

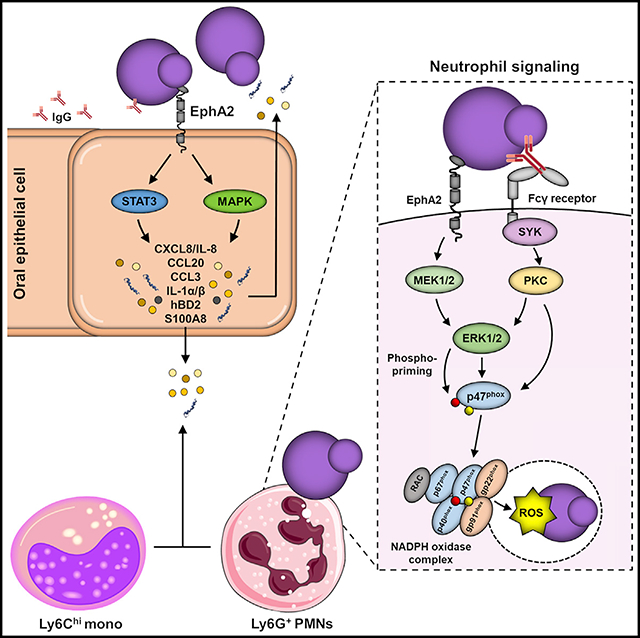
SUMMARY
During oropharyngeal candidiasis (OPC), Candida albicans proliferates and invades the superficial oral epithelium. Ephrin type-A receptor 2 (EphA2) functions as an oral epithelial cell β-glucan receptor that triggers the production of proinflammatory mediators in response to fungal infection. Because EphA2 is also expressed by neutrophils, we investigated its role in neutrophil candidacidal activity during OPC. We found that EphA2 on stromal cells is required for the accumulation of phagocytes in the oral mucosa of mice with OPC. EphA2 on neutrophils is also central to host defense against OPC. The interaction of neutrophil EphA2 with serum- opsonized C. albicans yeast activates the MEK-ERK signaling pathway, leading to NADPH subunit p47phox site-specific phospho-priming. This priming increases intracellular reactive oxygen species production and enhances fungal killing. Thus, in neutrophils, EphA2 serves as a receptor for β-glucans that augments Fcγ receptor-mediated antifungal activity and controls early fungal proliferation during OPC.
In Brief
In oral epithelial cells, EphA2 functions as a β-glucan receptor that triggers the production of proinflammatory mediators in response to oropharyngeal candidiasis. Here, Swidergall et al. show that, in neutrophils, EphA2 recognition of β-glucans augments Fcγ receptor-mediated antifungal activity and prevents fungal proliferation during the initiation of oropharyngeal infection.
Graphical Abstract
INTRODUCTION
The occurrence of fungal infections such as those caused by Candida albicans has progressively increased over the last decades (Bongomin et al., 2017; Brown et al., 2012). Invasive candidiasis is associated with mortality rates of 40% to 50%, while mucosal candidal infections such as oropharyngeal candidiasis (OPC) cause significant morbidity in millions of patients who are immunocompromised due to AIDS, neutropenia, diabetes mellitus, or the use of immunosuppressive drugs (Swidergall and Filler, 2017).
In healthy individuals, the collaborative antifungal activity of epithelial cells, phagocytes, and lymphocytes maintains mucosal immunity and prevents fungal proliferation (Verma et al., 2017a). Recognition of components of the fungal cell wall, such as exposed β-glucans, is crucial for inducing host antifungal responses (Dambuza et al., 2017; Levitz, 2010). Masking of β-glucans via mannan is a major mechanism by which fungi avoid recognition by the host (Ballou et al., 2016). However, immune attack and environmental pressures, such as exposure to hypoxia and echinocandin antifungal drugs, drive cell wall remodeling that unmasks β-glucans and enhances the host inflammatory responses (Hopke et al., 2016). Leukocytes recognize β-glucan via Dectin-1 and complement receptor 3 (CR3; CD11b/CD18), while oral epithelial cells sense exposed β-glucan via ephrin type-A receptor 2 (EphA2) and Dectin-1 (Brown et al., 2002; Swidergall, 2019; Swidergall et al., 2018; van Bruggen et al., 2009). Epithelial β-glucan sensing mediates protective immunity in the oral cavity by distinguishing between fungal colonization and fungal overgrowth (Swidergall et al., 2018). In response to fungal overgrowth, epithelial cells secrete cytokines and chemokines that recruit phagocytes to sites of infection, where they inhibit and eventually kill the proliferating and invading fungi. These phagocytes amplify the proinflammatory response by producing additional cytokines and chemokines (Lionakis and Levitz, 2018; Underhill and Pearlman, 2015). Although neutrophils are vital for host defense against OPC (Huppler et al., 2014), it is incompletely understood how these leukocytes recognize C. albicans in the oral cavity. Classical myeloid pattern recognition receptors (PRRs), such as Dectin-1 and TLR2, are dispensable for host defense against OPC (Verma et al., 2017b), suggesting that other neutrophil receptors must sense the presence of C. albicans in the oropharynx.
Previously, we reported that EphA2 is an oral epithelial cell receptor for β-glucans. Binding of fungal β-glucans to epithelial cell EphA2 initiates antifungal responses in the oral cavity by inducing cytokine and chemokine production (Swidergall et al., 2018). In the current study, we investigated the role of neutrophil EphA2 in host defense against OPC. We found that during OPC, EphA2−/− mice had delayed infiltration of neutrophils and inflammatory monocytes into the oral cavity, resulting in more severe disease compared with wild-type mice. Strikingly, we found that EphA2, in conjunction with Fcγ receptors (FcγRs), is required for neutrophils to limit C. albicans proliferation during OPC. The interaction of serum-opsonized yeast with neutrophil EphA2 activates mitogen-activated protein kinase (MAPK) signaling, leading to p47phox priming, increased intracellular reactive oxygen species (ROS) accumulation, and enhanced fungal killing. Thus, in neutrophils, EphA2 serves as a PRR for β-glucans that augments FcγR-mediated antifungal activity and is central to controlling early fungal proliferation during OPC.
RESULTS
EphA2 Signaling Induces Early Leukocyte Infiltration during OPC
The oral epithelium orchestrates the early antifungal response to infection by releasing cytokines and chemokines that recruit leukocytes to the infection site and then prime them for enhanced antifungal activity (Altmeier et al., 2016; Huppler et al., 2014; Swidergall and Ernst, 2014; Swidergall et al., 2018). To investigate the role of EphA2 in the host response to OPC, we determined the oral fungal burden, cytokine-chemokine response, and number of infiltrating neutrophils and inflammatory monocytes after 1 and 2 days of oral infection in wild-type and EphA2−/− mice. In agreement with our previous findings (Swidergall et al., 2018), after 1 day of infection, EphA2−/− mice had an increased oral fungal burden relative to that of wild-type mice (Figure 1A). This increase was associated with an impaired host response in the EphA2−/− mice, which had lower levels of CCL3, CXCL1/KC, interferon γ (IFN-γ), interleukin (IL)-23 p19, and IL-17A in their oral tissues relative to wild-type mice (Figure 1B). Furthermore, after 1 day of infection, the EphA2−/− mice had significantly fewer neutrophils and inflammatory monocytes in their oral tissues compared with the wild-type mice (Figures 1C and 1D; Figure S1). After 2 days of infection, the oral fungal burden of wild-type mice declined, whereas the oral fungal burden of EphA2−/− mice remained elevated (Figure 1A). In wild-type mice, the number of neutrophils and inflammatory monocytes declined to near basal levels by day 2, whereas in the EphA2−/− mice, the number of these leukocytes increased (Figures 1C and 1D). These results were verified by histopathological analysis, which demonstrated that the tongues of wild-type mice had few visible fungi or leukocytes at day 2 post-infection, whereas the tongues of EphA2−/− mice contained numerous fungi and leukocytes (Figure 1E). Thus, during OPC, EphA2 is required for the early recruitment of phagocytes and control of fungal growth.
Figure 1. EphA2 Signaling Orchestrates Early Phagocyte Recruitment during OPC.

(A) Oral fungal burden of immunocompetent wild-type and EphA2−/− mice after 1 and 2 days of OPC. Results are median ± interquartile range of six mice per group from two independent experiments. *p < 0.05 (Mann-Whitney test corrected for multiple comparisons). The y axis is set at the limit of detection (20 colony-forming units [CFUs] per gram of tissue).
(B) Level of chemokines and cytokines in the tongue homogenates of immunocompetent wild-type and EphA2−/− mice with OPC after 1 day of infection. Scatterplots show median and interquartile range of seven mice in each group, tested in duplicate in two independent experiments. *p < 0.05, **p < 0.01, ****p < 0.0001 (Mann-Whitney test corrected for multiple comparisons).
(C and D) Myeloid phagocyte infiltration in tongues of immunocompetent wild-type and EphA2−/− mice after 1 and 2 days of infection (n = 6 for infected mice, n = 4 for naive mice). Results are median ± interquartile range from combined results of two independent experiments. *p < 0.05, **p < 0.01 (Mann-Whitney test corrected for multiple comparisons). See Figure S1 for the gating strategy during flow cytometric analysis.
(E) Histopathology of the tongue of wild-type and EphA2−/− mice with OPC after 2 days of infection. Scale bar, 100 μm. Results are representative of 2 mice from the same experiment. Arrows indicate the C. albicans cells.
