

SUMMARY
In this study, we explore the diversity and its distribution along the wheat leaf rust resistance protein LR10 three‐dimensional structure. Lr10 is a leaf rust resistance gene encoding a coiled coil–nucleotide‐binding site–leucine‐rich repeat (CC–NBS–LRR) class of protein. Lr10 was cloned and sequenced from 58 accessions representing diverse habitats of wild emmer wheat in Israel. Nucleotide diversity was very high relative to other wild emmer wheat genes (π= 0.029). The CC domain was found to be the most diverse domain and subject to positive selection. Superimposition of the diversity on the CC three‐dimensional structure showed that some of the variable and positively selected residues were solvent exposed and may interact with other proteins. The LRR domain was relatively conserved, but showed a hotspot of amino acid variation between two haplotypes in the ninth repeat. This repeat was longer than the other LRRs, and three‐dimensional modelling suggested that an extensive α helix structure was formed in this region. The two haplotypes also differed in splicing regulation motifs. In genotypes with one haplotype, an intron was alternatively spliced in this region, whereas, in genotypes with the other haplotype, this intron did not splice at all. The two haplotypes are proposed to be ancient and maintained by balancing selection.
INTRODUCTION
The majority of disease resistance genes (R genes) isolated from plants, conferring resistance to bacterial, fungal, oomycete or viral pathogens, encode proteins containing a nucleotide‐binding site and a leucine‐rich repeat (NBS–LRR) domain (Dangl and Jones, 2001). The NBS (NB‐ARC) domain has a role in signal transduction mediated by nucleotide phosphorylation and is the most conserved part of the gene (van Ooijen et al., 2008; Pan et al., 2000). The LRR domain is involved in pathogen recognition and is subject to diversifying selection between paralogues and to balancing selection between alleles (Bakker et al., 2006; Bergelson et al., 2001; Jiang et al., 2007; Meyers et al., 1998). In monocots, only the coiled coil (CC)–NBS–LRR subclass is present, in which a CC domain is found at the N‐terminus of the NBS–LRR domain (Pan et al., 2000). The CC domain is involved in signalling and, in many cases, in pathogen recognition (Rairdan et al., 2008, and references cited therein).
R genes in plants are highly diverse and evolve rapidly (Dangl and Jones, 2001; Rose et al., 2004). Bakker et al. (2006) found that the nucleotide diversity and the number of segregating sites between alleles of R genes are higher than the whole genome average. In some R genes, diversity is maintained by frequency‐dependent selection (Tellier and Brown, 2007). In these genes, alleles are cycled between high and low frequency in a long co‐evolution with pathogens in a form of ‘trench warfare’. The trench warfare model predicts that the polymorphism will be old and under balancing selection (Holub, 2001; Stahl et al., 1999).
In some R genes, the LRR domain is alternatively spliced, where more than one alternative variant is required to confer resistance. In many cases, one of the alternative isoforms carries a premature stop codon that results in a shorter LRR domain or its complete truncation (Dinesh‐Kumar and Baker, 2000; Gassmann, 2008; Jordan et al., 2002). Splicing is regulated by splicing factors (SFs); these proteins bind to specific motifs on the pre‐mRNA and facilitate the splicing process. Some SFs can enhance and some can suppress splicing (Gromak et al., 2003).
The wheat leaf rust resistance gene Lr10, cloned from the A genome of cultivated bread wheat (Triticum aestivum), is one of 60 leaf rust resistance genes known in wheat, and one of four of these that have been cloned so far. Lr10 is one of three resistance genes with a known sequence in the wild emmer wheat genome (Cloutier et al., 2007; Feuillet et al., 2003; Fu et al., 2009; Huang et al., 2003; Krattinger et al., 2009). Lr10 is a single copy gene and a member of the CC–NBS–LRR subclass. Lr10 is an ancient and polymorphic gene present in all three ploidy levels in the Triticum genus. The Lr10 N‐terminus is the most diverse segment and is subject to diversifying selection (Feuillet et al., 2003; Isidore et al., 2005; Loutre et al., 2009; Sela et al., 2011). A recent study has shown that another gene, RGA 2, closely linked to Lr10, is required to mediate resistance by Lr10 (Loutre et al., 2009). Sela et al. (2011) have revealed that linkage disequilibrium rapidly decays along Lr10, except for the LRR domain, where a linkage disequilibrium block is found. This block is formed by two haplotypes, highly diverged, that do not recombine. These two haplotypes are found together in populations across Israel and in T. urartu, the ancestor of T. dicoccoides.
Wild emmer wheat (T. dicoccoides) is an allotetraploid (BBAA) species that emerged from the polyploidization event joining together the A genome from T. urartu and the B genome from an extinct member of the sitopsis section that is a close relative of Aegilops speltoides (Feldman et al., 1995). Wild emmer wheat is the progenitor of most cultivated wheat species, and its natural habitats are located in the Fertile Crescent of the Middle East. The centre of diversity of wild emmer wheat is found in the catchment area of the upper Jordan Valley in Israel and its vicinity (Luo et al., 2007; Nevo and Beiles, 1989). The wild emmer wheat gene pool is a valuable source of R genes that can be transferred into cultivated wheat (Knott et al., 2005; Marais et al., 2005; Mergoum et al., 2005; Rong et al., 2000; Uauy et al., 2005). The study of the genetic diversity of R genes can reveal novel alleles present in the wild emmer wheat population and their functional significance. This will help to plan efficient conservation and utilization of this important gene pool. Furthermore, a diversity study in a wild population that is related to an important crop has the advantage of capturing the long evolutionary history of wild populations in their natural habitats.
Leaf rust, caused by Puccinia triticina, is the most prevalent disease of wheat worldwide. It is highly specific and can be carried across thousands of kilometres by wind (Kolmer, 2005). Most of the T. dicoccoides accessions from Israel are susceptible to local and North American leaf rust isolates, but a small number are resistant (Anikster et al., 2005; Moseman et al., 1985; Sela et al., 2011). Nevertheless, leaf rust resistance genes from T. dicoccoides have been introgressed into cultivated wheat (Marais et al., 2005).
In a previous study (Sela et al., 2011), we investigated the distribution and diversity of Lr10 haplotypes in different populations and the recombination patterns of Lr10. In this article, we focus on the distribution of the diversity over the three‐dimensional structure of the LR10 protein, study the variation in alternative splicing regulation between genotypes and look for selection forces that have shaped the diversity.
RESULTS
Sequence diversity along Lr10
One hundred accessions of wild emmer wheat from 12 populations were tested for the presence of Lr10. In 95 accessions, Lr10 was present as determined by polymerase chain reaction (PCR) amplifications. The full‐length, 4‐kb Lr10 was cloned and sequenced from 58 accessions. Reads were first assembled into contigs for each accession, after which multiple alignments of the DNA sequences and the predicted proteins were generated from the contigs. Alignments of the most diverse predicted protein sequences are presented in Fig. 1. Four accessions had a 1.2‐kb deletion of the NBS domain. These sequences were not included in the diversity analysis. Three sequences had a premature stop codon in position 130. An alignment of the remaining 54 sequences revealed the presence of 33 haplotypes.
Figure 1.

Alignment of eight LR10 protein sequences from wild emmer wheat accessions representing the whole spectrum of diversity. Sequence 9 is a reference sequence from Thatcher Lr10 (GenBank AAQ01784). Similar and dissimilar substitutions relative to the consensus are shaded in light and dark grey, respectively. H1–H2 are helices in the coiled coil (CC) domain. Numbers 1–8 in the nucleotide‐binding site (NBS) domain are conserved motifs: 1, P‐loop; 2, Resistance NBS (RNBS)‐A; 3, kinase2; 4, RNBS‐B; 5, RNBS‐C; 6, GLPL; 7, RNBS‐D; 8, MHDV. R1–R16 in the leucine‐rich repeat (LRR) domain are the LxxLxL motifs. Exclamation marks denote positively selected codons. Letters above the exclamation marks are the positions of the residue in the helix (a and d, intramolecular dimerization positions; c and g, solvent‐exposed positions). X and Y are the hotspots of diversity indicated in 2, 7. Z is the hotspot of diversity indicated in 2, 6 and presented in 4, 5. CC, NBS, Linker and LRR mark the starting point from the C‐terminus to the N‐terminus of the domains. The black line above the linker marks a repeat in the sequence.
