

Abstract
Background
Angiotensin‐converting enzyme 2 (ACE2) is a homologue of angiotensin‐converting enzyme (ACE) and produces angiotensin peptides (APs), such as angiotensin 1‐9 and 1‐7 that are vasodilatory and natriuretic, and act to counterbalance angiotensin II.
Hypothesis
Evidence of ACE2 can be found in tissues and plasma of dogs. Equilibrium concentrations of renin angiotensin aldosterone system (RAAS) APs differ in dogs with heart disease compared to healthy dogs and recombinant human ACE2 (rhACE2) alters relative concentrations of APs.
Animals
Forty‐nine dogs with and 34 dogs without heart disease.
Methods
Immunohistochemistry and assays for tissue and plasma ACE2 activity and equilibrium concentrations of plasma RAAS APs were performed.
Results
Immunolabeling for ACE2 was present in kidney and myocardial tissue. Median plasma ACE2 activity was significantly increased in dogs with congestive heart failure (CHF; 6.9 mU/mg; interquartile range [IQR], 5.1‐12.1) as compared to control (2.2 mU/mg; IQR, 1.8‐3.0; P = .0003). Plasma equilibrium analysis of RAAS APs identified significant increases in the median concentrations of beneficial APs, such as angiotensin 1‐7, in dogs with CHF (486.7 pg/mL; IQR, 214.2‐1168) as compared to those with preclinical disease (41.0 pg/mL; IQR, 27.4‐45.1; P < .0001) or control (11.4 pg/mL; IQR, 7.1‐25.3; P = .01). Incubation of plasma samples from dogs with CHF with rhACE2 increased beneficial APs, such as angiotensin 1‐9 (preincubation, 10.3 pg/mL; IQR, 4.4‐37.2; postincubation, 2431 pg/mL; IQR, 1355‐3037; P = .02), while simultaneously decreasing maladaptive APs, such as angiotensin II (preincubation, 53.4 pg/mL; IQR, 28.6‐226.4; postincubation, 2.4 pg/mL; IQR, 0.50‐5.8; P = .02).
Conclusions and Clinical Importance
Recognition of the ACE2 system expands the conventional view of the RAAS in the dog and represents an important potential therapeutic target.
Keywords: angiotensin 1‐7, angiotensin II, degenerative mitral valve disease, renin‐angiotensin‐aldosterone system
Abbreviations
- 2D
2‐dimensional
- ACE
angiotensin‐converting enzyme
- ACE‐2
angiotensin‐converting enzyme 2
- ACEI
angiotensin‐converting enzyme inhibitor
- ACEsurr
surrogate measure of ACE activity
- AmP
aminopeptidase
- Ang1‐5
angiotensin 1‐5
- Ang1‐7
angiotensin 1‐7
- Ang1‐9
angiotensin 1‐9
- Ang2‐7
angiotensin 2‐7
- Ang2‐10
angiotensin 2‐10
- Ang3‐7
angiotensin 3‐7
- APs
angiotensin peptides
- AT1(1‐10)
angiotensin I
- AT2(1‐8)
angiotensin II
- AT3(2‐8)
angiotensin III
- AT4(3‐8)
angiotensin IV
- CHF
congestive heart failure
- DCM
dilated cardiomyopathy
- DMVD
degenerative mitral valve disease
- IHC
immunohistochemistry
- LA:Ao
left atrium to aortic root diameter ratio
- LVIDdN
normalized left ventricular internal diameter at end‐diastole
- LVIDsN
normalized left ventricular internal diameter at end‐systole
- MCA
4‐methylcoumaryl‐7‐amide
- PRAsurr
surrogate measure of plasma renin activity
- RAAS
renin‐angiotensin‐aldosterone system
- rhACE2
recombinant human angiotensin‐converting enzyme 2
- RT
room temperature
1. INTRODUCTION
The renin‐angiotensin‐aldosterone system (RAAS) plays an important role in the pathophysiology of heart disease and heart failure in dogs.1, 2, 3, 4 The traditional concept of the RAAS is as follows: cardiac disease, as well as treatments for cardiac disease, such as diuretics, decrease perfusion of the kidney, activating release of renin, which converts angiotensinogen to the 10‐amino acid peptide, angiotensin I (AT1[1‐10]). Subsequently, the dipeptidyl‐carboxypeptidase, angiotensin‐converting enzyme (ACE), cleaves 2 amino acids from the C‐terminus of AT1(1‐10) to form the octapeptide, angiotensin II (AT2[1‐8]). Angiotensin II acts primarily through angiotensin receptor type 1 and causes potent vasoconstriction, sodium and water conservation, aldosterone release, and myocardial remodeling.5 Thus, the traditional view of the RAAS is that of a linear and maladaptive pathological system that is overactive during heart failure and often suppressed using ACE inhibitors (ACEIs) and aldosterone antagonists. This understanding changed with the discovery of a homologue of ACE, called angiotensin‐converting enzyme 2 (ACE2).6, 7 Like ACE, ACE2 is a membrane‐bound zinc metalloproteinase; the catalytic site of which is capable of binding various angiotensin peptides (APs), such as AT2(1‐8). However, ACE2 is a mono‐carboxypeptidase and it removes only a single amino acid from the C‐terminus of its catalytic substrate.8, 9 For example, ACE2 acts on AT1(1‐10) and AT2(1‐8) to produce a 9‐amino acid peptide called angiotensin 1‐9 (Ang1‐9) and a 7‐amino acid peptide called angiotensin 1‐7 (Ang1‐7), respectively.10 Despite being different by only 1 amino acid, the biological effects of Ang1‐9 and Ang1‐7 are opposite of those associated with AT2(1‐8). Specifically, Ang1‐9 and Ang1‐7 are vasodilatory, natriuretic, diuretic, antifibrotic, and antiremodeling.9 These beneficial APs mediate the majority of their effects by binding to angiotensin receptor type 2 or an endogenous orphan Mas receptor (MasR), as opposed to binding to ATR1.11, 12 It is now known9, 13 that the ACE2 system provides an endogenous counterbalance to ACE and that an expanded view of the RAAS involves many different APs with structures differing by 1 to 5 amino acids. Thus, RAAS is considerably more complex than those traditionally viewed, and this discovery has uncovered a broad new set of molecules to explore, understand, and potentially modify in subjects with heart disease.
Very little is known about the ACE2 system and related APs in veterinary species. To the best of our knowledge, studies of ACE2 in dogs with naturally occurring heart disease are lacking. We sought to detect and describe the location of ACE2 immunoreactivity in the kidney and left ventricular (LV) myocardium to quantify the activity of plasma, kidney, and LV myocardial ACE2, to determine the equilibrium concentrations of 10 different circulating APs, and to determine the effect of recombinant human ACE2 on the relative amounts of beneficial versus maladaptive APs. Our hypotheses were that the evidence of ACE2 could be found in tissues and plasma of dogs, that equilibrium concentrations of RAAS APs would differ in dogs with heart disease versus healthy dogs, and recombinant human ACE2 (rhACE2) can alter relative concentrations of APs.
2. MATERIALS AND METHODS
The study protocol was reviewed and approved by the University of Pennsylvania Institutional Animal Care and Use Committee and informed owner consent was obtained.
2.1. Angiotensin‐converting enzyme 2 immunohistochemistry (IHC)
Cohorts of dogs that died or were euthanized for cardiac diseases and dogs that died from noncardiac reasons at the University of Pennsylvania were recruited. Sections of left kidney that included both cortical and medullary tissue and full thickness LV myocardial samples from the lateral wall of the ventricle just apical to the mid‐point of the paraconal coronary artery were obtained within 60 minutes of death. Kidney and myocardial samples were fixed in 10% neutral buffered formalin for up to 72 hours followed by sectioning. A rabbit polyclonal primary antibody against ACE2 (ARP53751‐P050, Aviva Systems Biology, San Diego, California) was used at a dilution of 1:1200 and incubated on the sections for 45 minutes at room temperature (RT). A biotin‐free polymeric IHC detection system consisting of horseradish peroxidase conjugated anti‐rabbit IgG then was applied for 25 minutes at RT. Finally, slides were counterstained in hematoxylin, scanned (Leica Aperio Versa slide scanner, Leica Biosystems, Buffalo Grove, Illinois), and images acquired (ImageScope, Leica Biosystems, Buffalo Grove, Illinois). Additional details regarding IHC methods can be found in the Supporting Information.
2.2. Plasma ACE2 activity
A cohort of dogs >3 years of age with active or previous congestive heart failure (CHF) caused by degenerative mitral valve disease (DMVD) was prospectively recruited. Criteria for DMVD included a left apical systolic murmur and echocardiographic evidence of thickened or prolapsing mitral valve leaflets and mitral regurgitation. Echocardiographic examinations (iE33, Philips Healthcare, Andover, Massachusetts) recorded left ventricular internal dimension at end diastole (LVIDdN) and end‐systole (LVIDsN) from the 2‐dimensional (2D) right short axis view, which were then normalized to body weight using previously reported formulas.14 The left atrium‐to‐aortic root ratio (LA:Ao) was calculated using measurements from the 2D right short axis.15 Echocardiographic values were the average value of 3 measured beats. Healthy dogs that primarily were owned by staff and employees of the veterinary hospital were recruited as a control group and included dogs without a heart murmur or clinical signs suggestive of heart disease. Control dogs weighing >25 kg had a 2D echocardiogram performed to exclude the potential for occult dilated cardiomyopathy (DCM). Venipuncture was performed, and lithium heparinized plasma samples were obtained. Samples were stored at −80°C until batch analysis. Activity of ACE2 in heparinized plasma was measured using a fluorometric assay kit (#K897‐100, Biovision Inc, Milpitas, California). The ACE2 activity results are reported in milliunits per mg protein (mU/mg). The assay has a lower detection limit of 0.4 mU. Additional details regarding the performance and validation of the assay are described in the Supporting Information.