Because EphA2 is expressed on both hematopoietic and nonhematopoietic cells (Finney et al., 2017; Heng and Painter, 2008; Petersen et al., 2012; Suram et al., 2013; Swidergall et al., 2018), we used bone marrow (BM) chimeric mice to dissect the role of EphA2 expressed by these different cell types in host defense against OPC. These mice were generated as diagrammed in Figure 2A and then orally infected with C. albicans 8 weeks after successful BM reconstitution (Figure S2). Wild-type mice that were reconstituted with EphA2−/− BM and EphA2−/− mice that were reconstituted with wild-type BM had significantly more severe OPC, as determined by increased oral fungal burden and greater weight loss, relative to wild-type mice that were reconstituted with wild-type BM (Figures 2B and 2C). In the BM chimeric mice that lacked EphA2 in either their stromal cells or their myeloid cells, the severity of OPC was similar to that in EphA2−/− BM mice that were reconstituted with EphA2−/−. These results suggest that EphA2 within both hematopoietic and nonhematopoietic compartments is required for protection against OPC.
Figure 2. EphA2 in Hematopoietic and Nonhematopoietic Cells Contributes to Resistance against Acute OPC.

(A) Scheme of bone marrow chimeric mice.
(B-E) Oral fungal burden (B), body weight (C), and innate phagocyte recruitment (D and E) after 1 day of oral C. albicans infection in the BM chimeras. Results are median ± interquartile range of six mice per group from a single experiment. **p < 0.01, NS, not significant (Mann-Whitney test corrected for multiple comparisons).
See also Figure S2.
To determine the biological function of EphA2 in these compartments, we next examined leukocyte accumulation in the oral tissues of the BM chimeric mice after 1 day of OPC. The accumulation of neutrophils in EphA2−/− mice reconstituted with wild-type BM was significantly reduced relative to the wild-type mice reconstituted with wild-type BM and similar to EphA2−/− mice reconstituted with EphA2−/− (Figure 2D). By contrast, the accumulation of neutrophils in wild-type mice reconstituted with EphA2−/− BM was similar to that of wild-type mice reconstituted with wild-type BM and significantly greater than EphA2−/− mice reconstituted with EphA2−/−. The accumulation of inflammatory monocytes in the EphA2−/− mice reconstituted with wild-type BM and the wild-type mice reconstituted with EphA2−/− BM was not significantly different from wild-type mice reconstituted with wild-type BM (Figure 2E). Collectively, these results indicate that EphA2 on stromal cells is required for the normal accumulation of neutrophils in the oral tissues during OPC. They also suggest that although EphA2−/− neutrophils are able to accumulate at wild-type levels at sites of OPC, they are unable to control fungal proliferation.
EphA2−/− Neutrophils Have Decreased Antifungal Activity In Vivo
The preceding findings suggested that EphA2 may be required for phagocytes to kill C. albicans. In addition to being expressed on epithelial cells, EphA2 is known to be expressed on myeloid phagocytes such neutrophils, macrophages, and monocytes (Heng and Painter, 2008; Petersen et al., 2012; Suram et al., 2013; Swidergall et al., 2018). To verify that EphA2 is expressed by murine neutrophils, we stained BM neutrophils with an anti-EphA2 monoclonal antibody. Using flow cytometry and indirect immunofluorescence, we found that EphA2 was expressed on the surface of unstimulated murine neutrophils (Figures 3A and 3B; Figure S3). Furthermore, EphA2 clustering was observed after phagocytosis of serum-opsonized yeast-phase C. albicans cells (Figure 3B). EphA2 is a receptor tyrosine kinase that is autophosphorylated on specific tyrosine residues when activated. In resting neutrophils, there was a low level of EphA2 autophosphorylation (Figure 3C; Figure S3). When the neutrophils interacted with C. albicans, EphA2 autophosphorylation increased and remained above basal levels for at least 60 min. Thus, neutrophils express EphA2 on their surface, and the receptor is activated by interaction with C. albicans.
Figure 3. EphA2−/− Neutrophils Have Decreased Capacity to Limit Fungal Proliferation In Vivo.

(A) Flow cytometry of EphA2 surface expression on wild-type neutrophils. Histograms of 5 × 105 events are shown.
(B) Wild-type neutrophils were either uninfected (top panel) or infected for 30 min with GFP-expressing C. albicans (bottom panel) and then stained with anti-mouse EphA2 (mEphA2) antibody (Ab) (red) and DAPI (blue). Scale bar, 10 μm.
(C) Immunoblot analysis showing the time course of EphA2 phosphorylation in wild-type neutrophils that had been infected with yeast-phase C. albicans SC5314 for the indicated time points.
(D) Oral fungal burden of wild-type and EphA2−/− mice that had been treated with either isotype control or anti-Gr-1 antibody and then orally infected with C. albicans for 2 days. Results are median ± interquartile range of five mice per group from two independent experiments. **p < 0.01 (Mann-Whitney test corrected for multiple comparisons). The y axis is set at the limit of detection.
(E) Body weight of EphA2−/− mice that had been treated with either isotype control or anti-Gr-1 antibody and then orally infected with C. albicans for 2 days. Results show median of three mice per group from two independent experiments. *p < 0.05 (Mann-Whitney test).
(F) Scheme of neutrophil adoptive transfer of orally infected CD18−/− mice. PMNs, polymorphonuclear neutrophils.
(G) Oral fungal burden 2 days post-infection of immunocompetent wild-type and CD18−/− mice after adoptive transfer of wild-type or EphA2−/− neutrophils. Results are median ± interquartile range of six mice per group from two independent experiments. **p < 0.01, ***p < 0.001 (Mann-Whitney test corrected for multiple comparisons). The y axis is set at the limit of detection (20 CFUs per gram of tissue).
(H) Neutrophil counts after 1 day of infection in the tongues of CD18−/− mice receiving wild-type or EphA2−/− neutrophils. Results are median ± interquartile in a single experiment (n = 4–5). NS, not significant (Mann-Whitney test).
See also Figure S3.
To investigate the role of EphA2 on phagocytes in host defense against OPC, we treated both wild-type and EphA2−/− mice with an anti-Gr1 monoclonal antibody to deplete neutrophils and monocytes. Antibody depletion in wild-type mice (Figure S3) increased their oral fungal burden to the same level as in neutrophil-sufficient EphA2−/− mice (Figure 3D; Figure S3). By contrast, phagocyte depletion in EphA2−/− mice did not result in a further increase in their already high oral fungal burden, but led to further body weight loss, a marker of disease severity (Figure 3E). These results suggest that innate leukocytes play a central role in the EphA2-mediated host response to OPC.
To determine whether EphA2 enhances the capacity of neutrophils to control the growth of C. albicans during OPC, we performed adoptive transfer experiments in which BM neutrophils from wild-type and EphA2−/− mice were injected into CD18−/− mice after they had been orally infected with C. albicans (Figure 3F). The neutrophils of CD18−/− mice cannot bind to intercellular adhesion molecule 1 (ICAM-1) on vascular endothelial cells and are therefore unable to migrate out of the bloodstream into infected tissues (Ding et al., 1999; Taylor et al., 2014). As expected, CD18−/− mice had more severe OPC than wild-type mice (Figure 3G). When wild-type neutrophils were adoptively transferred into the CD18−/− mice, there was a significant reduction in oral fungal burden. By contrast, transfer of EphA2−/− neutrophils into these mice did not significantly reduce their oral fungal burden. To determine whether the increased fungal burden in mice that received the EphA2−/− neutrophils was due to reduced trafficking of the cells into the infected tissue, we analyzed the accumulation of adoptively transferred neutrophils in the tongues of CD18−/− mice after 1 day of infection. We found that similar numbers of wild-type (WT) and EphA2−/− neutrophils accumulated in the tongues (Figure 3H; Figure S3), indicating that the inability of EphA2−/− neutrophils to control fungal growth during OPC is not due to a defect in migration. Collectively, these data suggest that EphA2 on neutrophils is required to limit fungal proliferation after the neutrophils have arrived at the site of infection.
EphA2 Is Required for Maximal Neutrophil Killing of Serum-Opsonized Yeast
Neutrophils interact with and kill C. albicans cells by different mechanisms, depending on the morphology of the organism and whether it is opsonized (Gazendam et al., 2014; Warnatsch et al., 2017). Although mice are immunologically naive to C. albicans, the organism can still be opsonized by anti-β-glucan antibodies induced by commensal fungi (Chiani et al., 2009; Ishibashi et al., 2005). We determined the capacity of wild-type and EphA2−/− neutrophils to kill C. albicans yeast and hyphae in the presence and absence of serum opsonization. Strikingly, EphA2−/− neutrophils isolated from either peripheral blood or BM were deficient in killing serum-opsonized C. albicans yeast (Figure 4A). However, EphA2−/− neutrophils were able to kill both unopsonized yeast and opsonized hyphae, similar to wild-type neutrophils (Figures 4B and 4C). Thus, EphA2 is required for neutrophils to kill opsonized yeast-phase C. albicans.
Figure 4. EphA2 Is Required for Maximal Fungal Killing of Serum-Opsonized Yeast-Phase C. albicans.

(A) Killing of serum-opsonized yeast-phase C. albicans by peripheral blood (PB) and bone marrow (BM) neutrophils isolated from wild-type and EphA2−/− mice. The percentage of organisms killed was determined by colony counting. Results are means ± SD from 4 experiments, each performed in triplicate. ****p < 0.0001 (Mann-Whitney test).
(B) Killing of unopsonized C. albicans yeast by neutrophils from the indicate mouse strains. NS, not significant.
(C) Neutrophil killing of opsonized C. albicans hyphae.
(D) Killing of serum-opsonized yeast-phase C. albicans by bone marrow-derived macrophages.
(E) Killing of opsonized yeast C. albicans by a mixed wild-type and EphA2−/− population. Results are means ± SD from 3 independent experiments, each performed in duplicate. ***p < 0.001; ****p < 0.0001 (Mann-Whitney test corrected for multiple comparisons).
(F) Phagocytosis of opsonized C. albicans yeast by wild-type and EphA2−/− neutrophils. NS, not significant. Data were normalized to the phagocytosis of wild-type neutrophils.
(G) Neutrophil killing of serum-opsonized S. aureus.
(H) Body weight of wild-type and EphA2−/− mice after oral infection with C. albicans. Results are median ± interquartile range of 5 mice per strain in a single experiment. *p < 0.05 (Mann-Whitney test for each day).
