

Abstract
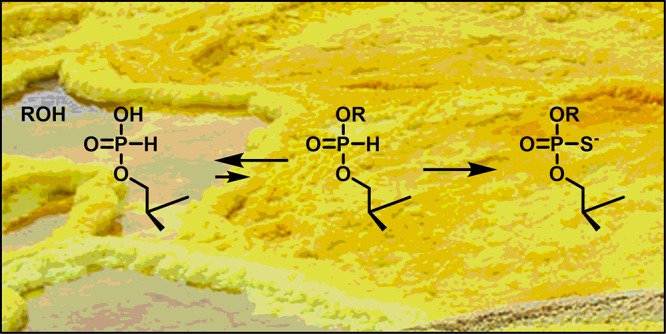
To assess the plausibility of prebiotic nucleic acid polymerization by a sequential phosphitylation–sulfurization mechanism, the rates of hydrolysis and sulfurization of bis(2′,3′-O-methyleneadenosin-5′-yl)-H-phosphonate, a dinucleoside H-phosphonate diester, have been determined over a wide pH range (0.52–7.25) and in the presence of varying amounts (0–30 mg) of elemental sulfur. The pH-rate profile of hydrolysis resembled the one previously reported for the H-phosphonate analogue of thymidylyl-3′,5′-thymidine, with a relatively wide pH-independent region flanked by acid- and base-catalyzed regions. Sulfurization to the respective phosphorothioate diester, in turn, was found to be base-catalyzed over the entire pH range studied. Despite the facile hydrolysis of H-phosphonate diesters and the extremely low solubility of elemental sulfur in water, sulfurization and hydrolysis proceeded at comparable rates under neutral and mildly acidic conditions.
Introduction
The emergence of an information-carrying polymer, possibly RNA, was a defining moment in the origin of life.1−6 The key step in the prebiotic polymerization of RNA, that is, nonenzymatic esterification of phosphoric acid, is hindered by significant kinetic and thermodynamic barriers, as well as the low solubility of phosphate minerals in water.7−9 With the notable exception of a recent report on in situ-activated polymerization of nucleoside 5′-monophosphates,10 the enzyme-free nucleic acid polymerizations described in the literature have employed preactivated monomers.11−14
The relatively facile reactions of trivalent phosphorus15 and high solubility of phosphite minerals16 make the involvement of phosphorous acid, rather than phosphoric acid, in prebiotic phosphorus chemistry an intriguing alternative.9,17−20 Trivalent phosphorus could have originated from either terrestrial21 or extraterrestrial22−25 sources, and in the Archean ocean, phosphite may have been more abundant than phosphate.26 Even in contemporary oceans, between 10 and 20% of the dissolved phosphorus bears an oxidation state lower than +5.27
In contrast to phosphoric acid, phosphorous acid undergoes rapid esterification under anhydrous conditions.28,29 The resulting H-phosphonate monoesters are stable in aqueous media, whereas the respective diesters are rapidly hydrolyzed.30 In the presence of a suitable oxidant, however, H-phosphonates are readily converted to the much more stable pentavalent counterparts, such as phosphates or phosphorothioates. Interestingly, this reaction is faster for diesters than for monoesters31 or inorganic phosphite32,33 and could, hence, drive polymerization. Oxidative coupling of H-phosphonate monoesters with alcohols has already been demonstrated in anhydrous media using elemental bromine34 or sulfur29 as the primary oxidant. Owing to the reversibility of the hydrolysis and esterification reactions of phosphorous acid derivatives, such a pathway could in principle work even in aqueous solution (Scheme 1). The plausibility of this hypothesis depends on the relative rates of the various hydrolysis, esterification, and oxidation steps.
Scheme 1. Oxidation, Esterification, and Hydrolysis Pathways for Phosphorous Acid and Its Mono- and Diesters.

In the present study, the rates of hydrolysis and sulfurization of bis(2′,3′-O-methyleneadenosin-5′-yl)-H-phosphonate (1h) were measured over a wide pH range and in the presence of various amounts of elemental sulfur. The symmetric H-phosphonate diester 1h was chosen as a model compound because of the relative simplicity of the expected product mixture. Fission of either of the P–O bonds yields the same alcohol and H-phosphonate monoester and sulfurization at the phosphorus atom does not give rise to a new chiral center. Finally, the methylene bridge between the 2′- and 3′-oxygens prevents H-phosphonate migration.
Results and Discussion
Preparation of the H-Phosphonate Diester and Monoester Model Compounds
The model H-phosphonate diester 1h was synthesized from 2′,3′-O-methyleneadenosine (2) following a procedure previously reported for various primary and secondary alcohols.35 Accordingly, the phenoxo ligands of diphenyl phosphite were displaced by 2′,3′-O-methyleneadenosine (2)36 in anhydrous pyridine (Scheme 2). The desired product 1h precipitated out of the reaction mixture and could be isolated by simple filtration. The respective monoester 3h was obtained through a similar process except that an excess of diphenyl phosphite was used, and the transesterification step was followed by hydrolytic cleavage of any remaining phenoxo ligands (Scheme 2). The same procedure has previously been used for the preparation of various nucleoside-5′-H-phosphonates.37
Scheme 2. Preparation of Bis(2′,3′-O-methyleneadenosin-5′-yl)-H-phosphonate (1h) and 2′,3′-O-Methyleneadenosin-5′-yl-H-phosphonate (3h).

