

Abstract
A new series of dicarboxylic derivatives of bispidines have been synthesized to develop novel copper(II) complexes suitable as imaging agents for positron emission tomography. For characterization purposes, copper complexes of bispidines were synthesized in the pure form and in quantitative yields by neutralization of ligands with malachite. The formation of complexes and their stoichiometries were studied by potentiometric titration, cyclic voltammetry, and spectroscopic methods. The stability constants were found to be fairly suitable for copper cation fixation inside dianionic chelate molecules.
Introduction
Positron emission tomography (PET) is one of the most widely used molecular imaging methods, which enables early and high-resolution diagnosis of various diseases, including oncological, neurological, and cardiological diseases. Among the PET radionuclides, fluorine-18 is regarded as an “ideal” PET radionuclide, with optimum nuclear-physical characteristics (97% β+, 3% E.C., E(β+)max = 0.635 MeV). However, in recent years a growing interest in new radiopharmaceuticals (RPs) with nonconventional PET radionuclides, for example, 86Y (14.7 h), 89Zr (78.4 h), 64Cu (12.7 h), and other radiometals, has been observed.1−5 This is motivated by their longer half-life periods, which enable tracing of processes with relatively slow pharmaceutical kinetics. These RPs could be applied to estimate the efficiency of radioimmunotherapy using labeled intact monoclonal antibodies and their fragments as well as so-called engineered protein (affibody, diabody, nanobody, etc.) antibodies and their fragments, whose biological action lasts from a few hours to a few days.6
Among copper isotopes, four (60Cu, 61Cu, 62Cu, 64Cu) are considered potential medical positron emitters.7,8 The results obtained using these agents for visualizing solid hypoxic tumors or their parts (60,62,64Cu-diacetyl-bis(N4-methylthiosemicarbazone) (60,62,64Cu-ATSM)) and choosing the therapy and estimating its efficiency, as well as in cardiologic (myocardial perfusion, 62Cu-pyruvaldehyde-bis(N4-methylthiosemicarbazone)) and, more rarely, neurological examinations are summarized in an overview.964Cu is the most interesting copper isotope because it manifests the smallest average energy of positrons, almost does not emit high-energy γ-quanta, and has a half-life that allows the radionuclide to be delivered to clinics within a region. 64Cu could be produced in 64Ni(p,n)64Cu with a high yield, of up to 5.9 GBq/μA (irradiation parameter 3 μA and 1 h of bombardment), and high purity using common medical cyclotrons.10 Alternative ways of 64Cu production are described in the recent review on nonconventional PET radionuclides.11
Unlike the incorporation conventional PET radionuclides, incorporation of metal isotopes requires the use of bifunctional chelating agents that, in turn, can form covalent bonds with various biomolecules, with a high specificity and selectivity toward certain targets.3 For this, elaboration of easily modified ligand systems (bifunctional chelators) with a high affinity toward metal cations as well as with wide possibilities of chemical modifications, including conjugation to biological vectors, is required. Only ligands that can provide an essential kinetic inertness combined with a high thermodynamic stability are interesting in this respect.4 The known examples of copper(II) chelates studied in vitro and in vivo lack either kinetic or thermodynamic stability.12 The task of attaching the vector group to the radiolabeling moiety is often difficult to solve synthetically.
For PET diagnostics with Cu isotopes, the most useful ligands are tetrazamacrocyclic compounds, such as ATSM, cyclen, cyclam, 1,4,7,10-tetra-azacyclododecane-1,4,7,10-tetrayltetra-acetic acid (DOTA), and diamsar (Figure 1). In this article, we start to explore the series of diazabicycles as a central component of the potentially useful ligand families. Some of our previous work on diazamonocycles and bicycles have already been published (piperazines,14−16 homopiperazines,17 bispidines,18,19). A promising approach is to explore chelate (Figure 2, top and middle) and macrocycle (Figure 2, bottom) effects. This could be achieved at the ligand-design stage, which includes the choice of the central diazacore, the nature (Q’s) and length (k’s) of the pendant arms, and that of the cyclizing bridge (n’s). In the context of this challenge, we noted with interest complexes of tetradentate bispidine-based ligands, studied mostly by Comba et al.20−36 Recently, bispidines themselves proved to be privileged structures in medicinal chemistry.37 The rigid framework of bispidines in double-chair conformation allows them to be used to study the binding of small cations for a long time.38 This class of preorganized acyclic ligands is similar to the well-known cyclohexyl-diethylenetriaminepentaacetic acid (CHX-DTPA) because rigidity of the acyclic fragment is achieved by partial cyclization. CHX-DTPA is used for complexation of trivalent cations like Bi3+, rare earth elements in conjugates with monoclonal antibodies39,40 because it provides an optimal set of thermodynamic and kinetic properties of the complexes formed.
Figure 1.

Selected structures of copper-chelating compounds.13
Figure 2.

Illustration of the ligand-design principles for metal-based PET experiments. Bidentate N2-chelating diazacores (top), tetradentate N2Q2-chelating systems with pendant arms (middle), and macrocyclic tetradentate N2Q2 systems (bottom).
Bispidines form very stable complexes with Cu2+, which can be modified to bind with biomolecules, such as enzymes, for target delivery.21,23,24,30,32 As a vast majority of these bispidines belong to chelating macrocycles of neutral N4, N5, and N6 types,24,26,33,36,41 they form positively charged complexes with Cu2+. This means that there is a necessity to use counteranions to compensate for the positive charge of the complex.
However, the simplest tetradentate ligand is bispidine of the N2O2 type (see Figure 2, middle right, Q = O), which possesses a rigid preorganized structure to form highly stable neutral complexes with Cu2+. 19
In the solid state, the metal ion, Cu2+, has a typical coordination number of 5 (corresponding to a distorted square-pyramidal coordination polyhedron), comprising two nitrogens from the bispidine backbone, two oxygens from the carboxylic residues, and an oxygen atom from the solvent molecule or a neighboring complex.
Bispidine-type ligands can be easily modified to fulfill a task of attaching to the desired vector of biological object. Indeed, these molecules possess at least two sites that can be used for conjugation with biomolecules (see Figure 1, bottom right): (a) if X = OH and Y = H, then the hydroxyl could be modified to carbamate33 or propargyl ether moieties; if XY = O, then the carbonyl could be reduced to a hydroxyl moiety42 and all of the possibilities for hydroxyl functionalization are then applied; moreover, the bispidinic carbonyl group is known to form hydantoins.43−45 (b) R groups could have linkable residues like SH, COOH, OH,46 and so on.
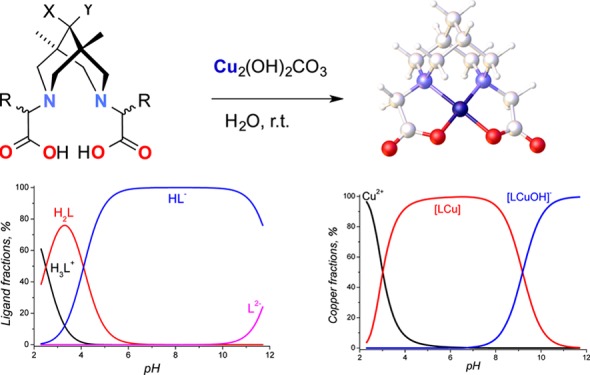
This article describes the synthesis of N2O2-type tetradentate bispidines differing in their substituents at position 9 (hereafter called “C9”) (Figure 3) and their respective copper complexes. The intention behind the synthesis of the target structures, 2a–c, 3b, and 3c, was as follows: (1) to study the influence of a substituent at C9 on the stability and synthetic availability of the complex and (2) to study the possible influence of a substituent in the pendant arm (H vs Me in pairs 2b/3b and 2c/3c) on complex stability. Basic copper carbonate (malachite) was applied as a copper source in the syntheses of complexes 4b, 4c, 5b, and 5c from ligands 2b, 2c, 3b, and 3c, respectively.
Figure 3.

Structures of organic compounds studied and/or discussed in this work, with their numbering. The inset shows the numbering of carbon atoms in 2a as an example.
The structures of the complexes were confirmed by UV–vis and IR spectroscopy and mass spectrometry. We compared the formation and stability of copper complexes of our target molecules, 2a–c, 3b, and 3c, with those of N,N′-unsubstituted analogues 1a and 1c and carboxyl-containing analogues 2d and 2e using potentiometric titration and cyclic voltammetry (CV).
Results and Discussion
Ligand Synthesis
Compounds 2a–c, 3b, and 3c were synthesized starting from urotropine and diethyl ketone by applying known sequences (Scheme 1).18,19,26,30,47−49 Alkylation of compounds 1a–c was performed using different alkylating agents. Whereas the reactions with chloroacetic acid for the preparation of unbranched compounds 2a–c proceeded readily, the reactions with racemic 2-chloropropionic acid for the preparation of products 3b–c were rather slow, that is, the conversions at 3 h did not exceed 50%. Thus, 2-bromopropionic acid was used instead of the chloro analogue, giving complete conversion after 3 h. Another point to be mentioned here is the excellent solubility of target compounds 2 and 3 in water, which is a very good property for administration to organisms but makes ligand separation from the reaction mixtures difficult. To overcome this problem, we used a known synthetic procedure for separation, which includes precipitation of the barium salt of the target compound and exclusion of the barium cation using diluted sulfuric acid.30 The overall yield of the alkylation step is quite moderate (30–50%). Considering the alkylation efficiency (by means of NMR) and efficiency of barium precipitation as barium sulfate (100%), one can conclude that the maximum loss of target ligands 2 and 3 occurred at the barium salt precipitation stage. Presumably, the main reason for this loss is the quite good solubility of barium salt of ligands in water especially in the case of ligands 3b and 3c.