2.3. Tissue ACE2 activity
A cohort of dogs that died or were euthanized as a consequence of cardiac disease and dogs that died of noncardiac diseases were recruited to obtain kidney and LV myocardial samples as previously described. Kidney and LV myocardium lysates were prepared by homogenizing approximately 100 μg tissue sections in ACE2 lysis buffer (Biovision Inc) using a disposable tissue homogenizer (#0254210, Fisher Thermo Scientific, Waltham, Massachusetts). Lysates were kept on ice for 10 minutes followed by gentle vortexing followed by another 5 minutes on ice. The tissue lysates then were centrifuged at 16 000g at 4°C for 10 minutes and the pellets discarded. Activity of ACE2 in tissue lysates was measured using the same fluorometric assay kit used to assay plasma ACE2 activity. The protein concentrations of the plasma and tissue lysates were measured using a bicinchoninic acid method and bovine serum albumin protein standard (#K813, Biovision Inc).
2.4. Renin‐angiotensin‐aldosterone system equilibrium AP analysis
A cohort of dogs >3 years of age with DMVD or DCM that were either preclinical (ie, asymptomatic) or had CHF were recruited. Criteria for diagnosis of DMVD were the same as those for dogs recruited for study of plasma ACE2 activity assay. Criteria for diagnosis of DCM were presence of systolic dysfunction and LV eccentric hypertrophy on echocardiography defined as LVIDsN and LVIDdN above the normal reference range in the absence of an identifiable cardiac cause. A previously reported clinical staging system for dogs with DMVD16 was modified for use in all dogs. Specifically, dogs were segregated into those with preclinical (asymptomatic) heart disease (stage B) and those with current or historical signs of CHF (stage C) or those with signs of CHF refractory to standard PO treatment with furosemide, ACEI, positive inotropes, and spironolactone, and therefore in need of additional diuretics, such as hydrochlorothiazide or torsemide (stage D). Further classification of dogs in stage B occurred depending on the absence (stage B1) or presence (stage B2) of heart enlargement on echocardiography defined as LVIDdN ≥ 1.7 and LA:Ao ≥ 1.6.17 Venipuncture, blood collection, and storage were performed as previously described. Healthy dogs were recruited as described above. To provide more uniformity to the equilibrium concentrations results in stages B and C, and to best reflect clinical practice at our institution, dogs in stage B were prohibited from receiving ACEI, whereas all dogs in stage C were required to receive ACEI. The plasma equilibrium concentration of 10 different RAAS APs, including AT1(1‐10), AT2(1‐8), Ang1‐9, Ang1‐7, angiotensin 1‐5 (Ang1‐5), angiotensin 2‐10 (Ang2‐10), angiotensin III (AT3[2‐8]), angiotensin IV (AT4[3‐8]), angiotensin 2‐7 (Ang2‐7), and angiotensin 3‐7 (Ang3‐7), were quantified by liquid chromatography‐mass spectrometry/mass spectroscopy (LC‐MS/MS) performed at a commercial laboratory (Attoquant Diagnostics, Vienna, Austria) using previously validated and described methods.18, 19 Testing samples from humans has shown similar qualitative outcomes when comparing the quantification of circulating and equilibrium AP concentrations.18, 19 Two additional calculations were performed using equilibrium AP concentrations. The first calculation was a surrogate measure of plasma renin activity (PRAsurr) calculated as the sum of the AT1(1‐10) and AT2(1‐8) concentrations and the second a surrogate measure of ACE activity (ACEsurr) calculated as the ratio of AT2(1‐8) to AT1(1‐10).20 To explore the effect of exogenous ACE2 on the relative concentrations of selected RAAS APs, plasma samples from stage B2 and stage C dogs also were incubated with 5 μg/mL of recombinant human ACE2 (rhACE2; R&D Systems, Minneapolis, Minnesota) at 37°C, followed by measurement of equilibrium concentrations of AT1(1‐10), AT2(1‐8), Ang1‐9, Ang1‐7, and Ang1‐5. Details regarding LC‐MS/MS methods and validation as well as the lower limit of quantification for each of the measured APs are presented in the Supporting Information.
2.5. Statistical methods
Descriptive statistics for the various patient groups were generated. Normality was tested using the Shapiro‐Wilk test. Nonparametric tests were used if data were not normally distributed. Data that were below the lower limit of quantification for any particular assay were entered as a value equal to 0.5 times the lower limit of quantification as previously recommended.21 The presence of any extreme outlying data points was identified by the Grubbs' test, which restricts potential removal to ≤1 data point from each data set.22 Differences between 2 unpaired groups were tested using Wilcoxon rank sum or unpaired t tests. Differences across ≥3 groups were tested using Kruskal‐Wallis or 1‐way analysis of variance (ANOVA) and post hoc pairwise comparisons were performed using Dunn's test with correction for multiple comparisons or Bonferroni correction, respectively. Box and whisker plots were constructed with the line representing the median value, the limits of the box representing the 25th and 75th percentiles, and the whiskers extending to the 5th and 95th percentiles. Correlations among various patient demographic data, echocardiographic variables, and assay results were explored using Pearson correlation coefficient or Spearman rho and unadjusted P values. Concentrations of APs before and after administration of rhACE2 were evaluated by Wilcoxon signed rank tests. Data are shown as median (interquartile range), unless otherwise specified. Statistical procedures were performed using statistical software (Prism 8 for OS X, GraphPad Software Inc, La Jolla, California). Significance was defined as P < .05.
3. RESULTS
3.1. Immunohistochemistry
Tissues from 6 dogs were obtained including 3 control dogs euthanized for noncardiac disease and 3 dogs euthanized because of CHF related to end‐stage DMVD. Control dogs did not have a heart murmur or any history or clinical signs of heart disease and included the following: 15‐year‐old female spayed mixed breed dog with a painful back, vomiting, and poor appetite; 11‐year‐old male neutered West Highland White Terrier with refractory seizures; and 10‐year‐old male neutered French Bulldog with upper airway obstruction. Dogs with heart disease included the following: 10‐year‐old female spayed mixed breed dog with severe CHF receiving furosemide, hydrochlorothiazide, enalapril, pimobendan, and spironolactone; 9‐year‐old male neutered Cocker Spaniel with severe CHF receiving furosemide, benazepril, hydrocodone, sildenafil, and pimobendan; and 10‐year‐old female spayed Chihuahua with severe CHF receiving furosemide and enalapril. Immunoreactivity for ACE2 was present in the kidneys of both control and DMVD dogs (Figure 1). Immunolabeling was especially evident and diffuse in the apical border of the epithelial cells of the proximal convoluted tubules. Granular cytoplasmic immunolabeling of variable intensity also was noted in the proximal convoluted tubules and other segments of the nephron. Immunoreactivity for ACE2 was present in the LV myocardium of both control and affected dogs (Figure 1). Staining was patchy with variably intense sarcoplasmic immunolabeling of the cardiomyocytes. Visual review of slides did not identify any readily apparent differences in the location or intensity of staining between groups.
Figure 1.
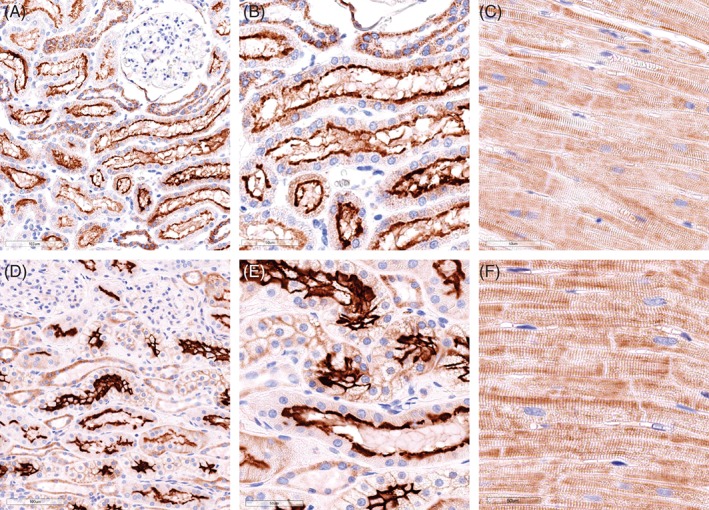
Representative photomicrographs of anti‐angiotensin‐converting enzyme 2 (ACE2) immunolabeling from a 10‐year‐old male neutered French Bulldog that was euthanized secondary to upper airway obstruction (A‐C), a 10‐year‐old female spayed mixed breed dog euthanized for severe congestive heart failure (CHF) because of degenerative mitral valve disease (DMVD) (D, E), and 10‐year‐old female spayed Chihuahua also euthanized for severe CHF due to DMVD (F). Photomicrographs of negative controls are presented in Supporting Information. ×20, A, D; ×40, B, C, E, F
3.2. Plasma ACE2 activity
A cohort of 34 dogs was recruited. The ACE2 activity (55 mU/mg) in 1 dog, a 9‐year‐old male healthy Miniature Poodle was found to be an extreme outlier and was excluded from the final analysis. Thus, the analysis set consisted of 33 dogs, including 18 healthy dogs and 15 dogs with either stage C or D CHF caused by DMVD. Signalment and treatments at the time of sampling are shown in Table 1. Validation data for the ACE2 assay are presented in the Supporting Information. The median plasma ACE2 activity in dogs with CHF (6.9 mU/mg [5.1‐12.1]) was significantly higher than that in healthy dogs (2.2 mU/mg [1.8‐3.0]; P = .0003; Figure 2). This result was consistent regardless of whether or not the outlier value was included in the analysis. No significant correlation was found between ACE2 activity and age (R 2 = .057, P = .34) or body weight (Spearman r = −.092, P = .72) in the healthy cohort.
Table 1.