Analysis of the sequence alignments showed that the CC domain was the most diverse (π= 0.083) and that the first intron was the most conserved segment (π= 0.005; Table 1, Fig. 2); the overall π value was 0.029. A sliding window diversity analysis revealed three hotspots of diversity (1, 2; X, Y, Z). These hotspots were further analysed by three‐dimensional modelling of the protein structure. The neutrality tests of Fu and Li (1993) and Tajima (1989) using a 100‐bp sliding window revealed significant deviation from neutrality in the region between nucleotide positions 3300 and 3500 in the middle of the LRR domain, which is the Z hotspot in Fig. 2 (D= 2.98, F= 2.48, P < 0.01). Negative D and F values were observed in the first intron and in the NBS and LRR regions (Fig. 2). At the protein level, analysis using average pairwise distances, determined by the Jones–Taylor–Thornton (JTT) substitution matrix (Jones et al., 1992), showed that the CC domain was the most diverse (average pairwise distance, 0.155) and the LRR domain was the most conserved (average pairwise distance, 0.033), with an overall protein diversity of 0.063 (Table 1). The fixed‐effects likelihood (FEL) method was used to screen for codons subject to positive and negative selection by testing the significance of the dN–dS difference (nonsynonymous versus synonymous substitutions) for each codon (Pond and Frost, 2005). Nine codons in the CC domain were significantly (P < 0.1) under positive selection (in three codons, P < 0.05), and 36 codons, scattered all over the gene, were under negative selection (1, 3). Amino acid replacements in the positively selected codons had, in most cases, negative BLOSUM62 scores, meaning that these replacements are rare and that the replaced residues are likely to have different physicochemical properties (Table 2) (Henikoff and Henikoff, 1992). The partition approach for robust inference of selection (PARRIS) (Scheffler et al., 2006), testingdN/dS ratios, detected signatures of positive selection in the CC domain and in the whole gene (Table 1, P= 0.006 and P= 7.4 × 10−6, respectively).
Table 1.
Nucleotide and protein sequence diversity along LR10.
| Domain | DNA, π | Protein, overall mean JTT distance | dN/dS | Signatures of positive selection P value* |
|---|---|---|---|---|
| CC | 0.083 | 0.155 | 1.25 | 0.006 |
| Intron 1 | 0.005 | – | – | – |
| NBS | 0.019 | 0.048 | 0.73 | 1 |
| LRR | 0.020 | 0.033 | 0.41 | 0.6 |
| Intron 2 | 0.010 | – | – | – |
| Coding sequence | 0.031 | 0.063 | 0.74 | 7.4 × 10−6 |
| Overall | 0.029 | – | – | – |
CC, coiled coil; dN/dS, nonsynonymous to synonymous substitution ratio; JTT, Jones–Taylor–Thornton; LRR, leucine‐rich repeat; NBS, nucleotide‐binding site.
P value is for ‘signatures of positive selection’ calculated by the partition approach for robust inference of selection (PARRIS) (Scheffler et al., 2006).
Figure 2.
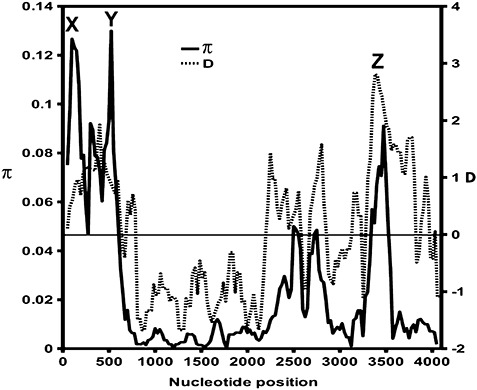
Nucleotide diversity (π) and D values of the Tajima neutrality test along Lr10. Graphs were calculated using a 100‐bp window with 25‐bp steps. X and Y are the hotspots of diversity indicated in 1, 7. Z is the hotspot of diversity indicated in 1, 6 and presented in 4, 5.
Figure 3.
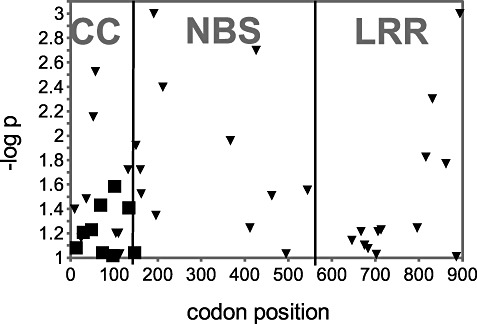
Distribution of codons under positive or negative selection along Lr10 as determined by the fixed‐effects likelihood (FEL) method. Positive (squares, dN > dS) and negative (triangles, dN < dS) selected codons in Lr10. The y axis represents –log of the one‐tailed significance level of the dN–dS difference. Significance levels of P= 0.1, 0.05, 0.01 and 0.001 equal –log P= 1, 1.3, 2 and 3, respectively. CC, coiled coil; LRR, leucine‐rich repeat; NBS, nucleotide‐binding site.
Table 2.
Codons subjected to positive selection in the coiled coil (CC) domain.
| Codon position | Amino acid substitutions* | BLOSUM62† scores range | Position in the helix‡ | FEL dN–dS§ | FEL P value¶ |
|---|---|---|---|---|---|
| 13 | M, V | 1 | a | 0.85 | 0.08 |
| 29 | I, K, R | −3 to 2 | c | 1.49 | 0.06 |
| 48 | D, M, I, A, V | −3 to 1 | g | 0.99 | 0.06 |
| 69 | C, L | −1 | d | 1.95 | 0.04 |
| 73 | V, M, A | −1 to 1 | a | 0.73 | 0.09 |
| 97 | P, S | −1 | 0.82 | 0.10 | |
| 101 | I, C, F | −2 to 0 | a | 2.74 | 0.03 |
| 134 | A, V | 0 | 1.37 | 0.04 | |
| 147 | C, H, F, E | −4 to 0 | 3.29 | 0.09 |
FEL, fixed‐effects likelihood.
Amino acid substitutions represented by the single letter code.
Range of BLOSUM62 score substitutions in the codon (Henikoff and Henikoff, 1992).
Positions of the codon in the helix. a and d, intramolecular dimerization positions; c and g, solvent‐exposed positions.
FEL dN–dS is the difference between nonsynonymous and synonymous substitutions as calculated by the DataMonkey server (Pond and Frost, 2005).
FEL P value is the probability that the codon is under positive selection.
Detection of a new alternatively spliced intron
cDNA was synthesized from eight accessions representing the whole spectrum of diversity of Lr10. In four accessions, two products were amplified for the LRR domain, one with the predicted size and the other 333 nucleotides shorter. Sequence analysis of these products revealed that the short product was the result of alternative splicing of an intron by ‘intron retention’ (Ner‐Gaon et al., 2004). The alternative splicing did not alter the reading frame, but deleted five repeats from the LRR domain. This second intron has not been detected previously in Lr10. A closer look at the 5′ splicing junction in all of the T. dicoccoides sequences revealed two haplotypes in this region: one had the GU junction required for splicing and the other lacked the junction (Fig. 4). Furthermore, a very high nucleotide and amino acid diversity was found in the adjacent region (Fig. 1, R9–R10; Fig. 2, hotspot Z), where 18 of 70 amino acids (26%) were replaced. Sequence analysis of this region in the A genome species T. urartu and T. monococcum (four accessions each) revealed that the two haplotypes were present in both species (data not shown). In order to determine whether the creation or deletion of the splicing site was more likely the outcome of a single event or of a longer process, a comparative analysis between the two haplotypes was conducted in a search for binding sites of SFs, using SFs that were known to be present in plants (Table 3) (Akerman et al., 2009). This screening was conducted on a segment of approximately 1 kb beginning 500 nucleotides upstream of the 5′ splice site of the second intron and ending at the stop codon. A summary of all binding sites, unique to each haplotype, is presented in Fig. 5. The most significant result was obtained for the CUG‐binding protein (CUG‐BP). The splicing haplotype had 19 CUG‐BP sites that were located upstream of the 5′ splice site. Five of them were unique to this haplotype. The nonsplicing haplotype had 14 binding sites, none of them unique.