Scheme 1. Preparation of Target Compounds.

(i) Et2CO, n-BuOH, AcOH, reflux, 2 h; (ii) N2H4, NaOH, ethylene glycol, reflux, 24 h; (iii) AcCl, NaHCO3, tetrahydrofuran (THF), H2O, room temperature (rt); (iv) Ac2O, rt, 8 h; (v) HCl, H2O, reflux; (vi) NaBH4, EtOH, rt, 2 h; (vii) HalCH(R)CO2H, NaOH, rt, 3 h; (viii) BaCl2, H2O, reflux; (ix) 2.3 M H2SO4, reflux.
Checking the spectroscopic properties of the ligands, we found the presence of ketone-gem-diol equilibrium for compounds 2b and 3b (Scheme 2). For example, compound 2b in dimethyl sulfoxide (DMSO)-d6 solution has only one set of resonances that is consistent with its structure. In contrast, in aqueous solution two sets of signals corresponding to ketone 2b (δ(13C(9)) = 212.03 ppm) and geminal diol 2b′ (δ(13C(9)) = 93.71 ppm) are observed (Figure S29).
Scheme 2. Ketone-gem-diol Equilibrium in Water.

In a room temperature equilibrium mixture, the 2b/2b′ ratio is about 0.9:1.0. Upon heating to 60 °C, the ratio changes to ∼1.8:1.0, reducing to the starting position after cooling to rt (Figure 4). An analogous hydration reaction was described for quaternary ammonium salts of 5,7-dimethyl-1,3-diazaadamantan-6-one in D2O50 and for bispidine platinum complexes.51 Commonly, the behavior of compound 3b in D2O is similar (Figure S31) to that of the glycine analogue, 2b, described above. The main difference is the presence of two pairs of diastereomers for 3b and 3b′, whose signals are clearly different in the 13C spectrum. In the rac-form (C2 symmetry) framework, the carbons of ketone and gem-diol that were located at opposite sites were pairwise equivalent (C2 and C6, C4 and C8, C1 and C5). This is also true for respective methyl groups and substituents at nitrogen atoms. For the meso-form (the symmetry plane passes through the C1, C9, and C5 atoms), carbon atoms C2 and C8 and C4 and C6 are pairwise equivalent but atoms C1 and C5 with methyl groups are nonequivalent and have paired signals of equal intensities in the 13C spectrum. Thus, the diastereomeric ratio rac-/meso- can be approximated as 1:0.8. The 1H NMR spectrum of this mixture is more complicated because of strong overlapping of signals (Figure S30). However, the CCH3 signals of 3b and 3b′ appear as singlets of the rac-form and a pair of singlets of the meso-form. If one considers the CCH3 groups, signals of 3b′ appear at a higher field (0.94 ppm) than those of 3b (1.02 ppm), and analogously to the 2b/2b′ mixture, the 3b/3b′ ratio in D2O solution is about 1.4:1.0. The protons of the framework of the methylene groups of compound 1c in D2O solution belong to the ABCD system, where only three spin–spin coupling constants (2JAB, 2JCD, and 4JAC) are not equal to zero. The difference in chemical shifts in the first approximation allows us to analyze this spin system as an AMPX system.
Figure 4.

1H NMR spectra of compound 2b in D2O (a), in D2O at 60 °C (b), and in DMSO-d6 (c).
Probably, long-range spin–spin coupling constants, 4JHH = 2.8 Hz, are visible for nonequivalent protons H2,4 and H6,8 (W-arrangement of chemical bonds). In the 1H spectrum of 2c, H2,4 and H6,8 proton interaction is invisible due to wider signals; so, NCH2 resonances can be considered as a superposition of two AB systems (or as two pairs of doublets). The 1H and 13C NMR spectra of compound 3c in D2O show the presence of a mixture of two diastereomers in an ∼5:1 ratio (Figure S34). Their signals are clearly visible in the 13C spectrum, but in the 1H spectrum, the major diastereomer masks the signal of the minor diastereomer. Because of the diastereotopy of NCH2 protons, their signals are a superposition of four AB systems rather than of two, as for 1c and 2c. All four framework CH2N carbons of both diastereomers (eight signals in 13C spectrum) are also nonequivalent.
Copper Complex Preparation
Generally, the synthesis and application of 64Cu complexes in PET means application of “wet” chemistry. Therefore one should concentrate on the synthesis and study of appropriate solutions. However, to obtain as much information about target complexes as possible, we have to isolate them in the pure form and collect information on several characteristics that are vital for their practical application, the first being their chemical and electrochemical stabilities. The main methods for isolation of the complex are (1) evaporation of the solvent after reaction of a ligand and copper inorganic salt, followed by subsequent crystallization of the copper complexes and (2) vapor diffusion of antisolvent into the copper complex solution.30,36 Unfortunately, these approaches were found to be ineffective for isolation of the compounds under investigation because of their high solubility in water. Another method for copper complex preparation is neutralization of freshly prepared copper(II) hydroxide with an acidic N2O2-chelate.19 We performed the same reaction with ligand 2b and proved that neutralization of acid 2b, followed by dissolution of Cu(OH)2, proceeds instantly under ambient conditions.
However, the main drawback of this method is the presence of inorganic salts, for example, KCl, which contaminate the copper complex solution after reaction completion. Thus, we suggest, for the first time, the use the malachite Cu(OH)2CO3 as a gravimetric form of Cu(OH)2 for copper–bispidine complex preparation, which has the advantages of the absence of dehydration in air and the absence of contamination products in the reaction (Scheme 3). Basic copper carbonate (malachite) reacts readily with bispidines 2b, 2c, 3b, and 3c in water at rt overnight (see the movie in the SI). All of the yields are quantitative. By this new approach, four copper salts of 2,2′-(1,5-dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl)dicarboxylic acids, 2b and 3b, and 2,2′-(1,5-dimethyl-9-hydroxy-3,7-diazabicyclo-[3.3.1]nonane-3,7-diyl)dicarboxylic acids, 2c and 3c, were synthesized for the first time.
Scheme 3. Copper Complex Preparation.

Spectroscopy of Copper Complexes
Because of the paramagnetic nature of Cu(II), no NMR spectra of sufficient quality can be registered for bispidine complexes. Therefore, complex formation was studied by comparison of the FTIR spectra of the complexes with those of the free ligands (Figure 5). All ligands have characteristic vibrations at 1610–1620 cm–1 and 1710–1720 cm–1 of the C=O group, which are typical of the carboxylic acid moiety30 (Figures 5 and S36). C=O vibrations of the keto group are likely positioned under the carboxylic C=O vibrations, giving coalescent peaks at 1610–1620 cm–1.30,52 It is interesting that all ligands have quite similar peaks at ca. 1698–1726 cm–1. This is probably due to the rotational freedom of carboxylic groups in free ligands, which leads to intramolecular hydrogen-bond formation between one carboxylic group and a nitrogen atom or oxygen atom of diol or secondary alcohol. The carboxylic groups became equivalent, being rigidly fixed in the copper complexes. This conclusion is supported by the presence of only one coalescent peak at 1595–1630 cm–1 for all copper complexes. The UV–vis spectra of all copper salts are almost identical, with a weak metal-to-ligand charge-transfer band at 602–612 nm and an adsorption band of the ligand at 247–252 nm (Figure S37). Copper complexes were also studied in a methanol–water mixture (1:1) by means of high-resolution mass spectrometry–electrospray ionization (HRMS–ESI) spectrometry. In a solution of the ketone-containing complex, 4b (Figure S38a), cations of mononuclear CuL(H2O)H+ (m/z 364.06977) and binuclear complexes Cu2L2H+ (m/z 691.11143), Cu2L2(H2O)H+ (m/z 709.12237), and Cu2L2(H2O)2H+ (m/z 727.13281) are present (consider free ligands as LH2). An analogous situation is observed for the hydroxyl-containing complex, 4c (Figure S38b): LH+ (m/z 287.16013), CuLH+ (m/z 348.07404), Cu2L2H+ (m/z 695.14155), and Cu2L2Na+ (m/z 717.12257). The presence of binuclear cations in the mass spectra coincides with the structure proposed by Comba et al. for complex 2a with copper(II) on the basis of quantum chemical calculations and ESP spectroscopy study.30
Figure 5.

Attenuated total reflection (ATR)-FTIR spectra of ligands 2b (a) and 2c (c) and copper complexes 4b (b) and 4c (d).