Signalment and existing treatments in 33 dogs undergoing assessment of plasma angiotensin‐converting enzyme 2 (ACE2) activity, including healthy dogs and dogs with congestive heart failure (CHF) because of degenerative mitral valve disease. Data shown as count or median (interquartile range)
| Healthy | CHF | P | |
|---|---|---|---|
| N | 18 | 15 | |
| Sex (F/M) | 10/8 | 9/6 | .62 |
| Age (years) | 8 (6.8‐10) | 12 (11‐14) | .0001 |
| Body weight (kg) | 11.1 (9.1‐24.5) | 8.1 (5.6‐11.4) | .02 |
| Breeds | Mixed breed (n, 5) | Mixed breed (n, 3) | |
| Boston Terrier (n, 2) | Shih Tzu (n, 2) | ||
| Doberman Pinscher (n, 2) | Other (n, 10 [1 each]) | ||
| Pug (n, 2) | |||
| Other (n, 7 [1 each]) | |||
| Cardiac medications at time of sampling | None | Furosemide (n, 14) | |
| Pimobendan (n, 14) | |||
| ACE inhibitor (n, 12) | |||
| Spironolactone (n, 10) | |||
| Sildenafil (n, 2) |
Figure 2.
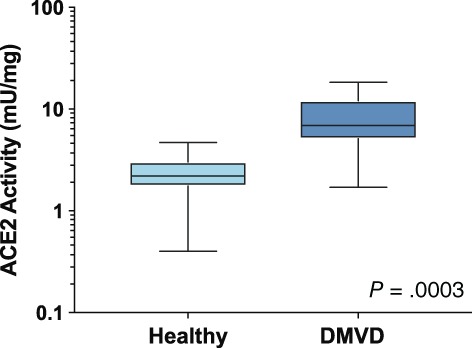
Plasma angiotensin‐converting enzyme 2 (ACE2) activity in 18 healthy dogs and 15 dogs with congestive heart failure because of degenerative mitral valve disease (DMVD)
3.3. Kidney and myocardial ACE2 activity
Kidney and LV myocardial tissues were obtained from 15 dogs: 7 with noncardiac disease and 8 with severe end‐stage DMVD. The 6 dogs the tissues of which were previously used for IHC, including 3 DMVD and 3 controls, were included in this cohort. All dogs euthanized because of cardiac disease were receiving ACEI. Noncardiac reasons for death in the 7 dogs with noncardiac disease included pulmonary or airway disease, neurological disease, and neoplasia (2/7 [28.6%]) and 1 dog (14.3%) with unspecified cause of back pain and vomiting. Four (4/7 [57.1%]) and 3/8 (37.5%) of dogs were male in the noncardiac and DMVD cohorts, respectively (P = .62). The mean (SD) age in years was 12.9 (1.9) and 11.4 (1.8) for dogs without cardiac disease and DMVD dogs, respectively (P = .15). The LV ACE2 activity (17.8 mU/mg) from 1 control dog, a 12 years old with splenic mass was an extreme outlier and this value was excluded from the LV analysis. The mean (SD) ACE2 activity in the kidneys (1552 mU/mg [1021]) and LV myocardium (2.3 mU/mg [2.0]) of the noncardiac group was not significantly different from results in the DMVD group (kidney, 1773 mU/mg [652]; LV, 3.4 mU/mg [2.1]; P between kidney results = .62; P between LV results = .36; Figure 3). This result was the same regardless of whether or not the outlier value was included in the analysis. The ACE2 activity per milligram of protein in kidney tissue was approximately 250 to 500 times higher than circulating or LV myocardial ACE2 activity. Assays for circulating and tissue ACE2 activity were performed on average 10 days (range, 3‐29) after the date of sample collection.
Figure 3.
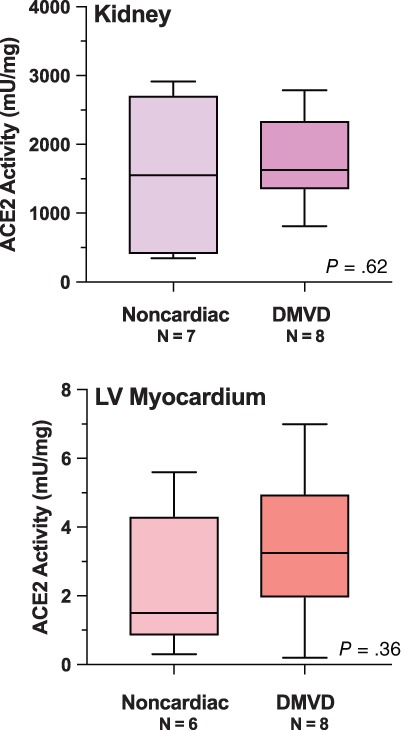
Kidney and left ventricular (LV) myocardium angiotensin‐converting enzyme 2 (ACE2) activity in dogs euthanized for or died of noncardiac disease versus end‐stage degenerative mitral valve disease (DMVD)
3.4. Renin‐angiotensin‐aldosterone system equilibrium AP analysis and effect of recombinant human ACE2
A cohort of 34 dogs consisting of 8 healthy dogs, 8 dogs with stage B1 DMVD, 9 dogs with stage B2 DMVD, and 9 dogs with stage C, including 6 dogs with DMVD and 3 dogs with DCM, was recruited. The signalment and treatments at the time of sampling are presented in Table 2. Differences in concentrations across and between groups were detected for a number of APs, including AT1(1‐10), Ang1‐7, Ang1‐9, Ang2‐10, Ang2‐7, and Ang3‐7 (Table 3). In general, the concentration of each of these APs was highest in dogs in stage C, intermediate in stage B1 or B2, and lowest in healthy dogs (Figure 4). A global overview of the expanded RAAS identified large differences in the overall activation and relative amounts of APs across patient groups (Figure 5). Of note, median AT1(1‐10) was >43 and 17 times higher in stage C than healthy and stage B2 dogs, respectively. Median Ang1‐7 in stage C was > 43 and 11 times higher than in healthy and stage B2 dogs, respectively, and median Ang1‐9 in stage C was >5 times higher than in both healthy and stage B1 or B2 dogs. No differences across groups were found for AT2(1‐8), AT3(2‐8), AT4(3‐8), or Ang1‐5. A number of APs, including AT1(1‐10), Ang1‐9, and Ang2‐10, had significant positive and modest correlation to LVIDdN and LA:Ao in affected dogs (see Supporting Information). The median surrogate plasma renin activity (PRAsurr) was significantly different across patient groups (Figure 6), and the median PRAsurr in stage C was approximately 27 and 12 times higher than that in healthy dogs or dogs in stage B2, respectively (Table 3). The median ACEsurr also was significantly different across patient groups (Figure 6). The median ACEsurr in stage C was 16 to 20 times lower than that in healthy dogs or dogs in stages B1 or B2 (Table 3). The addition of rhACE2 to plasma samples from 6 dogs in stage B2 and 7 dogs in stage C significantly increased paired median equilibrium concentrations of several APs while decreasing the concentration of others (Figure 7 and Supporting Information). Specifically, concentrations of AT1(1‐10) decreased in stage B2 dogs from 196.8 pg/mL (158.8‐292.8) to 75.4 pg/mL (48.9‐142.6; P = .03) and from 3373 pg/mL (2497‐4318) to 1187 pg/mL (492.8‐2201; P = .02) in dogs in stage C. Concentration of AT2(1‐8) in stage B2 dogs decreased from 80.0 pg/mL (71.1‐125.1) to 1.6 pg/mL (0.50‐3.38; P = .03) and from 53.4 pg/mL (28.6‐226.4) to 2.4 pg/mL (0.50‐5.8; P = .02) in dogs in stage C. Concentration of Ang1‐9 increased in stage B2 dogs from 1.8 pg/mL (1.8‐1.8) to 61.6 pg/mL (34.5‐89.0; P = .03) and from 10.3 pg/mL (4.4‐37.2) to 2431 pg/mL (1355‐3037; P = .02) in dogs in stage C. Concentration of Ang1‐7 in stage B2 dogs increased from 37.8 pg/mL (28.3‐62.5) to 131.5 pg/mL (80.9‐202.6; P = .03) and from 468.7 pg/mL (238.8‐1143) to 1004 pg/mL (373.7‐2594; P = .16) in dogs in stage C. Concentration of Ang1‐5 in stage B2 dogs increased from 22.0 pg/mL (19.7‐39.8) to 52.8 pg/mL (45.8‐113; P = .03) and from 25.7 pg/mL (3.4‐93.4) to 67.7 pg/mL (11.9‐254.2; P = .02) in dogs in stage C. Assays for RAAS AP equilibrium analysis and RAAS AP equilibrium analysis after incubation with rhACE2 were performed on average 30 days (range, 11‐45) and 50 days (range, 30‐64) after date of sample collection, respectively.
Table 2.