(I) Oral fungal burden of wild-type and EphA2−/− mice after 4 days of OPC. Results are median ± interquartile range of five mice per group in a single experiment. **p < 0.01 (Mann-Whitney test). The y axis is set at the limit of detection (20 CFUs per gram of tissue).
(J) FcγR signaling is required for killing of serum-opsonized C. albicans yeast. The neutrophil FcγRs, CD16, CD32, and CD64 were blocked using specific Abs, after which the killing of serum-opsonized yeast-phase C. albicans was determined by colony counting. Results are means ± SD from 4 experiments in triplicate. ****p < 0.0001, NS, not significant (Mann-Whitney test).
(K) FcγR signaling is required for yeast phagocytosis. The neutrophil FcγRs, CD16, CD32, and CD64 were blocked using specific Abs, after which phagocytosis of opsonized C. albicans yeast by wild-type was determined. Results are means ± SD from 3 independent experiments, each performed in duplicate. **p < 0.01 (Mann- Whitney test).
(L) Oral fungal burden of wild-type and FcyRI−/− mice after 2 days of OPC. Results are median ± interquartile range of five mice per group in a single experiment. **p < 0.01 (Mann-Whitney test). The y axis is set at the limit of detection.
See also Figure S4.
Because EphA2 is also expressed on macrophages (Finney et al., 2017; Suram et al., 2013), we tested the capacity of macrophages from EphA2−/− mice to kill opsonized C. albicans yeast. EphA2−/− BM-derived macrophages killed the organisms, similar to wild-type macrophages (Figure 4D), suggesting that EphA2 is dispensable for macrophage killing of C. albicans.
To investigate how EphA2 enhances neutrophil killing of C. albicans, we analyzed whether it influences the expression of other neutrophil cell surface receptors, extrinsic neutrophil priming, phagocytosis, and killing of opsonized Staphylococcus aureus. We found that EphA2-deficient neutrophils expressed similar levels of Dectin-1, CD11b, CD16/32, CD18, and CD64 compared with wild-type BM neutrophils (Figure S4). To determine whether EphA2 was required for extrinsic priming in which neutrophils release proinflammatory cytokines that prime other neutrophils for enhanced C. albicans killing (Summers et al., 2010), we incubated C. albicans with a mixture EphA2−/− and wild-type neutrophils and determined the extent of fungal killing. We found that adding wild-type neutrophils to EphA2−/− neutrophils did not significantly increase fungal killing (Figure 4E), suggesting that EphA2−/− neutrophils are not deficient in extrinsic priming. Furthermore, we determined that the phagocytosis of opsonized C. albicans yeast by EphA2−/− neutrophils was similar to that of wild-type neutrophils (Figure 4F), indicating that EphA2 is required for neutrophils to kill serum-opsonized yeast after they have been phagocytosed. Finally, EphA2−/− neutrophils were able to kill serum-opsonized S. aureus, similar to wild-type neutrophils (Figure 4G). Thus, the absence of EphA2 does not alter the surface expression of other neutrophil receptors or inhibit extrinsic neutrophil priming or phagocytosis. In addition, deletion of EphA2 does not result in a global defect in the capacity of neutrophils to kill opsonized microorganisms.
Hyphal cells are the predominant C. albicans morphotype during later time points of OPC (Solis and Filler, 2012; Solis et al., 2017). If lack of EphA2 only influences the capacity of neutrophils to kill opsonized yeast, then EphA2 deficiency should have less of an impact at later time points, when hyphae predominate and few yeast-phase organisms are present. To test this hypothesis, we orally infected wild-type and EphA2−/− mice and monitored their weight over time. We also determined their oral fungal burden after 4 days of infection. Relative to wild-type mice, EphA2−/− mice lost significantly more weight during the first 3 days of infection, but their weight returned to wild-type levels by day 4, at which time both strains of mice had essentially cleared the infection (Figures 4H and 4I). Thus, EphA2 is important for host defense against C. albicans during the initial phases of OPC but is dispensable after prolonged infection.
Serum-opsonized yeast are recognized by FcγRs expressed by neutrophils, monocytes, and macrophages, which trigger intracellular signaling pathways that result in the killing of phagocytosed C. albicans (Gazendam et al., 2016). To verify the importance of FcγRs in neutrophil killing of C. albicans, we blocked the FcγRs CD16, CD32, and CD64 using specific antibodies. In the absence of FcγR signaling, neither wild-type nor EphA2−/− neutrophils were able to kill serum-opsonized C. albicans, partly because of decreased phagocytosis (Figures 4J and 4K), thus highlighting the central role of FcγRs in mediating neutrophil killing of this organism. To determine whether early control of C. albicans in the oral cavity depends on FcγR signaling in phagocytes, we orally infected mice deficient in Fc-gamma receptor 1 (FcγRI; CD64). After 2 days of infection, FcγRI−/− mice had a significantly greater oral fungal burden relative to wild-type mice (Figure 4L). These results support the model that FcγR signaling is critical for controlling early fungal proliferation in the oral cavity.
The EphA2-MAPK Axis Primes p47phox to Enhance Intracellular ROS Production
Killing of serum-opsonized C. albicans occurs when FcγRs recognize antibodies on the fungal surface, thereby activating protein kinase C (PKC) and inducing the production of ROS (Gazendam et al., 2014). To investigate the post-phagocytic mechanisms of EphA2-dependent C. albicans killing, we measured intracellular ROS production by neutrophils as they interacted with opsonized C. albicans yeast in vitro. Wild-type neutrophils generated a large amount of intracellular ROS in response to C. albicans, whereas EphA2−/− neutrophils generated significantly less ROS (Figure 5A). ROS is produced by the NADPH oxidase enzyme complex, which is composed of two transmembrane proteins (p22phox and gp91phox) and four cytosolic proteins (p47phox, p67phox, p40phox, and the Rac guanosine triphosphatases [GTPases]) that assemble at membrane sites upon cell activation (El-Benna et al., 2009). By indirect immunofluorescence, we analyzed the localization of p47phox in wild-type and EphA2−/− neutrophils. We found that there was reduced accumulation of p47phox around the phagosomes of EphA2−/− neutrophils that had ingested serum-opsonized C. albicans yeast (Figures 5B and 5C). By contrast, the localization of membrane-bound gp91phox in the EphA2−/− neutrophils was similar to that of wild-type cells (Figure S5).
Figure 5. EphA2 Activates the MEK-ERK MAPK Module to Prime p47phox.

(A) Intracellular ROS accumulation measured by mean fluorescence (FL) intensity in wild-type and EphA2−/− neutrophils after45 min of infection with opsonized C. albicans yeast. Results are median ± interquartile range of neutrophils from 3 mice per strain, each tested in duplicate. ****p < 0.0001 (Mann-Whitney test corrected for multiple comparisons).
(B) Effects of EphA2 on localization of p47phox in neutrophils infected with C. albicans. Wild-type and EphA2−/− neutrophils were incubated with opsonized GFP-expressing C. albicans yeast for 30 min, fixed, and stained for p47phox (red) and F-actin (blue). Scale bar, 10 μm.
(C) Percentage of phagosomes containing C. albicans and surrounded by p47phox. Results are median ± interquartile of 60 neutrophils per mouse strain in three independent experiments. ****p < 0.01 (Mann-Whitney test).
(D) Representative immunoblot of MEK½ and ERK½ phosphorylation in wild-type and EphA2−/− neutrophils that had been infected with yeast-phase C. albicans for 30 min.
(E) Representative immunoblot of PKC-δ phosphorylation in wild-type and EphA2−/− neutrophils that had been infected with yeast-phase C. albicans SC5314 for 30 min with a 5:1 ratio.
(F) Representative immunoblot of ERK½ phosphorylation in wild-type and EphA2−/− neutrophils stimulated with 50 nM PMA for 30 min.
(G) Intracellular ROS accumulation (mean fluorescence [FL] intensity) of wild-type and EphA2−/− neutrophil stimulated with PMA for 45 min. Results are median ± interquartile range of neutrophils from 3 mice per strain, each tested in duplicate. ****p < 0.0001 (Mann-Whitney test corrected for multiple comparisons).
(H) Effects of inhibiting ERK with the specific inhibitor SCH7729884 on the killing of C. albicans by bone marrow neutrophils isolated from wild-type mice. The percentage of organisms killed was determined by colony counting. Results are median ± interquartile from 4 experiments in triplicate. **p < 0.01, ****p < 0.0001 (Mann-Whitney test).
(I) Representative immunoblot analysis of Ser345 p47phox phosphorylation in wild-type and EphA2−/− neutrophils that had been infected with yeast-phase C. albicans for 30 min.
(J) β-glucan induces p47phox priming. Wild-type BM neutrophils were incubated with 10 μg/mL of Sepharose IgG beads and/or 10 μg/mL of zymosan (depleted) for 30 min. Representative immunoblot analysis of Ser345 p47phox phosphorylation.
