested the importance of the vegetative to reproductive transition and carbohydrate allocation by examining symptom development in the id1/id1 mutant, which lacks a key regulator of floral induction (Coneva et al., 2007, 2012). We found that id1/id1 mutant plants were less susceptible to U. maydis, indicating that ID1 function normally contributes to symptom development on inoculation of stem tissue in 7‐day‐old plants. These results are consistent with the findings of Colasanti et al. (1998, 2006) that id1 is expressed primarily in the whorl of unexpanded leaves in the stem at similar time points. Interestingly, we also observed that heterozygous Id1/id1 plants had a susceptibility that was intermediate between wt (Id1/Id1) and homozygous mutant plants (id1/id1). This observation is consistent with gene expression studies which showed a relatively small number (55) of differentially expressed genes between the heterozygous (Id1/id1) and homozygous (id1/id1) lines compared with a larger number (460) between the wt (Id1/Id1) and homozygous (id1/id1) lines (Coneva et al., 2007, 2012). At the later stage of infection, when tumours have developed, our RNA‐Seq analysis revealed that the id1 transcript level was reduced relative to uninfected stem tissue. Thus, the subsequent dramatic reprogramming that results in tumour formation later in infection also impacts id1 expression, although transcript levels for the gene may not be reduced in other non‐tumour parts of the plant. We also observed that infected id1/id1 plants were not pigmented compared with the intensely purple phenotype of infected wt plants. ID1 may therefore influence secondary metabolism leading to anthocyanin production, and we note that a chalcone synthase (encoded by c2) is up‐regulated in id1/id1 plants (Coneva et al., 2007). Ustilago maydis is known to induce anthocyanin production by delivery of the fungal effector Tin2, perhaps as a mechanism to divert metabolites away from defence processes, such as lignin formation (Tanaka et al., 2014). Indeed, a maize mutant with altered lignin biosynthesis shows enhanced susceptibility to U. maydis (Tanaka et al., 2014). ID1 may contribute to the regulation of anthocyanin production, perhaps in conjunction with the maize kinase TTK1 which is targeted by Tin2 and activates genes for anthocyanin biosynthesis.
The influence of ID1 on susceptibility to U. maydis may also be caused in part by the altered carbohydrate metabolism and allocation, and this would be consistent with our observations on starch accumulation and sucrose metabolism in tumour tissue, as well as metabolite studies by other groups, as discussed below (Horst et al., 2010a, 2010b; Voll et al., 2011). A comparison of gene expression in id1/id1 versus wt (Id1/Id1) plants revealed that source leaves from id1/id1 plants show altered expression of genes for starch and sucrose metabolism (Coneva et al., 2012). For example, the sut1 gene encodes a transporter for phloem loading of sucrose in source leaves for export to sink tissues, and sut1 transcript levels were lower in id1/id1 leaves (Coneva et al., 2012; Ludewig and Flügge, 2013; Slewinski, 2012; Slewinski et al., 2009). However, the role of SUT1 in sucrose accumulation in sink stem tissue is not yet clear (Bihmidine et al., 2013). A comparison of metabolite levels indicated that id1/id1 mutant leaves also show higher levels of starch and sucrose than wt plants, and that the mutants show a decreased transitory starch to sucrose ratio. These observations led Coneva et al. (2012) to speculate that carbohydrate allocation in leaves is an important cue for the floral transition in maize. In particular, carbohydrate delivery to sink tissues is essential for floral induction because sucrose supports inflorescence growth. Therefore, the susceptibility of an id1/id1 mutant may reflect a broader impact of the mutation at the interface between metabolism and the floral transition.
A role for carbohydrate metabolism, as reflected by changes in starch and sucrose levels, is also supported by the impact of the su1 mutation and sugar and silver nitrate treatment on the U. maydis–maize interaction. SU1 is an isoamylase‐type starch‐debranching enzyme believed to participate in starch biosynthesis, and the su1/su1 mutation results in increased sucrose in maize kernels (resulting in commercially important sweetcorn). It is also known that su1/su1 plants are delayed in germination and have higher soluble sugars in stem tissue. Furthermore, the su1 mutation reduces amylopectin and increases phytoglycogen concentrations (James et al., 1995). Our observation of greatly reduced tumour formation on su1/su1 plants is consistent with perturbed starch formation and turnover (and altered sucrose levels), which may interfere with fungal proliferation. Starch and sucrose metabolism in infected tissue have been examined in some detail, although a clear picture has yet to emerge. For example, Horst et al. (2008) reported low levels of transient starch in infected leaves at an early stage of tumour formation (6 dpi). However, a chase experiment with radioactively labelled CO2 at 6 dpi in infected leaves indicated a slight accumulation of radioactively labelled carbon in the insoluble carbohydrate fraction (Horst et al., 2010b). In a metabolite analysis, no differences for starch accumulation at 8 dpi in leaf tumours were seen (Horst et al., 2010a). However, Doehlemann et al. (2008) observed the regulation of carbohydrate genes related to sucrose and starch metabolism in a transcriptome analysis, but did not report changes in starch accumulation at later stages (8 dpi) of leaf tumour formation. Voll et al. (2011) found that sucrose is elevated in infected maize tissue and noted from transcriptome analysis that a sucrose synthase gene was induced six‐fold in tumours. Horst et al. (2010b) also concluded that sucrose imported from systemic leaves was important for cell wall biosynthesis in tumour cells, the generation of hexoses to contribute osmotic pressure for tumour cell expansion and as a carbon source for U. maydis. In addition, Doehlemann et al. (2008) reported an increase of 30% at 8 dpi (see table 1 in Doehlemann et al., 2008). Sucrose is clearly important as a carbon source for U. maydis, as demonstrated by the role of the pathogen sucrose transporter Srt1 in virulence (Wahl et al., 2010).