Signalment, echocardiographic, and treatment data from 34 dogs undergoing analysis of equilibrium concentrations of renin angiotensin aldosterone system angiotensin peptides. Data shown as mean (SD) or median (interquartile range) unless otherwise specified. See text for description of the modified clinical staging system
| Healthy | Stage B1 | Stage B2 | Stage C | P | |
|---|---|---|---|---|---|
| N | 8 | 8 | 9 | 9 | |
| Sex (F/M) | 3/5 | 3/5 | 2/7 | 3/6 | .89 |
| Age (years) | 8.7 (2.9) | 10.8 (1.6) | 9.6 (1.7) | 9.6 (3.7) | .47 |
| Body weight (kg) | 20.0 (9.1‐30.5) | 7.5 (4.1‐10.8) | 8.4 (6.1‐10.3) | 9.4 (8.1‐32.0) | .17 |
| LVIDdN | 1.47 (0.10) | 1.91 (0.24) | 2.14 (0.36) | <.0001 | |
| B1 versus C, P < .0001 | |||||
| B2 versus C, P = .005 | |||||
| LVIDsN | 0.77 (0.64‐0.90) | 0.99 (0.96‐1.01) | 1.14 (0.94‐1.70) | .21 | |
| IVSdN | 0.40 (0.07) | 0.45 (0.04) | 0.45 (0.08) | .23 | |
| LVPWdN | 0.40 (0.36‐0.43) | 0.42 (0.41‐44) | 0.44 (0.42‐0.46) | .13 | |
| LADN | 1.09 (0.96‐1.15) | 1.21 (1.13‐1.38) | 1.62 (1.50‐1.71) | .003 | |
| B1 versus C, P = .002 | |||||
| AoDN | 0.73 (0.66‐0.81) | 0.70 (0.69‐0.75) | 0.69 (0.63‐0.76) | .75 | |
| LA:Ao | 1.43 (0.25) | 1.83 (0.18) | 2.37 (0.57) | .0002 | |
| B1 versus C, P < .0001 | |||||
| B2 versus C, P = .02 | |||||
| Furosemide (Y/N) | 0/8 | 0/9 | 9/0 | ||
| ACEI (Y/N) | 0/8 | 0/9 | 9/0 | ||
| Pimobendan (Y/N) | 0/8 | 5/4 | 9/0 | ||
| Spironolactone (Y/N) | 0/8 | 0/9 | 8/1 | ||
| Other medications | Sildenafil (n, 1) | ||||
| Hydrocodone (n, 2) | |||||
| Hydrochlorothiazide (n, 1) | |||||
| Diltiazem (n, 1) |
Abbreviations: ACEI, angiotensin‐converting enzyme inhibitor; AoDN, normalized aortic root diameter; IVSdN, normalized interventricular septum thickness at end‐diastole; LA:Ao, left atrium to aortic root diameter ratio; LADN, normalized left atrial diameter; LVIDdN, normalized left ventricular internal diameter at end‐diastole; LVIDsN, normalized left ventricular internal diameter at end‐systole; LVPWdN, normalized left ventricular posterior wall thickness at end‐diastole.
Table 3.
Median and interquartile value of plasma equilibrium concentrations of angiotensin peptides and surrogate measures of plasma renin activity (PRA‐surrogate) and angiotensin‐converting enzyme (ACE‐surrogate) activity in healthy dogs and dogs with heart disease. See text for description of the modified clinical staging system
| Healthy (n = 8) | Stage B1 (n = 8) | Stage B2 (n = 9) | Stage C (n = 9) | P | |
|---|---|---|---|---|---|
| AT1(1‐10) | 77.4 | 462.9 | 193.6 | 3372.9 | <.0001 |
| pg/mL | 53.8‐95.1 | 125.3‐662.6 | 159.6‐274.2 | 2132.0‐4060.0 | H versus C, P < .0001 |
| B2 versus C, P = .004 | |||||
| AT2(1‐8) | 39.9 | 135.7 | 75.9 | 56.2 | .10 |
| pg/mL | 26.7‐47.7 | 53.6‐298.0 | 60.9‐92.9 | 32.2‐319.2 | |
| Ang1‐9 | 1.8 | 1.8 | 1.8 | 10.3 | <.0001 |
| pg/mL | 1.8‐1.8 | 1.8‐1.8 | 1.8‐1.8 | 5.9‐27.0 | H versus C, P = .0002 |
| B1 versus C, P = .0002 | |||||
| B2 versus C, P < .0001 | |||||
| Ang1‐7 | 11.4 | 78.3 | 41.0 | 486.7 | <.0001 |
| pg/mL | 7.1‐25.3 | 19.2‐131.0 | 27.4‐45.1 | 214.2‐1168.0 | H versus C, P < .0001 |
| B2 versus C, P = .01 | |||||
| Ang1‐5 | 7.0 | 46.4 | 21.2 | 25.7 | .07 |
| pg/mL | 4.1‐10.7 | 18.3‐78.2 | 13.5‐26.5 | 4.6‐152.2 | |
| Ang2‐10 | 8.3 | 48.6 | 20.0 | 502.9 | <.0001 |
| pg/mL | 5.6‐10.9 | 16.3‐82.2 | 13.3‐36.5 | 354.1‐830.8 | H versus C, P < .0001 |
| B1 versus C, P = .05 | |||||
| B2 versus C, P = .004 | |||||
| AT3(2‐8) | 1.3 | 10.0 | 5.9 | 6.6 | .18 |
| pg/mL | 1.3‐1.3 | 3.3‐20.3 | 1.3‐10.0 | 1.3‐44.4 | |
| AT4(3‐8) | 3.6 | 18.3 | 6.7 | 15.7 | .19 |
| pg/mL | 2.9‐6.6 | 6.6‐2627.7 | 4.4‐11.1 | 1.1‐50.7 | |
| Ang2‐7 | 1.0 | 4.7 | 2.4 | 40.5 | <.0001 |
| pg/mL | 1.0‐1.0 | 1.7‐6.5 | 1.0‐4.5 | 21.7‐85.8 | H versus C, P < .0001 |
| B2 versus C, P = .007 | |||||
| Ang3‐7 | 0.5 | 4.3 | 0.5 | 42.5 | <.0001 |
| pg/mL | 0.5‐0.5 | 0.5‐8.8 | 0.5‐3.5 | 19.0‐93.6 | H versus C, P = .0002 |
| B2 versus C, P = .001 | |||||
| PRA‐surrogate | 121.6 | 658.1 | 277.1 | 3401.4 | <.0001 |
| pg/mL | 80.8‐135.3 | 178.9‐891.0 | 206.5‐359.5 | 2211.0‐4284.0 | H versus C, P < .0001 |
| B2 versus C, P = .006 | |||||
| ACE‐surrogate | 0.479 | 0.499 | 0.389 | 0.024 | .0001 |
| 0.468‐0.660 | 0.291‐0.540 | 0.311‐0.485 | 0.014‐0.094 | H versus C, P = .0002 | |
| B1 versus C, P = .003 | |||||
| B2 versus C, P = .02 |
Figure 4.
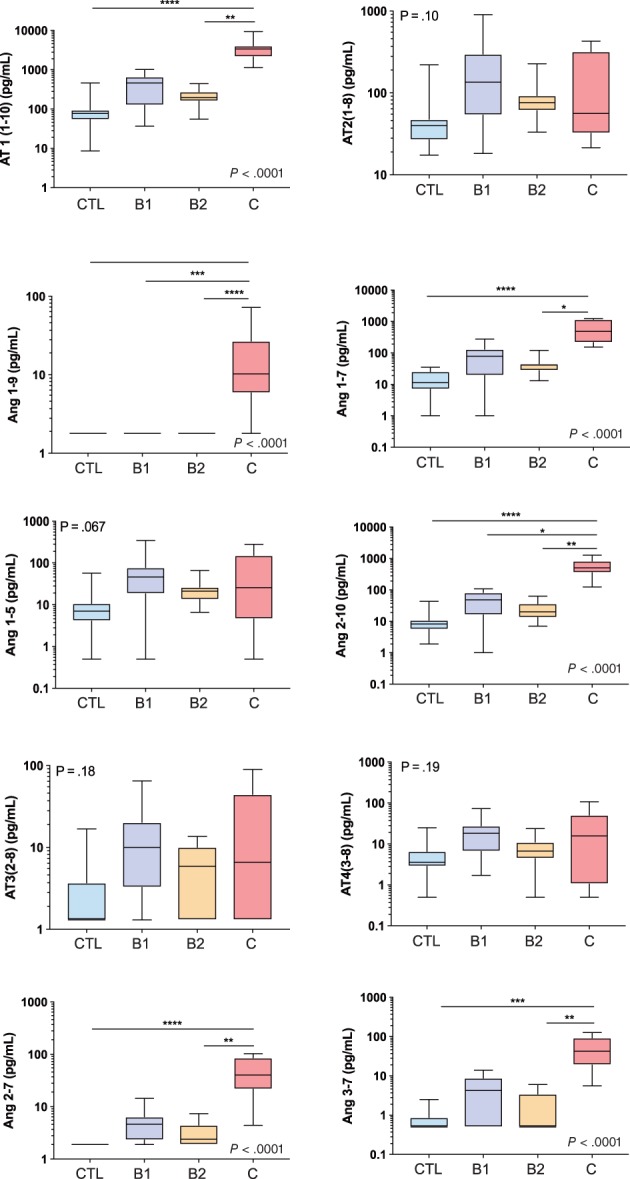
Equilibrium concentrations of angiotensin peptides (APs) of the renin‐angiotensin‐aldosterone system (RAAS) in control dogs (CTL) and dogs with stage B1, B2, and C heart disease. Ang1‐5, angiotensin 1‐5; Ang1‐7, angiotensin 1‐7; Ang1‐9, angiotensin 1‐9; Ang 2‐7, angiotensin 2‐7, Ang2‐10, angiotensin 2‐10; Ang3‐7, angiotensin 3‐7; AT1(1‐10), angiotensin I; AT2(1‐8), angiotensin II; AT3(2‐8), angiotensin III; AT4(3‐8), angiotensin IV. See text for description of the modified clinical staging system. *P < .05; **P < .01; ***P < .001; ****P < .0001
Figure 5.
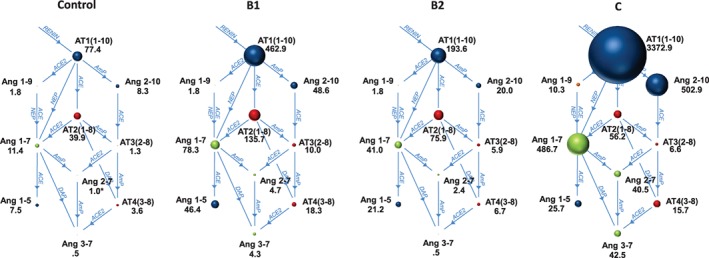
Equilibrium concentrations of angiotensin peptides (APs) and angiotensin converting enzyme (ACE) and angiotensin‐converting‐enzyme 2 (ACE2) pathways within a broader view of the renin‐angiotensin‐aldosterone system (RAAS) permits evaluation of the relative concentration and relationship between the various APs. The sizes of the circles are proportional to the median equilibrium concentration (pg/mL) of each AP. See text for description of the modified clinical staging system. AmP, aminopeptidase; DAP, aspartyl aminopeptidase; NEP, neutral endopeptidase. Other abbreviations same as in Figure 4
Figure 6.