Figure 4.

DNA and protein alignment of the two haplotypes in the second intron 5′ splicing site, hotspot Z in the leucine‐rich repeat (LRR) domain. Top sequence, no splicing haplotype. Bottom sequence, splicing haplotype. Mutations are in bold; ‘<‘ and underlined sequence represent a local frameshift. GU is the conserved motif for intron 5′ start. The BLOSUM62 scores are the scores of amino acid substitutions between the haplotypes.
Table 3.
Splicing factors and splicing factor binding site motifs (for reference, see Akerman et al., 2009).
| Splicing factor | Binding motifs |
|---|---|
| PTB | ucuu / cucucu |
| hnRNPA1 | uagggw / uagaca / uagagu |
| hnRNPAB | auagca |
| hnRNPH | uugggu / uguggg / ggcgg / gggug |
| SC35 | gryymcy / ugcygyy |
| SF2/ASF | crsmsgw / ugrwgvh |
| Srp40 | cuckucy / wcwwc |
| Srp55 | yywcwsg |
| 9G8 | wggacra / acgagagay |
| 9G8/SRp30c | gacgac |
| CUG‐BP | cugugb / cugcug / ugugug / yugcy / ycugy |
Nucleotide symbols used: b, c/u/g; k, g/u; r, a/g; s, c/g; w, a/u; y, c/u.
Figure 5.
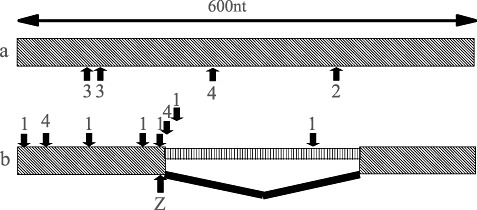
Comparison of unique splicing factor (SF) binding motifs between the two haplotypes in the second intron region, the hotspot Z in the leucine‐rich repeat (LRR) domain. Arrows mark mutations forming SF binding motifs. Numbers above/below the arrows are the splicing factors binding motifs 1 (CUG‐BP), 2 (hnRNPA1), 3 (hnRNPH/F) and 4 (PTB) (Akerman et al., 2009). Boxes with diagonal hatching are exons. Box with vertical hatching is the retained intron. Broken thick line is the spliced intron. (a) Nonsplicing haplotype. (b) Alternative splicing haplotype. nt, nucleotides.
Assessment of the LRR domain structure
In order to map the properties and variability of LR10 sequences onto the structure, a probabilistic three‐dimensional model of the LRR domain was built using the joint fragment remote homology modelling method described in Slootweg (2009). The diversity values of 31 T. dicoccoides LR10 sequences and the Thatcher LR10 (GenBank AAQ01784) sequence were mapped onto the model. The start position of the LRR domain was defined as the first LxxLxL motif after the MHD motif in the NBS domain (Fig. 1). Alternative candidates for LxxLxL motifs were present along the sequence. These were profiled for their local intrinsic disorder, accessibility and contact‐forming propensity, and then matched against the profiles of the documented LxxLxL motifs from our three‐dimensional LRR database (Slootweg, 2009). Only those LxxLxL motifs corresponding to the documented profiles were retained to define the LRR repeats. The LRR profile obtained in this manner had repeats with a constant length of about 23 amino acids, with only one exception, repeat number 9 (∼37 amino acids)—located in the centre of the structure. The reliability of the proposed LRR profile was confirmed by secondary structure prediction, and is entirely consistent in all 32 sequences, with variability restricted to the regions outside the defined LxxLxL.
Template analysis based on our LRR database (Slootweg, 2009) indicated that most of the LR10 LRR repeats were highly similar to counterpart repeats within the internalin C template from Listeria monocytogenes (PDB code: 1xeu; Ooi et al., 2006). The only exception was the LR10 ninth repeat, which was highly similar to the fifth repeat from RanGAP1 (PDB code: 1k5d; Seewald et al., 2002).
According to our method (Slootweg, 2009), the optimal global LRR frame of LR10 was built from four fragments of five, four, one and six repeats, respectively. Three of these fragments—one, two and four—were modelled starting from their best matching counterparts in 1xeu, whereas fragment three, consisting of repeat number 9 in LR10 (37 amino acids), was modelled starting from r5 from RanGAP1.
The overall integrated model is shown in Fig. 6. The hotspot of amino acid diversity is located around the ninth repeat. Amino acid substitutions in this region do not change the helix propensity and are predicted to be exposed to the surface. BLOSUM62 scores for the amino acid substitutions in this region were mainly negative, suggesting large differences in properties of the substituted residues. In the short variant of the protein, the five repeats R10–R15 are deleted, leaving only the acidic C‐terminus. The shorter LRR domain, according to this prediction, can still form a canonical horseshoe.
Figure 6.
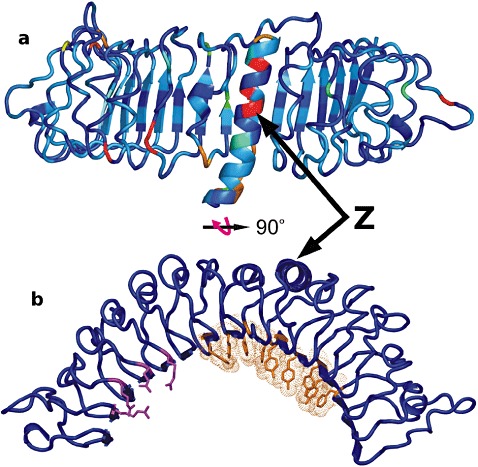
(a) Diagram of the LR10 leucine‐rich repeat (LRR) domain. Variability based on the BLOSUM62 matrix is represented using a colour scale from blue (conserved) to red (hypervariable). (b) The same object rotated by 90° on the x axis and coloured in blue. Amino acids forming the positive cluster are coloured magenta. Aromatic amino acids involved in stacking interactions on the concave part are coloured orange. Z is the hotspot of diversity, the ninth repeat, indicated in 1, 2 and presented in 4, 5.
Assessment of the CC domain structure
The boundaries of the CC domain were delineated with cdart, InterPro and predictors for secondary structure and intradomain loop sequences (see Experimental procedures). Combined, all these analyses indicated that, in LR10, the CC domain can be set in between amino acids 1 and 132 (Fig. 1). To spot structural differences between the CCs of LR10 sequences, an unrooted similarity tree was built with phylip, using BLOSUM62 (not shown). Using this structural similarity matrix, five main groups were identified with no obvious local structure differences. One gap‐free representative was retained for further analysis. In all cases, secondary structure predictors delineated three helical regions (H1, H2a and H2b) with high contact and low intrinsic disorder propensities, which suggest stable helical conformations.
Sequence analysis indicated that LR10 CC has 29.6% identity and over 60.0% similarity with the CC domain of Hordeum vulgare MLA10, which was recently crystallized in a dimeric form (Maekawa et al., 2011). In addition, independent secondary structure prediction of LR10 CC showed a perfect match with the MLA10 CC secondary structure pattern observed in the crystal structure. This very good match of secondary structure patterns and coiled coil‐specific heptads was further used to refine the alignment and place insertions within the loops that interconnect the secondary stretches (Fig. S1, see Supporting Information). To double check the alignment, threading of the LR10 CC sequence onto the template three‐dimensional structure was performed with slide (Hanganu et al., 2009), and resulted in a contiguous hydrophobic contact of H1 with the H2a–H2b strand. All of these steps indicate that homology modelling can be used to build a highly reliable model of LR10 CC starting from MLA CC. In sequence conserved regions, coordinates were transferred from the template, whereas connecting loops were generated ab initio and subjected to repeated rounds of simulated annealing and minimization.
