

SUMMARY
RPP13, a member of the cytoplasmic class of disease resistance genes, encodes one of the most variable Arabidopsis proteins so far identified. This variability is matched in ATR13, the protein from the oomycete downy mildew pathogen Hyaloperonospora parasitica recognized by RPP13, suggesting that these proteins are involved in tight reciprocal coevolution. ATR13 exhibits five domains: an N‐terminal signal peptide, an RXLR motif, a heptad leucine/isoleucine repeat, an 11‐amino‐acid repeated sequence and a C‐terminal domain. We show that the conserved RXLR‐containing domain is dispensable for ATR13‐mediated recognition, consistent with its role in transport into the plant cytoplasm. Sequencing ATR13 from 16 isolates of H. parasitica revealed high levels of amino acid diversity across the entire protein. The leucines/isoleucines of the heptad leucine repeat were conserved, and mutation of particular leucine or isoleucine residues altered recognition by RPP13. Natural variation has not exploited this route to detection avoidance, suggesting a key role of this domain in pathogenicity. The extensive variation in the 11‐amino‐acid repeat units did not affect RPP13 recognition. Domain swap analysis showed that recognition specificity lay in the C‐terminal domain of ATR13. Variation analyses combined with functional assays allowed the identification of four amino acid positions that may play a role in recognition specificity. Site‐directed mutagenesis confirmed that a threonine residue is absolutely required for RPP13 recognition and that recognition can be modulated by the presence of either an arginine or glutamic acid at other sites. Mutations in these three amino acids had no effect on the interaction of ATR13 with a resistance gene unlinked to RPP13, consistent with their critical role in determining RPP13‐Nd recognition specificity.
INTRODUCTION
Downy mildews are among the most important diseases of crop plants worldwide. In the UK they cause losses to commercially grown onions, leeks, brassicas, sugar beet and lettuces. They belong to the oomycete group within the kingdom Stramenopiles and, as such, are distinct from true fungi being more closely related to brown algae. One species in this kingdom is the obligate biotrophic oomycete Hyaloperonospora parasitica, which can infect the model plant Arabidopsis. This interaction has been studied extensively (Holub et al., 1994; McDowell et al., 2000; Parker et al., 1996) and has played a significant role in contributing to our understanding of the host genes involved in disease resistance responses. Resistance of Arabidopsis to this pathogen most often follows a ‘gene for gene’ model postulated by Flor (1971), whereby a plant resistance gene product (R) recognizes a product produced by a gene carried by the invading pathogen, the avirulence product (Avr). A hypersensitive response, manifest as localized cell death at the infection site, is mounted leading to the curtailment of pathogen growth. Plant R gene products have a conserved structure despite being active against a wide range of pathogens, including viruses, bacteria, fungi, oomycetes, nematodes and insects (Dangl and Jones, 2001). Nearly all R genes encode a leucine‐rich repeat domain, which can be extracellular but anchored in the plant membrane or be part of entirely cytoplasmically located proteins. The cytoplasmic class often contain a nucleotide‐binding site (NBS), which is preceded at the N‐terminus by either a coiled coil or TIR (Toll interleukin‐like) domain (Dangl and Jones, 2001). In contrast, avirulence gene products cloned from bacterial and fungal pathogens show few common features (Dodds et al., 2004; Joosten et al., 1994; van Kan et al., 1991; Luderer et al., 2002; Orbach et al., 2000). Given that these pathogen products probably play a role in pathogenicity and all face the same host target, these ‘effectors’ may have evolved independently. Furthermore, pathogens expressing effector molecules that interact directly with R proteins to mediate defence will be exposed to strong diversifying selection, whereby alternative forms that fail to trigger defence will be favoured.
Little is known of the molecular processes involved in suppression of host defence mechanisms by the pathogen and the role of pathogen proteins in the host (Abramovitch and Martin, 2004; Jones and Dangl, 2006). Such pathogen effector molecules are believed to be involved in pathogenicity, via modulation of host metabolism and physiological processes, as well as suppression of host defence mechanisms. For example, the Pseudomonas syringae effector protein AvrRPM1 phosphorylates the Arabidopsis RIN4 protein, which is likely to play a role in innate immunity (Kim et al., 2005). The RPM1 and RPS2 host resistance proteins detect the interaction of the AvrRPM1and AvrRpt2 effector proteins, respectively, with RIN4 (Mackey et al., 2002, 2003). Hence, the pathogen has developed two effector proteins targeted to RIN4, an important defence protein. This fascinating story is an example of many potential complex interactions between Arabidopsis and its pathogens. Therefore, such pathogen effectors are vulnerable to detection by host R gene‐dependent recognition and resistance systems.
The presence of leucine‐rich repeat (LRR) domains, which are implicated in protein–protein interactions, in R proteins suggests that R proteins would directly interact with the Avr targets. However, this has been demonstrated in only a few instances: AvrPto from P. syringae pv tomato was the first Avr protein shown to interact directly with a resistance gene product, namely Pto (Scofield et al., 1996; Tang et al., 1996), a cytoplasmically located protein kinase, not a member of the LRR‐containing class. Avr‐Pita from Magnaportha grisea and Pita, a cytoplasmically located NBS‐LRR R protein from rice, have also been shown to interact directly in yeast and in vitro (Jia et al., 2000). Avr‐Pita is predicted to be a zinc metalloprotease and a mutation in the protease motif caused loss of resistance and failure to interact with Pita. The flax rust avirulence protein AvrL567 has been shown to interact directly with the R protein, L, (in the NBS‐LRR class of R proteins) from flax in a yeast two‐hybrid system (Dodds et al., 2006). Naturally occurring variants of the AvrL567 genes exist and some escape recognition, but maintain structure and stability, indicating that they may overcome resistance by sequence diversity, not through loss of function.
This lack of ability to show direct interaction of Avr and R gene products in the majority of systems investigated has led to the formulation of the ‘Guard Hypothesis’ (van der Biezen and Jones, 1998) for R gene function, whereby the R gene product monitors the binding to or modification of other plant proteins by the Avr gene product. R proteins become activated through the indirect detection of the Avr molecule in plant cells, as exemplified by the RPM1 and Rps2 interaction with RIN4 (Mackey et al., 2002, 2003). Subsequently, the plant mounts a defence response. However, the failure to demonstrate an interaction may simply be due to limitations of currently available assay techniques.
Several avirulence genes have recently been cloned from the distinct group of oomycete organisms (Allen et al., 2004; Armstrong et al., 2005; Rehmany et al., 2005; Shan et al., 2004). These genes do not share homology either with fungal avirulence genes or with each other. However, all of these avirulence genes encode small, secreted proteins, which can be recognized intracellularly. Many secreted proteins from oomycetes possess a motif, RXLR, which occurs within 32 amino acids from the N terminal signal peptide (Rehmany et al., 2005). It has been shown that a similar motif can direct the delivery of Plasmodium proteins to erythrocytes (Hiller et al., 2004; Marti et al., 2004) and Whisson et al. (2007) have demonstrated that the RXLR motif is involved in the translocation of oomycete effectors into plant host cells.