Electrochemical Study
As the Cu(II)/Cu(I) redox reaction is suggested as a driving force for radiocopper loss,53,54 a CV study on the redox stability of our Cu(II) complexes was carried out. Earlier,19,55 we have studied the redox properties of a series of Cu(II) complexes with the general formula LCuCl2 (L is 3,7-dialkyldiazabicyclo[3.3.1]nonan-9-one) and of Cu(II) complexes with tetradentate N2O2 ligands derived from diazabicyclo[3.3.1]nonan-9-ones, including 2e and 3e. It was hypothesized that oxidation of the complexes with N2-type bidentate ligands occurs at the lone pairs of the nitrogen atoms, giving rise to CuCl2, whereas reduction leads to electron transfer from the electrode to the copper atom, both processes leading to decomposition of the complexes. In contrast, in the case of N2O2-type tetradentate ligands, high complementarity between the dianionic ligand and Cu(II) metal was observed, which is attributed to their high reduction potentials compared to those in the LCuCl2 series. Such complementarity leads to increased stability of the Cu(II) complexes toward reduction. In the present study, we examined the electrochemical properties of complexes 4b, 4c, 5b, and 5c by CV in dimethylformamide (DMF) at a Pt electrode (Table 1) and compared the results with the data published for complexes of ligands 2e and 3e.19
Table 1. Oxidation (EpOx) and Reduction (EpRed) Potentials of Complexes 4 and 5 at a Pt Electrodea.
Oxidation
All investigated copper complexes are irreversibly oxidized. Anodic behavior is similar within the set of compounds 4b and 4c and 5b and 5c.
At the same time, electron transfer from complexes 4b and 4c and 5b and 5c is approximately 500 mV easier in comparison to that from their phenyl analogue 5e. We assume that oxidation leads to the formation of cation radicals, [LCu]+Ÿ, with their consequent destruction to release free Cu2+ cations. This has been proven by the fact that peaks corresponding to Cu2+/Cu+ (0.31 V) and Cu+/Cu2+ (0.58 V) are observed after the first scan from 0 to 1.5 V and vice versa. Moreover, peaks at ∼1.0 V, probably corresponding to oxidation and destruction of free organic cation radicals that were formed after the release of Cu2+, are found in the anodic part of the CV curve after several scans (Figure S40). Comparison of the anodic electrochemical behavior of 4b and 4c and 5b and 5c to that of 4e and 5e clearly demonstrates the influence of substituents at positions 1 and 5 of the bicyclic core on the electrochemical oxidation of complexes. At the same time, substitution of a hydrogen atom for a methyl group in 4b/5b and 4c/5c does not influence the electrochemical behavior of the complexes.
Reduction
Electrochemical reduction of complexes 4b and 4c and 5b and 5c occurs in two single-electron stages with similar potentials (Table 1 and Figure 6). The first electron transfer is irreversible and corresponds to Cu2+/Cu+ transition within the chelate complex. The second quasireversible (EpRed – EpOx = 400 mV) peak corresponds to the formation of a [LCu0]−2 chelate complex similar to that of 4e and 5e. The absence of a desorption oxidation peak in the range from −100 to +100 mV proves the absence of oxidative desorption of copper from the electrode. This means that the intermediate dianionic complexes, [LCu0]−2, are rather stable on the electrochemical time scale. This result is in contrast to that obtained for complex 5e, for which the first step is quasireversible and the second occurs at −2.46 V.
Figure 6.

Cyclic voltammogram reduction of complex 4b (Pt electrode, DMF, Bu4NBF4, Ag/AgCl/KCl (sat.), 20 °C).
Potentiometric Study
It is known that N,N′-dimethylbispidine has pKa = 11.88 in water.56 Determination of the pKa of phenyl-substituted bispidines in acetonitrile or DMSO by titration using organic acids, such as p-toluenesulphonic or methanesulphonic acids, has been discussed in several papers.57,58 A correlation between these data and the data from aqueous solutions is established.59 According to these data, phenyl substituents on C1 and C5 lower the value of the acidity constant (pKa(DMSO) = 4.4) that have no further effects by carbomethoxy-functionalization of N3 and N7. However, replacement of phenyl by benzyl increases the pKa to 7.7, and addition of COOCH3 to C1 and C5 decrease the pKa by 2 orders (pKa(DMSO) = 5.3).57 Because the effects of functional groups at the different sites of bispidine backbone on the acidity are not additive, it seems reasonable to study the dependence of acidic properties as well as the complexing ability of our target molecules on the position of such substituents/groups.
Intensive, systematic research on the modifications of the backbone of bispidines on addition of pyridyl and picolyl groups to N3 and N7 and/or C2 and C4 has been carried out earlier.23,31,60,61 It was shown that different combinations of these substituents and sites of insertion can produce a wide range of acidities, ∑pKa = 11–17. These data could be explained by changes in the electron density on tertiary amines that are affected by interference of a withdrawing effect of pyridyl groups and donation by aliphatic substituents.31 However, the stability of Cu(II)L with these ligands is not correlated to their acidities because the stereochemistry of Cu(II) is mainly defined by the Jahn–Teller distortion: for example, methylation of the pyridine groups at the γ-position does not affect the acidity but has a crucial increase on the stability of Cu(II)L, by ∼7 orders of magnitude.23 Protonation of the ligands can be directly studied by potentiometric titration of aqueous solutions. Aqueous solutions of 0.001 M ligand have a pH of ∼3–4; addition of HClO4 with a known concentration is required to make the pH ∼2.5. This contribution of HClO4 was taken into account during calculation of constants. The results of potentiometric titration of the ligands are summarized in Table 2.
Table 2. Calculated Logarithms of Constants of Protonation and Complexation of Bispidine Ligands.
| log K |
||||||||
|---|---|---|---|---|---|---|---|---|
| cation | complex | 1b | 1c | 2a | 2b | 2c | 3b | 3c |
| H+ | L:3H+ | 18.5 ± 0.1 | 18.8 ± 0.1 | |||||
| L:2H+ | 12.6 ± 0.3 | 14.6 ± 0.3 | 15.9 ± 0.3 | 14.8 ± 0.1 | 16.0 ± 0.2 | 16.0 ± 0.1 | 16.3 ± 0.1 | |
| L:H+ | 8.6 ± 0.2 | 10.6 ± 0.1 | 11.3 ± 0.2 | 10.9 ± 0.1 | 11.7 ± 0.1 | 11.7 ± 0.1 | 12.2 ± 0.1 | |
| Cu2+ | 2L:Cu2+:2OH | 1.7 ± 0.4 | 2.4 ± 0.4 | |||||
| 2L:Cu2+:OH– | 11.3 ± 0.4 | 13.7 ± 0.4 | ||||||
| L:Cu2+:OH– | 3.1 ± 0.1 | 2.3 ± 0.1 | 2.8 ± 0.1 | 1.3 ± 0.2 | 1.9 ± 0.1 | |||
| L:Cu2+ | 9.9 ± 0.1 | 11.2 ± 0.1 | 14.1 ± 0.2 | 13.4 ± 0.1 | 14.2 ± 0.1 | 13.5 ± 0.1 | 13.7 ± 0.1 | |
| 2L:Cu2+ | 18.7 ± 0.1 | 20.9 ± 0.1 | ||||||
As has been mentioned in the introduction, polyaminopolycarboxylic acids have already proven their usefulness in the chelation of cationic radionuclides for the synthesis of RPs. However, information on bispidine-containing carboxylic acids is limited in the available literature;30,62 thus, our investigation is adequately justified. In general, pendant acetic groups increase the acidity of ligands, wherein larger carbon chains distinguish between bispidines and bispidinones. According to the calculated protonation constants (Table 2), the difference between 2b and 2c is 1 order and between 3b and 3c is the same, within the error. Literature data considering the acidity of carboxyl-containing bispidines report structural similarity between ligands 2d and 2e(62) and 2a and 2b. The protonation constants for 2d, log K1H = 11.93 and log K2H = 16.12, and 2e, log K1H = 10.38 and log K2H = 14.39, are virtually identical to those calculated in this work for 2a and 2b.
The protonation ability of water-soluble ligands is one of their most important properties, which defines their complexation strength with cations. At least a two-step protonation is established for all ligands. Ligand 1b, with a carbonyl group at C9, demonstrates the lowest value for the first step of protonation. Such an influence of keto groups is traditionally associated with interaction of nitrogen’s lone pairs of aminogroups and a carbonyl group, similarly to amides,63 due to through bond interaction between CO and N’s.64−68 This leads to a decrease in the Ka of bispidinones compared to that of bispidines by 4 orders of magnitude.31,57 The log K1H (1b) value calculated in this work (Table 2) is correlated with an earlier obtained value of log K1H = 7.0 for this compound taking in consideration the relation between the values determined in different solvents, log K1H(H2O) = log K1H(DMSO) ± 1.5.57,59 Substitution of the keto functional group by an alcohol group in 1c results in an increase in the log K1H value by 2 orders. The second proton addition, with log K1H – log K2H = 4 for all considered ligands, also correlates with the second protonation step of pyridyl-containing bispidines, log K1H – log K2H = 4.5–6.61,63 Protonation of the carboxylic groups in bispidines is expected to occur at an acidic pH, similar to that of glycine in 2 and α-alanine in 3. However, it becomes possible to determine the third step of protonation related to the carboxylic group only for 3, log K3H – log K2H = 2.5–2.6. The latter value is in good agreement with log KH(α-alanine) = 2.33.69 According to the ligand speciation diagrams, complete deprotonation of the considered bispidines occurs at pH’s 7–9 (Figure 7).
Figure 7.