Overall, a role for sucrose in the maize–U. maydis interaction is consistent with the emerging function of the sugar as a signal molecule in plants (reviewed by Tognetti et al., 2013). In particular, sucrose influences a number of metabolic processes relevant to disease, including anthocyanin synthesis, carbon and nitrogen assimilation/transport, photosynthesis, chlorophyll synthesis, amino acid biosynthesis and copper homeostasis. It also influences flowering, plant size, morphology, the circadian clock and senescence (Bolouri‐Moghaddam and Van den Ende, 2013; Roitsch et al., 2003; Roycewicz and Malamy, 2012; Tsai and Gazzarrini, 2014; Wingler and Roitsch, 2008). We noted that silver nitrate also interferes with sucrose and ethylene signalling, and anthocyanin production, in a light‐dependent manner in plants (Cournac et al., 1991; Jeong et al., 2010). Consistent with the observed transcriptional changes, silver nitrate treatment reduced the susceptibility to U. maydis, although additional experiments are needed to determine the mechanism, especially given the potential pleiotropic influence of silver. The impact of silver treatment on anthocyanin production and sucrose metabolism is consistent with known factors relevant to disease (Tanaka et al., 2014; Wahl et al., 2010). However, ethylene has numerous effects, including the promotion of the degradation of starch to single sugars and sucrose during fruit ripening (Thammawong and Arakawa, 2010). We also noted that transcripts for ethylene‐related functions were up‐regulated in tumours. For example, the ethylene synthase acs2 (GRMZM2G164405) transcript was up‐regulated 8.1‐fold, together with the ethylene‐responsive TFs (eil4, 17.9‐fold; eil6, 11.0‐fold) (Table S1).
Given our incomplete understanding of carbohydrate metabolism in maize and the complexities of connections with signalling and defence, it is clear that additional work is needed to fit the transcriptional changes observed in tumours into a metabolic model of the interaction. Key future experiments would include virulence assays with additional maize mutants perturbed in carbohydrate metabolism and ethylene signalling. For example, it would be particularly informative to examine mutants defective in sucrose transporters [e.g. sucrose transporter1 (sut1)], sucrose synthases [e.g. sucrose synthase1 (sus1) and sucrose synthase2 (sus2)], the ethylene synthase acs2 and the ethylene responsive TFs (eil4 and eil6). Subsequent studies could examine the potential targeting of these processes by U. maydis effectors, and the location and biochemical activities of the key functions revealed by virulence assays with maize mutants.
Experimental Procedures
Plant and fungal growth conditions
Maize seeds were planted in 340‐cm3 pots and grown in standard soil in a glasshouse with long‐day conditions. All experiments, except infection studies of Z. mays mutants, were conducted with the Golden Bantam variety. Other maize lines and mutants used in this study were su1 (W64a) and id1 (B73), and their respective wt lines. The maize lines were obtained from Drs Alan Myers and Joseph Colasanti and, in the case of the id1 mutants, the genotypes of each plant were confirmed by PCR. Primers for id1 were IdF (GAGCTCTGGGGGACTTGACTG), IdR4 (CTAGGTTTTCTCTCGATCCGTCCG) and DsR (GCTTTCTTGCATGGGATGGGCCTC). The su1 mutant line was the reference inbred line in the W64a background. The haploid U. maydis strains 001 (a2b2) and 002 (a1b1) were employed. Fungal strains were grown on potato dextrose agar (PDA) plates or in liquid cultures of potato dextrose broth (PDB) or MM at 30°C and 200 rpm.
RNA‐Seq analysis
Seven‐day‐old seedlings were infected with a mixture of 001 and 002 fungal cells. Ustilago cells were grown overnight in 5 ml of PDB and washed three times in distilled H2O. The cell number was determined with a haemocytometer and 5 × 106 cells/mL of each strain were mixed to achieve the mating culture of 1 × 107 cells/mL in distilled H2O. Golden Bantam seedlings were infected 7 days after planting at the VE (one to two leaf tips) stage by injecting mating culture into the crown area (0.5–1.0 cm above the soil level) into the centre of the seedling shoot. At day 17 (V3) after planting (i.e. 10 dpi), tumours which formed at the crown area between the soil and the first collar (i.e. up to 5 cm above the crown area in the seedling stem) were harvested from multiple plants (three plants per replicate). Corresponding tissue of uninfected plants was harvested. Infected plants showed slightly stunted growth compared with uninfected plants (∼33% reduced). Tumours started to form with the initial sign being the appearance of chlorosis. Three uninfected and infected biological replicates were used. The plant material was frozen in liquid nitrogen and ground to a fine powder in a liquid nitrogen‐cooled mortar with a pestle. The frozen powder of infected and uninfected corn plants was transferred to 1 mL of extraction buffer and RNA was extracted according to the manufacturer's recommendations (RNAeasy kit, Qiagen, Toronto, ON, Canada). An on‐column DNAse I treatment was conducted. RNA concentrations were determined with a Nanodrop spectrophotometer (Fisher Scientific, Ottawa, ON, Canada) and the integrity of the RNA was checked on a denaturing RNA gel.