The resistance gene RPP13 has been shown to be one of the most variable genes so far cloned from Arabidopsis (Bakker et al., 2006; Rose et al., 2004). This hypervariability, which resided within the LRR motifs, suggested that selective pressures were causing rapid evolution of novel forms of the RPP13 protein. The avirulence gene ATR13 from Hyaloperonospora parasitica, which triggers RPP13‐mediated resistance, also showed hyper‐variability between alleles (Allen et al., 2004). One explanation of such diversity is diversifying selection on ATR13 driven by coevolution with the RPP13 gene from Arabidopsis. Based on sequence comparisons of alleles that differed in their ability to trigger an RPP13‐dependent response, we were able to localize a major determinant of this recognition by RPP13 to the C‐terminal domain of ATR13. Here we used domain swap experiments to confirm the role of the C‐terminal domain in specifying RPP13‐dependent recognition. Analysis of natural variation amongst 15 variants of ATR13 allowed us to define key amino acid residues that are functionally important for this interaction. Using site‐directed mutagenesis within the hyper‐variable C‐terminal domain we defined one amino acid essential for triggering RPP13‐mediated resistance and two amino acids that modulate the response.
RESULTS
Natural selection on ATR13 genes
To access sufficient natural variation to identify amino acids involved in RPP13‐Nd recognition, ATR13 alleles were cloned and sequenced from 16 isolates of H. parasitica and their predicted proteins were compared (Fig. 1). There were 15 protein variants. All alleles (except ATR13‐Goco1‐B) were tested in the biolistic assay, which has been described previously (Allen et al., 2004), for their ability to be recognized by the resistance gene RPP13‐Nd. In addition to the alleles previously described as being recognized, namely ATR13‐Maks9, ATR13‐Aswa1, ATR13‐Goco1‐A and ATR13‐Emco5, another allele, ATR13‐Bico1, was also recognized. ATR13 genes from the 11 other isolates were not recognized by RPP13‐Nd.
Figure 1.


Amino acid alignment and domains of ATR13 proteins encoded by 18 alleles from 16 isolates of Hyaloperonospora parasitica. Dots indicate synonymous amino acids to ATR13‐Maks9. Dashes indicate amino acids not present in variant form. The RXLR motif is underlined. Leucines/isoleucines in heptad repeat region are indicated by asterisks. Alleles whose encoded protein is above the black line are recognized by RPP13‐Nd in the biolistic assay and those below the line are not (# ATR13‐Goco1‐B is identical to ATR13‐Goco1‐A with one synonymous difference between ATR13‐Goco alleles, but not tested in the biolistic assay). In the C‐terminal region amino acids indicated by asterisks are those predicted to be key in the recognition response by RPP13‐Nd.
We evaluated the levels of non‐synonymous and synonymous polymorphism at the ATR13 locus and the Ppat5 locus. Ppat5 encodes a dnaK‐type molecular chaperone (Bittner‐Eddy et al., 2003), which would not be expected to be under the same selective pressure as ATR13, and served as a reference gene in these analyses. ATR13 showed higher levels of polymorphism overall (πtotal = 0.042) compared with Ppat5 (πtotal = 0.002) (Table 1a). From three individuals, isolates Emwa1, Goco1 and Cand5, we amplified two variants of ATR13, demonstrating that they were heterozygous at the ATR13 locus or a duplication of ATR13 had occurred, while the rest were homozygous at ATR13. Proteins encoded by the two ATR13‐Emwa1 alleles differ by one amino acid. The polymorphism between the Goco1 alleles does not alter the predicted protein. Only one of the Cand5 alleles was cloned and used in the analysis. In the collection of 18 ATR13 gene sequences from 16 isolates, 15 allelic forms were detected. ATR13‐Hind2 showed considerable divergence in the C‐terminal domain of the predicted protein and contributed 28 non‐synonymous differences to the dataset in this. Therefore, all analyses were conducted including and excluding this allele, to evaluate whether this single, highly divergent allele accounted for our statistically significant results.
Table 1.
Polymorphism at ATR13 and Ppat5.
| (a) Average pairwise differences per site (π) | |||||
|---|---|---|---|---|---|
| Gene | Isolates | No. of alleles | π total | π syn* | π non† |
| ATR13 | 18 | 15 | 0.042 | 0.015 | 0.05 |
| ATR13 ‡ | 17 | 14 | 0.033 | 0.014 | 0.039 |
| Ppat5 | 16 | 11 | 0.002 | 0.010 | 0.00008 |
| (b) Chi‐squared test comparing observed and expected polymorphism at ATR13 and Ppat5 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Gene | Total no. of sites | Syn. obs.§ | Syn. exp.¶ | Non‐obs.§ | Non‐exp.¶ | χ2 | P‐value | |
| ATR13 | 528 | 7 | 21 | 84 | 70 | 12.04 | 0.0005 | Ka >> Ks |
| ATR13 ‡ | 531 | 7 | 14 | 55 | 48 | 4.80 | 0.0285 | Ka > Ks |
| Ppat5 | 1983 | 12 | 3 | 1 | 10 | 4.80 | 0.0285 | Ka > Ks |
Pairwise differences at synonymous sites.
Pairwise differences at non‐synonymous sites.
Excluding Hind2 allele.
Number of polymorphisms observed at synonymous (or non‐synonymous) sites.
Expected number of polymorphisms at synonymous (or non‐synonymous) sites assuming neutral evolution.
The vast majority of the nucleotide polymorphisms in ATR13 would result in an amino acid variation: in this sample of 15 ATR13 alleles, 84 out of 91 nucleotide polymorphisms resulted in an amino acid variation between encoded proteins. This level of non‐synonymous polymorphism is significantly greater than expected under neutrality, Ka » Ks (Table 1B). When the ATR13‐Hind2 allele is excluded, this result is still significant, indicating that the level of non‐synonymous variation at this locus is significantly greater than synonymous variation and not simply due to the inclusion of one highly divergent allele.
In contrast, the Ppat5 locus showed the opposite pattern, i.e. 12 out of 13 of the observed nucleotide polymorphisms were synonymous and only one polymorphism resulted in an amino acid difference between encoded proteins (supplementary Table S1). As with the ATR13 analysis, a chi‐squared test of independence was used to determine whether the proportion of synonymous and non‐synonymous variation was consistent with neutral evolution at this locus (Table 1b). Here, too, we were able to reject the hypothesis of neutrality; however, rather than observing too many non‐synonymous changes, we observed too many synonymous changes at this locus indicating the action of purifying selection operating at Ppat5.
Codon usage analysis provided an independent assessment of the action of natural selection at these loci. For each amino acid, a certain synonymous codon may be preferred or used more frequently than others, resulting in codon usage bias. Betancourt and Presgraves (2002) observed a negative correlation between the frequency of optimal (preferred) codon usage and the rate of non‐synonymous evolution. They hypothesized that interference between selection acting on codon usage bias and amino acid substitutions caused the decline of optimal codon usage with increasing rate of non‐synonymous evolution. Subsequently, theoretical simulations showed that, for reasonable parameter estimates, the level of codon bias declines with an increasing rate of substitution at the strongly selected locus (Kim, 2004). We used the effective number of codons (ENC) as a measure of codon usage bias at these two loci. At ATR13, ENC was 59.23 (out of a possible total of 61), indicating that codon usage bias is negligible at this locus (because nearly all possible codons are used). At Ppat5, ENC was 37.51, indicating a large codon usage bias. These results corroborate the inference that strong positive selection may be acting at ATR13, resulting in the maintenance of amino acid polymorphism, and a relaxation of codon usage bias.
