

Abstract
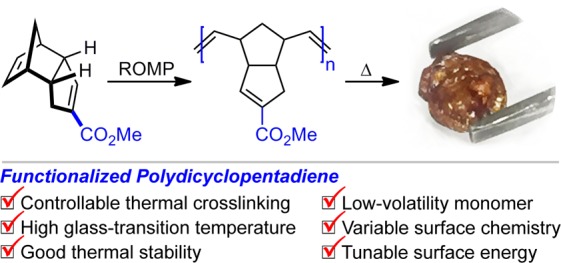
Polydicyclopentadiene (PDCPD) is a tough, heavily crosslinked thermoset polymer that has high heat, chemical, and impact resistance coupled with a low density. Current limitations to the broader industrial application of PDCPD include its low surface energy and lack of chemical tunability. Here, we report the first example of a polymer derived from a carboxyl-functionalized dicyclopentadiene monomer and its subsequent thermal crosslinking. The resulting material has the highest glass-transition temperature reported for a polydicyclopentadiene and allows for the facile manipulation of the surface chemistry through alteration of the embedded functional group. We also report the first observation by differential scanning calorimetry of the crosslinking step as a discreet thermal event.
Introduction
Polydicyclopentadiene (PDCPD, Scheme 1A) is an industrially important material that is produced via ring-opening metathesis polymerization (ROMP) from an abundant dicyclopentadiene (DCPD) monomer feedstock.1,2 The resulting polymer is extensively crosslinked when made under typical manufacturing conditions, which results in a very high impact resistance, coupled with a good resistance to chemical corrosion and a high heat deflection temperature. These properties make PDCPD attractive for use in the automotive industry. Initially used to make cowlings for snowmobiles (due to its high impact resistance at low temperatures), PDCPD is now used to make body panels, bumpers, and other components for trucks, buses, tractors, and construction equipment.3,4 Other prospective applications include the creation of porous materials for tissue engineering or gas storage applications5 as well as microencapsulated dicyclopentadiene for use in self-healing polymers.6 The polymerization of DCPD can be accomplished using a number of different transition metal catalysts (e.g., Ru, Mo, W, Ti)1,2 and has recently been reported under metal-free conditions via photoredox catalysis.7 The exact structure of the resulting material depends to some extent on the precise reaction conditions used for the polymerization. Whereas the crosslinked polymer is typically suggested to have arisen from metathesis of both alkenes in the parent monomer (to give the structure shown in gray in Scheme 1A),2,8 Wagener showed that this is often not representative of the true structure of the crosslink. Instead, for many polymerization conditions at least, it is more likely that only the strained norbornene ring in the monomer undergoes olefin metathesis under the conditions of the reaction. Subsequent crosslinking steps result from thermal (probably radical) condensation of the remaining olefins in the linear polymer.9,1b
Scheme 1. (A) Conventional Synthesis of PDCPD, (B) 1-Hydroxydicyclopentadiene-Based fPDCPD, (C) Proposed fPDCPD Formed via Controllable Thermal Crosslinking.

Despite the numerous advantages outlined above, polydicyclopentadiene has several disadvantages that have limited its broader application as an industrial material. Many of these originate from the lack of chemical functionality present on the polymer. For example, PDCPD has a low surface energy when freshly prepared; this can make it difficult to paint PDCPD parts or to attach them to other objects without a time- and space-intensive ageing step to oxidize the surface.10 Also, the polymer is not chemically tunable since the parent monomer (itself a homodimer of cyclopentadiene) cannot be easily functionalized without disrupting its ability to participate in the metathesis reaction. Thus, whereas polymers of functionalized ethylene (e.g., propylene, styrene, acrylic acid, methyl acrylate, acrylonitrile, methyl methacrylate, etc.) exhibit a broad range of very distinct and very useful material properties, no such variability can be readily obtained for DCPD-based homopolymers. Finally, PDCPD often inherits an acrid, camphor-like odor from its monomer DCPD, limiting its indoor utilization.
Each of these issues could presumably be solved by bringing functionality to PDCPD. The resulting functionalized polydicyclopentadiene (fPDCPD) will naturally have a higher surface energy (owing to the presence of a functional group), tunable properties (since the functional group could be modified), and a reduced odor (owing to a higher molecular weight for the monomer). Indeed, many groups have developed postpolymerization strategies to functionalize the residual double bonds in PDCPD by bromination,11 epoxidation,12 inverse-demand Diels–Alder,13 radical-initiated thiol–ene addition,14 etc. However, to the best of our knowledge, there are only two publications describing the polymerization of a prefunctionalized DCPD monomer (Scheme 1B).15,16 Both reports (by Xu in 201515 and Lemcoff in 201616) took advantage of a known SeO2-mediated allylic oxidation on DCPD,17 followed by further esterification or etherification and Ru-catalyzed ROMP to achieve a new family of odorless fPDCPD polymers. These novel fPDCPDs mostly possessed glass-transition temperatures (Tg) ranging from 80 to 143 °C,16,18 which is between those of linear PDCPD (Tg ∼ 53 °C)19 and crosslinked PDCPD (Tg ∼ 155–165 °C).3a,20 The glass-transition temperature is closely related to the maximum service temperature for PDCPD.21 It has a significant correlation with both the thermal properties of the polymer and the physicochemical and mechanical properties (such as molecular weight, degree of crosslinking and crystallinity, and shear modulus).22 The relatively low Tg values for the fPDCPDs summarized in Scheme 1B (most of which were <100 °C) may suggest a low degree of crosslinking relative to the parent material; indeed, the 1-hydroxydicylopentadiene-derived polymers were originally designed to disfavor crosslinking.15 In addition, both reports described substantial mass losses occurring at temperatures above ∼220 °C,15,16 presumably owing to loss of the allylic acetate or ether leaving groups. Thus, while existing fPDCPDs have interesting properties that may be useful in certain applications, the low Tg values and poor thermal stability may undercut their industrial utility.
