

SUMMARY
Sweetpotato chlorotic stunt virus (SPCSV; genus Crinivirus, family Closteroviridae) is one of the most important pathogens of sweetpotato (Ipomoea batatas L.). It can reduce yields by 50% by itself and cause various synergistic disease complexes when co‐infecting with other viruses, including sweetpotato feathery mottle virus (SPFMV; genus Potyvirus, family Potyviridae). Because no sources of true resistance to SPCSV are available in sweetpotato germplasm, a pathogen‐derived transgenic resistance strategy was tested as an alternative solution in this study. A Peruvian sweetpotato landrace ‘Huachano’ was transformed with an intron‐spliced hairpin construct targeting the replicase encoding sequences of SPCSV and SPFMV using an improved genetic transformation procedure with reproducible efficiency. Twenty‐eight independent transgenic events were obtained in three transformation experiments using a highly virulent Agrobacterium tumefaciens strain and regeneration through embryogenesis. Molecular analysis indicated that all regenerants were transgenic, with 1–7 transgene loci. Accumulation of transgene‐specific siRNA was detected in most of them. None of the transgenic events was immune to SPCSV, but ten of the 20 tested transgenic events exhibited mild or no symptoms following infection, and accumulation of SPCSV was significantly reduced. There are few previous reports of RNA silencing‐mediated transgenic resistance to viruses of Closteroviridae in cultivated plants. However, the high levels of resistance to accumulation of SPCSV could not prevent development of synergistic sweet potato virus disease in those transgenic plants also infected with SPFMV.
INTRODUCTION
Sweetpotato chlorotic stunt virus (SPCSV; genus Crinivirus, family Closteroviridae) is one of the most damaging viruses of sweetpotato (Ipomoea batatas [L.] Lam), especially in Africa. By itself it can reduce yields by 50% (Gutiérrez et al., 2003; Mukasa et al., 2006; Untiveros et al., 2007). However, what makes SPCSV most harmful is its ability to break down the natural resistance of sweetpotato to other viruses and mediate synergistic viral diseases in co‐infected plants (Karyeija et al., 2000a; Kokkinos and Clark, 2006; Mukasa et al., 2006; Untiveros et al., 2007). The most common and severe of these diseases is known as sweetpotato virus disease (SPVD) and is caused by co‐infection with SPCSV and sweetpotato feathery mottle virus (SPFMV; genus Potyvirus, family Potyviridae) (Carey et al., 1999; Gibson et al., 1998). Yield losses of 70–100% are regularly observed in infected plants (Guttiérez et al., 2003; Milgram et al., 1996; Mukasa et al., 2006; Njeru et al., 2004). Whereas most sweetpotato cultivars are extremely resistant to SPFMV infecting alone, searches of sweetpotato germplasm have revealed no true resistance and only low levels of tolerance to SPCSV or SPVD. Although SPVD‐resistant landraces characterized by low incidence of infection in the field occur in East Africa (Karyeija et al., 2000b), most of them have various inadequacies such as poor and late yield (Aritua et al., 1998). Furthermore, their resistance appears to be governed by multiple recessive genes (Mwanga et al., 2002). Therefore, alternative means of generating resistance to SPCSV need to be pursued.
Genetic engineering of crop plants holds promise for introducing traits for which no conventional solutions are available or which are governed by complex genetics. This is particularly true for sweetpotato, a clonally propagated, highly heterozygous, polyploid and out‐crossing species. Furthermore, low fertility of sweetpotatoes makes even the introgression of dominant single gene traits challenging. Transformation of sweetpotato is, however, not a trivial task. The protocols for Agrobacterium tumefaciens‐mediated transformation and regeneration tend to function only with few genotypes and at a low efficiency, and results often show low reproducibility (Cipriani et al., 1999, 2001; Dodds et al., 1991; Gama et al., 1996; Kimura et al., 2001; Luo et al., 2006; Morán et al., 1998; Newell et al., 1995; Otani et al., 2003; Sheng‐Jun et al., 2004; Shimada et al., 2006; Song et al., 2004).
Pathogen‐derived resistance (PDR) by transforming plants to express viral sequences has been used for engineering resistance to viruses in crop plants for two decades (reviewed by Prins et al., 2008). The discovery of RNA silencing as a natural defence mechanism against viruses in plants and the double‐stranded RNA (dsRNA) as the inducer of RNA silencing (reviewed by Wang and Metzlaff, 2005) led to the development of plant transformation constructs that express dsRNA to activate and direct RNA silencing specifically to one or several viruses (Bucher et al., 2006; Smith et al., 2000). The high frequency of resistant plants reportedly obtained using such constructs (Bucher et al., 2006; Smith et al., 2000) makes this technique an attractive choice for crops in which it is difficult to obtain large numbers of transformed events, such as sweetpotato. Furthermore, this technology does not result in accumulation of transgene transcripts, as they are quickly degraded to small interfering RNA (siRNA), nor expression of a foreign protein, which makes it attractive from the biosafety point of view (reviewed by Latham and Wilson, 2008).
The aim of this study was to develop a reproducible method for transforming the Peruvian sweetpotato landrace ‘Huachano’ and engineer this variety to express RNA silencing‐mediated resistance to SPCSV and SPVD using an intron‐spliced hairpin construct (Smith et al., 2000).
RESULTS
Plant transformation and analysis of regenerants
The sweetpotato transformation protocol of Cipriani et al. (2001) was modified by using the hypervirulent A. tumefaciens strain EHA105, previously shown to improve transformation efficiency in sweetpotato (Otani et al., 1998), and introducing minor changes in hormone concentrations based on unpublished experiences gained at the International Potato Center (CIP) in Peru in efforts to improve embryogenesis in sweetpotato. This A. tumefaciens strain containing the binary vector pCIP41 (Fig. 1A) was used for inoculation of explants. Callus formation started 3 weeks after transplanting the explants to F9 callus induction medium. Several calli emerging from the leaf lamina and petiole developed from each explant, but only those emerging from the base of the petiole survived repeated subculture and formed embryogenic callus on selective media. At this stage residual contamination with A. tumefaciens was a problem, despite the presence of an appropriate antibiotic. Many explants (5–18% in different experiments) had to be discarded due to excessive bacterial growth. Green calli were harvested following 3–9 weeks of growth on F9 and transferred to G24D embryo induction medium for embyogenesis. Embryogenic structures formed on G24D after 1 month and were transferred to F25 embryo maturation medium. The first mature embryos appeared after 1 month on F25 and were transferred to F9 without antibiotics to allow development into plantlets.
Figure 1.