The RNA quality control, library preparation, sequencing and part of the bioinformatic analysis for the six samples (three infected and three uninfected) was conducted by GENEWIZ with a 1 × 50 configuration on an Illumina HighSeq (Genewiz, South Plainfield, NJ, USA) platform according to the manufacturer's recommended protocols. Raw reads were quality trimmed using Trimmomatic v. 0.32 employing the following settings (Bolger et al., 2014): ‘ILLUMINACLIP:TruSeq3‐PE.fa:2:30:10 LEADING:10 TRAILING:10 SLIDINGWINDOW:5:10 MINLEN:50’. Filtered RNA‐Seq reads were aligned to the Z. mays B73 genome at http://www.maizegdb.org/version 2 using TopHat v. 2.0.13. Transcript assembly based on aligned reads was performed using Cufflinks 2.1.1 (Trapnell et al., 2009, 2010). Read counts per gene were normalized amongst libraries by calculating the reads per kilobase of exon per million mapped reads (RPKM; Mortazavi et al., 2008). RPKM, differential expression fold changes and significance thresholds were calculated using the R package cummeRbund version 0.1.3 using the method of Trapnell et al., 2009, 2010 (Method 1). Additional data analyses were conducted by GENEWIZ (Method 2) and in house with a differential expression analysis as described previously (Method 3; Anders and Huber, 2010). In addition to reference genome‐guided differential expression analyses, transcripts were assembled de novo directly from filtered RNA‐Seq reads. For this, Trinity version 2.1.0 was used in default settings to generate contigs of individual transcripts (Method 1; Grabherr et al., 2011).
The results from all three analyses were combined to determine the final expression values, fold changes and P values for the top regulated maize genes and the classical gene list. Furthermore, q‐values were also determined for analyses 1 and 3. In general, expression changes of the three uninfected samples compared with the three infected samples were considered to be significantly different if the P value was below 0.05. More specifically, several conditions were used for the categorization of the genes into the highest up‐ and down‐regulated categories. First, a gene was only included in the analysis if it was expressed in all three biological replicates of a given condition (e.g., infected or uninfected). Second, a gene had to have an average RPKM of ∼1 (10 times the detection limit, as determined by qPCR). Third, a gene had to have the same trend in all three biological replicates; it was excluded if it was only highly expressed in one replicate of the condition, but the other replicates were similar to the three replicates of the second condition. Genes that showed different expression between the different analysis methods were also excluded, as they indicate gene annotation problems between the different analysis methods. The expression values of all known maize transcripts are shown in Table S1 (parts 1 and 2). Expression changes were calculated with infected divided by uninfected values: 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively. Additional information on the bioinformatics analysis of the RNA‐Seq data is given in Text S1. The data have been deposited in the National Center for Biotechnology Information (NCBI) Short Read Archive under accession numbers SRR3203312 to SRR3203318.
Validation of RNA‐Seq data
RNA was extracted from seedlings at 10 dpi, tassels at 15 dpi and ears at 14 dpi to validate the RNA‐Seq data and to check the expression of the chosen genes in other infected organs (tassel and ear). Tassels and emerging ears were infected as described previously (Gao et al., 2013; Walbot and Skibbe, 2010). RNA was extracted with a citrate–sodium dodecylsulphate (SDS)‐based buffer system, as described previously (Kretschmer et al., 2014). RNA was DNAse I treated with the Ambion DNAse free turbo kit and 1 µg of total RNA was transcribed into cDNA with the verso cDNA kit (Fisher Scientific, Ottawa, ON, Canada). Relative quantification of gene expression was performed with elongation‐factor 1 and actin‐depolymerization‐factor alpha as control genes. Expression changes were determined with the ΔΔCT method for the genes in Table S7 (see Supporting Information).
Infection of Z. mays mutants to determine susceptibility to U. maydis
One‐week‐old seedlings of the different mutants and corresponding wt plants were infected with 50 µL of mating culture, prepared as described above for the RNASeq analysis, except that the final cell concentration was 5 × 106 cells/mL to generate a wider range of symptoms. This approach allows the scoring of increased and decreased symptoms of the infected plants. The infections were scored after 14 days, as described previously (Kretschmer et al., 2012). The su1 mutant showed delayed germination. Thus, seedlings of the mutant and the control wt line were infected at 10 days after planting.
Glucose and sucrose treatment during infection
Fourteen‐day‐old seedlings were injected with a mixture of 001 and 002 wt cells at 5 × 106 cells/mL using a 26‐gauge needle. The infection was allowed to progress for 60 h before the different treatments were started. Fifty microlitres of water at pH 7.0 and containing 0.438 m sucrose or 1 m glucose were injected at the site of infection with a 30‐gauge needle at each time point (Peng et al., 2013). The treatments were conducted in two 5‐day blocks with a 1‐day break. Controls included uninfected and untreated plants, and uninfected plants that were treated with the different solutions (water pH 7.0, 0.438 m sucrose or 1 m glucose). Infected plants included the untreated but infected control and infected plants treated with the solutions. Weak and small plants were eliminated at the beginning of the treatments. Secondary infections were also eliminated. Control plants and infected plants were evaluated at 14 dpi.
Silver nitrate treatment
Silver nitrate was added to the soil before planting the seeds at 0 g, 1 g (0.165 g/L soil), 2.5 g (0.412 g/L soil) and 5 g (0.823 g/L soil) per tray. Seeds were planted directly in the soil and the pots were transferred to trays and hand watered during the treatment. Ustilago maydis strains were grown and used for infection as described above in the section on Plant and fungal growth conditions. Control plants and infected plants were evaluated at 14 dpi.
Supporting information
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 RNA‐sequencing (RNA‐Seq) read quality control, P value distribution and top 50 up‐ and down‐regulated genes shown in a heat map. (A) q‐value distribution of the three uninfected and three infected libraries. (B) Distribution of P values of expressed genes of the three uninfected relative to three infected libraries. P values below 0.05 are considered to be significantly different. (C) Heat map of the top 50 up‐ and down‐regulated host genes during tumour formation.
Fig. S2 Expression of key carbohydrate metabolism genes during tumour formation. Blue arrows indicate significant down‐regulation of genes or entire gene families at the indicated steps by at least two‐fold. Red arrows indicate significant up‐regulation of genes or entire gene families at the indicated steps by at least two‐fold. Red (up‐regulation) or blue (down‐regulation) text highlights the changes in key aspects of the pathways. Leaf or flower indicates the preferred expression of the genes in different maize tissues.