Results and Discussion
Rather than functionalizing DCPD through allylic oxidation (which seems to lower the Tg of the resulting polymer while also reducing its thermal stability), we envisioned that it would be more beneficial to install a C-linked ester on the unstrained olefin of dicyclopentadiene (Scheme 1C). We hypothesized that this strategic change to the chemical structure of the monomer would engender several desirable properties in the resulting polymeric material:
-
(1)
Esters typically have pleasant fruity odors,23 which should limit any concern about residual monomer in the final material.
-
(2)
Unlike the allylically disposed acetates and ethers used previously (Scheme 1B), the C-linked ester cannot function as a leaving group. As a result, our polymers should have a thermal stability closer to that of the parent PDCPD.
-
(3)
Addition of an extra functional group (particularly an electron-withdrawing group) to the unstrained olefin in DCPD should completely block olefin metathesis from happening at this site (at least for modest reaction temperatures).28b This should limit any confusion about the mechanism of the crosslinking step since thermal curing will now be fully decoupled from metathesis processes.
-
(4)
Indeed, the positioning of an ester at C2 of DCPD effectively embeds a methyl methacrylate motif into the chemical framework of the postmetathesis linear polymer. The thermal polymerization of methyl methacrylate (via self-initiated radical processes) is well known to afford predominantly a head-to-tail linkage in the resulting polymer.24 We would expect our material to behave similarly, generating regiochemically predictable chemical crosslinks, as shown in Scheme 1C. This is in stark contrast to PDCPD itself, where presumably all of the residual olefins can react in the thermal crosslinking step, with little to no regiochemical control.
-
(5)
The result of our predicted chemical crosslinking step would position the ester group at a quaternary center. This increased steric hindrance to bond rotation should result in an increased Tg.25
-
(6)
The identity of the ester group could be changed (either before or after polymerization), providing for the facile tuning of properties in the final polymer material.
With these advantages in mind, we targeted compound 4 (Scheme 2) as our monomer of choice. The heterodimerization of carboxylated cyclopentadiene and unmodified cyclopentadiene to give a mixture of regioisomers 3 and 4 is well known.26 Building upon these earlier reports and taking advantage of our recently reported cyclopentadienylide salt-based route to ester-containing derivatives of dicyclopentadiene,27 we were able to achieve the synthesis of 3 and 4 (in a ∼1.5:1 ratio) in 60% yield. The reaction behaved consistently upon scale-up and was easily increased to 45 g scale. In contrast to unmodified DCPD, the mixture of 3 and 4 has an agreeable fruity smell.
Scheme 2. Synthesis of the Ester-Containing Monomer.

At first glance, the formation of a mixture of regioisomers (which are difficult to separate on preparative scale) might seem to be problematic. But we hypothesized that of the four olefins in 3 and 4, only one alkene, the strained norbornene-type olefin in 4, would be a good substrate for olefin metathesis using a ruthenium-based catalyst. The other three alkenes are either unstrained or else bear an additional electron-withdrawing group, making them less likely to react with the catalyst.28 This was quickly proven by treating a mixture of 3 and 4 with the Grubbs second-generation catalyst at room temperature (Figure 1). As expected, compound 4 underwent selective polymerization to afford the desired linear polymer 5. Polymer formation was indicated by broadening of the NMR signals (particularly for the downfield signal corresponding to the electron-deficient alkene C–H) and an upfield shift for the protons on the unfunctionalized olefin (corresponding to the formation of a less-strained alkene). Compound 3 was completely unreactive under the conditions employed. Indeed, even when the reaction was repeated at higher temperatures (refluxing benzene or toluene), we observed no polymer arising from monomer 3.
Figure 1.

Selective polymerization of monomer 4. (A) Initial monomer mixture. (B) Crude mixture of polymer product and unreacted monomer following selective polymerization. (C) Polymer product isolated by precipitation from ether. (D) Recovered unreacted monomer from the supernatant.
After quenching the catalyst with ethyl vinyl ether, the two products were separated by the addition of diethyl ether to precipitate the polymer. Centrifugation provided pure polymer 5 (Figure 1C) and left behind a supernatant that contained only unreacted 3 (Figure 1D). The cracking of ester-functionalized dicyclopentadienes like 3 has already been reported by Franklin, which means that the unwanted monomer can be recycled back to the original mixture of 3 and 4 with heating.29
The molecular weight of 5 was estimated at ∼45 000 g/mol by comparing the 1H NMR integration of the phenyl end-group and the internal vinyl protons. 1H and 13C NMR data, together with the high solubility of the freshly prepared polymer, indicated the absence of any appreciable degree of crosslinking. However, like all polydicyclopentadienes14,30 compound 5 was somewhat air-sensitive and underwent slow oxidation upon exposure to atmospheric conditions. This resulted in a growth of molecular weight (presumably owing to chemical crosslinking initiated by molecular oxygen) that we were able to monitor by dynamic light scattering (DLS; Figure 2). Similar trends were observed for three separate preparations of polymer. Oxidative crosslinking was further supported by the determination from gel permeation chromatography (GPC) of increasing molecular weights following sample storage (up to ∼30 000 000 Da for a 2-day-old sample; see Figure S1 in the Supporting Information for details) as well as by the observation that after 2–3 days the samples became distinctly less soluble (see Figure S2).
Figure 2.
