

Abstract

A series of dyes 2–5 based on 5-thienyl-2,1,3-benzothiadiazole and 5-thienyl-2,1,3-benzoselenadiazole cores were synthesized as near-infrared-emitting two-photon-absorbing fluorophores. Fluorescence maxima wavelengths as long as 714 nm and quantum yields as high as 0.67 were realized. The fluorescence quantum yields of dyes 2–4 were nearly constant, regardless of solvent polarity. These diazoles exhibited large Stokes shifts (>110 nm) and high two-photon figure of merit. Cells incubated on a 3D scaffold with probe 4 (encapsulated in Pluronic micelles) exhibited bright fluorescence, enabling 3D two-photon fluorescence imaging to a depth of 100 μm.
Introduction
Imaging biological samples via two-photon fluorescence microscopy (2PFM) has a unique advantage of realizing high-contrast 3D subcellular images up to several millimeters depth.1−7 Hence, 2PFM has been widely used for various imaging applications, such as for investigating brain or neural network dynamics,8−12 visualizing kidney dynamics,13 studying the chemistry of vision and the structure of skin,14,15 and for imaging vascular morphology and density.16,17 However, 2PFM has not migrated into clinical applications. One possible explanation for the lack of clinical use of 2PFM is the relatively shallow depth (micrometers to millimeters) it can achieve relative to other deep tissue imaging techniques, such as photoacoustic imaging or even computed tomography and magnetic resonance imaging in clinical applications, although its resolution is generally superior. Recently, progress has been reported in the application of near-infrared (NIR) fluorescent probes to overcome depth challenges in intraoperative microscopic image-guided cancer surgery.18−20 Biomaterials in general have an optical window in the NIR between 700 and 900 nm, in which the absorption of many biological materials is at a minimum;21 hence, light can penetrate deeper, up to centimeters through blood and tissues.22 Several NIR imaging systems have been developed for intraoperative NIR fluorescence imaging.23−31 One of them, a system called fluorescence-assisted resection and exploration (developed by Harvard’s Beth Israel Deaconess Medical Center) is under clinical trials for use in the colon and breast cancer surgery.30 This technique employs conventional (one-photon) fluorescence, whereas two-photon fluorescence imaging possesses some advantages with its higher fluorescence signal-to-noise ratio because of its unique highly localized excitation.
Two-photon excitation in 2PFM is typically conducted in the NIR. Therefore, probes that undergo two-photon absorption (2PA) in the NIR and fluoresce in the far-red to NIR region with high quantum yields are particularly desirable to achieve deep tissue imaging. In addition, to realize high-quality 2PFM images, other properties need to be optimized, such as high 2PA cross section and high photostability. Molecules that can fulfill these requirements are fairly limited. Most of the reported NIR fluorescent probes for one-photon NIR imaging have not been thoroughly evaluated in two-photon applications. Among those that have been examined, the most commonly used are cyanine dyes, such as commercially available indocyanine green, Cy5, or Cy7. These have been used as fluorophores for some time, and they possess relatively high 2PA cross sections (hundreds to thousands of Göppert Mayer (GM) units, 1 GM = 1 × 10–50 cm4 s/photon), although they are photochemically labile.
One particular type of cyanine dye, squaraine, possesses a 2PA cross section of tens of thousands of GM units.32−37 However, most squaraines suffer from low photochemical and nucleophile stability and have strong adsorption on plasma proteins. When applied in aqueous solution, strong dye aggregation often quenches fluorescence. For example, a squaraine dye reported in our previous study has 2PA cross sections of >10 000 GM, but in water, the fluorescence was quenched, and when encapsulated in Pluronic micelles, its fluorescence quantum yield was only about 4%.38
Boron-dipyrromethene (BODIPY) dyes are photostable, and their fluorescence quantum yields are high. Most reported BODIPY compounds for 2PFM bioimaging have low 2PA cross sections, ranging from 10 to <300 GM.39−42 Some of our recent efforts resulted in compounds with much improved 2PA cross sections (to >1000 GM), and their application in deep tissue imaging is still under evaluation.43 Other NIR dyes can be found in the literature, although only a few were reported for 2PFM bioimaging,44 including one that functioned better when encapsulated in silica nanoparticles, by aggregation-enhanced emission.45
To increase the candidate pool of highly efficient NIR probes for NIR 2PFM imaging and image-guided surgical applications, a class of chromophores with a 5-thienyl-2,1,3-benzothiadiazole core designed for organic photovoltaic devices and fluorescence bioprobes46,47 caught our attention. A typical derivative, compound 1 (Figure 1), exhibited strong red fluorescence. Recently, it has been reported that an analogue of this dye (with alkyl groups attached to thiophene rings) exhibited a high fluorescence quantum yield, for example, 0.85 in chloroform, with an emission maximum of 651 nm.48 In that study, an effort was taken to study a series of analogues but with limited success on finding dyes with longer emission wavelengths and reasonably high fluorescence quantum yields. Herein, we report a series of dyes (2–5, Figure 1) based on parent 1 that were designed to achieve emission in the NIR while maintaining high fluorescence quantum yields and high 2PA absorptivity. Preliminary investigation for the potential of one of these dyes for 2PFM NIR imaging is disclosed.49
Figure 1.

Structures of dyes 1–5.
Results and Discussion
Bis-thienylbenzothiadiazole 2 was prepared by Stille coupling of 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole 16, which was synthesized according to a method described in the literature,50 with 9,9-didecyl-2-iodo-7-(phenylsulfonyl)-9H-fluorene 10 in 63% yield (Figure 2). Intermediate 10 was synthesized by alkylation of 2-iodo-7-(phenylsulfonyl)-9H-fluorene 9 using bromodecane in the presence of KI, powdered KOH, and dimethyl sulfoxide (DMSO), following a similar procedure used in our previous study (Figures 2 and 3).51 The preparation of sulfone 9 was accomplished by iodination of the known compound 2-(phenylsulfonyl)-9H-fluorene 8(52) with periodic acid and iodine in HOAc/H2O/H2SO4 (100:20:3), giving a high yield of 85%. Dye 3 was prepared (Figure 3) in moderate yield (35%) by Stille coupling of 16 with 2-bromo-5-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 13, which was in turn synthesized by brominating 2-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 12 with N-bromosuccinimide (NBS) in AcOH/CHCl3 (1:5). The thiophenyl group in 12 was introduced into compound 10 by another Stille coupling with 2-(tributylstannyl)thiophene 11 (Figure 2).
Figure 2.

Synthesis of intermediates 10, 13, and 15. (a) SnCl4, 140 °C, 30 min, 63%; (b) periodic acid, iodine, HOAc/H2O/H2SO4 (v/v 100:20:3), 50–55 °C, 20 h, 85%; (c) BrC10H21, KOH, KI, DMSO, r.t., 20 h, 79%; (d) Pd(PPh3)4, DMF, reflux, 16 h, 85%; (e) NBS, HOAc/CHCl3 (v/v 1:5), r.t., 16 h, 83%; (f) trimethylsilylacetylene, PdCl2(PPh3)2, CuI, i-Pr2NH, toluene, reflux, 24 h, 67%; and (g) K2CO3, ether/MeOH (v/v 1:1), r.t., 16 h, 89%.
