

Summary
Viroids are small single‐stranded RNA pathogens which cause significant damage to plants. As their nucleic acids do not encode for any proteins, they are dependant solely on their structure for their propagation. The elucidation of the secondary structures of viroids has been limited because of the exhaustive and time‐consuming nature of classic approaches. Here, the method of high‐throughput selective 2′‐hydroxyl acylation analysed by primer extension (hSHAPE) has been adapted to probe the viroid structure. The data obtained using this method were then used as input for computer‐assisted structure prediction using RNAstructure software in order to determine the secondary structures of the RNA strands of both (+) and (–) polarities of all Avsunviroidae members, one of the two families of viroids. The resolution of the structures of all of the members of the family provides a global view of the complexity of these RNAs. The structural differences between the two polarities, and any plausible tertiary interactions, were also analysed. Interestingly, the structures of the (+) and (–) strands were found to be different for each viroid species. The structures of the recently isolated grapevine hammerhead viroid‐like RNA strands were also solved. This species shares several structural features with the Avsunviroidae family, although its infectious potential remains to be determined. To our knowledge, this article represents the first report of the structural elucidation of a complete family of viroids.
Keywords: Avsunviroidae, RNA structure, SHAPE, viroid
Introduction
Viroids are single‐stranded, noncoding RNAs that infect plants. Although they are small (245–401 nucleotides), surprisingly, they may cause significant damage affecting crop quality and causing important economic loses (Flores et al., 2005). To date, more than 30 species of viroid have been discovered. Viroids are classified into two families: the Pospiviroidae and the Avsunviroidae. The Pospiviroidae family is composed of viroids that replicate in the nucleus through an asymmetric rolling‐circle mechanism and fold into a rod‐like structure. The Potato spindle tuber viroid (PSTVd) is the type species of the Pospiviroidae. The Avsunviroidae family is composed of viroids that replicate in chloroplasts via a symmetric rolling‐circle mechanism that involves a self‐cleaving hammerhead motif. This family includes four members, and the type species is the Avocado sunblotch viroid (ASBVd). That said, recently, the Peach latent mosaic viroid (PLMVd) has received more attention (Herranz et al., 2013; Navarro et al., 2012; Wang et al., 2013).
As viroids do not code for any protein, their structures are of the utmost importance for their survival on infection. More precisely, their structure must be the cornerstone of their life cycle, and must dictate all interactions with the host in order to ensure the transportation, replication, accumulation and pathogenesis of the viroid. Unfortunately, the structures of most viroid species remain unknown. The only structural information available includes proposed secondary structures obtained by computer‐assisted predictions (Bussiere et al., 1996; Fadda et al., 2003; Navarro and Flores, 1997; Symons, 1981).
Most of the software used for the prediction of the secondary structures of viroids is based on thermodynamic approaches that employ dynamic programming algorithms, examples being RNAstructure (Reuter and Mathews, 2010) and mfold (Zuker, 2003). Such programs have been highly used in recent years, and have had interesting success for RNAs shorter than 200 nucleotides (Masquida et al., 2010). Unfortunately, the prediction of the folding of longer RNA species remains restricted, as more information is required to produce an accurate structural model. Comparative approaches exploiting both sequence and structure conservation information provide more reliable results, as does the integration of structural probing information into the folding of the RNA. Regardless, the structural models of viroids would benefit significantly from receiving physical support before the initiation of any biological study.
Prior to 2010, the secondary structures of only three viroid species had been solved in solution: PSTVd (Domdey et al., 1978; Gast et al., 1996; Gross et al., 1978) and the Citrus exocortis viroid (CEVd) (Gross et al., 1982) of the Pospiviroidae, and PLMVd (Bussiere et al., 2000; Dube et al., 2010) of the Avsunviroidae. Classic RNA probing approaches, such as RNAse treatment, are simply too laborious, and have not been adapted to probe RNA species the size of viroids. However, the recently developed technique of selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) (Merino et al., 2005) has been applied to probe the structures of viroids. More specifically, the PLMVd secondary structures of two variants have been determined using this method (Dube et al., 2011). These two structures were in relatively good agreement with those reported previously (Bussiere et al., 2000; Dube et al., 2011). Subsequently, the secondary structures of five distinct viroid species from the Pospiviroidae were determined using the SHAPE technique combined with computer‐assisted RNA structure prediction with RNAstructure software (Xu et al., 2012). In brief, the data from the probing of viroids in solution were used as input for the computer‐based structure prediction. This process improves significantly the reliability of the structure prediction, and it has been proposed that the coupling of these techniques represents the gold standard for the determination of the structure of viroids (Xu et al., 2012).
The approach described above involves revealing the results of the probing of the RNA strand by performing primer extension using radiolabelled oligonucleotides, a process which requires authorizations and specific precautions. Consequently, it is of limited help for many laboratories. Moreover, the sequencing‐gel electrophoresis resolution of radiolabelled cDNA molecules is limited to ∼100 nucleotides, which therefore requires the probing of several overlapping strands derived from a given viroid to obtain a complete structure. The final step involves the normalization of the data, a process which is also challenging. In order to overcome all of these hurdles, a high‐throughput SHAPE (hSHAPE) technique involving the use of fluorescent oligonucleotides and analysis by capillary electrophoresis was adopted (Vasa et al., 2008). This method is significantly faster, and can resolve fragments of up to 500 nucleotides in a single experiment. In this study, the probing by hSHAPE, coupled with secondary structure prediction, was used for the elucidation of the structures of the RNA strands of both the (+) and (–) polarities of the four Avsunviroidae species, as well as for the grapevine hammerhead viroid‐like RNAs. To our knowledge, this represents the only report of the structures of all of the known members of the Avsunviroidae family.
Results and Discussion
Reprobing PLMVd strands
To illustrate the accuracy of the hSHAPE technique, the RNA strand of (+) polarity of a PLMVd variant [sequence variant PLMVd.282 (+) according to the subviral database classification (Rocheleau and Pelchat, 2006) and accession number DQ680690 according to the National Center for Biotechnology Information (NCBI)] was probed by SHAPE. More specifically, the monomeric linear conformer, the most abundant species found in chloroplasts (Bussiere et al., 1999), was probed. This RNA strand had been probed previously by both conventional SHAPE, using a radiolabelled primer, and by a classic enzymatic approach (Bussiere et al., 2000; Dube et al., 2011). Full‐length PLMVd transcripts (338 nucleotides) were produced by in vitro transcription from a plasmid containing a dimeric, head‐to‐tail copy of PLMVd.282. The particular clone used possessed two mutations, U9A and A329U, which prevented hammerhead self‐cleavage. Using three different primer sets, it was possible, by polymerase chain reaction (PCR), to produce DNA templates which could then be subjected to T7 RNA polymerase run‐off transcription. This resulted in the generation of RNA strands starting at positions 90, 156 or 255 (Fig. 1A), and permitted multiple probing data per nucleotide as well as ensuring the generation of data for all positions. Following transcription, the RNA samples were fractionated by denaturing polyacrylamide gel electrophoresis (PAGE), and the one‐unit‐length RNA strands were recovered. The RNA strands were then heat denatured at 95 °C for 2 min, snap‐cooled on ice for 5 min, slowly warmed to 37 °C (in order to permit renaturation) and incubated for an additional 30 min at 37 °C with or without MgCl2. The presence of MgCl2 may stabilize some tertiary interactions, such as pseudoknots. Subsequently, SHAPE probing was performed using benzoyl cyanide (BzCN), a procedure which offers the advantage of a rapid reaction ensuring that any excess reagent is hydrolysed within a few seconds (Mortimer and Weeks, 2008). The resulting RNA samples were employed as templates in primer extension reactions using 5′ fluorescent‐labelled oligonucleotides complementary to the 3′ ends of the viroid strands. The reverse transcriptase stops one nucleotide prior to the modification formed by the SHAPE reagent at the 2′‐hydroxyl of the ribose moiety of the flexible nucleotides. Folded, but not SHAPE‐treated, RNA strands were also reverse transcribed as negative controls to establish the background produced by both the imperfect processing of the polymerase and the intrinsic probability that the RNA was spontaneously hydrolysed. In addition, a sequencing reaction using dideoxynucleotides (ddNTP) and the same primers labelled with a different fluorophore was performed to assign a reactivity to each nucleotide of the RNA sequence. The results of the probing data were revealed by capillary electrophoresis. The cDNAs from both the SHAPE and the sequencing reactions were eluted in the same capillary from a capillary electrophoresis instrument. Afterwards, both the negative control and the sequencing reactions were eluted into a second capillary. Using this approach, two electropherograms containing either a SHAPE (+) or SHAPE (–) reaction, as well as a sequencing reaction, were obtained. The use of this ‘two‐capillaries’ method ensured that the subsequent analysis was easier than that required with other hSHAPE techniques. All reactions were performed at least in duplicate, starting from the production of the DNA templates.