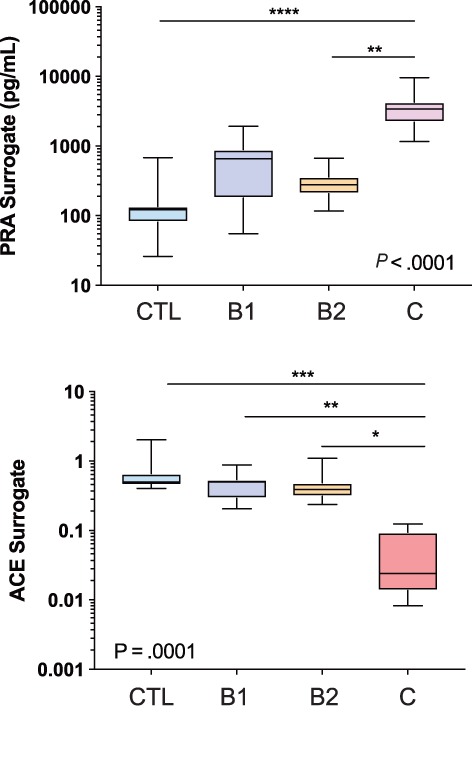
Surrogate measures of plasma renin activity (PRAsurr) and angiotensin converting enzyme (ACE) activity (ACEsurr) in healthy control (CTL) and dogs with stage B1, B2, and C heart disease. See text for description of the modified clinical staging system. *P < .05; **P < .01; ***P < .001; ****P < .0001
Figure 7.
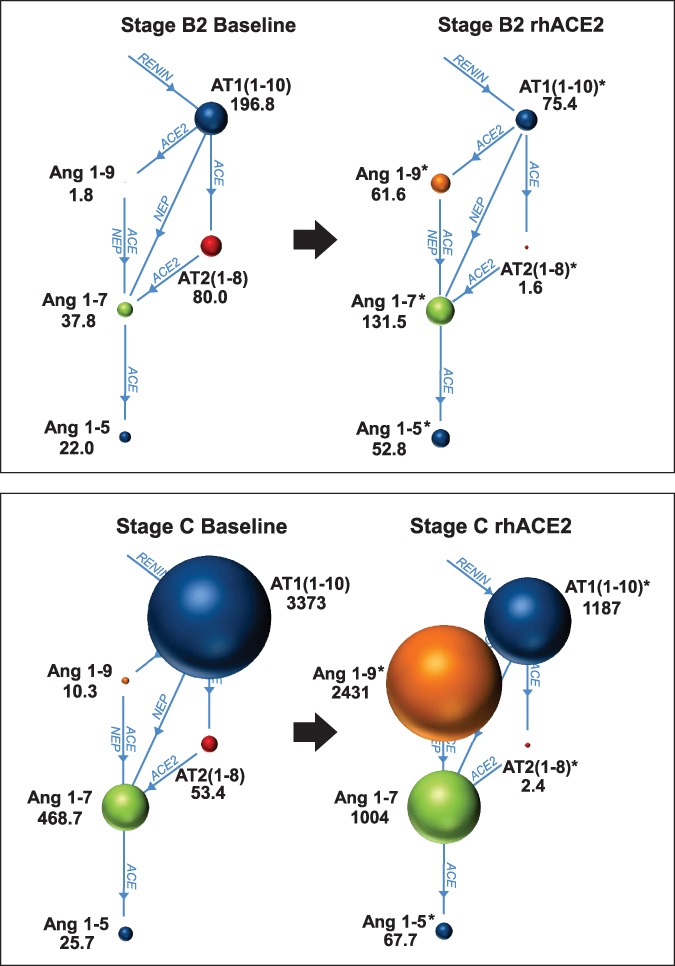
Plasma equilibrium concentrations of select angiotensin peptides (APs) before and after incubation with 5 μg/mL recombinant human ACE2 (rhACE2) from 6 dogs with stage B2 (top panel) and 7 dogs with stage C (bottom panel) degenerative mitral valve disease. The sizes of the circles are proportional to the median equilibrium concentration (pg/mL) of each AP. See text for description of the modified clinical staging system. *P < .05 versus baseline
4. DISCUSSION
The discovery of ACE2 and its related APs (Figure 8) is a subject of intense study in humans with heart disease,23, 24, 25, 26, 27 and to our knowledge, our study is the first to provide an expanded view of RAAS in dogs with heart disease. Our results identify ACE2 immunolabeling and activity in canine LV myocardium. Previous reports in rats28 and humans6, 28 indicated similar findings, suggesting an important role for ACE2 in cardiac function. Strong immunolabeling and a high degree of ACE2 activity also was found in the renal tubular epithelium, consistent with previous studies in dogs,29 mice,30 and humans.6, 7 Our results indicate that the ACE2 system is regulated differently in dogs with heart disease as compared to healthy dogs. First, plasma ACE2 activity was significantly increased in dogs with CHF, similar to previous reports in human patients with heart disease. In 1 such study,31 serum ACE2 activity was significantly increased secondary to ischemic and nonischemic heart disease and was independent of medications or comorbidities. In other studies,32, 33, 34 serum ACE2 activity was positively correlated with echocardiographic heart size, negatively correlated with heart function, and was independently associated with important clinical events, including death or hospitalization. The role of circulating or soluble ACE2, as opposed to tissue‐bound ACE2, is not fully understood. As previously mentioned, ACE2 is an integral membrane‐bound protein and the balance of tissue versus circulating ACE2 is mediated by membrane metalloproteinases that cleave portions of the cell membrane, which then are released into the surrounding extracellular space.33, 35, 36 The resultant ectosomes contain ACE2 as well as the immediate surrounding extracellular environment and enter the circulation or urine. This process is described as ectodomain shedding.36, 37 Previous studies35 indicate that shedding of ACE2 is more frequent than shedding of ACE, which also is highly membrane bound, and that AT2(1‐8), tumor necrosis factor, interleukin 1β, and disintegrin metalloproteases are potent stimuli for ACE2 shedding in mice and humans.37, 38, 39 Thus, the increase in circulating ACE2 might represent pathological displacement of ACE2 from its normal tissue membrane location, mediated in part by a feedback loop in which various substances decrease the activity of its negative regulator.39 As a result, circulating ACE2, which is removed from its normal location and ideal environment, is hypothesized to represent wasted or ineffective ACE2.31, 39 In our study, activity of circulating ACE2 normalized to protein amount was approximately 100‐fold less than activity of kidney ACE2, which supports this hypothesis. Our results, however, also indicated that both the kidney and LV ACE2 activity were not significantly different in dogs with CHF as compared to healthy dogs, suggesting that ACE2 shedding was not an important factor in the total extent of tissue‐bound ACE2 activity, but a loss of tissue ACE2 into the circulation would tend to decrease the overall compensatory potential of ACE2.31, 33, 39 Further elucidation of a seemingly complex relationship between circulating and tissue ACE2 is needed.
Figure 8.
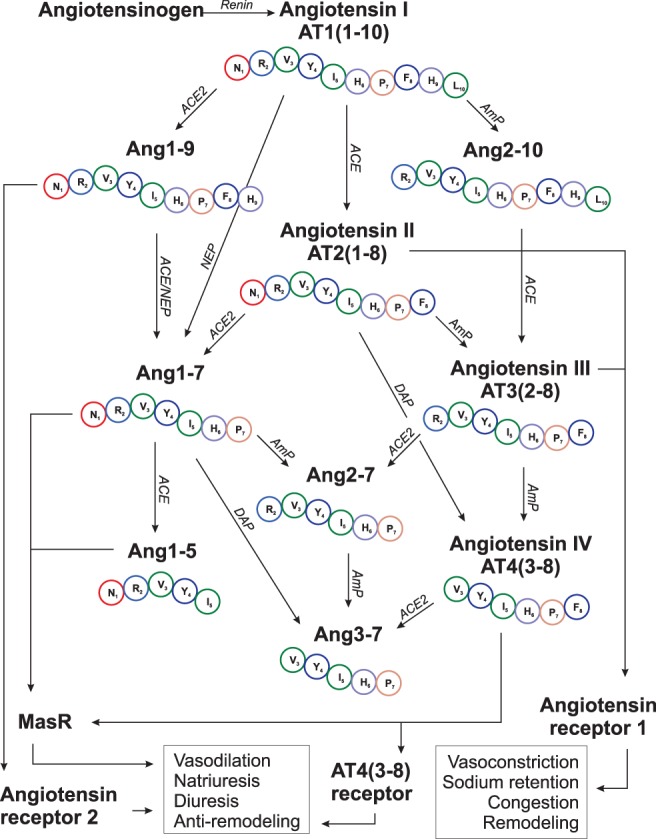
The activity of angiotensin converting enzyme (ACE) and angiotensin‐converting enzyme 2 (ACE2) and other peptidases on various angiotensin peptides (APs) within a more comprehensive renin‐angiotensin‐aldosterone system (RAAS) than what is traditionally considered. Each amino acid's identifying letter and number with respect to sequence within the parent molecule, angiotensin I (AT1[1‐10]), is shown. Other abbreviations same as in Figures 4 and 5
A second important finding of our study is the relationships and relative concentrations of various APs within the expanded RAAS, which provides insight into the pathophysiology of disease. Beginning at the first portion of RAAS (Figure 8), our results indicate that heart disease in the dog is associated with increased plasma renin activity. This finding is consistent with previous reports in dogs1, 40 and leads to higher concentrations of AT1(1‐10). In stages B1 and B2, downstream activation of RAAS and generation of AT2(1‐8) appears only mild, which also is consistent with a previous study of neurohormonal activation in dogs.2 Specifically, our data indicate higher concentrations of AT1(1‐10) and AT2(1‐8) in stages B1 and B2 as compared to healthy dogs, but these changes failed to reach statistical significance. Yet, once CHF develops (ie, stage C), PRA and Ang1(1‐10) are greatly increased and serve as potential substrate for generation of downstream APs, such as AT2(1‐8) (Figure 5). In our study, all dogs in stage C were receiving ACEI, which prevented conversion of AT1(1‐10) into large amounts of AT2(1‐8). Thus, concentrations of AT2(1‐8) were not significantly different in stage C as compared to other patient groups. Instead, our results indicated that in dogs with CHF, AT1(1‐10) presumably was cleaved by the proteases aminopeptidase (AmP), ACE2, and neutral endopeptidase into significantly higher concentrations of Ang2‐10, Ang1‐9, and Ang1‐7, respectively.12 The actions of these APs, and in particular Ang1‐9 and Ang1‐7, have been studied in a variety of species, including humans, dogs, and rats, and include vasodilatation, natriuresis, activation of nitric oxide, antagonism of ATR1, protection against ischemia and reperfusion injury, simulation of natriuretic peptide release, and improvement in glycolysis and high energy phosphate production.11, 12, 41, 42, 43, 44, 45, 46, 47 Thus, the reported benefits48, 49 of ACEI in dogs with CHF might be a result of increased concentrations and salutary actions of Ang1‐9 and Ang1‐7, as well as inhibition of AT2(1‐8) formation. Our findings within an expanded RAAS indicate the existence of closely related but biologically distinct groups of APs, 1 of which promotes the CHF phenotype while the other acts as a counterbalance working to shift the balance within RAAS back toward the beneficial APs.