Sequence variability mapping is shown in Fig. 7 as a colour gradient running from blue to red, corresponding to a decrease in similarity from high to low, respectively. As can be readily seen in Fig. 7, two apparently unrelated regions of the surface show significant variability: X, the exposed surface of the first strand (H1) and W, the linkers between H2a and H2b. These linkers have a significant propensity for intrinsic disorder; thus, a certain degree of variability can be expected in these locations without disturbing the structure. Two of the nine positively selected codons (Table 2) were located on the helix surface outside the hydrophobic zipper (intramolecular contact surface) and had BLOSUM62 scores of −3 (Table 2). Four positively selected codons were located in the hydrophobic zipper and had mainly positive BLOSUM62 scores. The rest of the positively selected codons were located outside the helices.
Figure 7.
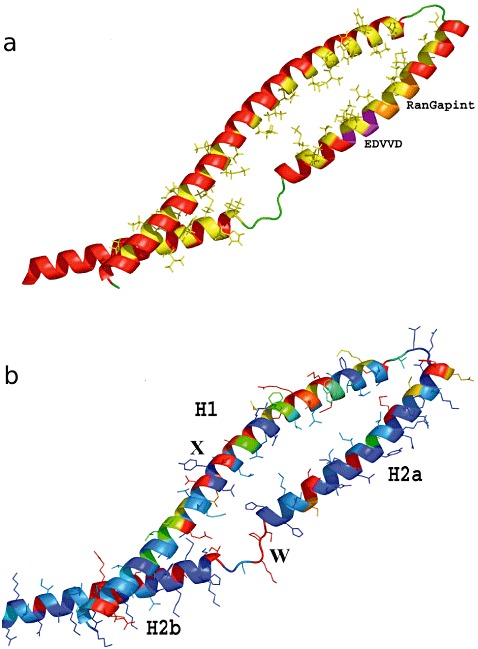
LR10 coiled coil (CC) model. (a) Diagram coloured by secondary structure; amino acids involved in hydrophobic zipping are coloured yellow to pinpoint the hydrophobic matching between helices. The RanGAP interacting motif and the EDVVD motif are coloured in orange and magenta, respectively (Rairdan et al., 2008; Slootweg et al., 2010; Tameling and Baulcombe, 2007). (b) Diagram with variability mapped on a scale from blue (conserved) to red (hypervariable); X, the exposed surface of the first strand (H1); W, the linkers between H2a and H2b.
DISCUSSION
R genes in plants are highly diverse and evolve rapidly (Dangl and Jones, 2001; Rose et al., 2004). The rapid rate of evolution in R genes makes them a good model to study the processes of co‐evolution.
In this study, we found high nucleotide diversity of the leaf rust resistance gene, Lr10, among wild emmer wheat populations originating from Israel, and differences in the level of nucleotide diversity between the CC, NBS and LRR domains of this gene. The high nucleotide diversity revealed in this study allowed us to detect the outcome of several evolutionary processes that shaped this diversity, and to determine how the diversity is distributed on the protein three‐dimensional structure.
Nucleotide diversity of Lr10 domains among natural wild emmer wheat populations
The Lr10 nucleotide diversity (π= 0.029) within a collection of wild emmer wheat accessions from a relatively small area in Israel (12 locations) was found to be 10 times higher than the average π value (0.0027) calculated for 21 gene loci (21 kb) in a collection of 28 wild emmer wheat accessions from all over the Fertile Crescent (Haudry et al., 2007). In contrast with the majority of plant R genes analysed so far, where the LRR domain is the most diverse domain, LRR in Lr10 is the most conserved domain (Jiang et al., 2007; Mauricio et al., 2003; Rose et al., 2004; Yahiaoui et al., 2009). These results suggest a different mechanism of interaction between LR10 and pathogen molecules than the mechanism in most of the other studied R genes. The hypothesis that such a mechanistic difference exists is also supported by the high diversity observed in the CC domain compared with the low or absent diversity in CC domains of other R genes, such as barley MLA, Solanum Rx and Gpa2, and wheat Pm3 (Bieri et al., 2004; Butterbach, 2007; Yahiaoui et al., 2006). Lr10 does not act alone; it requires the closely linked RGA2 (Loutre et al., 2009). There is a growing body of evidence to indicate that some R genes function in pairs and that NBS‐LRR genes vary in their mode of action (Eitas and Dangl, 2010) Thus, the mode of action of Lr10 could be different from that of other R genes, which act alone and have a conserved CC domain.
The three‐dimensional modelling of the CC domain shows two anti‐parallel coils (H1 and H2). The high variability in the CC domain is concentrated in the H1 region. The variable amino acids in this region are predicted to be solvent exposed (1, 7). However, H2a is highly conserved; this region is known to interact with NBS and with RanGap in Rx from Solanum, a CC–NBS–LRR gene (Rairdan et al., 2008; Slootweg et al., 2010; Tameling and Baulcombe, 2007). Therefore, H2a has similar properties to conserved CC domains in other CC–NBS–LRR genes, whereas H1 is different because of its high variability. In Rx, this region may be involved in cell localization (Slootweg et al., 2010). The diversity in the CC domain is linked, in some codons, to positive selection, retaining a variation of amino acids with different properties, and could point to the interaction of this domain with pathogen effectors, as also suggested by Loutre et al. (2009) and Sela et al. (2011). Several studies have reported an indirect interaction of CC domains with pathogen effectors (Ade et al., 2007; Burch‐Smith et al., 2007; Mackey et al., 2002; Rairdan et al., 2008). Others have shown direct interaction of another N‐terminus, the Toll and interleukin‐1 receptor (TIR) domain, with pathogen effectors (Ellis et al., 2007; Luck et al., 2000). To our knowledge, there are no reports of CC domains interacting directly with pathogen effectors, as suggested for the LR10 CC domain. In Sela et al. (2011), we revealed that the linker between the NBS and CC domains of Lr10 showed high indel polymorphism (1, 2, hotspot Y). We postulated that the high variability is created by recombination. This region is intriguing, because of the large differences in length between the sequences and the spread of the length variants in all of the Triticum species (Loutre et al., 2009; Sela et al., 2011). It is not clear whether or how this region may affect the conformation and function of the Lr10‐coded protein. Several studies have shown that the CC domain in plant R genes can function even when expressed separately from the rest of the protein (Moffett et al., 2002; Rairdan et al., 2008), making the effect of indel variability within the linker on gene function questionable. In this study, we found that the three‐dimensional structure predicted for this region has no intrinsic disorder. Therefore, the region can modulate the structural properties of the hinge between the CC and NBS domains.
The Lr10 NBS region has relatively high amino acid sequence diversity, but, in most cases, the diversity is found only outside of the NBS motifs (Fig. 1). A surprising observation was the high conservation of the first intron. Some studies have demonstrated that first introns may contain regulatory elements, and therefore may be subject to purifying selection (Bornstein et al., 1987; Fu et al., 2005).
Alternative splicing and ancient haplotypes in the LRR domain
Although the Lr10 LRR domain is relatively conserved, the region of the ninth LRR repeat has a very high nucleotide and protein diversity between the two main haplotypes found in the wild emmer wheat accessions. Each haplotype is highly conserved and can be traced back to T. monoccocum and T. urartu, the putative donors of the A genome to T. dicoccoides (Loutre et al., 2009). The positive values of neutrality tests (Fu and Li, 1993; Tajima, 1989) suggest that this region is under balancing selection. Negative values in nearby loci minimize the probability that demographic factors are the cause of the positive values. Signatures of balancing selection have also been observed in many NBS–LRR genes in Arabidopsis (Bakker et al., 2006). The two haplotypes found in this study are also present in durum wheat, and both confer resistance to leaf rust (Loutre et al., 2009). The three‐dimensional modelling of the LRR showed that the ninth repeat is unique because it forms an extensive α helix, whereas the other repeats form only coils (Fig. 6). The concentration of many amino acid replacements in this particularly well‐defined structure does not seem to be random. The varying residues in this region are also solvent exposed and have negative BLOSUM62 scores. Therefore, they may have significant effects on the domain's interaction with other plant proteins or pathogen effectors, and may affect specificity (Palomino et al., 2002). The testing of these haplotypes against many leaf rust races may reveal differences in their specificity.