Figure 3.

Synthesis of dyes 2–5. (a) Pd(PPh3)4, DMF, reflux, 16 h, 63% for 2, 70% for 3; (b) PdCl2(PPh3)2, CuI, toluene/Et3N (v/v 4:1), 100 °C, 20 h, 35%; (c) (i) 13, n-BuLi, trimethylborate, −78 °C, r.t., 24 h and (ii) 18, ethylene glycol, Pd(PPh3)4, 2 N Na2CO3, reflux, 36 h, 28%.
Bis-ethynylthienylbenzothiadiazole 4 was prepared (Figure 3) by Sonogashira reaction of commercially available bis(2-bromo-5-thienyl)-2,1,3-benzothiadiazole 17 with 9,9-didecyl-2-ethynyl-7-(phenylsulfonyl)-9H-fluorene 15 (Figure 2) in a toluene/Et3N mixture, catalyzed by PdCl2(PPh3)2 and CuI, in 35% yield. Alkynyl fluorene 15 was synthesized by deprotection of 14 using K2CO3 as a base. Compound 14is the product obtained from the reaction of 9,9-didecyl-2-iodo-7-(phenylsulfonyl)-9H-fluorene (10) with trimethylsilylacetylene in toluene, using PdCl2(PPh3)2 and CuI as a catalyst and N,N-diisopropylamine as a base.
Bis-thienylbenzoselenadiazole 5 (Figure 3) was prepared by a one-pot Suzuki reaction. 2-Bromo-5-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 13 was converted to a borate ester in situ by first adding n-BuLi and then trimethylborate at −78 °C. The resulting solution was used directly for Suzuki coupling with 4,7-dibromobenzo[c][1,2,5]selenadiazole 18 in the presence of ethylene glycol and Pd(PPh3)4 in 28% overall yield.
The absorption spectra, fluorescence spectra, and fluorescence quantum yields of dyes 2–5 were measured in three solvents, that is, in toluene, tetrahydrofuran (THF), and methylene chloride. Figure 4 shows the spectra measured in methylene chloride (see Figures S1–S4 for spectra measured in all three solvents). In all of these solvents, the absorption spectra of all four dyes exhibit no significant changes in their spectral profiles or absorption maxima. With increasing solvent polarity, bathochromic shifts in fluorescence were observed for all four dyes, although only moderately, from 21 nm for dye 4 to 36 nm for dye 5. The fluorescence quantum yields of 0.60–0.67, 0.27–0.28, and 0.64–0.66 for dyes 2–4, respectively, were fairly constant in the three solvents investigated, implying that these dyes may maintain a relatively high fluorescence quantum yield in water when used as fluorescence bioimaging probes. This is a very interesting topic currently driving the synthesis of hydrophilic versions of these dyes. Excitation anisotropy spectra of all four dyes look similar, indicating an S0–S1 transition in the long wavelength band and another major transition at approximately 400 nm, corresponding to a two-photon allowed transition. These dyes also showed Stokes shifts greater than 110 nm, suggesting little reabsorption of fluorescence even when the local concentration is high.
Figure 4.

Absorption spectra (1, red, in CH2Cl2), fluorescence spectra (2, excited at the maximum absorption wavelengths, black, in CH2Cl2), 2PA spectra (3, blue, in CH2Cl2), and excitation anisotropy (4, gray, in polyTHF) of dyes 2–5.
Figure 4 also shows the 2PA spectra and excitation anisotropy spectra of dyes 2–5.49 The fluorescence maximum of 3 was red-shifted 36 nm (to 683 nm) relative to that of 2, as an extra thiophenyl group was introduced at each side of the benzothiadiazole core. While this red shift is desirable for NIR imaging; unfortunately there was a significant decrease in the fluorescence quantum yield, from 0.60 for 2 to 0.27 for 3. Here, it is reasonable to assume that the extra partially rotatable thiophenyl groups in dye 3 reduced the molecular rigidity and hence lowered the fluorescence quantum yield. On the other hand, the 2PA cross section of 2800 GM at 700 nm for 3 was twice that of 2 (1400 GM at 700 nm) because of its extended conjugation (more polarizable) system.
To evaluate the overall performance of a two-photon fluorescent (2PF) probe, a figure of merit (FM, the product of fluorescence quantum yield ΦFL and 2PA cross section δ2PA normalized by the photodecomposition quantum yield η), introduced in our previous study,53 was used. The photodecomposition quantum yields of 2 and 3 in methylene chloride were 1.4 × 10–6 and 7.0 × 10–6, respectively. The higher η and lower ΦFL of dye 3 resulted in a FM of 1.08 × 10–4 GM, about 6-fold lower than that of 2 at 6.00 × 10–4 GM, despite its higher δ2PA, making it clear that dye 2 should be a more efficient 2PF probe than dye 3, even though its δ2PA is lower. For this reason, dye 4 was designed and synthesized. The conjugation length of dye 4 with respect to that of dye 2 was extended by inserting an acetylene group between the fluorenyl group and the thiophenyl group to provide a more polarizable chromophore. The use of acetylene groups in dye 4 was also aimed to maintain some rigidity and to help maintain a high fluorescence quantum yield. Indeed, the fluorescence quantum yield of 0.64 for 4 in methylene chloride was comparable to that for 2. The fluorescence maximum of 634 nm for 4 was slightly blue-shifted relative to that of 2. The δ2PA of 4 in methylene chloride was 1100 GM at 700 nm, slightly lower than that of 2. Considering the shorter absorption wavelength of 4, 498 versus 506 nm for 2, it is reasonable to assume that the higher δ2PA value will appear at wavelengths shorter than 700 nm, which in this study was not measured owing to possible interference of the tail from the one-photon absorption. The photodecomposition quantum yield of dye 4 was decreased to 6.72 × 10–8, resulting in a FM of 1.04 × 10–2 GM, a 17-fold improvement compared with that of dye 2.
In 5, selenium replaced sulfur in 2 to further shift the absorption toward longer wavelength (538 nm), whereas the fluorescence emission maximum moved to 714 nm. The longer fluorescence wavelength should be beneficial for deep tissue imaging. Selenadiazole 5 had a fluorescence quantum yield ΦFL of 0.37, photodecomposition quantum yield η of 2.47 × 10−7, and δ2PA of 1150 GM at 740 nm, affording a FM of 1.72 × 10−3 GM, making this a promising 2PF probe that exhibits longer wavelength emission that the sulfur analog.
Because 4 exhibited the highest FM, it was selected for evaluation in 2PFM imaging. Cells were incubated with 4 (encapsulated in Pluronic micelles) and exhibited bright fluorescence in specific positions around the cell nucleus area (Figure 5C,D) similar to our previous study using the same Pluronic micelles with different dyes.54,55 Little fluorescence was observed in a negative control (Figure 5A). To ascertain possible applications for tissue imaging, cells cultured on a 3D scaffold were used as a model system. As shown in Figure 6, bright two-photon fluorescence images were collected that reached 100 μm in depth. Thus, this dye shows great potential for deep tissue imaging, a subject of further investigation.
Figure 5.