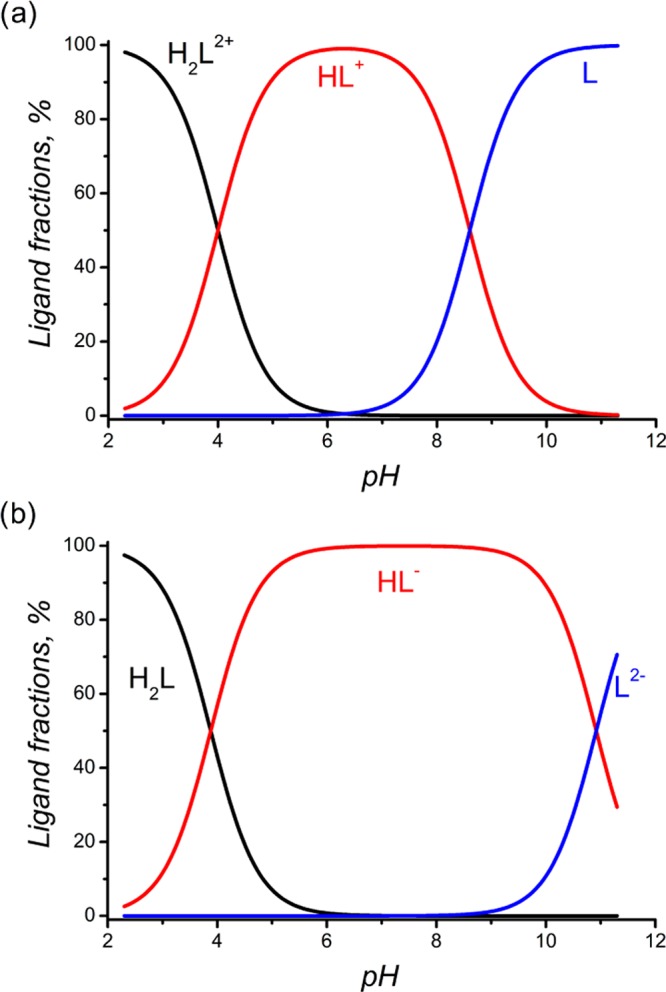
Distribution of protonated and deprotonated forms of 1b (a) and 2b (b) in aqueous solution as a function of pH.
For complexation studies, a titrated solution containing ligand and Cu2+ at concentrations of 5–10 × 10–4 M was used. Hyperquad 2013 software was used to calculate the complexation constants of CuL and CuL(OH)m. For calculation of the complexation constants, the protonation constants of the ligands were obtained in this study and their hydrolysis constants were taken from the literature.70
All considered ligands form complexes with Cu2+, which is evident from the different titration curves obtained for the ligands and the ligands in the presence of Cu2+ (Figure 8). The complexation constants are correlated with the protonation constants, as seen from Table 2. The maximal equilibration time for each experimental point was set as 5 min and was achieved only at pH > 10, when insoluble CuO started to form.
Figure 8.

Typical titration curves of L (0.001 M) acidified by excess HClO4 (0.004 M) (A) and L (0.001 M) with Cu2+ (0.0005 M) acidified by excess HClO4 (0.004 M) (B); μ = 0.1 M NaClO4 at 25 °C. (a) L = 1b, (b) L = 2b.
Changes in colors and changes in titration curves proceed very fast (no more than 5 minutes). This fact indirectly confirms fast complexation of Cu2+ by bispidines. Ligands 1b and 1c form two types of complexes, with M/L = 1:1 and 1:2 stoichiometries. The latter is confirmed by spectrophotometric titration at pH 5.0 ± 0.1 (Figure 9a). On addition of Cu2+ to the ligand solution, the absorption at 555–556 nm increases. The latter corresponds to a complex with stoichiometry [L2Cu]2+, and then, a bathochromic shift of the maximum occurs from c(Cu2+)/c(L) = 0.5 to c(Cu2+)/c(L) = 1 (Figure 9a inset). Moreover, the change in the maximum absorption (Figure 9b) proves the formation of two types of complexes, [L2Cu]2+ and [LCu]2+. The possibility of formation of [L2Cu]2+complexes was confirmed by us recently by means of an X-ray study and an ESI mass-spectral study.18
Figure 9.

(a) Absorption spectra upon spectrophotometric titration and shift in the maximum with Cu2+/L variation (inset), L = 1b. (b) Change in the absorption maximum with Cu2+/L variation, L = 1b.
During potentiometric titration, initially (at lower pH values), the formation of [LCu]2+ takes place. Then, with an increase in pH, a corresponding ligand deprotonation complex, [L2Cu]2+, forms, which at an alkaline pH transforms to the hydroxoforms of the complex, [L2CuOH]+ and [L2Cu(OH)2] (Figure 10a). After titration, the solution is characterized by the same purple color and possesses the same spectrum, with a maximum at 555 nm, as those for [L2Cu]2+, which also supports the idea of keeping the 1:2 stoichiometry for the hydroxide forms. The same results are shown for 1c (Figures S21–S22 of the Supporting Information). Recently,30 single crystals of Cu-2d have been characterized, indicative of the formation of a 1:1 stoichiometry, whereas by electron paramagnetic resonance spectroscopy and molecular mechanic structure optimization, the formation of dinuclear complexes with a 2:2 stoichiometry has been suggested. Our results, that is, 1:1 complexation of copper with 2a–c, 3b, and 3c, are in line with those obtained from single-crystal X-ray data in ref (30) and from the complexation study in ref (62). On the other hand, for ligands having carboxylic groups as part of their structures, complex CuL is stable over a wide pH range (Figure 10b). Furthermore, the value calculated for complex 4a coincides with that obtained from the literature, log K(2dCu) = 14.45.62 However, 4b is almost 3 orders less stable than 4e (log K = 16.02). Apparently, the higher stability of 4e is attributed to phenyl substituents at C1 and C5, which is in line with the results of our electrochemistry study. It is noteworthy that substitution of glycine by α-alanine does not influence the complexation with Cu2+, and despite the difference in the protonation of carboxyl-containing ligands, the generally stability of [LCu] remains unchanged (Table 2).
Figure 10.

Distribution of Cu2+ (c = 5 × 10–4 M) species in aqueous solutions (μ = 0.1 M NaClO4) in the presence of L (c = 1 × 10–3 M), as a function of pH: (a) L = 1b, (b) L = 2b.
Logarithms of the obtained stability constants are in the ranges of 13–14 and 19–21, which proves a high affinity of bispidines to copper cations. The log KML values with carboxylate-containing bispidine are less than that for known chelators like triethylenetetramine, DOTA, and 1,4,7-triazacyclononane-1,4,7-triyltriacetic acid (log K = 21–234) but the known systems suffer from hard complexation conditions71−73 that are not always appropriate for vectors and despite high values of log KCuL the known examples lack the stability in the biologic medium.72,74,75 So, it is not evident that the overall stability directly coincides with log KCuL. Complexation data for Sar-ligands with Cu2+ are limited in the literature. For the first approximation, the obtained values, especially for 1b and 1c, for “soft” cations such as Cu2+ might be high enough for stability in a biologic medium. As a following step, in vitro and in vivo stability would be checked.
According to this study, carboxylic pendant arms affect not only the stoichiometry but also the stability of complexes of bispidines with Cu2+. However, the stability of LCu tends to decrease with growth of the carbon backbone in the pendant arm; thus, glycine substituents seem to be optimal for further investigation.
Conclusions
In this article, we have shown that dicarboxylic derivatives of bispidines are promising ligand systems for copper chelation, which have potential applications in RPs for PET. The copper complexes with N2O2-type ligands, 2 and 3, possess sufficient thermodynamic stability in aqueous solutions and fast complexation kinetics, that is, they are formed at low concentrations, at ambient temperatures, and in minimal time. Electrochemical study shows good redox stability for complexes with N2O2 tetradentate dianionic ligands toward Cu(II)/Cu(I) reduction, which makes them promising RP candidates against the redox systems present in vivo. Another goal for our future investigations is the discovery of methods to attach copper-binding ligands to the desired peptide vectors or biological objects. For example, the presence of a hydroxyl group at C9 in 2c and 3c allows the synthetic modification of the ligands with short linkers, such as propargyl, paving the way for triazole linker chemistry. So, the reactivity of 9-OH is currently under investigation in our labs. For their use in RPs, bispidines will be studied from the point of view of radiolytic stability to ensure that the conjugates retain their desired properties during the entire procedure.
Materials and Methods
1H and 13C NMR spectra were recorded at 25 °C on Bruker Avance-400 and Bruker Avance-600 spectrometers. Chemical shifts were referred to the signals of the deuterated solvents (4.79 ppm for D2O, 7.26 and 77.0 ppm for CDCl3, 2.49 and 39.5 ppm for (CD3)2SO). Elemental analysis was carried out on a Eurovector EA 3000 automated analyzer. Electrochemical measurements were performed using an IPC 100 potentiostat, with a platinum electrode (3.5 mm in diameter). A silver chloride electrode was used as the reference electrode, and Bu4NBF4 was used as the supporting electrolyte. All measurements were made in DMF. The potentials are recorded taking into account iR compensation. The number of electrons was determined by comparing the oxidation peaks of ferrocene obtained at equal concentrations. HRMS spectra were measured on an Orbitrap Elite instrument using ESI. A syringe injection was used for solutions in methanol/water (1:1; 10 μL/min flow rate). Nitrogen was applied as a dry gas. ATR–FTIR spectra were obtained on an iS5 spectrometer (Thermo Fisher Scientific) with ZnSe element (45°) (20 scans, resolution of 4 cm–1).