Figure 1.
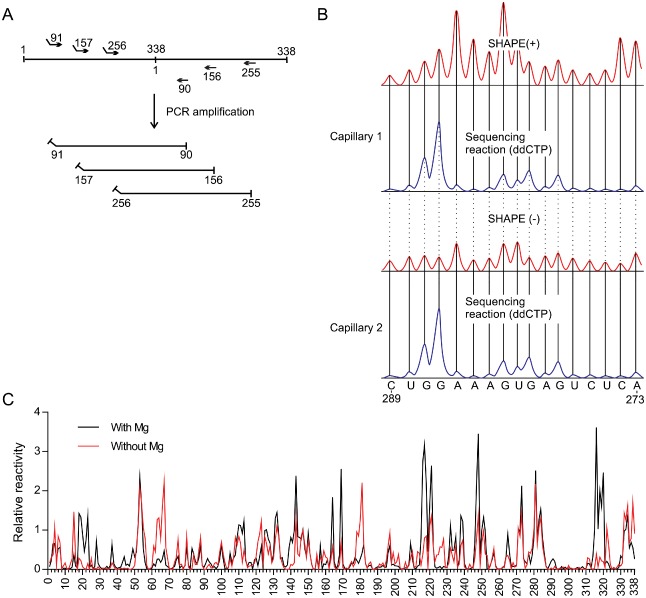
Example of a selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) experiment adapted to probe the structure of Peach latent mosaic viroid (PLMVd) (+). (A) Representation of the head‐to‐tail dimer of PLMVd and of the primers used to produce the monomeric RNA transcripts of PLMVd starting from different sites. Noncomplementary regions represent the T7 promoter used for the run‐off transcription. (B) The cDNAs obtained from the primer extensions of the SHAPE (+) and SHAPE (–) reactions were electrophoresed on two different capillaries, both of which were accompanied by a sequencing reaction. Both the SHAPE and sequencing reactions were performed with the same primer, except that the fluorophore bound in 5′ was changed (this is represented by the blue and red traces). ddCTP, dideoxycytidine 5′‐triphosphate. (C) The normalized SHAPE reactivities for the reactions performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Figure 1B illustrates both a typical SHAPE trace and the dideoxycytidine 5′‐triphosphate (ddCTP) sequencing reaction obtained for the nucleotides located between positions 273 and 289. The traces were then analysed using the default parameters of QuSHAPE software (Karabiber et al., 2013). The software permitted the integration of the peaks, subtraction of the background and normalization of the data so that almost all of the values fall between ‘0’ and ‘3’ (Fig. 1C). A normalized SHAPE value lower than 0.40 indicates that the nucleotide is most probably constrained, whereas a value higher than 0.85 denotes a flexible nucleotide (Karabiber et al., 2013). The averaged normalized SHAPE data were then finally added to the RNAstructure 5.5 nearest‐neighbour parameters of the Fold tool, as a pseudo‐free energy change, in order to predict the secondary structure of a given viroid (Bellaousov et al., 2013). Only the most stable structures were retained for further analysis.
The strategy of using viroid RNA with various 5′ extremities, or starting sites, permitted the generation of information for all positions. Previously, this was not possible because no information from the sequence complementary to the primer involved in the reverse transcription reaction could be obtained. That said, it is important to verify whether or not the location of the 5′ extremity in the probed RNA may alter its structure. To address this question, the proportions of both ambiguous and unambiguous positions were determined. If the reactivity of a nucleotide transits from a highly reactive to a low reactive state depending on the 5′ extremity present, it is deemed to be ambiguous. In this case, as well as for all subsequent structures, the SHAPE probing was performed at least twice for each starting site, and the data for each of the starting sites were averaged before determining the ambiguity of a given nucleotide. The percentage of the number of unambiguous nucleotides was calculated in order to evaluate the final probing data. Overall, 92.9% of all the nucleotides were considered to be unambiguous. In other words, 314 of 338 nucleotides were identical, regardless of the RNA strand probed.
Although the natural linear viroid starts at the cleavage/ligation site following the hammerhead self‐cleavage, the probing could not be performed with an RNA starting at this site because the RNA polymerase requires the presence of at least one guanine in order to initiate the in vitro run‐off transcription, and it was desired to avoid the introduction of any new mutations. To evaluate whether or not the location of the 5′ extremity had an important influence on the structure predicted by RNAstructure software, the averaged SHAPE data obtained for the probing of the three RNAs possessing different 5′ extremities were loaded into the Fold tool of the software package. A percentage change relative to the RNA strand's length was then calculated by comparing the structure of PLMVd in which the 5′ extremity is located at the cleavage/ligation site with PLMVd RNA strands with the different 5′ extremities. In order to do this, all of the nucleotides, varying from a paired to an unpaired situation and vice versa, and those in changing pairs, were added. The strand starting at position 91 showed no change, that starting at position 255 showed only a 0.01% change and that starting at position 156 showed the greatest percentage of changed positions (10%). However, most of these changes were located near the 5′ and 3′ ends of the RNA strand. This corresponds to a region that shows limited stability (Dube et al., 2011), and clearly demonstrates the importance of selecting a 5′ extremity that is located in an organized and stable region, so as to limit the possibility of variation in the secondary structure. Together, the results obtained from the three RNA strands provided physical data for all positions, something which had not been achieved previously (Dube et al., 2011).
