

Abstract
FBN1 encodes the gene for fibrillin-1, a structural macromolecule that polymerizes into microfibrils. Fibrillin microfibrils are morphologically distinctive fibrils, present in all connective tissues and assembled into tissue-specific architectural frameworks. FBN1 is the causative gene for Marfan syndrome, an inherited disorder of connective tissue whose major features include tall stature and arachnodactyly, ectopia lentis, and thoracic aortic aneurysm and dissection. More than one thousand individual mutations in FBN1 are associated with Marfan syndrome, making genotype-phenotype correlations difficult. Moreover, mutations in specific regions of FBN1 can result in the opposite features of short stature and brachydactyly characteristic of Weill-Marchesani syndrome and other acromelic dysplasias. How can mutations in one molecule result in disparate clinical syndromes? Current concepts of the fibrillinopathies require an appreciation of tissue-specific fibrillin microfibril microenvironments and the collaborative relationship between the structures of fibrillin microfibril networks and biological functions such as regulation of growth factor signaling.
1. Introduction
1.1 Fibrillin Microfibrils
FBN1 encodes the gene for fibrillin-1. In humans, there are three different genes (FBN1, FBN2, and FBN3) encoding fibrillins. Fibrillins are large (~350,000 MW) structural macromolecules that contribute to the integrity and function of all connective tissues. They are considered to be “structural macromolecules” because, like the collagens, the fibrillins form fibers that are visible in transmission electron micrographs. Unlike the collagens, fibrillins form “microfibrils” with uniform diameters (10–12 nm) that are not periodically cross-striated or “banded”. Fibrillin microfibrils display a characteristic morphology consisting of light and dark or hollow areas that give the appearance of railroad tracks. Fibrillin microfibrils exist as large bundles of microfibrils, as short individual microfibrils (usually in close proximity to basement membranes, for example on the endothelial cell side of the glomerular basement membrane), or as the peripheral microfibril mantle around elastin in all elastic fibers. Typical morphological features of fibrillin microfibrils are shown in Figure 1. In the various types of connective tissue, fibrillin microfibrils are organized to best suit the functional integrity of the tissue: for example, in skin, elastic fibers form a loose network of interconnecting highways; in the dermis, these highways run parallel to the epidermis with turn-offs coursing perpendicularly up from the deeper elastic fibers to the basement membrane at the dermal-epidermal junction, where bundles of microfibrils intersect the lamina densa; in tendons and perichondrium/periosteum, elastic fibers run parallel to the long axis; in muscular arteries, elastic fibers encircle the lumen.
Figure 1. Ultrastructure of Fibrillin Microfibrils.
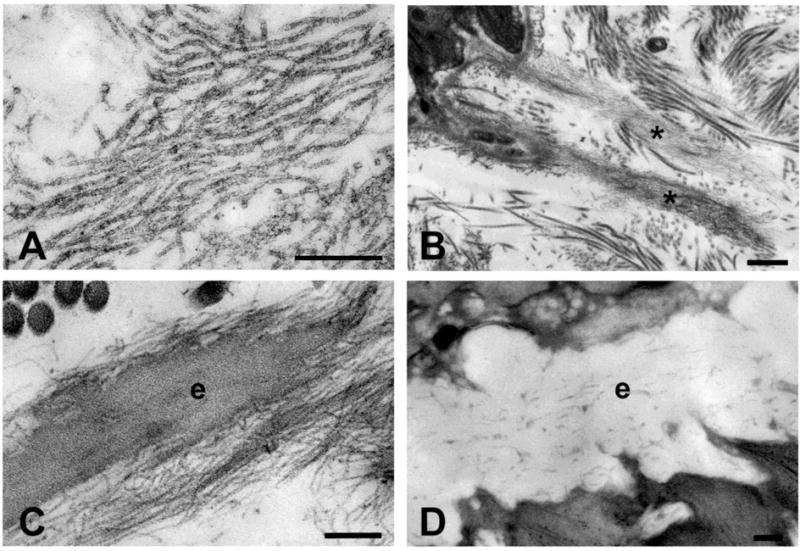
A. High magnification images of fibrillin microfibrils in human amnion show fibrils of uniform diameter with alternating hollow and filled (light and dark) regions. B. Fibrillin microfibrils exist in bundles (*), especially in close proximity to basement membranes. Here are two bundles of microfibrils intersecting the lamina densa at the dermal-epidermal junction in human skin. C. Fibrillin microfibrils surround amorphous elastin (e) in all elastic fibers. Shown here is an elastic fiber in human skin. D. In the aorta, elastic fibers are organized circumferentially in lamellae around the lumen of the vessel. Using high pressure freezing techniques, cell processes are seen directly adjacent to and even within the elastic fiber. In the mouse aorta, microfibrils are barely visible around the amorphous elastin. Scale bars = 200 nm (A,C,D); 500 nm (B).
Although “10 nm microfibrils” had been described as ultrastructural entities, the molecular components of these microfibrils were not known until 1986. Protocols to extract microfibrillar molecules used harsh denaturing conditions as well as disulfide bond reducing agents (1). Reductive guanidine extractions of fetal bovine nuchal ligament, an elastic fiber rich tissue, yielded a 31,000 MW glycoprotein, which was named MAGP (“microfibril associated glycoprotein”) (2). MAGP antiserum localized to elastin-associated microfibrils (2). Today, a number of additional molecules are known to be associated with microfibrils. These molecules have been both immunolocalized to microfibrils and shown to bind directly to fibrillin. These include the fibulins (3,4), the LTBPs (Latent TGFβ Binding Proteins) (5–7), and members of the Adamtslike (8–10) and Adamts (11) family of proteins.
Initially, it was thought that the main function of microfibrils is to serve as the scaffold for elastic fiber formation. This function was based on morphological studies of developing elastic tissues, which documented that microfibrils appeared first in the embryo, followed by the deposition of amorphous elastin onto the microfibril scaffold (12). Biochemical investigations as well as genetic evidence from both humans and mice have uncovered many more functions of fibrillin microfibrils. Today we know that fibrillin microfibrils perform important tissue-specific architectural functions, beyond serving as scaffolds for elastin deposition. For example, fibrillin microfibrils are specifically required for the structural integrity of both the aortic wall (which contains elastin) and the suspensory ligament of the lens (which does not contain elastin). In addition, over the last decade, a novel and highly significant function of fibrillin microfibrils has emerged: fibrillin microfibrils target and sequester members of the TGFβ superfamily of growth factors. Because this superfamily of growth factors includes more than 30 different members, this function diversifies the biological roles performed by fibrillin microfibrils, even though the microfibrils themselves are ubiquitous elements of all connective tissues. Using tissue-specific architectures, fibrillin microfibrils pattern the targeting and sequestration of a variety of growth factors and contribute to organ formation and repair. In this manner, the structures of fibrillin microfibrils collaborate with biological functions to shape and maintain connective tissues, and mutations in fibrillins exert powerful, even opposing, forces on tissue growth and homeostasis.
1.2 The Discovery of Fibrillin, the Major Structural Component of Microfibrils
In the early 1980’s, the era of protein discovery was in full swing. Monoclonal antibody technology, developed in 1975 by Kӧhler and Milstein (who shared the 1984 Nobel Prize), was employed to generate specific antibodies that yielded novel immunofluorescence staining patterns on human fetal membrane (amnion and chorion) and skin sections. In a large screen of antibody clones, two yielded patterns suggestive of the microfibrillar component of elastic fibers. These were selected and used to immunoprecipitate a large intra-chain disulfide bonded molecule from the medium of cultured human fibroblasts. The molecule was named “fibrillin” because electron microscopic immunolocalization experiments demonstrated periodic labeling along the lengths of 10 nm diameter microfibrils (13). Monomeric fibrillin molecules, purified from fibroblast medium, were shown to be flexible extended strings with lengths of 148 nm and diameters of 2.2 nm (Figure 2) (14).
Figure 2. Rotary-shadowed Images of Fibrillin and Extracted Microfibrils.
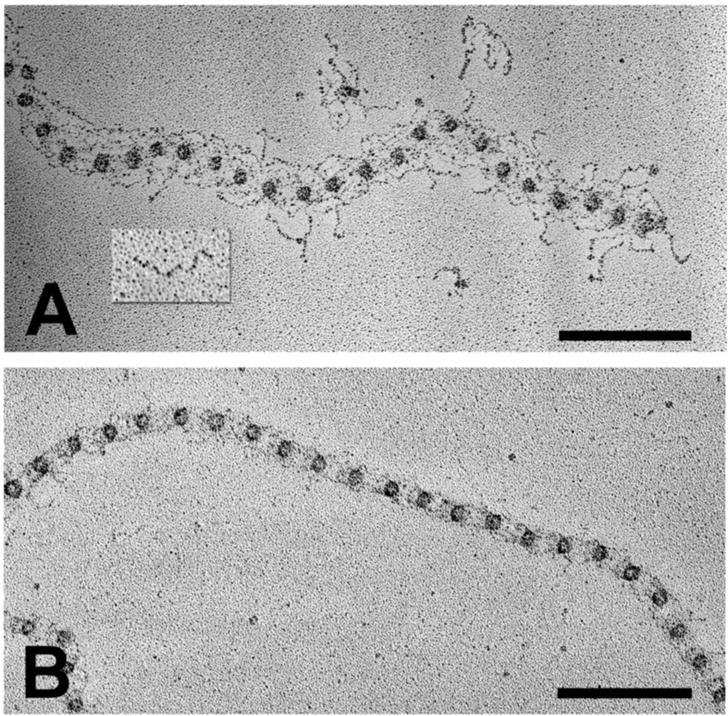
Fibrillin microfibrils were extracted from human fetal membranes with guanidine (A) or with collagenase (B). A fibrillin monomer isolated from the medium of fibroblast cultures is shown as an inset (A). Scale bars = 200 nm.
After rotary shadowing and electron microscopy, microfibrils extracted from tissues were visualized as “beaded strings” (Figure 2) (15). These images suggested that monomeric fibrillin molecules likely contribute to the many strings present in the beaded string structure. Based on epitope mapping studies, it was proposed that single molecules of fibrillin-1 are organized in a parallel, head-to-tail fashion within individual microfibrils (16). Eight fibrillin molecules were estimated in each individual microfibril (17).
Since monomeric fibrillin molecules assembled very quickly into disulfide-bonded aggregates (18), it is possible that fibrillin also contributes to the globular bead structure. However, other proteins, particularly small globular molecules like MAGP, might contribute to the bead. Immunolocalization of MAGP was shown to be periodic along the length of individual microfibrils, suggesting that it might also be a structural component of the microfibril (19). Extraction of connective tissue microfibrils with collagenase resulted in extended periods between beads (17). These extended lengths were sometimes greater than the length of single fibrillin molecules. Therefore, molecules in addition to fibrillins may be required to form the backbone structure of microfibrils. Alternatively, a model of staggered fibrillin molecules folding back on themselves was proposed to fit the ultrastructural images (17). However, collagenase cleaved fibrillin molecules at specific sites, and extraction of tissue microfibrils with guanidine yielded microfibrils with much shorter periods (20), suggesting that the long periods seen between beads in collagenase digested microfibrils were due to cleavage events rather than to unfolding or an unwinding of the microfibril structure. Together with additional antibody epitope mapping, a working model with fibrillin-1 molecules arranged head-to-tail and staggered in the beaded string microfibril was proposed (20). Figure 3A depicts this model.
Figure 3. Model of Fibrillin Molecules Within the Microfibril.

A. In this model (taken from reference 20), single fibrillin molecules span two bead lengths and are staggered. The N- and C- termini are marked for 4 molecules with colored domains (yellow=calcium-binding EGF-like domains; red=8-cysteine domains; blue=hybrid domains; green=generic EGF-like domains; purple=proline-rich domain). The dark green ovals represent the beads seen in beaded string microfibrils (Figure 2). B. Domains in which mutations cause Weill-Marchesani syndrome (WMS), geleophysic dysplasia (GD), acromicric dysplasia (AD), and Stiff Skin Syndrome (SSkS) are boxed. These domains are predicted to be close together within the microfibril and may form a special microenvironment. The region containing mutations leading to neonatal Marfan syndrome is also boxed. C. Pro-BMP complexes (BMP), large latent TGFβ complexes (LTBP), and perlecan with an associated pro-GDF-8 complex (myostatin) are shown bound to fibrillin.