Potentiometric and Spectrophotometric Titration
Cu(ClO4)2·6H2O (Aldrich), 70% HClO4 aqueous solution (Aldrich), KNO3, NaClO4·H2O, and NaOH were of analytical grade and were used as received, without further purification.
In all of the experiments, deionized water (18.2 MΩ) was used. The Cu(ClO4)2 solution was standardized by titration with ethylenediaminetetraacetic acid, using xylenol orange as an indicator, as described in detail earlier.76 The 0.1 M NaOH solution was standardized potentiometrically with oxalic acid, which was used as a primary standard. The 0.1 M HClO4 solution was prepared by dilution of the 70% aqueous solution and was standardized by titration with NaOH. Stock solutions of 0.01 M ligand (L), 1 M KNO3, and 1 M NaClO4 were prepared by dissolution of weighed amounts of these compounds in volumetric flasks.
For potentiometric titration, autotitrator 848 Titrino plus, equipped with a 20 mL autoburette and a Metrohm combined glass electrode (model 60262100), was used. A constant temperature of 25.0 ± 0.1 °C was maintained in the cell using a thermostat. A combined glass electrode was calibrated by titration of a preliminarily standardized solution of HClO4 with a NaOH solution of known concentration and calculation of the equivalent point using Gans’ method.77 The latter allowed us to define the standard electrode potential, E0, and the slope of the electrode function. The ionic product of water in 0.1 M KNO3/NaClO4 was obtained, pKw = 13.78. For determination of the ligand’s protonation constants, potentiometric titration of a 20 mL solution containing 8–10 × 10–4 M ligand, 5–6 × 10–3 M HClO4, and 0.1 M KNO3/NaClO4 was carried out in a glass cell in a water-jacketed thermostated vessel using 9.5–11 × 10–2 M NaOH as the titrant. Titration was performed over a pH range of 2.5–10.5. The electrode’s electromotive force values were measured after the addition of 0.03 mL increments of standard NaOH solution. After each addition of titrant, equilibrium was considered to have been established if the variation in the potential was <0.2 mV/min. The protonation constants of the ligands and the complexation constants were calculated using Hyperquad software.78 At least two replicates of the titration procedure per L and Cu2+ + L were performed.
For spectrophotometric titration, titrate solutions of 8 × 10–3 M ligand were prepared at pH 5.0 ± 0.1. Absorption spectra were recorded in the wavelength range 400–1000 nm (Shimadzu UV-1800 spectrophotometer) after the addition of 0.01 mL increments of standard Cu(ClO4)2 solution with a Hamilton microsyringe (V = 0.05 mL) and adjusting the pH to 5.0 ± 0.1.
Synthetic Procedures
5,7-Dimethyl-1,3-diazaadamantan-9-one
A mixture of 20 g (0.143 mol) of urotropin, 14.61 g (0.169 mol) of pentanone-3, 18.77 mL (0.328 mol) of glacial acetic acid, and 82 mL of butanol-1 was refluxed for 2.5 h. The red solution obtained was concentrated, and the residual viscous oil was extracted several times with 4 × 100 mL of hot n-heptane. The supernatant was stirred with 20 g of alumina for 30 min at 80 °C. The hot suspension was filtered, the filtrate was evaporated to dryness, and the dry residue was washed with a mixture of 135 mL of dichloromethane (DCM) and 50 mL of water. The organic phase was separated, dried over sodium sulfate, and evaporated to dryness. Yield: 13.4 g (52%) of pink crystals.
The 1H and 13C NMR spectra in CDCl3 coincide with the literature data.30
5,7-Dimethyl-1,3-diazaadamantane
A mixture of 2.44 g (13.5 mmol) of 5,7-dimethyl-1,3-diazaadamantan-9-one, 20.41 mL (0.416 mol) of hydrazine hydrate, and 2.04 g (51 mmol) of sodium hydroxide in 10.2 mL of ethylene glycol was refluxed for 24 h. The reaction mixture was distilled at 120 °C. The distillate was extracted with 4 × 25 mL of petroleum ether and dried over sodium sulfate. The solvent was evaporated. Yield: 1.44 g (64%) of white powder.
The 1H and 13C NMR spectra in CDCl3 coincide with the literature data.30
3,7-Diacetyl-1,5-dimethyl-3,7-diazabicyclo[3.3.1]nonane
To a solution of 1.44 g (8.7 mmol) of 5,7-dimethyl-1,3-diazadamantane in 17 mL of THF was added an aqueous solution (8.7 mL) of 1.53 g (18.2 mmol) of sodium hydrogen carbonate. The reaction mixture was cooled to 4 °C, and a solution of 1.3 mL (18.2 mmol) of acetyl chloride in 6 mL of THF was added dropwise with stirring. The reaction mixture was stirred at rt overnight. The organic solvent was evaporated, and the residual aqueous solution was adjusted to pH 7 with sodium hydrogen carbonate solution. After extraction with 4 × 15 mL of DCM, the organic phase was dried over sodium sulfate and evaporated to dryness. The dry residue was recrystallized from 1,4-dioxane. Yield: 0.897 g (43%) of colorless crystals.
The 1H and 13C NMR spectra in CDCl3 coincide with the literature data.30
1,5-Dimethyl-3,7-diazabicyclo[3.3.1]nonane (1a)
A solution of 0.84 g (5 mmol) of 3,7-diacetyl-1,5-dimethyl-3,7-diazabicyclo[3.3.1]-nonane in a mixture of 1.35 mL of concentrated hydrochloric acid and 6.77 mL of water was refluxed for 24 h. The solution was evaporated to dryness. The residue was mixed with 5 mL of water and 15 mL of diethyl ether and saponified to pH 13 with aqueous potassium hydroxide solution. The organic phase was separated, and the water phase was extracted with 3 × 15 mL of diethyl ether. The organic phases were combined and dried over sodium sulfate. The solvent was evaporated. Yield: 0.365 g (47%) of white powder.
The 1H and 13C NMR spectra in D2O coincide with the literature data.30
3,7-Diacetyl-1,5-dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane
A mixture of 1.55 g (8.64 mmol) of 5,7-dimethyl-1,3-diazaadamantanone and 7.77 mL (82.32 mmol) of acetic anhydride was stirred for 8 h at rt. The reaction mixture was diluted with 7.77 mL of water and stirred at rt for 30 min. The solvent was evaporated. The residue was suspended in 4.66 mL of water and filtered. The product was dried in air. Yield: 1.88 g (86%) of white powder.
The 1H and 13C NMR spectra in CDCl3 coincide with the literature data.18
1,5-Dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane (1b)
A solution of 6 g (23.88 mmol) of 3,7-diacetyl-1,5-dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane in 59.7 mL of 5 M aqueous hydrochloric acid was refluxed for 5 h. The solution was concentrated up to one-third of the volume. The precipitated crystals were filtered and dried. The crystals and filtrate were separately saponified to pH 10 with 50% aqueous sodium hydroxide, extracted with 3 × 30 mL of chloroform, and dried over sodium sulfate. The solvent was evaporated and the residue was recrystallized from petroleum ether. Yield: 3.22 g (80%) of colorless crystals. Mp 70–71 °C. Lit. mp18 68–69 °C.
The 1H and 13C NMR spectra in CDCl3 coincide with the literature data.18
1,5-Dimethyl-9-hydroxy-3,7-diazabicyclo[3.3.1]nonane (1c)
To a solution of 1.5 g (8.9 mmol) of 1,5-dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane in 44 mL of ethanol, 0.34 g (8.9 mmol) of sodium borohydride was added in small portions, with stirring. The reaction mixture was stirred for 2 h at rt. The suspension was evaporated to dryness. The dry residue was dissolved in 22 mL of water and evaporated again. The product was extracted with 3 × 50 mL of hot chloroform. The solvent was evaporated, and the residue was recrystallized from toluene. Yield: 1.075 g (71%) of colorless crystals. Mp 213.4–215.0 °C. Lit. mp42 190–192 °C. Found: C, 63.45; H, 10.33; N, 16.27. Calcd for C9H18N2O: C, 63.49; H, 10.66; N, 16.45. 1H NMR (D2O, δ/ppm, J/Hz): 0.73 (s, 6H, CH3); 2.60 (dd, 2H, 2JHH = 14.0, 4JHH = 2.8), 2.95 (d, 2H, 2JHH = 14.0) (NCH22,4 or 6,8); 2.69 (d, 2H, 2JHH = 13.6), 2.80 (dd, 2H, 2JHH = 13.6, 4JHH = 2.8) (NCH26,8 or 2,4); 3.33 (s, 1H, CH(OH)). 13C NMR (D2O, δ/ppm): 20.28 (CH3); 33.89 (CCH3); 48.12, 56.41 (CH2N); 77.87 (CH(OH)).
General Procedure for Preparation of 2,2′-(1,5-Dimethyl-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl)dicarboxylic Acids (2 and 3)
An aqueous solution of 4 mmol of sodium hydroxide was added dropwise to a solution of 2 mmol of halogencarboxylic acid cooled in an ice–water bath. Then, an aqueous solution of 2 mmol of the appropriate 3,7-diazabicyclo[3.3.1]nonane was added, and the reaction mixture was stirred for 3 h at rt. To the hot reaction mixture, an aqueous solution of 1 equiv of barium chloride dihydrate was added, and the suspension was refluxed for 30 min. The obtained white precipitate was filtered, washed with hot water, and dried for 2 h at 120 °C. The barium salt was suspended in water, heated to reflux, and 1 equiv of 2.27 M sulfuric acid was added to it dropwise. The suspension was refluxed for 15 min and filtered. The precipitate was washed with hot water. The filtrate was evaporated to dryness. The white foam obtained was dried at 70 °C for 2 h at a pressure of 5 mmHg.