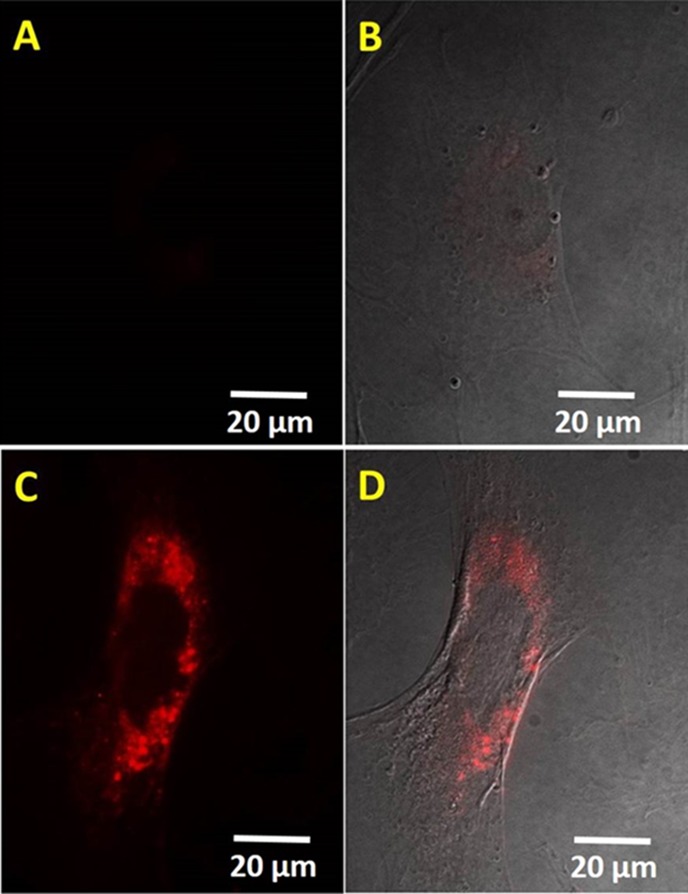
Fluorescent (A,C) and differential interference contrast overlay (B,D) images of 3T3 cells incubated without (A,B) or with (C,D) dye 4 (concentration 10 μM) encapsulated in Pluronic micelles. Scale bars show 20 μm.
Figure 6.
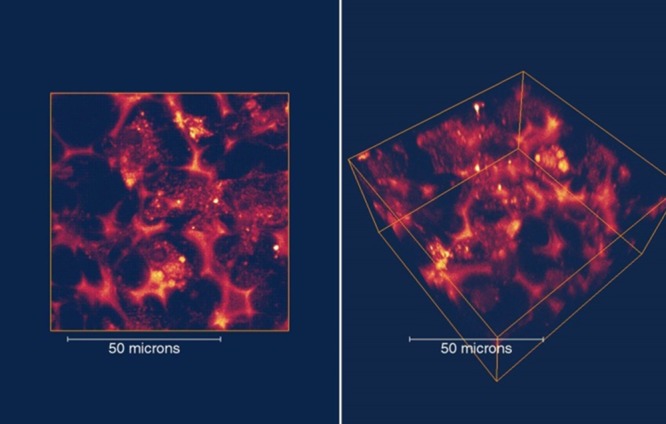
2PFM images of 3D cultured 3T3 cells with dye 4 (10 μM) in the X–Y top–down view (left) and 3D side view (right).
Conclusions
In summary, a series of dyes (2–5) based on 5-thienyl-2,1,3-benzothiadiazole and 5-thienyl-2,1,3-benzoselenadiazole cores designed to achieve NIR emission and high 2PF figure of merit were successfully synthesized and characterized. The fluorescence quantum yields of dyes 2 and 4 were quite good at 0.60–0.67. Interestingly, the fluorescence quantum yields for dyes 2–4 were almost constant regardless of the solvents used. These dyes also showed Stokes shifts larger than 110 nm, suggesting minimum reabsorption of fluorescence even when the local concentration is high (e.g., in cells). Among these dyes, the highest 2PF FM of 1.04 × 10–2 GM was determined for 4 when excited at 700 nm, whereas dye 2, with a longer emission wavelength, possessed a very high FM of 6.0 × 10–4 GM (excited at 740 nm), making both promising 2PF probes. Cells incubated on a 3D scaffold with 4 (encapsulated in Pluronic micelles) exhibited bright fluorescence, enabling high-contrast 3D 2PFM images up to 100 μm in depth. On the basis of these preliminary results, dyes 2–5 are promising candidates for intraoperative NIR fluorescence microscopic image-guided surgery, using either one-photon or two-photon excitation while the development of hydrophilic analogues is currently under way.
Experimental Section
General
2-(Phenylsulfonyl)-9H-fluorene 8(52) and 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole 16(50) were prepared according to literature methods. 2-(Tri-n-butylstannyl)thiophene 11, 4,7-bis(2-bromo-5-thienyl)-2,1,3-benzothiadiazole 17, and 4,7-dibromobenzo[c][1,2,5]selenadiazole 18 were purchased from Alfa Aesar, Apic Laboratories, and TCI America Fine Chemicals, respectively. All other reagents and solvents were used as received from commercial suppliers. Melting points are uncorrected. 1H NMR and 13C NMR spectra were recorded at 300 or 500 MHz and at 75 or 125 MHz, respectively. Mass spectroscopy (MS), high-resolution MS (HRMS), and matrix-assisted laser desorption/ionization-time of flight MS (MALDI-TOF MS) were measured on gas chromatography/MS, HRMS, and MALDI-TOF MS instruments, respectively, in the Department of Chemistry at the University of Florida.
Preparation of 2-Iodo-7-(phenylsulfonyl)-9H-fluorene 9
2-(Phenylsulfonyl)-9H-fluorene 8 (1.50 g, 4.90 mmol) was dissolved in 30 mL of solvent HOAc/H2O/H2SO4 (100:20:3) by heating to reflux. Upon cooling to 50–55 °C, periodic acid (0.37 g, 1.62 mmol) and iodine (0.83 g, 3.27 mmol) were added. The mixture was kept at this temperature for 20 h, during which the yellow precipitate was formed. Upon cooling, the precipitate was collected by filtration and thoroughly washed with water to give 1.80 g of product (85% yield). No further purification was needed. Mp 212–213 °C; 1H NMR (500 MHz, CDCl3-d) δ 8.09 (s, 1H), 7.98 (m, 3H), 7.92 (s, 1H), 7.83 (d, J = 7.8 Hz, 1H), 7.73 (d, J = 8.1 Hz, 1H), 7.49–7.57 (m, 4H), 3.90 (s, 2H); 13C NMR (125 MHz, CDCl3-d) δ 146.2, 145.7, 143.4, 141.9, 139.9, 139.3, 136.3, 134.5, 133.1, 129.3, 127.6, 127.1, 124.4, 122.4, 120.5, 94.2, 36.6; HRMS (ESI) for C19H13IO2S theoretical m/z [M + H]+ = 432.9754, found m/z [M + H]+ = 432.9756.