1.3 The Fibrillin Gene Family
Fibrillin was first cloned from human placental cDNA libraries using a mixed pool of oligonucleotides representing all coding possibilities for a fibrillin peptide sequence (CEDIDEC) (21). Fibrillin peptide sequences were determined by amino acid sequencing of pepsin-resistant peptides extracted from human tissues and identified using fibrillin monoclonal antibodies (22). The complete deduced amino acid sequence revealed a modular domain structure consisting primarily of Epidermal Growth Factor (EGF) -like domains (with 6 cysteines per domain) and novel domains containing 8-cysteines (21,23). Each fibrillin molecule is composed of 47 EGF-like domains, 43 of which are predicted to bind calcium (cbEGF), 7 8-cysteine containing domains (8-cys), 2 “hybrid” domains that share features of both the 8-cysteine domain and the EGF-like domain, a proline-rich domain, and amino- and carboxyl- terminal domains (Figure 4).
Figure 4. Domain Structures of the Fibrillin-LTBP Family of Proteins.
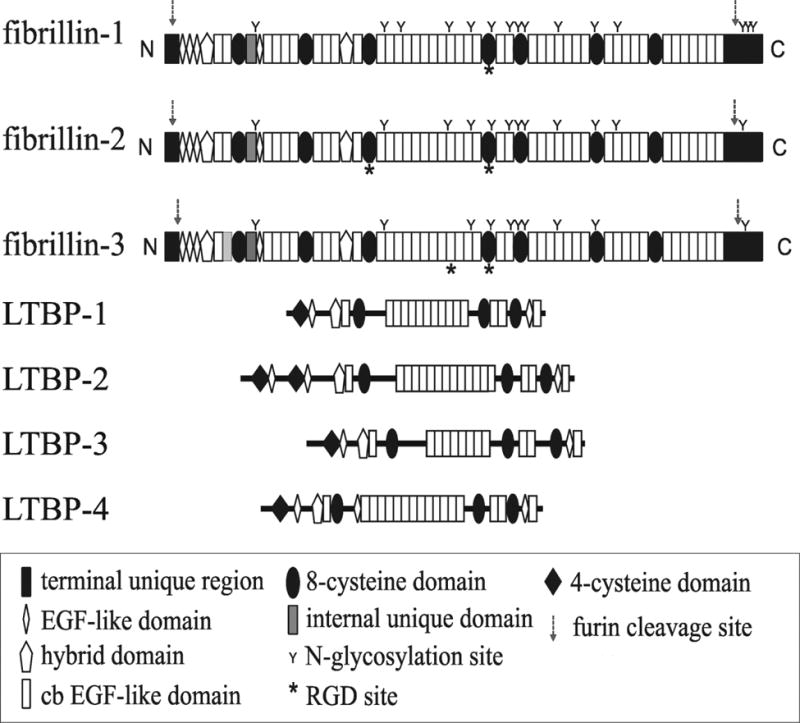
Fibrillins and LTBPs are composed of the same types of domains. The 8-cysteine domain is present only in this family of proteins. The fibrillins are the same size, whereas the LTBPs vary in size.
Cloning methods led to the discovery of a second fibrillin (24). It turned out that the monoclonal antibodies that were used to first characterize and clone fibrillin had identified fibrillin-1 (21,22). Expression of the gene for fibrillin-2 was largely limited to fetal development (25). Both fibrillin-1 and fibrillin-2 molecules were present within individual fetal microfibrils, indicating that fibrillin microfibrils are heteropolymers (26). Fibrillin-2 molecules were found in postnatal tissues but epitopes were masked by fibrillin-1 (27), suggesting that mature microfibril structure involves the accretion of fibrillin-1 molecules around a core of fibrillin-2 molecules that were assembled during fetal development. Mutations in FBN2 were discovered to cause congenital contractural arachnodactyly (now called Distal Arthrogryposis, Type 9). These mutations clustered between exons 23 and 34 (28).
A third fibrillin, fibrillin-3, was also found by cloning methods (29,30). Expression of FBN3 was also limited to fetal development, and fibrillin-3 was immunolocalized to fetal microfibrils (30). All three fibrillins share the same overall organization of modular domains (Figure 4). However, in place of the proline-rich domain in fibrillin-1, there is a glycine-rich domain in fibrillin-2 and a proline- and glycine-rich domain in fibrillin-3. Specific functions for these distinctive domains are not known. The second calcium-binding EGF-like domain is missing in fibrillin-3. In mouse, the gene for fibrillin-3 was inactivated during evolution.
Figure 4 depicts the molecular structures of the three fibrillins and the LTBPs, molecules structurally and functionally related to fibrillins. There are 4 LTBPs, and all four are composed of domain modules found in fibrillins. The 8-cysteine module is present only in the LTBPs and in the fibrillins. Interestingly, the 8-cysteine module in LTBPs is used to form covalent bonds with the small latent complex of TGFβ (31,32). Unlike the fibrillins, which are restricted to a very similar size, the LTBPs vary in size and are smaller than fibrillin. LTBPs have been immunolocalized to microfibrils (5,6), and LTBP-1, -2 and -4 bind directly to fibrillin (6,7,33). Periodic localization of LTBPs along the lengths of microfibrils has not been reported. Furthermore, because the LTBPs are smaller than fibrillins and variable in size, it seems unlikely that these molecules contribute in the same way as fibrillins to the backbone beaded string structure of microfibrils.
2. The Marfan Syndrome
The Marfan syndrome is a relatively common (1 case in every 3–5,000 people) dominantly inherited disorder of connective tissue with variable clinical features in the musculoskeletal, cardiovascular and ocular systems (34). Individuals with the Marfan syndrome are usually very tall with long limbs, long face, and long fingers and toes, hypomusculature, and chest, spine, hip, and foot deformities. A cardinal and potentially life-threatening aspect of Marfan syndrome is aortic root aneurysm with subsequent dissection and rupture. Other important clinical features include mitral valve prolapse, pneumothorax, dural ectasia, and myopia. Tissues affected in the Marfan syndrome (the aorta, musculoskeletal tissues, the dura, lung, and cornea) are fibrillin-rich.
Ectopia lentis is another cardinal characteristic of the Marfan syndrome, present in about 60% of patients. It is caused by a weakness of the ciliary zonule (the suspensory ligament of the lens). Immunolocalization of fibrillin to the ciliary zonule (13) prompted interest in fibrillin as the candidate gene for Marfan syndrome. Screening of skin biopsies and cultured fibroblasts demonstrated that fibrillin immunostaining patterns could distinguish Marfan samples from control samples (35), supporting the candidacy of fibrillin as the Marfan gene. At the same time, genetic mapping of the Marfan gene locus identified a region on chromosome 15 (36). Very quickly thereafter, once the gene for fibrillin-1 had been cloned (21), it was mapped to chromosome 15,q21.1 (37), and the first mutation in a patient with Marfan syndrome was identified (38). Today, identification of a mutation in FBN1 is not mandatory for the diagnosis of Marfan syndrome, but a mutation can be very useful in cases of incomplete phenotypes and for subsequent screening of family members.
2.1 FBN1 Mutations in Marfan syndrome
Since the identification of FBN1 as the causal gene for Marfan syndrome, 1847 different mutations in 3044 DNA samples have been reported, according to the currently most extensive FBN1 mutation database (http://www.umd.be/FBN1/- last updated 8/28/2014). Mutations are found throughout the entire length of the gene, and only 12% of all reported FBN1 mutations are recurrent (39). It is likely that the numbers of known mutations are actually much larger, since not all FBN1 mutations that have been identified worldwide have been reported or entered into this database. Penetrance of FBN1 mutations is extremely high, but age-dependent, and no example of nonpenetrance has been documented in hundreds of pedigrees (40). About 25% of cases are caused by new or spontaneous mutations arising during zygote formation. Gonadal mosaicism has been reported rarely in Marfan syndrome (41,42). Paternal age is advanced (36 versus 29 years) at the time of conception of isolated cases, consistent with the knowledge that a new mutation in a spermatagonium is a frequent cause of sporadic Marfan syndrome (43).
Mutations in the FBN1 gene include many different types (44). Missense mutations are most frequently encountered (about 2/3 of cases). These commonly substitute cysteine residues that form disulfide bonds within one of the cbEGF or 8-Cys domains, but missense mutations creating novel cysteine residues in these modules are also common. About one quarter of missense mutations affect modules other than cbEGF domains. Small insertions, deletions, or duplications represent around 10–15% of all reported mutations, the majority of which will create a premature termination codon (PTC). Another 10–15% of reported mutations consist of various classes of splicing errors, most commonly affecting canonical splice sequences at exon/intron boundaries. Many FBN1 splice site mutations result in in-frame exon skipping, such that the mutant fibrillin-1 lacks an entire cbEGF domain. Some exon-skipping mutations in FBN1 result in a frameshift and reduced mutant RNA levels caused by nonsense-mediated decay of the mutant transcript (45,46). Larger rearrangements, including both deletions and insertions, have been reported in a minority of Marfan patients (47–50); entire gene deletions are rare (51). In view of this large array of types of mutations found in FBN1, mutation screening of the FBN1 gene should not only include regular techniques for the detection of exonic mutations, but should also be supplemented by techniques for the detection of large deletions/insertions (for example, multiplex ligation-dependent probe amplification, MLPA) (52) and even for deep-intronic mutations (cDNA analysis, whole-genome sequencing) (53).
2.2 Genotype-Phenotype Correlations in Marfan Syndrome
Genotype-phenotype correlations have been explored, in order to investigate whether certain types of mutations in FBN1 are associated with specific clinical features of Marfan syndrome. These efforts have been made difficult by the known clinical variability in Marfan syndrome. Both inter-familial and intra-familial variability is well appreciated in Marfan syndrome, such that the established marked phenotypic variability among individuals carrying the same mutation tends to blur distinctions between different mutations (54). Other factors to explain the high clinical variability in Marfan syndrome include the level of expression of the normal FBN1 allele (55,56) and the role of hyperhomocysteinemia (57). It is likely that genetic modifiers, or epigenetic and environmental factors also play important roles in clinical variability in Marfan syndrome.
Table 1 provides an overview of the main genotype-phenotype correlations that have been reported in studies of large numbers of subjects. Case reports are not included in this list.
Table 1.
Genotype-phenotype Correlations in Marfan syndrome
| Type of Mutation | Phenotype | Comments | Reference |
|---|---|---|---|
| Cysteine substitutions in EGF-like domains | High incidence of ectopia lentis; severe early onset in exons 26–32 | 24 distinct mutations; 44 subjects | (58) |
| Premature termination codons (PTC) | Low incidence of ectopia lentis; high incidence of large joint hypermobility; high incidence of skin striae; higher incidence of aortic dissection? | 33 distinct mutations; 60 subjects | (59) |
| First 15 exons; arginine to cysteine mutations? | Predominant ectopia lentis | 11 subjects; younger EL subjects may develop further symptoms of Marfan syndrome | (60) |
| Full spectrum of mutation types | Cysteine mutations correlate strongly with ectopia lentis; PTC mutations are associated with severe skeletal and skin phenotypes; mutations in exons 24–32 are associated with severe disease | 803 mutations in 1,013 probands | (61) |
| 75% of mutations were in- frame; 25% resulted in PTCs; 33% of mutations occurred in exons 24–32; incidence of PTCs was smaller than in adult cohort | Lethal neonatal Marfan syndrome is a genuine clinical entity; clinical manifestations increase with age | 320 subjects <18 years old; 15% were diagnosed with neonatal Marfan syndrome | (62) |
| Full range of mutation types | “Incomplete” or mild Marfan syndrome was associated with mutations in exons 59–65; mutations at the ends (in exons 1–15 and 59–65) may be milder than mutations in between | 193 mutations in 503 subjects | (63) |
| Full range of mutation types | Truncating and splicing mutations were associated with aortic events | 179 probands with FBN1 mutations | (64) |
Ectopia lentis (EL) has been reported more frequently in cysteine missense mutations (58,61). In patients diagnosed by age 25, EL was present in 59% for those with a missense mutation involving a cysteine, compared with 32% for those with other missense mutations (61). In contrast, EL in patients diagnosed by age 25 was 23% for those with a mutation leading to a PTC, compared with 50% for those with inframe mutations (61). Isolated EL has been associated with mutations located in the first 15 exons of the gene (60,63). This latter association is however only significant if young patients are included, so it cannot be excluded that some of the young EL patients might develop additional features of Marfan syndrome in their adult life, necessitating reclassification out of the EL group. A recent review of all published cases over the last 20 years indicated that 57/123 (46.3%) should now be classified as Marfan, according to the revised Ghent nosology, since 37/96 mutations (38.5%) reported to cause isolated EL have also been found in patients with aortic dilation/dissection (65). Based on these findings, it was suggested that EL caused by mutations in FBN1 is part of a spectrum of Marfan syndrome, and the term “Isolated Ectopia Lentis” should be avoided in these cases (65).