Distribution of variation across the protein
All alleles encode a signal peptide that is remarkably conserved between them with only a single position, F15, being replaced by M or L in nine alleles. All alleles recognized by RPP13‐Nd contain the phenylalanine residue at this polymorphic position. The RXLR motif region (comprising 30 amino acids) is completely conserved with the exception of three polymorphisms: H28 to Y encoded by seven alleles, and A42 to V (ATR13‐Waco5) and V47 to D (ATR13‐Hind2) transitions encoded by single alleles.
In contrast to the preceding domains, the leucine heptad repeat region contains five protein variants with 12 amino acid polymorphisms. Three of the different variants were recognized by RPP13‐Nd, suggesting that alteration of the amino acid sequence in this region is tolerated. Leucine heptad repeats are a feature of coiled coils where the leucines are embedded in an alpha helical region. The existence of a leucine/isoleucine heptad repeat in this region suggested the presence of a coiled coil. Secondary structure prediction (SSP program) of ATR13 revealed that this protein is predicted to be highly alpha helical, suggesting a globular, tightly folded structure. In the region of the leucine heptad repeat, only one small stretch of alpha helix is predicted, spanning the central leucine, suggesting the absence of a coiled coil. However, only one of the isoleucine residues (I71), among the 18 sequences, is substituted (a conservative substitution with leucine), perhaps due to strong selective constraint in this region. Conservation of the protein sequence in this region may indicate an important role of these residues in the function of this effector protein.
All protein variants contain at least one 11‐amino‐acid direct repeat region. A single repeat unit is found in ATR13‐Aswa1, ATR13‐Emco5, ATR13‐Goco1‐A and ATR13‐Goco1‐B (ATR13‐Hind2 has one repeat unit followed by an additional 3 amino acids) whereas three units are present in ATR13‐Bico1, ATR13‐Waco5 and ATR13‐Wela3, and four units are present in ATR13‐Maks9, ATR13‐Hiks1, ATR13‐Cala2, ATR13‐Cand5, ATR13‐Hind4, ATR13‐Emwa1‐A, ATR13‐Emwa1‐B, ATR13‐Emoy2, ATR13‐Noks1 and ATR13‐Ahnd1. In addition there are 14 polymorphic positions, resulting in a total of seven variant forms within this domain. No association between the number or sequence identity of these repeats and recognition by RPP13‐Nd could be detected.
The C‐terminus of the predicted protein is the most highly variable domain between variants with 62% of polymorphic sites (49 out of 79) in the protein residing in this domain, which itself constitutes 27% of the protein. ATR13‐Maks9, which is recognized by RPP13‐Nd, and ATR13‐Emoy2, which is not recognised by RPP13‐Nd, exhibit 11 amino acid differences between them and all these lie in the C‐terminus and are due to only 12 nucleotide polymorphisms (two being present in one codon). This suggests that specificity for the recognition by RPP13‐Nd lies in this C terminal domain.
Importance of the heptad repeat region in the recognition response
The leucine/isoleucines of the heptad repeat are highly conserved, with a single conservative amino acid substitution in one allele, in all 15 protein variants of ATR13 (Fig. 1), which suggests that they may be required for structure or function. We mutated these amino acids to determine whether they play a role in the recognition of ATR13‐Maks9 by RPP13‐Nd (supplementary Table S2). Mutations L50G, L57G or I71G did not affect recognition. Mutation I78G resulted in a distinctly different recognition response, which we term intermediate (see Fig. 2 for an example and definition of this phenotype). The biological relevance of this phenotype is unknown but is consistent in all experiments. Mutation L64G abolished recognition. We considered whether this change in phenotype was due to lack of protein stability of the mutant ATR13 proteins. We expressed ATR13‐Maks9 and the mutants L64G and I78G in an Escherichia coli expression system and found that they were produced in comparable amounts to the wild‐type protein (data not shown).
Figure 2.
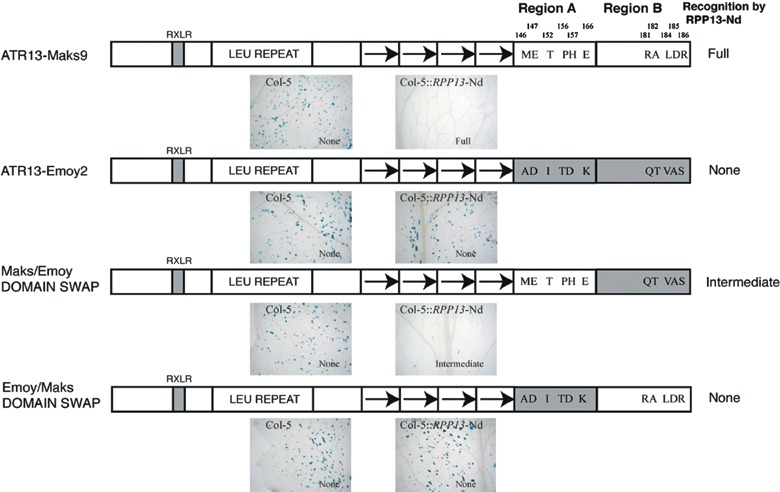
ATR13 domain swap constructs and their recognition response by RPP13‐Nd in the biolistic assay. Region A and Region B are the two domains of the C‐terminal region and amino acids shown are the 11 amino acids that differ between ATR13‐Maks9 and ATR13‐Emoy2. Photographs (representative images) show the three distinct recognition response phenotypes in Col‐5 and Col‐5::RPP13‐Nd; no response results in 300–1000 blue‐stained cells per leaf, a full response results in fewer than ten per leaf and an intermediate response results in 50–150 per leaf.
Heptad leucine repeats are components of coiled coils, which mediate protein–protein interactions. This region of ATR13 is not predicted to be a coiled coil as the leucine repeats are not embedded in an alpha helical region. However, there is a short stretch of alpha helix in this region, which spans L64. The L64G mutation, which would disrupt the alpha helix, caused a loss of recognition of ATR13‐Maks9 by RPP13‐Nd. However, a further mutation, L64A, which would retain the alpha helix, also resulted in a loss of recognition by RPP13‐Nd. A mutation, K66G, made between the conserved leucines or isoleucines, which would also disrupt the alpha helix, had no effect on the recognition response in the biolistic assay. These results suggest that L64 is absolutely required for the resistance response, but the presence of an alpha helix in this region is dispensable.
C‐terminal domain can be subdivided in terms of RPP13‐Nd recognition
As the avirulence allele ATR13‐Maks9 and the virulence allele ATR13‐Emoy2 only encode differences in the C‐terminal domain (in 11 amino acids), we constructed domain swaps within this region to delineate further the positions involved in recognition. The domain could be divided into two regions: with six amino acid differences between these variants in Region A (positions 146, 147, 152, 156, 157 and 166) and five amino acids differences in Region B (181, 182, 184, 185 and 186) (Fig. 2). Domain swaps between these were made producing an Emoy/Maks (Region A Emoy2, Region B Maks9) domain swap and a Maks/Emoy (Region A Maks9, Region B Emoy2) domain swap. These domain swaps were identical apart from the C‐terminus. The Emoy/Maks domain swap was not recognized by RPP13‐Nd in the biolistic assay, but the corresponding Maks/Emoy domain swap was recognized, but resulted in an intermediate response (Fig. 2). These results show that one or more of the amino acids in Region A are required for recognition and one or more in Region B are required to elicit a full recognition phenotype.