The resulting structure for the PLMVd strand of (+) polarity is composed of 14 stems, including two pseudoknots (i.e. P8 and P14), as well as a cruciform formed with stems P12 and P13 (Fig. 2A). This proposed structure is in good agreement with that reported previously for the PLMVd.282 variant (Dube et al., 2011). The only differences between the two structures are located between the P4 and P9 stems, which are slightly reorganized (compare Fig. 2A and its inset). The P4 stem is somewhat longer and the L4 loop is larger in the revised structure. The consequence of this is that the nucleotide pairing is modified in the P5 to P9 stems. Even so, the P6 and P7 stems remain unchanged, as does the pseudoknot P8. In order to evaluate the consequences of this new folding for the region located between positions 131 and 261, the Gibbs free energy (ΔG) was estimated using the RNAstructure program's Fold tool. The novel structure was more thermodynamically stable, possessing a ΔG value of −51.4 kcal/mol relative to −36.7 kcal/mol for the earlier one (depicted in the inset). Both of the pseudoknots (P8 and P14), and the cruciform structure involving the P12 and P13 stems, were detected only in the presence of MgCl2 (Fig. 1C). The presence of magnesium did not permit the detection of any other significant differences. In the absence of magnesium, the P11 stem was longer, as the nucleotides located in the loops of the cruciform (18–23 and 315–320) became unreactive to the BzCN reagent.
Figure 2.
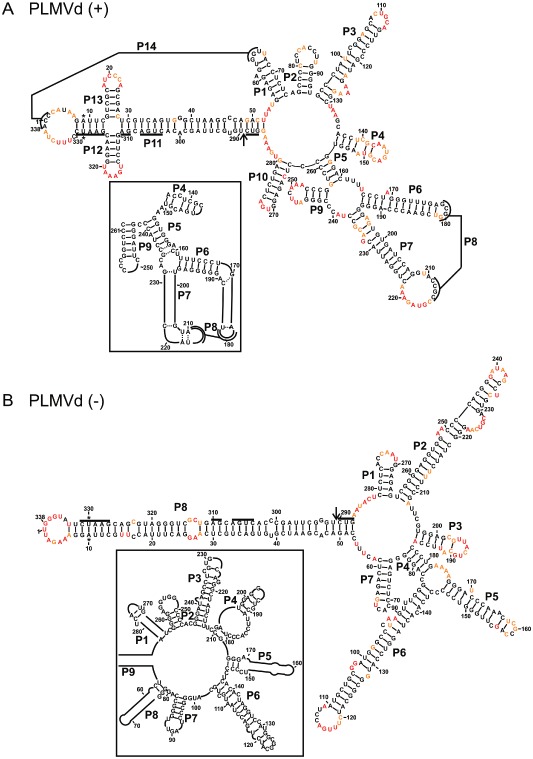
The most stable structures for both polarities of Peach latent mosaic viroid (PLMVd). The final structural models of both PLMVd (+) (A) and PLMVd (–) (B) obtained by selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) and folded by RNAstructure. The nucleotides in black denote low SHAPE reactivities (0–0.40), those in orange intermediate reactivities (0.40–0.85) and those in red highly reactive nucleotides (>0.85). The nucleotides forming the hammerhead are underlined, the cleavage site is indicated by an arrow and the mutated nucleotides are identified by stars. The insets show the differences observed when structures are compared with previously accepted models. Only regions with variations are shown in the insets. The numbering of the stems is in accordance with the first reported PLMVd structure (Bussiere et al., 2000). The numbering of the stems of the (–) polarity structure is in accordance with that used for the (+) polarity.
Next, the PLMVd strand of (–) polarity was probed. The SHAPE‐based method was applied using two distinct RNA strands starting at positions 96 and 268. In this case, 97.9% of the positions were found to be unambiguous (i.e. 331 of 338 positions), suggesting that the results from both probing experiments were in excellent agreement. The resulting structure of (–) polarity is clearly more linear (rod‐like) than that of (+) polarity (Fig. 2B). It is composed of only eight stems, and several of these are relatively long (i.e. P2, P6 and P8). The experiment was also performed both with and without MgCl2 and no pseudoknot was found in this structure (Fig. S1, see Supporting Information). This is in good agreement with previous experiments showing that the (–) strand is soluble in the presence of LiCl (Dube et al., 2010), a property associated with rod‐like structures (Diener, 2003). The structure of the (–) strand variant of PLMVd.282 (NCBI accession number DQ680690) presented here corresponds relatively well with those reported previously for two variants (Dube et al., 2011). For example, the P1, P5, P7 and P8 (P8 and P9 in the inset, respectively) stems, and their corresponding stem‐loops, are very much alike. However, the most important differences reside in the merging of the stems corresponding to the previous P2 and P3 stems, which are now shown to form a long stem. Although the nucleotides of the L3 loop are single stranded, they are located in an internal loop. Nucleotides 236–241, which were part of the P3 stem, are very reactive and are now shown to be in the end loop of this long stem. In addition, the former stems P7 and P9 that contained nucleotides 84–144 now form a single long stem. Furthermore, this long stem is part of a multi‐branched structure that spans the nucleotides 78–184. Overall, the hSHAPE technique combined with the bioinformatic prediction provides a very sound structural model for the PLMVd strands of both the (+) and (–) polarities.
Structure of the Chrysanthemum chlorotic mottle viroid (CChMVd)
CChMVd is the longest known member (398–401 nucleotides) of the Avsunviroidae family. Some of its molecular features have been studied in detail (Gago et al., 2005). For example, a UUUC tetra‐loop has been shown to be important for the appearance of symptoms in infected plants (de la Pena et al., 1999). However, to date, only a computer‐assisted prediction of the secondary structure of the (+) polarity strand, which appears to be a branched structure like that of PLMVd, has been reported. Consequently, it was decided to probe the strands of both polarities of the CChMVd.022 variant (NCBI accession number AJ878085).
For the probing of the (+) strand, RNA templates starting at positions 71 and 205 were synthesized. The resulting RNA structure, which is branched, included 99.5% unambiguous positions, indicating a very high level of definition. The CChMVd (+) strand structure obtained is composed of 11 stems, including one potential pseudoknot (P10; see below) (Fig. 3A). It is characterized by the presence of three four‐way junctions that are located in the left, centre and right domains of the molecule. Moreover, between stems P6 and P7 an internal loop that is composed of nucleotides highly reactive in SHAPE was found (i.e. positions 169–174 and 353–359). A pseudoknot, called P10, was found in the four‐way junction of the right domain. This pseudoknot is most probably formed in only a fraction of the RNA strands. Specifically, the nucleotides located in positions 255–259 were reactive to the BzCN reagent in the absence of MgCl2, but not in its presence, suggesting that these five nucleotides form a pseudoknot. Conversely, the nucleotides located in positions 219–223 appeared to be reactive towards BzCN, suggesting that, if the formation of the pseudoknot is promoted by the presence of Mg2+, only a fraction of the RNA strands will adopt this structural rearrangement. This hypothesis also received support from the analysis of the transcribed CChMVd (+) strand following separation on a native gel, as more than one band was detected. This is reminiscent of that observed with PLMVd (+), suggesting the presence of more than one structural conformation (data not shown). This was not the case for any of the other RNA strands probed below. Moreover, for the CChMVd strand of (+) polarity, the most stable predicted structure did not include the formation of the pseudoknot, but the second most stable structure did (Fig. S2, see Supporting Information). There was no significant difference in terms of Gibbs free energy between these two predicted structures (ΔG = −342.6 and −340.9 kcal/mol for the first and second most stable structures, respectively). Finally, the structure described here is less branched than that predicted previously, a structure which was not supported by any physical data. The left domains are identical in both structures, including the tetra‐loop found at the end of the P3 stem (Gago et al., 2005; de la Pena et al., 1999). The most important differences occurred in the right domain, and primarily involved the replacement of smaller stems by longer ones. The internal loops located between the P6 and P7 stems, and those located between the P8 and P9 stems, are the main causes of the rearrangement of CChMVd, because most of these nucleotides were previously found to be located in stems and could not be predicted to be single stranded without SHAPE probing.