A similar distribution, but at the 3′ end of the Fbn1 gene, was observed in patients presenting an “incomplete Marfan” phenotype, with a statistically significant clustering of their mutations in exons 59–65, whether or not patients under 18 were included (63). The “incomplete” Marfan group did not fulfill the criteria for a diagnosis of classic Marfan syndrome, suggesting that this group might represent a milder phenotype. However, it should be noted that the association of milder cardiovascular features with exons 59–65 was present, but not significant (63). Previously, it was suggested that mutations in this region was associated with mild phenotypes lacking significant aortic pathology (66), but numbers in this earlier study were small.
The most consistent genotype-phenotype correlation that has emerged is the association of mutations in the middle region of the gene (exons 24–32) with all cases of “neonatal” Marfan syndrome and with other severe forms of Marfan syndrome (58,61,62). Even if the mutation does not result in lethal neonatal Marfan syndrome, the presence of a mutation occurring in this region of the gene appears to be the best indicator of early onset aortic risk (61).
Recently, truncating and splicing mutations were found to occur more frequently in Marfan patients who have had aortic events (64). A higher frequency of truncating or splicing mutations (79%) were found in patients who had had aortic events than in those without an aortic event (39%), and those who had an aortic event before age 30 (100%) or before age 40 (95%) had truncating or splicing mutations (64). A trend toward higher frequency of truncating or splicing mutations was found in those who had dissections compared to those who had prophylactic surgery. The latter observation was also previously reported (59). Similarly, it was previously suggested that splicing mutations resulting in exon skipping of a cbEGF domain can be associated with particularly severe phenotypes, including lethal neonatal Marfan syndrome (4 of 11 cases) or with aortic involvement early in life (67).
Nonsense, frameshift, and splicing mutations were considerably underrepresented (27.8%) in exons 24–32, compared to all other exons (44.4%), and nonsense mutations were never reported in cases of neonatal Marfan syndrome (61). Therefore, the collective data indicate that a higher risk for an early aortic event is associated with mutations in exons 24–32 (61) and with truncating and splicing mutations (64). Nevertheless, in spite of these genotype-phenotype correlations, it is fair to say that we are not able to predict the phenotype for any given FBN1 mutation.
3. FBN1 Mutations in Other Disorders
3.1 Marfan-related Disorders
In addition to the interfamilial, as well as intrafamilial, clinical variability known in Marfan syndrome, there is extensive clinical variability in individuals harboring mutations in the FBN1 gene that do not give rise to Marfan syndrome. For the Marfan syndrome, the clinical spectrum ranges from mild (“incomplete” Marfan syndrome) to severe disease (lethal neonatal Marfan syndrome). For other disorders, mutations in FBN1 have been reported in patients with mild or isolated Marfan-related phenotypes including MASS phenotype (myopia, mitral valve prolapse, mild aortic dilatation, skeletal features, skin striae) (68), familial ectopia lentis, familial kyphoscoliosis (69), adolescent idiopathic scoliosis (70), and in familial thoracic aortic aneurysms (71–73).
Familial thoracic aortic aneurysms or heritable thoracic aortic disorders (H-TAD) can present with syndromic or isolated (nonsyndromic) clinical manifestations. The prototype for syndromic disease is the Marfan syndrome. Other forms of syndromic H-TAD include Loeys-Dietz syndrome (caused by mutations in the receptors for TGFβ, TGFBR1 and TGFBR2) and aneurysm-osteoarthritis syndrome (caused by mutations in SMAD3, an intracellular component of the TGFβ signaling pathway). Clinical diagnosis of these syndromes has been difficult due to overlapping manifestations. Gene panel screening of individuals with H-TAD demonstrates clinical overlap in patients diagnosed with Marfan syndrome: in a study of 175 patient samples, 19 patients clinically diagnosed with Marfan syndrome yielded 10 pathogenic mutations in FBN1 and 1 in TGFBR1 and variants of unknown significance in FBN2, MYH11, MLK, SMAD3, and COL3A1; 3 patients clinically diagnosed with Loeys-Dietz syndrome yielded 1 pathogenic mutation in FBN1 and variants of unknown significance in FBN1 and SMAD3 (73). Gene panel screening of 233 TAD patient samples, in which 19 patients were clinically diagnosed with Marfan syndrome but had no mutation in FBN1, SMAD3 mutations were revealed in 6 patients and 1 in TGFB2 (72).
These emerging data demonstrate the difficult clinical diagnosis of syndromic forms of H-TAD. Gene screening for nonsyndromic forms of TAD also demonstrate difficulties in clinical diagnosis. FBN1 mutations were revealed in 12 patients who were not clinically diagnosed with Marfan syndrome, but who had some Marfanoid features; some Marfanoid features were also present in TAD patients in whom SMAD3 and TGFB2 mutations were detected (72). Recently, the frequency of FBN1 mutations causing H-TAD was estimated to be about 3% (5/183) (74). None of these affected individuals fulfilled the clinical criteria of Marfan syndrome, however, two of the FBN1 mutations identified in these H-TAD patients had been previously reported in patients with Marfan syndrome.
3.2 Acromelic Dysplasias
Since clinical manifestations of Marfan syndrome are pleiotropic, it may not be surprising that mutations in FBN1 were found in nonsyndromic Marfan-related disorders. It was suprising, however, when mutations in FBN1 were found to cause acromelic dysplasias, which manifest short stature, short hands and feet, stiff joints, and a hypermuscular build. Acromelic dysplasias include Weill-Marchesani syndrome, geleophysic dysplasia, acromicric dysplasia, and Myhre syndrome (75). McKusick described Weill-Marchesani syndrome as the “opposite” of Marfan syndrome (76). Although ectopia lentis is common to both Marfan syndrome and Weill-Marchesani syndrome, individuals with Marfan syndrome normally present with tall stature, arachnodactyly, hypermobile joints and a thin hypomuscular build. Mutations in FBN1 were first discovered in autosomal dominant Weill-Marchesani (77,78) and then in geleophysic and acromicric dysplasias (79).
Mutations in FBN1 that underlie the acromelic dysplasias are predominantly limited to a hot spot in fibrillin-1. An in-frame deletion of 24 nucleotides in exon 41 causes Weill-Marchesani syndrome (77), and missense mutations in exons 41 and 42 cause geleophysic dysplasia and acromicric dysplasia (79). Exons 41 and 42 encode the 5th 8-cysteine domain in fibrillin-1. A second mutation causing Weill-Marchesani syndrome was found to be an in-frame deletion of exons 9–11, encoding the first 8-cysteine domain, the proline-rich region and the fourth EGF-like domain (78). While this mutation is distinct from the other acromelic dysplasia mutations which are all in the 5th 8-cysteine domain, it was proposed that the three-dimensional structure of fibrillin molecules within a microfibril may create a microenvironment consisting of these domains in close proximity (78). Figure 3B maps the proximity of domains in fibrillin-1 that cause acromelic dysplasias.
3.3 “Stiff Skin Syndrome”
Mutations in FBN1 causing “stiff skin syndrome” occur in the 4th 8-cysteine domain (80). This domain contains the single Arginine-Glycine-Aspartic Acid (RGD) sequence present in fibrillin-1. Loss of integrin binding to this site is implicated as the cause of stiff skin syndrome, since a mouse in which the RGD site is mutated to RGE develops stiff skin (81). The 4th 8-cysteine domain is predicted to be in close proximity to the microenvironment formed by the domains which contain the known mutations for the acromelic dysplasias (see Figure 3B). Interestingly, one of the clinical features of Weill-Marchesani syndrome and geleophysic dysplasia is thick skin. Therefore, it is possible that any of these mutations within this microenvironment may perturb integrin binding to fibrillin-1, leading to thick or very stiff skin.
3.4 Progeroid Fibrillinopathies
Mutations in FBN1 causing progeroid fibrillinopathies cluster in a region encoding the C-terminus of fibrillin-1 (82). Clinical manifestations resembled those in Marfan syndrome, but in addition, patients presented with a neonatal progeroid appearance. Five mutations involving nucleotide deletions or insertions and one missense mutation were described.
4. Mouse Models
There are multiple mouse models in which mutations have been generated in mouse Fbn1 by homologous recombination. These mouse models include a complete Fbn1 null, a homozygous hypomorph (mgR/mgR), several deletions (mgΔ/+; GT-8/+; WMΔ/+; H1Δ/+), and several missense mutations (C1039G/+; W1572C/+; D1545E/+). Table 2 summarizes the phenotypes of these mouse models.
Table 2.
Fbn1 Mutant Mouse Models
| Type of Mutation | Phenotypes | Comments | Reference1 |
|---|---|---|---|
| Fbn1 mgΔ | Homozygous mgΔ die in the early postnatal period; heterozygous mice are similar to mgR/+ and mgN/+ | In-frame deletion of exons 19–24; presence of Neo represses expression of the deletion | (83) |
| Fbn1 mgΔloxPneo | Heterozygous mice show Marfan phenotypes | By removing Neo, the deletion of exons 19–24 is fully expressed | (85) |
| Fbn1 mgR | Homozygous mgR die from aortic rupture during early adulthood; heterozygous mice appear normal | Presence of Neo represses expression of wildtype fibrillin-1 to around 20% of normal; homozygous mgR model severe aortic disease | (84) |
| Fbn1 mgN | Homozygous mgN die in the early postnatal period; heterozygous mgN appear normal | The complete null establishes early postnatal death around P14, phenocopied by homozygous mgΔ, C1039G, and GT-8; establishes haploinsufficent phenotypes | (88) |
| Fbn1 C1039G | Homozygous C1039G mice die in the early postnatal period; heterozygous mice show aortic dilatation but have normal life spans | A heterozygous missense mutation causes Marfan phenotypes in mice | (86) |
| Fbn1 GT-8 | Homozygous GT-8 mice die in the early postnatal period; heterozygous mice show aortic dilatation but have normal life spans | Cre-inversion of eGFP truncates fibrillin-1 after cbEGF18 (exon 32), tagging fibrillin-1 with eGFP (preserving the “neonatal region”); presence of GFP tag shows assembly of mutant fibrillin-1, followed by proteolysis of microfibrils | (87) |
| Fbn1 H1Δ | Homozygous and heterozygous H1Δ mice live long lives with no signs of aortic disease and no structural defects in microfibrils | In-frame deletion of exon 7, encoding the first hybrid domain | (87) |
| Fbn1 WMΔ | Homozygous and heterozygous WMΔ mice live long lives with no signs of aortic disease; thick skin and reduced bone growth; microfibrils are abnormally aggregated | In-frame deletion of exons 9–11, phenocopying a human family with autosomal dominant Weill-Marchesani syndrome | (78) |
| Fbn1 W1507C | Heterozygous W1507C mice show dermal fibrosis and loss of subcutaneous fat; homozygous W1507C show accelerated skin fibrosis but remain viable | This missense mutation phenocopies a human mutation causing Stiff Skin Syndrome | (81) |
| Fbn1 D1545E | Like heterozygous W1507C, heterozygous D1545E develop dermal fibrosis and loss of subcutaneous fat; homozygous D1545E are embryonic lethal | This missense mutation alters the RGD integrin binding site to RGE | (81) |
| Tight skin (Tsk) | Heterozygous tsk mice are a model for sclerodema; hets also display emphysema and myocardial hypertrophy; homozygous tsk die at E7–8 | The tsk mutation results in a large tandem duplication of fibrillin-1 (the region encoding cbEGF7- cbEGF24 is duplicated after cbEGF24) | (92) |
Only the original reference is cited.