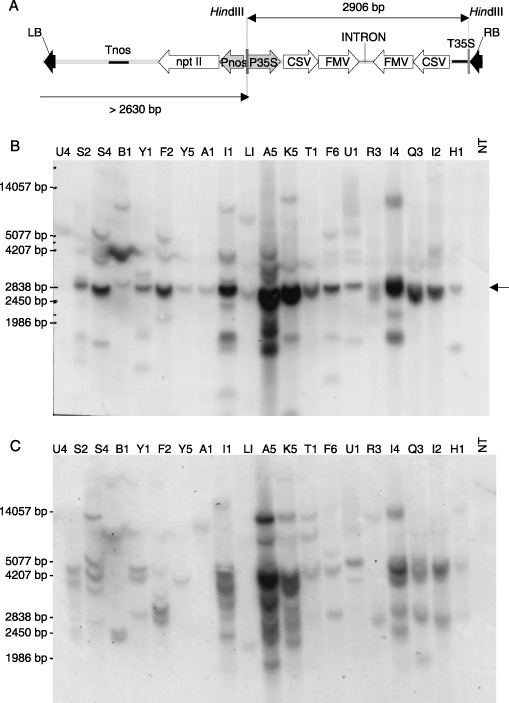
(A) Schematic representation of the T‐DNA region of the pCIP41 binary vector plasmid used for plant transformation indicates the HindIII restriction sites in the T‐DNA and the expected restriction fragment sizes to be detected with the probes used in B and C. CSV: SPCSV replicase, FMV: SPFMV replicase. (B) Southern blot analysis of HindIII‐digested total DNA from transgenic events and the non‐transgenic control (NT) hybridized with a probe corresponding to the 35S promoter. The expected band of ~2900 bp is detected in all lines, but additional bands are also detected. (C) Southern blot analysis of HindIII‐digested total DNA as in B but using a probe corresponding to the NptII gene to estimate transgene copy numbers.
The first fully developed plants were obtained 5 months from the beginning of the transformation experiment. Additional plants continued to be harvested for up to a year. In the three experiments, performed with ~200 explants each, 12, eight and eight independent kanamycin‐resistant events were regenerated, respectively. All these regenerated plants were transgenic as tested by PCR (data not shown) and confirmed by Southern blot (see below). The overall transformation efficiency (transgenic events/inoculated explants) was 4.7% and the efficiency of selection was 100%. The 28 independent transformed plants were subsequently transferred to a biosafety greenhouse for further evaluation. All plants appeared to be normal and were indistinguishable from the non‐transgenic plants of ‘Huachano’. Southern blot analysis on total DNA extracted from leaves confirmed that all 28 events were transgenic (Fig. 1B). Southern blot analysis also indicated that the plants developed from different embryos of the same callus were identical (data not shown), which suggested that each callus originated from a single transformation event.
The restriction fragment length polymorphism patterns obtained in Southern blot analysis indicated that the plants regenerated from different calli represented independent events (Fig. 1C) and contained 1–7 or possibly more loci of the transgene. The high number of transgene loci observed is in our experience unusual for A. tumefaciens‐mediated transformation in sweetpotato. Southern blot analyses revealed unexpected bands. Restriction of DNA with HindIII and using the 35S promoter sequence as a probe was expected to release the transgene cassette and reveal a single band, but a number of additional unexpected bands of various sizes were also observed (Fig. 1B). In particular, those bands corresponding to shorter restriction fragments than expected could not be explained by incomplete digestion of the DNA. It is possible that secondary structures, formed due to the large size of the hairpin construct (997 bp), induced partial degradation and recombination. Although random insertion of incomplete T‐DNA copies might explain the observed extra bands, there was a noticeable correlation between a higher transgene locus number and the number and complexity of unexpected bands. Additional bands of unexpected sizes were also observed with the digested hairpin construct‐containing binary plasmids used as controls in Southern blots (data not shown), even though they could not be seen in the agarose gel stained with ethidium bromide following electrophoresis. These observations suggest that the additional bands may represent a minor population of restriction fragments that form secondary structures after digestion.
Northern blot analysis of the low‐molecular‐weight RNA revealed variable amounts of siRNA homologous to the transgene hairpin construct in most of the transgenic events (Fig. 2), which indicated expression of the transgene. The SPVD‐affected non‐transgenic plants that were co‐infected with SPCSV and SPFMV also accumulated high amounts of virus‐specific siRNA, whereas the siRNA amounts were non‐detectable and barely detectable in the plants infected only with SPCSV or SPFMV, respectively (Fig. 2).
Figure 2.
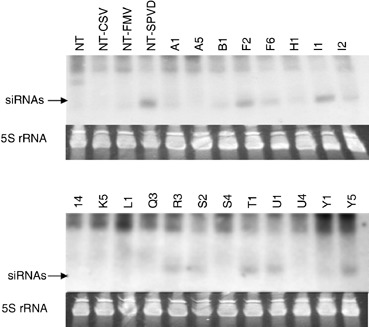
Northern blot analysis of low‐molecular‐weight RNA in 20 events using a probe specific to the transgene that includes sequences of SPCSV and SPFMV. The 5S rRNA stained by ethidium bromide and visualized under UV light in the lower panel is shown to control equal loading of RNA. Position of the signal corresponding to the 21‐nt siRNA is indicated with an arrowhead. NT, non‐transgenic healthy control; NT‐CSV, NT‐FMV and NT‐SPVD: non‐transgenic plant infected with SPCSV, SPFMV and both viruses, respectively.
Virus resistance in transgenic events
Inoculation of sweetpotatoes with SPCSV using the vector whiteflies was rather inefficient and resulted in infection of only 50% in the non‐transgenic control plants (NT). All of the 20 tested events were, however, eventually infected following 1–3 repeated inoculations. The ELISA absorbance values at 405 nm (A 405) were determined by triple‐antibody sandwich ELISA (TAS‐ELISA) for eight infected plants of each event at 21, 42 and 84 days post‐inoculation (dpi). Data were subjected to the non‐parametric Van der Waerden test to detect significant differences in virus titres between non‐transgenic controls and transgenic events. Results indicated significantly lower levels of virus accumulation in ten transgenic events (A1, U1, Y5, I2, T1, U4, R3, Q3, K5 and Y1) as compared with the control (P < 0.05) (Table 1, Fig. 3). The events with the lowest A 405 values (A1, U1, Y5 and I2) were selected for re‐evaluation in an additional experiment that included 12 plants of each event. Events A1, Y5 and I2 again generated significantly lower A 405 values than the control, whereas the values of U1 were not significantly different (data not shown).
Table 1.