Our results also provide new information about less well‐known APs, such as AT3(2‐8) and AT4(3‐8). The exact biologic actions of certain APs are incompletely understood and are predicted based on extension from studies in animal models. Previous studies in mice50, 51 indicated that AT3(2‐8) binds to ATR1, thereby eliciting effects similar to AT2(1‐8). Within the central nervous system, AT3(2‐8) is the final effector molecule that mediates effects of AT2(1‐8), such as release of vasopressin from the pituitary gland.50 In contrast, despite differing from AT3(2‐8) by only a single amino acid, AT4(3‐8) binds to MasR and a novel AT4(3‐8) receptor, eliciting effects similar to Ang1‐7, including prevention of apoptosis, vasodilatation, and anti‐remodeling (Figure 8).52, 53, 54 In our study, equilibrium concentrations of AT3(2‐8) and AT4(3‐8) in dogs with heart disease were not significantly different from those of healthy dogs. The reason for this finding in dogs with preclinical disease might stem from the relatively low overall activity of RAAS whereas in dogs with severe disease and CHF, formation of AT3(2‐8) from its precursor, Ang2‐10, is primarily mediated by ACE and would have been blocked by the presence of ACEI. Subsequently, low amounts of AT3(2‐8) then would limit downstream formation of AT4(3‐8; Figure 8). Relatively little is known about other APs in the expanded RAAS. A previous study47 reported that Ang2‐10 is vasodilatory and antiproliferative whereas another study55 reported that Ang2‐10 was vasoconstrictive, but likely only after conversion to AT3(2‐8). As for the remaining APs, in humans, Ang(2‐7) induces mild vasoconstriction,56 Ang(3‐7) is thought to be a centrally acting vasoconstrictor,57 and Ang1‐5 binds MasR and causes vasodilatation and increased natriuretic peptide release.58 Thus, results of RAAS AP equilibrium analysis in dogs with CHF receiving ACEI indicated an increase in beneficial APs including Ang1‐9, Ang1‐7, and Ang2‐10; no change in beneficial APs including AT4(3‐8) and Ang1‐5; an increase in maladaptive APs, including Ang2‐7 and Ang3‐7; and no change in maladaptive APs including AT2(1‐8) and AT3(2‐8). These data offer insight into the pathophysiology of disease and suggest novel treatment approaches as discussed below.
A third important finding of our study is the ability to shift the balance of APs within the RAAS using rhACE2. The counter regulatory role of ACE2 on the maladaptive effects of ACE and AT2(1‐8) make ACE2 and its related APs attractive targets for pharmacotherapy.23, 25, 26, 27, 45, 59, 60, 61 Treatment with exogenous ACE2 is particularly interesting in light of the fact that the affinity of ACE2 to cleave AT2(1‐8) into beneficial Ang1‐7 is 12‐fold greater than the affinity of ACE to cleave AT1(1‐10) to AT2(1‐8),62 suggesting that exogenous ACE2 or ACE2 activators would be capable of converting even large amounts of AT2(1‐8). In our study, incubation of plasma samples with rhACE2 from dogs with stage B2 or stage C heart disease resulted in significantly higher equilibrium concentrations of beneficial APs, including Ang1‐9, Ang1‐7, and Ang1‐5, while simultaneously decreasing the concentration of maladaptive APs, including AT1(1‐10) and AT2(1‐8). Previous studies18, 63 of rhACE2 involving human serum or healthy volunteers reported similar increases in beneficial APs and suppression of maladaptive APs, and have led to early trials64 in humans with pulmonary hypertension. Collectively, these data provide conceptual support of therapies targeting the ACE2 system. Our data also suggest that the methodology employed in our study, namely equilibrium analysis of APs, is a useful preclinical analytical tool.
Our study had a number of limitations. Larger studies are needed to confirm findings from the relatively small number of animals in our study. Plasma ACE2 activity was quantified only in dogs with CHF secondary to advanced DMVD and important differences might exist in dogs with early DMVD or in animals with other causes of CHF. The potential correlation among circulating ACE2 activity, duration of CHF and administration of medications, and clinical outcome requires longitudinal studies. The clinical implication of circulating ACE2 appears complex. For example, although previous observational studies32, 33, 34 determined that increased ACE2 activity was associated with poor outcome, in a study involving patients after intensive treatment for acute CHF,65 increased ACE2 activity over baseline was associated with improved survival. The number of samples obtained for kidney and LV myocardial ACE2 activity was small because of difficulty in procuring such samples, and future studies of larger populations might identify important differences. Evaluation of the expanded RAAS was performed using equilibrium analysis and the reported concentrations are higher than the actual circulating concentrations, but the 2 values are highly correlated,18 which preserve the ability to ascertain relative differences among APs. The expanded model of RAAS remains a simple representation of a highly complex pathophysiological network with many additional (both identified and unidentified) components and relationships. Regardless, the proposed RAAS model offers new insights and proof of concept for a novel treatment strategy. Dogs were receiving a variety of cardiac medications including diuretics and anti‐RAAS agents such as ACEI and spironolactone that might have influenced the results. Dogs in the RAAS AP equilibrium analysis included both dogs with DMVD and DCM, and important differences in the magnitude of RAAS AP concentrations might exist. Studies that specifically examine the effect of various treatments on RAAS are of interest. For example, a previous study (Ames MK, Potter BK, Hess A. Effect of loop diuretics on novel components of the renin‐angiotensin‐aldosterone system in healthy dogs [Abstract]. Proceedings of the 2018 ACVIM Forum, Seattle, WA, 2018) in dogs compared the relative extent of RAAS activation after administration of furosemide as compared to torsemide by analyzing the equilibrium concentrations of Ang1‐7, Ang1‐3, AT3(2‐8), and AT4(3‐8), whereas others18 have reported increased Ang1‐9, Ang1‐7, and Ang1‐5 mediated by combined treatment with angiotensin receptor blockers and rhACE2 in serum samples of humans. Finally, healthy dogs weighing <25 kg were not required to undergo echocardiographic studies, but the incidence of asymptomatic DCM in this population of dogs likely was low.
In summary, our results provide novel information about the presence, distribution, activity, and equilibrium concentrations of components of the ACE2 system and an expanded view of the RAAS in dogs with heart disease. Activity of both the traditional and ACE2‐related components of RAAS was significantly different in affected dogs, and potential new targets for treatment were identified. Our study provided proof of concept that rhACE2 could modify RAAS in such a manner that would increase newly described beneficial APs while suppressing maladaptive APs. Consideration of an expanded RAAS that includes ACE2 and related APs opens new opportunities to understand and treat heart disease in dogs.
CONFLICT OF INTEREST DECLARATION
Authors declare no conflict of interest.
OFF‐LABEL ANTIMICROBIAL DECLARATION
Authors declare no off‐label use of antimicrobials.
INSTITUTIONAL ANIMAL CARE AND USE COMMITTEE (IACUC) OR OTHER APPROVAL DECLARATION
University of Pennsylvania IACUC approval: #803099.
HUMAN ETHICS APPROVAL DECLARATION
Authors declare human ethics approval was not needed for this study.
Supporting information
Supplemental Figure 1 Representative photomicrographs of anti‐angiotensin converting enzyme 2 (ACE2) immunolabeling negative controls from a 10 year old male neutered French bulldog that was euthanized secondary to upper airway obstruction (Kidney, A, B; left ventricular myocardium, C), a 10 year old female spayed mixed breed dog euthanized for severe heart failure due to degenerative mitral valve disease (DMVD) (Kidney, D, E), and a 10 year old female spayed Chihuahua also euthanized for severe heart failure due to DMVD (LV myocardium, F). There is an absence of immunolabeling in all sections. 20x, A, D; 40X, B, C, E, F.
Supplemental Figure 2 Linearity of the 4‐methylcoumaryl‐7‐amide (MCA) standard curve used for measurement of angiotensin converting enzyme 2 activity.
Supplemental Figure 3 Fluorescent activity curves using different volumes of canine kidney supernatant fluid.