It was known in 1991 that the Marfan gene is FBN1. Therefore, initial experiments were directed toward generating a mouse model of Marfan syndrome. The first attempt resulted in two hypomorphic mouse models, in which phenotypes were only revealed in homozygosity (mgΔ/mgΔ and mgR/mgR) (83,84). These mice were phenotypically normal in heterozygosity (mgΔ/+ and mgR/+). The deletion of exons 19–24, originally called mgΔ (83), was re-engineered in order to remove the neo gene which had caused decreased expression of Fbn1 in the original mgΔ and mgR mice. The re-engineered mgΔloxPneo mouse, in heterozygosity, displayed phenotypes of Marfan syndrome (85). Heterozygous C1039G/+ (86) and GT-8/+ (87) also demonstrated features of Marfan syndrome.
Homozygous C1039G and homozygous GT-8, like homozygous null mice (88), die in the early postnatal period. In humans, only three cases of compound heterozygosity at the FBN1 locus have been reported. In one case of compound heterozygosity (W217G/G2627R), the affected child had a severe phenotype leading to early death (89). Compound heterozygous probands (R2726W/C1928S in one family and R240H/c.3861delC in a second family) consistently presented with more severe phenotypes compared to their heterozygous family members (90). In these two families, the mutations R2726W and R240H are not considered pathogenic by themselves, but in combination with a second pathogenic mutation, these mutations were not neutral, suggesting the possibility that certain trans-located FBN1 mutations might act as intrafamilial modifiers of phenotype (90). A single allele of FBN1 with two mutations (I1071S and E1073D) was reported in an infant with neonatal Marfan syndrome (91). The homozygous mutations in mouse Fbn1 leading to early postnatal death are not equivalent to these compound mutations in humans. Although early death of homozygous Fbn1 mouse mutants have been suggested to model neonatal Marfan syndrome, clearly these homozygous mutants are not equivalent to the heterozygous FBN1 mutations leading to neonatal Marfan syndrome and early lethality in humans.
In contrast to the first Fbn1 mutant mouse models, which showed signs of Marfan syndrome and died in homozygosity, homozygous H1Δ (87) and homozygous WMΔ (78) lived long lives. This was somewhat surprising at the time. However, it is now clear in both mouse and human that not all fibrillin-1 mutations result in Marfan syndrome and not all homozygous mouse mutations lead to early death. The WMΔ Fbn1 mouse was generated in order to test whether the mutation found in a human family with Weill-Marchesani syndrome would display features of Weill-Marchesani syndrome or Marfan syndrome (78). The WMΔ Fbn1 mice, in both heterozygosity and homozygosity, replicated features (thick skin and short bones) of Weill-Marchesani syndrome and did not show any signs of aortic disease (78). Similarly, heterozygous W1570C/+ mice phenocopied stiff skin syndrome, and homozygous W1507C mice showed accelerated skin fibrosis but remained viable (81).
Heterozygous RGE mice (D1545E/+) also phenocopied stiff skin syndrome, but homozygous RGE mice were embryonic lethal before E10.5 (81). This finding suggests that, in homozygosity, RGE fibrillin-1 acts differently than W1570C fibrillin-1. RGE fibrillin-1 must significantly perturb important interactions with molecules in addition to integrins. Since the complete Fbn1 null mouse dies around P14 (88), loss of fibrillin-1 binding to integrins is compatible with life until that time. Mechanisms to explain the early embryonic lethality (a “gain of function” phenotype) of homozygous RGE mice are unknown. It is interesting that one allele of tight skin (Tsk) Fbn1 results in a scleroderma-like phenotype, but homozygous tsk mice also die during early embryonic development (92). In Tsk fibrillin-1, the RGD site is duplicated. Functional differences between W1570C, RGE, and tsk fibrillin-1 clearly exist, but the molecular bases for these differences are not understood.
5. Fibrillin, Growth Factors, and Genetic Pathways
The fibrillins and the LTBPs are composed of very similar domain modules. The 8-cysteine domain, which is used by LTBPs to bind covalently to the propeptide of TGFβs (31,32), is present in only 7 proteins in humans: three fibrillins and 4 LTBPs. Fibrillins do not bind to TGFβ propeptides (93). However, fibrillins do bind to propeptides of BMPs and GDFs. So far, biochemical studies have demonstrated interactions between fibrillins and the propeptides of BMP-2, -4, -5, -7 and GDF-5, but not the propeptide of GDF-8, which binds to a glycosaminoglycan side chain present on perlecan (94–96).
Extracellular regulation of TGFβ super-family members involves interactions between propeptides of growth factor complexes and fibrillin microfibril networks. Figure 3C shows binding sites in fibrillins for pro-BMP complexes, for LTBPs, and for perlecan/GDF-8. The functions of these interactions include targeting of growth factors to appropriate locations in specific tissues and sequestration of latent growth factor complexes. In addition, for TGFβ complexes, LTBPs facilitate the correct folding of TGFβ (functioning as intracellular chaperones) and perform roles in the activation of latent TGFβ (97). Less is known about the function of interactions between fibrillins and propeptides of BMPs and GDFs. Biochemical investigations of pro-BMP-7 (98) and comparison of the structures of pro-TGFβ and pro-BMP-9 (99) demonstrate that, unlike pro-TGFβ, pro-BMP-7 and -9 are not latent. Therefore, the functions of interactions between fibrillins and pro-BMPs may be somewhat different from the functions of LTBP interactions with pro-TGFβ. Recently, abnormal activation of BMP signaling was found in limb skeletal muscle in Fbn2 null mice, and loss of inhibition of BMP signaling normally conferred by interaction of fibrillin with pro-BMP was suggested as a potential mechanism for this abnormal activation (100). However, these and other studies of fibrillin or LTBP function in mice are complicated because of the overlapping functions of fibrillin-1 and -2 and of the LTBPs in vivo. Moreover, teasing out regulation of growth factor signaling from the structural functions of these microfibrillar proteins is not an easy task.
LTBPs bind directly to fibrillins, presumably targeting large latent TGFβ complexes to fibrillin microfibrils (6,7). Therefore, when TGFβ signaling was found to be abnormally activated in the lungs of Fbn1 mgΔ/mgΔ mice (101), it was hypothesized that loss of fibrillin-1 in this severely hypomorphic mouse model might result in a specific failure to target and sequester large latent TGFβ complexes, which would then lead to abnormally activated TGFβ signaling. Abnormal activation of TGFβ signaling was also observed in Fbn1 C1039G/+ aorta (102) and skeletal muscle (103). At the same time, mutations in a gene encoding a receptor for TGFβ (TGFBR2) were identified, and TGFBR2 was suggested to be a second locus for Marfan syndrome (104). Mutations in TGFBR1 and TGFBR2 were subsequently reclassified as a new syndrome, which was named Loeys-Dietz syndrome (105). Originally, TGFBR2 mutation constructs were shown to result in reduced TGFβ signaling in luciferase reporter assays (104). However, examination of cells and tissues from individuals with mutations in TGFβ receptors showed evidence of increased, rather than decreased, TGFβ signaling (105). All together, these results led to the concept that abnormally activated TGFβ signaling is the main driver of pathogenesis in Marfan syndrome and related disorders (106). Clinical trials of losartan, an angiotensin II type I receptor blocker, which also inhibited TGFβ signaling and rescued aortic disease in Fbn1 C1039G mice (102), were carried out in children (107) and adults (108) with Marfan syndrome. Results from multiple other trials are currently being reported.
Currently, the concept that abnormally activated TGFβ signaling causes Marfan syndrome and related disorders like Loeys-Dietz syndrome is not so clear. It is clear, however, that FBN1 and components of the TGFβ signaling pathway (TGFBR1 and TGFBR2 (104,105), SMAD3 (109), TGFB2 (110,111), and TGFB3 (112)) share similar genetic pathways. Thoracic aortic aneurysm and dissection can be the common result from a mutation in any of these genes. However, recent findings that mutations in the ligands, TGFβ-2 and TGFβ-3, are likely to be loss-of-function mutations, together with the original proposal that mutations in TGFBR2 were loss-of-function mutations (104), now suggest that the role of TGFβ signaling in H-TAD is “paradoxical” (111,112). The “paradox” is that loss-of-function mutations co-exist with evidence for abnormal activation of TGFβ signaling. This evidence consists primarily of immunocytochemical staining for phosphorylated Smad2/3 (102,105,111,112), a reporter for intracellular signaling initiated by TGFβ.
Proof, however, that abnormal activation of TGFβ signaling is the main driver of pathogenesis in Marfan syndrome did not come simply from reporters showing activation of intracellular TGFβ signaling pathways. Proof came from the rescue of aortic disease in Marfan mice after administration of TGFβ neutralizing antibodies (102). More recent studies in mice demonstrated that inhibiting TGFβ signaling by either TGFβ neutralizing antibodies (113) or by genetic ablation of Tgfbr2 (114) can cause severe aortic disease, consistent with mutations in TGFβ receptors and ligands causing H-TAD through loss-of-function mechanisms.
Instead of performing a primary role in pathogenesis, activated TGFβ signaling in Marfan syndrome is likely a compensatory response by aortic wall cells to the altered fibrillin extracellular matrix. TGFβ is a context-dependent “switch” that provides mechanisms whereby a cell can respond appropriately to its environment (115). As a switch, TGFβ can be either activating or inhibiting; stimulating or repressing; promoting or preventing, depending on the context. Biochemical and genetic studies establish a role for fibrillin in determining the extracellular context for TGFβ signaling.
Genetic studies implicate the structure of fibrillin microfibrils as an important determinant in clinical phenotypes. Fibrilllin-1 and fibrillin-2 perform roles in genetic pathways that control musculoskeletal phenotypes such as joint mobility, arachnodactyly, and muscularity. Mutations in FBN1 and or FBN2 result in arachnodactyly and hypomuscularity, features of Marfan syndrome and congenital contractural arachnodactyly. However, mutations in FBN1 usually cause joint hypermobility, while mutations in FBN2 are associated with contractures of the small and large joints. FBN1 mutations that cause Weill-Marchesani syndrome result in stiff joints. In Weill-Marchesani syndrome, unusual aggregates of fibrillin microfibrils were associated with thick skin (78). Similarly, aggregated masses of fibrillin microfibrils were found in skin samples from patients with stiff skin syndrome (80). These unusual structures of fibrillin microfibrils contrast with apparently normal microfibrils which undergo gradual degradation in skin from human or mouse Marfan syndrome (35,87). Proteolysis of fibrillin microfibrils in patients with thoracic aortic aneurysm and dissection is supported by the presence of circulating fibrillin-1 fragments in these individuals compared with controls (116). The effects of mutations in fibrillin-2 on the fine structure of fibrillin microfibrils are currently unknown.
An emerging genetic pathway involves FBN1 and genes that encode members of the Adamts and Adamtslike family of proteins. In addition to dominant Weill-Marchesani syndrome, caused by mutations in FBN1, recessive Weill-Marchesani syndrome is caused by mutations in ADAMTS10 (117). Clinical features of both the dominant and recessive forms are similar. Related acromelic dysplasias are associated with mutations in FBN1 (acromicric dysplasia and geleophysic dysplasia) (79) and in ADAMTSL2 (geleophysic dysplasia) (118). A Weill-Marchesani related syndrome is caused by mutations in ADAMTS17 (119), and mutations in ADAMTSL4 have been reported in autosomal recessive isolated ectopia lentis (120), a clinical feature common to Marfan syndrome and Weill-Marchesani syndrome.