Comparison of the means and normal score means of the ELISA absorbance values of 20 transgenic events and the non‐transgenic control (NT) in plants infected with SPCSV (n = 8) at 21, 42 and 84 days after planting the infected cuttings (all measurements combined).
| Event | Absorbance means | Score means* | Significance† |
|---|---|---|---|
| F6 | 0.65 | 1.189 | a |
| B1 | 0.54 | 0.989 | ab |
| A5 | 0.44 | 0.780 | abc |
| I4 | 0.43 | 0.567 | bcd |
| NT | 0.29 | 0.430 | cd |
| L1 | 0.28 | 0.307 | de |
| S4 | 0.24 | 0.283 | def |
| S2 | 0.25 | 0.271 | def |
| H1 | 0.26 | 0.258 | def |
| i1 | 0.25 | 0.239 | def |
| F2 | 0.22 | 0.173 | def |
| Y1 | 0.22 | –0.034 | efg |
| K5 | 0.20 | –0.131 | fg |
| Q3 | 0.18 | –0.260 | gh |
| R3 | 0.18 | –0.433 | ghi |
| U4 | 0.06 | –0.570 | hij |
| T1 | 0.04 | –0.689 | ijk |
| Y5 | 0.04 | –0.888 | jk |
| U1 | 0.02 | –0.901 | jk |
| I2 | 0.02 | –0.928 | jk |
| A1 | 0.02 | –1.051 | k |
Means of the normal scores obtained after converting the ranks of absorbances to quantiles of the standard normal distribution in the non‐parametric Van der Waerden test.
Within columns, values followed by the same letter do not differ significantly at P < 0.05 in Student's t‐test.
Figure 3.
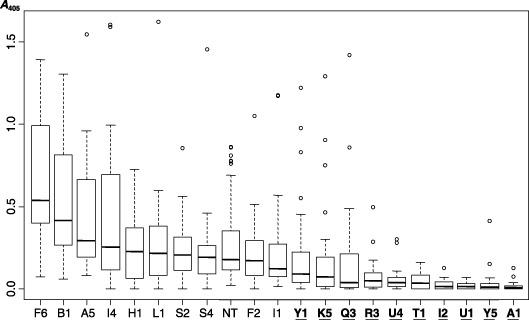
Box‐plot graphics showing dispersions and medians of the TAS‐ELISA absorbance values (A 405) combined from three time points at which plants were tested for SPCSV (21, 48 and 84 days) after planting eight SPCSV‐infected cuttings of each of the 20 transgenic events in soil. Absorbance values were corrected by subtracting a constant value resulting from the mean value of non‐infected samples. Central boxes indicate distribution of 50% of central data, and includes the median (horizontal bold line), and the upper (75% percentile) and lower (25% percentile) hinges. Limits of the dashed lines depict minimum and maximum values containing 95% of the central data. Open dots indicate extreme values. Underlined events had significantly reduced SPCSV titres as compared with the non‐transgenic (NT) control as determined by Van der Waerdens test (see Table 1).
Co‐infection with SPCSV and SPFMV resulted in SPVD‐like symptoms in all 28 transgenic events (eight plants tested per event) by 21 dpi. All plants generated similarly high A 405 values for SPFMV in ELISA (data not shown). Detection of SPCSV in the plants of the two transgenic events (Y5 and I2) which generated the lowest A 405 values when inoculated with SPCSV alone indicated that SPCSV titres in Y5 were lower than in NT controls also in the plants co‐infected with SPCSV and SPFMV, whereas the difference between I2 and NT was not significant (data not shown).
DISCUSSION
The results of the present study show that resistance to SPCSV, manifested as reduced virus titres and alleviated symptom severity, can be obtained by engineering sweetpotato plants to express virus‐specific dsRNA. Many transgenic events of the sweetpotato variety ‘Huachano’ expressed detectable amounts of transgene‐derived siRNA and half of the tested transgenic events accumulated lower titres of SPCSV than the non‐transgenic controls. There are few data available on resistance to SPCSV in sweetpotato, which suggests that sources of natural resistance to SPCSV are rare in sweetpotato germplasm. The present data demonstrate that engineered transgenic resistance may offer an alternative to the currently missing sources of resistance to SPCSV. Furthermore, the sweetpotato transformation protocol of Cipriani et al. (2001) modified in this study provided low but reproducible transformation efficiency with similar numbers of transgenic events in different experiments on the variety ‘Huachano’. Low numbers of transgenic events of sweetpotato have been reported also in many previous studies (Cipriani et al., 1999, 2001; Dodds et al., 1991; Gama et al., 1996; Kimura et al., 2001; Luo et al., 2006; Morán et al., 1998; Newell et al., 1995; Otani et al., 2003; Sheng‐Jun et al., 2004; Shimada et al., 2006; Song et al., 2004) but the reproducibility observed in the three independent transformation experiments carried out by two different scientists was promising.
The results of the present study are even more significant given the lack of previous success in attempts to engineer resistance to any Closteroviridae members in cultivated plants. Some studies have achieved pathogen‐derived transgenic resistance against chimeric viruses carrying a sequence from citrus tristeza virus (CTV, genus Closterovirus) in the experimental host Nicotiana benthamiana transformed with the corresponding CTV sequence (Batuman et al., 2006; Roy et al., 2006). However, no reproducible and durable RNA silencing‐based resistance to CTV has been obtained in citrus despite many attempts (Batuman et al., 2006; Domíngues et al., 2002; Fagoaga et al., 2006; Febres et al., 2003; Moreno et al., 2008, and references therein). The only exception appears to be a recent report by Febres et al. (2008) who identified a single transgenic event with apparently reproducible levels of resistance to CTV throughout clonal propagations. However, the mechanism is unclear as no mRNA or siRNAs were produced in the resistant event and the entire transgene appeared to be methylated.
Data suggested that the transgene construct used in this study was relatively efficient in conferring resistance to SPCSV. Detection of transgene‐specific siRNA showed that the transgene was expressed and the expected double‐stranded (hairpin) RNA transcript induced RNA silencing. Following inoculation with SPCSV all transgenic events were detectably infected with SPCSV, but many of them had significantly reduced virus titres and displayed only mild interveinal chlorosis in younger leaves and reddening of lower leaves if any symptoms were observed. Reassessment of resistance in the transgenic events A1, I2, U1 and Y5 that accumulated the lowest titres of SPCSV confirmed the high level of resistance in events A1, I2 and Y5, whereas the event U1 contained higher virus titres than in the initial experiments. The change in the response of event U1 may be attributed to environmental conditions (light and temperature). This event accumulated detectable amounts of transgene‐specific siRNA when tested, but minor changes in leaf temperature may affect accumulation of siRNA (Kalanditis et al., 2002) and the efficiency of RNA silencing in general (Szittya et al., 2003). Furthermore, the observed levels of siRNA accumulation and resistance to SPCSV showed no apparent correlation, as reported in previous studies with other viruses (Kalanditis et al., 2002; Missiou et al., 2004), which indicates that other as yet unknown factors may affect the final outcome of resistance. This seems to be typical with viruses of Closteroviridae (Batuman et al., 2006; Domíngues et al., 2002; Fagoaga et al., 2006; Febres et al., 2003, 2008; Moreno et al., 2008, and references therein).