See also Figure S5.
p47phox is activated on selective sites by several protein kinases, such as PKC, protein kinase A (PKA), and the MAPK ERK, which in turn leads to localization of the protein to the phagocyte membrane (El Benna et al., 1996, 2009; Fontayne et al., 2002). In oral epithelial cells, fungal recognition by EphA2 activates MEK½, an upstream element of ERK signaling (Roberts and Der, 2007; Swidergall et al., 2018). We therefore investigated the extent of activation of MEK½, and ERK½ in neutrophils in response to serum-opsonized C. albicans yeast and found that the fungus strongly induced MEK and ERK phosphorylation in wild-type neutrophils, but not EphA2−/− neutrophils (Figure 5D; Figure S5). The spleen tyrosine kinase (SYK) is essential for FcγR signaling in macrophages and neutrophils (Kiefer et al., 1998). PKC-δ, a known downstream target of FcγR signaling, activates both ERK and p47phox (Bey et al., 2004; Gazendam et al., 2014; Limnander et al., 2011; Ueda et al., 1996). Therefore, we tested whether EphA2 is required for activation of SYK and PKC-δ in response to serum-opsonized C. albicans. Exposure to C. albicans induced the phosphorylation of SYK and PKC-δ in both wild-type and EphA2−/− neutrophils (Figure 5E; Figure S5), suggesting that EphA2 influences the activity of p47phox independent of the SYK-PKC axis. PKC can be activated via a receptor-independent mechanism using phorbol myristate acetate (PMA) to induce ERK activation and subsequent ROS production (Karlsson et al., 2000). To verify that ERK can be activated and ROS can be produced in the absence of EphA2, we stimulated wild-type and EphA2−/− neutrophils with PMA and found that the magnitude of ERK phosphorylation in EphA2−/− neutrophils was similar to that in wild-type cells (Figure 5F; Figure S5). Moreover, addition of PMA to both wild-type and EphA2−/− neutrophils induced similar amounts of intracellular ROS accumulation (Figure 5G). Collectively, these results indicate that EphA2 signals in parallel to PKC through MEK½ to enhance ERK activation. Activation of ERK is required for neutrophils to respond to serum-opsonized C. albicans, because inhibition of ERK abolished neutrophil killing of these organisms (Figure 5H).
Neutrophils can be primed by host factors, as well as microbial products such as lipopolysaccharide (LPS), for enhanced antimicrobial responses (Dang et al., 1999; Forehand et al., 1989). ERK½ primes the neutrophil oxidative burst by phosphorylating p47phox at Ser345 (Dang et al., 2006). Immunoblot analysis showed that although exposure to opsonized C. albicans yeast induced phosphorylation of the p47phox Ser345 priming site in wild-type neutrophils, this response was markedly reduced in EphA2−/− neutrophils (Figure 5I; Figure S5). By contrast, phosphorylation of Ser304, which is required for NADPH oxidase activation (El-Benna et al., 2009), was not diminished in the absence of EphA2 (Figure S5). These data indicate that the impaired capacity of EphA2−/− neutrophils to kill serum-opsonized C. albicans is due to failure of p47phox priming, which results in reduced production of intracellular ROS.
On oral epithelial cells, EphA2 specifically senses surface-exposed β-glucan on fungi (Swidergall et al., 2018). Next, we determined whether β-glucan, the fungal ligand of EphA2, is able to induce priming of p47phox in murine neutrophils. Immunoglobulin G (IgG)-coated Sepharose beads were not able to induce phosphorylation Ser345, while zymosan, the particulate form of β-glucan, strongly activated phosphorylation of this residue, both by itself and especially in combination with IgG beads (Figure 5J; Figure S5). Collectively, these results support the model that β-glucan recognition via EphA2 is required for maximal activation of the MEK-ERK signaling pathway, which in turn primes p47phox, leading to enhanced intracellular ROS production and oxidant-mediated killing of opsonized C. albicans yeast.
DISCUSSION
Neutrophils play a key role in host defense against OPC (Altmeier et al., 2016; Huppler et al., 2014). In vitro, neutrophils kill C. albicans by two distinct mechanisms, depending on whether the organism has been opsonized (Gazendam et al., 2014). Phagolysosomal killing of unopsonized C. albicans depends on CR3 recognition and signaling via SYK, phosphatidylinositol 3-kinase (PI3K), and caspase recruitment domain-containing protein 9 (CARD9) but is independent of NADPH oxidase activity. By contrast, phagolysosomal killing of serum-opsonized C. albicans depends on the FcγRs, PKC, and ROS generated by the NADPH oxidase system. Our studies reveal that EphA2 not only is required for recruitment of phagocytes to foci of oral infection but also is a critical enhancer of the neutrophil defense against OPC by priming p47phox to boost FcγR-mediated ROS production and subsequent killing of phagocytosed serum-opsonized yeast.
When C. albicans proliferates on and invades oral epithelial cells, it activates distinct host signaling pathways that lead to an antifungal response consisting of production of antimicrobial peptides and secretion of chemokines and proinflammatory cytokines (Conti et al., 2016; Moyes et al., 2010; Swidergall et al., 2018). We have reported that exposed β-glucan on the C. albicans surface binds to EphA2 on oral epithelial cells, stimulating these cells to mount an antifungal response (Swidergall et al., 2018). Our current finding that EphA2−/− mice, and EphA2−/− mice reconstituted with wild-type BM, had delayed accumulation of neutrophils and inflammatory monocytes in the oropharynx during OPC indicates that EphA2 signaling is required for the early recruitment of these phagocytes. This defect in phagocyte recruitment was likely due to the absence of chemotactic signals produced by the stromal cells. It was not due to defective chemotaxis of EphA2−/− phagocytes, because neutrophils from EphA2−/− mice accumulated in the oral cavity, similar to wild-type neutrophils during OPC when they were adoptively transferred into CD18−/− mice and when wild-type mice were reconstituted with EphA2−/− BM.
Two lines of data indicate that EphA2 is central to the capacity of neutrophils to prevent C. albicans overgrowth during OPC. First, the oral fungal burden of EphA2−/− mice was similar to that of mice depleted of phagocytes. Second, the adoptive transfer of EphA2−/− neutrophils into CD18−/− mice did not reduce their oral fungal burden. Our in vitro studies demonstrated that although EphA2−/− neutrophils had reduced capacity to kill opsonized yeast-phase C. albicans, they retained the ability to kill both opsonized hyphae and unopsonized yeast. This finding suggests that during OPC, opsonized yeast-phase organisms are present, at least during the initial stages of infection. These organisms are likely opsonized by either secretory immunoglobulin A (sIgA) or IgG, the two principal antibody classes present in the saliva in the oral cavity (Brandtzaeg, 2013).
Our data show that in neutrophils, EphA2 signaling plays a key role in augmenting FcγR-mediated antifungal activity. We found that FcγR is required for neutrophils to kill C. albicans in vitro and that mice lacking the FcγRI were also highly susceptible to OPC. These results are consistent with those of Gazendam and colleagues, who found that blocking FcγR greatly impaired the capacity of neutrophils to inhibit the growth of C. albicans in vitro (Gazendam et al., 2014). Here we show that the early neutrophilic antifungal responses in the oral cavity depend on FcγR signaling. When serum-opsonized C. albicans yeast interacted with FcγR in EphA2−/− neutrophils, there was virtually no phosphorylation of MEK½, reduced phosphorylation of ERK½, and absent phosphorylation of p47phox on Ser345. As a result, p47phox was not primed, leading to reduced intracellular ROS accumulation and decreased C. albicans killing. Thus, EphA2 functions in parallel to FcγR to enhance ERK½ phosphorylation, which augments p47phox priming, leading to increased intracellular ROS production and neutrophil killing of opsonized C. albicans yeast. In epithelial cells, EphA2 binds to and is activated by fungal β-glucans, leading to enhanced secretion of proinflammatory cytokines (Swidergall et al., 2018). Our current data show that in neutrophils, EphA2 recognition of fungal β-glucans is necessary for p47phox priming and production of fungicidal levels of ROS in response to FcγR ligation.
The finding that EphA2−/− neutrophils could still kill opsonized S. aureus suggests that p47phox can also be primed by an EphA2-independent pathway. Extracellular host factors, such as granulocyte-macrophage colony-stimulating factor (GM-CSF) and tumor necrosis factor alpha (TNF-α), can also prime p47phox and enhance intracellular ROS production (Dang et al., 2006). How EphA2 interacts with these host factors to prime neutrophils for enhanced microbicidal activity is unknown and a topic of future investigation.
Although we found that the EphA2-FcγR-MAPK pathway is required for neutrophils to protect against OPC, studies of CARD9 knockout mice and humans with CARD9 deficiency suggest that the CR3-SYK-CARD9 pathway is dispensable for host defense against this mucosal infection. Both mice and humans with CARD9 deficiency are resistant to OPC (Bishu et al., 2014; Drewniak et al., 2013; Drummond et al., 2018). However, CARD9 is required for host defense against disseminated candidal infections, particularly in the brain (Drummond et al., 2015). These results indicate that distinct sets of neutrophil receptors recognize C. albicans at different anatomic sites.
Although EphA2 was required for maximal neutrophil killing of opsonized C. albicans yeast, it was dispensable for the killing of opsonized hyphae. When neutrophils ingest yeast, the organisms are contained within the phagolysosome and trigger intracellular ROS production. However, when neutrophils encounter larger organisms, such as hyphae, that are too large to be contained within the phagolysosome, ROS is produced extracellularly, amplifying IL-1β expression and leading to neutrophil swarming (Warnatsch et al., 2017). In C. albicans yeast, β-glucans are exposed at the sites of bud scars, whereas in hyphae, β-glucans are largely masked by mannans (Gantner et al., 2005). The lack of β-glucan exposure by hyphae provides a potential explanation for why EphA2−/− neutrophils retained wild-type capacity to kill this C. albicans morphotype. Collectively, these results reinforce the concept that neutrophils use different receptors to sense and respond to different growth forms of C. albicans in different niches within the host.
Because EphA2 recognizes β-glucans on pathogenic fungi other than C. albicans, such as Aspergillus fumigatus and Rhizopus oryzae (Swidergall et al., 2018), it is probable that it enhances the neutrophil response to these organisms too. Moreover, EphA2 has been found to recognize Plasmodium, Chlamydia trachomatis, and several viruses (Chen et al., 2018; Kaushansky et al., 2015; Lupberger et al., 2011; Subbarayal et al., 2015; TerBush et al., 2018; Zhang et al., 2018). However, the studies with these organisms were performed using normally nonphagocytic host cells, such as hepatocytes or epithelial cells. Our discovery that EphA2 enhances FcγR-mediated activity during C. albicans infection suggests that this protein may also mediate the response of myeloid cells to other microbial pathogens.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Scott G. Filler (sfiller@ucla.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
For in vivo animal studies, age-and sex matched mice were used. Animals were bred/housed under pathogen-free conditions at the Los Angeles Biomedical Research Institute, and UC Irvine. For strain specific information please see section “Mouse Model of Oropharyngeal Candidiasis” and “Generation of BM chimeric mice.” In all of the mouse studies, the animals were randomly assigned to the different treatment groups. The researchers were not blinded to the experimental groups because the primary endpoint (oral fungal burden) was an objective measure of disease severity.