Adamtslike proteins and Adamts enzymes have been shown to bind to fibrillin and to promote fibrillin fibril formation in vitro (8–11). Therefore, it might have been expected that mutations in the genes encoding these molecules would result in pathologies similar to the fibrillinopathies. However, this is a large family of molecules, and the fibrillinopathies are diverse. Genetic pathways provide invaluable information. In the cases known to date, mutations in ADAMTSL and ADAMTS genes result in phenotypes related primarily to the fibrillinopathy, Weill-Marchesani syndrome, rather than to Marfan syndrome. Mutations in specific members of the ADAMTSL and ADAMTS family and the resulting clinical features may be the combined results of effects on the fine structure of fibrillin microfibrils as well as tissue-specific gene expression. The fine structure of fibrillin microfibrils, as seen in Weill-Marchesani syndrome (78), may be common to the Weill-Marchesani-related disorders and may be the result of defects in Adamtslike-Adamts modifications on fibrillin fibril formation.
It is very interesting that Myhre syndrome, a syndrome related to the acromelic dysplasias, is caused by heterozygous mutations in SMAD4 (121). Smad-4 partners with intracellular signaling molecules (Smad-2, -3 or Smad-1, -5, -8) to transduce signals initiated by TGFβs or by BMPs. Therefore, the finding that mutations in SMAD4 cause a syndrome related to the acromelic dysplasias implicates growth factor signaling in these related disorders. However, it should also be noted that heterozygous mutations in SMAD4 cause juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome. Differences between these syndromes may be due to gain-of-function mutations associated with Myhre syndrome and loss-of-function mutations associated with juvenile polyposis/hereditary hemorrhagic telangiectasia syndrome.
6. Future Directions: Cellular Interactions with Fibrillin-1
Biochemical and genetic evidence demonstrate important roles for fibrillin-1.. In this review, we have underscored collaborative interactions between the architectural and biological functions of fibrillin-1. From this perspective of collaborative interactions, roles for the cell have not received sufficient attention. It is the cell that assembles fibrillin microfibrils during growth and development and that senses and responds to defects in the fibrillin microenvironment. Moreover, following a fundamental concept elucidated in work by Elizabeth D. Hay (122), the cell’s extracellular environment controls cell shape and gene expression. This concept has been recently used to provide a framework unifying the finding that mutations in both intracellular cytoskeletal molecules and extracellular molecules cause thoracic aortic aneurysm and dissection (123,124). The cellular mechanosensor, coordinating the extracellular environment with the intracellular cytoskeleton, and with TGFβ signaling, in the thoracic aorta is currently unknown. Whether mechanosensing is the job of one receptor or multiple receptors is also unknown. Here genetic evidence would be very informative. Presumably, when mutated, such mechanosensors would cause thoracic aortic disease.
An important role for integrin sensing of fibrillin-1 has been established in skin (80,81). Integrin sensing of fibrillin-1 has also been implicated in the heart, since dilated cardiomyopathy is the sole clinical feature resulting from combined haploinsufficiency of fibrillin-1 and β1 integrins (125). From these data, it seems likely that cellular sensors for fibrillin will vary depending on the tissue and that the cellular sensor for fibrillin-1 in the thoracic aorta may not be an integrin.
As suggested by Jurgen Engel (126), the fibrillin microfibril network may work like a machine to control growth factor signaling. In addition to the microfibrillar molecules, growth factors, inhibitors, and activators that are components of the microfibril network, cellular receptors may perform critical roles in the supramolecular assemblies that are required to make the machine work properly. In order to capture the dynamics of how the fibrillin microfibril machine works, new technological approaches to unraveling microfibril fine structure and microenvironments will be required.
Highlights.
FBN1 encodes fibrillin-1, a structural macromolecule for extracellular microfibrils.
Mutations in FBN1 cause the Marfan syndrome and related disorders.
Mutations in FBN1 also cause acromelic dysplasias and stiff skin syndrome.
Abnormal growth factor signaling is implicated in these fibrillinopathies.
Acknowledgments
This review and the corresponding Gene Wiki article are written as part of the Cardiac Gene Wiki Review series—a series resulting from a collaboration between the journal GENE, the Gene Wiki initiative, and the BD2K initiative. The Cardiac Gene Wiki Initiative is supported by National Institutes of Health (GM089820 and GM114833). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. The authors are especially grateful to the Shriners Hospital for Children for supporting investigations of fibrillin and fibrillinopathies for 32 years. We also acknowledge funding from the National Institutes of Health and from the Fund for Scientific Research, Flanders (Belgium).
List of Abbreviations
- FBN
fibrillin gene
- MAGP
microfibril associated glycoprotein
- LTBP
latent TGFβ binding protein
- TGFβ
transforming growth factor β
- TGFBR
TGFβ receptor gene
- EGF
epidermal growth factor
- cbEGF
calcium-binding EGF
- PTC
premature termination codon
- EL
ectopia lentis
- H-TAD
heritable thoracic aortic disorders
- BMP
bone morphogenetic protein
- GDF
growth and differentiation factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross R, Bornstein P. The elastic fiber. I. The separation and partial characterization of its macromolecular components. J Cell Biol. 1969;40:366–381. doi: 10.1083/jcb.40.2.366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gibson MA, Hughes JL, Fanning JC, Cleary EG. The major antigen of elastin-associated microfibrils is a 31-kDa glycoprotein. J Biol Chem. 1986;261:11429–11436. [PubMed] [Google Scholar]
- 3.Reinhardt DP, Sasaki T, Dzamba BJ, Keene DR, Chu ML, Gӧhring W, Timpl R, Sakai LY. Fibrillin-1 and fibulin-2 interact and are colocalized in some tissues. J Biol Chem. 1996;271:19489–19496. doi: 10.1074/jbc.271.32.19489. [DOI] [PubMed] [Google Scholar]
- 4.El-Hallous E, Sasaki T, Hubmacher D, Getie M, Tiedemann K, Brinckmann J, Bӓtge B, Davis EC, Reinhardt DP. Fibrillin-1 interactions with fibulins depend on the first hybrid domain and provide an adaptor function to tropoelastin. J Biol Chem. 2007;282:8935–8946. doi: 10.1074/jbc.M608204200. [DOI] [PubMed] [Google Scholar]
- 5.Dallas SL, Miyazono K, Skerry TM, Mundy GR, Bonewald LF. Dual role for the latent transforming growth factor-beta binding protein in storage of latent TGF-beta in the extracellular matrix and as a structural matrix protein. J Cell Biol. 1995;131:539–549. doi: 10.1083/jcb.131.2.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 7.Ono RN, Sengle G, Charbonneau NL, Carlberg V, Bӓchinger HP, Sasaki T, Lee-Arteaga S, Zilberberg L, Rifkin DB, Ramirez F, Chu ML, Sakai LY. Latent transforming growth factor beta-binding proteins and fibulins compete for fibrillin-1 and exhibit exquisite specificities in binding sites. J Biol Chem. 2009;284:16872–16881. doi: 10.1074/jbc.M809348200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsutsui K, Manabe R, Yamada T, Nakano I, Oguri Y, Keene DR, Sengle G, Sakai LY, Sekiguchi K. ADAMTSL-6 is a novel extracellular matrix protein that binds to fibrillin-1 and promotes fibrillin-1 fibril formation. J Biol Chem. 2010;285:4870–4882. doi: 10.1074/jbc.M109.076919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gabriel LA, Wang LW, Bader H, Ho JC, Majors AK, Hollyfield JG, Traboulsi EI, Apte SS. ADAMTSL4, a secreted glycoprotein widely distributed in the eye, binds fibrillin-1 microfibrils and accelerates microfibril biogenesis. Invest Ophthalmol Vis Sci. 2012;53:461–469. doi: 10.1167/iovs.10-5955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bader HL, Wang LW, Ho JC, Tran T, Holden P, Fitzgerald J, Atit RP, Reinhardt DP, Apte SS. A disintegrin-like and metalloprotease domain containing thrombospondin type 1 motif-like 5 (ADAMTSL5) is a novel fibrillin-1, fibrillin-2, and heparin-binding member of the ADAMTS superfamily containing a netrin-like module. Matrix Biol. 2012;31:398–411. doi: 10.1016/j.matbio.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kutz WE, Wang LW, Bader HL, Majors AK, Iwata K, Traboulsi EI, Sakai LY, Keene DR, Apte SS. ADAMTS10 protein interacts with fibrillin-1 and promotes its deposition in extracellular matrix of cultured fibroblasts. J Biol Chem. 2011;286:17156–17167. doi: 10.1074/jbc.M111.231571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fahrenbach WH, Sandberg LB, Cleary EG. Ultrastructural studies on early elastogenesis. Anat Rec. 1966;155:563–575. [Google Scholar]
- 13.Sakai LY, Keene DR, Engvall E. Fibrillin, a new 350-kD glycoprotein, is a component of extracellular microfibrils. J Cell Biol. 1986;103:2499–24509. doi: 10.1083/jcb.103.6.2499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sakai LY, Keene DR, Glanville RW, Bӓchinger HP. Purification and partial characterization of fibrillin, a cysteine-rich structural component of connective tissue microfibrils. J Biol Chem. 1991;266:14763–14770. [PubMed] [Google Scholar]
- 15.Keene DR, Maddox BK, Kuo HJ, Sakai LY, Glanville RW. Extraction of extendable beaded structures and their identification as fibrillin-containing extracellular matrix microfibrils. J Histochem Cytochem. 1991;39:441–449. doi: 10.1177/39.4.2005373. [DOI] [PubMed] [Google Scholar]
- 16.Reinhardt DP, Keene DR, Corson GM, Pӧschl E, Bӓchinger HP, Gambee JE, Sakai LY. Fibrillin-1: organization in microfibrils and structural properties. J Mol Biol. 1996;258:104–116. doi: 10.1006/jmbi.1996.0237. [DOI] [PubMed] [Google Scholar]
- 17.Baldock C, Koster AJ, Ziese U, Rock MJ, Sherratt MJ, Kadler KE, Shuttleworth CA, Kielty CM. The supramolecular organization of fibrillin-rich microfibrils. J Cell Biol. 2001;152:1045–1056. doi: 10.1083/jcb.152.5.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reinhardt DP, Gambee JE, Ono RN, Bӓchinger HP, Sakai LY. Initial steps in assembly of microfibrils. Formation of disulfide-cross-linked multimers containing fibrillin-1 J Biol Chem. 2000;275:2205–2210. doi: 10.1074/jbc.275.3.2205. [DOI] [PubMed] [Google Scholar]