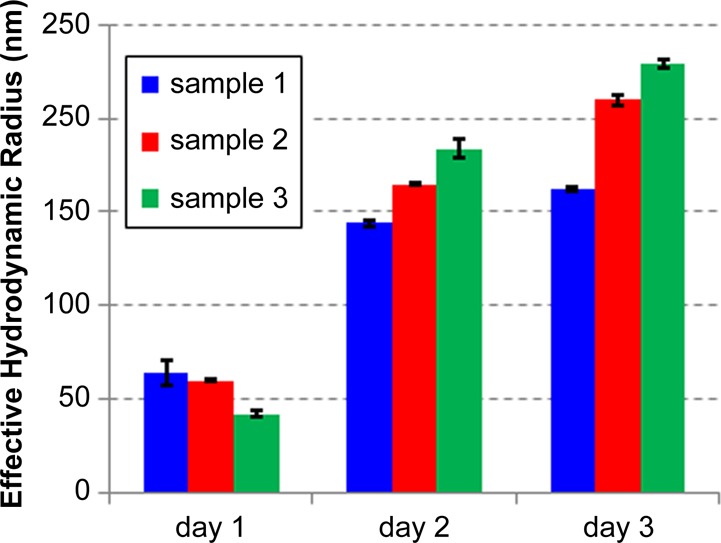
Oxidative crosslinking leads to a slow growth in effective hydrodynamic radii.
Compound 5 was thermally cured at 180 °C to afford a hard, completely insoluble material (6), for which we tentatively assign the structure shown in Figure 3.
Figure 3.

Thermal Curing of fPDCPD. The red asterisk indicates the measured glass-transition temperature for the crosslinked polymer.
The glass-transition temperature for crosslinked polymer 6 was reproducibly measured to be 200 °C (inset to Figure 3). To the best of our knowledge, this is the highest Tg ever reported for an unaged20 polydicyclopentadiene and is consistent with our hypothesis that the increased steric hindrance (and therefore reduced rotational freedom) that arises from our strategic positioning of the carboxyl substituent on the alkene that participates in the crosslinking step will serve to increase the Tg.
Since the crosslinking reaction evidently occurred at a temperature that was below the Tg, we wondered whether we might be able to observe the crosslinking event directly by differential scanning calorimetry (DSC). To this end, freshly prepared linear polymer (5) was dried under vacuum and transferred directly to the DSC under a nitrogen atmosphere. Upon heating, we observed a distinct endothermic event at ∼140 °C, prior to the usual glass transition at 200 °C (Figure 4A). After reaching a final temperature of 300 °C, the sample was cooled to room temperature and then immediately rerun. In the second analysis (of the now-crosslinked polymer, 6), the 140 °C endothermic event was not observed, but a sharp glass transition occurred at the same temperature as before. These data allowed us to assign the 140 °C transition as the crosslinking step and also demonstrate that the crosslinks in 6 are not thermally reversible.
Figure 4.
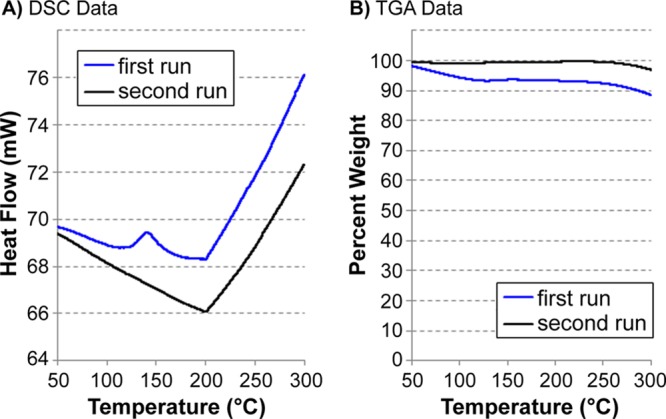
TGA/DSC analysis from linear polymer 5, showing in situ crosslinking to 6 and good thermal stability. (A) DSC data used to observe crosslinking and glass-transition temperature. (B) TGA used to evaluate thermal stability.
At the same time that we were monitoring the DSC data, we also measured the percent mass loss by thermogravimetric analysis (TGA). As shown in Figure 4B, we saw only a small mass loss during our first run (mostly below 100 °C), which we attribute to removal of residual solvent under the experimental conditions. Other than this, the polymer appears to be extremely stable up to 300 °C. This observation is consistent with our earlier hypothesis that positioning the functional group at the C2 position of the dicyclopentadiene monomer (rather than at C1, as other groups have done) limits the chemistry available to the system, thereby improving the thermal stability.
Having thus addressed the thermal properties for 5 and 6, we next focused on the controllable modification of the surface chemistry. To accomplish this, we spin-coated the linear polymer 5 onto a series of glass slides and then incubated each slide in a 180 °C oven to facilitate crosslinking. The slides were then suspended in a solution of methanolic sodium hydroxide, followed by a solution of aqueous HCl.
For each slide, we first measured the mean surface roughness (Ra) by atomic force microscopy (AFM) to establish that our hydrolysis protocol had not caused substantial pitting or otherwise introduced surface defects that would invalidate our subsequent contact angle measurements. These data (Figure 5C) confirmed that each of our samples was smoother than the unmodified glass slide used as a control. With the sample quality thereby established, we proceeded to measure surface energy through a two-solvent contact angle protocol.31
Figure 5.

Hydrolysis of fPDCPD 6 and representative water contact angle measurements. (A) Saponification/acidification protocol used to alter surface energy. (B) Representative contact angle measurements for each sample. (C) Surface roughnesses determined for each sample, as well as a control.
As shown in Figure 5B, we found a marked difference in the water contact angle of the original methyl-ester-containing polymer (6) compared to that of the (partially) hydrolyzed carboxylate, 7, and free acid, 8. Water contact angles ranged from nearly 90° for the unmodified polymer (compared with 120° for an authentic sample of the parent PDCPD32) down to ∼30° for the carboxylic acid. The fact that the contact angle continued to change with additional operations suggests that only partial hydrolysis was effected at each stage of the protocol. This is unsurprising given that many of the ester groups in the polymer will not be surface-accessible.