Site‐directed mutagenesis of key amino acids in the C‐terminal domain
Of the 11 amino acid polymorphisms in the C terminus, variants at positions 146, 157, 182, 185 and 186 were not associated with recognition by RPP13‐Nd. This allowed us to narrow down the possible candidates that affect isolate‐specific recognition by RPP13‐Nd to six positions (147, 152, 156, 166, 181 and 184). Having determined that the C‐terminal domain plays a key role in RPP13 recognition we carried out site‐directed mutagenesis of these six amino acids. As both regions of the domain swap clones initiated different recognition responses, we mutated these regions independently. Mutation of the clone carrying Emoy2 Region A and Maks9 Region B at the four amino acids putatively involved in RPP13‐mediated recognition, in Region A (clones Emoy/Maks.1 to Emoy/Maks.4), revealed that a threonine at position 152 was absolutely required and that these other amino acid positions had no effect on recognition (Fig. 3). To test if T152 was required for protein stability, a T152I ATR13‐Maks9 mutation was made and expressed in an E. coli expression system. The encoded protein was expressed at similar levels to that of ATR13‐Maks9 wild‐type (data not shown). Mutation of the two candidate amino acids in the B region of ATR13‐Maks9 revealed that L184V had no effect on recognition whereas R181Q clearly demonstrated that even with T152, lack of an arginine at position 181 results in an intermediate recognition phenotype seen in the original domain swap experiment (Fig. 2). Hence, T152 is absolutely required for RPP13‐Nd recognition and this phenotype is modulated by R181.
Figure 3.
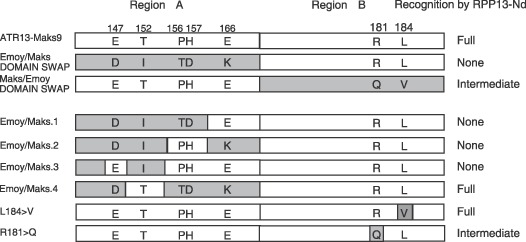
C‐terminal regions of Emoy/Maks and Maks/Emoy domain swaps and mutants of domain swaps and their recognition responses by RPP13‐Nd in the biolistic assay. Grey shaded boxes indicate sequence from the ATR13‐Emoy2 allele; unshaded boxes indicate sequence from the ATR13‐Maks9 allele.
Mutation of ATR13‐Emoy2 to gain of recognition
The results from site‐directed mutagenesis suggested that ATR13‐Emoy2 is not recognized by RPP13‐Nd because it has I152 and Q181 rather than T152 and R181. We tested whether the substitutions of I152T and Q181R would convert a non‐recognised variant into a recognised variant. When the I152T substitution was introduced into the ATR13‐Emoy2 protein, this mutant showed intermediate recognition by RPP13‐Nd (Fig. 4), as expected based on the domain swap experiments and confirmed our finding that a threonine at position 152 is critical for recognition. When the Q181R substitution was introduced into the ATR13‐Emoy2 protein no recognition was observed in the biolistic assay, as the clone lacked T152. However, surprisingly, when these mutations were combined, the construct resulted in only an intermediate recognition phenotype. This implies that the context of the amino acids, which are involved in recognition, affects recognition and that another amino acid position (or positions) may contribute to recognition.
Figure 4.

(A) C‐terminal region of mutant forms of ATR13‐Emoy2 and their recognition response by RPP13‐Nd in the biolistic assay. (B) C‐terminal region of ATR13‐Maks9, ATR13‐Wela3 and mutants of ATR13‐Wela3 and their recognition response by RPP13‐Nd. All differing amino acids between ATR13‐Maks9 and ATR13‐Wela3 are shown in the wild‐type constructs. Grey shaded boxes indicate sequence from ATR13‐Wela3; unshaded boxes indicate sequence from the ATR13‐Maks9 allele.
Mutation of ATR13‐Wela3 confirms role of C‐terminal domain in RPP13‐Nd recognition
The ATR13‐Wela3 variant is not recognized by RPP13‐Nd and exhibits polymorphisms in all the domains when compared with ATR13‐Maks9. We reasoned therefore that if the structure of the C‐terminal domain was key to RPP13‐Nd‐mediated recognition then only polymorphisms within this domain in ATR13‐Wela3 would be significant in the interaction. There are six amino acid differences between the recognized ATR13‐Maks9 variant and that of ATR13‐Wela3 within the C‐terminus in addition to differences outside of the C‐terminus (Fig. 4). Four of these polymorphisms (D157, T182, A185, S186) are present in variants, which are recognized by RPP13‐Nd (Fig. 1). ATR13‐Wela3 has an isoleucine at position 152 rather than the essential threonine and K147 instead of E147 which is present in all variants recognized by RPP13‐Nd. In common with ATR13‐Maks9, ATR13‐Wela3 has R181. Therefore, we predicted that to convert ATR13‐Wela3 from an unrecognized to a recognized form would require the two substitutions I152T and K147E. These two substitutions independently did not result in recognition by RPP13‐Nd (clones ATR13‐Wela3.1 and ATR‐Wela3.2), but a combination of the two substitutions, K147E and I152T (clone ATR13‐Wela3.3) resulted in full recognition (Fig. 4). This demonstrates the importance of E147 and T152 and confirms that the polymorphisms outside the C‐terminal domain were not significant to the absence of recognition of ATR13‐Wela3 by RPP13‐Nd.
To test that the proteins encoded by the ATR13‐Wela3 mutants were produced in a functional form in the biolistic assay we assessed their recognition capability on Arabdiopsis accessions UKID71. We will show in other work in preparation that accession UKID71 shows a resistance response in the biolistic assay to wild‐type ATR13‐Wela3 and that this interaction is not mediated via RPP13 but by an unlinked Arabidopsis resistance gene. Therefore, we hypothesized that if the proteins were produced in planta in the biolistic assay and the mutations were specific to the RPP13 interaction then all of the variant forms would be recognized by UKID71. Hence, we tested the response of this accession to ATR13‐Wela3 and to the K147E, I152T and K147E I152T mutants of ATR13‐Wela3. We found that all of the mutants were recognized by UKID71 in the same manner as ATR13‐Wela3 (Fig. 5). A control variant, ATR13‐Waco5, showed no recognition by this accession as expected. This shows that these mutant protein variants must be stably produced in the detached leaves of the assay and that the mutations did not affect recognition by the R gene unlinked to RPP13.
Figure 5.
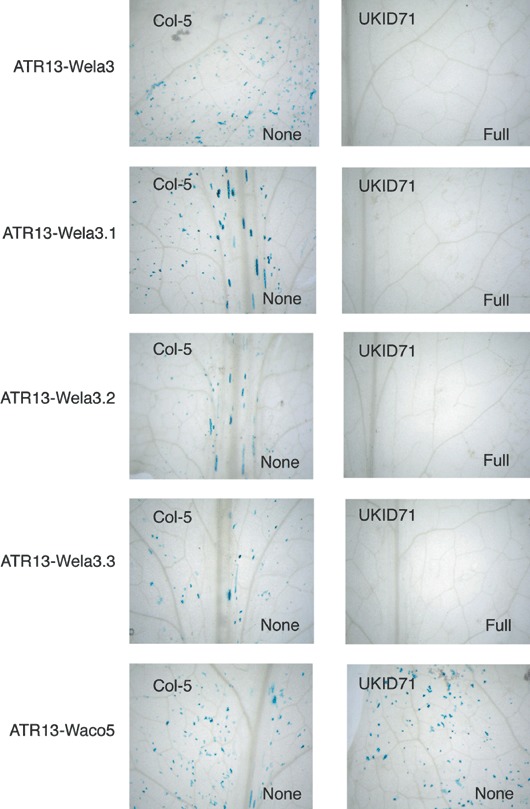
Recognition response of ATR13‐Wela3 mutants by UKID71 and Col‐5 in the biolistic assay.