2,2′-(1,5-Dimethyl-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl)diacetic Acid (2a)
The starting compound was chloroacetic acid. Yield: 53%. Mp 235–237 °C (dec). The 1H and 13C NMR spectra in D2O coincide with the literature data.30
2,2′-(1,5-Dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane-3,7-di-yl)diacetic Acid (2b)
The starting compound was chloroacetic acid. Yield: 41%. Mp 237–238 °C (dec). Lit. mp47 225–227 °C. Found: C, 51.94; H, 7.61; N, 9.10. Calcd for C13H20N2O5·0.9H2O: C, 51.96; H, 7.31; N, 9.32. 1H NMR ((CD3)2SO, δ/ppm, J/Hz): 0.86 (s, 6H, CH3); 3.03, 3.47 (each d, 4H, 2JHH = 11.4, CH2N); 3.32 (s, 4H, CH2COOH). 13C NMR ((CD3)2SO, δ/ppm): 16.41 (CH3); 45.85 (CCH3); 56.83 (CH2COOH); 63.31 (CH2N); 169.15 (COOH); 210.65 (C=O).
Mixture of 2b and 2b′ (∼0.9:1.0)
1H NMR (D2O, δ/ppm, J/Hz): 0.97 (s, CH3 (2b′)); 1.04 (s, CH3 (2b)); 3.15, 3.21 (AB-system, 2JHH = 11.6, CH2N (2b′)); 3.24, 3.74 (each d, 2JHH = 12.0, CH2N (2b)); 3.51 (s, CH2COOH (2b′)); 3.63(s, CH2COOH (2b)). 13C NMR (D2O, δ/ppm): 14.22, 14.95 (CH3 (2b, 2b′)); 39.80 (CCH3 (2b′)); 46.24 (CCH3 (2b)); 56.57, 57.13 (CH2COOH (2b, 2b′)); 60.07, 63.67 (CH2N (2b, 2b′)); 93.71 (C(OH)2 (2b′); 172.12, 172.36 (COOH (2b, 2b′)); 212.03 (C=O (2b)).
2,2′-(1,5-Dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane-3,7-di-yl)dipropionic Acid (3b)
The starting compound was 2-bromopropionic acid. Yield: 33%. Mixture of 3b and 3b′ (∼2.3:1.0) (2 diastereomers for each compound). Mp 155–160 °C (dec). Found: C, 54.05; H, 7.64; N, 8.47. Calcd for C15H24N2O5·1.1H2O: C, 54.24; H, 7.95; N, 8.43. 1H NMR (D2O, δ/ppm, J/Hz): 0.93, 0.94 (each s, CCH3 (3b′)); 1.01, 1.02 (each s, CCH3 (3b)); 1.35–1.40 (set of d, CHCH3 (3b, 3b′)); 3.04–3.24 (set of m, CH2N (3b, 3b′)); 3.47–3.70 (set of m, CHCH3 (3b, 3b′), CH2N (3b)).
2,2′-(1,5-Dimethyl-9-hydroxy-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl)diacetic Acid (2c)
The starting compound was chloroacetic acid. Yield: 56%. Mp 235–237 °C (dec). Found: C, 53.14; H, 7.63; N, 9.19. Calcd for C13H22N2O5·0.4H2O: C, 53.19; H, 7.83; N, 9.54. 1H NMR (D2O, δ/ppm, J/Hz): 0.91 (s, 6H, CH3); 2.71 (d, 2H, 2JHH = 11.3), 3.16 (d, 2H, 2JHH = 11.3) (NCH22,4 or 6,8); 3.00 (d, 2H, 2JHH = 11.6), 3.17 (d, 2H, 2JHH = 11.6) (NCH26,8 or 2,4); 3.35, 3.50 (each s, 2H, CH2COOH); 3.39 (s, 1H, CH(OH)). 13C NMR (D2O, δ/ppm): 21.75 (CH3); 38.74 (CCH3); 58.96, 65.22 (CH2N); 60.37, 61.01 (CH2COOH); 77.15 (CH(OH)); 174.69, 176.75 (COOH).
2,2′-(1,5-Dimethyl-9-hydroxy-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl)dipropionic Acid (3c)
The starting compound was 2-bromopropionic acid. Yield: 43%. Mixture of two diastereomers, A/B ∼ 5:1. Mp 239–241 °C (dec). Found: C: 53.58; H: 8.43; N: 8.01. Calcd for C15H24N2O5·1.46H2O: C: 53.20; H: 8.03; N: 8.27. Mixture of 3b and 3b′ (∼1.4:1.0) (two diastereomers for each compound (rac-:meso ∼ 1:0.8)). 1H NMR (D2O, δ/ppm, J/Hz): 0.935, 0.952 (each s, CCH3, meso-3b′); 0.944 (s, CCH3, rac-3b′); 1.010, 1.025 (each s, CCH3, meso-3b); 1.018 (s, CCH3, rac-3b); 1.35–1.40 (set of overlapping d, CHCH3 (3b, 3b′)); 3.04–3.26 (set of m, CH2N (3b, 3b′)); 3.47–3.70 (set of m, CHCH3 (3b, 3b′), CH2N (3b)). 13C NMR (D2O, δ/ppm): 11.68 (CHCH3, rac-3b); 11.83 (CHCH3, meso-3b′); 11.96 (CHCH3, rac-3b′); 12.12 (CHCH3, meso-3b); 14.34, 14.54 (CCH3, meso-3b′); 14.44 (CCH3, rac-3b′); 15.04, 15.26 (CCH3, meso-3b); 15.15 (CCH3, rac-3b); 39.36, 39.73 (CCH3, meso-3b′); 39.51 (CCH3, rac-3b′); 45.87, 46.29 (CCH3, meso-3b); 46.04 (CCH3, rac-3b); 55.56, 58.27, 62.64 (CH2N, CHCH3, rac-3b′); 55.67, 58.35, 62.76 (CH2N, CHCH3, meso-3b′); 59.26, 61.93, 62.39 (CH2N, CHCH3, rac-3b); 59.54, 62.14, 62.70 (CH2N, CHCH3, meso-3b); 94.03 (C(OH)2, rac-3b′); 94.06 (C(OH)2, meso-3b′); 175.71 (COOH, rac-, meso-3b); 175.95 (COOH, meso-3b′); 176.05 (COOH, rac-3b′); 212.20 (C=O, rac-3b); 212.25 (C=O, meso-3b).
General Procedure for the Preparation of Copper 2,2′-(1,5-Dimethyl-3,7-diazabicyclo[3.3.1]-nonane-3,7-diyl) Dicarboxylates
Solutions of ligands 2 and 3 (0.1 mmol each) in 5 mL of water were stirred with 0.05 mmol of basic copper carbonate (malachite) at rt overnight. The blue solution obtained was evaporated to dryness and dried at 70 °C for 2 h at a pressure of 5 mmHg. The yields are quantitative.
Copper 2,2′-(1,5-Dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl) Diacetate (4b)
Mp 260–270 °C (dec). λmax (MeOH/H2O 1:1)/nm 247 (ε/dm3 mol–1 cm–1 4800), 610 (150). Found: C, 40.76; H, 5.65; N, 7.36. Calcd for C13H18CuN2O5·2H2O: C, 40.89; H, 5.81; N, 7.34. High-resolution ESIMS: m/z 364.06977; 691.11143; 709.12237; 727.13281 (calcd for C13H21N2O6Cu+m/z 364.0696, calcd for C26H37N4O10Cu2+m/z 691.1102, calcd for C26H39N4O11Cu2+m/z 709.1207, calcd for C26H41N4O12Cu2+m/z 727.1313).
Copper 2,2′-(1,5-Dimethyl-9-oxo-3,7-diazabicyclo[3.3.1]nonane-3,7-diyl) Dipropionate (5b)
Mp 250–260 °C (dec). λmax (MeOH/H2O 1:1)/nm 247 (ε/dm3 mol–1 cm–1 4800), 610 (160). Found: C, 43.33; H, 6.18; N, 7.13. Calcd for C15H22CuN2O5·2.3H2O: C, 43.32; H, 6.46; N, 6.74. High-resolution ESIMS: m/z 392.10002; 747.17226; 765.18296; 783.19373 (calcd for C15H25N2O6Cu+m/z 392.1009, calcd for C30H25N4O10Cu2+m/z 747.1728, calcd for C30H27N4O11Cu2+m/z 765.1833, calcd for C30H29N4O12Cu2+m/z 783.1939).
Copper 2,2′-(1,5-Dimethyl-9-hydroxy-3,7-diazabicyclo[3.3.1]-nonane-3,7-diyl) Diacetate (4c)
Mp 260–270 °C (dec). λmax (MeOH/H2O 1:1)/nm 250 (ε/dm3 mol–1 cm–1 5000), 612 (160). Found: C, 42.01; H, 6.15; N, 7.56. Calcd for C13H20CuN2O5·1.3H2O: C, 42.06; H, 6.14; N, 7.55. High-resolution ESIMS: m/z 348.07404; 695.14155 (calcd for C13H21N2O5Cu+m/z 348.0746, calcd for C26H41N4O10Cu2+m/z 695.1415).