Fig. S3 Silver nitrate treatment in the soil reduces fungal virulence. Zero grams, 1 g (0.165 g/L soil), 2.5 g (0.412 g/L soil) or 5 g (0.823 g/L soil) of AgNO3 per tray were added to the soil before planting the maize seed. The small seedlings were infected 7 days after planting. After 14 days, the disease symptoms were scored (DI, disease index) (A). Control plants showed a silver dose‐dependent reduction in growth and increased anthocyanin formation in the stem (B). Increasing silver concentrations reduced the disease symptoms (C). Infection studies were repeated three times with different concentrations. Significance levels: *P < 0.05; **P < 0.01. Standard deviations are shown.
Fig. S4 Starch as single carbon source does not support fungal growth. The growth of Ustilago maydis was supported with 1% glucose or 1% starch with 1% α‐amylase as single carbon source in minimal medium (MM). 1% starch alone did not support the growth of the haploid strain or the solo‐pathogenic strain. 1% α‐amylase alone led to minor fungal growth, most likely because of carbon sources in the enzyme storage buffer (e.g. glycerol).
Fig. S5 Summary of the major metabolic changes predicted by transcriptome analysis of maize genes during tumour formation by Ustilago maydis. Functions in red were up‐regulated, whereas those in blue were down‐regulated.
Table S1 Expression of all B73 transcripts, highest up‐ and down‐regulated genes, regulation of transcription factors and regulation of pathway genes during tumour formation. The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S2 Expression of Golden Bantam‐specific transcripts and highest up‐ and down‐regulated genes. The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S3 Confirmation of the RNA‐sequencing (RNA‐Seq) expression changes for selected genes in seedling stems and in tumours in tassels and ears. The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S4 Gene ontology (GO) term enrichment analysis of up‐ and down‐regulated functions during tumour formation.
Table S5 Regulation of genes from the classical gene list of Zea mays during tumour formation induced by Ustilago maydis in seedling stems at 10 days post‐inoculation (dpi). The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S6 Categorization of genes from the classical gene list into functional groups and their regulation during tumour formation.
Table S7 Primers used in this study.
Text S1 Supporting experimental procedures, and results and discussion.
Acknowledgements
This work was supported by the Natural Sciences and Engineering Research Council of Canada (RGPIN 41758) to JWK. DC was supported by grant PA00P3_145360 from the Swiss National Science Foundation. We thank Sanja Rogic and Paul Pavlidis for assistance with the RNA‐Seq data analysis and constructive discussions. The authors thank George Chuck for useful discussions and Dr Alan Myers and Joseph Colasanti for generously supplying the maize mutants. We also acknowledge Genewiz (Genewiz, South Plainfield, NJ, USA), Inc. for support with the RNA‐Seq analysis.
References
- Anders, S. and Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse, C.W. (2005) Dissecting defense‐related and developmental transcriptional responses of maize during Ustilago maydis infection and subsequent tumor formation. Plant Physiol. 138, 1774–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basse, C.W. and Steinberg, G. (2004) Ustilago maydis, model system for analysis of the molecular basis of fungal pathogenicity. Mol. Plant Pathol. 5, 83–92. [DOI] [PubMed] [Google Scholar]
- Bihmidine, S. , Hunter, C.T., III , Johns, C.E. , Koch, K.E. and Braun, D.M. (2013) Regulation of assimilate import into sink organs: update on molecular drivers of sink strength. Front Plant Sci. 4, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M. , Lohse, M. and Usadel, B. (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolouri‐Moghaddam, M.R. and Van den Ende, W. (2013) Sweet immunity in the plant circadian regulatory network. J. Exp. Bot. 64, 1439–1449. [DOI] [PubMed] [Google Scholar]
- Bortiri, E. and Hake, S. (2007) Flowering and determinacy in maize. J. Exp. Bot. 58, 909–916. [DOI] [PubMed] [Google Scholar]
- Brefort, T. , Doehlemann, G. , Mendoza‐Mendoza, A. , Reissmann, S. , Djamei, A. and Kahmann, R. (2009) Ustilago maydis as a pathogen. Annu. Rev. Phytopathol. 47, 423–445. [DOI] [PubMed] [Google Scholar]
- Colasanti, J. , Yuan, Z. and Sundaresan, V. (1998) The indeterminate gene encodes a zinc finger protein and regulates a leaf‐generated signal required for the transition to flowering in maize. Cell, 93, 593–603. [DOI] [PubMed] [Google Scholar]
- Colasanti, J. , Tremblay, R. , Wong, A.Y. , Coneva, V. , Kozaki, A. and Mable, B.K. (2006) The maize INDETERMINATE1 flowering time regulator defines a highly conserved zinc finger protein family in higher plants. BMC Genomics, 7, 158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coneva, V. , Zhu, T. and Colasanti, J. (2007) Expression differences between normal and indeterminate1 maize suggest downstream targets of ID1, a floral transition regulator in maize. J. Exp. Bot. 58, 3679–3693. [DOI] [PubMed] [Google Scholar]