- 19.Hanssen E, Hew FH, Moore E, Gibson MA. MAGP-2 has multiple binding regions on fibrillins and has a covalent periodic association with fibrillin-containing microfibrils. J Biol Chem. 2004;279:29185–29194. doi: 10.1074/jbc.M313672200. [DOI] [PubMed] [Google Scholar]
- 20.Kuo CL, Isogai Z, Keene DR, Hazeki N, Ono RN, Sengle G, Bӓchinger HP, Sakai LY. Effects of fibrillin-1 degradation on microfibril ultrastructure. J Biol Chem. 2007;282:4007–4020. doi: 10.1074/jbc.M606370200. [DOI] [PubMed] [Google Scholar]
- 21.Maslen CL, Corson GM, Maddox BK, Glanville RW, Sakai LY. Partial sequence of a candidate gene for the Marfan syndrome. Nature. 1991;352:334–337. doi: 10.1038/352334a0. [DOI] [PubMed] [Google Scholar]
- 22.Maddox BK, Sakai LY, Keene DR, Glanville RW. Connective tissue microfibrils. Isolation and characterization of three large pepsin-resistant domains of fibrillin J Biol Chem. 1989;264:21381–21385. [PubMed] [Google Scholar]
- 23.Corson GM, Chalberg SC, Dietz HC, Charbonneau NL, Sakai LY. Fibrillin binds calcium and is coded by cDNAs that reveal a multidomain structure and alternatively spliced exons at the 5′ end. Genomics. 1993;17:476–484. doi: 10.1006/geno.1993.1350. [DOI] [PubMed] [Google Scholar]
- 24.Lee B, Godfrey M, Vitale E, Hori H, Mattei MG, Sarfarazi M, Tsipouras P, Ramirez F, Hollister DW. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature. 1991;352:330–334. doi: 10.1038/352330a0. [DOI] [PubMed] [Google Scholar]
- 25.Zhang H, Apfelroth SD, Hu W, Davis EC, Sanguineti C, Bonadio J, Mecham RP, Ramirez F. Structure and expression of fibrillin-2, a novel microfibrillar component preferentially located in elastic matrices. J Cell Biol. 1994;124:855–863. doi: 10.1083/jcb.124.5.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Charbonneau NL, Dzamba BJ, Ono RN, Keene DR, Corson GM, Reinhardt DP, Sakai LY. Fibrillins can co-assemble in fibrils, but fibrillin fibril composition displays cell-specific differences. J Biol Chem. 2003;278:2740–2749. doi: 10.1074/jbc.M209201200. [DOI] [PubMed] [Google Scholar]
- 27.Charbonneau NL, Jordan CD, Keene DR, Lee-Arteaga S, Dietz HC, Rifkin DB, Ramirez F, Sakai LY. Microfibril structure masks fibrillin-2 in postnatal tissues. J Biol Chem. 2010;285:20242–20251. doi: 10.1074/jbc.M109.087031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gupta PA, Putnam EA, Carmical SG, Kaitila I, Steinmann B, Child A, Danesino C, Metcalfe K, Berry SA, Chen E, Delome CV, Thong MK, Adѐs LC, Milewicz DM. Ten novel FBN2 mutations in congenital contractural arachnodactyly: delineation of the molecular pathogenesis and clinical phenotype. Hum Mutat. 2002;19:39–48. doi: 10.1002/humu.10017. [DOI] [PubMed] [Google Scholar]
- 29.Nagase T, Nakayama M, Nakajima D, Kikuno R, Ohara O. Prediction of the coding sequences of unidentified human genes. XX. The complete sequences of 100 new cDNA clones from brain which code for large proteins in vitro. DNA Res. 2001;8:85–95. doi: 10.1093/dnares/8.2.85. [DOI] [PubMed] [Google Scholar]
- 30.Corson GM, Charbonneau NL, Keene DR, Sakai LY. Differential expression of fibrillin-3 adds to microfibril variety in human and avian, but not rodent, connective tissues. Genomics. 2004;83:461–472. doi: 10.1016/j.ygeno.2003.08.023. [DOI] [PubMed] [Google Scholar]
- 31.Saharinen J, Taipale J, Keski-Oja J. Association of the small latent transforming growth factor-beta with an eight cysteine repeat of its binding protein LTBP-1. EMBO J. 1996;15:245–253. [PMC free article] [PubMed] [Google Scholar]
- 32.Gleizes PE, Beavis RC, Mazzieri R, Shen B, Rifkin DB. Identification and characterization of an eight-cysteine repeat of the latent transforming growth factor-beta binding protein-1 that mediates bonding to the latent transforming growth factor-beta1. J Biol Chem. 1996;271:29891–29896. doi: 10.1074/jbc.271.47.29891. [DOI] [PubMed] [Google Scholar]
- 33.Hirani R, Hanssen E, Gibson MA. LTBP-2 specifically interacts with the amino-terminal region of fibrillin-1 and competes with LTBP-1 for binding to this microfibrillar protein. Matrix Biol. 2007;26:213–223. doi: 10.1016/j.matbio.2006.12.006. [DOI] [PubMed] [Google Scholar]
- 34.Keane MG, Pyeritz RE. Medical management of Marfan syndrome. Circulation. 2008;117:2802–2813. doi: 10.1161/CIRCULATIONAHA.107.693523. [DOI] [PubMed] [Google Scholar]
- 35.Hollister DW, Godfrey M, Sakai LY, Pyeritz RE. Immunohistologic abnormalities of the microfibrillar-fiber system in the Marfan syndrome. N Engl J Med. 1990;323:152–159. doi: 10.1056/NEJM199007193230303. [DOI] [PubMed] [Google Scholar]
- 36.Kainulainen K, Pulkkinen L, Savolainen A, Kaitila I, Peltonen L. Location on chromosome 15 of the gene defect causing Marfan syndrome. N Engl J Med. 1990;323:935–939. doi: 10.1056/NEJM199010043231402. [DOI] [PubMed] [Google Scholar]
- 37.Magenis RE, Maslen CL, Smith L, Allen L, Sakai LY. Localization of the fibrillin (FBN) gene to chromosome 15, band q21.1. Genomics. 1991;11:346–351. doi: 10.1016/0888-7543(91)90142-2. [DOI] [PubMed] [Google Scholar]
- 38.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, Stetten G, Meyers DA, Francomano CA. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337–339. doi: 10.1038/352337a0. [DOI] [PubMed] [Google Scholar]
- 39.Collod-Béroud G, Le Bourdelles S, Ades L, Ala-Kokko L, Booms P, Boxer M, Child A, Comeglio P, De Paepe A, Hyland JC, Holman K, Kaitila I, Loeys B, Matyas G, Nuytinck L, Peltonen L, Rantamaki T, Robinson P, Steinmann B, Junien C, Béroud C, Boileau C. Update of the UMD-FBN1 mutation database and creation of an FBN1 polymorphism database. Hum Mutat. 2003;22:199–208. doi: 10.1002/humu.10249. [DOI] [PubMed] [Google Scholar]
- 40.Pyeritz RE. The Marfan syndrome. Annu Rev Med. 2000;51:481–510. doi: 10.1146/annurev.med.51.1.481. [DOI] [PubMed] [Google Scholar]
- 41.Collod-Béroud G, Lackmy-Port-Lys M, Jondeau G, Mathieu M, Maingourd Y, Coulon M, Guillotel M, Junien C, Boileau C. Demonstration of the recurrence of Marfan-like skeletal and cardiovascular manifestations due to germline mosaicism for an FBN1 mutation. Am J Hum Genet. 1999;65:917–921. doi: 10.1086/302545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tekin M, Cengiz FB, Ayberkin E, Kendirli T, Fitoz S, Tutar E, Ciftçi E, Conba A. Familial neonatal Marfan syndrome due to parental mosaicism of a missense mutation in the FBN1 gene. Am J Med Genet A. 2007;143A:875–880. doi: 10.1002/ajmg.a.31660. [DOI] [PubMed] [Google Scholar]
- 43.Murdoch JL, Walker BA, McKusick VA. Parental age effects on the occurrence of new mutations for the Marfan syndrome. Ann Hum Genet. 1972;35:331–336. doi: 10.1111/j.1469-1809.1957.tb01406.x. [DOI] [PubMed] [Google Scholar]
- 44.Robinson PN, Arteaga-Solis E, Baldock C, Collod-Béroud G, Booms P, De Paepe A, Dietz HC, Guo G, Handford PA, Judge DP, Kielty CM, Loeys B, Milewicz DM, Ney A, Ramirez F, Reinhardt DP, Tiedemann K, Whiteman P, Godfrey M. The molecular genetics of Marfan syndrome and related disorders. J Med Genet. 2006;43:769–787. doi: 10.1136/jmg.2005.039669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo D, Tan FK, Cantu A, Plon SE, Milewicz DM. FBN1 exon 2 splicing error in a patient with Marfan syndrome. Am J Med Genet. 2001;101:130–134. doi: 10.1002/1096-8628(20010615)101:2<130::aid-ajmg1333>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 46.Frischmeyer PA, Dietz HC. Nonsense-mediated mRNA decay in health and disease. Hum Mol Genet. 1999;8:1893–1900. doi: 10.1093/hmg/8.10.1893. [DOI] [PubMed] [Google Scholar]
- 47.Blyth M, Foulds N, Turner C, Bunyan D. Severe Marfan syndrome due to FBN1 exon deletions. Am J Med Genet A. 2008;146A:1320–1324. doi: 10.1002/ajmg.a.32229. [DOI] [PubMed] [Google Scholar]
- 48.Liu W, Schrijver I, Brenn T, Furthmayr H, Francke U. Multi-exon deletions of the FBN1 gene in Marfan syndrome. BMC Med Genet. 2001;2:11. doi: 10.1186/1471-2350-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mátyás G, Alonso S, Patrignani A, Marti M, Arnold E, Magyar I, Henggeler C, Carrel T, Steinmann B, Berger W. Large genomic fibrillin-1 (FBN1) gene deletions provide evidence for true haploinsufficiency in Marfan syndrome. Hum Genet. 2007;122:23–32. doi: 10.1007/s00439-007-0371-x. [DOI] [PubMed] [Google Scholar]
- 50.Singh KK, Elligsen D, Liersch R, Schubert S, Pabst B, Arslan-Kirchner M, Schmidtke J. Multi-exon out of frame deletion of the FBN1 gene leading to a severe juvenile onset cardiovascular phenotype in Marfan syndrome. J Mol Cell Cardiol. 2007;42:352–356. doi: 10.1016/j.yjmcc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- 51.Hilhorst-Hofstee Y, Hamel BC, Verheij JB, Rijlaarsdam ME, Mancini GM, Cobben JM, Giroth C, Ruivenkamp CA, Hansson KB, Timmermans J, Moll HA, Breuning MH, Pals G. The clinical spectrum of complete FBN1 allele deletions. Eur J Hum Genet. 2011;19:247–252. doi: 10.1038/ejhg.2010.174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Furtado LV, Wooderchak-Donahue W, Rope AF, Yetman AT, Lewis T, Plant P, Bayrak-Toydemir P. Characterization of large genomic deletions in the FBN1 gene using multiplex ligation-dependent probe amplification. BMC Med Genet. 2011;12:119. doi: 10.1186/1471-2350-12-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gillis E, Kempers M, Salemink S, Timmermans J, Cheriex EC, Bekkers SC, Fransen E, De Die-Smulders CE, Loeys BL, Van Laer L. An FBN1 deep intronic mutation in a familial case of Marfan syndrome: an explanation for genetically unsolved cases? Hum Mutat. 2014;35:571–574. doi: 10.1002/humu.22540. [DOI] [PubMed] [Google Scholar]
- 54.De Backer J, Loeys B, Leroy B, Coucke P, Dietz H, De Paepe A. Utility of molecular analyses in the exploration of extreme intrafamilial variability in the Marfan syndrome. Clin Genet. 2007;72:188–198. doi: 10.1111/j.1399-0004.2007.00845.x. [DOI] [PubMed] [Google Scholar]
- 55.Hutchinson S, Furger A, Halliday D, Judge DP, Jefferson A, Dietz HC, Firth H, Handford PA. Allelic variation in normal human FBN1 expression in a family with Marfan syndrome: a potential modifier of phenotype? Hum Mol Genet. 2003;12:2269–2276. doi: 10.1093/hmg/ddg241. [DOI] [PubMed] [Google Scholar]
- 56.Aubart M, Gross MS, Hanna N, Zabot MT, Sznajder M, Detaint D, Gouya L, Jondeau G, Boileau C, Stheneur C. The clinical presentation of Marfan syndrome is modulated by expression of wild-type FBN1 allele. Hum Mol Genet. 2015;24:2764–2770. doi: 10.1093/hmg/ddv037. [DOI] [PubMed] [Google Scholar]