Preparation of 9,9-Didecyl-2-iodo-7-(phenylsulfonyl)-9H-fluorene 10
A mixture of 2-iodo-7-(phenylsulfonyl)-9H-fluorene 9 (1.80 g, 4.16 mmol), 1-bromodecane (1.84 g, 8.32 mmol), and KI (0.07 g, 0.42 mmol) in 25 mL of DMSO was degassed by Ar for 20 min, to which powdered KOH (0.99 g, 17.65 mmol) was added under Ar. The reaction mixture was reacted at room temperature for 20 h, poured into water, and extracted with hexanes. The organic extract was washed with water, dried over MgSO4, and concentrated. The crude product was purified by column chromatography using silica gel (4:1 hexanes/methylene chloride), providing 2.33 g of product (79% yield). Mp 42–43 °C; 1H NMR (500 MHz, CDCl3-d) δ 7.95 (m, 3H), 7.89 (m, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.70 (m, 2H), 7.46–7.57 (m, 4H), 1.96 (m, 4H), 1.14 (m, 28H), 0.87 (t, J = 7.2 Hz, 6H), 0.52 (m, 4H). 13C NMR (125 MHz, CDCl3-d) δ 154.0, 151.2, 145.1, 142.2, 140.0, 138.6, 136.4, 133.0, 132.4, 129.2, 127.3, 127.2, 122.4, 122.2, 120.4, 95.0, 55.9, 39.8, 31.9, 31.6, 29.8, 29.5, 29.3, 29.2, 23.7, 22.7, 14.1; HRMS (ESI) for C39H53IO2S theoretical m/z [M + H]+ = 713.2889, found m/z [M + H]+ = 713.2868.
Preparation of 2-(9,9-Didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 12
9,9-Didecyl-2-iodo-7-(phenylsulfonyl)-9H-fluorene 10 (1.62 g, 2.27 mmol) and Pd(PPh3)4 (0.13 g, 0.11 mmol) in dry dimethylformamide (DMF) were degassed by Ar for 20 min, then 2-(tributylstannyl)thiophene 11 (0.72 mL, 2.28 mmol) was injected into the mixture, which was then heated at reflux for 16 h. Upon cooling, water was added and the product was extracted with methylene chloride (3 × 20 mL). The combined organic phase was washed with water and dried over MgSO4. The solvent was removed, and the crude product was purified by column chromatography using silica gel (1:1.5 hexanes/CH2Cl2), providing 1.29 g of product (85% yield) as a viscous oil. 1H NMR (500 MHz, CDCl3-d) δ 7.96 (m, 3H), 7.90 (m, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.64 (m, 1H), 7.48–7.59 (m, 4H), 7.42 (m, 1H), 7.33 (m, 1H), 7.13 (m, 1H), 2.02 (m, 4H), 1.00–1.28 (m, 28H), 0.86 (t, J = 7.2 Hz, 6H), 0.67 (m, 4H). 13C NMR (125 MHz, CDCl3-d) δ 152.7, 151.9, 145.7, 144.4, 142.4, 139.3, 138.4, 135.1, 132.9, 129.2, 128.2, 127.3, 127.2, 125.3, 125.2, 123.5, 122.2, 121.3, 120.3, 120.2, 55.8, 40.0, 31.9, 29.8, 29.5, 29.5, 29.3, 29.2, 23.8, 22.7, 14.1. HRMS (ESI) for C43H56O2S2 theoretical m/z [M + NH4]+ = 686.4060, found m/z [M + NH4]+ = 686.4072.
Preparation of 2-Bromo-5-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 13
2-(9,9-Didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 12 (2.40 g, 3.59 mmol) and NBS (0.64 g, 3.60 mmol) in HOAc/CHCl3 (1:5, 30 mL) were stirred under Ar at room temperature in dark for 16 h. Water was added, and the organic phase was separated, washed with diluted Na2CO3 solution, and dried over MgSO4. The solvent was removed, and the crude product was purified by column chromatography (1:1 hexanes/CH2Cl2), providing 2.24 g of product (83% yield) as a viscous oil. 1H NMR (500 MHz, CDCl3-d) δ 7.96 (m, 3H), 7.90 (m, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.64 (m, 1H), 7.48–7.59 (m, 4H), 7.42 (m, 1H), 7.33 (m, 1H), 7.13 (m, 1H), 2.02 (m, 4H), 1.00–1.28 (m, 28H), 0.86 (t, J = 7.2 Hz, 6H), 0.67 (m, 4H). 13C NMR (125 MHz, CDCl3-d) δ 152.7, 151.9, 145.7, 144.4, 142.4, 139.3, 138.4, 135.1, 132.9, 129.2, 128.2, 127.3, 127.2, 125.3, 125.2, 123.5, 122.2, 121.3, 120.3, 120.2, 55.8, 40.0, 31.9, 29.8, 29.5, 29.5, 29.3, 29.2, 23.8, 22.7, 14.1. HRMS (APCI) for C43H55BrO2S2 theoretical m/z [M + H]+ = 747.2900, found m/z [M + H]+ = 747.2900.
Preparation of 4,7-Bis(5-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole 2
9,9-Didecyl-2-iodo-7-(phenylsulfonyl)-9H-fluorene 10 (0.26 g, 0.365 mmol), 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole 16 (0.11 g, 0.176 mmol), and Pd(PPh3)4 (20 mg, 0.017 mmol) in dry DMF were degassed by Ar for 20 min and heated at reflux for 16 h. Upon cooling, water was added, and the product was extracted with CH2Cl2 (3 × 20 mL). The combined organic phase was washed with water and dried over MgSO4. The solvent was removed, and the crude product was purified by column chromatography using silica gel (1:2 hexanes/CH2Cl2), providing 0.16 g of product (63% yield). Mp 64–65 °C. 1H NMR (500 MHz, CDCl3-d) δ 8.14 (d, J = 3.9 Hz, 2H), 7.97 (m, 6H), 7.91 (m, 4H), 7.70–7.78 (m, 6H), 7.68 (s, 2H), 7.48–7.56 (m, 8H), 2.05 (m, 8H), 1.01–1.25 (m, 56H), 0.83 (t, J = 7.1 Hz, 12H), 0.59 (m, 8H). 13C NMR (12.5 MHz, CDCl3-d) δ 152.8, 152.6, 152.0, 145.7, 145.6, 142.3, 139.4, 139.0, 138.8, 134.7, 132.9, 129.2, 128.7, 127.3, 127.3, 125.8, 125.4, 125.1, 124.6, 122.2, 121.4, 120.3, 120.0, 55.8, 40.0, 31.9, 29.9, 29.5, 29.5, 29.3, 29.2, 23.8, 22.7, 14.1. HRMS (ESI) for C92H112N2O4S5 theoretical m/z [M + H]+ = 1469.7252, found m/z [M + H]+ = 1469.7308.