Figure 3.
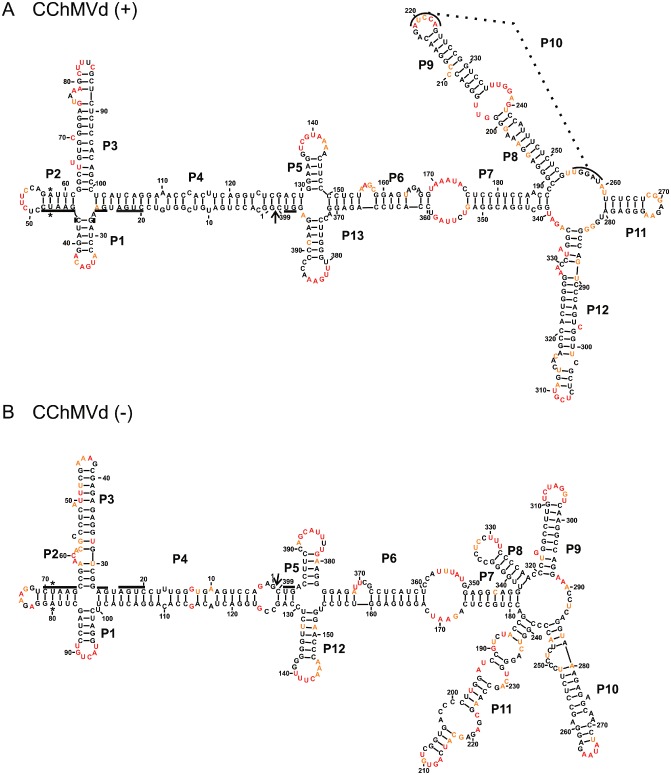
The most stable structures for both polarities of Chrysanthemum chlorotic mottle viroid (CChMVd). The final structural models of both CChMVd (+) (A) and CChMVd (–) (B) obtained by selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) and folded by RNAstructure. The nucleotides in black denote low SHAPE reactivities (0–0.40), those in orange intermediate reactivities (0.40–0.85) and those in red highly reactive nucleotides (>0.85). The nucleotides forming the hammerhead are underlined, the cleavage site is indicated by an arrow and the mutated nucleotides are identified by stars. The numbering of the stems is in accordance with the first full stem encountered clockwise of the first nucleotide of the (+) polarity strand. The numbering of the stems of the (–) polarity structure is in accordance with that used for the (+) polarity structure.
The CChMVd strand of (–) polarity was probed using RNA species starting at either position 78 or 347. Overall, this structure, which is supported by 97.2% of the positions being unambiguous, is relatively similar to that of its counterpart of (+) polarity (Fig. 3). The two four‐way junctions of the left domain, as well as the larger internal loop located between the P6 and P7 stems, are conserved, The right domain is also a multi‐branched region; however, in the case of the (–) polarity molecule, this involves five stems forming the branches, whereas only four are involved in the case of the (+) polarity molecule. In brief, the CChMVd (–) strand appears to be slightly less structured than its (+) counterpart, mainly because of the absence of the pseudoknot (Fig. S3, see Supporting Information). Importantly, to our knowledge, this is the first time that probing in solution has been performed in order to support the secondary structure adopted by strands of both polarities of CChMVd.
ASBVd folds into rod‐like stems
Both strands of ASBVd were probed. This species is the smallest known viroid (247 nucleotides) and is characterized by its relatively high content of both adenosine and uridine residues (Hutchins et al., 1986). The variant ASBVd.084 (NCBI accession number X52041) was probed using RNA strands starting at positions 26 and 114. The resulting structures of the (+) polarity strands included 97.6% unambiguous nucleotides. This RNA strand folded into a rod‐like structure that included a large loop located in the left domain, a central loop that included the conserved hammerhead nucleotides and a relatively large right terminal loop (Fig. 4A). These loops were not present in the previously proposed bioinformatic predicted structure, and the analysis of the SHAPE traces strongly supported their existence (Fig. S4, see Supporting Information). To obtain additional support, the SHAPE experiments were also performed at 25 °C and virtually the same structure was obtained (data not shown). The structure of the ASBVd strand of (–) polarity was also probed on RNAs starting at positions 2 and 158. The final structure included 98.4% unambiguous positions and was relatively similar to that of (+) polarity, with the exception that the central domain appeared to be more like a double‐stranded domain. The presence of MgCl2 did not permit the detection of any distinct structures for the strands of either (+) or (–) polarity (Fig. S4).
Figure 4.
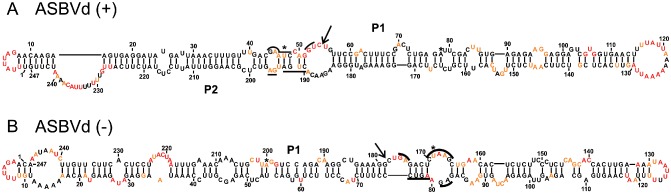
The most stable structures for both polarities of Avocado sunblotch viroid (ASBVd). The final structural models of ASBVd (+) (A) and ASBVd (–) (B) obtained by selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) and folded by RNAstructure. The nucleotides in black denote low SHAPE reactivities (0–0.40), those in orange intermediate reactivities (0.40–0.85) and those in red are highly reactive nucleotides (>0.85). The nucleotides forming the hammerhead are underlined, the cleavage site is indicated by an arrow and the mutated nucleotides are identified by stars.
Structures of the Eggplant latent viroid (ELVd)
ELVd was the last member of the Avsunviroidae to be formally characterized (ELV.001 and NCBI accession number NC_004728). The SHAPE probing of both the (+) (starting sites 140 and 268) and (–) (starting sites 84 and 229) strands revealed secondary structures that are relatively similar and defined by 98.5% and 99.4% unambiguous positions, respectively (Fig. 5). Both are composed of a relatively long rod‐like centre domain that ends with a three‐way junction in the left domain. The RNA strand of (+) polarity also possesses a three‐way junction that forms its right domain, whereas the right domain of the (–) strand ends with five stems. Two of these, P5 and P8, are reminiscent of the P4 and P5 stems found in the structure of the (+) polarity strand. In both cases, the presence of MgCl2 did not lead to the detection of any significant differences (Fig. S5, see Supporting Information), suggesting that these structures, as for ASBVd, lack a pseudoknot. Moreover, the presence of a larger region rich in adenosine and uridine residues located near the left terminal loop [i.e. in the regions of positions 70 and 130 for the (+) and (–) polarities, respectively] is also reminiscent of the ASBVd structure, and different from the observations for both PLMVd and CChMVd.
Figure 5.