Sequences of the RNA‐dependent RNA polymerase (RdRp)‐encoding region of an SPFMV isolate from Uganda that belongs to strain C (Tairo et al., 2005) were included in the transgene construct. However, the isolate of strain C available at CIP (Moyer, 1986; Tairo et al., 2005) was only 92% identical to the sequence used in the transgene and it was uncertain whether the transgene would confer resistance to the isolate used for inoculation. This was also not necessary because ‘Huachano’ is extremely resistant to SPFMV‐C and the infected plants usually do not accumulate detectable amounts of the virus, even after graft‐inoculation (our unpublished data). However, all plants of the transgenic events and non‐transgenic ‘Huachano’ co‐infected with SPCSV and SPFMV‐C developed similarly high titres of SPFMV and severe symptoms of SPVD. Even the plants of events A1, U1 and Y5 with high levels of resistance to accumulation of SPCSV could not avoid development of severe SPVD symptoms when co‐infected with SPFMV. SPCSV mediates synergism with SPFMV and many other viruses by suppressing the natural resistance of sweetpotato to them, which allows accumulation of the other viruses to high titres (Karyeija et al., 2000a; Kokkinos and Clark, 2006; Mukasa et al., 2006; Untiveros et al., 2007). In contrast, the titres of SPCSV are not altered or they are reduced in co‐infected plants (Cuellar et al., 2008; Mukasa et al., 2006). In this respect it was not unexpected that the event Y5 retained significantly lower SPCSV titres also during co‐infection with SPFMV, albeit suppression of RNA silencing by SPFMV could have lowered the transgene‐mediated resistance to SPCSV. SPCSV produces two novel proteins, an RNaseIII enzyme (RNase3) and a p22 protein, that co‐operatively suppress RNA silencing (Kreuze et al., 2005). These proteins were thought to be responsible for the observed synergisms with other viruses, but recent findings indicate that most isolates of SPCSV, including M2‐47 used in the present study, lack the p22 gene and are still able to induce SPVD in plants co‐infected with SPFMV (Cuellar et al., 2008). Therefore, synergism must be attributable to RNase3 and/or other proteins encoded by SPCSV. The fact that even low levels of SPCSV, as found in the three transgenic events of ‘Huachano’, were sufficient to break down resistance to SPFMV indicates that immunity to SPCSV may be required to prevent the synergistic diseases caused by SPCSV in plants co‐infected with other viruses.
EXPERIMENTAL PROCEDURES
Molecular cloning
The RdRp‐encoding regions of SPCSV and SPFMV were chosen as targets for an intron spliced hairpin construct, as the RdRp represents the most conserved region in viruses and thus increased the likelihood of creating resistance to different virus strains. To create the construct pCIP41, a 479‐nt fragment of the RdRp gene of SPCSV was amplified by reverse transcriptase (RT)‐PCR from SPCSV‐Ug (EA‐strain; Kreuze et al., 2002) with primers including restriction sites for NotI and EcoRI, or FseI and EcoRI. A 532‐nt fragment of the RdRp‐encoding sequence (NIb) of SPFMV was amplified from SPFMV‐Nam1 (Karyeija et al., 2000a; Kreuze et al., 2000) with primers containing sites for EcoRI or BamHI. The 201‐nt intron IV2 of the ST‐LS1 gene with border sequences optimized in respect to the consensus sequence for plant introns was amplified from the vector p35S‐GUS‐INT (Vancanneyt et al., 1990) with primers containing restriction sites for XbaI and BclI or BamHI. All PCR products except that of IV2 (intron) were first cloned into the TA‐cloning vector pCRII (Invitrogen, Carlsbad, CA) where they were checked by sequencing. The IV2 sequence was checked by sequencing in the plasmid pRdNibInt (see below). Subsequently, the RdRp and NIb fragments were cut from pCRII and cloned into the vector pKOH122 (Kreuze et al., 2005) in three steps. NotI‐RdRp‐EcoRI + EcoRI‐NIb‐BamHI + BclI‐IV2‐BamHI‐XbaI were ligated into pKOH122 using NotI and XbaI sites. The resulting plasmid was designated as pRdNIbInt. FseI‐RdRp‐EcoRI + EcoRI‐NIb‐BamHI were ligated into pKOH122 at FseI and BamHI sites. The resulting plasmid was designated as pRdNIb. The RdRp‐NIb‐IV2 fragment was cut out of pRdNIbInt by NotI and BamHI, and the RdRp‐NIb fragment was cut out of pRdNIb by FseI and BamHI. NotI‐RdRp‐NIb‐IV2‐BamHI + FseI‐RdRp‐NIb‐BamHI were then ligated into pKOH122 to the NotI and FseI sites. The resulting plasmid was designated as pCSFMhr. To enable directional ligation of the IV2 intron the construct was designed so as to produce a 2‐nt loop after excision of the intron. As a final step the CSFMhr fragment was transferred to the binary plasmid pMOG800 (derived from pMOG23, Sijmons et al., 1990) under the control of the cauliflower mosaic virus 35S promoter and terminator sequences. The resulting plasmid was designated as pCIP41 (Fig. 1A).
Plant transformation and regeneration
Pathogen‐free in vitro plants of the Peruvian sweetpotato landrace ‘Huachano’ were obtained from the germplasm bank of the CIP and cultured on propagation medium [MPB: 4.3 g/L MS salts (Gibco BRL, Invitrogen), 30 g/L sucrose, 0.2 g/L ascorbic acid, 0.1 g/L arginine, 0.02 g/L 1,4‐diaminobutane, 0.01 mg/L gibberellic acid, 2 mg/L calcium pantothenate and 3 g/L Phytagel (Sigma, St Louis, MO), pH 5.8] and grown for 4 weeks in a growth room at 25–27 °C, 70% relative humidity and 16‐h photoperiod (3000 lux).
Three transformation experiments with ca. 200 explants in each were done following the protocol of Cipriani et al., (2001) with a few modifications. A single bacterial colony of the A. tumefaciens strain EHA105 harbouring the binary vector pCIP41 was used to inoculate 3 mL of standard LB liquid media that contained kanamycin (100 mg/L) and rifampicin (100 mg/L) and the culture was grown in a water bath shaker (200 r.p.m.) at 28 °C overnight. A 30‐µL aliquot of this overnight culture was transferred into a 100‐mL Erlenmeyer flask containing 30 mL LB medium with kanamycin and rifampicin and grown as above until the optical density reached 0.4–0.6 at 600 nm. Bacterial cells were spun at 2000 g for 10 min at 4 °C and resuspended in 30 mL of the bacterial infection media [MIB: 4.6 g/L MS salts (Gibco BRL), 0.34 g/L sucrose, 40 mg/L acetosyringone, and 1× vitamins stock (0.5 mg/L nicotinic acid, 0.1 mg/L thiamine, 0.5 mg/L pyridoxine, 2 mg/L glycine), adjusted to pH 5.5].