Together with an equivalent set of contact angles measured with diiodomethane (see Table S1 in the Supplementary Information for a summary of all individual contact angle measurements), these data allowed us to calculate the dispersion surface tension (γsvd), polar surface tension (γsv), and overall surface tension using the Owens, Wendt, Rabel, and Kaelble (OWRK) equations31 (Table 1).
Table 1. Summary of Surface Tension Measurements.
| polymer | contact angles (deg) | γsvd (mN/m) | γsvp (mN/m) | γsv (mN/m) |
|---|---|---|---|---|
| 6 | H2O: 87.2 ± 0.9 | 36.6 ± 0.5 | 1.9 ± 0.1 | 38.5 ± 0.6 |
| CH2I2: 45.8 ± 0.9 | ||||
| 7 | H2O: 63.6 ± 2.5 | 36.2 ± 0.8 | 11.8 ± 1.1 | 48.0 ± 1.9 |
| CH2I2: 46.4 ± 1.5 | ||||
| 8 | H2O: 29.0 ± 1.1 | 33.0 ± 0.5 | 33.6 ± 0.1 | 66.6 ± 0.7 |
| CH2I2: 52.2 ± 1.4 |
Gratifyingly, we observed a clear increase of γsvp and decrease of γsv with increasing ester hydrolysis. More importantly, the overall γsv values were enhanced from 38.5 mN/m (for the methyl ester) to 66.6 mN/m (for the carboxylic acid). This is a much greater range of surface energies than is available to unfunctionalized PDCPD, which has a γsv value of 36–38 mN/m when freshly made,10 increasing to 48–52 mN/m following oxidation.10,33 Indeed, to the best of our knowledge, polymers 6–8 represent the largest (and therefore most tunable) range of surface energies known for any polydicyclopentadiene-based homopolymer.34
To illustrate the dramatic change in hydrophobicity between 6 and 7/8 on a macroscopic scale, we used a Pasteur pipette to add droplets of water (containing a colored dye to aid visibility) to the surface of a glass slide that had been coated with polymer 6 by the spin-coating/crosslinking method described above and then half-immersed in a solution of methanolic NaOH (see Figure S3 in the Supporting Information for a picture of the experimental setup).
We observed the water droplets to rapidly shift from the hydrophobic side of the sample (coated with unmodified 6) toward the hydrolyzed (and therefore hydrophilic) side of the slide (refer to the Supporting Information for a video of this process). Eventually, all of the water accumulated on the portion of the sample that had been exposed to saponification conditions, as shown in Figure 6. This exceptional distinction between hydrophilic and hydrophobic regions of a single spin-coated sample may open the door toward the use of fPDCPD in microfluidic applications.
Figure 6.
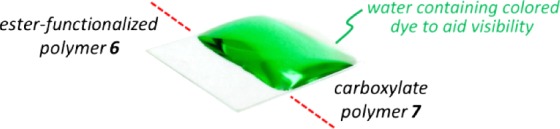
Macroscopic observation of changes in surface hydrophobicity following ester hydrolysis.
Conclusions
In summary, we have achieved the first high-Tg functionalized polydicyclopentadiene polymer with good thermal stability, controllable surface energy, and no unpleasant odor. The thermal crosslinking process was observed directly by DSC and shown to be irreversible under the conditions employed. In future work, we plan to study the mechanical properties of our novel fPDCPDs in greater detail and to exploit their inherent functionality to create a chemically recyclable thermoset.
Experimental Section
General Experimental Methods
Liquid reagents were transferred via glass microsyringe. Solvents were transferred via syringe with a stainless steel needle. Organic solutions were concentrated at 40 °C by rotary evaporation under vacuum. Analytical thin-layer chromatography (TLC) was performed using aluminum plates precoated with silica gel (0.20 mm, 60 Å pore size, 230–400 mesh, Macherey-Nagel) impregnated with a fluorescent indicator (254 nm). TLC plates were visualized by exposure to ultraviolet light. Flash-column chromatography was performed over SiliaFlash F60 (40–63 μm). Centrifugation was performed by Beckman Coulter Allegra X-12R benchtop centrifuge.
Commercial solvents and reagents were used as received with the following exceptions. Tetrahydrofuran (THF) was dried by distillation over sodium and benzophenone. Dichloromethane was dried by passage through alumina in a commercial solvent purification system.
Proton nuclear magnetic resonance spectra (1H NMR) were recorded at 300 or 500 MHz at ambient temperature. Proton chemical shifts are expressed in parts per million (ppm, δ scale) downfield from tetramethylsilane and are referenced to residual protium in the NMR solvent (CDCl3, δ 7.26; CD2Cl2, δ 5.32). Carbon NMR spectra (13C NMR) were recorded at 75 or 125 MHz at ambient temperature. Carbon chemical shifts are reported in ppm downfield from tetramethylsilane and are referenced to the carbon resonances of the solvent (CDCl3, δ 77.16; CD2Cl2, δ 53.84). Infrared (IR) spectra were obtained using a Fourier transform IR spectrometer referenced to a polystyrene standard. Accurate masses were obtained using an orbitrap mass spectrometer. Gas chromatography–mass spectrometry (GC–MS) data was collected using a Clarus 680 GC coupled to an AxIon iQT MS with cold-EI ionization.