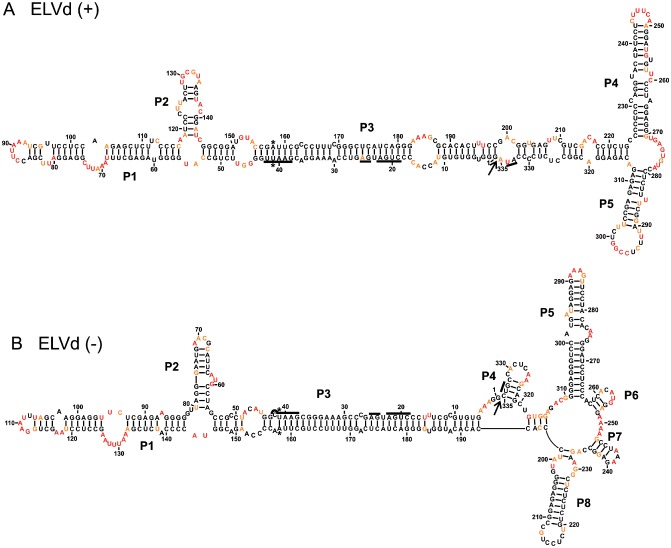
The most stable structures for both polarities of Eggplant latent viroid (ELVd). The final structural models of ELVd (+) (A) and ELVd (–) (B) obtained by selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) and folded by RNAstructure. The nucleotides in black denote low SHAPE reactivities (0–0.40), those in orange intermediate reactivities (0.40–0.85) and those in red are highly reactive nucleotides (>0.85). The nucleotides forming the hammerhead are underlined, the cleavage site is indicated by an arrow and the mutated nucleotides are identified by stars. The numbering of the stems is according to the first full stem encountered clockwise of the first nucleotide of the (+) polarity strand. The numbering of the stems of the (–) polarity structure was performed in accordance with that used for the (+) polarity structure.
Structure of the grapevine hammerhead viroid‐like RNAs
Recently, a novel viroid‐like sequence was identified by the deep sequencing of a library of total small RNAs isolated from a grapevine (Wu et al., 2012). The circular sequence of 375 nucleotides possesses an active hammerhead ribozyme motif in both (+) and (–) polarities. However, to our knowledge, there are no reported experiments demonstrating that it is infectious. In other words, the Koch's postulates have yet to be satisfied. Consequently, it is not possible to formally conclude that it is a viroid species. For now, this RNA molecule is tentatively named ‘grapevine hammerhead viroid‐like RNA’ (GHVd) (Wu et al., 2012). Nevertheless, species of both polarities were probed because they are reminiscent of the species that belong to the Avsunviroidae.
Figure 6 illustrates the structures of both the (+) and (–) polarity strands. That of (+) polarity includes 97.9% unambiguous positions, whereas that of (–) polarity includes 98.4% unambiguous positions. Both structures include virtually identical rod‐like domains in the left region. The right domains also show some similarities, although that of (–) polarity appears to be more complex. No significant differences were observed in the probing step in either the absence or presence of magnesium, with one exception (Fig. S6, see Supporting Information). More specifically, a five‐nucleotide (184–188) region of the (+) polarity strand appeared to be unreactive in the presence of magnesium (i.e. these might be double‐stranded residues) and more reactive in its absence (Fig. 6A). However, it was not possible to detect five complementary nucleotides that also demonstrated an identical probing difference. These differences may be the result of an increase in the reactivity of the nucleotides composing the core of the four‐way junction (P4, P5, P6 and P7), which, in turn, causes a rearrangement of the residues of the whole right domain. The predicted structures of the RNA strands of both polarities appear to be similar to that of PLMVd: a long stem in the left domain and a branched right domain.
Figure 6.
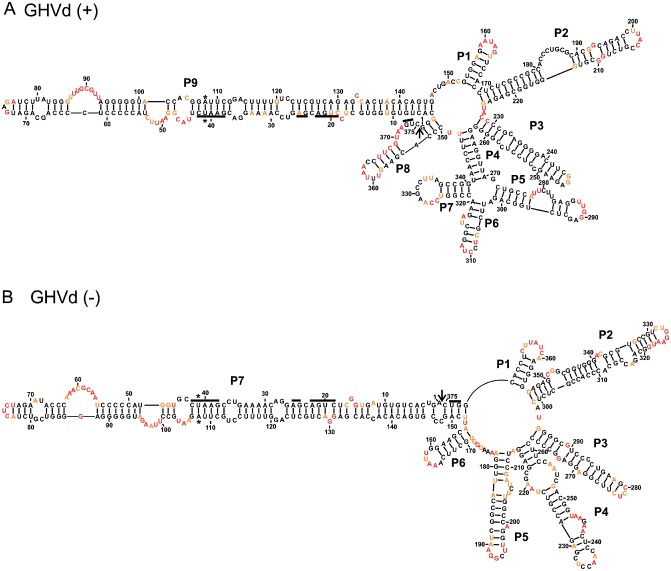
The most stable structures for both polarities of grapevine hammerhead viroid‐like RNA (GHVd). The final structural models of the strands of both (+) (A) and (–) (B) polarities obtained by selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) and folded by RNAstructure. The nucleotides in black denote low SHAPE reactivities (0–0.40), those in orange intermediate reactivities (0.40–0.85) and those in red are highly reactive nucleotides (>0.85). The nucleotides forming the hammerhead are underlined, the cleavage site is indicated by an arrow and the mutated nucleotides are identified by stars. The numbering of the stems is in accordance with the first full stem encountered clockwise of the first nucleotide of the (+) polarity strand. The numbering of the stems of the (–) polarity structure is in accordance with that used for the (+) polarity structure.
The complexity of the A vsunviroidae viroids
The different RNA species can be ordered into a schematic representation of their complexities (Fig. 7). ASBVd appears to possess the least complex structure of the family (i.e. it contains only rod‐like structures). ELVd and GHVd exhibit a greater degree of complexity as their structures include more stem‐loop domains. Finally, PLMVd and CChMVd possess the most complex structures because they include both multi‐branched loops and pseudoknots. Of the two, CChMVd is the more complex because of the presence of three multi‐branched domains located at the right, centre and left of the molecule. The analysis of the different structures did not permit the development of a method for distinguishing the structures of both polarities of a given strand. For example, the percentage of base pairs and the complexity of the structure of one strand are not indications of its polarity. More importantly, it seems possible to follow the evolution of the Avsunviroidae members by looking at their probed secondary structures.
Figure 7.
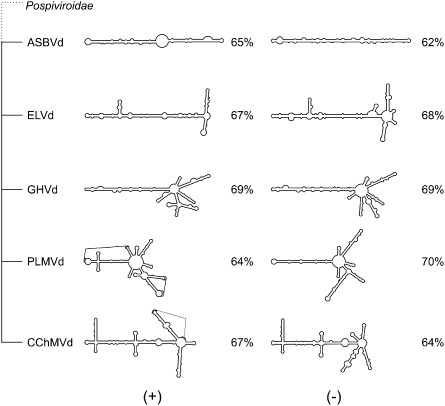
Structural outline of the Avsunviroidae family. A schematic representation of the most stable structures for both polarities of each species of the Avsunviroidae family. The structures are classified in order to illustrate the progression in complexity from the most rod‐like to the most complex structure. The numbers shown represent the percentage of base pairs. ASBVd, Avocado sunblotch viroid; CChMVd, Chrysanthemum chlorotic mottle viroid; ELVd, Eggplant latent viroid; GHVd, grapevine hammerhead viroid‐like RNA; PLMVd, Peach latent mosaic viroid.