Leaves with the petiole (1.5–2 cm) were cut from the top third of the in vitro plantlets and used as explants in transformation experiments. They were harvested into MRE medium [4.6 g/L MS salts (Gibco BRL), 30 g/L sucrose, 0.5 g/L MES, 0.1 mg/L naphtalene acetic acid, 1 mg/L benzilaminopurine, 0.01 mg/L gibberellic acid, 40 mg/L acetosyringone, 1× vitamins stock (adjusted to pH 5.5)], submerged into MIB with the strain EHA105 harbouring the binary vector pCIP41, and kept in the dark at 25–27 °C without agitation for 1 h. After this co‐culture step, explants were blotted onto sterile filter paper and transferred on to a semisolid medium (0.43 g/L MS salts, 20 g/L d‐glucose, 7 g/L agar, 0.5 g/L MES, 0.1× vitamins stock, 0.1 mg/L naphtalene acetic acid, 1 mg/L benzylaminopurine, 2 mg/L gibberellic acid, 0.2 mg/L acetosyringone, adjusted to pH 5.5) and incubated at 28 °C for 2 days. Regeneration of the putative transformed plants was achieved following somatic embryogenesis. The explants were first transferred to F15 medium (4.3 g/L MS salts, 0.05 mg/L 2,4‐d, 0.2 mg/L zeatin riboside, 0.1 g/L myo‐inosytol, 50 mg/L kanamycin, 200 mg/L cefotaxime, 30 g/L sucrose, 1× vitamins stock and 3.0 g/L phytagel) for 3 days, then transferred to F9 medium (same as F15 but without 2,4‐d) and further to fresh media once in 15 days. Necrotic tissue was carefully removed at each transfer. All calli were transferred to G24D medium (similar to F15 but contains 0.1 mg/L gibberellic acid instead of zeatin riboside) for development of embryogenic calli and transferred to fresh G24D medium once in 15 days. As soon as embryogenic structures were obtained, they were transferred to F25 medium (similar to F15 but contains 1 mg/L ABA instead of 2,4‐d and zeatine riboside) without antibiotics for embryo maturation and development and further transferred to fresh medium once in 15 days. Somatic embryos obtained on this medium were transferred to F9 medium without antibiotics for elongation of plantlets. The plantlets were multiplied on MPB propagation medium and transferred to a biosafety greenhouse at CIP for characterization.
Extraction of nucleic acids
For DNA isolation from leaves, 4 g of plant tissue was ground in liquid nitrogen, added to 20 mL of CTAB extraction buffer (100 mM Tris‐HCl, 25 mM EDTA, 1.4 M NaCl and 2% CTAB) with 200 µL of 2‐mercaptoethanol in a 50‐mL polyethylene tube and incubated at 65 °C in a water bath for 20 min. An equal volume of chloroform/isoamyl alcohol (24 : 1) was mixed with the extract by inverting the tubes for 5 min and spun at 16 000 g for 5 min. The upper phase was transferred to a new 50‐mL tube and 0.6 volumes of isopropanol (20 °C) was added. DNA was precipitated by placing tubes at 20 °C for 10 min and spun at 16 000 g for 5 min. Supernatant was discarded. The pellet was washed twice with 75% ethanol and dried. The pellet was then dissolved in 30 µL Nuclease‐free water and digested with 1 µL of 10 mg/mL RNase for 15 min at 37 °C.
Total RNA was extracted from leaves with the Trizol reagent (Invitrogen) according to the manufacturer's recommendations. The low‐molecular‐weight fraction of RNA was separated by the LiCl4 precipitation method as described (Kreuze et al., 2005) and used for detection of siRNA. The quality of extracted nucleic acids was checked by TBE agarose gel electrophoresis and observation under UV light following staining with ethidium bromide (Sambrook et al., 2001). Concentration and purity of nucleic acids were estimated by measuring the A 260 and A 280 values with a UV spectrophotometer (Sambrook et al., 2001).
PCR analysis was used for rapid initial detection of the putative transgenic plants and for analysis of integrity of the transgene constructs in the plant. Primers were designed (Table 2) to amplify various regions of the transgene construct, including the 35S promoter, SPCSV, SPFMV and intron sequences. PCR reactions (15 µL) contained 10 ng of DNA, 1× PCR buffer (Gibco‐BRL), 0.4 µM of each primer, 0.5 µM dNTP and 1 unit Taq DNA polymerase (Gibco‐BRL). The annealing temperature varied according to the primer combination used, but the general amplification programme was as follows: denaturation at 95 °C for 1 min followed by 35 cycles of 95 °C for 15 s, 50–60 °C for 30 s and 72 °C for 1 min, and a final elongation step at 72 °C for 5 min. PCR products (10 µL) were analysed by agarose gel electrophoresis as described above.
Table 2.
Primers used.
| Primer | Sequence |
|---|---|
| CSV1 sense | 5′‐TTTCTGCAAATTTGACAAGTCC‐3′ |
| CSV2 antisense | 5′‐TGCTTCAAACCCGAAATT‐3′ |
| CSV3 antisense | 5′‐CGAAAAACCTCACAGGGTCAGGC‐3′ |
| FMV1 antisense | 5′‐TTCCACCATTGCAGCACA‐3′ |
| FMV2 sense | 5′‐TTCAAYAGTGGKCAGCCG‐3′ |
| FMV3 antisense | 5′‐CAAGCTCAGCAAAGCACTCACCG‐3′ |
| P35S1 sense | 5′‐CGCACAATCCCACTATCCTTCGC‐3′ |
| P35S2 sense | 5′‐GGCCGGCCTCAACATGGTGGAGCACGAC‐3′ |
| P35S2 antisense | 5′‐GGTACCTCCTCTCCAAATGAAATGAAC‐3′ |
| INT antisense | 5′‐GGATCCTGCACATCAACAAATTTTG‐3′ |
| INT sense | 5′‐GAAAATGATCAGGTAAGTTTCTGCTTCTAC‐3′ |
| T35S sense | 5′‐CTAGAGTCGTCCGCAAATCACC‐3′ |
| RB antisense | 5′‐GAAGGCGGGAAACGACAATC‐3′ |
For Southern blot analysis (Sambrook et al., 2001), total plant DNA (20 µg) was digested with HindIII and BamHI (20 units) to assess the integrity of the transgene, and with DraIII, SacI and EcoRI to determine the number of transgene copies. Each digestion was done in a 100‐µL reaction volume and incubated at 37 °C overnight. DNA fragments were separated by agarose gel (1%) electrophoresis overnight and moved to a nylon membrane (Hybond™– N+, Amersham, Piscataway, NJ) by capillary transfer. DNA fragments were bound to the membrane by UV cross‐linking (Stratalinker 2400, Stratagene, La Jolla, CA). Probes were developed from several regions of the transgene construct amplified by PCR from the plasmid pCIP41. They were labelled with [α32P]dCTP using Gene Images Random Prime Labeling kit or Gene Images AlkPhos Direct Labeling kit (Amersham) according to the manufacturer's recommendation. The probe was hybridized at 65 °C for 18 h. The filters hybridized with radioactive probes were washed with 1× SSC, 0.1% SDS (w/v) and 0.5× SSC, 0.1% SDS (w/v) at 65 °C for 15 min each with gentle agitation. In the case of the Gene Images kit, membranes were hybridized and washed according to the manufacturer's recommendation. The hybridized membrane was wrapped in polyethylene plastic and exposed to a radiographic film (Kodak).