Preparation of 4,7-Bis(5′-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)-2,2′-bithiophen-5-yl)benzo[c][1,2,5]thiadiazole 3
2-Bromo-5-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 13 (0.11 g, 0.147 mmol), 4,7-bis(5-(trimethylstannyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole 16 (0.045 g, 0.072 mmol), and Pd(PPh3)4 (9 mg, 0.008 mmol) in dry DMF were degassed by Ar for 20 min and heated at reflux for 16 h. Upon cooling, water was added, and the product was extracted with CH2Cl2 (3 × 15 mL). The combined organic phase was washed with water and dried over MgSO4. The solvent was removed, and the crude product was purified by column chromatography using silica gel (1:2 to 1:2.5 hexanes/CH2Cl2). The product was further purified by recrystallization from hexanes/CH2Cl2 to provide 0.082 g of product (70% yield) as a dark red solid. Mp 217–218 °C; 1H NMR (500 MHz, CDCl3-d) δ 8.09 (d, J = 4.0 Hz, 2H), 7.97 (m, 6H), 7.90 (m, 4H), 7.78 (d, J = 8.1 Hz, 2H), 7.74 (d, J = 7.9 Hz, 2H), 7.64 (m, 2H), 7.60 (m, 2H), 7.49–7.57 (m, 6H), 7.38 (d, J = 3.9 Hz, 2H), 7.34 (d, J = 3.9 Hz, 2H), 7.32 (d, J = 3.7 Hz, 2H), 2.04 (m, 8H), 1.02–1.27 (m, 56H), 0.86 (t, J = 7.1 Hz, 12H), 0.59 (m, 8H). 13C NMR (125 MHz, CDCl3-d) δ 151.8, 151.4, 150.9, 144.5, 142.5, 141.3, 138.3, 137.7, 137.6, 137.2, 135.9, 133.5, 131.9, 128.2, 127.3, 126.3, 126.3, 124.5, 124.1, 124.0, 123.9, 123.5, 123.4, 121.2, 120.4, 119.3, 118.8, 54.8, 39.0, 30.9, 28.8, 28.5, 28.5, 28.2, 28.2, 22.8, 21.6, 13.1. HRMS (ESI) for C100H116N2O4S7 theoretical m/z [M + H]+ = 1633.7004, found m/z [M + H]+ = 1633.6968.
Preparation of ((9,9-Didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)ethynyl)trimethylsilane 14
9,9-Didecyl-2-iodo-7-(phenylsulfonyl)-9H-fluorene 10 (0.78 g, 1.09 mmol), trimethylsilylacetylene (0.186 mL, 1.30 mmol), PdCl2(PPh3)2 (42 mg, 0.06 mmol), CuI (23 mg, 0.12 mmol), and N,N-diisopropylamine (1.0 mL, 7.13 mmol) in 40 mL of dry toluene were mixed under Ar and heated at slight reflux for 24 h. Upon cooling, saturated NH4Cl solution was added, and the product was extracted with hexane (3 × 25 mL). The combined organic phase was washed with water and dried over MgSO4. The removal of solvent and purification by column chromatography (2.5:1 hexane/CH2Cl2) afforded 0.63 g of product (67% yield) as an oil. 1H NMR (300 MHz, CDCl3-d) δ 7.90 (m, 4H), 7.75 (d, J = 8.2 Hz, 1H), 7.65 (d, J = 7.6 Hz, 1H), 7.54–7.45 (m, 5H), 1.96 (m, 4H), 0.98–1.25 (m, 28H), 0.86 (t, J = 6.7 Hz, 6H), 0.48 (m, 4H), 0.28 (s, 9H). 13C NMR (125 MHz, CDCl3-d) δ 152.1, 151.7, 145.3, 142.3, 139.7, 139.3, 132.9, 131.4, 129.2, 127.3, 127.2, 126.4, 123.3, 122.2, 120.6, 120.6, 105.5, 95.3, 55.7, 40.0, 31.9, 29.8, 29.5, 29.5, 29.2, 29.2, 23.7, 22.7, 14.1, 0.0. HRMS (MALDI-TOF) for C44H62O2SSi theoretical m/z [M + H]+ = 683.4312, found m/z [M + H]+ = 683.4308.
Preparation of 9,9-Didecyl-2-ethynyl-7-(phenylsulfonyl)-9H-fluorene 15
9,9-Didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)ethynyl)trimethylsilane 14 (0.51 g, 0.75 mmol) and K2CO3 (0.21 g, 1.52 mmol) in 20 mL of ether/MeOH (1:1) were degassed by Ar for 20 min and reacted at room temperature for 16 h. Then, saturated NH4Cl aqueous solution (50 mL) was added to the reaction mixture, and the resultant solution was stirred at room temperature for 0.5 h. The above-mentioned solution was extracted with ethyl acetate (30 mL × 3). The combined organic phase was washed with water and dried over MgSO4. The solvent was evaporated, and the residue was purified by column chromatography (3:1 hexane/CH2Cl2) to give 0.48 g of product (89% yield) as an oil. 1H NMR (500 MHz, CDCl3-d) δ 7.95 (m, 3H), 7.90 (m, 1H), 7.77 (m, 1H), 7.68 (m, 1H), 7.48–7.57 (m, 5H), 3.19 (s, 1H), 1.98 (m, 4H), 0.97–1.29 (m, 28H), 0.87 (t, J = 7.2 Hz, 6H), 0.51 (m, 4H). 13C NMR (125 MHz, CDCl3-d) δ 152.1, 151.8, 145.2, 142.2, 139.9, 139.6, 133.0, 131.5, 129.2, 127.3, 127.2, 126.7, 122.3, 122.2, 120.7, 120.7, 84.1, 78.1, 55.7, 39.9, 31.9, 29.8, 29.5, 29.5, 29.2, 29.2, 23.8, 22.7, 14.1. HRMS (MALDI-TOF) for C41H54O2S theoretical m/z [M + Na]+ = 633.3737, found m/z [M + Na]+ = 633.3757.
Preparation of 4,7-Bis(5-((9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)ethynyl)thiophen-2-yl)benzo[c][1,2,5]thiadiazole 4
9,9-Didecyl-2-ethynyl-7-(phenylsulfonyl)-9H-fluorene 15 (0.53 g, 0.87 mmol), 4,7-bis(2-bromo-5-thienyl)-2,1,3-benzothiadiazole 17 (0.15 mL, 0.43 mmol), PdCl2(PPh3)2 (30 mg, 0.043 mmol), and CuI (16 mg, 0.084 mmol) in 25 mL of toluene/Et3N (4/1) were heated at 100 °C for 20 h under Ar. The color of the mixture turned red, and orange fluorescence was observed. Water was added, and the organic phase was separated. The organic phase was washed with water and dried over sodium sulfate. The solvent was removed, and the crude product was purified by column chromatography using CH2Cl2/hexanes (2:1) as an eluant. Recrystallization from CH2Cl2 hexanes afforded 0.23 g of product as red needle crystals (35% yield). Mp 142–143 °C; 1H NMR (500 MHz, CDCl3-d) δ 8.07 (d, J = 3.9 Hz, 2H), 7.97 (m, 6H), 7.91 (m, 4H), 7.79 (d, J = 7.9 Hz, 2H), 7.73 (d, J = 7.8 Hz, 2H), 7.49–7.57 (m, 10H), 7.41 (d, J = 3.9 Hz, 2H), 2.01 (m, 8H), 0.97–1.29 (m, 56H), 0.86 (t, J = 7.2 Hz, 12H), 0.55 (m, 8H). 13C NMR (125 MHz, CDCl3-d) δ 152.1, 151.8, 145.2, 142.2, 139.9, 139.6, 133.0, 131.5, 129.2, 127.3, 127.2, 126.7, 122.3, 122.2, 120.7, 120.7, 84.1, 78.1, 55.7, 39.9, 31.9, 29.8, 29.5, 29.5, 29.2, 29.2, 23.8, 22.7, 14.1. HRMS (MALDI-TOF) for C96H112N2O4S5 theoretical m/z [M + H]+ = 1517.7251, found m/z [M + H]+ = 1517.7233.