- Coneva, V. , Guevara, D. , Rothstein, S.J. and Colasanti, J. (2012) Transcript and metabolite signature of maize source leaves suggests a link between transitory starch to sucrose balance and the autonomous floral transition. J. Exp. Bot. 63, 5079–5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cournac, L. , Dimon, B. , Carrier, P. , Lohou, A. and Chagvardieff, P. (1991) Growth and photosynthetic characteristics of Solanum tuberosum plantlets cultivated in vitro in different conditions of aeration, sucrose supply, and CO(2) enrichment. Plant Physiol. 97, 112–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djamei, A. , Schipper, K. , Rabe, F. , Ghosh, A. , Vincon, V. , Kahnt, J. , Osorio, S. , Tohge, T. , Fernie, A.R. , Feussner, I. , Feussner, K. , Meinicke, P. , Stierhof, Y.D. , Schwarz, H. , Macek, B. , Mann, M. and Kahmann, R. (2011) Metabolic priming by a secreted fungal effector. Nature, 478, 395–398. [DOI] [PubMed] [Google Scholar]
- Doehlemann, G. , Wahl, R. , Horst, R.J. , Voll, L.M. , Usadel, B. , Poree, F. , Stitt, M. , Pons‐Kühnemann, J. , Sonnewald, U. , Kahmann, R. and Kämper, J. (2008) Reprogramming a maize plant: transcriptional and metabolic changes induced by the fungal biotroph Ustilago maydis . Plant J. 56, 181–195. [DOI] [PubMed] [Google Scholar]
- Duan, K.J. , Sheu, D.C. and Chen, J.S. (1993) Purification and characterization of beta‐fructofuranosidase from Aspergillus japonicus TIT‐KJ1. Biosci. Biotechnol. Biochem. 57, 1811–1815. [DOI] [PubMed] [Google Scholar]
- Eveland, A.L. , Goldshmidt, A. , Pautler, M. , Morohashi, K. , Liseron‐Monfils, C. , Lewis, M.W. , Kumari, S. , Hiraga, S. , Yang, F. , Unger‐Wallace, E. , Olson, A. , Hake, S. , Vollbrecht, E. , Grotewold, E. , Ware, D. and Jackson, D. (2014) Regulatory modules controlling maize inflorescence architecture. Genome Res. 24, 431–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, L. , Kelliher, T. , Nguyen, L. and Walbot, V. (2013) Ustilago maydis reprograms cell proliferation in maize anthers. Plant J. 75, 903–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr, M.G. , Haas, B.J. , Yassour, M. , Levin, J.Z. , Thompson, D.A. , Amit, I. , Adiconis, X. , Fan, L. , Raychowdhury, R. , Zeng, Q. , Chen, Z. , Mauceli, E. , Hacohen, N. , Gnirke, A. , Rhind, N. , di Palma, F. , Birren, B.W. , Nusbaum, C. , Lindblad‐Toh, K. , Friedman, N. and Regev, A. (2011) Full‐length transcriptome assembly from RNA‐Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandbastien, M.A. , Lucas, H. , Morel, J.B. , Mhiri, C. , Vernhettes, S. and Casacuberta, J.M. (1997) The expression of the tobacco Tnt1 retrotransposon is linked to plant defense responses. Genetica, 100, 241–252. [PubMed] [Google Scholar]
- Hemetsberger, C. , Herrberger, C. , Zechmann, B. , Hillmer, M. and Doehlemann, G. (2012) The Ustilago maydis effector Pep1 suppresses plant immunity by inhibition of host peroxidase activity. PLoS Pathog. 8, e1002684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry, R. (2009) Plant Resources for Food, Fuel and Conservation. London: Earthscan; ISBN‐13: 978–1844077212. [Google Scholar]
- Hoeberichts, F.A. , van Doorn, W.G. , Vorst, O. , Hall, R.D. and van Wordragen, M.F. (2007) Sucrose prevents up‐regulation of senescence‐associated genes in carnation petals. J. Exp. Bot. 58, 2873–2885. [DOI] [PubMed] [Google Scholar]
- Horst, R.J. , Engelsdorf, T. , Sonnewald, U. and Voll, L.M. (2008) Infection of maize leaves with Ustilago maydis prevents establishment of C4 photosynthesis. J. Plant Physiol. 165, 19–28. [DOI] [PubMed] [Google Scholar]
- Horst, R.J. , Doehlemann, G. , Wahl, R. , Hofmann, J. , Schmiedl, A. , Kahmann, R. , Kämper, J. , Sonnewald, U. and Voll, L.M. (2010a) Ustilago maydis infection strongly alters organic nitrogen allocation in maize and stimulates productivity of systemic source leaves. Plant Physiol. 152, 293–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horst, R.J. , Doehlemann, G. , Wahl, R. , Hofmann, J. , Schmiedl, A. , Kahmann, R. , Kämper, J. and Voll, L.M. (2010b) A model of Ustilago maydis leaf tumor metabolism. Plant Signal Behav. 5, 1446–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishimoto, M. and Nakamura, A. (1997) Purification and properties of beta‐fructofuranosidase from Clostridium perfringens . Biosci. Biotechnol. Biochem. 61, 599–603. [DOI] [PubMed] [Google Scholar]
- James, M.G. , Robertson, D.S. and Myers, A.M. (1995) Characterization of the maize gene sugary1, a determinant of starch composition in kernels. Plant Cell, 7, 417–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong, S.W. , Das, P.K. , Jeoung, S.C. , Song, J.Y. , Lee, H.K. , Kim, Y.K. , Kim, W.J. , Park, Y.I. , Yoo, S.D. , Choi, S.B. , Choi, G. and Park, Y.I. (2010) Ethylene suppression of sugar‐induced anthocyanin pigmentation in Arabidopsis. Plant Physiol. 