For siRNA analysis, 8 µg of the low‐molecular‐weight RNA was separated on a 15% polyacrylamide Tris‐borate–EDTA–urea gel as described (Kreuze et al., 2005) and blotted to a nylon membrane (Hybond‐NX, Amersham) using a Trans‐Blot semi‐dry electrophoretic transfer cell (Bio‐Rad, Hercules, CA) for 30 min. RNA was UV cross‐linked as described above. Sense and antisense [α32P]UTP‐labelled RNA probes specific to the transgene were synthesized with T7 RNA polymerase (Promega, Madison, WI, USA) using the plasmid pCSFMhr linearized with XhoI. For hybridization to siRNA, the probe was cleaved by alkaline hydrolysis in carbonate buffer (120 mm Na2CO3, 80 mm NaHCO3, pH 10.2) to fragments of an average length of 50 bp and hybridized, washed and developed as described (Kreuze et al., 2005).
Virus inoculations and testing
Isolate M2‐47 (Gutiérez et al., 2003), which belongs to the East African strain of SPCSV (Cuellar et al., 2008), was inoculated to sweetpotato plants with the vector whitefly (Bemisia tabaci Bellows and Perring; biotype B) or graft‐inoculation. Whiteflies were reared on sweetpotato plants in an insect‐proof cage at 23–26 °C. For virus acquisition, whiteflies were stunned at 4 °C for 15 min after which whitefly‐infested leaves were transferred to a new cage containing SPCSV‐infected sweetpotato plants for 3 days. Subsequently, plants were inoculated with 50 putatively viruliferous whiteflies by placing whitefly‐infested leaves into cages each containing a single plant to be inoculated. Three days later whiteflies were killed by spraying with 1% Vertimec and 0.5% Applaud. SPCSV was tested at 3‐week intervals for 2 months by TAS‐ELISA (see below). Plants that were negative in ELISA were top‐grafted with scions of healthy I. setosa, an indicator plant for sweetpotato viruses, and the growing I. setosa shoots monitored for symptoms and tested by ELISA at 3 weeks post grafting. Inoculation of SPCSV with whiteflies was done only with the 20 first transgenic events and repeated up to three times. In the first experiment two cuttings of each event were inoculated. The 12 events for which plants remained negative for SPCSV were tested in the second experiment in which three plants of each event were inoculated. The four events that still tested negative were forwarded to the third experiment with eight cuttings inoculated per event. Four, six and eight non‐transgenic control cuttings were used in these experiments, respectively. Plants found to be infected were multiplied by taking and rooting cuttings in soil (eight plants per event) for quantitative analysis of virus titres by TAS‐ELISA (see below) at 21, 48 and 84 days after planting (dap). The four events with the lowest A 405 values were re‐tested by inoculating 12 plants and testing them for SPCSV at 21 and 48 dap.
All transgenic events were also challenged with SPFMV (isolate C; Moyer, 1986) and SPCSV, which was done by grafting a scion from SPVD‐affected non‐transgenic ‘Huachano’ on to the plant to be tested (eight plants per event). These plants were tested for both viruses by ELISA directly from the sweetpotato plants at 21 and 48 dpi.
Virus detection by ELISA
Double‐antibody sandwich ELISA and TAS‐ELISA were used for detection and quantification of SPFMV and SPCSV, respectively, as described (Karyeija et al., 2000a). One disc (diameter 1 cm) was taken from the 5th and 6th leaf of each plant and ground together in a 1.5‐mL Eppendorf tube with 1 mL ELISA extraction buffer. Virus‐specific polyclonal antibodies and monocolonal antibodies to the coat proteins of SPFMV and SPCSV were provided by the Laboratory of Virology at CIP. Anti‐mouse alkaline phosphatase‐conjugated goat IgG used in TAS‐ELISA was obtained from Bio‐Rad. Absorbances were measured at 405 nm 4–16 h after adding the substrate (p‐nitrophenyl phosphate) using a Microplate reader (Bio‐Rad). Results were subjected to statistical analysis using the statistical package R (R Development Core Team, 2006) using analysis of variance (ANOVA) or the non‐parametric Van der Waerden test (Van der Waerden, 1952).
ACKNOWLEDGEMENTS
We are grateful to Segundo Fuentes for providing virus antibodies and Heidi Gamara and Elio Campbell for help and advice with whitefly rearing and SPCSV transmission. We thank Felipe de Mendiburu for help with statistical analysis of ELISA results, and Maximo Fernandez for invaluable advice and assistance with sweetpotato transformation. J.F.K. was funded by the Swedish International Development Agency (Sida). Financial support from the Belgium Directorate General for Development Cooperation and the Academy of Finland (grants 1102134 and 1110797) is gratefully acknowledged.