Preparation of 4,7-Bis(5-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophen-2-yl)benzo[c][1,2,5]selenadiazole 5
To 2-bromo-5-(9,9-didecyl-7-(phenylsulfonyl)-9H-fluoren-2-yl)thiophene 13 (0.72 g, 0.96 mmol) in dry THF (25 mL) was added 1.6 M n-BuLi in hexane (0.60 mL, 0.96 mmol) at −78 °C and reacted at this temperature for 30 min. Trimethylborate (0.11 mL, 0.96 mmol) was injected into the mixture at −78 °C, and the mixture was stirred for 24 h while the temperature was allowed to warm to room temperature. The resultant mixture was then refluxed with 4,7-dibromobenzo[c][1,2,5]selenadiazole (0.13 g, 0.38 mmol), three drops of ethylene glycol, Pd(PPh3)4 (56 mg, 0.05 mmol), and 2 N aqueous sodium carbonate solution (0.48 mL, 0.96 mmol) in Ar degassed THF (20 mL) for 36 h. Upon cooling, water was added, and the product was extracted with hexanes and purified by column chromatography (1:1.5 hexanes/CH2Cl2) to give 0.16 g of product (28% yield). Mp 160–162 °C; 1H NMR (500 MHz, CDCl3-d) δ 8.05 (d, J = 4.0 Hz, 2H), 7.97 (m, 4H), 7.95 (m, 2H), 7.90 (m, 2H), 7.87 (s, 2H), 7.78 (d, J = 8.1 Hz, 2H), 7.74 (m, 4H), 7.68 (m, 2H), 7.48–7.56 (m, 8H), 3.19 (s, 1H), 1.98 (m, 4H), 0.97–1.29 (m, 28H), 0.87 (t, J = 7.2 Hz, 6H), 0.51 (m, 4H). 13C NMR (125 MHz, CDCl3-d) δ 158.1, 152.8, 152.0, 146.1, 145.6, 142.4, 139.4, 139.3, 138.7, 134.8, 132.9, 129.2, 128.5, 127.3, 127.3, 125.6, 125.1, 124.3, 122.2, 121.4, 120.3, 120.0, 55.8, 40.0, 31.9, 29.9, 29.5, 29.5, 29.3, 29.2, 23.8, 22.7, 14.1. HRMS (MALDI-TOF) for C92H112N2O4S4Se theoretical m/z [M + H]+ = 1516.6681, found m/z [M + H]+ = 1516.6676.
Linear Optical Properties
All steady-state absorption, fluorescence emission, excitation, and excitation anisotropy spectra of dyes 2–5 were investigated in 10 mm path length quartz cuvettes at room temperature. The steady-state absorption was measured with an Agilent 8453 UV–visible spectrophotometer. Fluorescence emission and excitation spectra were obtained using an Edinburgh Photonics FLS980 spectrometer equipped with a thermoelectric cooled photomultiplier detector (Hamamatsu) and a liquid-nitrogen cooled NIR-photomultiplier detector (Hamamatsu). All measurements were carried out with the optical density below 0.12 at the excitation wavelength to avoid reabsorption. The excitation and fluorescence emission spectra were corrected for the spectral sensitivity of Edinburgh Photonics excitation and detection system using factory-measured correction files. Excitation anisotropy spectra were measured with two computer-controlled polarizers in the FLS980 spectrometer in a high viscous solvent (polyTHF).
Fluorescence quantum yields were determined by a standard relative method with rhodamine 6G (Φ ≈ 0.94 in ethanol) and cresyl violet (Φ ≈ 0.54 in methanol) as a reference.56 The equation used is as follows
where Φ is the quantum yield, the subscript R refers to the reference, I is the integrated emission signal, OD is the optical density at the excitation wavelength, n is the refractive index of the solvent, and RP is the relative power of the light source of the spectrofluorimeter at the excitation wavelength. Photochemical stability measurements of all materials were carried out using continuous-wave (cw) diode laser irradiation (λexc ≈ 532 nm for dye 2–5), and photodecomposition quantum yields, η, were calculated using absorption methodology.57
Nonlinear Optical Properties
2PA spectra of dyes 2–5 were determined using an open-aperture Z-scan method using a femtosecond laser system (Coherent, Inc.). The output of a Ti/sapphire laser (Mira 900-F, tuned to 800 nm, with a repetition rate of 76 MHz, average power ≈ 1.1 W, and pulse duration ≈ 200 fs), pumped by the second harmonic of Nd3+/YAG laser (Verdi-10), was regeneratively amplified with a 1 kHz repetition rate (Legend Elite USP) providing ≈100 fs pulses (FWHM) with energy ≈ 3.6 mJ/pulse. This output at 800 nm was split into two separate beams and pumped two ultrafast optical parametric amplifiers (OPAs) (OPerA Solo, Coherent Inc.) with a tuning range of 0.24–20 μm, ≈100 fs (FWHM), and pulse energies up to ≈100 μJ. A single laser beam from the first OPA was used for the open aperture Z-scan method.58 2PA measurements were carried out in 1 mm spectrofluorometric quartz cuvettes with a concentration of 10–3 M ≤ C ≤ 10–2 M at room temperature.
Preparing Micelle with Dye 4
A stock solution of dye 4 was prepared in CH2Cl2 at a concentration of 0.1 mM. The stock solution was then mixed with 0.25% Pluronic F-127 solution in a 1:1 volume ratio. The solution was then sonicated, and the organic solvent was evaporated. The stock micelle solution of 4 was maintained at 4 °C.
Cell Imaging
For one-photon absorption cell imaging, 3T3 cells (ATCC) were seeded on poly-d-lysine-coated coverslips at a concentration of 5 × 104 cells/mL and incubated for 48 h. The micelle solution of dye 4 was then diluted to 10 μM with minimum essential media (MEM) (Corning, Cellgro) and added to the cells. The cells were incubated for 30 min and then fixed with 4% formaldehyde. NaBH4 was added twice at 1 mg/mL for 5 min to reduce autofluorescence. Coverslips were mounted on slides with ProLong Gold antifade reagent and then imaged using an Olympus IX70 disk scanning unit microscope fitted with a Texas Red filter cube.
For 2PFM cell imaging, 3T3 cells were cultured on 3D scaffolds (Reinnervate, Alvetex) at a concentration of 8 × 106 cells/mL and incubated for 5 days. The micelle solution of dye 4 was then diluted to 10 μM with MEM and added to the cells. The cells were incubated for 2 h and then fixed with 4% formaldehyde. Scaffolds were mounted on slides with ProLong Gold antifade reagent and then imaged using a Leica SP5 II microscope equipped with a Coherent Chameleon Vision S Laser source (prechirped compensated, 70 fs, 80 MHz) tuned to 800 nm. Scanned images were processed with Amira for 3D reconstruction.
Acknowledgments
We acknowledge the National Science Foundation (CBET-1517273 and CHE-0832622), the U.S. National Academy of Sciences (PGAP210877), the National Academy of Sciences of Ukraine (grants 1.4.1.B/153 and VC/157), and the People Programme (Marie Curie Actions) of the European Union’s Seventh Framework Programme (FP7/2007-2013/under REA grant agreement no. 607721) for financial support.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00289.