These two haplotypes also differed in their DNA signals for the regulation of alternative splicing, with one haplotype not having the 5′ GU splice junction, and the other haplotype having the junction and five additional CUG‐BP motifs. However, these motifs can also be recognized by the polypyrimidine tract‐binding protein (PTB), which has an antagonistic regulatory effect, and so the effect of the motifs on splicing cannot be determined in silico (Gromak et al., 2003). The creation or deletion of the splicing regulation in the Lr10 LRR domain is not a single evolutionary event, but a process that includes many mutation steps. These mutations are positioned in exonic regions, and therefore they also affect the amino acid sequence. Hence, it would be interesting to find which selection force maintains the variation. Is it the selection that acts on the splicing regulation? Or the selection that acts on the amino acid sequence. Or, do they both have equal weight? Unlike other plant R genes, it is clear that alternative splicing is not essential to confer resistance (Dinesh‐Kumar and Baker, 2000; Gassmann, 2008; Jordan et al., 2002; Zhang and Gassmann, 2003), because the resistant Thatcher Lr10 does not have the splicing junction (Feuillet et al., 2003). However, these haplotypes are ancient; they evolved in the ancestor of the A genome wheat species, prior to the species’ radiation 0.5–3 million years ago (Wicker et al., 2003). The haplotypes represent a long evolutionary history of host–parasite interactions. This observation indicates that this alternative splicing has an important adaptive value that preserved their existence side‐by‐side.
To date, shorter LRR domains, or complete truncation of LRR domains in splice variants, have been observed mainly in TIR–NBS–LRR genes and not in CC–NBS–LRR genes, with the exception of JA1tr in the bean Phaseolus vulgaris (Ferrier‐Cana et al., 2005). Unlike alternative splicing reported for other NBS–LRR genes, alternative splicing in Lr10 does not result in a premature stop codon, but rather yields an in‐frame deletion of five repeats. Shorter LRRs can release the suppression of the LRR domain on NBS activity (Gassmann, 2008; Jordan et al., 2002). The short splice variant may play a role in expression of regulation via the nonsense‐mediated RNA decay pathway, but it does not have the premature stop codon normally associated with this process (Chang et al., 2007). However, the shorter alternative variant still forms a canonical horseshoe, and so it may be involved in the recognition of different races or pathogens.
The polymorphism represented by the two haplotypes in the LRR domain is evolutionarily stable; the two haplotypes co‐existed before the A genome species’ radiation 0.5–3 million years ago (Wicker et al., 2003), suggesting a balanced polymorphism. This is further supported by Tajima's test indicating balancing selection. Furthermore, in Sela et al. (2011), we observed that the two haplotypes do not recombine and form a linkage disequilibrium block, and that they are present side‐by‐side in many populations across Israel. Moreover, the two haplotypes were also persevered during the bottleneck of domestication, as they are both present in the cultivated wheat T. durum (Loutre et al., 2009). The ancient diversity observed in both studies supports a long evolutionary history of Lr10–pathogen effector interaction in the ‘trench warfare’ model. In this model, polymorphism is maintained by alleles that fluctuate between high and low frequency shifted by frequency‐dependent selection (Holub, 2001; Stahl et al., 1999). The findings of this study reject the ‘arms race’ model for Lr10, which predicts young genes with low variation (Holub, 2001). The observation that the diversity is stable suggests that it may have an impact on the gene function or specificity.
Genetic diversity has been narrowed down in wheat cultivars as a result of modern breeding practices and selection for high yield. The signatures of balancing selection that were revealed between Lr10 haplotypes suggest that, even though many of the alleles may not confer resistance to the current prevailing pathogen races, they are maintained in the population by frequency‐dependent selection and may be functional against rare pathogen races (Stahl et al., 1999). Furthermore, approximately 80% of Lr10 alleles were translated into full‐length proteins; hence, they may be functional against unknown leaf rust races. Therefore, it is important to preserve as much diversity as possible, as genotypes that are susceptible to the current prevailing pathogen races may be resistant to new emerging races. These insights have immediate relevance for wheat germplasm collection and in situ conservation that can serve for the breeding of crop plants against pathogens for the security of world food production. The hotspots of diversity that were revealed in this study in the CC domain and LRR domain may serve as starting points for in planta or in vitro structure–function studies of LR10, which seems to function somewhat differently from other CC–NBS–LRR genes studied so far.
EXPERIMENTAL PROCEDURES
Plant material
One hundred T. dicoccoides accessions, from 12 collection sites in Israel, were used in this study, as described in Sela et al. (2011). The collection sites represent diverse habitats across the distribution range of T. dicoccoides in Israel. A full description of their geographical origin and climatic conditions can be found in Nevo and Beiles (1989).
Lr10 gene isolation and sequencing
The Lr10 gene was isolated as described in Sela et al. (2011). Briefly, the full‐length, 4‐kb Lr10 gene was amplified as one PCR product using the primers ThLR10_V (CGGAACTATGGAGAGTGAAC) and ThLR10_U (GGGAAATGTAGACAGGTACAT) (Feuillet et al., 2003). The PCR products were separated on agarose gels, extracted, cloned into the pSMART vector (Lucigen, Middleton, WI, USA) and sequenced. The Lr10 DNA sequences were aligned using muscle (Edgar, 2004) and manually corrected using BioEdit (Hall, 2007).
Statistical analysis
DNA sequence alignments were analysed for nucleotide diversity (π) (Nei, 1987) using DNAsp (Rozas et al., 2003). This software was also used to conduct Tajima (Tajima, 1989) and Fu and Li (Fu and Li, 1993) neutrality tests. Protein diversity was calculated as the average distance between pairwise sequences using the JTT amino acid substitution model (Jones et al., 1992) in mega software (Kumar et al., 2004). Tests for positive selection were performed using the fel method on the DataMonkey server (Pond and Frost, 2005). The method uses likelihood‐based analysis to identify sites at which the rate of nonsynonymous substitutions is greater than the rate of synonymous substitutions. Recombination break points in the alignment were detected with a genetic algorithm for recombination (gard) tool on the same server, and were used in fel analysis in order to correct for the effect of recombination. parris (Scheffler et al., 2006) on the DataMonkey server detected overall signatures of positive selection in the different domains. Screening for protein‐binding motifs involved in splicing regulation (SFs) was conducted by the method described in Akerman et al. (2009). Briefly, the nucleotide sequence was screened with a set (Table 3) of known splicing regulatory motifs (Lorkovic and Barta, 2002) using a sliding window moved in steps of one nucleotide. At every position, a score was calculated for each motif based on the similarity between the motif at that position and a known motif (Table 3). Subsequently, each motif was given a score from zero to one, representing the reliability of the prediction. The magnitude of the score depends on the similarity of the queried sequence to a given motif and the genome content around the motif. The screen searched for new binding sites for SFs formed by mutations between two conserved haplotypes in the LRR domain.
mRNA analysis
RNA was extracted from 10‐day‐old seedlings using the Aurum RNA Extraction Kit (Bio‐Rad, Hercules, CA, USA). The cDNA first strand was synthesized using the Reverse‐iT™ First Strand Synthesis Kit (ABgene, Portsmouth, NH, USA). PCR amplification was conducted using primers LRR‐Lr10‐L (TGCTCGACGTACAAGATTGC) and LRR‐Lr10‐R (CTCCACATAGGCAGCACTGA) derived from the LRR region of the cloned Lr10 sequence (Feuillet et al., 2003). PCR products were separated on agarose gels, extracted and sequenced. The sequences were aligned and screened for splicing junctions using the bdgp splice predictor (Reese et al., 1997).
Three‐dimensional structure modelling
Sequence similarity searches were performed with Blast using BLOSUM62 (Henikoff and Henikoff, 1992). Patterns, profiles and domain recognition were scanned with InterPro (Quevillon et al., 2005) and cdart (Geer et al., 2002). Secondary structure predictions were performed with sopma (Geourjon and Deleage, 1995), gor iv (Garnier et al., 1996), PsiPred (Jones, 1999), Jpred (Cole et al., 2008), hnn (Guermeur, 1997) and Prof (Ouali and King, 2000). Linkers were predicted by dlp‐svm (Miyazaki et al., 2002).