Supplemental Figure 4 Performance of the kit‐supplied angiotensin converting enzyme 2 (ACE2) inhibitor on total ACE2 activity with varying canine tissue samples. Columns show the mean percent inhibition +/− the SE of the measurement.
Supplemental Figure 5 Equilibrium concentrations of various individual angiotensin peptides (APs) at baseline and after incubation with recombinant human angiotensin converting enzyme 2 (rhACE2). Treatment of plasma from dogs with stage B2 or stage C heart disease decreased maladaptive APs such as angiotensin I (AT1 [1–10]) and angiotensin II (AT2 [1–8]) and increased cardioprotective APs such as angiotensin 1‐9 (Ang1‐9), angiotensin 1‐7 (Ang1‐7), and angiotensin 1‐5 (Ang1‐5). Data suggest that the balance between the beneficial and maladaptive APs can be made more favorable by rhACE2. See text for description of the modified clinical staging system.
Appendix S1: Supporting information
ACKNOWLEDGMENT
The authors thank Charles Bradley and Enrico Radaelli and Patrick Savickas and Lindsey Citron for technical assistance.
Larouche‐Lebel É, Loughran KA, Oyama MA, et al. Plasma and tissue angiotensin‐converting enzyme 2 activity and plasma equilibrium concentrations of angiotensin peptides in dogs with heart disease. J Vet Intern Med. 2019;33:1571–1584. 10.1111/jvim.15548
Present address Danielle S. Laughlin, BluePearl Specialty and Emergency Pet Hospital, 455 Abernathy Road NE, Sandy Springs, GA 30328. Melissa D. Sánchez, Antech Diagnostics, 410 Union Ave, Framingham, MA 01702.
Éva Larouche‐Lebel and Kerry A. Loughran contributed equally to this study.
REFERENCES
- 1. Pedersen HD, Koch J, Poulsen K, Jensen AL, Flagstad A. Activation of the renin‐angiotensin system in dogs with asymptomatic and mildly symptomatic mitral valvular insufficiency. J Vet Intern Med. 1995;9:328‐331. [DOI] [PubMed] [Google Scholar]
- 2. Haggstrom J, Hansson K, Kvart C, et al. Effects of naturally acquired decompensated mitral valve regurgitation on the renin‐angiotensin‐aldosterone system and atrial natriuretic peptide concentration in dogs. Am J Vet Res. 1997;58:77‐82. [PubMed] [Google Scholar]
- 3. Koch J, Pedersen HD, Jensen AL, Flagstad A, Poulsen K. Activation of the renin‐angiotensin system in dogs with asymptomatic and symptomatic dilated cardiomyopathy. Res Vet Sci. 1995;59:172‐175. [DOI] [PubMed] [Google Scholar]
- 4. Tidholm A, Haggstrom J, Hansson K. Effects of dilated cardiomyopathy on the renin‐angiotensin‐aldosterone system, atrial natriuretic peptide activity, and thyroid hormone concentrations in dogs. Am J Vet Res. 2001;62:961‐967. [DOI] [PubMed] [Google Scholar]
- 5. Mehta PK, Griendling KK. Angiotensin II cell signaling: physiological and pathological effects in the cardiovascular system. Am J Physiol Cell Physiol. 2007;292:C82‐C97. [DOI] [PubMed] [Google Scholar]
- 6. Donoghue M, Hsieh F, Baronas E, et al. A novel angiotensin‐converting enzyme‐related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1‐9. Circ Res. 2000;87:E1‐E9. [DOI] [PubMed] [Google Scholar]
- 7. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. A human homolog of angiotensin‐converting enzyme. Cloning and functional expression as a captopril‐insensitive carboxypeptidase. J Biol Chem. 2000;275:33238‐33243. [DOI] [PubMed] [Google Scholar]
- 8. Warner FJ, Smith AI, Hooper NM, Turner AJ. Angiotensin‐converting enzyme‐2: a molecular and cellular perspective. Cell Mol Life Sci. 2004;61:2704‐2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Patel VB, Zhong JC, Grant MB, Oudit GY. Role of the ACE2/angiotensin 1‐7 axis of the renin‐angiotensin system in heart failure. Circ Res. 2016;118:1313‐1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Danilczyk U, Eriksson U, Oudit GY, et al. Physiological roles of angiotensin‐converting enzyme 2. Cell Mol Life Sci. 2004;61:2714‐2719. [DOI] [PubMed] [Google Scholar]
- 11. Santos RAS, Sampaio WO, Alzamora AC, et al. The ACE2/angiotensin‐(1‐7)/MAS axis of the renin‐angiotensin system: focus on angiotensin‐(1‐7). Physiol Rev. 2018;98:505‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ocaranza MP, Michea L, Chiong M, et al. Recent insights and therapeutic perspectives of angiotensin‐(1‐9) in the cardiovascular system. Clin Sci (Lond). 2014;127:549‐557. [DOI] [PubMed] [Google Scholar]
- 13. Koka V, Huang XR, Chung AC, et al. Angiotensin II up‐regulates angiotensin I‐converting enzyme (ACE), but down‐regulates ACE2 via the AT1‐ERK/p38 MAP kinase pathway. Am J Pathol. 2008;172:1174‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Cornell CC, Kittleson MD, Della TP, et al. Allometric scaling of M‐mode cardiac measurements in normal adult dogs. J Vet Intern Med. 2004;18:311‐321. [DOI] [PubMed] [Google Scholar]
- 15. Hansson K, Haggstrom J, Kvart C, Lord P. Left atrial to aortic root indices using two‐dimensional and M‐mode echocardiography in cavalier King Charles spaniels with and without left atrial enlargement. Vet Radiol Ultrasound. 2002;43:568‐575. [DOI] [PubMed] [Google Scholar]
- 16. Atkins C, Bonagura J, Ettinger S, et al. Guidelines for the diagnosis and treatment of canine chronic valvular heart disease. J Vet Intern Med. 2009;23:1142‐1150. [DOI] [PubMed] [Google Scholar]
- 17. Boswood A, Haggstom J, Gordon SG, et al. Effect of pimobendan in dogs with preclinical myxomatous mitral valve disease and cardiomegaly: the EPIC study‐a randomized clinical trial. J Vet Intern Med. 2016;30:1765‐1779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Basu R, Poglitsch M, Yogasundaram H, Thomas J, Rowe BH, Oudit GY. Roles of angiotensin peptides and recombinant human ACE2 in heart failure. J Am Coll Cardiol. 2017;69:805‐819. [DOI] [PubMed] [Google Scholar]
- 19. Kovarik JJ, Antlanger M, Domenig O, et al. Molecular regulation of the renin‐angiotensin system in haemodialysis patients. Nephrol Dial Transplant. 2015;30:115‐123. [DOI] [PubMed] [Google Scholar]
- 20. Pavo N, Goliasch G, Wurm R, et al. Low‐ and high‐renin heart failure phenotypes with clinical implications. Clin Chem. 2018;64:597‐608. [DOI] [PubMed] [Google Scholar]
- 21. Keizer RJ, Jansen RS, Rosing H, et al. Incorporation of concentration data below the limit of quantification in population pharmacokinetic analyses. Pharmacol Res Perspect. 2015;3:e00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Grubbs F. Procedures for detecting outlying observations in samples. Dent Tech. 1969;11:1‐21. [Google Scholar]
- 23. Jiang F, Yang J, Zhang Y, et al. Angiotensin‐converting enzyme 2 and angiotensin 1‐7: novel therapeutic targets. Nat Rev Cardiol. 2014;11:413‐426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kuba K, Imai Y, Penninger JM. Multiple functions of angiotensin‐converting enzyme 2 and its relevance in cardiovascular diseases. Circ J. 2013;77:301‐308. [DOI] [PubMed] [Google Scholar]
- 25. Patel VB, Lezutekong JN, Chen X, Oudit GY. Recombinant human ACE2 and the angiotensin 1‐7 axis as potential new therapies for heart failure. Can J Cardiol. 2017;33:943‐946. [DOI] [PubMed] [Google Scholar]
- 26. Tamargo M, Tamargo J. Future drug discovery in renin‐angiotensin‐aldosterone system intervention. Expert Opin Drug Discov. 2017;12:827‐848. [DOI] [PubMed] [Google Scholar]
- 27. Kittana N. Angiotensin‐converting enzyme 2‐angiotensin 1‐7/1‐9 system: novel promising targets for heart failure treatment. Fundam Clin Pharmacol. 2018;32:14‐25. [DOI] [PubMed] [Google Scholar]
- 28. Burrell LM, Risvanis J, Kubota E, et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur Heart J. 2005;26:369‐375. [DOI] [PubMed] [Google Scholar]
- 29. Mitani S, Yabuki A, Sawa M, et al. Intrarenal distributions and changes of angiotensin‐converting enzyme and angiotensin‐converting enzyme 2 in feline and canine chronic kidney disease. J Vet Med Sci. 2014;76:45‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Munoz MC, Burghi V, Miquet JG, et al. Downregulation of the ACE2/Ang‐(1‐7)/Mas axis in transgenic mice overexpressing GH. J Endocrinol. 2014;221:215‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Epelman S, Tang WH, Chen SY, et al. Detection of soluble angiotensin‐converting enzyme 2 in heart failure: insights into the endogenous counter‐regulatory pathway of the renin‐angiotensin‐aldosterone system. J Am Coll Cardiol. 2008;52:750‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Uri K, Fagyas M, Kertesz A, et al. Circulating ACE2 activity correlates with cardiovascular disease development. J Renin Angiotensin Aldosterone Syst. 2016;17:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Epelman S, Shrestha K, Troughton RW, et al. Soluble angiotensin‐converting enzyme 2 in human heart failure: relation with myocardial function and clinical outcomes. J Card Fail. 2009;15:565‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ramchand J, Patel SK, Srivastava PM, et al. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS One. 2018;13:e0198144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Warner FJ, Lew RA, Smith AI, Lambert DW, Hooper NM, Turner AJ. Angiotensin‐converting enzyme 2 (ACE2), but not ACE, is preferentially localized to the apical surface of polarized kidney cells. J Biol Chem. 2005;280:39353‐39362. [DOI] [PubMed] [Google Scholar]