Contact angles were obtained with a Holmarc contact angle meter (HO-IAD-CAM-01). The polar liquid was deionized water. The dispersive liquid was diiodomethane. DSC and TGA measurements were conducted on a TA Instruments Q600 SDT simultaneous thermal analyzer with samples being placed in an aluminum oxide crucible, referenced against an empty aluminum oxide crucible. Data was collected with a ramp rate of 5 °C/min following temperature equalization at 50 °C under a nitrogen atmosphere flowing at 100 mL/min. For spin casting of polymer films, a Best Tools, LLC model SC110-B Spin Coater was used. AFM measurements of the glass coverslips were performed on an Agilent Technologies 5500 Scanning Probe Microscope equipped with a Ted Pella TAP190-G AFM probe operating in tapping mode.
GPC measurements were performed using a Viscotek Model 302 liquid chromatography system (Viscotek GPCmax + TDA 302 triple detector array) equipped with refractive index (RI), low-angle light scattering (θ = 7°), and right-angle light scattering (θ = 90°) detection. THF was used as the mobile phase at a flow rate of 1 mL/min, and the column temperature was set at 35 °C. All polymer solutions were filtered through membrane filters with a nominal pore size of 0.45 μm prior to injection into the GPC columns. The data was collected and analyzed using appropriate GPC software from Viscotek. The system was installed with a Tosoh Biosciences, LLC TSKgel HHR series guard and two separation columns in series; specifically, HHR-H guard column and G3000HHR and GMHHR-M columns, respectively. Molecular weights were calculated from GPC data using an algorithm from Viscotek.
DLS measurements were collected on a Brookhaven Instruments BI-200SM goniometer equipped with a BI-9000AT digital autocorrelator and a Brookhaven Instruments Mini-L30 compact diode laser (637 nm) with a 30 mW output. Samples were diluted with filtered THF into thoroughly washed glass cells. Measurements were carried out in cylindrical glass cells, thereby simplifying corrections of variations in RI. Cells were immersed in a vat of decalin to minimize light refraction. Triplicate measurements collected at 90° were analyzed through the use of a second order cumulant expansion, which provides the hydrodynamic radius (Rg) calculated from the translational diffusion coefficient.35
Synthesis of Monomer Mixture (i.e., 3 and 4)
A flame-dried round-bottom flask fitted with an oven-dried condenser was charged with 7 mL sodium cyclopentadienylide solution (2 M in THF, 14 mmol). To this solution was added 5.9 mL dimethylcarbonate (70 mmol) at room temperature with stirring. The reaction mixture was heated to reflux for 6 h and then cooled to room temperature. The mixture was concentrated in vacuo. To the resulting solid was added iPrOH (to 0.33 M), 0.41 mL sulfuric acid (0.55 equiv, 7.7 mmol), and 2.35 mL of freshly cracked cyclopentadiene (28 mmol) at room temperature with stirring. Acidification was marked by a brown to orange color change. The solution was heated to 50 °C overnight. The reaction mixture was concentrated in vacuo, and the resulting oil was dissolved in toluene and loaded onto a silica gel column. Elution with hexanes–ethyl acetate provided 1.60 g of a mixture of 3 and 4 (60%). Major: 1H NMR (300 MHz, CDCl3) δ 6.84 (d, J = 3.5 Hz ,1H), 5.45–5.50 (m, 2H), 3.72 (s, 3H), 3.35–3.40 (m, 1H), 3.28–3.31 (m, 1H), 3.01–3.04 (m, 1H), 2.23 (ddq, J = 18.3, 10.3, 1.9 Hz, 1H), 1.70–1.77 (m, 1H), 1.64 (dt, J = 8.2, 1.8 Hz, 1H), 1.30 (d, J = 8.2 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 166.9, 148.5, 133.1, 133.1, 130.8, 54.2, 51.4, 50.6, 47.1, 46.4, 40.8, 34.2. Minor: 1H NMR (300 MHz, CDCl3) δ 6.54 (d, J = 2.3 Hz ,1H), 6.03 (dd, J = 5.7, 3.0 Hz, 1H), 5.93 (dd, J = 5.7, 3.0 Hz, 1H), 3.68 (s, 3H), 2.92–2.96 (m, 1H), 2.89–2.92 (m, 1H), 2.80–2.88 (m, 2H), 2.42 (ddt, J = 17.3, 10.3, 2.0 Hz, 1H), 1.91 (dtd, J = 17.3, 4.0, 2.0 Hz, 1H), 1.49 (dt, J = 8.2, 1.7 Hz, 1H), 1.30 (d, J = 8.2 Hz, 1H). 13C NMR (75 MHz, CDCl3) δ 166.7, 144.5, 137.1, 135.7, 133.0, 55.0, 51.3, 50.3, 46.3, 45.6, 41.3, 33.6; IR (cm–1, film) 2955, 1732, 1717, 1634, 1439, 1268, 1096, 735. GC–MS 190, 125, 93, 66 m/z observed for four isomeric species in a ratio of 1:0.6:0.17:0.015. HRMS (ESI): [M + Na]+ calcd for C12H14O2Na, 213.08861; found, 213.08864.
Synthesis of Polymer 5
To a mixture of 3 and 4 (190 mg, 1 mmol) in 3 mL of DCM was added 8 mg Grubbs second-generation catalyst (monomer/catalyst = 40:1). The mixture was allowed to stir at room temperature for 40 min. To this solution was added 1 mL ethyl vinyl ether. The reaction was stirred for an additional 1 h, after which 10 mL of diethyl ether was added. White precipitate indicated formation of the polymer. The resulting mixture was centrifuged at 3000 rpm and 4 °C for 5 min. Polymer 5 was isolated as a precipitate (64 mg, 90% based upon the amount of 4 in the starting mixture), and unreacted 3 was obtained in the ether supernatant. 1H NMR (500 MHz, CDCl3) δ 6.52–6.65 (br, 1H), 5.22–5.55 (br, 2H), 3.69–3.75 (br, 3H), 3.33–3.42 (br, 1H), 2.85–3.02 (br, 2H), 2.47–2.73 (br, 2H), 1.59–1.76 (br, 1H), 1.20–1.34 (br, 1H). 13C NMR (75 MHz, CDCl3) δ 165.3, 143.9, 136.6, 131.5, 130.7, 56.0, 51.4 (the remaining carbon resonances appeared as overlapping signals from 47.3 to 34.2 ppm).