Putative LxxLxL motifs in the LRR domain were checked for local intrinsic disorder, accessibility and contact‐forming propensity against a full LRR structure database built by us on the basis of the Protein Data Bank (http://www.wwpdb.org) (Slootweg, 2009). Homology modelling was performed with slide (Hanganu et al., 2009), a software for interactive threading, and Insight II (Accelrys, San Diego, CA, USA). Variability at a given site along the multiple alignments of LR10 protein sequences was defined as the average of the BLOSUM62 substitution matrix values between every sequence. This was mapped onto the three‐dimensional structure using a Python script written for PyMol (DeLano Scientific LLC, Palo Alto, CA, USA).
Supporting information
Fig. S1 Alignment of MLA10 coiled coil (CC) domain and Lr10 (P522) CC domain. Disorder, level of disorder, with high values indicating less order; KIH, positions in the helix structure; MLA, MLA amino acid sequence; Similar MLA, the level of similarity between MLA and Lr10; SS, secondary structure. Charge distribution on the sequence is represented on a scale from 0 (negative, red) to 9 (positive, blue), where 4 is neutral. Yellow shading represents amino acids in the hydrophobic zipper.
Supporting info item
ACKNOWLEDGEMENTS
This work was supported by grants from the European Union sixth framework programme (FP6) in the BioExploit project (No. CT‐2005–513949), The Israel Science Foundation (ISF) grant #205/08, and ISF equipment grants #1478/04, and #1719/08 HS acknowledge the binational agricaltural research and devlopment fund (BARD) grant No. US4323‐10C. LNS and A‐JP also acknowledge the support from Consiliul National al Cercetarii Stiintifice din Invatamantul Superior (CNCSIS) grant PN‐II‐ID‐PCE 249, 168/2007 and POSDRU/89/1.5/S/60746. We would like to thank Mrs Ortal Mergi and Mrs Anne‐Mari Narvanto for technical assistance.
The sequences obtained in this study were deposited in GenBank (accessions GU393247–GU393304).
REFERENCES
- Ade, J. , DeYoung, B.J. , Golstein, C. and Innes, R.W. (2007) Indirect activation of a plant nucleotide binding site–leucine‐rich repeat protein by a bacterial protease. Proc. Natl Acad Sci. U.S.A., 104, 2531–2536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akerman, M. , David‐Eden, H. , Pinter, R.Y. and Mandel‐Gutfreund, Y. (2009) A computational approach for genome‐wide mapping of splicing factors binding sites. Genome Biol. 10, R30. DOI: 10.1186/gb‐2009‐10‐3‐r30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anikster, Y. , Manisterski, J. , Long, D.L. and Leonard, K.J. (2005) Leaf rust and stem rust resistance in Triticum dicoccoides populations in Israel. Plant Dis. 89, 55–62. [DOI] [PubMed] [Google Scholar]
- Bakker, E.G. , Toomajian, C. , Kreitman, M. and Bergelson, J. (2006) A genome‐wide survey of R gene polymorphisms in Arabidopsis . Plant Cell, 18, 1803–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergelson, J. , Kreitman, M. , Stahl, E.A. and Tian, D. (2001) Evolutionary dynamics of plant R‐genes. Science, 292, 2281–2285. [DOI] [PubMed] [Google Scholar]
- Bieri, S. , Mauch, S. , Shen, Q.H. , Peart, J. , Devoto, A. , Casais, C. , Ceron, F. , Schulze, S. , Steinbiss, H.H. and Shirasu, K. (2004) RAR1 positively controls steady state levels of barley MLA resistance proteins and enables sufficient MLA6 accumulation for effective resistance. Plant Cell, 16, 3480–3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bornstein, P. , McKay, J. , Morishima, J.K. , Devarayalu, S. and Gelinas, R.E. (1987) Regulatory elements in the first intron contribute to transcriptional control of the human 1 (I) collagen gene. Proc. Natl Acad Sci. U.S.A., 84, 8869–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burch‐Smith, T.M. , Schiff, M. , Caplan, J.L. , Tsao, J. , Czymmek, K. and Dinesh‐Kumar, S.P. (2007) A novel role for the TIR domain in association with pathogen‐derived elicitors. PLoS Biol. 5, e68. DOI: 10.1371/journal.pbio.0050068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butterbach, P. (2007) Molecular evolution of the disease resistance gene Rx in Solanum. PhD Dissertation, Wageningen University, Wageningen.
- Chang, Y. , Imam, J.S. and Wilkinson, M.F. (2007) The nonsense‐mediated decay RNA surveillance pathway. Annu. Rev. Biochem. 76, 51–74. [DOI] [PubMed] [Google Scholar]
- Cloutier, S. , McCallum, B.D. , Loutrem, C. , Banksm, T.W. , Wicker, T. , Feuillet, C. , Keller, B. and Jordan, M.C. (2007) Leaf rust resistance gene Lr1 isolated from bread wheat (Triticum aestivum L.) is a member of the large psr567 gene family. Plant Mol. Biol. 65, 93–106. [DOI] [PubMed] [Google Scholar]
- Cole, C. , Barber, J.D. and Barton, G.J. (2008) The Jpred 3 secondary structure prediction server. Nucleic Acids Res. 36, W197–W201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dangl, J.L. and Jones, J.D.G. (2001) Plant pathogens and integrated defence responses to infection. Nature, 411, 826–833. [DOI] [PubMed] [Google Scholar]
- Dinesh‐Kumar, S.P. and Baker, B.J. (2000) Alternatively spliced N resistance gene transcripts: their possible role in tobacco mosaic virus resistance. Proc. Natl Acad. Sci. U.S.A., 97, 1908–1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eitas, T.K. and Dangl, J.L. (2010) NB‐LRR proteins: pairs, pieces, perception, partners, and pathways. Curr. Opin. Plant Biol. 13, 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis, J.G. , Dodds, P.N. and Lawrence, G.J. (2007) Flax rust resistance gene specificity is based on direct resistance–avirulence protein interactions. Annu. Rev. Phytopathol. 45, 289–306. [DOI] [PubMed] [Google Scholar]
- Feldman, M. , Lupton, F.G.H. , Miller, T.E. and Feldman, M. (1995) Wheats In: Evolution of Crops, 2nd edn. (Smartt J. and Simmonds N.W., eds), pp. 184–192. London: Longman Scientific. [Google Scholar]
- Ferrier‐Cana, E. , Macadré, C. , Sévignac, M. , David, P. , Langin, T. and Geffroy, V. (2005) Distinct post‐transcriptional modifications result in seven alternative transcripts of the CC–NBS–LRR gene JA1tr of Phaseolus vulgaris . Theor. Appl. Genet. 110, 895–905. [DOI] [PubMed] [Google Scholar]
- Feuillet, C. , Travella, S. , Stein, N. , Albar, L. , Nublat, A. and Keller, B. (2003) Map‐based isolation of the leaf rust disease resistance gene Lr10 from the hexaploid wheat (Triticum aestivum L.) genome. Proc. Natl Acad. Sci. U.S.A., 100, 15 253–15 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, D. , Szucs, P. , Yan, L. , Helguera, M. , Skinner, J.S. , von Zitzewitz, J. , Hayes, P.M. and Dubcovsky, J. (2005) Large deletions within the first intron in VRN‐1 are associated with spring growth habit in barley and wheat. Mol. Genet. Genomics, 273, 54–65. [DOI] [PubMed] [Google Scholar]
- Fu, D. , Uauy, C. , Distelfeld, A. , Blechl, A. , Epstein, L. , Chen, X. , Sela, H. , Fahima, T. and Dubcovsky, J. (2009) A Kinase‐START gene confers temperature‐dependent resistance to wheat stripe rust. Science, 323, 1357–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Y.X. and Li, W.H. (1993) Statistical tests of neutrality of mutations. Genetics, 133, 693–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garnier, J. , Gibrat, J.F. and Robson, B. (1996) GOR secondary structure prediction method version IV. Methods Enzymol. 266, 540–553. [DOI] [PubMed] [Google Scholar]
- Gassmann, W. (2008) Alternative splicing in plant defense In: Nuclear Pre‐mRNA Processing in Plants (Reddy A.S.N. and Golovkin M., eds), pp. 219–233. Berlin: Springer. [Google Scholar]
- Geer, L.Y. , Domrachev, M. , Lipman, D.J. and Bryant, S.H. (2002) CDART: protein homology by domain architecture. Genome Res. 12, 1619–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geourjon, C. and Deleage, G. (1995) SOPMA: significant improvements in protein secondary structure prediction by consensus prediction from multiple alignments. Bioinformatics, 11, 681–684. [DOI] [PubMed] [Google Scholar]
- Gromak, N. , Matlin, A.J. , Cooper, T.A. and Smith, C.W.J. (2003) Antagonistic regulation of α‐actinin alternative splicing by CELF proteins and polypyrimidine tract binding protein. RNA, 9, 443–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermeur, Y. (1997) Combinaison de classifieurs statistiques, application à la prédiction de la structure secondaire des protéines. PhD Dissertation, University of Paris, Paris.