- 36. Lambert DW, Yarski M, Warner FJ, et al. Tumor necrosis factor‐alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe‐acute respiratory syndrome‐coronavirus (SARS‐CoV) receptor, angiotensin‐converting enzyme‐2 (ACE2). J Biol Chem. 2005;280:30113‐30119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Xiao F, Zimpelmann J, Agaybi S, Gurley SB, Puente L, Burns KD. Characterization of angiotensin‐converting enzyme 2 ectodomain shedding from mouse proximal tubular cells. PLoS One. 2014;9:e85958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jia HP, Look DC, Tan P, et al. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2009;297:L84‐L96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Patel VB, Clarke N, Wang Z, et al. Angiotensin II induced proteolytic cleavage of myocardial ACE2 is mediated by TACE/ADAM‐17: a positive feedback mechanism in the RAS. J Mol Cell Cardiol. 2014;66:167‐176. [DOI] [PubMed] [Google Scholar]
- 40. Pedersen HD. Effects of mild mitral valve insufficiency, sodium intake, and place of blood sampling on the renin‐angiotensin system in dogs. Acta Vet Scand. 1996;37:109‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keidar S, Kaplan M, Gamliel‐Lazarovich A. ACE2 of the heart: from angiotensin I to angiotensin (1‐7). Cardiovasc Res. 2007;73:463‐469. [DOI] [PubMed] [Google Scholar]
- 42. Ferrario CM, Trask AJ, Jessup JA. Advances in the biochemical and functional roles of angiotensin converting enzyme 2 and angiotensin‐(1‐7) in the regulation of cardiovascular function. Am J Physiol Heart Circ Physiol. 2005;289:H3019‐H3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu E, Yang S, Xu Z, Li J, Yang W, Li G. Angiotensin‐(1‐7) prevents atrial fibrosis and atrial fibrillation in long‐term atrial tachycardia dogs. Regul Pept. 2010;162:73‐78. [DOI] [PubMed] [Google Scholar]
- 44. Zhao J, Liu E, Li G, Qi L, Li J, Yang W. Effects of the angiotensin‐(1‐7)/Mas/PI3K/Akt/nitric oxide axis and the possible role of atrial natriuretic peptide in an acute atrial tachycardia canine model. J Renin Angiotensin Aldosterone Syst. 2015;16:1069‐1077. [DOI] [PubMed] [Google Scholar]
- 45. Iwata M, Cowling RT, Yeo SJ, et al. Targeting the ACE2‐Ang‐(1‐7) pathway in cardiac fibroblasts to treat cardiac remodeling and heart failure. J Mol Cell Cardiol. 2010;51:542‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ocaranza MP, Diaz‐Araya G, Carreno JE, et al. Polymorphism in gene coding for ACE determines different development of myocardial fibrosis in rats. Am J Physiol Heart Circ Physiol. 2004;286:H498‐H506. [DOI] [PubMed] [Google Scholar]
- 47. Mustafa MR, Dharmani M, Kunheen NK, Sim MK. Effects of des‐aspartate‐angiotensin I on the actions of angiotensin III in the renal and mesenteric vasculature of normo‐ and hypertensive rats. Regul Pept. 2004;120:15‐22. [DOI] [PubMed] [Google Scholar]
- 48. BENCH (BENazepril in Canine Heart disease) Study Group . The effect of benazepril on survival times and clinical signs of dogs with congestive heart failure: results of a multicenter, prospective, randomized, double‐blinded, placebo‐controlled, long‐term clinical trial. J Vet Cardiol. 1999;1:7‐18. [DOI] [PubMed] [Google Scholar]
- 49. Ettinger SJ, Benitz AM, Ericsson GF, et al. Effects of enalapril maleate on survival of dogs with naturally acquired heart failure. The Long‐Term Investigation of Veterinary Enalapril (LIVE) study group. J Am Vet Med Assoc. 1998;213:1573‐1577. [PubMed] [Google Scholar]
- 50. Zini S, Fournie‐Zaluski MC, Chauvel E, Roques BP, Corvol P, Llorens‐Cortes C. Identification of metabolic pathways of brain angiotensin II and III using specific aminopeptidase inhibitors: predominant role of angiotensin III in the control of vasopressin release. Proc Natl Acad Sci U S A. 1996;93:11968‐11973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yugandhar VG, Clark MA. Angiotensin III: a physiological relevant peptide of the renin angiotensin system. Peptides. 2013;46:26‐32. [DOI] [PubMed] [Google Scholar]
- 52. Yang H, Zeng XJ, Wang HX, et al. Angiotensin IV protects against angiotensin II‐induced cardiac injury via AT4 receptor. Peptides. 2011;32:2108‐2115. [DOI] [PubMed] [Google Scholar]
- 53. Park BM, Cha SA, Han BR, Kim SH. Angiotensin IV stimulates high atrial stretch‐induced ANP secretion via insulin regulated aminopeptidase. Peptides. 2015;63:30‐37. [DOI] [PubMed] [Google Scholar]
- 54. Mustafa T, Lee JH, Chai SY, et al. Bioactive angiotensin peptides: focus on angiotensin IV. J Renin Angiotensin Aldosterone Syst. 2001;2:205‐210. [DOI] [PubMed] [Google Scholar]
- 55. Campbell WB, Schmitz JM, Itskovitz HD. (des‐Asp1) angiotensin I: a study of its pressor and steroidogenic activities in conscious rats. Endocrinology. 1977;100:46‐51. [DOI] [PubMed] [Google Scholar]
- 56. Kono T, Taniguchi A, Imura H, et al. Pressor activity of angiotensin II‐(2‐7)‐hexapeptide in man. Endocrinol Jpn. 1985;32:767‐769. [DOI] [PubMed] [Google Scholar]
- 57. Ferreira PM, Souza Dos Santos RA, Campagnole‐Santos MJ. Angiotensin‐(3‐7) pressor effect at the rostral ventrolateral medulla. Regul Pept. 2007;141:168‐174. [DOI] [PubMed] [Google Scholar]
- 58. Yu L, Yuan K, Phuong HT, et al. Angiotensin‐(1‐5), an active mediator of renin‐angiotensin system, stimulates ANP secretion via Mas receptor. Peptides. 2016;86:33‐41. [DOI] [PubMed] [Google Scholar]
- 59. Kazemi‐Bajestani SM, Patel VB, Wang W, et al. Targeting the ACE2 and apelin pathways are novel therapies for heart failure: opportunities and challenges. Cardiol Res Pract. 2012;2012:823193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weber KT, Diez J. Targeting the cardiac myofibroblast secretome to treat myocardial fibrosis in heart failure. Circ Heart Fail. 2016;9:e003315. [DOI] [PubMed] [Google Scholar]
- 61. Chamsi‐Pasha MA, Shao Z, Tang WH. Angiotensin‐converting enzyme 2 as a therapeutic target for heart failure. Curr Heart Fail Rep. 2014;11:58‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Rice GI, Thomas DA, Grant PJ, et al. Evaluation of angiotensin‐converting enzyme (ACE), its homologue ACE2 and neprilysin in angiotensin peptide metabolism. Biochem J. 2004;383:45‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haschke M, Schuster M, Poglitsch M, et al. Pharmacokinetics and pharmacodynamics of recombinant human angiotensin‐converting enzyme 2 in healthy human subjects. Clin Pharmacokinet. 2013;52:783‐792. [DOI] [PubMed] [Google Scholar]
- 64. Hemnes AR, Rathinasabapathy A, Austin EA, et al. A potential therapeutic role for angiotensin‐converting enzyme 2 in human pulmonary arterial hypertension. Eur Respir J. 2018;51:1702638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Shao Z, Shrestha K, Borowski AG, et al. Increasing serum soluble angiotensin‐converting enzyme 2 activity after intensive medical therapy is associated with better prognosis in acute decompensated heart failure. J Card Fail. 2013;19:605‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1 Representative photomicrographs of anti‐angiotensin converting enzyme 2 (ACE2) immunolabeling negative controls from a 10 year old male neutered French bulldog that was euthanized secondary to upper airway obstruction (Kidney, A, B; left ventricular myocardium, C), a 10 year old female spayed mixed breed dog euthanized for severe heart failure due to degenerative mitral valve disease (DMVD) (Kidney, D, E), and a 10 year old female spayed Chihuahua also euthanized for severe heart failure due to DMVD (LV myocardium, F). There is an absence of immunolabeling in all sections. 20x, A, D; 40X, B, C, E, F.
Supplemental Figure 2 Linearity of the 4‐methylcoumaryl‐7‐amide (MCA) standard curve used for measurement of angiotensin converting enzyme 2 activity.
Supplemental Figure 3 Fluorescent activity curves using different volumes of canine kidney supernatant fluid.
Supplemental Figure 4 Performance of the kit‐supplied angiotensin converting enzyme 2 (ACE2) inhibitor on total ACE2 activity with varying canine tissue samples. Columns show the mean percent inhibition +/− the SE of the measurement.
Supplemental Figure 5 Equilibrium concentrations of various individual angiotensin peptides (APs) at baseline and after incubation with recombinant human angiotensin converting enzyme 2 (rhACE2). Treatment of plasma from dogs with stage B2 or stage C heart disease decreased maladaptive APs such as angiotensin I (AT1 [1–10]) and angiotensin II (AT2 [1–8]) and increased cardioprotective APs such as angiotensin 1‐9 (Ang1‐9), angiotensin 1‐7 (Ang1‐7), and angiotensin 1‐5 (Ang1‐5). Data suggest that the balance between the beneficial and maladaptive APs can be made more favorable by rhACE2. See text for description of the modified clinical staging system.
Appendix S1: Supporting information