DISCUSSION
ATR13 is a hyper‐variable gene from the oomycete pathogen H. parasitica that can be recognized by the Arabidopsis surveillance protein RPP13‐Nd (Allen et al., 2004). This recognition triggers localized cell death, and prevents further growth and reproduction of the pathogen. Here we carried out an in‐depth study of natural variation and evaluated the relationship between the observed amino acid polymorphism and protein function of ATR13. The high level of diversity and the distribution of the variation allowed us to identify specific amino acid positions that are associated with recognition by RPP13‐Nd. We tested the functional importance of these positions by generating novel variants through domain swaps and site‐directed mutagenesis and expressing these in planta.
In this extended dataset, we found additional evidence for an elevated rate of protein evolution, corroborating our previous evolutionary studies of ATR13. Remarkably, none of the alleles are predicted to encode truncated proteins, implying that an intact ATR13 protein may be important for host infection. In contrast to Ppat5 (a reference gene), ATR13 has a greatly expanded codon usage profile, utilizing 59.2 out of 61 possible codons. The lack of codon bias and the excess of non‐synonymous polymorphism at ATR13 is consistent with interference between natural selection for optimal codon usage and the maintenance of a large repertoire of different ATR variants within this species.
The Hind2 allele of ATR13 shows a large number of non‐synonymous differences (an extra 28 compared with all other variants) and most of these are present within the C‐terminal region. This level of divergence may have resulted from recombination with another gene or an indel (insertion or deletion) leading to a frame‐shift mutation. However, in the C‐terminal region, ATR13‐Hind2 shares several polymorphisms with other variants, especially with ATR13‐Ahnd1. The divergent positions within ATR13‐Hind2 are intercalated into a well‐conserved in‐frame backbone sequence. Isolates Hind2, Hind4 and Hiks1 were all isolated from Hilliers Arboretum, Romsey, UK, and while ATR13‐Hiks1 and ATR13‐Hind4 only differ from each other in the signal peptide and RXLR domains, ATR13‐Hind2 shows differences throughout the protein. The diversity uncovered thus far from this local population suggests additional collections from this population may reveal even more diversity.
The signal peptide and RXLR region showed the greatest sequence conservation. The amino acid at position 15 is variant between ATR13 alleles, resulting in three different forms of the signal peptide. The variant containing phenylalanine (F) at position 15 is encoded by alleles that trigger a resistance response. However, a functional association with RPP13‐Nd‐dependent recognition is unlikely as this domain will be lost on secretion from the pathogen. The RXLR region reveals only three amino acid polymorphisms resulting in four variant types. All protein variants recognized by RPP13‐Nd share a common form but this is also present in several unrecognized alleles, suggesting no association with recognition by the R protein. All variants that contain an M15 also contain Y28, suggesting an historical linkage between the signal peptide and RXLR domain. The signal peptide is likely to be involved in effector secretion and the RXLR domain is involved in uptake into the host cytoplasm (Whisson et al., 2007) and may be under strong selective constraint, resulting in the lower rate of protein evolution in these regions as proposed by Win et al. (2007). Consistent with this, deletion of these domains in ATR13 has no effect on RPP13‐mediated recognition (data not shown), which has also been reported by Bos et al. (2006) in the interaction between the Phytophthora infestans AVR3a effector and potato R3a resistance protein.
In the leucine heptad repeat region of ATR13 the leucines/isoleucines that define the repeat are conserved amongst the proteins encoded by all the alleles. Variation exists within the heptad repeat region, but this is not associated with differential recognition specificity by RPP13‐Nd. Many ATR13 variants that do not trigger the resistance response are identical in this region to the ATR13‐Maks9 variant that does trigger recognition. Also, there are three variant forms of ATR13 in this region that do trigger RPP13‐Nd‐dependent recognition. We mutated each of the leucines and isoleucines forming the heptad repeat structure in turn. Mutation of L64 led to a loss of recognition by RPP13‐Nd and mutation of I78 resulted in an intermediate recognition phenotype. No other mutation affected the recognition response. Therefore, conservation of these amino acids is essential to RPP13‐Nd recognition; however, the specificity of the interaction is not determined strictly by this region. The conservation of the leucines/isoleucines within the leucine heptad repeat may imply a key role in the primary function of this protein. One possibility is that this region is involved in the interaction with a host component that is the target of the virulence function of ATR13 and that this interaction is a prerequisite for RPP13‐Nd recognition of ATR13. A mutation of L64 or I78 may prevent interaction of ATR13 with its host target and, hence, ATR13 is not perceived by RPP13.
In the direct repeat region three variant forms containing either one, three or four direct repeat units are able to trigger RPP13‐Nd‐dependent recognition. Eight variants that fail to trigger resistance are sequence identical within this region to that of ATR13‐Maks9, which induces RPP13‐Nd‐dependent recognition. Therefore, despite variation in repeat number and protein sequence no association with RPP13‐Nd recognition was detected. This is in stark contrast to the situation in the avirulence protein AvrBs3 from Xanthomonas campestris where the number and nature of the repeats determines resistance specificity (van den Ackerveken et al., 1996). In the case of AvrBs3, altered repeat structures changed the recognition profile of the AVRBs3 protein by the Bs3 resistance protein and led to novel recognition by different plant proteins. Diversity in the repeat region of ATR13 may be maintained due to recognition by another resistance gene not yet identified, as our results indicate that this variation is not associated with RPP13.
Two observations suggest that the C‐terminal region plays a key role in RPP13‐Nd‐dependent recognition; the large amount of non‐synonymous polymorphism in this region suggests it is experiencing diversifying selection and ATR13‐Maks9 (recognized) and ATR13‐Emoy2 (not recognized) are sequence identical throughout the protein except for 11 amino acid polymorphisms in the C‐terminal domain. Therefore, amino acid changes in this region affect RPP13‐Nd recognition. Our integrated evaluation of protein variation and recognition specificity allowed us to identify six candidate amino acids (E147, T152, P156, E166, R181, L184) that were potentially associated with recognition specificity. Using domain swap and site‐directed mutagenesis we demonstrated that T152 was absolutely required for RPP13‐Nd‐dependent recognition and that R181 modulated this response as an R181Q mutation caused an intermediate recognition phenotype. To test our predictions functionally, we attempted to mutate the unrecognized ATR13‐Emoy2 allele to a form that could be recognized. Mutation I152T resulted in intermediate recognition. Likewise, the substitution of Q181R did not result in recognition due to the absence of T152. Unexpectedly, a combination of both I152T and Q181R resulted in only an intermediate resistance response, implicating further amino acids involved in triggering RPP13‐Nd recognition. Analysis of the unrecognized ATR13‐Wela3 allele showed that it lacked the essential T152 and suggested that position 147 may also play a significant role. Therefore, the substitutions I152T and K147E were made individually in ATR13‐Wela3 and, as predicted, neither produced an allele capable of inducing an RPP13‐Nd resistance response. When these substitutions were combined, however, the resulting protein was recognized. Interestingly all the mutations that alter recognition results in a change in physical property of the amino acids involved: I152T changes from hydrophobic to hydrophilic; K147E changes from basic to acidic; and Q181R changes from basic to uncharged. Therefore, we conclude that specific recognition of RPP13‐Nd is dependent on the structure of the C‐terminal region and a recognized ATR13 variant requires E147, T152 and R181. Similarly, Wang et al. (2007) report that specific amino acids can affect the interaction of the AVRL567 avirulence proteins with L5 and L6 resistance proteins in flax. As in the AVRL567/L interactions the context of the key amino acids in ATR13 altered the specific interactions.