- Hall, T. (2007) BioEdit, Version 7.0. 9. Computer program and documentation. Carlsbad, CA: Ibis Biosciences. [Google Scholar]
- Hanganu, A. , Micluta, M. , Popa, B. , Spiridon, L. and Tacutu, R. (2009) “Slide”: an interactive threading refinement tool for homology modeling. Romanian Journal of Biochemistry, 46, 123–127. [Google Scholar]
- Haudry, A. , Cenci, A. , Ravel, C. , Bataillon, T. , Brunel, D. , Poncet, C. , Hochu, I. , Poirier, S. , Santoni, S. and Glemin, S. (2007) Grinding up wheat: a massive loss of nucleotide diversity since domestication. Mol. Biol. Evol. 24, 1506–1517. [DOI] [PubMed] [Google Scholar]
- Henikoff, S. and Henikoff, J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl Acad Sci. U.S.A., 89, 10 915–10 919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holub, E.B. (2001) The arms race is ancient history in arabidopsis, the wildflower. Nat. Rev. Genet. 2, 516–527. [DOI] [PubMed] [Google Scholar]
- Huang, L. , Brooks, S.A. , Li, W. , Fellers, J.P. , Trick, H.N. and Gill, B.S. (2003) Map‐based cloning of leaf rust resistance gene Lr21 from the large and polyploid genome of bread wheat. Genetics, 164, 655–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isidore, E. , Scherrer, B. , Chalhoub, B. , Feuillet, C. and Keller, B. (2005) Ancient haplotypes resulting from extensive molecular rearrangements in the wheat A genome have been maintained in species of three different ploidy levels. Genome Res. 15, 526–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H. , Wang, C. , Ping, L. , Tian, D. and Yang, S. (2007) Pattern of LRR nucleotide variation in plant resistance genes. Plant Sci. 173, 253–261. [Google Scholar]
- Jones, D.T. (1999) Protein secondary structure prediction based on position‐specific scoring matrices. J. Mol. Biol. 292, 195–202. [DOI] [PubMed] [Google Scholar]
- Jones, D.T. , Taylor, W.R. and Thornton, J.M. (1992) The rapid generation of mutation data matrices from protein sequences. Bioinformatics, 8, 275–282. [DOI] [PubMed] [Google Scholar]
- Jordan, T. , Schornack, S. and Lahaye, T. (2002) Alternative splicing of transcripts encoding Toll‐like plant resistance proteins—what's the functional relevance to innate immunity? Trends Plant Sci. 7, 392–398. [DOI] [PubMed] [Google Scholar]
- Knott, D.R. , DaPeng, B. and Zale, J. (2005) The transfer of leaf and stem rust resistance from wild emmer wheats to durum and common wheat. Can. J. Plant. Sci. 85, 49–57. [Google Scholar]
- Kolmer, J.A. (2005) Tracking wheat rust on a continental scale. Curr. Opin. Plant Biol. 8, 441–449. [DOI] [PubMed] [Google Scholar]
- Krattinger, S.G. , Lagudah, E.S. , Spielmeyer, W. , Singh, R.P. , Huerta‐Espino, J. , McFadden, H. , Bossolini, E. , Selter, L.L. and Keller, B. (2009) A putative ABC transporter confers durable resistance to multiple fungal pathogens in wheat. Science, 323, 1360–1363. [DOI] [PubMed] [Google Scholar]
- Kumar, S. , Tamura, K. and Nei, M. (2004) mega3: integrated software for molecular evolutionary genetics analysis and sequence alignment. Brief. Bioinform. 5, 150–163. [DOI] [PubMed] [Google Scholar]
- Lorkovic, Z.J. and Barta, A. (2002) Genome analysis: RNA recognition motif (RRM) and K homology (KH) domain RNA‐binding proteins from the flowering plant Arabidopsis thaliana . Nucleic Acids Res. 30, 623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loutre, C. , Wicker, T. , Travella, S. , Galli, P. , Scofield, S. , Fahima, T. , Feuillet, C. and Keller, B. (2009) Two different CC‐NBS‐LRR genes are required for Lr10‐mediated leaf rust resistance in tetraploid and hexaploid wheat. Plant J. 60, 1043–1054. [DOI] [PubMed] [Google Scholar]
- Luck, J.E. , Lawrence, G.J. , Dodds, P.N. , Shepherd, K.W. and Ellis, J.G. (2000) Regions outside of the leucine‐rich repeats of flax rust resistance proteins play a role in specificity determination. Plant Cell, 12, 1367–1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, M.C. , Yang, Z.L. , You, F.M. , Kawahara, T. , Waines, J.G. and Dvorak, J. (2007) The structure of wild and domesticated emmer wheat populations, gene flow between them, and the site of emmer domestication. Theor. Appl. Genet. 114, 947–959. [DOI] [PubMed] [Google Scholar]
- Mackey, D. , Holt, B.F. , Wiig, A. and Dangl, J.L. (2002) RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1‐mediated resistance in Arabidopsis . Cell, 108, 743–754. [DOI] [PubMed] [Google Scholar]
- Maekawa, T. , Cheng, W. , Spiridon, L.N. , Töller, A. , Lukasik, E. , Saijo, Y. , Liu, P. , Shen, Q.H. , Micluta, M.A. and Somssich, I.E. (2011) Coiled‐coil domain‐dependent homodimerization of intracellular barley immune receptors defines a minimal functional module for triggering cell death. Cell Host & Microbe, 9, 187–199. [DOI] [PubMed] [Google Scholar]
- Marais, G.F. , Pretorius, Z.A. , Wellings, C.R. , McCallum, B. and Marais, A.S. (2005) Leaf rust and stripe rust resistance genes transferred to common wheat from Triticum dicoccoides . Euphytica, 143, 115–123. [Google Scholar]
- Mauricio, R. , Stahl, E.A. , Korves, T. , Tian, D. , Kreitman, M. and Bergelson, J. (2003) Natural selection for polymorphism in the disease resistance gene Rps2 of Arabidopsis thaliana . Genetics, 163, 735–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mergoum, M. , Frohberg, R.C. , Miller, J.D. and Stack, R.W. (2005) Registration of ‘Steele‐ND’ wheat registration by CSSA. Crop Sci. 45, 1163–1164. [Google Scholar]
- Meyers, B.C. , Shen, K.A. , Rohani, P. , Gaut, B.S. and Michelmore, R.W. (1998) Receptor‐like genes in the major resistance locus of lettuce are subject to divergent selection. Plant Cell, 10, 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyazaki, S. , Kuroda, Y. and Yokoyama, S. (2002) Characterization and prediction of linker sequences of multi‐domain proteins by a neural network. J. Struct. Funct. Genomics, 2, 37–51. [DOI] [PubMed] [Google Scholar]
- Moffett, P. , Farnham, G. , Peart, J. and Baulcombe, D.C. (2002) Interaction between domains of a plant NBS−LRR protein in disease resistance‐related cell death. EMBO J. 21, 4511–4519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moseman, J.G. , Nevo, E. , Gerechter‐Amitai, Z.K. , El‐Morshidy, M.A. and Zohary, D. (1985) Resistance of Triticum dicoccoides collected in Israel to infection with Puccinia recondita tritici . Crop Sci. 25, 262–265. [Google Scholar]
- Nei, M. (1987) Molecular Evolutionary Genetics. New York: Columbia University Press. [Google Scholar]
- Ner‐Gaon, H. , Halachmi, R. , Savaldi‐Goldstein, S. , Rubin, E. , Ophir, R. and Fluhr, R. (2004) Intron retention is a major phenomenon in alternative splicing in Arabidopsis. Plant J. 39, 877–885. [DOI] [PubMed] [Google Scholar]