Concluding Remarks
This study has elucidated the largest number of viroid secondary structures of any study to date. Specifically, the structures of both the (+) and (–) polarity strands of all viroids belonging to the Avsunviroidae family were determined. In addition, the secondary structures of GHVd were also determined. In this report, a ‘representative’ sequence variant previously studied and used by most investigators for each viroid species was probed. Because, many variants of viroids exist, a structural study of a population of PLMVd variants, which should permit us to learn from the notion of consensus structures, is already underway. Importantly, a knowledge of the secondary structures in solution should now be the starting point of the design of experiments to provide a better understanding of the life cycles of these viroid species.
This study also describes an efficient procedure for the determination of the secondary structures of viroids in particular, and of long RNA molecules in general. Importantly, this procedure does not use radioactive primer extension and, consequently, can be performed in almost any laboratory. This procedure should be considered as the new gold standard for the determination of a viroid's structure. That said, it is important to remember that this method includes, at the final stage, a prediction of the most stable secondary structure. This process includes the notion of RNA structural homogeneity in solution. For example, the structural homogeneity can be supported by experiments such as the analysis of RNA species by native PAGE, which should reveal the presence of a single band. However, when this experiment was performed, this was not always found to be the case. For example, it has been reported that the PLMVd and CChMVd strands of (+) polarity yield more than one band (Dube et al., 2010; Gago et al., 2005). Most probably, these RNA species adopt more than one conformation. PLMVd possesses several suboptimal structures that were folded by the Fold tool of RNAstructure software. These structures differed from one another in several small ways, although they shared energy values that were similar. This group of structures can be represented by a principal component analysis, a mathematical procedure that can provide a general notion of the differences between two structures (Halvorsen et al., 2010). It is also useful to examine the regions that seem to be the most stable or that adopt different structures (Fig. S7, see Supporting Information). Importantly, this reminds us that, even if the structures proposed following such a probing are unique, it remains possible that structural heterogeneity may occur. Indeed, in most, if not all, cases one is dealing with a population of structures for a unique RNA species. An additional limitation of this study is the fact that the secondary structure of a given viroid might also be influenced by interactions with the host proteins in cellulo. Nevertheless, this study is significant in that it provides secondary structures for each viroid of the Avsunviroidae family that are not based solely on computer‐based predictions. Indeed, all of the secondary structures proposed in this report exhibited differences from those obtained by computer prediction, supporting the notion that the use of probing leads to more accurate structures.
Experimental Procedures
Viroid sources
PLMVd.282a (NCBI accession number DQ680690), CChMVd.022 (NCBI accession number AJ878085), ASBVd.084 (NCBI accession number X52041), ELVd.001 (NCBI accession number NC_004728) and GHVd (Wu et al., 2012) head‐to‐tail dimers were commercially synthesized and inserted into plasmids by Life Technologies (GENEART Inc. gene synthesis platform, Burlington, ON, Canada). All possessed two mutations preventing the hammerhead ribozyme cleavage. All plasmids were transformed in Escherichia coli DH5α and subsequently sequenced in order to identify which clones contained the reported sequences.
Viroid preparation
The amplification of each DNA clone was performed with purified Pfu DNA polymerase using a pair of oligonucleotides (see Fig. S8, Supporting Information, for the complete list of primers used). The forward primer contained a T7 promoter for the production of monomeric (+) polarity strands and a T3 promoter for the production of (–) polarity strands. PCR amplification of monomeric viroids involved an initial 1 min of denaturation at 94 °C, followed by 35 cycles of 1 min at 94 °C, 1 min at 60 °C and 1 min at 72 °C in buffer containing 20 mm tris(hydroxymethyl)aminomethane (Tris)‐HCl, pH 8.8, 10 mm (NH4)2SO4, 10 mm KCl, 0.1% Triton X‐100, 20 mm dNTPs, 200 mm MgSO4, 200 μm of each primer and 2 μL of purified Pfu DNA polymerase. After the final cycle, the samples were heated at 72 °C for 5 min. The amplification products were precipitated with ethanol and verified by electrophoresis on a 1% agarose gel.
The transcription reactions were performed as described previously using the DNA produced in the previous step and either T7 or T3 polymerase depending on the desired polarity of the viroid (Dube et al., 2011). Briefly, the transcription reactions were performed for 90 min at 37 °C in a final volume of 100 μL containing 80 mm N‐2‐hydroxyethylpiperazine‐N′‐2‐ethanesulphonic acid (HEPES)–KOH (pH 7.5), 24 mm MgCl2, 2 mm spermidine, 40 mm dithiothreitol (DTT), 5 mm of each NTP, 0.004 units/mL pyrophosphatase (Roche Diagnostics, Laval, QC, Canada), 40 units of RNAseOUT (Life Technologies) and 2 μL of purified T7 or T3 RNA polymerase. The transcription reactions were stopped by the addition of 2 volumes of gel loading buffer [0.03% w/v each of bromophenol blue and xylene cyanol, 10 mm ethylenediaminetetraacetic acid (EDTA), pH 7.5, and 97.5% v/v deionized formamide]. Transcription reactions were purified on 5% PAGE (8 m urea). The RNA was visualized by UV shadowing, the bands corresponding to each viroid were excised and the RNA was eluted overnight in elution buffer [500 mm NH4OAc, 10 mm EDTA and 0.1% sodium dodecylsulphate (SDS)]. The eluted RNA was then precipitated with ethanol, the pellet was washed with 70% ethanol and the RNA was dissolved in 0.5 × TE buffer (1 × stock: 10 mm Tris‐HCl (pH 7.5), 1 mm EDTA) prior to quantification by spectrophotometry at 260 nm.
hSHAPE
For each SHAPE reaction, 5 pmol of RNA was dissolved in 8 μL of 0.5 × TE buffer and then heated at 95 °C for 3 min prior to being snap‐cooled for 5 min on ice. To each reaction, 1 μL of folding mix (500 mm Tris‐HCl, pH 7.5, 500 mm NaCl) was added, followed by 5 min of incubation at 37 °C. Subsequently, 1 μL of either 100 mm MgCl2 or water was added to the mixtures, which were then incubated at 37 °C for 30 min prior to the addition of 1 μL of either BzCN [600 mm dissolved in dimethylsulphoxide (DMSO)] or DMSO alone. The RNA molecules were then precipitated with ethanol in the presence of 1 μL of glycogen. The resulting RNA pellets were washed with 70% ethanol, dried and dissolved in 10 μL of 0.5 × TE buffer. The dissolved RNA was denatured by heating at 95 °C for 2 min, followed by placing on ice for 5 min. Fluorescently 5′‐labelled primers (6‐FAM, 1 pmol) were annealed (65 °C for 5 min, 37 °C for 5 min and 1 min on ice), 8 μL of reverse transcriptase mix [4 μL of 5 × first‐strand buffer (Invitrogen), 1 μL of DTT (100 mm), 1 μL of dNTPs (10 mm) and 2 μL of DMSO] were added and the mixtures were heated at 52 °C for 1 min. SuperScript III (0.7 μL, Life Technologies) was added and the mixtures were incubated at 37 °C for 30 min, prior to the reactions being stopped by the addition of 1 μL of 2 M NaOH and heating at 95 °C for 5 min. The cDNAs were then precipitated with ethanol in the presence of 1 μL of glycogen, washed with 70% ethanol and finally dried (all of these steps in the dark). In parallel, 5 pmol of RNA standard was prepared for a sequencing reaction. The only differences between the sequencing and SHAPE reactions were that a fluorescent primer of identical sequence, but possessing a different fluorophore (NED) at the 5′ end, was annealed and 0.75 mm of ddCTP was added to the reverse transcriptase mix. The pellets were sent to a sequencing and genotyping facility (Plateforme de séquençage et de génotypage des genomes, Centre de Recherche du CHUL (CHUQ), QC, Canada), where they were dissolved in 10 μL of H2O. Then, 10 μL of formamide containing a Lyz‐labelled control DNA ladder (Invitrogen) was added. To this mix, a diluted sample of the sequencing reaction was added to both the SHAPE and negative control reactions. The volumes of the samples used were dependent on the strength of the fluorescence signal read on an ABI 3100 Genetic Analyzer (Life Technologies).