Copper 2,2′-(1,5-Dimethyl-9-hydroxy-3,7-diazabicyclo[3.3.1]-nonane-3,7-diyl) Dipropionate (5c)
Mp 250–260 °C (dec). λmax (MeOH/H2O 1:1)/nm 252 (ε/dm3 mol–1 cm–1 5300), 602 (170). Found: C, 44.59; H, 6.30; N, 7.12. Calcd for C15H24CuN2O5·1.7H2O: C: 44.54; H: 6.33; N: 6.93. High-resolution ESIMS: m/z 376.1056; 751.2039 (calcd for C15H25N2O5Cu+m/z 376.1059, calcd for C30H49N4O10Cu2+m/z 751.2041).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00237.
This work was supported by Russian Science Foundation (grant no 16-33-00114).
The authors declare no competing financial interest.
Supplementary Material
References
- Jødal L.; Le Loirec C.; Champion C. Phys. Med. Biol. 2014, 59, 7419. 10.1088/0031-9155/59/23/7419. [DOI] [PubMed] [Google Scholar]
- Holland J. P.; Williamson M. J.; Lewis J. S. Mol. Imaging 2010, 9, 1. 10.2310/7290.2010.00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch M. J.; Laforest R.; Lewis J. S.. Production of Non-Standard PET Radionuclides and the Application of Radiopharmaceuticals Labeled with These Nuclides. In PET Chemistry: The Driving Force in Molecular Imaging; Schubiger P. A., Lehmann L., Friebe M., Eds.; Springer: Berlin, 2007; pp 159–181. [DOI] [PubMed] [Google Scholar]
- Price E. W.; Orvig C. Chem. Soc. Rev. 2014, 43, 260. 10.1039/C3CS60304K. [DOI] [PubMed] [Google Scholar]
- Cutler C. S.; Hennkens H. M.; Sisay N.; Huclier-Markai S.; Jurisson S. S. Chem. Rev. 2013, 113, 858. 10.1021/cr3003104. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Baidoo K. E.; Brechbiel M. W. Adv. Drug Delivery Rev. 2013, 65, 1098. 10.1016/j.addr.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. S.; Herrero P.; Sharp T. L.; Engelbach J. A.; Fujibayashi Y.; Laforest R.; Kovacs A.; Gropler R. J.; Welch M. J. J. Nucl. Med. 2002, 43, 1557. [PubMed] [Google Scholar]
- Lewis J. S.; Laforest R.; Dehdashti F.; Grigsby P. W.; Welch M. J.; Siegel B. A. J. Nucl. Med. 2008, 49, 1177. 10.2967/jnumed.108.051326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujibayashi Y.; Cutler C. S.; Anderson C. J.; McCarthy D. W.; Jones L. A.; Sharp T.; Yonekura Y.; Welch M. J. Nucl. Med. Biol. 1999, 26, 117. 10.1016/S0969-8051(98)00049-3. [DOI] [PubMed] [Google Scholar]
- Avila-Rodriguez M. A.; Nye J. A.; Nickles R. J. Appl. Radiat. Isot. 2007, 65, 1115. 10.1016/j.apradiso.2007.05.012. [DOI] [PubMed] [Google Scholar]
- Krasikova R. N.; Aliev R. A.; Kalmykov S. N. Mendeleev Commun. 2016, 26, 85. 10.1016/j.mencom.2016.03.001. [DOI] [Google Scholar]
- Ligand Design in Medicinal Inorganic Chemistry; Tim S., Ed.; John Wiley & Sons: Chichester, 2014; pp 62–65. [Google Scholar]
- Smith S. V. Expert Opin. Drug Discovery 2007, 2, 659. 10.1517/17460441.2.5.659. [DOI] [PubMed] [Google Scholar]
- Li W.-S.; Vatsadze S. V.; Blake A. J.; Mountford P. Acta Crystallogr., Sect. C: Struct. Chem. 1998, 54, IUC9800019 10.1107/S0108270198099661. [DOI] [Google Scholar]
- Rakhimov R. D.; Vatsadze S. Z.; Butin K. P.; Zyk N. V. Russ. J. Electrochem. 2003, 39, 1253. 10.1023/B:RUEL.0000003455.12268.e2. [DOI] [Google Scholar]
- Lloyd J.; Vatsadze S. Z.; Robson D. A.; Blake A. J.; Mountford P. J. Organomet. Chem. 1999, 591, 114. 10.1016/S0022-328X(99)00447-7. [DOI] [Google Scholar]
- Vatsadze S. Z.; Krainova Y. V.; Kovalkina M. A.; Zyk N. V. Chem. Heterocycl. Compd. 2000, 36, 1185. 10.1023/A:1002820816850. [DOI] [Google Scholar]
- Vatsadze S. Z.; Semashko V. S.; Manaenkova M. A.; Krut’ko D. P.; Nuriev V. N.; Rakhimov R. D.; Davlyatshin D. I.; Churakov A. V.; Howard J. A. K.; Maksimov A. L.; Li W.; Yu H. Russ. Chem. Bull. 2014, 63, 895. 10.1007/s11172-014-0526-6. [DOI] [Google Scholar]
- Vatsadze S. Z.; Tyurin V. S.; Zyk N. V.; Churakov A. V.; Kuz’mina L. G.; Avtomonov E. V.; Rakhimov R. D.; Butin K. P. Russ. Chem. Bull. 2005, 54, 1825. 10.1007/s11172-006-0044-2. [DOI] [Google Scholar]
- Comba P.; Nuber B.; Ramlow A. J. Chem. Soc., Dalton Trans. 1997, 347. 10.1039/a603635j. [DOI] [Google Scholar]
- Brox D.; Comba P.; Herten D. P.; Kimmle E.; Morgen M.; Ruhl C. L.; Rybina A.; Stephan H.; Storch G.; Wadepohl H. J. Inorg. Biochem. 2015, 148, 78. 10.1016/j.jinorgbio.2015.05.009. [DOI] [PubMed] [Google Scholar]
- Comba P.; Lang C.; de Laorden C. L.; Muruganantham A.; Rajaraman G.; Wadepohl H.; Zajaczkowski M. Chem. - Eur. J. 2008, 14, 5313. 10.1002/chem.200701910. [DOI] [PubMed] [Google Scholar]
- Born K.; Comba P.; Ferrari R.; Lawrance G. A.; Wadepohl H. Inorg. Chem. 2007, 46, 458. 10.1021/ic061501+. [DOI] [PubMed] [Google Scholar]
- Comba P.; Rudolf H.; Wadepohl H. Dalton Trans. 2015, 44, 2724. 10.1039/C4DT03262D. [DOI] [PubMed] [Google Scholar]
- Atanasov M.; Comba P.; Hanson G. R.; Hausberg S.; Helmle S.; Wadepohl H. Inorg. Chem. 2011, 50, 6890. 10.1021/ic102430a. [DOI] [PubMed] [Google Scholar]
- Comba P.; Kubeil M.; Pietzsch J.; Rudolf H.; Stephan H.; Zarschler K. Inorg. Chem. 2014, 53, 6698. 10.1021/ic500476u. [DOI] [PubMed] [Google Scholar]
- Comba P.; Kerscher M.; Lawrance G. A.; Martin B.; Wadepohl H.; Wunderlich S. Angew. Chem., Int. Ed. 2008, 47, 4740. 10.1002/anie.200800515. [DOI] [PubMed] [Google Scholar]
- Comba P.; Haaf C.; Helmle S.; Karlin K. D.; Pandian S.; Waleska A. Inorg. Chem. 2012, 51, 2841. 10.1021/ic2019296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born K.; Comba P.; Daubinet A.; Fuchs A.; Wadepohl H. JBIC, J. Biol. Inorg. Chem. 2007, 12, 36. 10.1007/s00775-006-0161-2. [DOI] [PubMed] [Google Scholar]
- Comba P.; Daumann L.; Lefebvre J.; Linti G.; Martin B.; Straub J.; Zessin T. Aust. J. Chem. 2009, 62, 1238. 10.1071/CH09321. [DOI] [Google Scholar]
- Bleiholder C.; Börzel H.; Comba P.; Ferrari R.; Heydt M.; Kerscher M.; Kuwata S.; Laurenczy G.; Lawrance G. A.; Lienke A.; Martin B.; Merz M.; Nuber B.; Pritzkow H. Inorg. Chem. 2005, 44, 8145. 10.1021/ic0513383. [DOI] [PubMed] [Google Scholar]
- Börzel H.; Comba P.; Hagen K. S.; Kerscher M.; Pritzkow H.; Schatz M.; Schindler S.; Walter O. Inorg. Chem. 2002, 41, 5440. 10.1021/ic011114u. [DOI] [PubMed] [Google Scholar]
- Stephan H.; Walther M.; Fähnemann S.; Ceroni P.; Molloy J. K.; Bergamini G.; Heisig F.; Müller C. E.; Kraus W.; Comba P. Chem. - Eur. J. 2014, 20, 17011. 10.1002/chem.201404086. [DOI] [PubMed] [Google Scholar]
- Comba P.; Kerscher M.; Schiek W. In Progress in Inorganic Chemistry; Karlin K. D., Ed.; Wiley-Interscience Publications, 2007; pp 613–704. [Google Scholar]