- Nevo, E. and Beiles, A. (1989) Genetic diversity of wild emmer wheat in Israel and Turkey—structure, evolution, and application in breeding. Theor. Appl. Genet. 77, 421–455. [DOI] [PubMed] [Google Scholar]
- Ooi, A. , Hussain, S. , Seyedarabi, A. and Pickersgill, R.W. (2006) Structure of internalin C from Listeria monocytogenes . Acta Crystallogr. Sect. D, 62, 1287–1293. [DOI] [PubMed] [Google Scholar]
- van Ooijen, G. , Mayr, G. , Kasiem, M. , Albrecht, M. , Cornelissen, B.J.C. and Takken, F.L.W. (2008) Structure–function analysis of the NB‐ARC domain of plant disease resistance proteins. J. Exp. Bot. 59, 1383. [DOI] [PubMed] [Google Scholar]
- Ouali, M. and King, R.D. (2000) Cascaded multiple classifiers for secondary structure prediction. Protein Sci. 9, 1162–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomino, M.M. , Meyers, B.C. , Michelmore, R.W. and Gaut, B.S. (2002) Patterns of positive selection in the complete NBS‐LRR gene family of Arabidopsis thaliana . Genome Res. 12, 1305–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Q. , Wendel, J. and Fluhr, R. (2000) Divergent evolution of plant NBS‐LRR resistance gene homologues in dicot and cereal genomes. J. Mol. Evol. 50, 203–213. [DOI] [PubMed] [Google Scholar]
- Pond, S.L.K. and Frost, S.D.W. (2005) DataMonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics, 21, 2531–2533. [DOI] [PubMed] [Google Scholar]
- Quevillon, E. , Silventoinen, V. , Pillai, S. , Harte, N. , Mulder, N. , Apweiler, R. and Lopez, R. (2005) InterProScan: protein domains identifier. Nucleic Acids Res. 33, 116–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rairdan, G.J. , Collier, S.M. , Sacco, M.A. , Baldwin, T.T. , Boettrich, T. and Moffett, P. (2008) The coiled‐coil and nucleotide binding domains of the potato RX disease resistance protein function in pathogen recognition and signaling. Plant Cell, 20, 739–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese, M.G. , Eeckman, F.H. , Kulp, D. and Haussler, D. (1997) Improved splice site detection in Genie. J. Comput. Biol. 4, 311–323. [DOI] [PubMed] [Google Scholar]
- Rong, J. , Millet, E. , Manisterski, J. and Feldman, M. (2000) A new powdery mildew resistance gene: introgression from wild emmer into common wheat and RFLP‐based mapping. Euphytica, 115, 121–126. [Google Scholar]
- Rose, L.E. , Bittner‐Eddy, P.D. , Langley, C.H. , Holub, E.B. , Michelmore, R.W. and Beynon, J.L. (2004) The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana . Genetics, 166, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J. , Sanchez‐DelBarrio, J.C. , Messegue, R.X. and Rozas, R. (2003) DnaSP. DNA polymorphism analyses by the coalescent and other methods. Bioinformatics, 19, 2496–2497. [DOI] [PubMed] [Google Scholar]
- Scheffler, K. , Martin, D.P. and Seoighe, C. (2006) Robust inference of positive selection from recombining coding sequences. Bioinformatics, 22, 2493–2499. [DOI] [PubMed] [Google Scholar]
- Seewald, M.J. , Korner, C. , Wittinghofer, A. and Vetter, I.R. (2002) RanGAP mediates GTP hydrolysis without an arginine finger. Nature, 415, 662–666. [DOI] [PubMed] [Google Scholar]
- Sela, H. , Loutre, C. , Keller, B. , Schulman, A. , Nevo, E. , Korol, A. and Fahima, T. (2011) Rapid linkage disequilibrium decay in the Lr10 gene in wild emmer wheat (Triticum dicoccoides) populations. Theor. Appl. Genet. 122, 157–187. [DOI] [PubMed] [Google Scholar]
- Slootweg, E.J. (2009) Structure, function and subcellular localization of the potato resistance protein Rx1. PhD Dissertation, Wageningen University, Wageningen.
- Slootweg, E. , Roosien, J. , Spiridon, L.N. , Petrescu, A.J. , Tameling, W. , Joosten, M. , Pomp, R. , van Schaik, C. , Dees, R. , Borst, J.W. , Smant, G. , Schots, A. , Bakker, J. and Goverse, A. (2010) Nucleocytoplasmic distribution is required for activation of resistance by the potato NB‐LRR receptor Rx1 and is balanced by its functional domains. Plant Cell, 22, 4195–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl, E.A. , Dwyer, G. , Mauricio, R. , Kreitman, M. and Bergelson, J. (1999) Dynamics of disease resistance polymorphism at the Rpm1 locus of arabidopsis. Nature, 400, 667–671. [DOI] [PubMed] [Google Scholar]
- Tajima, F. (1989) Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tameling, W.I.L. and Baulcombe, D.C. (2007) Physical association of the NB‐LRR resistance protein rx with a ran GTPase‐activating protein is required for extreme resistance to potato virus X. Plant Cell, 19, 1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier, A. and Brown, J.K.M. (2007) Stability of genetic polymorphism in host–parasite interactions. Proc. R. Soc. Lond. B, Biol. Sci. 274, 809–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uauy, C. , Brevis, J.C. , Chen, X. , Khan, I. , Jackson, L. , Chicaiza, O. , Distelfeld, A. , Fahima, T. and Dubcovsky, J. (2005) High‐temperature adult‐plant (HTAP) stripe rust resistance gene Yr36 from Triticum turgidum ssp. dicoccoides is closely linked to the grain protein content locus Gpc‐B1 . Theor. Appl. Genet. 112, 97–105. [DOI] [PubMed] [Google Scholar]
- Wicker, T. , Yahiaoui, N. , Guyot, R. , Schlagenhauf, E. , Liu, Z. , Dubcovsky, J. and Keller, B. (2003) Rapid genome divergence at orthologous low molecular weight glutenin loci of the A and am genomes of wheat. Plant Cell, 15, 1186–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yahiaoui, N. , Brunner, S. and Keller, B. (2006) Rapid generation of new powdery mildew resistance genes after wheat domestication. Plant J. 47, 85–98. [DOI] [PubMed] [Google Scholar]
- Yahiaoui, N. , Kaur, N. and Keller, B. (2009) Independent evolution of functional Pm3 resistance genes in wild tetraploid wheat and domesticated bread wheat. Plant J. 57, 846–856. [DOI] [PubMed] [Google Scholar]
- Zhang, X.C. and Gassmann, W. (2003) RPS4‐mediated disease resistance requires the combined presence of RPS4 transcripts with full‐length and truncated open reading frames. Plant Cell, 15, 2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 Alignment of MLA10 coiled coil (CC) domain and Lr10 (P522) CC domain. Disorder, level of disorder, with high values indicating less order; KIH, positions in the helix structure; MLA, MLA amino acid sequence; Similar MLA, the level of similarity between MLA and Lr10; SS, secondary structure. Charge distribution on the sequence is represented on a scale from 0 (negative, red) to 9 (positive, blue), where 4 is neutral. Yellow shading represents amino acids in the hydrophobic zipper.
Supporting info item