154, 1514–1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kämper, J. , Kahmann, R. , Bölker, M. , Ma, L.J. , Brefort, T. , Saville, B.J. , Banuett, F. , Kronstad, J.W. , Gold, S.E. , Müller, O. , Perlin, M.H. , Wösten, H.A. , de Vries, R. , Ruiz‐Herrera, J. , Reynaga‐Peña, C.G. , Snetselaar, K. , McCann, M. , Pérez‐Martín, J. , Feldbrügge, M. , Basse, C.W. , Steinberg, G. , Ibeas, J.I. , Holloman, W. , Guzman, P. , Farman, M. , Stajich, J.E. , Sentandreu, R. , González‐Prieto, J.M. , Kennell, J.C. , Molina, L. , Schirawski, J. , Mendoza‐Mendoza, A. , Greilinger, D. , Münch, K. , Rössel, N. , Scherer, M. , Vranes, M. , Ladendorf, O. , Vincon, V. , Fuchs, U. , Sandrock, B. , Meng, S. , Ho, E.C. , Cahill, M.J. , Boyce, K.J. , Klose, J. , Klosterman, S.J. , Deelstra, H.J. , Ortiz‐Castellanos, L. , Li, W. , Sanchez‐Alonso, P. , Schreier, P.H. , Häuser‐Hahn, I. , Vaupel, M. , Koopmann, E. , Friedrich, G. , Voss, H. , Schlüter, T. , Margolis, J. , Platt, D. , Swimmer, C. , Gnirke, A. , Chen, F. , Vysotskaia, V. , Mannhaupt, G. , Güldener, U. , Münsterkötter, M. , Haase, D. , Oesterheld, M. , Mewes, H.W. , Mauceli, E.W. , DeCaprio, D. , Wade, C.M. , Butler, J. , Young, S. , Jaffe, D.B. , Calvo, S. , Nusbaum, C. , Galagan, J. and Birren, B.W. (2006) Insights from the genome of the biotrophic fungal plant pathogen Ustilago maydis . Nature, 444, 97–101. [DOI] [PubMed] [Google Scholar]
- Kimura, Y. , Tosa, Y. , Shimada, S. , Sogo, R. , Kusaba, M. , Sunaga, T., Betsuyaku, S., Eto, Y., Nakayashiki, H. and Mayama, S. (2001) OARE‐1, a Ty1‐copia retrotransposon in oat activated by abiotic and biotic stresses. Plant Cell Physiol. 42, 1345–1354. [DOI] [PubMed] [Google Scholar]
- Koeck, M. , Hardham, A.R. and Dodds, P.N. (2011) The role of effectors of biotrophic and hemibiotrophic fungi in infection. Cell Microbiol. 13, 1849–1857. doi: 10.1111/j.1462-5822.2011.01665.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer, M. , Klose, J. and Kronstad, J.,W. (2012) Defects in mitochondrial and peroxisomal β‐oxidation influence virulence in the maize pathogen Ustilago maydis . Eukaryot. Cell, 11, 1055–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer, M. , Reiner, E. , Hu, G. , Tam, N. , Oliveira, D.L. , Caza, M. , Yeon, J.H. , Kim, J. , Kastrup, C.J. , Jung, W.H. and Kronstad, J.W. (2014) Defects in phosphate acquisition and storage influence virulence of Cryptococcus neoformans . Infect. Immun. 82, 2697–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretschmer, M. , Croll, D. and Kronstad, J.W. (2016) Chloroplast‐associated metabolic functions influence the susceptibility of maize to Ustilago maydis . Mol. Plant Pathol. doi: 10.1111/mpp.12485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan, H.B. , Blanchette, J.T. and Okita, T.W. (1985) Wheat invertases. Characterization of cell wall‐bound and soluble forms. Plant Physiol. 78, 241–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronstad, J.W. and Leong, S.A. (1990) The b mating‐type locus of Ustilago maydis contains variable and constant regions. Genes Dev. 4, 1384–1395. [DOI] [PubMed] [Google Scholar]
- Kwon, Y. , Oh, J.E. , Noh, H. , Hong, S.W. , Bhoo, S.H. and Lee, H. (2011) The ethylene signaling pathway has a negative impact on sucrose‐induced anthocyanin accumulation in Arabidopsis. J. Plant Res. 124, 193–200. [DOI] [PubMed] [Google Scholar]
- Leuthner, B. , Aichinger, C. , Oehmen, E. , Koopmann, E. , Müller, O. , Müller, P. , Kahmann, R. , Bölker, M. and Schreier, P.H. (2005) H2O2‐producing glyoxal oxidase is required for filamentous growth and pathogenicity in Ustilago maydis . Mol. Genet. Genomics, 272, 639–650. [DOI] [PubMed] [Google Scholar]
- van der Linde, K. , Hemetsberger, C. , Kastner, C. , Kaschani, F. , van der Hoorn, R.A. , Kumlehn, J. and Doehlemann G. (2012) A maize cystatin suppresses host immunity by inhibiting apoplastic cysteine proteases. Plant Cell, 24, 1285–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, C. , Huang, L. , Chang, C. and Sung, H. (2006) Purification and characterization of soluble invertases from suspension‐cultured bamboo (Bambusa edulis) cells. Food Chem. 96, 621–631. [Google Scholar]
- Ludewig, F. and Flügge, U.I. (2013) Role of metabolite transporters in source–sink carbon allocation. Front. Plant Sci. 4, 231. doi: 10.3389/fpls.2013.00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monaco, M.K. , Sen, T.Z. , Dharmawardhana, P.D. , Ren, L. , Schaeffer, M. , Naithani, S. , Amarasinghed, V. , Thomason, J. , Harper, L. , Gardiner, J. , Cannon, E.K.S. , Lawrence, C.J. , Ware, D. and Jaiswal, P. (2013) Maize metabolic network construction and transcriptome analysis. Plant Genome. 9, 1–12. [Google Scholar]
- Mortazavi, A. , Williams, B.A. , McCue, K. , Schaeffer, L. and Wold, B. (2008) Mapping and quantifying mammalian transcriptomes by RNA‐Seq. Nat. Methods, 5, 621–628. [DOI] [PubMed] [Google Scholar]
- Mueller, A.N. , Ziemann, S. , Treitschke, S. , Aßmann, D. and Doehlemann, G. (2013) Compatibility in the Ustilago maydis–maize interaction requires inhibition of host cysteine proteases by the fungal effector Pit2. PLoS Pathog. 9, e1003177. doi: 10.1371/journal.ppat.1003177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller, O. , Kahmann, R. , Aguilar, G. , Trejo‐Aguilar, B. , Wu, A. and de Vries, R.P. (2008) The secretome of the maize pathogen Ustilago maydis . Fungal Genet. Biol. 45, 63–70. doi: 10.1016/j.fgb.2008.03.012 [DOI] [PubMed] [Google Scholar]
- Nishizawa, M. , Maruyama, Y. and Nakamura, M. (1980) Purification and characterization of invertase isoenzymes from Fusarium oxysporum . Agric. Biol. Chem. 44, 489–498. [Google Scholar]
- Pautler, M. , Tanaka, W. , Hirano, H.Y. and Jackson, D. (2013) Grass meristems I: shoot apical meristem maintenance, axillary meristem determinacy and the floral transition. Plant Cell Physiol. 54, 302–312. [DOI] [PubMed] [Google Scholar]