The functional analyses of ATR13 reported here are based on the interaction with RPP13‐Nd and the mutations we describe altered recognition by that protein. However, the extensive amino acid variation seen amongst ATR13 alleles suggests interaction with more than one resistance protein. Consistent with that model, we demonstrate that recognition of ATR13‐Wela3 by an Arabidopsis resistance gene unlinked to RPP13 is not affected by mutations that alter recognition by RPP13‐Nd. ATR13‐Wela3 has amino acid variation throughout the protein, when compared with ATR13‐Maks9 and it will be interesting to determine where specificity for this novel interaction lies.
This survey of ATR13 alleles has allowed us to use natural variation to predict key amino acids required for the recognition response by RPP13‐Nd. Using domain swap and site‐directed mutagenesis we confirmed those predictions and showed that three specific amino acids in the C‐terminus, E147, T152 and R181, play a key role. No other natural variation could be associated with recognition by RPP13‐Nd. Altering these amino acids did not affect recognition by a resistance gene unlinked to RPP13, raising the intriguing possibility that the extended variation in ATR13 is due to interaction with other resistance genes not yet identified.
EXPERIMENTAL PROCEDURES
H. parasitica isolates and Arabidopsis thaliana accessions
All H. parasitica isolates used in this study were collected from naturally infected Arabidopsis populations within the UK. The location of these populations was described in Bittner‐Eddy et al. (1999) and Holub et al. (1994). The Col‐5::RPP13‐Nd transgenic line was described in Bittner‐Eddy et al. (2000).
Cloning the ATR13 alleles
Genomic DNA preparations from Arabidopsis infected with H. parasitica were generated as described in Allen et al. (2004). As the recognition response of ATR13 by RPP13‐Nd was shown to be unaffected by the presence or absence of the signal peptide the alleles were cloned in their native form (with the signal peptide) using Gateway recombination technology (Invitrogen, Carlsbad, CA). Cloning of ATR13‐Aswa1, ATR13‐Emco5, ATR13‐Goco1 and ATR13‐Hind4 has been previously described in Allen et al. (2004). ATR13‐Maks9, ATR13‐Emoy2, ATR13‐Wela3, ATR13‐Cala2, ATR13‐Noks1, ATR13‐Hiks1, ATR13‐Hind2, ATR13‐Emwa1, ATR13‐Ahnd1, ATR13‐Bico1 and ATR13‐Waco5 were cloned with ATR13‐7F as a 5′ primer and the 3′ primers: ATR13‐8R for ATR13‐Cand5, ATR13‐Emwa1 and ATR13‐Noks1; ATR13‐7R for ATR13‐Maks9 and ATR13‐Waco5; ATR13‐13R for ATR13‐Hind2; and ATR13‐14R for ATR13‐Wela3. These primers contained half the Gateway recombination sites attB1 (for 5′ primer) and attB2 (for 3′ primer) followed by the ATR13‐specific sequence. The products of these primary amplifications were used in a secondary PCR reaction using the Gateway full‐length attB1 (Invitrogen) and attB2 (Invitrogen) primers. The PCR products were recombined with the entry vector pDONR221 (Invitrogen) and the inserted DNA was transferred to the Gateway destination vector pK2GW7 (Plant Systems Biology, University of Ghent, Belgium) by LR recombination and the sequence verified.
Analysis of ATR13 alleles
The DNA sequences from 16 isolates of H. parasitica were analysed using the DnaSP program (v. 4.0, Rozas and Rozas) (Fig. 1). These sequences were aligned using ClustalX (v. 1.81) and phylogenetic analyses were performed using PAUP (v. 4.0 b10). The sequence evolution at ATR13 was compared with that of the Ppat5 locus, a reference gene sequenced from these same isolates. A chi‐squared test of independence was used to evaluate if the ratio of non‐synonymous polymorphism per non‐synonymous sites to synonymous polymorphism per synonymous site was significantly different from unity, i.e. the expectation under neutrality. The numbers of non‐synonymous and synonymous sites were calculated using the SITES program (Hey and Wakeley, 1997). We analysed codon usage at these genes using the program codonW (http://bioweb.pasteur.fr/seqanal/interfaces/codonw.html). Prediction of alpha helix was by the SSP program (http://softberry.com/berry.phtml?topic=ssp&group=programs&subgroup=propt).
Sequence data from this article have been deposited with the EMBL/Genbank data libraries under accession numbers EU549820, EU549821, EU549822, EU549823, EU549824, EU549825, EU549826, EU549827, EU549828, EU549829 and EU549830.
Generation of reporter gene construct
The GUS expression clone used as a reporter was constructed by amplifying the 35S promoter from the plasmid pHellsgate (Helliwell et al., 2002) using primers 35SHells1F and 35SHells1R. The PCR product was re‐amplified with 5′ and 3′ primers containing half of the Gateway recombination sites attB1 and atttB2, respectively, and the 35S‐specific sequences, 35SHells1Fgate and 35SHells1Rgate. This PCR product was re‐amplified using full‐length attB1 and attB2 primers. The 35S promoter entry clone was created using BP recombination of this PCR product with pDONR207 (Invitrogen) and the 35S promoter was transferred to the promoterless GUS destination vectors pKGWS7 and pBGWFS7 (Plant Systems Biology, University of Ghent, Belgium) by LR recombination. Primer sequences are given in supplementary Table S3.
Biolistic analysis
Biolistic assays were carried out as described by Allen et al. (2004). Assays were repeated several times and at least four replicate shots per construct per experiment were carried out. Leaves were incubated for 16 h before staining for β‐glucuronidase.
Generation of domain swap clones
As the six amino acids in Region B could be straddled by a single primer, the domain swap clones could be generated using a 3′ primer designed to incorporate portions of these different ATR13 genes from different isolates using a single round of PCR. For instance, to swap the Emoy2 ATR13 Region B into the ATR13‐Maks9, a 3′ primer designed to the Emoy2 sequence of this region was used to amplify from Maks9 DNA. These domain swapped alleles were cloned without the signal peptide and incorporated the Kozak consensus translational start sequence (Kozak, 1987) and a half Gateway recombination site attB1 at the 5′ end by using a primer ATR13‐11F together with the 3′ primer ATR13‐12R designed for generating the Maks9/Emoy2 domain swap product. The primary amplifications were carried out with an annealing temperature of 45 °C to allow for the reduced specificity of the 3′ primer. A secondary amplification was carried out using the full‐length attB1 and attB2 primers and these PCR products were cloned by Gateway recombination via entry vector pDONR221 into the vector pK2GW7 (Plant Systems Biology, University of Ghent, Belgium). Similarly the primer ATR13‐11F was used together with ATR13‐11R for generating the Emoy2/Maks9 domain swap product and this was cloned by Gateway recombination into the vector pK2GW7. Primer sequences are given in supplementary Table S3. The sequence of all clones was confirmed by cycle sequencing using ABI Big Dye Technology (ABI, Applied Biosystems, Foster City, CA).