The traces were then analysed with QuSHAPE software using the default parameters described previously (Karabiber et al., 2013). Briefly, (+) SHAPE and (–) SHAPE traces obtained from the ABI 3100 Genetic Analyzer were input into the program. The peaks from the traces, which were determined with the help of the sequencing ladders, were aligned with the sequence of the viroid. At this point, the software integrates each peak and then subtracts the value of the (–) SHAPE trace to provide a final reactivity value (between ‘0’ and almost ‘2’) for each nucleotide. All of the SHAPE experiments were repeated at least twice from the beginning of the protocol. The normalized results were analysed using Microsoft Office Excel. For each primer, the results were averaged separately, and the reactivity of each nucleotide was compared. Nucleotides that changed from a highly reactive to low reactive state, and vice versa, were noted. The reactivities of each nucleotide with the different primers were averaged and used as the final SHAPE reactivity values for the secondary structure predictions.
RNAstructure software
The Fold tool of the RNAstructure 5.5 program was used to fold the RNA as described previously (Dube et al., 2011). Briefly, the SHAPE data were used as a pseudo‐energy constraint to fold the RNA. A dot‐braket (.dbn) file was prepared with the resulting structures, and the figures presented in this article were made using the Varna program, followed by adjustment with the CorelDraw suite (Darty et al., 2009).
Supporting information
Fig. S1 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Peach latent mosaic viroid (PLMVd) (–). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S2 Comparison of the two most stable structures of (+) polarity Chrysanthemum chlorotic mottle viroid (CChMVd). Plausible structural models of the most stable structure (A) and the second most stable structure (B) of CChMVd (+) obtained by selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) and folded by RNAstructure. The nucleotides in black possess low SHAPE reactivities (0–0.40), those in orange intermediate reactivities (0.40–0.85) and those in red are highly reactive (>0.85). The nucleotides forming the hammerhead are underlined, the cleavage site is indicated by an arrow and the mutated nucleotides are identified by stars.
Fig. S3 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Chrysanthemum chlorotic mottle viroid (CChMVd) (+) (A) and CChMVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S4 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Avocado sunblotch viroid (ASBVd) (+) (A) and ASBVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S5 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Eggplant latent viroid (ELVd) (+) (A) and ELVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S6 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of grapevine hammerhead viroid‐like RNA (GHVd) (+) (A) and GHVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S7 Principal component analysis (PCA) of the suboptimal structures proposed for Peach latent mosaic viroid (PLMVd) (+) by RNAstructure. The graph shows the differences between the structures obtained after folding of the PLMVd (+) structure. Each point represents a distinct structure, and they are numbered in decreasing order of stability. The distance between any two points correlates with the differences between the two structures in question. The outline for each structure is shown, with the region that is different from the most stable structure being depicted in red.
Fig. S8 Primers used for the amplification of the monomeric DNA and subsequent transcription. The underlined sequences are either the T7 or T3 promoter. Unless specified, oligonucleotides of the same sequences as used in the amplification, and that do not contain the T7 or T3 promoter, were used for the primer extension reactions performed during the selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) experiments.
ACKNOWLEDGEMENTS
This work was supported by a grant from the Natural Sciences and Engineering Research Council of Canada (NSERC, grant number 155219‐07) to J‐PP. The RNA group is supported by grants from the Université de Sherbrooke. JPP holds the Research Chair of Université de Sherbrooke in RNA Structure and Genomics and is a member of the Centre de Recherche Clinique Étienne‐Lebel.
References
- Bellaousov, S. , Reuter, J.S. , Seetin, M.G. and Mathews, D.H. (2013) RNAstructure: web servers for RNA secondary structure prediction and analysis. Nucleic Acids Res. 41, W471–W474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere, F. , Lafontaine, D. and Perreault, J.P. (1996) Compilation and analysis of viroid and viroid‐like RNA sequences. Nucleic Acids Res. 24, 1793–1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere, F. , Lehoux, J. , Thompson, D.A. , Skrzeczkowski, L.J. and Perreault, J. (1999) Subcellular localization and rolling circle replication of peach latent mosaic viroid: hallmarks of group A viroids. J. Virol. 73, 6353–6360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussiere, F. , Ouellet, J. , Cote, F. , Levesque, D. and Perreault, J.P. (2000) Mapping in solution shows the peach latent mosaic viroid to possess a new pseudoknot in a complex, branched secondary structure. J. Virol. 74, 2647–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darty, K. , Denise, A. and Ponty, Y. (2009) VARNA: interactive drawing and editing of the RNA secondary structure. Bioinformatics, 25, 1974–1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diener, T.O. (2003) Discovering viroids—a personal perspective. Nat. Rev. Microbiol. 1, 75–80. [DOI] [PubMed] [Google Scholar]
- Domdey, H. , Jank, P. , Sanger, L. and Gross, H.J. (1978) Studies on the primary and secondary structure of potato spindle tuber viroid: products of digestion with ribonuclease A and ribonuclease T1, and modification with bisulfite. Nucleic Acids Res. 5, 1221–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube, A. , Baumstark, T. , Bisaillon, M. and Perreault, J.P. (2010) The RNA strands of the plus and minus polarities of peach latent mosaic viroid fold into different structures. RNA, 16, 463–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube, A. , Bolduc, F. , Bisaillon, M. and Perreault, J.P. (2011) Mapping studies of the peach latent mosaic viroid reveal novel structural features. Mol. Plant Pathol. 12, 688–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadda, Z. , Daros, J.A. , Fagoaga, C. , Flores, R. and Duran‐Vila, N. (2003) Eggplant latent viroid, the candidate type species for a new genus within the family avsunviroidae (hammerhead viroids). J. Virol. 77, 6528–6532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores, R. , Hernandez, C. , Martinez de Alba, A.E. , Daros, J.A. and Di Serio, F. (2005) Viroids and viroid–host interactions. Annu. Rev. Phytopathol. 43, 117–139. [DOI] [PubMed] [Google Scholar]
- Gago, S. , De la Pena, M. and Flores, R. (2005) A kissing‐loop interaction in a hammerhead viroid RNA critical for its in vitro folding and in vivo viability. RNA, 11, 1073–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gast, F.U. , Kempe, D. , Spieker, R.L. and Sanger, H.L. (1996) Secondary structure probing of potato spindle tuber viroid (PSTVd) and sequence comparison with other small pathogenic RNA replicons provides evidence for central non‐canonical base‐pairs, large A‐rich loops, and a terminal branch. J. Mol. Biol. 262, 652–670. [DOI] [PubMed] [Google Scholar]