Ethics Statement
All animal work was approved by the Institutional Animal Care and Use Committee (IACUC) of the Los Angeles Biomedical Research Institute and UC Irvine.
Strains
C. albicans SC5314 (Fonzi and Irwin, 1993), a GFP expressing strain (Levitin et al., 2007), and S. aureus RN6390 (Kahl et al., 2000) were used. For use in the experiments, the C. albicans cells were grown for 18 h in yeast extract-peptone dextrose (YPD) at 30°C and the S. aureus cells were grown in brain heart infusion broth at 37°C. The next day, the cells were harvested by centrifugation, washed twice with phosphate-buffered saline (PBS), and counted using a hemacytometer (C. albicans) or a spectrophotometer (S. aureus).
METHOD DETAILS
Mouse Model of Oropharyngeal Candidiasis
EphA2−/− (B6.129S6-Epha2tm1Jrui/J) mice were provided by A. Wayne Orr, LSU Health Sciences Center, Shreveport, LA (Finney et al., 2017), and CD18−/− (B6.129S7-Itgb2tm1Bay/J) mice were purchased from The Jackson Laboratory. Deficient mice were bred under pathogen-free conditions at the Los Angeles Biomedical Research Institute and UC Irvine. C57BL/6 control mice were obtained from The Jackson Laboratory. The EphA2−/− and C57BL/6 control mice were cohoused for at least 1 week before the experiments. Male 7 week old Fcer1g (FcγRI−/−) mice and age-and sex matched C57BL/6 mice were purchased from Taconics. OPC was induced in all strains of mice as described previously (Solis and Filler, 2012; Solis et al., 2017). For inoculation, the animals were sedated, and a swab saturated with 2 × 107 C. albicans cells was placed sublingually for 75 min. Mice were sacrificed after 1, 2 and 4 days of infection, respectively. The tongues were harvested, weighed, homogenized and quantitatively cultured. For histopathologic analysis, thin sections of paraffin-embedded tongues were stained with periodic acid-Schiff stain (PAS). The researchers were not blinded to the experimental groups because the primary endpoint (oral fungal burden) was an objective measure of disease severity.
Generation of BM chimeric mice
Bone marrow chimeric mice were generated as previously described (Lionakis et al., 2013). Briefly, for BM cell transfers, femurs and tibias from 6- to 8-week-old donor wild-type (EphA2+/+; CD45.1; or CD45.2) and EphA2−/− (CD45.2) mice were removed aseptically and BM was flushed using cold PBS supplemented with 2 mM EDTA. Six week-old recipient wild-type (CD45.1) and EphA2−/− (CD45.2) mice were irradiated with 9 Gy and were reconstituted 6 hours after irradiation with 2.5 × 106 EphA2+/+ CD45.2 (WT→WT), EphA2+/+ CD45.1 (wild-type→EphA2−/−), or EphA2−/− CD45.2 (EphA2−/−→wild-type CD45.1 or EphA2−/−) BM cells by lateral tail-vein injection. Mice were given enrofloxacin (Victor Medical) in the drinking water for the first 4 weeks of reconstitution before being switched to antibiotic-free water. Chimeras were infected with C. albicans 8 weeks after transplantation. Prior to infection, we confirmed that mice reconstituted with congenic BM stem cells had achieved a satisfactory level of chimerism by assessing the number of CD45.1 and CD45.2 leukocytes in the blood, using flow cytometry (Figure S2).
Cytokine and Chemokine Measurements In Vivo
To determine the whole tongue cytokine and chemokine protein concentrations, the mice were orally infected with C. albicans. The mice were sacrificed after one day post-infection, and their tongues were harvested, weighed and homogenized. The homogenates were cleared by centrifugation as previously described (Break et al., 2015; Swidergall et al., 2018) and the concentration of inflammatory mediators was measured using a multiplex bead array assay (R&D Systems).
Flow Cytometry of Infiltrating Leukocytes
Immune cells in the mouse tongues were characterized as described elsewhere (Sparber and LeibundGut-Landmann, 2017). Briefly, mice were orally infected with C. albicans strain SC5314. After 1 and 2 days of infection, the animals were administered a sub lethal anesthetic mix intraperitoneally. The thorax was opened, and a part of the rib cage removed to gain access to the heart. The vena cava was cut open and the blood was washed out by slowly injecting 10 mL PBS into the right ventricle. The tongue was harvested and cut into small pieces in 100 μL of ice-cold PBS. 1 mL digestion mix (4.8 mg/ml Collagenase IV; Worthington Biochem, and 200 μg/ml DNase I; Roche Diagnostics, in 1x PBS) was added after which the tissue was incubated at 37°C for 45 min. The resulting tissue suspension was then passed through a 100 μm cell strainer. The single-cell suspensions were then incubated with rat anti-mouse CD16/32 (2.4G2; BD Biosciences) for 10 minutes (1:100) in FACS buffer at 4°C to block Fc receptors. For staining of surface antigens, cells were incubated with fluorochrome-conjugated (FITC, PE, PE-Cy7, allophycocyanin [APC], APC-eFluor 780,) antibodies against mouse CD45 (30-F11; BD Biosciences), Ly6C (AL-21; BD Biosciences), Ly6G (1A8, BioLegend), CD11b (M1/70; eBioscience), and CD90.2 (30-H12; BioLegend). After washing with FACS buffer, the cell suspension was stained with a LIVE/DEAD fluorescent dye (7-AAD; BD Biosciences) for 10 minutes (1:500). The stained cells were analyzed on a 2-laser LSRII flow cytometer (BD Biosciences), and the data were analyzed using FACS Diva (BD Biosciences) and FlowJo software (Treestar). Only single cells were analyzed, and cell numbers were quantified using PE-conjugated fluorescent counting beads (Spherotech).
Antibody Depletion
Mice were injected intraperitoneally with 80 μg anti-Gr-1 (RB6–8C5, eBiosciences) or isotype control (BioXcell) monoclonal antibodies 24 h prior to inoculation with C. albicans (Day 0). The degree of depletion was assessed in peripheral blood that was obtained by cardiac puncture. The percentage of neutrophils was determined by enumeration of a blood smear stained with modified Giemsa stain solution (Sigma-Aldrich), counting 100–200 cells per sample. The absolute neutrophil count (ANC) was determined by multiplying the percentage of neutrophils (mature and band forms) by total leukocyte counts, which were determined by counting tryptophan blue stained cells using a hemacytometer.
Neutrophil Isolation from Mouse Bone Marrow
Mouse neutrophils were purified from bone marrow cells using negative magnetic bead selection (MojoSort, BioLegend). In brief, bone marrow cells were flushed from femurs and tibias using sterile RPMI 1640 medium supplemented with 10% FBS and 2 mM EDTA onto a 50 mL screw top Falcon tube fitted with a 100 μm filter (Swamydas et al., 2015). Cells were washed with 1X MojoSort buffer (1X PBS, 0.5% BSA, 2mM EDTA). Neutrophils were isolated according to the manufacturer’s instructions. Bone marrow-enriched neutrophils had > 90% purity and > 90% viability as determined by flow cytometry (in vitro experiments). In the adoptive transfer experiments, the purity exceeded 94%.The remaining ~10% of cells were identified as CD45+ CD19+ (B cells) < 2.72%, CD45+ CD3+ (T cells) < 0.24%, CD45+ Ly6Chi (inflammatory monocytes) < 0.31%, CD45+ MHC II+ (dendritic cells) < 2.37%, and CD45− (stromal cells) < 4.00%.
Neutrophil Isolation from Mouse Peripheral Blood
Peripheral blood was collected via cardiac puncture, anticoagulated with EDTA, and then layered over Ficoll-Histopaque 1077 (Sigma Aldrich). After centrifugation (4°C, 1500 rpm) the supernatant was discarded, and red blood cells were lysed using ACK lysis buffer (GIBCO). After 5 min cold PBS was added and cells were washed twice. The isolated neutrophils were resuspended in RPMI 1640 medium + 2% inactivated mouse serum (Gemini Bio-Products).
Adoptive Transfer of Neutrophils
Adoptive transfer of neutrophils was performed as described elsewhere (Leal et al., 2012). Briefly, male and female CD18−/− mice aged 6–12 weeks were orally infected as described above. After 6 hours post infection 5 × 106 naive donor bone marrow neutrophils from EphA2−/− and C57BL/6 (wild-type) mice were injected into the tail vein of CD18−/− recipient mice. After 1 and 2 days of infection, mice were euthanized, and the oral fungal burden was determined.
Surface Staining of Neutrophil Receptors
Mouse neutrophils were isolated as described above. Purified neutrophils were incubated with anti-mouse CD16/32 (2.4G2; BD Biosciences) blocking antibody, followed by anti-mEphA2 (PE conjugated; FAB639P; R&D Systems), anti-Dectin-1 (PE conjugated; Clone RH1; BioLegend), anti-CD11b (PE conjugated; Clone M1/70; BioLegend), anti-CD18 (PE conjugated; Clone M18/2; BioLegend), anti-CD64 (PE conjugated; Clone X54–5/7.1; BioLegend), and surface expression was measured using flow cytometry. For CD16/32 staining neutrophils were stained with anti-CD16/32 (PE conjugated; Clone 93; BioLegend) without Fc-block.