- 57.Giusti B, Marcucci R, Lapini I, Sestini I, Lenti M, Yacoub M, Pepe G. Role of hyperhomocysteinemia in aortic disease. Cell Mol Biol. 2004;50:945–952. [PubMed] [Google Scholar]
- 58.Schrijver I, Liu W, Brenn T, Furthmayr H, Francke U. Cysteine substitutions in epidermal growth factor-like domains of fibrillin-1: distinct effects on biochemical and clinical phenotype. Am J Hum Genet. 1999;65:1007–20. doi: 10.1086/302582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schrijver I, Liu W, Odom R, Brenn T, Oefner P, Furthmayr H, Francke U. Premature termination mutations in FBN1: distinct effects on differential allelic expression and on protein and clinical phenotypes. Am J Hum Genet. 2002;71:223–237. doi: 10.1086/341581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Comeglio P, Evans AL, Brice G, Cooling RJ, Child AH. Identification of FBN1 gene mutations in patients with ectopia lentis and marfanoid habitus. Br J Ophtalmol. 2002;86:1359–1362. doi: 10.1136/bjo.86.12.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Faivre L, Collod-Béroud G, Loeys BL, Child A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Marziliano N, Dietz HC, Halliday D, Béroud C, Bonithon-Kopp C, Claustres M, Muti C, Plauchu H, Robinson PN, Adѐs LC, Biggin A, Benetts B, Brett M, Holman KJ, De Backer J, Coucke P, Francke U, De Paepe A, Jondeau G, Boileau C. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454–466. doi: 10.1086/520125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Faivre L, Masurel-Paulet A, Collod-Béroud G, Callewaert BL, Child AH, Stheneur C, Binquet C, Gautier E, Chevallier B, Huet F, Loeys BL, Arbustini E, Mayer K, Arslan-Kirchner M, Kiotsekoglou A, Comeglio P, Grasso M, Halliday DJ, Béroud C, Bonithon-Kopp C, Claustres M, Robinson PN, Adѐs L, De Backer J, Coucke P, Francke U, De Paepe A, Boileau C, Jondeau G. Clinical and molecular study of 320 children with Marfan syndrome and related type I fibrillinopathies in a series of 1009 probands with pathogenic FBN1 mutations. Pediatrics. 2009;123:391–398. doi: 10.1542/peds.2008-0703. [DOI] [PubMed] [Google Scholar]
- 63.Comeglio P, Johnson P, Arno G, Brice G, Evans A, Aragon-Martin J, da Silva FP, Kiotsekoglou A, Child A. The importance of mutation detection in Marfan syndrome and Marfan-related disorders: report of 193 FBN1 mutations. Hum Mutat. 2007;28:928. doi: 10.1002/humu.9505. [DOI] [PubMed] [Google Scholar]
- 64.Baudhuin LM, Kotzer KE, Lagerstedt SA. Increased frequency of FBN1 truncating and splicing variants in Marfan syndrome patients with aortic events. Genet Med. 2015;17:177–187. doi: 10.1038/gim.2014.91. [DOI] [PubMed] [Google Scholar]
- 65.Chandra A, Patel D, Aragon-Martin JA, Pinard A, Collod-Béroud G, Comeglio P, Boileau C, Faivre L, Charteris D, Child AH, Arno G. The revised ghent nosology; reclassifying isolated ectopia lentis. Clin Genet. 2015;87:284–287. doi: 10.1111/cge.12358. [DOI] [PubMed] [Google Scholar]
- 66.Palz M, Tiecke F, Booms P, Gӧldner B, Rosenberg T, Fuchs J, Skovby F, Schumacher H, Kaufmann UC, von Kodolitsch Y, Nienaber CA, Leitner C, Katzke S, Vetter B, Hagemeier C, Robinson PN. Clustering of mutations associated with mild Marfan-like phenotypes in the 3′ region of FBN1 suggests a potential genotype-phenotype correlation. Am J Med Genet. 2000;91:212–221. doi: 10.1002/(sici)1096-8628(20000320)91:3<212::aid-ajmg12>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 67.Liu W, Qian C, Comeau K, Brenn T, Furthmayr H, Francke U. Hum. Mol Genet. 1996;5:1581–1587. doi: 10.1093/hmg/5.10.1581. [DOI] [PubMed] [Google Scholar]
- 68.Dietz HC, Pyeritz RE. Mutations in the human gene for fibrillin-1 (FBN1) in the Marfan syndrome and related disorders. Hum Mol Genet. 1995;4:1799–1809. doi: 10.1093/hmg/4.suppl_1.1799. [DOI] [PubMed] [Google Scholar]
- 69.Adѐs LC, Sreetharan D, Onikul E, Stockton V, Watson KC, Holman KJ. Segregation of a novel FBN1 gene mutation, G1796E, with kyphoscoliosis and radiographic evidence of vertebral dysplasia in three generations. Am J Med Genet. 2002;109:261–270. doi: 10.1002/ajmg.10333. [DOI] [PubMed] [Google Scholar]
- 70.Buchan JG, Alvarado DM, Haller GE, Cruchaga C, Harms MB, Zhang T, Willing MC, Grange DK, Braverman AC, Miller NH, Morcuende JA, Tang NL, Lam TP, Ng BK, Cheng JC, Dobbs MB, Gurnett CA. Rare variants in FBN1 and FBN2 are associated with severe adolescent idiopathic scoliosis. Hum Mol Genet. 2014;23:5271–5282. doi: 10.1093/hmg/ddu224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Francke U, Berg MA, Tynan K, Brenn T, Liu W, Aoyama T, Gasner C, Miller DC, Furthmayr H. A Gly1127Ser mutation in an EGF-like domain of the fibrillin-1 gene is a risk factor for ascending aortic aneurysm and dissection. Am J Hum Genet. 1995;56:1287–1296. [PMC free article] [PubMed] [Google Scholar]
- 72.Campens L, Callewaert B, Muiño Mosquera L, Renard M, Symoens S, De Paepe A, Coucke P, De Backer J. Gene panel sequencing in heritable thoracic aortic disorders and related entities—results of comprehensive testing in a cohort of 264 patients. Orphanet J Rare Dis. 2015;10:9. doi: 10.1186/s13023-014-0221-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wooderchak-Donahue W, VanSant-Webb C, Tvrdik T, Plant P, Lewis T, Stocks J, Raney JA, Meyers L, Berg A, Rope AF, Yetman AT, Bleyl SB, Mesley R, Bull DA, Collins RT, Ojeda MM, Roberts A, Lacro R, Woerner A, Stoler J, Bayrak-Toydemir P. Clinical utility of a next generation sequencing panel assay for Marfan and Marfan-like syndromes featuring aortopathy. Am J Med Genet A. 2015;167A:1747–1757. doi: 10.1002/ajmg.a.37085. [DOI] [PubMed] [Google Scholar]
- 74.Regalado ES, Guo DC, Santos-Cortez RL, Hostetler E, Bensend TA, Pannu H, Estrera A, Safi H, Mitchell AL, Evans JP, Leal SM, Bamshad M, Shendure J, Nickerson DA, University of Washington for Center for Mendelian Genomics. Milewicz DM. Pathogenic FBN1 variants in familial thoracic aortic aneurysms and dissections. Clin Genet. 2015 Dec 1; doi: 10.1111/cge.12702. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Le Goff C, Cormier-Daire V. From tall to short: the role of TGFβ signaling in growth and its disorders. Am J Med Genet C Semin Med Genet. 2012;160C:145–153. doi: 10.1002/ajmg.c.31337. [DOI] [PubMed] [Google Scholar]
- 76.McKusick VA. The Weill-Marchesani syndrome. In: McKusick VA, editor. Heritable disorders of connective tissue. 4th. St. Louis, MO: CV Mosby Company; 1972. pp. 282–291. [Google Scholar]
- 77.Faivre L, Gorlin RJ, Wirtz MK, Godfrey M, Dagoneau N, Samples JR, Le Merrer M, Collod-Bѐroud G, Boileau C, Munnich A, Cormier-Daire V. In frame fibrillin-1 gene deletion in autosomal dominant Weill-Marchesani syndrome. J Med Genet. 2003;40:34–36. doi: 10.1136/jmg.40.1.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sengle G, Tsutsui K, Keene DR, Tufa SF, Carlson EJ, Charbonneau NL, Ono RN, Sasaki T, Wirtz MK, Samples JR, Fessler LI, Fessler JH, Sekiguchi K, Hayflick SJ, Sakai LY. Microenvironmental regulation by fibrillin-1. PLoS Genet. 2012;8:e1002425. doi: 10.1371/journal.pgen.1002425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Le Goff C, Mahaut C, Wang LW, Allali S, Abhyankar A, Jensen S, Zylberberg L, Collod-Bѐroud G, Bonnet D, Alanay Y, Brady AF, Cordier MP, Devriendt K, Genevieve D, Kiper PO, Kitoh H, Krakow D, Lynch SA, Le Merrer M, Mégarbane A, Mortier G, Odent S, Polak M, Rohrbach M, Sillence D, Stolte-Dijkstra I, Superti-Furga A, Rimoin DL, Topouchian V, Unger S, Zabel B, Bole-Feysot C, Nitschke P, Handford P, Casanova JL, Boileau C, Apte SS, Munnich A, Cormier-Daire V. Mutations in the TGFβ binding-protein-like domain 5 of FBN1 are responsible for acromicric and geleophysic dysplasias. Am J Hum Genet. 2011;89:7–14. doi: 10.1016/j.ajhg.2011.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Loeys BL, Gerber EE, Riegert-Johnson D, Iqbal S, Whiteman P, McConnell V, Chillakuri CR, Macaya D, Coucke PJ, De Paepe A, Judge DP, Wigley F, Davis EC, Mardon HJ, Handford P, Keene DR, Sakai LY, Dietz HC. Mutations in fibrillin-1 cause congenital scleroderma: stiff skin syndrome. Sci Transl Med. 2010;2:23ra20. doi: 10.1126/scitranslmed.3000488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gerber EE, Gallo EM, Fontana SC, Davis EC, Wigley FM, Huso DL, Dietz HC. Integrin-modulating therapy prevents fibrosis and autoimmunity in mouse models of scleroderma. Nature. 2013;503:126–130. doi: 10.1038/nature12614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Garg A, Xing C. De novo heterozygous FBN1 mutations in the extreme C-terminal region cause progeroid fibrillinopathy. Am J Med Genet A. 2014;164A:1341–1345. doi: 10.1002/ajmg.a.36449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Pereira L, Andrikopoulos K, Tian J, Lee SY, Keene DR, Ono R, Reinhardt DP, Sakai LY, Biery NJ, Bunton T, Dietz HC, Ramirez F. Targetting of the gene encoding fibrillin-1 recapitulates the vascular aspect of Marfan syndrome. Nat Genet. 1997;17:218–222. doi: 10.1038/ng1097-218. [DOI] [PubMed] [Google Scholar]
- 84.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Lima BL, Santos EJ, Fernandes GR, Merkel C, Mello MR, Gomes JP, Soukoyan M, Kerkis A, Massironi SM, Visintin JA, Pereira LV. A new mouse model for marfan syndrome presents phenotypic variability associated with the genetic background and overall levels of Fbn1 expression. PLoS One. 2010;5:e14136. doi: 10.1371/journal.pone.0014136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114:172–181. doi: 10.1172/JCI20641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Charbonneau NL, Carlson EJ, Tufa S, Sengle G, Manalo EC, Carlberg VM, Ramirez F, Keene DR, Sakai LY. In vivo studies of mutant fibrillin-1 microfibrils. J Biol Chem. 2010;285:24943–24955. doi: 10.1074/jbc.M110.130021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Carta L, Pereira L, Arteaga-Solis E, Lee-Arteaga SY, Lenart B, Starcher B, Merkel CA, Sukoyan M, Kerkis A, Hazeki N, Keene DR, Sakai LY, Ramirez F. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281:8016–8023. doi: 10.1074/jbc.M511599200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Karttunen L, Raghunath M, Lӧnnqvist L, Peltonen L. A compound-heterozygous Marfan patient: two defective fibrillin alleles result in a lethal phenotype. Am J Hum Genet. 1994:1083–1091. [PMC free article] [PubMed] [Google Scholar]