- Peng, Y. , Li, C. and Fritschi, F.B. (2013) Apoplastic infusion of sucrose into stem internodes during female flowering does not increase grain yield in maize plants grown under nitrogen‐limiting conditions. Physiol. Plant. 148, 470–480. [DOI] [PubMed] [Google Scholar]
- Pressey, R. and Avants, J.K. (1980) Invertases in oat seedlings. Plant Physiol. 65, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redkar, A. , Hoser, R. , Schilling, L. , Zechmann, B. , Krzymowska, M. , Walbot, V. and Doehlemann, G. (2015) A secreted effector protein of Ustilago maydis guides maize leaf cells to form tumors. Plant Cell, 27, 1332–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rengel, Z. and Kordan, H.A. (1987) Effects of growth regulators on light‐dependent anthocyanin production in Zea mays seedlings. Physiol. Plant. 69, 511–516. [Google Scholar]
- Roitsch, T. , Balibrea, M.E. , Hofmann, M. , Proels, R. and Sinha, A.K. (2003) Extracellular invertase: key metabolic enzyme and PR protein. J. Exp. Bot. 54, 513–524. [DOI] [PubMed] [Google Scholar]
- Roycewicz, P. and Malamy, J.E. (2012) Dissecting the effects of nitrate, sucrose and osmotic potential on Arabidopsis root and shoot system growth in laboratory assays. Philos. Trans. R. Soc. London B: Biol. Sci. 367, 1489–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvi, S. , Sponza, G. , Morgante, M. , Tomes, D. , Niu, X. , Fengler, K.A. , Meeley, R. , Ananiev, E.V. , Svitashev, S. , Bruggemann, E. , Li, B. , Hainey, C.F. , Radovic, S. , Zaina, G. , Rafalski, J.A. , Tingey, S.V. , Miao, G.H. , Phillips, R.L. and Tuberosa, R. (2007) Conserved noncoding genomic sequences associated with a flowering‐time quantitative trait locus in maize. Proc. Natl. Acad. Sci. USA, 104, 11 376–11 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvo, S.A. , Hirsch, C.N. , Buell, C.R. , Kaeppler, S.M. and Kaeppler, H.F. (2014) Whole transcriptome profiling of maize during early somatic embryogenesis reveals altered expression of stress factors and embryogenesis‐related genes. PLoS One, 9, e111407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schilling, L. , Matei, A. , Redkar, A. , Walbot, V. and Doehlemann, G. (2014) Virulence of the maize smut Ustilago maydis is shaped by organ‐specific effectors. Mol. Plant Pathol. 15, 780–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnable, J. and Freeling, M. (2011) Genes identified by visible mutant phenotypes show increased bias toward one of two subgenomes of maize. PLoS One, 6, e17855. doi: 10.1371/journal.pone.0017855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skibbe, D.S. , Doehlemann, G. , Fernandes, J. and Walbot, V. (2010) Maize tumors caused by Ustilago maydis require organ‐specific genes in host and pathogen. Science, 328, 89–92. [DOI] [PubMed] [Google Scholar]
- Slewinski, T.L. (2012) Non‐structural carbohydrate partitioning in grass stems: a target to increase yield stability, stress tolerance, and biofuel production. J. Exp. Bot. 63, 4647–4670. [DOI] [PubMed] [Google Scholar]
- Slewinski, T.L. , Meeley, R. and Braun, D.M. (2009) Sucrose transporter1 functions in phloem loading in maize leaves. J. Exp. Bot. 60, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung, H.Y. and Su, J.C. (1977) Sucrose synthetase. II. Sucrose synthetase isozymes of pea seedlings – purification and general properties. J. Chin. Biochem. Soc. 6, 22–37. [Google Scholar]
- Supek, F. , Bošnjak, M. , Škunca, N. and Šmuc, T. (2011) REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One, 6, e21800. doi: 10.1371/journal.pone.0021800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, S. , Brefort, T. , Neidig, N. , Djamei, A. , Kahnt, J. , Vermerris, W. , Koenig, S. , Feussner, K. , Feussner, I. and Kahmann, R. (2014) A secreted Ustilago maydis effector promotes virulence by targeting anthocyanin biosynthesis in maize. Elife, 3, e01355. doi: 10.7554/eLife.01355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka, W. , Pautler, M. , Jackson, D. and Hirano, H.Y. (2013) Grass meristems II – inflorescence architecture, flower development and meristem fate. Plant Cell Physiol. 54, 313–324. [DOI] [PubMed] [Google Scholar]
- Thammawong, M. and Arakawa, O. (2010) Starch to sugar conversion in “Tsugaru” apples under ethylene and 1‐methylcyclopropene treatments. J. Agric. Sci. Technol. 12, 617–626. [Google Scholar]
- Tognetti, J.A. , Pontis, H.G. and Martínez‐Noël, G.M. (2013) Sucrose signaling in plants: a world yet to be explored. Plant Signal Behav. 8, e23316. doi: 10.4161/psb.23316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , van Baren, M.J. , Salzberg, S.L. , Wold, B.J. and Pachter, L. (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai, A.Y. and Gazzarrini, S. (2014) Trehalose‐6‐phosphate and SnRK1 kinases in plant development and signaling: the emerging picture. Front. Plant Sci. 5, 119. doi: 10.3389/fpls.2014.00119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voll, L.M. , Horst, R.J. , Voitsik, A.M. , Zajic, D. , Samans, B. , Pons‐Kühnemann, J. , Doehlemann, G. , Münch, S. , Wahl, R. , Molitor, A. , Hofmann, J. , Schmiedl, A. , Waller, F. , Deising, H.B. , Kahmann, R. , Kämper, J. , Kogel, K.H. and Sonnewald, U. (2011) Common motifs in the response of cereal primary metabolism to fungal pathogens are not based on similar transcriptional reprogramming. Front. Plant Sci. 2, 39. doi: 10.3389/fpls.2011.00039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahl, R. , Wippel, K. , Goos, S. , Kämper, J. and Sauer, N. (2010) A novel high‐affinity sucrose transporter is required for virulence of the plant pathogen Ustilago maydis . PLoS Biol. 8, e1000303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walbot, V. and Skibbe, D.S. (2010) Maize host requirements for Ustilago maydis tumor induction. Sex Plant Reprod. 23, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warchol, M. , Perrin, S. , Grill, J.P. and Schneider, F. (2002) Characterization of a purified beta‐fructofuranosidase from Bifidobacterium infantis ATCC 15697. Lett. Appl. Microbiol. 35, 462–467. [DOI] [PubMed] [Google Scholar]
- Wenzler, H. and Meins, F. (1987) Persistent changes in the proliferative capacity of maize leaf tissues induced by Ustilago infection. Physiol. Mol. Plant Pathol. 30, 309–319. [Google Scholar]
- Wingler, A. and Roitsch, T. (2008) Metabolic regulation of leaf senescence: interactions of sugar signalling with biotic and abiotic stress responses. Plant Biol. 10, 50–62. [DOI] [PubMed] [Google Scholar]
- Wong, A.Y. and Colasanti, J. (2007) Maize floral regulator protein INDETERMINATE1 is localized to developing leaves and is not altered by light or the sink/source transition. J. Exp. Bot. 58, 403–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional Supporting Information may be found in the online version of this article at the publisher's website:
Fig. S1 RNA‐sequencing (RNA‐Seq) read quality control, P value distribution and top 50 up‐ and down‐regulated genes shown in a heat map. (A) q‐value distribution of the three uninfected and three infected libraries. (B) Distribution of P values of expressed genes of the three uninfected relative to three infected libraries. P values below 0.05 are considered to be significantly different. (C) Heat map of the top 50 up‐ and down‐regulated host genes during tumour formation.
Fig. S2 Expression of key carbohydrate metabolism genes during tumour formation. Blue arrows indicate significant down‐regulation of genes or entire gene families at the indicated steps by at least two‐fold. Red arrows indicate significant up‐regulation of genes or entire gene families at the indicated steps by at least two‐fold. Red (up‐regulation) or blue (down‐regulation) text highlights the changes in key aspects of the pathways. Leaf or flower indicates the preferred expression of the genes in different maize tissues.
Fig. S3 Silver nitrate treatment in the soil reduces fungal virulence. Zero grams, 1 g (0.165 g/L soil), 2.5 g (0.412 g/L soil) or 5 g (0.823 g/L soil) of AgNO3 per tray were added to the soil before planting the maize seed. The small seedlings were infected 7 days after planting. After 14 days, the disease symptoms were scored (DI, disease index) (A). Control plants showed a silver dose‐dependent reduction in growth and increased anthocyanin formation in the stem (B). Increasing silver concentrations reduced the disease symptoms (C). Infection studies were repeated three times with different concentrations. Significance levels: *P < 0.05; **P < 0.01. Standard deviations are shown.
Fig. S4 Starch as single carbon source does not support fungal growth. The growth of Ustilago maydis was supported with 1% glucose or 1% starch with 1% α‐amylase as single carbon source in minimal medium (MM). 1% starch alone did not support the growth of the haploid strain or the solo‐pathogenic strain. 1% α‐amylase alone led to minor fungal growth, most likely because of carbon sources in the enzyme storage buffer (e.g. glycerol).
Fig. S5 Summary of the major metabolic changes predicted by transcriptome analysis of maize genes during tumour formation by Ustilago maydis. Functions in red were up‐regulated, whereas those in blue were down‐regulated.
Table S1 Expression of all B73 transcripts, highest up‐ and down‐regulated genes, regulation of transcription factors and regulation of pathway genes during tumour formation. The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S2 Expression of Golden Bantam‐specific transcripts and highest up‐ and down‐regulated genes. The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S3 Confirmation of the RNA‐sequencing (RNA‐Seq) expression changes for selected genes in seedling stems and in tumours in tassels and ears. The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S4 Gene ontology (GO) term enrichment analysis of up‐ and down‐regulated functions during tumour formation.
Table S5 Regulation of genes from the classical gene list of Zea mays during tumour formation induced by Ustilago maydis in seedling stems at 10 days post‐inoculation (dpi). The fold changes reflect the level in infected tissue divided by that in uninfected tissue, e.g. 2 and 0.5 equal up‐regulated two‐fold and down‐regulated two‐fold, respectively.
Table S6 Categorization of genes from the classical gene list into functional groups and their regulation during tumour formation.
Table S7 Primers used in this study.
Text S1 Supporting experimental procedures, and results and discussion.