Neutrophil Killing Assays
Neutrophils from mice were isolated as described above. Neutrophil killing of C. albicans yeast was determined by CFU enumeration. Briefly, unopsonized or serum-opsonized (2% heat-inactivated mouse serum; Gemini Bio-Products) C. albicans SC5314 yeast cells were incubated with isolated neutrophils in a 1:20 ratio for 3 hours. Neutrophils were lysed with 0.02% Triton X-100 in ice-cold water for 5 minutes, diluted and remaining Candida cells were quantitatively cultured. In some experiments, Fcγ receptors were blocked using antibodies against CD16/32 (2.4G2; BD Biosciences), and CD64 (clone X54–5/7.1; BioLegend). To determine the effect of ERK inhibition during killing BM-neutrophils were incubated with 1 μM SCH772984 (Cayman Chemical Company) for 45 min prior to infection. The capacity of neutrophils to kill serum-opsonized S. aureus RN6390 was also determined by quantitative culture using a S. aureus to neutrophil ratio of 10:1 and an incubation time of 30 minutes. The capacity of neutrophils to kill C. albicans hyphae was determined using the alamarBlue (Invitrogen) reduction as a measure of fungal inactivation. Neutrophils were incubated in duplicate wells of flat bottom 96-well plates containing hyphae that had been grown for 3 hours with serum opsonization, at a neutrophil to C. albicans ratio of 1:4 at 37°C. After 2.5 hours, the neutrophils were lysed with 0.02% Triton X-100 in water for 5 minutes, after which the C. albicans hyphae were washed twice with PBS and incubated with 1 × alamarBlue (Invitrogen) for 18 hours at 37°C. Optical density at a wavelength of 570nm and 600nm was determined. Neutrophil killing capacity was calculated as the amount of alamarBlue reduced by wells containing C. albicans hyphae incubated with and without neutrophils.
BM-derived macrophages
BM-derived macrophages were generated by culturing BM cells obtained as described above from wild-type and EphA2−/− mice in RPMI 1640 supplemented with 10% FBS, 2 mM L-glutamine (Mediatech), 20 mM HEPES, 1% penicillin/streptomycin and 40 ng/ml M-CSF (Peprotech Inc.) for 5 to 7 days.
Neutrophil Phagocytosis
For phagocytosis assays, 5 × 105 neutrophils from wild-type or EphA2−/− mice were incubated with 5 × 105 serum-opsonized C. albicans yeast cells for 30 minutes at 37°C on fibronectin-coated circular glass coverslips in 24-well tissue culture plates. Phagocytosed yeast cells were quantified using a differential fluorescence assay as outlined elsewhere (Liu et al., 2016; Phan et al., 2007). Briefly, 5 × 105 BM neutrophils were incubated with 5 × 105 C. albicans expressing GFP. After 30 min cells were fixed using 3% paraformaldehyde for 10 min. Coverslips were washed 3x in PBS and incubated for 30 min with anti-Candida Alexa 568 Ab. The coverslips were viewed with an epifluorescence microscope, and the number of phagocytosed organisms per high-power field was determined, counting at least 100 organisms per coverslip. Phagocytosed C. albicans appeared green (GFP), while non-phagocytosed fungal cells were both green (GFP) and red (anti-Candida Alexa 568).
Indirect Immunofluorescence
To determine protein localization within the neutrophils, the cells were incubated in RPMI 1640 medium containing 2% heat-inactivated mouse serum for 30 min on fibronectin coated coverslips. The cells were then incubated with opsonized C. albicans CAI4-GFP for 30 min. Next, they were fixed with 3% paraformaldehyde, blocked with 10% BSA, followed by anti-mouse CD16/32 (2.4G2; BD Biosciences), and incubated with antibodies against EphA2 (D4A2, #6997; Cell Signaling), and p47phox (A-7; Santa Cruz), followed by an Alexa 488-conjugated mouse anti-rabbit antibody. To visualize F-actin, the cells were also stained with Phalloidin-IFluor 405 (Abcam). The cells were then imaged by confocal microscopy.
Immunoblotting
Neutrophils were incubated with opsonized C. albicans at a multiplicity of infection of 5 or with 10 μg/mL IgG Sepharose beads (#3420; Cell Signaling) and/or 10 μg/mL zymosan (depleted, tlrl-zyd; InvivoGen). At various time points, the cells were washed with cold PBS, collected by centrifugation, and boiled in sample buffer. The lysates were separated by SDS-PAGE, and phosphorylation was detected by immunoblotting with specific antibodies against pEphA2 (#6347, Cell Signaling), pSer345 p47phox (orb126026; biorbyt), pSer304 p47phox (PA5–36773; Thermo Fisher), pp44–42 (#4370; Cell Signaling), pPKC (#2055; Cell Signaling). Next, the blot was stripped, and the total amount of each protein was detected by immunoblotting with antibodies against EphA2 (D4A2, Cell Signaling, or clone 233720; R&D Systems), p47phox (A-7; Santa Cruz), p44/42 (#3085R; BioVision), and PKC (#9616; Cell Signaling). Each experiment was performed at least 3 times.
QUANTIFICATION AND STATISTICAL ANALYSIS
At least three biological replicates were performed for all in vitro experiments unless otherwise indicated. Data were compared by Mann-Whitney corrected for multiple comparisons using GraphPad Prism (v. 8) software. P values < 0.05 were considered statistically significant.
DATA AND CODE AVAILABILITY
The raw data that support the findings of this study are available from the corresponding authors upon request.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Mouse CD16/CD32 (Mouse BD Fc Block) | BD Biosciences | Cat# 553142 RRID:AB_394657 |
| PE anti-mouse CD16/32 antibody | BioLegend | Cat# 101308, RRID:AB_312807 |
| Rat Anti-CD45 Monoclonal Antibody, Allophycocyanin Conjugated, Clone 30-F11 | BD Biosciences | Cat# 559864, RRID:AB_398672 |
| Rat Anti-Ly-6C Monoclonal Antibody, FITC Conjugated, Clone AL-21 | BD Biosciences | Cat# 553104 RRID: AB_394628 |
| PE/Cy7 anti-mouse Ly-6G antibody | BioLegend | Cat# 127618, RRID:AB_1877261 |
| CD11b Monoclonal Antibody (M1/70), APC-Cyanine7 | eBioscience | Cat# A15390 RRID:AB_2534404 |
| PE anti-mouse/human CD11b antibody | BioLegend | Cat# 101207, RRID:AB_312790 |
| PE anti-mouse CD18 antibody | BioLegend | Cat# 101407, RRID:AB_312816 |
| PE anti-mouse CD90.2 antibody | BioLegend | Cat# 105308, RRID:AB_313179 |
| PE anti-mouse CD369 (Dectin-1, CLEC7A) antibody | BioLegend | Cat# 144304, RRID:AB_2561501 |
| APC/Cyanine7 anti-mouse CD45.1 antibody | BioLegend | Cat# 110715, RRID:AB_313504 |
| FITC anti-mouse CD45.2 antibody | BioLegend | Cat# 109805, RRID:AB_313442 |
| Ly-6G (Gr-1) Monoclonal Antibody (RB6–8C5) | eBiosciences | Cat# 14–5931–82, RRID:AB_467730 |
| InVivoMab rat IgG2b isotype control antibody | Bio X Cell | Cat# BE0090, RRID:AB_1107780 |
| PE anti-mouse CD64 (FcRI) antibody | BioLegend | Cat# 139303, RRID:AB_10613467 |
| Purified anti-mouse CD64 (FcRI) antibody | BioLegend | Cat# 139301, RRID:AB_10612757 |
| MHC Class II (I-A/I-E) Monoclonal Antibody (M5/114.15.2), FITC | eBiosciences | Cat# 11–5321–82, RRID:AB_465232 |
| APC anti-mouse CD19 antibody | BioLegend | Cat# 115511, RRID:AB_313646 |
| APC anti-mouse CD3 antibody | BioLegend | Cat# 100235, RRID:AB_2561455 |
| Mouse EphA2 Phycoerythrin mAb (Clone 233720) antibody | R&D | Cat# FAB639P, RRID:AB_2099100 |
| EphA2 (D4A2) XP Rabbit mAb antibody | Cell Signaling Technology | Cat# 6997, RRID:AB_10827743 |
| Phospho-EphA2 (Ser897) (D9A1) Rabbit mAb antibody | Cell Signaling Technology | Cat# 6347, RRID:AB_11220420 |
| MEK1/2 Antibody | Cell Signaling Technology | Cat# 9122, RRID:AB_823567 |
| Anti-MEK1 / 2, phospho (Ser217 / Ser221) Antibody, Unconjugated | Cell Signaling Technology | Cat# 9121, RRID:AB_331648 |
| p44/42 MAPK (Erk1/2) Antibody | BioVision | Cat# 3085R-100, RRID:AB_10989057 |
| Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) antibody | Cell Signaling Technology | Cat# 4370, RRID:AB_2315112 |
| PKCdelta (D10E2) Rabbit mAb antibody | Cell Signaling Technology | Cat# 9616, RRID:AB_10949973 |
| Phospho-Zap-70 (Tyr319)/Syk (Tyr352) (65E4) Rabbit mAb antibody | Cell Signaling Technology | Cat# 2717F RRID: AB_2218658 |
| Syk (D3Z1E) XP Rabbit Antibody | Cell Signaling Technology | Cat# 13198 RRID: AB_2687924 |
| Phospho-PKCdelta (Tyr311) Antibody | Cell Signaling Technology | Cat# 2055, RRID:AB_330876 |
| p47 phox (phospho-Ser345) antibody | Biorbyt | orb126026 |
| Phospho-p47phox (Ser304) Polyclonal Antibody | Thermo Fisher Scientific | Cat# PA5–36773, RRID:AB_2553720 |
| p47-phox (A-7) antibody | Santa Cruz Biotechnology | Cat# sc-17844, RRID:AB_627987 |
| Mouse IgG (Sepharose Bead Conjugate) antibody | Cell Signaling Technology | Cat# 3420, RRID:AB_1549744 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 7-AAD | BD Biosciences | Cat# 559925 |
| 2% heat-inactivated mouse serum | Gemini Bio-Products | Cat#100–113 |
| SCH772984 | Cayman Chemical Company | Ca# 19166 CAS# 942183–80–4 |
| alamarBlue | Invitrogen | Cat# DAL1025 |
| Zymosan (depleted) | InvivoGen | Cat# tlrl-zyd Lot# DZN-40–01 |
| Recombinant Murine M-CSF | Peprotech Inc | Cat# 315–02 |
| H2DCFDA | Invitrogen | Cat# D399 |
| Critical Commercial Assays | ||
| MojoSort Mouse Neutrophil Isolation Kit | BioLegend | Cat# 480058 |
| Luminex Mouse Magnetic Assay | R&D | Cat# LXSAMSM |
| Experimental Models: Organisms/Strains | ||
| C. albicans SC5314 | Fonzi and Irwin, 1993 | N/A |
| C. albicans-GFP | Levitin et al., 2007 | N/A |
| S. aureus RN6390 | Kahl et al., 2000 | N/A |
| Epha2trn1Jrui/J | Finney et al., 2017 | N/A |
| C57BL/6J | The Jackson Laboratory | RRID: IMSR_JAX:000664 |
| B6.129S7-Itgb2trn1Bay/J | The Jackson Laboratory | RRID:IMSR_JAX:002128 |
| B6.129P2-Fcer1gtrn1Rav N12 | Taconics | RRID:IMSR_TAC:583 |
| Software and Algorithms | ||
| FlowJo V10 | Treestar | https://www.flowjo.com/ |
| GraphPad Prism V8 | GraphPad | https://www.graphpad.com/ |
| Other | ||
| Luminex multiplex analyzer | Luminex | https://www.luminexcorp.com |
| BD LSR II | BD Biosciences | https://www.bd.com |
| Leica TCS SP8 Confocal system | Leica | https://www.leica-microsystems.com |
Highlights.