- 90.Van Dijk FS, Hamel BC, Hilhorst-Hofstee Y, Mulder BJ, Timmermans J, Pals G, Cobben JM. Compound-heterozygous Marfan syndrome. Eur J Med Genet. 2009;52:1–5. doi: 10.1016/j.ejmg.2008.11.004. [DOI] [PubMed] [Google Scholar]
- 91.Wang M, Kishnani P, Decker-Phillips M, Kahler SG, Chen YT, Godfrey M. Double mutant fibrillin-1 (FBN1) allele in a patient with neonatal Marfan syndrome. J Med Genet. 1996;33:760–763. doi: 10.1136/jmg.33.9.760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Siracusa LD, McGrath R, Ma Q, Moskow JJ, Manne J, Christner PJ, Buchberg AM, Jimenez SA. A tandem duplication within the fibrillin-1 gene is associated with the mouse tight skin mutation. Genome Res. 1996;6:300–313. doi: 10.1101/gr.6.4.300. [DOI] [PubMed] [Google Scholar]
- 93.Saharinen J, Keski-Oja J. Specific sequence motif of 8-Cys repeats of TGF-beta binding proteins, LTBPs, creates a hydrophobic interaction surface for binding of small latent TGF-beta. Mol Biol Cell. 2000;11:2691–2704. doi: 10.1091/mbc.11.8.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gregory KE, Ono RN, Charbonneau NL, Kuo CL, Keene DR, Bӓchinger HP, Sakai LY. The prodomain of BMP-7 targets the BMP-7 complex to the extracellular matrix. J Biol Chem. 2005;280:27970–27980. doi: 10.1074/jbc.M504270200. [DOI] [PubMed] [Google Scholar]
- 95.Sengle G, Charbonneau NL, Ono RN, Sasaki T, Alvarez J, Keene DR, Bӓchinger HP, Sakai LY. Targeting of bone morphogenetic protein growth factor complexes to fibrillin. J Biol Chem. 2008;283:13874–13888. doi: 10.1074/jbc.M707820200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Sengle G, Ono RN, Sasaki T, Sakai LY. Prodomains of transforming growth factor beta (TGFbeta) superfamily members specify different functions: extracellular matrix interactions and growth factor bioavailability. J Biol Chem. 2011;286:5087–5099. doi: 10.1074/jbc.M110.188615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rifkin DB. Latent transforming growth factor-beta (TGF-beta) binding proteins: orchestrators of TGF-beta availability. J Biol Chem. 2005;280:7409–7412. doi: 10.1074/jbc.R400029200. [DOI] [PubMed] [Google Scholar]
- 98.Sengle G, Ono RN, Lyons KM, Bӓchinger HP, Sakai LY. A new model for growth factor activation: type II receptors compete with the prodomain for BMP-7. J Mol Biol. 2008;381:1025–1039. doi: 10.1016/j.jmb.2008.06.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mi LZ, Brown CT, Gao Y, Tian Y, Le VQ, Walz T, Springer TA. Structure of bone morphogenetic protein 9 procomplex. Proc Natl Acad Sci USA. 2015;112:3710–3715. doi: 10.1073/pnas.1501303112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sengle G, Carlberg V, Tufa SF, Charbonneau NL, Smaldone S, Carlson EJ, Ramirez F, Keene DR, Sakai LY. Abnormal activation of BMP signaling causes myopathy in Fbn2 null mice. PLoS Genet. 2015;11:e1005340. doi: 10.1371/journal.pgen.1005340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 102.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT-1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Cohn RD, van Erp C, Habashi JP, Soleimani AA, Klein EC, Lisi MT, Gamradt M, ap Rhys CM, Holm TM, Loeys BL, Ramirez F, Judge DP, Ward CW, Dietz HC. Angiotensin II type 1 receptor blockade attenuates TGF-beta- induced failure of muscle regeneration in multiple myopathic states. Nat Med. 2007;13:204–210. doi: 10.1038/nm1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mizuguchi T, Collod-Béroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M, Morisaki H, Ihara M, Kinoshita A, Yoshiura K, Junien C, Kajii T, Jondeau G, Ohta T, Kishino T, Furukawa Y, Nakamura Y, Niikawa N, Boileau C, Matsumoto N. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 106.Doyle JJ, Gerber EE, Dietz HC. Matrix-dependent perturbation of TGFβ signaling and disease. FEBS Lett. 2012;586:2003–2015. doi: 10.1016/j.febslet.2012.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Lacro RV, Dietz HC, Sleeper LA, Yetman AT, Bradley TJ, Colan SD, Pearson GD, Selamet Tierney ES, Levine JC, Atz AM, Benson DW, Braverman AC, Chen S, De Backer J, Gelb BD, Grossfeld PD, Klein GL, Lai WW, Liou A, Loeys BL, Markham LW, Olson AK, Paridon SM, Pemberton VL, Pierpont ME, Pyeritz RE, Radojewski E, Roman MJ, Sharkey AM, Stylianou MP, Wechsler SB, Young LT, Mahony L, Pediatric Heart Network Investigators Atenolol versus losartan in children and young adults with Marfan’s syndrome. N Engl J Med. 2014;371:2061–2071. doi: 10.1056/NEJMoa1404731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Milleron O, Arnoult F, Ropers J, Aegerter P, Detaint D, Delorme G, Attias D, Tubach F, Dupuis-Girod S, Plauchu H, Barthelet M, Sassolas F, Pangaud N, Naudion S, Thomas-Chabaneix J, Dulac Y, Edouard T, Wolf JE, Faivre L, Odent S, Basquin A, Habib G, Collignon P, Boileau C, Jondeau G. Marfan sartan: a randomized, double-blind placebo-controlled trial. Eur Hear J. 2015;36:2160–2166. doi: 10.1093/eurheartj/ehv151. [DOI] [PubMed] [Google Scholar]
- 109.van de Laar IM, Odenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, Hoedemaekers YM, Willemsen R, Severijnen LA, Venselaar H, Vriend G, Pattynama PM, Collée M, Majoor-Krakauer D, Poldermans D, Frohn-Mulder IM, Micha D, Timmermans J, Hilhorst-Hofstee Y, Bierma-Zeinstra SM, Willems PJ, Kros JM, Oei EH, Oostra BA, Wessels MW, Bertoli-Avella AM. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43:121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 110.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, Varret M, Prakash SK, Li AH, d’Indy H, Braverman AC, Grandchamp B, Kwartier CS, Guoya L, Santos-Cortez RL, Abifadel M, Leal SM, Muti C, Shendure J, Gross MS, Rieder MJ, Vahanian A, Nickerson DA, Michel JB, Lung, and Blood Institute (NHLBI) Go Exome Sequencing Project. Jondeau G, Milewicz DM. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44:916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, Kempers MJ, Fishman EK, Chen Y, Myers L, Bjeda D, Oswald G, Elias AF, Levy HP, Anderlid BM, Yang MH, Bongers EM, Timmermans J, Braverman AC, Canham N, Mortier GR, Brunner HG, Byers PH, Van Eyk J, Van Laer L, Dietz HC, Loeys BL. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44:922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JM, de Graaf BM, van de Beek G, Gallo E, Kruithof BP, Venselaar H, Myers LA, Laga S, Doyle AJ, Oswald G, van Cappellen GW, Yamanaka I, van der Helm RM, Beverloo B, de Klein A, Pardo L, Lammens M, Evers C, Devriendt K, Dumoulein M, Timmermans J, Bruggenwirth HT, Verheijen F, Rodrigus I, Baynam G, Kempers M, Saenen J, Van Craenenbroeck EM, Minatoya K, Matsukawa R, Tsukube T, Kubo N, Hofstra R, Goumans MJ, Bekkers JA, Roos-Hesselink JW, van de Laar IM, Dietz HC, Van Laer L, Morisaki T, Wessels MW, Loeys BL. Mutations in a TGF-β ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65:1324–1336. doi: 10.1016/j.jacc.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor-β signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35:911–917. doi: 10.1161/ATVBAHA.114.305150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–767. doi: 10.1172/JCI69942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sporn MB. The early history of TGF-beta, and a brief glimpse of its future. Cytokine Growth Factor Rev. 2006;17:3–7. doi: 10.1016/j.cytogfr.2005.09.012. [DOI] [PubMed] [Google Scholar]
- 116.Marshall LM, Carlson EJ, O’Malley J, Snyder CK, Charbonneau NL, Hayflick SJ, Coselli JS, Lemaire SA, Sakai LY. Thoracic aortic aneurysm frequency and dissection are associated with fibrillin-1 fragment concentrations in circulation. Circ Res. 2013;113:1159–1168. doi: 10.1161/CIRCRESAHA.113.301498. [DOI] [PubMed] [Google Scholar]
- 117.Dagoneau N, Benoist-Lasselin C, Huber C, Faivre L, Mégarbané A, Alswaid A, Dolifus H, Alembik Y, Munnich A, Leqeai-Mallet L, Cormier-Daire V. ADAMTS10 mutations in autosomal recessive Weill-Marchesani syndrome. Am J Hum Genet. 2004;75:801–806. doi: 10.1086/425231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Le Goff C, Morice-Picard F, Dagoneau N, Wang LW, Perrot C, Crow YJ, Bauer F, Flori E, Prost-Squarcioni C, Krakow D, Ge G, Greenspan DS, Bonnet D, Le Merrer M, Munnich A, Apte SS, Cormier-Daire V. ADAMTSL2 mutations in geleophysic dysplasia demonstrate a role for ADAMTS-like proteins in TGF-beta bioavailability regulation. Nat Genet. 2008;40:1119–1123. doi: 10.1038/ng.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Morales J, Al-Sharif L, Khalil DS, Shinwari JM, Bavi P, Al-Mahrougi RA, Al-Rajhi A, Alkuraya FS, Meyer BF, Al Tassan N. Homozygous mutations in ADAMTS10 and ADAMTS17 cause lenticular myopia, ectopia lentis, glaucoma, spherophakia, and short stature. Am J Hum Genet. 2009;85:558–568. doi: 10.1016/j.ajhg.2009.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ahram D, Sato TS, Kohilan A, Tayeh M, Chen S, Leal S, Al-Salem M, El-Shanti H. A homozygous mutation in ADAMTSL4 causes autosomal-recessive isolated ectopia lentis. Am J Hum Genet. 2009;84:274–278. doi: 10.1016/j.ajhg.2009.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Le Goff C, Mahaut C, Abhyankar A, Le Goff W, Serre V, Afenjar A, Destrée A, di Rocco M, Héron D, Jacquemont S, Marlin S, Simon M, Tolmie J, Verloes A, Casanova JL, Munnich A, Cormier-Daire V. Mutations at a single codon in Mad homology 2 domain of SMAD4 cause Myhre syndrome. Nat Genet. 2011;44:85–88. doi: 10.1038/ng.1016. [DOI] [PubMed] [Google Scholar]
- 122.Hay ED. Extracellular matrix, cell skeletons, and embryonic development. Am J Med Genet. 1989;34:14–29. doi: 10.1002/ajmg.1320340107. [DOI] [PubMed] [Google Scholar]
- 123.Humphrey JD, Milewicz DM, Tellides G, Schwartz MA. Cell biology. Dysfunctional mechanosensing in aneurysms. Science. 2014;344:477–479. doi: 10.1126/science.1253026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Humphrey JD, Schwartz MA, Tellides G, Milewicz DM. Role of mechanotransduction in vascular biology: focus on thoracic aortic aneurysms and dissections. Circ Res. 2015;116:1448–1461. doi: 10.1161/CIRCRESAHA.114.304936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Cook JR, Carta L, Bénard L, Chemaly ER, Chiu E, Rao SK, Hampton TG, Yurchenco P, GenTAC Registry Consortium. Costa KD, Hajjar RJ, Ramirez F. Abnormal muscle mechanosignaling triggers cardiomyopathy in mice with Marfan syndrome. J Clin Invest. 2014;124:1329–1339. doi: 10.1172/JCI71059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Engel J. Guest editorial letter: molecular machines in the matrix? Matrix Biol. 2006;25:200–201. doi: 10.1016/j.matbio.2006.04.001. [DOI] [PubMed] [Google Scholar]