Thermal Crosslinking
An oven-dried vial was charged with polymer 5 under an argon atmosphere. The reaction was heated to 180 °C overnight. A tough, insoluble material 6 was obtained. Refer to Figure 3 for DSC characterization data.
Spin Casting and Functionalization of Polymers
Spin casting was performed on freshly cleaned, 18 × 18 mm2 glass coverslips. Cleaning was performed as follows: 10 min of sonication in chloroform and 10 min in methanol followed by overnight drying under vacuum. Polymer samples were dissolved to 4 wt % in CH2Cl2 and a 50 μL droplet was dropped onto a coverslip spinning at 2000 rpm. Following deposition, the film was allowed to spin for 60 s to ensure that the majority of solvent had been removed.
Linear polymer 5 was spin-coated on precleaned glass slides. These coated glass slides were kept in a 180 °C oven under vacuum overnight to give crosslinked polymer 6. These polymer-6-coated slides were immersed in a solution of methanolic NaOH (1:1 MeOH/10% aqueous NaOH) under vacuum. After 8 h, the slides were washed with water and MeOH and then dried in a 70 °C oven under vacuum overnight. The resulting polymer-7-coated slides were acidified with 10% HCl solution under vacuum for 30 min. All the slides were then washed with water and MeOH and dried in a 70 °C oven under vacuum overnight to give polymer-8-coated slides.
Slides half-coated with polymer 6 and half with polymer 7 were prepared by suspending half of each polymer-6-coated slide in a solution of methanolic NaOH (1:1 MeOH/10% aqueous NaOH) for 8 h under argon (see Figure S3). The resulting slides were washed with water and MeOH and then dried at 70 °C oven under vacuum overnight.
AFM Measurements
AFM measurements of the coated glass coverslips were performed on an Agilent Technologies 5500 Scanning Probe Microscope equipped with a Ted Pella TAP190-G AFM probe operating in tapping mode. To minimize vibrations, the microscope was covered in a vibration-resistant case on a vibration isolation platform maintained at 80 psi. Each sample was imaged at three separate locations on the slide, surface roughness measurements being collected over a 10 μm × 10 μm area. Data was analyzed with the use of the Gwyddion data analysis software package. All images underwent slight modification to remove experimental artifacts such as sloped background, contrast alteration for ease of viewing (this was performed after measurements), and in some cases performing Fourier filtering of an unknown 10 Hz noise.
Contact Angle Measurements
A drop of liquid (2 μL) was deposited on the freshly prepared substrate using a Hamilton microsyringe with a mechanical dispenser. Side-view images of the drop on the substrate were taken by a high-performance aberration-corrected imaging lens with precise manual focus adjustment (CMOS sensor). Advancing contact angles were measured on these images. Two glass chips were prepared for each substrate. Three drops of liquid were deposited at three different regions of each film. A mean contact angle and standard deviation were thus determined from the resulting measurements. A sample of conventional PDCPD was obtained from Product Rescue BVBA, Waarschoot, Belgium. Prior to analysis, the sample was polished with 400 grit sandpaper, then washed with water and MeOH, and dried at 70 °C in a vacuum oven overnight.
Surface tensions were calculated by the combination of the OWRK equations (eqs 1 and 2).
| 1 |
| 2 |
H2O γlv = 72.8 mN/m, H2O γlvd = 21.8 mN/m, H2O γlv = 50.0 mN/m. CH2I2 γlv = 50.8 mN/m, CH2I2 γlvd = 50.8 mN/m, CH2I2 γlv = 00.0 mN/m. θ = contact angle
Acknowledgments
We are grateful to Dirk Vervacke (Product Rescue BVBA) for helpful discussions on the utility and current limitations for PDCPD. We also thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for operating funds, and the Michael Smith Foundation for Health Research and the Canada Research Chairs program for salary support to J.W. as well as incentive funds used to support this research. Finally, we thank Prof. Martin Jun for the use of his contact angle measurement apparatus, and Dr. Ori Granot for analytical support and GC–MS measurements.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.6b00193.