REFERENCES
- Aritua, V. , Adipala, E. , Carey, E.E. and Gibson, R.W. (1998) The incidence of sweet potato virus disease and virus resistance of sweet potato grown in Uganda. Ann. Appl. Biol. 132, 399–411. [Google Scholar]
- Batuman, O. , Mawassi, M. and Bar‐Joseph, M. (2006) Transgenes consisting of a dsRNA of an RNAi suppressor plus the 3′ UTR provide resistance to Citrus tristeza virus sequences in Nicotiana benthamiana but not in citrus. Virus Genes, 33, 319–327. [DOI] [PubMed] [Google Scholar]
- Bucher, E. , Lohuis, D. , Van Poppel, P.M.J.A. , Geerts‐Dimtriadou, C. , Goldbach, R. and Prins, M. (2006) Multiple virus resistance at a high frequency using a single transgene construct. J. Gen. Virol. 87, 3697–3701. [DOI] [PubMed] [Google Scholar]
- Carey, E.E. , Gibson, R.W. , Fuentes, S. , Machmud, M. , Mwanga, R.O.M. , Turyamureeba, G. , Zhang, L. , Ma, D. , Abo El‐Abbas, F. , El‐Bedewy, R. and Salazar, L.F. (1999) The causes and control of virus diseases of sweetpotato in developing countries: is sweetpotato virus disease the main problem? In: CIP Program Report 1997–98 (Arthur C., Fergusson P., Smith B., eds.), pp. 241–248. Lima: International Potato Center. [Google Scholar]
- Cipriani, G. , Michaud, D. , Brunelle, F. , Golmirzaie, A. and Zhang, D.P. (1999) Expression of Soybean Proteinase inhibitor in Sweetpotato In: CIP Program Report 1997–1998 (Arthur C., Fergusson P., Smith B., eds.), pp. 271–277. Lima: International Potato Center. [Google Scholar]
- Cipriani, G. , Fuentes, S. , Bello, V. , Salazar, L.F. , Ghislain, M. and Zhang, D.P. (2001) Transgene expression of rice cysteine proteinase inhibitors for the development of resistance against sweetpotato feathery mottle virus In: CIP Program Report 1999–2000 (Fergusson P., Parrott S., Sheridan K., Smith B., Stares J., eds.), pp. 267–271. Lima: International Potato Center. [Google Scholar]
- Cuellar, W.J. , Tairo, F. , Kreuze, J.F. and Valkonen, J.P.T. (2008) Analysis of gene content in sweet potato chlorotic stunt virus RNA1 reveals the presence of the p22 RNA silencing suppressor in only a few isolates: implications for viral evolution and synergism. J. Gen. Virol. 89, 573–582. [DOI] [PubMed] [Google Scholar]
- Dodds, J.H. , Merzdorf, C. , Zambrano, V. , Sigüeñas, C. and Jaynes, J. (1991) Potential use of agrobacterium‐mediated gene transfer to confer insect resistance in sweet potato In: Sweet Potato Pest Management: a Global Perspective (Jansson R.K., Raman K.V., eds), pp. 203–219. Oxford: West View Press. [Google Scholar]
- Domínguez, A. , De Mendoza, A.H. , Guerri, J. , Cambra, M. , Navarro, L. , Moreno, P. and Peña, P. (2002) Pathogen‐derived resistance to Citrus tristeza virus (CTV) in transgenic mexican lime (Citrus aurantifolia (Christ.) Swing.) plants expressing its p25 coat protein gene. Mol. Breed. 10, 1–10. [Google Scholar]
- Fagoaga, C. , Lopez, C. , De Mendoza, A.H. , Moreno, P. , Navaroo, L. , Flores, R. and Peña, L. (2006) Post‐transcriptional gene silencing of the p23 silencing suppressor of Citrus tristeza virus confers resistance to the virus in transgenic Mexican lime. Plant Mol. Biol. 60, 153–165. [DOI] [PubMed] [Google Scholar]
- Febres, V.J. , Niblett, C.L. , Lee, R.F. and Moore, G.A. (2003) Characterization of grapefruit plants (Citrus paradisi Macf.) transformed with citrus tristeza closterovirus genes. Plant Cell Rep. 21, 421–428. [DOI] [PubMed] [Google Scholar]
- Febres, V.J. , Lee, R.F. and Moore, G.A. (2008) Transgenic resístanse to Citrus tristeza virus in grapefruit. Plant Cell Rep. 27, 93–104. [DOI] [PubMed] [Google Scholar]
- Gama, M. , Leite, R.P. , Cordeiro, A.R. and Cantliffe, D.J. (1996). Transgenic sweet potato plants obtained by Agrobacterium tumefaciens‐mediated transformation. Plant Cell Tissue Organ. Cult. 46, 237–244. [Google Scholar]
- Gibson, R.W. , Mpembe, I. , Alicai, T. , Carey, E.E. , Mwanga, R.O.M. , Seal, S.E. and Vetten, H.J. (1998) Symptoms, aetiology and serological analysis of sweet potato virus disease in Uganda. Plant Pathol. 47, 95–102. [Google Scholar]
- Gutiérrez, D. , Fuentes, S. and Salazar, L.F. (2003) Sweetpotato virus disease (SPVD): Distribution, incidence, and effect on sweetpotato yield in Peru. Plant Dis. 87, 297–302. [DOI] [PubMed] [Google Scholar]
- Kalanditis, K. , Psaradakis, S. , Tabler, M. and Tsagris, M. (2002) The occurrence of CMV‐specific short RNAs in transgenic tobacco expressing virus‐derived double stranded RNA is indicative of resistance to the virus. Mol. Plant–Microbe Interact. 8, 826–833. [DOI] [PubMed] [Google Scholar]
- Karyeija, R.F. , Kreuze, J.F. , Gibson, R.W. and Valkonen, P.T. (2000a) Synergistic interactions of a potyvirus and a phloem‐limited crinivirus in sweet potato plants. Virology, 268, 26–36. [DOI] [PubMed] [Google Scholar]
- Karyeija, R.F. , Kreuze, J.F. , Gibson, R.W. and Valkonen, J.PT . (2000b). Two serotypes of Sweetpotato feathery mottle virus in Uganda and their interaction with resistant sweetpotato cultivars. Phytopathology, 90, 1250–1255. [DOI] [PubMed] [Google Scholar]
- Kimura, T. , Otani, M. , Noda, T. , Ideta, O. , Shimada, T. and Saito, A. (2001) Absence of amylose in sweet potato [Ipomoea batatas (L.) Lam.] following the introduction of granule‐bound starch synthase I cDNA. Plant Cell Rep. 20, 663–666. [Google Scholar]
- Kokkinos, C.D. and Clark, C.A. (2006) Interactions among Sweet potato chlorotic stunt virus and different potyviruses and potyvirus strains infecting sweet potato in the United States. Plant Dis. 90, 1347–1352. [DOI] [PubMed] [Google Scholar]
- Kreuze, J.F. , Karyeija, R.F. , Gibson, R.W. and Valkonen, J.P.T. (2000) Comparisons of coat protein gene sequences show that East African isolates of Sweet potato feathery mottle virus form a genetically distinct group. Arch. Virol. 145, 567–574. [DOI] [PubMed] [Google Scholar]
- Kreuze, J.F. , Savenkov, E.I. and Valkonen, J.P.T. (2002) Complete genome sequence and analyses of the subgenomic RNAs of Sweet potato chlorotic stunt virus reveals several new features for the genus Crinivirus. J. Virol. 76, 9260–9270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreuze, J.F. , Savenkov, E.I. , Cuellar, W. , Li, X. and Valkonen, J.P.T. (2005) Viral Class 1 RNase III involved in suppression of RNA silencing. J. Virol. 79, 7227–7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham, J.R. and Wilson, A.K. (2008) Transcomplementation and synergism in plants: implications for viral transgenes? Mol. Plant Pathol. 9, 85–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, H.R. , Santa Maria, M. , Benavides, J. , Zhang, D.P. , Zhang, Y.Z. and Ghislain, M. (2006) Rapid genetic transformation of sweetpotato (Ipomoea batatas (L.) Lam) via organogenesis. Afr. J. Biotech. 5, 1851–1857. [Google Scholar]