Site‐directed mutagenesis
Site‐directed mutagenesis of Gateway entry clones was carried out using the Quik‐change mutagenesis kit by Stratagene (La Jolla, CA), as directed by the manufacturer. Primers for site‐directed mutagenesis were synthesized by Invitrogen (Carlsbad, CA) and HPLC‐purified. Entry clones were mutated and the mutated inserted DNA was transferred to the destination vector pK2GW7 by LR recombination.
Expression of ATR13 mutants in E.coli
The mutant clones were transferred to the E. coli expression vector, pDest17 (Invitrogen) by Gateway technology and E. coli BL21(DE3) were transformed with them. The resulting transformants were grown at 30 °C for 18 h in Luria broth supplemented with 0.2%l‐arabinose to induce protein expression. The cell pellets were re‐suspended in 2 mL buffer containing 500 mm NaCl; 50 mm NaH2PO4, pH 8.0. The cell pellets were sonicated on ice at 22 µ peak to peak for five 30‐s bursts with 30‐s intervals. Insoluble material was removed by centrifugation. Expression was analysed by SDS‐PAGE, staining with Coomassie Brilliant Blue R (Sigma) and de‐staining with 40% (v/v) methanol, 10% (v/v) acetic acid.
Supporting information
Table S1 Ppat5 polymorphisms in 16 isolates of Hyaloperonospora parastitica.
Table S2 Recognition responses of leucine heptad repeat mutants of ATR13‐Maks9.
Table S3 Primers used in this work.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item
ACKNOWLEDGEMENTS
This work was funded by the Biotechnology and Biological Science Research Council. We thank Eric Holub for kindly providing the H. parasitica isolates used in this work.
REFERENCES
- Abramovitch, R.B. and Martin, G.B. (2004) Strategies used by bacterial pathogens to suppress plant defenses. Curr. Opin. Plant. Biol. 7, 356–364. [DOI] [PubMed] [Google Scholar]
- Van Den Ackerveken, G. , Marois, E. and Bonas, U. (1996) Recognition of the bacterial avirulence protein AvrBs3 occurs inside the host plant cell. Cell, 87, 1307–1316. [DOI] [PubMed] [Google Scholar]
- Allen, R.L. , Bittner‐Eddy, P.D. , Grenville‐Briggs, L.J. , Meitz, J.C. , Rehmany, A.P. , Rose, L.E. and Beynon, J.L. (2004). Host–parasite coevolutionary conflict between Arabidopsis and downy mildew. Science, 306, 1957–1960. [DOI] [PubMed] [Google Scholar]
- Armstrong, M.R. , Whisson, S.C. , Pritchard, L. , Bos, J.I.B. , Venter, E. , Avrova, A.O. , Rehmany, A.P. , Bohme, U. , Brooks, K. , Cherevach, I. , Hamlin, N. , White, B. , Frasers, A. , Lord, A. , Quail, M.A. , Churcher, C. , Hall, N. , Berriman, M. , Huang, S. , Kamoun, S. , Beynon, J.L. and Birch, P.R.J. (2005). An ancestral oomycete locus contains late blight avirulence gene Avr3a, encoding a protein that is recognized in the host cytoplasm. Proc. Natl. Acad. Sci. USA, 102, 7766–7771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakker, E.G. , Toomajian, C. , Kreitman, M. and Bergelson, J. (2006) A genome‐wide survey of R gene polymorphisms. Plant Cell, 18, 1803–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancourt, AJ , Presgraves, DC (2002). Linkage limits the power of natural selection in Drosophila. Proc. Natl Acad. Sci. USA, 99, 13616–13620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Biezen, E.A. and Jones, J.D.G. (1998) Plant disease‐resistance proteins and the gene‐for‐gene concept. Trends Biochem. Sci. 23, 454–456 [DOI] [PubMed] [Google Scholar]
- Bittner‐Eddy, P.D. , Canan, C. , Gunn, N. , Pinel, M. , Tor, M. , Crute, I.R. , Holub, E.B. and Beynon, J.L. (1999). Genetic and physical mapping of the RPP13 locus, in Arabidopsis, responsible for specific recognition of several Peronospora parasitica (downy mildew) isolates. Mol. Plant–Microbe Interact. 12, 792–802. [DOI] [PubMed] [Google Scholar]
- Bittner‐Eddy, P.D. , Crute, I.R. , Holub, E.B. and Beynon, J.L. (2000). RPP13 is a simple locus in Arabidopsis thaliana for alleles that specify downy mildew resistance to different avirulence determinants in Peronospora parasitica . Plant J. 21, 177–188. [DOI] [PubMed] [Google Scholar]
- Bittner‐Eddy, P.D. , Allen, R.L. , Rehmany, A.P. , Birch, P. and Beynon, J.L. (2003). Use of suppression subtractive hybridization to identify downy mildew genes expressed during infection of Arabidopsis thaliana . Mol. Plant Pathol. 4, 501–507. [DOI] [PubMed] [Google Scholar]
- Bos, J.I.B. , Kanneganti, T‐D. , Young, C. , Cakir, C. , Huitema, E. , Win, J. , Armstrong, M.R. , Birch, P.R.J. and Kamoun, S. (2006) The C‐terminal half of Phytophthora infestans RXLR effector AVR3a is sufficient to trigger R3a‐mediated hypersensitivity and suppress INF1‐induced cell death in Nicotiana benthamiana . Plant J. 48, 165–176. [DOI] [PubMed] [Google Scholar]
- Dangl, J.L. and Jones, J.D.G. (2001) Plant pathogens and integrated defence responses to infection. Nature, 411, 826–833. [DOI] [PubMed] [Google Scholar]
- Dodds, P.N. , Lawrence, G.J. , Catanzariti, A. , Aycliffe, M.A. and Ellis, J.G. (2004) The Melampsora lini AvrL567 avirulence genes are expressed in haustoria and their products are recognised inside plant cells. Plant Cell, 16, 755–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodds, P.N. , Lawrence, G.J. , Catanzariti, A.M. , Teh, T. , Wang, C.I.A. , Aycliffe, M.A. , Kobe, B. and Ellis, J.G. (2006) Direct protein interaction underlies gene‐for‐gene specificity and coevolution of the flax resistance genes and flax rust avirulence genes. Proc. Natl Acad. Sci. USA, 103, 8888–8893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flor, H.H. (1971) Current status of the gene‐for‐gene concept. Annu. Rev. Phytopathol. 9, 275–296 [Google Scholar]
- Helliwell, C.A. , Wesley, S.V. , Wielopolska, A.J. and Waterhouse, P.M. (2002) High‐throughput vectors for efficient gene silencing in plants. Func. Plant Biol. 29, 1217–1225. [DOI] [PubMed] [Google Scholar]
- Hey, J. and Wakeley, J. (1997) A coalescent estimator of the population recombination rate. Genetics, 145, 833–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiller, N.L. , Bhattacharjee, S. , Van Ooij, C. , Liolios, K. , Harrison, T. , Lopez‐Estrano, C. and Haldar, K. (2004) A host‐targeting signal in virulence proteins reveals a secretome in malarial infection. Science, 269, 843–846. [DOI] [PubMed] [Google Scholar]