- Comba P.; Morgen M.; Wadepohl H. Inorg. Chem. 2013, 52, 6481. 10.1021/ic4004214. [DOI] [PubMed] [Google Scholar]
- Comba P.; Hunoldt S.; Morgen M.; Pietzsch J.; Stephan H.; Wadepohl H. Inorg. Chem. 2013, 52, 8131. 10.1021/ic4008685. [DOI] [PubMed] [Google Scholar]
- Tomassoli I.; Guendisch D. Curr. Top. Med. Chem. 2016, 16, 1314. 10.2174/1568026615666150915111434. [DOI] [PubMed] [Google Scholar]
- Hancock R. D.; Melton D. L.; Harrington J. M.; McDonald F. C.; Gephart R. T.; Boone L. L.; Jones S. B.; Dean N. E.; Whitehead J. R.; Cockrell G. M. Coord. Chem. Rev. 2007, 251, 1678. 10.1016/j.ccr.2006.10.007. [DOI] [Google Scholar]
- Rosenblat T. L.; McDevitt M. R.; Mulford D. A.; Pandit-Taskar N.; Divgi C. R.; Panageas K. S.; Heaney M. L.; Chanel S.; Morgenstern A.; Sgouros G.; Larson S. M.; Scheinberg D. A.; Jurcic J. G. Clin. Cancer Res. 2010, 16, 5303. 10.1158/1078-0432.CCR-10-0382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer G.-J.; Miederer M.; Vranješ-Đurić S.; Čomor J. J.; Künzi G.; Hartley O.; Senekowitsch-Schmidtke R.; Soloviev D.; Buchegger F. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 547. 10.1007/s00259-003-1413-9. [DOI] [PubMed] [Google Scholar]
- Roux A.; Nonat A. M.; Brandel J.; Hubscher-Bruder V.; Charbonnière L. J. Inorg. Chem. 2015, 54, 4431. 10.1021/acs.inorgchem.5b00207. [DOI] [PubMed] [Google Scholar]
- Kuznetsov A. I.; Senan I. M.; Razenko I. O.; Serova T. M. Russ. Chem. Bull. 2014, 63, 2689. 10.1007/s11172-014-0800-7. [DOI] [Google Scholar]
- Trigo G. G.; Galvez E.; Avendaño C. J. Heterocycl. Chem. 1978, 15, 907. 10.1002/jhet.5570150603. [DOI] [Google Scholar]
- Kuznetsov A. I.; Sokolova T. D.; Vladimirova I. A.; Serova T. M.; D’yakov M. Y.; Shundrin L. A.; Moskovkin A. S.; Unkovskii B. V. Chem. Heterocycl. Compd. 1992, 28, 546. 10.1007/BF00475254. [DOI] [Google Scholar]
- Yamashita H.; Demizu Y.; Misawa T.; Shoda T.; Kurihara M. Tetrahedron 2015, 71, 2241. 10.1016/j.tet.2015.02.076. [DOI] [Google Scholar]
- Chekhlov A. N. J. Struct. Chem. 2000, 41, 294. 10.1007/BF02741595. [DOI] [Google Scholar]
- Minasyan G. G.; Agadzhanyan K. E.; Admyan G. G. Chem. Heterocycl. Compd. 1994, 30, 94. 10.1007/BF01164742. [DOI] [Google Scholar]
- Kuznetsov A. I.; Basargin E. B.; Moskovkin A. S.; Ba M. K.; Miroshnichenko I. V.; Botnikov M. Y.; Unkovskii B. V. Chem. Heterocycl. Compd. 1985, 21, 1382. 10.1007/BF00842964. [DOI] [Google Scholar]
- Agadzhanyan T. E.; Arutyunyan A. D.; Arutyunyan G. L. Chem. Heterocycl. Compd. 1992, 28, 772. 10.1007/BF00474490. [DOI] [Google Scholar]
- Semashko V. S.; Vatsadze S. Z.; Zyk N. V.; Godovikov I. A. Russ. Chem. Bull. 2008, 57, 2207. 10.1007/s11172-008-0303-5. [DOI] [Google Scholar]
- Cui H.; Goddard R.; Pörschke K.-R.; Hamacher A.; Kassack M. U. Inorg. Chem. 2016, 55, 2986. 10.1021/acs.inorgchem.5b02855. [DOI] [PubMed] [Google Scholar]
- Black D. S. C.; Deacon G. B.; Rose M. Tetrahedron 1995, 51, 2055. 10.1016/0040-4020(94)01069-C. [DOI] [Google Scholar]
- Bhattacharyya S.; Dixit M. Dalton Trans. 2011, 40, 6112. 10.1039/c1dt10379b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomä M. D. Inorg. Chim. Acta 2012, 389, 36. 10.1016/j.ica.2012.01.061. [DOI] [Google Scholar]
- Vatsadze S. Z.; Zyk N. V.; Rakhimov R. D.; Butin K. P.; Zefirov N. S. Russ. Chem. Bull. 1995, 44, 440. 10.1007/BF00702383. [DOI] [Google Scholar]
- Douglass J. E.; Ratliff T. B. J. Org. Chem. 1968, 33, 355. 10.1021/jo01265a073. [DOI] [Google Scholar]
- Gogoll A.; Grennberg H.; Axén A. Organometallics 1997, 16, 1167. 10.1021/om9609527. [DOI] [Google Scholar]
- Toom L.; Kütt A.; Kaljurand I.; Leito I.; Ottosson H.; Grennberg H.; Gogoll A. J. Org. Chem. 2006, 71, 7155. 10.1021/jo0604991. [DOI] [PubMed] [Google Scholar]
- Bos M.; van der Linden W. E. Anal. Chim. Acta 1996, 332, 201. 10.1016/0003-2670(96)00200-0. [DOI] [Google Scholar]
- Hosken G. D.; Hancock R. D. J. Chem. Soc., Chem. Commun. 1994, 1363. 10.1039/c39940001363. [DOI] [Google Scholar]
- Hosken G. D.; Allan C. C.; Boeyens J. C. A.; Hancock R. D. J. Chem. Soc., Dalton Trans. 1995, 3705. 10.1039/dt9950003705. [DOI] [Google Scholar]
- Stetter H.; Dieminger K. Chem. Ber. 1959, 92, 2658. 10.1002/cber.19590921042. [DOI] [Google Scholar]
- Sasaki T.; Eguchi S.; Kiriyama T.; Sakito Y. J. Org. Chem. 1973, 38, 1648. 10.1021/jo00949a006. [DOI] [Google Scholar]
- Kuthan J.; Paleček J.; Musil L. Collect. Czech. Chem. Commun. 1973, 38, 3491. 10.1135/cccc19733491. [DOI] [Google Scholar]
- Miller B. J. Chem. Soc., Chem. Commun. 1974, 750. 10.1039/c39740000750. [DOI] [Google Scholar]
- Sasaki T.; Eguchi S.; Kiriyama T.; Sakito Y.; Kato H. J. Chem. Soc., Chem. Commun. 1974, 725. 10.1039/C39740000725. [DOI] [Google Scholar]
- Dekkers A. W. J. D.; Verhoeven J. W.; Speckamp W. N. Tetrahedron 1973, 29, 1691. 10.1016/0040-4020(73)80114-0. [DOI] [Google Scholar]
- Gleiter R.; Kobayashi M.; Kuthan J. Tetrahedron 1976, 32, 2775. 10.1016/0040-4020(76)80122-6. [DOI] [Google Scholar]
- CRC Handbook of Chemistry and Physics, 94th ed.; Haynes W. M., Ed.; CRC Press, 2013; pp 7-1. [Google Scholar]
- Beverskog B. J. Electrochem. Soc. 1997, 144, 3476. 10.1149/1.1838036. [DOI] [Google Scholar]
- Wadas T. J.; Anderson C. J. Nat. Protoc. 2006, 1, 3062. 10.1038/nprot.2006.431. [DOI] [PubMed] [Google Scholar]
- Garrison J. C.; Rold T. L.; Sieckman G. L.; Figueroa S. D.; Volkert W. A.; Jurisson S. S.; Hoffman T. J. J. Nucl. Med. 2007, 48, 1327. 10.2967/jnumed.107.039487. [DOI] [PubMed] [Google Scholar]
- Prasanphanich A. F.; Nanda P. K.; Rold T. L.; Ma L.; Lewis M. R.; Garrison J. C.; Hoffman T. J.; Sieckman G. L.; Figueroa S. D.; Smith C. J. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 12462. 10.1073/pnas.0705347104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass L. A.; Wang M.; Welch M. J.; Anderson C. J. Bioconjugate Chem. 2000, 11, 527. 10.1021/bc990167l. [DOI] [PubMed] [Google Scholar]
- Boswell C. A.; Sun X.; Niu W.; Weisman G. R.; Wong E. H.; Rheingold A. L.; Anderson C. J. J. Med. Chem. 2004, 47, 1465. 10.1021/jm030383m. [DOI] [PubMed] [Google Scholar]
- Novick S. G. J. Chem. Educ. 1997, 74, 1463. 10.1021/ed074p1463. [DOI] [Google Scholar]
- Gans P.; O’Sullivan B. Talanta 2000, 51, 33. 10.1016/S0039-9140(99)00245-3. [DOI] [PubMed] [Google Scholar]
- Gans P.; Sabatini A.; Vacca A. Talanta 1996, 43, 1739. 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.