EphA2 is a receptor for β-glucans on oral epithelial cells and neutrophils
Epithelial cell EphA2 recruits phagocytes to foci of oral C. albicans infection
Neutrophil EphA2 primes p47phox to enhance production of reactive oxygen species
Neutrophil EphA2 augments killing of opsonized C. albicans yeast
ACKNOWLEDGMENTS
This work was supported in part by NIH grants R01DE022600 and R01AI124566 to S.G.F. and K99DE026856 to M.S. Research in the laboratory of E.P. is supported by NIH grant R01EY018612. Research by M.S.L. was supported by the Intramural Research Program of the National Institute of Allergy and Infectious Diseases. We are grateful to A.W. Orr for providing the EphA2−/− mice and L. Li for providing bacterial strains. We thank K. Iwamoto for irradiation assistance, T. Hohl for assistance with the BM chimera studies, S.W. French and E. Vitocruz for histopathology, and members of the Division of Infectious Diseases at the Harbor-UCLA Medical Center for critical suggestions. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
DECLARATION OF INTERESTS
S.G.F. is a co-founder of and shareholder in NovaDigm Therapeutics, Inc., a company that is developing a vaccine against mucosal and invasive Candida infections.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.06.020.
REFERENCES
- Altmeier S, Toska A, Sparber F, Teijeira A, Halin C, and LeibundGut-Landmann S (2016). IL-1 Coordinates the Neutrophil Response to C. albicans in the Oral Mucosa. PLoS Pathog 12, e1005882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballou ER, Avelar GM, Childers DS, Mackie J, Bain JM, Wagener J, Kastora SL, Panea MD, Hardison SE, Walker LA, et al. (2016). Lactate signalling regulates fungal β-glucan masking and immune evasion. Nat. Microbiol 2, 16238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bey EA, Xu B, Bhattacharjee A, Oldfield CM, Zhao X, Li Q, Subbulakshmi V, Feldman GM, Wientjes FB, and Cathcart MK (2004). Protein kinase C delta is required for p47phox phosphorylation and translocation in activated human monocytes. J. Immunol 173, 5730–5738. [DOI] [PubMed] [Google Scholar]
- Bishu S, Hernández-Santos N, Simpson-Abelson MR, Huppler AR, Conti HR, Ghilardi N, Mamo AJ, and Gaffen SL (2014). The adaptor CARD9 is required for adaptive but not innate immunity to oral mucosal Candida albicans infections. Infect. Immun 82, 1173–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bongomin F, Gago S, Oladele RO, and Denning DW (2017). Global and Multi-National Prevalence of Fungal Diseases-Estimate Precision. J. Fungi (Basel) 3, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandtzaeg P (2013). Secretory immunity with special reference to the oral cavity. J. Oral Microbiol 5 10.3402/jom.v3405i3400.20401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Break TJ, Jaeger M, Solis NV, Filler SG, Rodriguez CA, Lim JK, Lee CC, Sobel JD, Netea MG, and Lionakis MS (2015). CX3CR1 is dispensable for control of mucosal Candida albicans infections in mice and humans. Infect. Immun 83, 958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SYC, and Gordon S (2002). Dectin-1 is a major β-glucan receptor on macrophages. J. Exp. Med 196, 407–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD, Denning DW, Gow NA, Levitz SM, Netea MG, and White TC (2012). Hidden killers: human fungal infections. Sci. Transl. Med 4, 165rv13. [DOI] [PubMed] [Google Scholar]
- Chen J, Sathiyamoorthy K, Zhang X, Schaller S, Perez White BE, Jardetzky TS, and Longnecker R (2018). Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus. Nat. Microbiol 3, 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiani P, Bromuro C, Cassone A, and Torosantucci A (2009). Anti-beta-glucan antibodies in healthy human subjects. Vaccine 27, 513–519. [DOI] [PubMed] [Google Scholar]
- Conti HR, Bruno VM, Childs EE, Daugherty S, Hunter JP, Mengesha BG, Saevig DL, Hendricks MR, Coleman BM, Brane L, et al. (2016). IL-17 Receptor Signaling in Oral Epithelial Cells Is Critical for Protection against Oropharyngeal Candidiasis. Cell Host Microbe 20, 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dambuza IM, Levitz SM, Netea MG, and Brown GD (2017). Fungal Recognition and Host Defense Mechanisms. Microbiol. Spectr 5, [DOI] [PubMed] [Google Scholar]
- Dang PM, Dewas C, Gaudry M, Fay M, Pedruzzi E, Gougerot-Pocidalo MA, and El Benna J (1999). Priming of human neutrophil respiratory burst by granulocyte/macrophage colony-stimulating factor (GM-CSF) involves partial phosphorylation of p47(phox). J. Biol. Chem 274, 20704–20708. [DOI] [PubMed] [Google Scholar]
- Dang PM-C, Stensballe A, Boussetta T, Raad H, Dewas C, Kroviarski Y, Hayem G, Jensen ON, Gougerot-Pocidalo M-A, and El-Benna J (2006). A specific p47phox-serine phosphorylated by convergent MAPKs mediates neutrophil NADPH oxidase priming at inflammatory sites. J. Clin. Invest 116, 2033–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZM, Babensee JE, Simon SI, Lu H, Perrard JL, Bullard DC, Dai XY, Bromley SK, Dustin ML, Entman ML, et al. (1999). Relative contribution of LFA-1 and Mac-1 to neutrophil adhesion and migration. J. Immunol 163, 5029–5038. [PubMed] [Google Scholar]
- Drewniak A, Gazendam RP, Tool AT, van Houdt M, Jansen MH, van Hamme JL, van Leeuwen EM, Roos D, Scalais E, de Beaufort C, et al. (2013). Invasive fungal infection and impaired neutrophil killing in human CARD9 deficiency. Blood 121, 2385–2392. [DOI] [PubMed] [Google Scholar]
- Drummond RA, Collar AL, Swamydas M, Rodriguez CA, Lim JK, Mendez LM, Fink DL, Hsu AP, Zhai B, Karauzum H, et al. (2015). CARD9-Dependent Neutrophil Recruitment Protects against Fungal Invasion of the Central Nervous System. PLoS Pathog. 11, e1005293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond RA, Franco LM, and Lionakis MS (2018). Human CARD9: A Critical Molecule of Fungal Immune Surveillance. Front. Immunol 9, 1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El Benna J, Han J, Park JW, Schmid E, Ulevitch RJ, and Babior BM (1996). Activation of p38 in stimulated human neutrophils: phosphorylation of the oxidase component p47phox by p38 and ERK but not by JNK. Arch. Biochem. Biophys 334, 395–400. [DOI] [PubMed] [Google Scholar]
- El-Benna J, Dang PM, Gougerot-Pocidalo MA, Marie JC, and Braut-Boucher F (2009). p47phox, the phagocyte NADPH oxidase/NOX2 organizer: structure, phosphorylation and implication in diseases. Exp. Mol. Med 41, 217–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finney AC, Funk SD, Green JM, Yurdagul A Jr., Rana MA, Pistorius R, Henry M, Yurochko A, Pattillo CB, Traylor JG, et al. (2017). EphA2 Expression Regulates Inflammation and Fibroproliferative Remodeling in Atherosclerosis. Circulation 136, 566–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontayne A, Dang PM, Gougerot-Pocidalo MA, and El-Benna J (2002). Phosphorylation of p47phox sites by PKC alpha, beta II, delta, and zeta: effect on binding to p22phox and on NADPH oxidase activation. Biochemistry 41, 7743–7750. [DOI] [PubMed] [Google Scholar]
- Fonzi WA, and Irwin MY (1993). Isogenic strain construction and gene mapping in Candida albicans. Genetics 134, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forehand JR, Pabst MJ, Phillips WA, and Johnston RB Jr. (1989). Lipopolysaccharide priming of human neutrophils for an enhanced respiratory burst. Role of intracellular free calcium. J. Clin. Invest 83, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gantner BN, Simmons RM, and Underhill DM (2005). Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. EMBO J. 24, 1277–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazendam RP, van Hamme JL, Tool AT, van Houdt M, Verkuijlen PJ, Herbst M, Liese JG, van de Veerdonk FL, Roos D, van den Berg TK, and Kuijpers TW (2014). Two independent killing mechanisms of Candida albicans by human neutrophils: evidence from innate immunity defects. Blood 124, 590–597. [DOI] [PubMed] [Google Scholar]
- Gazendam RP, van de Geer A, Roos D, van den Berg TK, and Kuijpers TW (2016). How neutrophils kill fungi. Immunol. Rev 273, 299–311. [DOI] [PubMed] [Google Scholar]
- Heng TS, and Painter MW; Immunological Genome Project Consortium (2008). The Immunological Genome Project: networks of gene expression in immune cells. Nat. Immunol 9, 1091–1094. [DOI] [PubMed] [Google Scholar]
- Hopke A, Nicke N, Hidu EE, Degani G, Popolo L, and Wheeler RT (2016). Neutrophil Attack Triggers Extracellular Trap-Dependent Candida Cell Wall Remodeling and Altered Immune Recognition. PLoS Pathog. 12, e1005644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppler AR, Conti HR, Hernández-Santos N, Darville T, Biswas PS, and Gaffen SL (2014). Role of neutrophils in IL-17-dependent immunity to mucosal candidiasis. J. Immunol 192, 1745–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishibashi K, Yoshida M, Nakabayashi I, Shinohara H, Miura NN, Adachi Y, and Ohno N (2005). Role of anti-beta-glucan antibody in host defense against fungi. FEMS Immunol. Med. Microbiol 44, 99–109. [DOI] [PubMed] [Google Scholar]