- Holub, E.B. , Beynon, J.L. and Crute, I.R. (1994) Phenotypic and genotypic characterization of interactions between isolates of Peronospora parasitica and accessions of Arabidopsis thaliana . Mol. Plant–Microbe Interact. 7, 223–239. [Google Scholar]
- Jia, Y. , McAdams, S.A. , Bryan, G.T. , Hershey, H.P. and Valent, B. (2000) Direct interaction of resistance gene and avirulence gene products confers rice blast resistance. EMBO J. 19, 4004–4014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, J.D.G. and Dangl, J.L. (2006) The plant immune system. Nature, 444, 323–329. [DOI] [PubMed] [Google Scholar]
- Joosten, M.H.A.J. , Cozijnsen, T.J. and De Wit, P.J.M. (1994) Host‐resistance to a fungal tomato pathogen lost by a single base‐pair change in an avirulence gene. Nature, 367, 384–386. [DOI] [PubMed] [Google Scholar]
- Van Kan, J.A.L. , Van Der Ackerveken, G.F. and De Wit, P.J. (1991) Cloning and characterization of cDNA of avirulence gene AVR9 of the fungal pathogen Cladosporium fulvum, causal agent of tomato leaf mold. Mol. Plant–Microbe Interact. 4, 52–59. [DOI] [PubMed] [Google Scholar]
- Kim, Y. (2004) Effect of strong directional selection on weakly selected mutations at linked sites for synonymous codon usage. Mol. Biol. Evol. 21, 286–294. [DOI] [PubMed] [Google Scholar]
- Kim, M.G. , Da Cunha, L. , McFall, A.J. , Belkhadir, Y. , DebRoy, S. , Dangl, J.L. and Mackey, D. (2005) Two pseudomonas syringae type III effectors inhibit RIN4‐regulated basal defense in arabidopsis. Cell, 121, 749–759. [DOI] [PubMed] [Google Scholar]
- Kozak, M. (1987) An analysis of 5′‐noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res. 15, 8125–8148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luderer, R. , Takken, F.L.W. , De Wit, P.J.G.M. and Joosten, M.H.A.J. (2002) Cladosporium fulvum overcomes Cf‐2 mediated resistance by producing truncated AVR2 elicitor proteins. Mol. Microbiol. 45, 875–884. [DOI] [PubMed] [Google Scholar]
- Mackey, D. , Holt, B.F. , Wiig, A. and Dangl, J.L. (2002) RIN4 interacts with Pseudomonas syringae type III effector molecules and is required for RPM1‐mediated resistance in Arabidopsis. Cell, 108, 743–754. [DOI] [PubMed] [Google Scholar]
- Mackey, D. , Belkhadir, Y. , Alonso, J.M. , Ecker, J.R. and Dangl, J. (2003) Arabidopsis RIN4 is a target of the type III virulence effector AvrRpt2 and modulates Rps2‐mediated resistance. Cell, 112, 379–389. [DOI] [PubMed] [Google Scholar]
- Marti, M. , Good, R.T. , Rug, M. , Kneuper, E. and Cowman, A.F. (2004) Targeting malaria virulence and remodeling proteins to the host erythrocyte. Science, 306, 1930–1933. [DOI] [PubMed] [Google Scholar]
- McDowell, J.M. , Cuzick, A. , Canan, C. , Beynon, J. , Dangl, J.L. and Holub, E.B. (2000) Downy mildew (Peronospora parasitica) resistance genes in Arabidopsis vary in functional requirements for NDR1, EDS1, NPR1 and salicylic acid accumulation. Plant J. 22, 523–529. [DOI] [PubMed] [Google Scholar]
- Orbach, M.J. , Farrall, L. , Sweigard, J.A. , Chumley, F.G. and Valent, B. (2000) A telomeric avirulence gene determines efficacy for the rice blast resistance gene Pi‐ta. Plant Cell, 12, 2019–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, J.E. , Holub, E.B. , Frost, L.N. , Falk, A. , Gunn, N.D. and Daniels, M.J. (1996) Characterization of eds1, a mutation in Arabidopsis suppressing resistance to Peronospora parasitica specified by several different RPP genes. Plant Cell, 8, 2033–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rehmany, A.P. , Gordon, A. , Rose, L.E. , Allen, R.L. , Armstrong, M.R. , Whisson, S.C. , Kamoun, S. , Tyler, B.M. , Birch, P.R.J. and Beynon, J.L. (2005) Differential recognition of highly divergent downy mildew avirulence alleles by RPP1 resistance genes from two Arabidopsis lines. Plant Cell, 17, 1839–1850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose, L.E. , Bittner‐Eddy, P.D. Langley, C.H. , Holub, E.B. Michelmore, R.W. and Beynon, J.L. (2004) Maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana . Genetics, 166, 1517–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scofield, S.R. , Tobias, C.M. , Rathjen, J.P. , Chang, J.H. , Lavelle, D.T. , Michelmore, R.W. and Staskawicz, B.J. (1996) Molecular basis of gene‐for‐gene specificity in bacterial speck disease of tomato. Science, 274, 2063–2065. [DOI] [PubMed] [Google Scholar]
- Shan, W. , Cao, M. , Leung, D. and Tyler, B.M. (2004) The Avr1b locus of Phytophthora sojae encodes an elicitor and a regulator required for avirulence on soybean plants carrying resistance gene Rps1b. Mol Plant–Microbe Interact. 17, 394–403. [DOI] [PubMed] [Google Scholar]
- Tang, X. , Frederick, R.D. , Zhou, J. , Halterman, D.A. , Jia, Y. and Martin, G.B. (1996) Initiation of plant disease resistance by physical interaction of AvrPto and Pto kinase. Science, 274, 2060–2063. [DOI] [PubMed] [Google Scholar]
- Wang, C‐I.A. , Guncar, G , Forwood, J.K. , The, T. , Catanzariti, A‐M. , Lawrence, G.J. , Loughlin, F.E. , Mackay, J.P. , Horst, J.S. , Anderson, P.A. , Ellis, J.G. , Dodds, P.N. and Kobe, B. (2007) Crystal structures of flax rust avirulence proteins AvrL567‐A and ‐D reveal details of the structural basis for flax disease resistance specificity. Plant Cell, 19, 2898–2912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whisson, S.C. , Boevink, P.C. , Moleleki, L. , Avrova, A.O. , Morales, J.G. , Gilroy, E.M. , Armstrong, M.R. , Grouffaud, S. , Van West, P. , Chapman, S. , Hein, I. , Toth, I.K. , Pritchard, L. and Birch, P.R.J. (2007) A translocation signal for delivery of oomycete effector proteins into host plant cells. Nature, 450, 115–118. [DOI] [PubMed] [Google Scholar]
- Win, J. , Morgan, W. , Bos, J. , Krasileva, K.V. , Cano, L.M. , Chaparro‐Garcia, A. , Ammar, R. , Staskawicz, B.J. and Kamoun, S. (2007) Adaptive evolution has targeted the C‐terminal domain of the RXLR effectors of plant pathogenic oomycetes. Plant Cell, 19, 2349–2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1 Ppat5 polymorphisms in 16 isolates of Hyaloperonospora parastitica.
Table S2 Recognition responses of leucine heptad repeat mutants of ATR13‐Maks9.
Table S3 Primers used in this work.
Please note: Blackwell Publishing are not responsible for the content or functionality of any supplementary materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
Supporting info item
Supporting info item
Supporting info item