Copies of 1H and 13C NMR and high resolution mass spectra for all new compounds as well as for absorption spectra, fluorescence spectra, and excitation anisotropy for dyes 2–5 in toluene, THF, and CH2Cl2 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Dong C.-Y.; Kim K. H.; Buehler C.; Hsu L.; Kim H.; So P. T. C.; Masters B. R.; Gratton E.; Kochevar I. E. In Emerging Tools for Single-Cell Analysis; John Wiley & Sons, Inc., 2002; p 221. [Google Scholar]
- Helmchen F.; Denk W. Nat. Methods 2005, 2, 932. 10.1038/nmeth818. [DOI] [PubMed] [Google Scholar]
- Nakamura O. Microsc. Res. Tech. 1999, 47, 165.. [DOI] [PubMed] [Google Scholar]
- Oheim M.; Michael D. J.; Geisbauer M.; Madsen D.; Chow R. H. Adv. Drug Delivery Rev. 2006, 58, 788. 10.1016/j.addr.2006.07.005. [DOI] [PubMed] [Google Scholar]
- Rubart M. Circ. Res. 2004, 95, 1154. 10.1161/01.RES.0000150593.30324.42. [DOI] [PubMed] [Google Scholar]
- So P. T. C.; Dong C. Y.; Masters B. R.; Berland K. M. Annu. Rev. Biomed. Eng. 2000, 2, 399. 10.1146/annurev.bioeng.2.1.399. [DOI] [PubMed] [Google Scholar]
- Wallace V. P.; Dunn A. K.; Coleno M. L.; Tromberg B. J. In Methods in Cellular Imaging; Periasamy A., Ed.; Springer: New York, 2001; p 180. [Google Scholar]
- Fumagalli S.; Ortolano F.; De Simoni M.-G. Prog. Neurobiol. 2014, 121, 36. 10.1016/j.pneurobio.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Homma R.; Baker B. J.; Jin L.; Garaschuk O.; Konnerth A.; Cohen L. B.; Zecevic D. Philos. Trans. R. Soc., B 2009, 364, 2453–2467. 10.1098/rstb.2009.0084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lütcke H.; Helmchen F. Rep. Prog. Phys. 2011, 74, 086602. 10.1088/0034-4885/74/8/086602. [DOI] [Google Scholar]
- Denk W. Proc. SPIE 2002, 4620, 62–63. 10.1117/12.470676. [DOI] [Google Scholar]
- Ellis-Davies G. C. R. ACS Chem. Neurosci. 2011, 2, 185. 10.1021/cn100111a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashworth S. L.; Sandoval R. M.; Tanner G. A.; Molitoris B. A. Kidney Int. 2007, 72, 416. 10.1038/sj.ki.5002315. [DOI] [PubMed] [Google Scholar]
- Imanishi Y.; Lodowski K. H.; Koutalos Y. Biochemistry 2007, 46, 9674. 10.1021/bi701055g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park K. J. Controlled Release 2008, 132, 1. 10.1016/j.jconrel.2008.09.077. [DOI] [PubMed] [Google Scholar]
- Yanez C. O.; Morales A. R.; Yue X.; Urakami T.; Komatsu M.; Järvinen T. A. H.; Belfield K. D. PLoS One 2013, 8, e67559 10.1371/journal.pone.0067559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue X.; Morales A. R.; Githaiga G. W.; Woodward A. W.; Tang S.; Sawada J.; Komatsu M.; Liu X.; Belfield K. D. Org. Biomol. Chem. 2015, 13, 10716. 10.1039/c5ob01536g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibbs S. L. Quant. Imag. Med. Surg. 2012, 2, 177. 10.3978/j.issn.2223-4292.2012.09.04. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keereweer S.; Van Driel P. B. A. A.; Snoeks T. J. A.; Kerrebijn J. D. F.; de Jong R. J. B.; Vahrmeijer A. L.; Sterenborg H. J. C. M.; Lowik C. W. G. M. Clin. Cancer Res. 2013, 19, 3745. 10.1158/1078-0432.CCR-12-3598. [DOI] [PubMed] [Google Scholar]
- Vahrmeijer A. L.; Hutteman M.; van der Vorst J. R.; van de Velde C. J. H.; Frangioni J. V. Nat. Rev. Clin. Oncol. 2013, 10, 507. 10.1038/nrclinonc.2013.123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weissleder R. Nat. Biotechnol. 2001, 19, 316. 10.1038/86684. [DOI] [PubMed] [Google Scholar]
- Chance B. Ann. N.Y. Acad. Sci. 1998, 838, 29. 10.1111/j.1749-6632.1998.tb08185.x. [DOI] [PubMed] [Google Scholar]
- Handgraaf H. J. M.; Verbeek F. P. R.; Tummers Q. R. J. G.; Boogerd L. S. F.; van de Velde C. J. H.; Vahrmeijer A. L.; Gaarenstroom K. N. Gynecol. Oncol. 2014, 135, 606. 10.1016/j.ygyno.2014.08.005. [DOI] [PubMed] [Google Scholar]
- Rasmussen J. C.; Tan I.-C.; Marshall M. V.; Fife C. E.; Sevick-Muraca E. M. Curr. Opin. Biotechnol. 2009, 20, 74. 10.1016/j.copbio.2009.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieog J. S. D.; Hutteman M.; van der Vorst J. R.; Kuppen P. J. K.; Que I.; Dijkstra J.; Kaijzel E. L.; Prins F.; Löwik C. W. G. M.; Smit V. T. H. B. M.; van de Velde C. J. H.; Vahrmeijer A. L. Breast Cancer Res. Treat. 2011, 128, 679. 10.1007/s10549-010-1130-6. [DOI] [PubMed] [Google Scholar]
- Yamashita S.-i.; Tokuishi K.; Anami K.; Miyawaki M.; Moroga T.; Kamei M.; Suehiro S.; Ono K.; Takeno S.; Chujo M.; Yamamoto S.; Kawahara K. J. Thorac. Cardiovasc. Surg. 2011, 141, 141. 10.1016/j.jtcvs.2010.01.028. [DOI] [PubMed] [Google Scholar]
- Cahill R. A.; Anderson M.; Wang L. M.; Lindsey I.; Cunningham C.; Mortensen N. J. Surg. Endosc. 2012, 26, 197. 10.1007/s00464-011-1854-3. [DOI] [PubMed] [Google Scholar]
- van der Poel H. G.; Buckle T.; Brouwer O. R.; Olmos R. A. V.; van Leeuwen F. W. B. Eur. Urol. 2011, 60, 826. 10.1016/j.eururo.2011.03.024. [DOI] [PubMed] [Google Scholar]
- Yamauchi K.; Nagafuji H.; Nakamura T.; Sato T.; Kohno N. Ann. Surg. Oncol. 2011, 18, 2042. 10.1245/s10434-010-1499-9. [DOI] [PubMed] [Google Scholar]