- Kahl BC, Goulian M, van Wamel W, Herrmann M, Simon SM, Kaplan G, Peters G, and Cheung AL (2000). Staphylococcus aureus RN6390 replicates and induces apoptosis in a pulmonary epithelial cell line. Infect. Immun 68, 5385–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karlsson A, Nixon JB, and McPhail LC (2000). Phorbol myristate acetate induces neutrophil NADPH-oxidase activity by two separate signal transduction pathways: dependent or independent of phosphatidylinositol 3-kinase. J. Leukoc. Biol 67, 396–404. [DOI] [PubMed] [Google Scholar]
- Kaushansky A, Douglass AN, Arang N, Vigdorovich V, Dambrauskas N, Kain HS, Austin LS, Sather DN, and Kappe SHI (2015). Malaria parasites target the hepatocyte receptor EphA2 for successful host infection. Science 350, 1089–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiefer F, Brumell J, Al-Alawi N, Latour S, Cheng A, Veillette A, Grinstein S, and Pawson T (1998). The Syk protein tyrosine kinase is essential for Fcgamma receptor signaling in macrophages and neutrophils. Mol. Cell. Biol 18, 4209–4220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leal SM Jr., Vareechon C, Cowden S, Cobb BA, Latgé JP, Momany M, and Pearlman E (2012). Fungal antioxidant pathways promote survival against neutrophils during infection. J. Clin. Invest. 122, 2482–2498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitin A, Marcil A, Tettweiler G, Laforest MJ, Oberholzer U, Alarco AM, Thomas DY, Lasko P, and Whiteway M (2007). Drosophila melanogaster Thor and response to Candida albicans infection. Eukaryot. Cell 6, 658–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levitz SM (2010). Innate recognition of fungal cell walls. PLoS Pathog. 6, e1000758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limnander A, Depeille P, Freedman TS, Liou J, Leitges M, Kurosaki T, Roose JP, and Weiss A (2011). STIM1, PKC-δ and RasGRP set a threshold for proapoptotic Erk signaling during B cell development. Nat. Immunol 12, 425–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lionakis MS, and Levitz SM (2018). Host Control of Fungal Infections: Lessons from Basic Studies and Human Cohorts. Annu. Rev. Immunol 36, 157–191. [DOI] [PubMed] [Google Scholar]
- Lionakis MS, Swamydas M, Fischer BG, Plantinga TS, Johnson MD, Jaeger M, Green NM, Masedunskas A, Weigert R, Mikelis C, et al. (2013). CX3CR1-dependent renal macrophage survival promotes Candida control and host survival. J. Clin. Invest 123, 5035–5051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Lee MJ, Solis NV, Phan QT, Swidergall M, Ralph B, Ibrahim AS, Sheppard DC, and Filler SG (2016). Aspergillus fumigatus CalA binds to integrin α5β1 and mediates host cell invasion. Nat. Microbiol 2, 16211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, et al. (2011). EGFR and EphA2 are host-factors for hepatitis C virus entry and possible targets for antiviral therapy. Nat. Med 17, 589–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyes DL, Runglall M, Murciano C, Shen C, Nayar D, Thavaraj S, Kohli A, Islam A, Mora-Montes H, Challacombe SJ, and Naglik JR (2010). A biphasic innate immune MAPK response discriminates between the yeast and hyphal forms of Candida albicans in epithelial cells. Cell Host Microbe 8, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen BC, Budelsky AL, Baptist AP, Schaller MA, and Lukacs NW (2012). Interleukin-25 induces type 2 cytokine production in a steroid-resistant interleukin-17RB+ myeloid population that exacerbates asthmatic pathology. Nat. Med 18, 751–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan QT, Myers CL, Fu Y, Sheppard DC, Yeaman MR, Welch WH, Ibrahim AS, Edwards JE Jr., and Filler SG (2007). Als3 is a Candida albicans invasin that binds to cadherins and induces endocytosis by host cells. PLoS Biol. 5, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts PJ, and Der CJ (2007). Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26, 3291–3310. [DOI] [PubMed] [Google Scholar]
- Solis NV, and Filler SG (2012). Mouse model of oropharyngeal candidiasis. Nat. Protoc 7, 637–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solis NV, Swidergall M, Bruno VM, Gaffen SL, and Filler SG (2017). The Aryl Hydrocarbon Receptor Governs Epithelial Cell Invasion during Oropharyngeal Candidiasis. MBio 8, e00025–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparber F, and LeibundGut-Landmann S (2017). Assessment of Immune Responses to Fungal Infections: Identification and Characterization of Immune Cells in the Infected Tissue. Methods Mol. Biol 1508, 167–182. [DOI] [PubMed] [Google Scholar]
- Subbarayal P, Karunakaran K, Winkler AC, Rother M, Gonzalez E, Meyer TF, and Rudel T (2015). EphrinA2 receptor (EphA2) is an invasion and intracellular signaling receptor for Chlamydia trachomatis. PLoS Pathog. 11, e1004846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, and Chilvers ER (2010). Neutrophil kinetics in health and disease. Trends Immunol. 31, 318–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suram S, Silveira LJ, Mahaffey S, Brown GD, Bonventre JV, Williams DL, Gow NAR, Bratton DL, Murphy RC, and Leslie CC (2013). Cytosolic phospholipase A(2)α and eicosanoids regulate expression of genes in macrophages involved in host defense and inflammation. PLoS ONE 8, e69002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamydas M, Luo Y, Dorf ME, and Lionakis MS (2015). Isolation of mouse neutrophils. Curr. Protoc. Immunol 110, 3.20.1–3.20.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidergall M (2019). Candida albicans at Host Barrier Sites: Pattern Recognition Receptors and Beyond. Pathogens 8, E40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidergall M, and Ernst JF (2014). Interplay between Candida albicans and the antimicrobial peptide armory. Eukaryot. Cell 13, 950–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidergall M, and Filler SG (2017). Oropharyngeal Candidiasis: Fungal Invasion and Epithelial Cell Responses. PLoS Pathog. 13, e1006056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swidergall M, Solis NV, Lionakis MS, and Filler SG (2018). EphA2 is an epithelial cell pattern recognition receptor for fungal β-glucans. Nat. Microbiol 3, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor PR, Roy S, Leal SM Jr., Sun Y, Howell SJ, Cobb BA, Li X, and Pearlman E (2014). Activation of neutrophils by autocrine IL-17A-IL-17RC neutrophil activation in fungal infections is regulated by IL-6, IL-23, RORγt and Dectin-2. Nat. Immunol 15, 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TerBush AA, Hafkamp F, Lee HJ, and Coscoy L (2018). A Kaposi’s Sarcoma-Associated Herpesvirus Infection Mechanism Is Independent of Integrins α3β1, αVβ3, and aVp5. J. Virol 92, e00803–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueda Y, Hirai Si., Osada Si., Suzuki A, Mizuno K, and Ohno S (1996). Protein kinase C activates the MEK-ERK pathway in a manner independent of Ras and dependent on Raf. J. Biol. Chem 271, 23512–23519. [DOI] [PubMed] [Google Scholar]
- Underhill DM, and Pearlman E (2015). Immune Interactions with Pathogenic and Commensal Fungi: A Two-Way Street. Immunity 43, 845–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Bruggen R, Drewniak A, Jansen M, van Houdt M, Roos D, Chapel H, Verhoeven AJ, and Kuijpers TW (2009). Complement receptor 3, not Dectin-1, is the major receptor on human neutrophils for beta-glucan-bearing particles. Mol. Immunol 47, 575–581. [DOI] [PubMed] [Google Scholar]
- Verma A, Gaffen SL, and Swidergall M (2017a). Innate immunity to mucosal Candida infections. J. Fungi (Basel) 3, E60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verma AH, Richardson JP, Zhou C, Coleman BM, Moyes DL, Ho J, Huppler AR, Ramani K, McGeachy MJ, Mufazalov IA, et al. (2017b). Oral epithelial cells orchestrate innate type 17 responses to Candida albicans through the virulence factor candidalysin. Sci. Immunol 2, eaam8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnatsch A, Tsourouktsoglou TD, Branzk N, Wang Q, Reincke S, Herbst S, Gutierrez M, and Papayannopoulos V (2017). Reactive Oxygen Species Localization Programs Inflammation to Clear Microbes of Different Size. Immunity 46, 421–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li Y, Wang HB, Zhang A, Chen ML, Fang ZX, Dong XD, Li SB, Du Y, Xiong D, et al. (2018). Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol 3, 1–8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data that support the findings of this study are available from the corresponding authors upon request.