GPC traces for polymer 5 (Figure S1); visual comparison between freshly prepared polymer 5 solution and 3-day-old suspension (Figure S2); apparatus used for the preparation of coated slides (Figure S3); measured contact angles for fPDCPD and PDCPD (Table S1); collected NMR spectra (Figures S4–S12) (PDF)
Video showing water on a modified fPDCPD surface (GIF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Autenrieth B.; Jeong H.; Forrest W. P.; Axtell J. C.; Ota A.; Lehr T.; Buchmeiser M. R.; Schrock R. R. Stereospecific Ring-Opening Metathesis Polymerization (ROMP) of endo-Dicyclopentadiene by Molybdenum and Tungsten Catalysis. Macromolecules 2015, 48, 2480–2492. 10.1021/acs.macromol.5b00123. [DOI] [Google Scholar]; b Davidson T. A.; Wagener K. B. The Polymerization of Dicyclopentadiene: an Investigation of Mechanism. J. Mol. Catal. A: Chem. 1998, 133, 67–74. 10.1016/S1381-1169(98)00091-0. [DOI] [Google Scholar]; c Grubbs R. H.; Gilliom L. R., Siove A. U.S. Patent 4,607,112, August 19, 1986;; d Woodson C. S.; Grubbs R. H. U.S. Patent 6,020,443, February 1, 2000;; e Pacreau A.; Fontanille M. Linear Polymerization of endo-Dicyclopentadiene Initiated by Metathesis Catalysts. Makromol. Chem. 1987, 188, 2585–2595. 10.1002/macp.1987.021881110. [DOI] [Google Scholar]; f Rule J. D.; Moore J. S. ROMP Reactivity of endo-and exo-Dicyclopentadiene. Macromolecules 2002, 35, 7878–7882. 10.1021/ma0209489. [DOI] [Google Scholar]
- Ivin K. J.; Mol J. C.. Olefin Metathesis and Metathesis Polymerization; Academic Press: San Diego, CA, 1997. [Google Scholar]
- a Vervacke D.An Introduction to PDCPD; Product Rescue: Waarschoot, Belgium, 2008; [Google Scholar]; b Della Martina A.; Garamszegi L.; Hilborn J. G. J. Polym. Sci., Part A: Polym. Chem. 2003, 41, 2036. 10.1002/pola.10749. [DOI] [Google Scholar]
- Mol J. C. Industrial Applications of Olefin Metathesis. J. Mol. Catal. A: Chem. 2004, 213, 39. 10.1016/j.molcata.2003.10.049. [DOI] [Google Scholar]
- Kovačič S.; Jeřábek K.; Krajnc P.; Slugovc C. Ring Opening Metathesis Polymerisation of Emulsion Templated Dicyclopentadiene Giving Open Porous Materials with Excellent Mechanical Properties. Polym. Chem. 2012, 3, 325–328. 10.1039/C2PY00518B. [DOI] [Google Scholar]
- White S. R.; Sottos N. R.; Geubelle P. H.; Moore J. S.; Kessler M. R.; Sriram S. R.; Brown E. N.; Viswanathan S. Autonomic Healing of Polymer Composites. Nature 2001, 409, 794–797. 10.1038/35057232. [DOI] [PubMed] [Google Scholar]
- Goetz A. E.; Boydston A. J. Metal-Free Preparation of Linear and Cross-Linked Polydicyclopentadiene. J. Am. Chem. Soc. 2015, 137, 7572–7575. 10.1021/jacs.5b03665. [DOI] [PubMed] [Google Scholar]
- Fisher R. A.; Grubbs R. H. Ring-opening Metathesis Polymerization of exo-Dicyclopentadiene: Reversible Crosslinking by a Metathesis Catalyst. Makromol. Chem., Macromol. Symp. 1992, 63, 271–277. 10.1002/masy.19920630120. [DOI] [Google Scholar]
- Davidson T. A.; Wagener K. B.; Priddy D. B. Polymerization of Dicyclopentadiene: A Tale of Two Mechanisms. Macromolecules 1996, 29, 786–788. 10.1021/ma950852x. [DOI] [Google Scholar]
- Vervacke D. Product Rescue BVBA, Waarschoot, Belgium. Personal communication, 2016.
- Perring M.; Bowden N. B. Assembly of Organic Monolayers on Polydicyclopentadiene. Langmuir 2008, 24, 10480–10487. 10.1021/la8015782. [DOI] [PubMed] [Google Scholar]
- Perring M.; Long T. R.; Bowden N. B. Epoxidation of the Surface of Polydicyclopentadiene for the Self-Assembly of Organic Monolayers. J. Mater. Chem. 2010, 20, 8679–8685. 10.1039/c0jm01999b. [DOI] [Google Scholar]
- Knall A.-C.; Kovačič S.; Hollauf M.; Reishofer D.; Saf R.; Slugovc C. Inverse Electron Demand Diels–Alder (iEDDA) Functionalisation of Macroporous Poly(dicyclopentadiene) Foams. Chem. Commun. 2013, 49, 7325–7327. 10.1039/c3cc42925c. [DOI] [PubMed] [Google Scholar]
- Kovačič S.; Krajnc P.; Slugovc C. Inherently Reactive PolyHIPE Material from Dicyclopentadiene. Chem. Commun. 2010, 46, 7504–7506. 10.1039/c0cc02610g. [DOI] [PubMed] [Google Scholar]
- Gong L.; Liu K.; Ou E.; Xu F.; Lu Y.; Wang Z.; Gao T.; Yang Z.; Xu W. ROMP of Acetoxy-Substituted Dicyclopentadiene to a Linear Polymer with a High Tg. RSC Adv. 2015, 5, 26185–26188. 10.1039/C5RA01855B. [DOI] [Google Scholar]
- Saha S.; Ginzburg Y.; Rozenberg I.; Iliashevsky O.; Ben-Asuly A.; Lemcoff N. G. Cross-linked ROMP Polymers Based on Odourless Dicyclopentadiene Derivatives. Polym. Chem. 2016, 7, 3071–3075. 10.1039/C6PY00378H. [DOI] [Google Scholar]
- a Rosenblum M. Preparation and Thermal Rearrangement of Several Dicyclopentadiene Derivatives. J. Am. Chem. Soc. 1957, 79, 3179–3181. 10.1021/ja01569a050. [DOI] [Google Scholar]; b Woodward R. B.; Katz T. J. The Mechanism of the Diels-Alder Reaction. Tetrahedron 1959, 5, 70–89. 10.1016/0040-4020(59)80072-7. [DOI] [Google Scholar]; c Mironov V. A.; Fadeeva T. M.; Stepanyants A. U.; Akhrem A. A. Thermal Isomerization of 1-Methylcyclopentadiene. Russ. Chem. Bull. 1967, 16, 418–420. 10.1007/BF00912463. [DOI] [Google Scholar]
- Xu reported a relatively high Tg (136–159 °C) for linear acetoxy-functionalized PDCPD (see ref (15)); this is higher than most of the Tg values that Lemcoff reported for the crosslinked materials (see ref (16)) and may reflect a difference in the measurement protocol or possibly even crosslinking under the conditions of the Tg measurement. In any event, Xu’s polymer experienced a substantial loss of mass at temperatures above 221 °C, which brings up similar questions regarding its thermal stability.