- Gross, H.J. , Domdey, H. , Lossow, C. , Jank, P. , Raba, M. , Alberty, H. and Sanger, H.L. (1978) Nucleotide sequence and secondary structure of potato spindle tuber viroid. Nature, 273, 203–208. [DOI] [PubMed] [Google Scholar]
- Gross, H.J. , Krupp, G. , Domdey, H. , Raba, M. , Jank, P. , Lossow, C. , Alberty, H. , Ramm, K. and Sanger, H.L. (1982) Nucleotide sequence and secondary structure of citrus exocortis and chrysanthemum stunt viroid. Eur. J. Biochem. 121, 249–257. [DOI] [PubMed] [Google Scholar]
- Halvorsen, M. , Martin, J.S. , Broadaway, S. and Laederach, A. (2010) Disease‐associated mutations that alter the RNA structural ensemble. PLoS Genet. 6, e1001074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herranz, M.C. , Niehl, A. , Rosales, M. , Fiore, N. , Zamorano, A. , Granell, A. and Pallas, V. (2013) A remarkable synergistic effect at the transcriptomic level in peach fruits doubly infected by prunus necrotic ringspot virus and peach latent mosaic viroid. Virol. J. 10, 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchins, C.J. , Rathjen, P.D. , Forster, A.C. and Symons, R.H. (1986) Self‐cleavage of plus and minus RNA transcripts of avocado sunblotch viroid. Nucleic Acids Res. 14, 3627–3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karabiber, F. , McGinnis, J.L. , Favorov, O.V. and Weeks, K.M. (2013) QuShape: rapid, accurate, and best‐practices quantification of nucleic acid probing information, resolved by capillary electrophoresis. RNA, 19, 63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masquida, B. , Beckert, B. and Jossinet, F. (2010) Exploring RNA structure by integrative molecular modelling. Nat. Biotechnol. 27, 170–183. [DOI] [PubMed] [Google Scholar]
- Merino, E.J. , Wilkinson, K.A. , Coughlan, J.L. and Weeks, K.M. (2005) RNA structure analysis at single nucleotide resolution by selective 2′‐hydroxyl acylation and primer extension (SHAPE). J. Am. Chem. Soc. 127, 4223–4231. [DOI] [PubMed] [Google Scholar]
- Mortimer, S.A. and Weeks, K.M. (2008) Time‐resolved RNA SHAPE chemistry. J. Am. Chem. Soc. 130, 16 178–16 180. [DOI] [PubMed] [Google Scholar]
- Navarro, B. and Flores, R. (1997) Chrysanthemum chlorotic mottle viroid: unusual structural properties of a subgroup of self‐cleaving viroids with hammerhead ribozymes. Proc. Natl. Acad. Sci. USA, 94, 11 262–11 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarro, B. , Gisel, A. , Rodio, M.E. , Delgado, S. , Flores, R. and Di Serio, F. (2012) Small RNAs containing the pathogenic determinant of a chloroplast‐replicating viroid guide the degradation of a host mRNA as predicted by RNA silencing. Plant J. 70, 991–1003. [DOI] [PubMed] [Google Scholar]
- de la Pena, M. , Navarro, B. and Flores, R. (1999) Mapping the molecular determinant of pathogenicity in a hammerhead viroid: a tetraloop within the in vivo branched RNA conformation. Proc. Natl. Acad. Sci. USA, 96, 9960–9965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter, J.S. and Mathews, D.H. (2010) RNAstructure: software for RNA secondary structure prediction and analysis. BMC Bioinformatics 11, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocheleau, L. and Pelchat, M. (2006) The subviral RNA database: a toolbox for viroids, the hepatitis delta virus and satellite RNAs research. BMC Microbiol. 6, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Symons, R.H. (1981) Avocado sunblotch viroid: primary sequence and proposed secondary structure. Nucleic Acids Res. 9, 6527–6537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasa, S.M. , Guex, N. , Wilkinson, K.A. , Weeks, K.M. and Giddings, M.C. (2008) ShapeFinder: a software system for high‐throughput quantitative analysis of nucleic acid reactivity information resolved by capillary electrophoresis. RNA, 14, 1979–1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, L. , He, Y. , Kang, Y. , Hong, N. , Farooq, A.B. , Wang, G. and Xu, W. (2013) Virulence determination and molecular features of peach latent mosaic viroid isolates derived from phenotypically different peach leaves: a nucleotide polymorphism in L11 contributes to symptom alteration. Virus Res. 177, 171–178. [DOI] [PubMed] [Google Scholar]
- Wu, Q. , Wang, Y. , Cao, M. , Pantaleo, V. , Burgyan, J. , Li, W.X. and Ding, S.W. (2012) Homology‐independent discovery of replicating pathogenic circular RNAs by deep sequencing and a new computational algorithm. Proc. Natl. Acad. Sci. USA, 109, 3938–3943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, W. , Bolduc, F. , Hong, N. and Perreault, J.P. (2012) The use of a combination of computer‐assisted structure prediction and SHAPE probing to elucidate the secondary structures of five viroids. Mol. Plant Pathol. 13, 666–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuker, M. (2003) Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Peach latent mosaic viroid (PLMVd) (–). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S2 Comparison of the two most stable structures of (+) polarity Chrysanthemum chlorotic mottle viroid (CChMVd). Plausible structural models of the most stable structure (A) and the second most stable structure (B) of CChMVd (+) obtained by selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) and folded by RNAstructure. The nucleotides in black possess low SHAPE reactivities (0–0.40), those in orange intermediate reactivities (0.40–0.85) and those in red are highly reactive (>0.85). The nucleotides forming the hammerhead are underlined, the cleavage site is indicated by an arrow and the mutated nucleotides are identified by stars.
Fig. S3 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Chrysanthemum chlorotic mottle viroid (CChMVd) (+) (A) and CChMVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S4 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Avocado sunblotch viroid (ASBVd) (+) (A) and ASBVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S5 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of Eggplant latent viroid (ELVd) (+) (A) and ELVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S6 The normalized selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) reactivities for the nucleotides of grapevine hammerhead viroid‐like RNA (GHVd) (+) (A) and GHVd (–) (B). The reactions were performed in either the presence (black) or absence (red) of MgCl2. The results are presented as a function of nucleotide position.
Fig. S7 Principal component analysis (PCA) of the suboptimal structures proposed for Peach latent mosaic viroid (PLMVd) (+) by RNAstructure. The graph shows the differences between the structures obtained after folding of the PLMVd (+) structure. Each point represents a distinct structure, and they are numbered in decreasing order of stability. The distance between any two points correlates with the differences between the two structures in question. The outline for each structure is shown, with the region that is different from the most stable structure being depicted in red.
Fig. S8 Primers used for the amplification of the monomeric DNA and subsequent transcription. The underlined sequences are either the T7 or T3 promoter. Unless specified, oligonucleotides of the same sequences as used in the amplification, and that do not contain the T7 or T3 promoter, were used for the primer extension reactions performed during the selective 2′‐hydroxyl acylation analysed by primer extension (SHAPE) experiments.