- Milgram, M. , Cohen, J. and Loebenstein, G. (1996) Effects of Sweet potato feathery mottle virus and Sweet potato sunken vein virus on sweet potato yields and rates of reinfection of virus‐free planting material in Israel. Phytoparasitica, 24, 189–193. [Google Scholar]
- Missiou, A. , Kalantidis, K. , Boutla, A. , Tzortzakaki, S. , Tabler, M. and Tsagris, M. (2004) Generation of transgenic potato plants highly resistant to potato virus Y (PVY) through RNA silencing. Mol. Breeding, 14, 185–197. [Google Scholar]
- Morán, R. , García, R. , López, A. , Zaldúa, Z. , Mena, J. , García, M. , Armas, R. , Somonte, D. , Rodríguez, J. , Gómez, M. and Pimentel, E. (1998) Transgenic sweet potato plants carrying the delta‐endotoxin gene from Bacillus thuringiensis var. tenebrionis . Plant Sci. 139, 175–184. [Google Scholar]
- Moreno, P. , Ambrós, S. , Albiach‐Martí, M.R. , Guerri, J. and Peña, L. (2008) Citrus tristeza virus: a pathogen that changed the course of the citrus industry. Mol. Plant Pathol. 9, 251–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer, J.W. (1986) Variability among strains of sweet potato feathery mottle virus. Phytopathology, 76, 1126. [Google Scholar]
- Mukasa, S.B. , Rubaihayo, P.R. and Valkonen, J.P.T. (2006) Interactions between a crinivirus, an ipomovirus and a potyvirus in coinfected sweetpotato plants. Plant Pathol. 55, 458–467. [Google Scholar]
- Mwanga, R.O.M. , Kriegner, A. , Cervantes‐Flores, J.C. , Zhang, D.P. , Moyer, J.W. and Yencho, G.C. (2002) Resistance to sweetpotato chlorotic stunt virus and sweetpotato feathery mottle virus is mediated by two separate recessive genes in sweetpotato. J. Amer. Soc. Hort. Sci. 127, 798–806. [Google Scholar]
- Newell, C.A. , Lowe, J.M. , Merryweather, A. , Rooke, L.M. and Hamilton, W.D.O. (1995) Transformation of sweet potato (Ipomoea batatas (L.) Lam.) with Agrobacterium tumefaciens and regeneration of plants expressing cowpea trypsin inhibitor and snowdrop lectin. Plant Sci. 107, 215–227. [Google Scholar]
- Njeru, R.W. , Mburu, M.W.K. , Cheramgoi, E. , Gibson, R.G. , Kiburi, Z.M. , Obudho, E. and Yobera, D. (2004) Studies on the physiological effects of viruses on sweet potato yield in Kenya. Ann. Appl. Biol. 145, 71–76. [Google Scholar]
- Otani, M. , Shimada, T. , Kimura, T. and Saito, A. (1998) Transgenic plant production from embryogenic callus of sweet potato (Ipomoea batatas (L.) Lam.) using Agrobacterium tumefaciens . Plant Biotech. 15, 11–16. [Google Scholar]
- Otani, M. , Wakita, Y. and Shimada, T. (2003) Production of herbicide‐resistant sweetpotato (Ipomoea batatas (L.) Lam.) Plants by agrobacterium tumefaciens‐mediated transformation. Breeding Science 53, 145–148. [Google Scholar]
- Prins, M. , Laimer, M. , Noris, E. , Schubert, J. , Wassenegger, M. and Tepfer, M. (2008) Strategies for antiviral resistance in transgenic plants. Mol. Plant Pathol. 9, 73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2006) R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. [Google Scholar]
- Roy, G. , Sudarshana, M.R. , Ullman, D.E. , Ding, S.W. , Dandekar, A.M. and Falk, B.W. (2006) Chimeric cDNA sequences from Citrus tristeza virus confer RNA silencing‐mediated resistance in transgenic Nicotiana benthamiana plants. Phytopathology 96, 819–827. [DOI] [PubMed] [Google Scholar]
- Sambrook, J. , Fristh, E.F. and Maniatis, T. (2001) Molecular Cloning: a Laboratory Manual, 3rd edn. New York: Cold Spring Harbor Press. [Google Scholar]
- Sheng‐Jun, J. , Qing‐Chang, L. , Hong, Z. , Li‐Sha, W. and Yu‐Ping, W. (2004). Regeneration of sweet potato transgenic plants with oryzacystatin‐I (OCI) gene. Chinese J. Agri. Biotech. 1, 99–102. [Google Scholar]
- Shimada, T. , Otani, M. , Hamada, T. and Kim, S.H. (2006) Increase of amylose content of sweetpotato starch by RNA interference of the starch branching enzyme II gene (IbSBEII). Plant Biotech. 23, 85–90. [Google Scholar]
- Sijmons, P.C. , Dekker, B.M. , Schrammeijer, B. , Verwoerd, T.C. , Van Den Elzen, P.J. and Hoekema, A. (1990) Production of correctly processed human serum albumin in transgenic plants. Biotechnology (NY), 8, 217–221. [DOI] [PubMed] [Google Scholar]
- Smith, N.A. , Singh, S.P. , Wang, M.B. , Stoutjesdijk, P.A. , Green, A.G. and Waterhouse, P.M. (2000). Total silencing by intron‐spliced hairpin RNAs. Nature, 407, 319–320. [DOI] [PubMed] [Google Scholar]
- Song, G.Q. , Honda, H. and Yamaguchi, K.H. (2004) Efficient Agrobacterium tumefaciens‐mediated transformation of sweet potato (Ipomoea batatas (L.) Lam.) from stem explants using a two‐step Kanamycin–Hygromycin selection method. In-Vitro Cell. Dev. Biol. Plant, 40, 359–365. [Google Scholar]
- Szittya, G. , Silhavy, D. , Molnar, A. , Havelda, Z. , Lovas, A. , Lakatos, L. , Banfalvi, Z. and Burgyan, J. (2003) Low temperature inhibits RNA silencing‐mediated defence by the control of siRNA generation. EMBO J. 22, 633–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tairo, F. , Mukasa, S.B. , Jones, R.A.C. , Kullaya, A. , Rubaihayo, P.B. and Valkonen, J.P.T. (2005) Unraveling the genetic diversity of the three main viruses involved in Sweet Potato Virus Disease (SPVD), and its practical implications. Mol. Plant. Pathol. 6, 199–211. [DOI] [PubMed] [Google Scholar]
- Untiveros, M. , Fuentes, S. and Salazar, L.F. (2007) Synergistic interaction of Sweet potato chlorotic stunt virus (Crinivirus) with carla‐, cucumo‐, ipomo‐ and potyviruses infecting sweet potato. Plant Dis. 91, 669–676. [DOI] [PubMed] [Google Scholar]
- Van der Waerden, B.L. (1952) Order tests for the two‐sample problem and their power. Proc. Kon. Nederl. Akad. Wetensch. 55, 453–458. [Google Scholar]
- Vancanneyt, G. , Schmidt, R. , O’Connor‐Sanchez, A. , Willmitze, L. and Rocha‐Sosa, M. (1990) Construction of an intron‐containing marker gene: splicing of the intron in transgenic plants and its use in monitoring early events in Agrobacterium‐mediated plant transformation. Mol. Gen. Genet. 220, 245–250. [DOI] [PubMed] [Google Scholar]
- Wang, M.B. and Metzlaff, M. (2005) RNA silencing and antiviral defense in plants. Curr. Opin. Plant Biol. 8, 216–222. [DOI] [PubMed] [Google Scholar]