- Abadie M. J.; Dimonie M.; Couve C.; Dragutan V. New Catalysts for Linear Polydicyclopentadiene Synthesis. Eur. Polym. J. 2000, 36, 1213–1219. 10.1016/S0014-3057(99)00185-8. [DOI] [Google Scholar]
- Vidavsky Y.; Navon Y.; Ginzburg Y.; Gottlieb M.; Lemcoff N. G. Thermal Properties of Ruthenium Alkylidene-Polymerized Dicyclopentadiene. Beilstein J. Org. Chem. 2015, 11, 1469–1474. 10.3762/bjoc.11.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Gac P. Y.; Choqueuse D.; Paris M.; Recher G.; Zimmer C.; Melot D. Durability of Polydicyclopentadiene Under High Temperature, High Pressure and Seawater (Offshore Oil Production Conditions). Polym. Degrad. Stab. 2013, 98, 809–817. 10.1016/j.polymdegradstab.2012.12.023. [DOI] [Google Scholar]
- Ebewele R. O.Polymer Science and Technology; CRC Press, 2000. [Google Scholar]
- Surburg H.; Panten J.. Common Fragrance and Flavor Materials; Wiley-VCH, 2006. [Google Scholar]
- a Srinivasan S.; Lee M. W.; Grady M. C.; Soroush M.; Rappe A. M. Computational Evidence for Self-Initiation in Spontaneous High-Temperature Polymerization of Methyl Methacrylate. J. Phys. Chem. A 2011, 115, 1125–1132. 10.1021/jp107704h. [DOI] [PubMed] [Google Scholar]; b Walling C.; Briggs E. R. The Thermal Polymerization of Methyl Methacrylate. J. Am. Chem. Soc. 1946, 68, 1141–1145. 10.1021/ja01211a001. [DOI] [PubMed] [Google Scholar]; c Stickler M.; Meyerhoff G. Die Thermische Polymerisation von Methylmethacrylat, 1. Polymerisation in Substanz. Makromol. Chem. 1978, 179, 2729–2745. 10.1002/macp.1978.021791115. [DOI] [Google Scholar]
- Mathias L. J.; Lewis C. M.; Wiegel K. N. Poly(ether ether ketone)s and Poly(ether sulfones) with Pendent Adamantyl Groups. Macromolecules 1997, 30, 5970–5975. 10.1021/ma960012y. [DOI] [Google Scholar]
- a Peters D. Simple Derivatives of Cyclopentadiene. Part III. The Adducts of Methyl Cyclopentadienecarboxylate and Cyclopentadiene. J. Chem. Soc. 1961, 1037–1042. 10.1039/jr9610001037. [DOI] [Google Scholar]; b Alder K.; Flock F. H.; Hausweiler A.; Reeber R. Über die Konstitution der Cyclopentadien-carbonsäure aus Cyclopentadien-kalium. Chem. Ber. 1954, 87, 1752–1759. 10.1002/cber.19540871125. [DOI] [Google Scholar]
- Chen J.; Kilpatrick B.; Oliver A. G.; Wulff J. E. Expansion of Thiele’s Acid Chemistry in Pursuit of a Suite of Conformationally Constrained Scaffolds. J. Org. Chem. 2015, 80, 8979–8989. 10.1021/acs.joc.5b01332. [DOI] [PubMed] [Google Scholar]
- a Hejl A.; Scherman O. A.; Grubbs R. H. Ring-Opening Metathesis Polymerization of Functionalized Low-Strain Monomers with Ruthenium-Based Catalysts. Macromolecules 2005, 38, 7214–7218. 10.1021/ma0501287. [DOI] [Google Scholar]; b Chatterjee A. K.; Choi T.-L.; Sanders D. P.; Grubbs R. H. A General Model for Selectivity in Olefin Cross Metathesis. J. Am. Chem. Soc. 2003, 125, 11360–11370. 10.1021/ja0214882. [DOI] [PubMed] [Google Scholar]
- Franklin W. E. Kinetics of the Reverse Diels-Alder Dissociation of Substituted Dicyclopentadienes. J. Org. Chem. 1970, 35, 1794–1796. 10.1021/jo00831a016. [DOI] [Google Scholar]
- Yang Y.-S.; Lafontaine E.; Mortaigne B. NMR Characterisation of Dicyclopentadiene Resins and Polydicyclopentadienes. J. Appl. Polymer Sci. 1996, 60, 2419–2435. . [DOI] [Google Scholar]
- Kwok D. Y.; Neumann A. W. Contact Angle Measurement and Contact Angle Interpretation. Adv. Colloid Interface Sci. 1999, 81, 167–249. 10.1016/S0001-8686(98)00087-6. [DOI] [Google Scholar]
- A sample of PDCPD was obtained from Product Rescue BVBA.
- Perwuelz A.; Campagne C.; Lam T. M. Caractérisation de la Surface du Polydicyclopentadiène (Poly-DCPD): Influence du Vieillissement sur le Mouillage et l’Adhésion de Peinture. J. Chim. Phys. 1999, 96, 904–922. 10.1051/jcp:1999178. [DOI] [Google Scholar]
- Lemcoff and co-workers measured contact angles for three allylically functionalized PDCPDs (refer to the Supplementary Information associated with ref (16)), but surface energies were not explicitly determined.
- Marcelo G.; Prazeres T. J. V.; Charreyre M.-T.; Martinho J. M. G.; Farinha J. P. S. Thermoresponsive Micelles of Phenanthrene-α-end-labeled Poly(N-decylacrylamide-b-N,N-diethylacrylamide) in Water. Macromolecules 2010, 43, 501–510. 10.1021/ma902103q. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.