- Troyan S. L.; Kianzad V.; Gibbs-Strauss S. L.; Gioux S.; Matsui A.; Oketokoun R.; Ngo L.; Khamene A.; Azar F.; Frangioni J. V. Ann. Surg. Oncol. 2009, 16, 2943. 10.1245/s10434-009-0594-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinoglio G.; Priora F.; Bianchi P. P.; Lucido F. S.; Licciardello A.; Maglione V.; Grosso F.; Quarati R.; Ravazzoni F.; Lenti L. M. Surg. Endosc. 2013, 27, 2156. 10.1007/s00464-012-2733-2. [DOI] [PubMed] [Google Scholar]
- Ohira S.; Rudra I.; Schmidt K.; Barlow S.; Chung S.-J.; Zhang Q.; Matichak J.; Marder S. R.; Brédas J.-L. Chem.—Eur. J. 2008, 14, 11082. 10.1002/chem.200801055. [DOI] [PubMed] [Google Scholar]
- Chung S.-J.; Zheng S.; Odani T.; Beverina L.; Fu J.; Padilha L. A.; Biesso A.; Hales J. M.; Zhan X.; Schmidt K.; Ye A.; Zojer E.; Barlow S.; Hagan D. J.; Van Stryland E. W.; Yi Y.; Shuai Z.; Pagani G. A.; Brédas J.-L.; Perry J. W.; Marder S. R. J. Am. Chem. Soc. 2006, 128, 14444. 10.1021/ja065556m. [DOI] [PubMed] [Google Scholar]
- Fu J.; Padilha L. A.; Hagan D. J.; Van Stryland E. W.; Przhonska O. V.; Bondar M. V.; Slominsky Y. L.; Kachkovski A. D. J. Opt. Soc. Am. B 2007, 24, 67–76. 10.1364/josab.24.000067. [DOI] [Google Scholar]
- Belfield K. D.; Bondar M. V.; Haniff H. S.; Mikhailov I. A.; Luchita G.; Przhonska O. V. ChemPhysChem 2013, 14, 3532. 10.1002/cphc.201300447. [DOI] [PubMed] [Google Scholar]
- Collini E.; Carlotto S.; Ferrante C.; Bozio R.; Polimeno A.; Bloino J.; Barone V.; Ronchi E.; Beverina L.; Pagani G. A. Phys. Chem. Chem. Phys. 2011, 13, 12087. 10.1039/c1cp20945k. [DOI] [PubMed] [Google Scholar]
- Liu T.; Bondar M. V.; Belfield K. D.; Anderson D.; Masunov A. E.; Hagan D. J.; Van Stryland E. W. J. Phys. Chem. C 2016, 120, 11099. 10.1021/acs.jpcc.6b02446. [DOI] [Google Scholar]
- Ahn H.-Y.; Yao S.; Wang X.; Belfield K. D. ACS Appl. Mater. Interfaces 2012, 4, 2847. 10.1021/am300467w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z.; Chen B.; Geng J.; Chang Z.; Aparicio-Ixta L.; Nie H.; Goh C. C.; Ng L. G.; Qin A.; Ramos-Ortiz G.; Liu B.; Tang B. Z. Part. Part. Syst. Charact. 2014, 31, 481. 10.1002/ppsc.201300223. [DOI] [Google Scholar]
- Didier P.; Ulrich G.; Mély Y.; Ziessel R. Org. Biomol. Chem. 2009, 7, 3639. 10.1039/b911587k. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Xu G.; Prasad P. N. Chem.—Eur. J. 2008, 14, 5812. 10.1002/chem.200800309. [DOI] [PubMed] [Google Scholar]
- Collado D.; Remón P.; Vida Y.; Najera F.; Sen P.; Pischel U.; Perez-Inestrosa E. Chem.—Asian J. 2014, 9, 797. 10.1002/asia.201301334. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Xiao Y.; Qi J.; Qu J.; Kim B.; Yue X.; Belfield K. D. J. Org. Chem. 2013, 78, 9153. 10.1021/jo401379g. [DOI] [PubMed] [Google Scholar]
- Yao S.; Belfield K. D. Eur. J. Org. Chem. 2012, 3199. 10.1002/ejoc.201200281. [DOI] [Google Scholar]
- Wang X.; Morales A. R.; Urakami T.; Zhang L.; Bondar M. V.; Komatsu M.; Belfield K. D. Bioconjugate Chem. 2011, 22, 1438. 10.1021/bc2002506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neto B. A. D.; Carvalho P. H. P. R.; Correa J. R. Acc. Chem. Res. 2015, 48, 1560. 10.1021/ar500468p. [DOI] [PubMed] [Google Scholar]
- Neto B. A. D.; Lapis A. A. M.; da Silva E. N. Jr.; Dupont J. Eur. J. Org. Chem. 2013, 228. 10.1002/ejoc.201201161. [DOI] [Google Scholar]
- Ellinger S.; Graham K. R.; Shi P.; Farley R. T.; Steckler T. T.; Brookins R. N.; Taranekar P.; Mei J.; Padilha L. A.; Ensley T. R.; Hu H.; Webster S.; Hagan D. J.; Van Stryland E. W.; Schanze K. S.; Reynolds J. R. Chem. Mater. 2011, 23, 3805. 10.1021/cm201424a. [DOI] [Google Scholar]
- Belfield K. D.; Bondar M. V.; Yao S.; Mikhailov I. A.; Polikanov V. S.; Przhonska O. V. J. Phys. Chem. C 2014, 118, 13790. 10.1021/jp503106k. [DOI] [Google Scholar]
- Biniek L.; Chochos C. L.; Leclerc N.; Boyron O.; Fall S.; Lévèque P.; Heiser T. J. Polym. Sci., Part A: Polym. Chem. 2012, 50, 1861. 10.1002/pola.25961. [DOI] [Google Scholar]
- Belfield K. D.; Schafer K. J.; Mourad W.; Reinhardt B. A. J. Org. Chem. 2000, 65, 4475. 10.1021/jo991950+. [DOI] [PubMed] [Google Scholar]
- Aleykutty A. A.; Baliah V. J. J. Indian Chem. Soc. 1955, 32, 702. [Google Scholar]
- Wang X.; Nguyen D. M.; Yanez C. O.; Rodriguez L.; Ahn H.-Y.; Bondar M. V.; Belfield K. D. J. Am. Chem. Soc. 2010, 132, 12237. 10.1021/ja1057423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao S.; Ahn H.-Y.; Wang X.; Fu J.; Van Stryland E. W.; Hagan D. J.; Belfield K. D. J. Org. Chem. 2010, 75, 3965. 10.1021/jo100554j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrade C. D.; Yanez C. O.; Qaddoura M. A.; Wang X.; Arnett C. L.; Coombs S. A.; Yu J.; Bassiouni R.; Bondar M. V.; Belfield K. D. J. Fluoresc. 2011, 21, 1223. 10.1007/s10895-010-0801-3. [DOI] [PubMed] [Google Scholar]
- Lakowicz J. R. In Principles of Fluorescence Spectroscopy; Springer: New York, 1999. [Google Scholar]
- Corredor C. C.; Belfield K. D.; Bondar M. V.; Przhonska O. V.; Yao S. J. Photochem. Photobiol., A 2006, 184, 105. 10.1016/j.jphotochem.2006.03.036. [DOI] [Google Scholar]
- Sheik-Bahae M.; Said A. A.; Wei T.-H.; Hagan D. J.; Van Stryland E. W. IEEE J. Quantum Electron. 1990, 26, 760. 10.1109/3.53394. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.