Reagents and conditions: (a) diphenyl phosphite, pyridine, 25 °C; (b) 1. Diphenyl phosphite, pyridine, 25 °C and 2. H2O, Et3N, pyridine, 25 °C.
H-Phosphonate Diester Hydrolysis and Sulfurization
The reactions of the H-phosphonate diester 1h were first studied in the presence of 0–30 mg of sublimed elemental sulfur to find out whether sulfurization can compete with hydrolysis in aqueous solution. The reaction mixtures were thermostated to 60.0 ± 0.1 °C, and their pH was adjusted to 4.56 with a 100 mM acetic acid buffer and ionic strength to 1.0 M with sodium chloride. The volume of the reaction mixtures was 272 μL, and the starting concentration of 1h was 10.0 μM. In other words, the amount of sulfur vastly exceeded the amount of 1h in most of the experiments. However, owing to the extremely low solubility of sulfur in water,38 the situation was reversed in the solution phase. To obtain a reproducible dispersion of the insoluble sulfur, the reaction mixtures were sonicated before starting the kinetic runs. Samples were withdrawn from the reaction mixtures at appropriate time intervals, and their compositions were determined by reversed-phase high-performance liquid chromatography (RP-HPLC). Only the expected reactions, viz., hydrolysis of 1h to 2′,3′-O-methyleneadenosine (2) and its 5′-H-phosphonate (3h) (Scheme 3, k1) and sulfurization of 1h to the corresponding phosphorothioate (1s) (Scheme 3, k2) were observed during a typical kinetic run. A representative time–concentration profile is depicted in Figure 1. Pseudo-first-order rate constants for the overall disappearance of 1h were obtained by applying the first-order rate law to the time-dependent concentration of 1h. The rate constants thus obtained were then broken down to contributions of hydrolysis and sulfurization based on the relative concentrations of 3h and 1s, respectively.
Scheme 3. Reaction Pathways for the Hydrolysis and Sulfurization of the H-Phosphonate Diester 1h.

Figure 1.

Time-dependent concentration of 1h (□), its hydrolysis products 3h (●) and 2 (○), and the sulfurization product 1s (■) in the presence of 3 mg of sublimed elemental sulfur; T = 60.0 °C, pH = 4.56 (200 mM acetate buffer), and I(NaCl) = 1.0 M.
The observed pseudo-first-order rate constants for the hydrolysis and sulfurization of 1h as a function of the amount of sulfur in the reaction mixtures are presented in Figure 2. As expected, the hydrolysis rate was essentially independent on m(S8). The sulfurization rate, in turn, exhibited a clear dependence on m(S8) in the presence of small amounts of sulfur, with leveling-off in the presence of larger amounts. The sulfurization rate exceeded the hydrolysis rate when approximately 15 mg of S8 was added to the reaction mixture.
Figure 2.

Observed rate constants for the hydrolysis (●) and sulfurization (○) of 1h as a function of the amount of elemental sulfur; T = 60 °C, pH = 4.56 (100 mM acetate buffer), and I(NaCl) = 1.0 M.
Given the very low (and constant) concentration of elemental sulfur in the solution phase, it appears likely that sulfurization of 1h takes place at the interface between the solution and the insoluble sulfur particles. This interpretation is further borne out by the observation that the rate of sulfurization was dependent on the particle size of the sulfur used: a single flake of sulfur was approximately an order of magnitude less efficient as a sulfurizing agent than an equal amount of sublimed sulfur (data not shown). Although some of the reaction components, notably the hydrolysis product 3h, are amphiphilic and could, in principle, solubilize sulfur, such a process is not likely to play a major role at the low concentration of the potential surfactants.39 The saturation of the sulfurization rate observed at larger amounts of sulfur could stem from aggregation of the particles and the scattering of the data from difficulties in maintaining a uniform particle size distribution throughout the kinetic runs.
Rate constants for the hydrolysis and sulfurization of 1h were also determined over a wide pH range (0.52–7.25) in the presence of a fixed amount (3.0 mg) of sublimed sulfur (Figure 3). The pH of the reaction solutions was adjusted with hydrogen chloride and formic, acetic, and cacodylic acid buffers. Hydrolysis was subject to a very modest buffer catalysis in formic and acetic acid buffers. For the pH-rate profile, rate constants at zero buffer concentration were obtained by linear extrapolation from rate constants measured in 20, 50, 100, and 200 mM buffers. No buffer catalysis was observed with hydrolysis in cacodylate buffer or with sulfurization under any of the conditions used. In these cases, the rate constants presented in Figure 2 are averages of at least three measurements carried out at different buffer concentrations. All of the observed rate constants are summarized in the Supporting Information.
Figure 3.

pH-rate profiles for the hydrolysis (●) and sulfurization (○) of 1h in the presence of 3.0 mg of sublimed elemental sulfur; T = 60 °C and I(NaCl) = 1.0 M.
The pH-rate profile for hydrolysis closely resembles the one previously reported for the H-phosphonate analogue of thymidylyl-3′,5′-thymidine.30 Accordingly, hydrolysis of 1h was first order in hydronium ion under strongly acidic conditions and first order in hydroxide ion under neutral and mildly acidic conditions. The intervening pH-independent region, however, was considerably wider than with thymidylyl-3′,5′-thymidine. The observed rate constant may be expressed by eq 1
| 1 |
where k1H and k1 are the second-order rate constants for the hydronium- and hydroxide-ion-catalyzed hydrolysis, respectively, k1W is the first-order rate constant for the pH-independent hydrolysis, and KW is the ion product of water under the experimental conditions (1.58 × 10–13 M2). The values obtained for k1, k1W, and k1 by nonlinear least-squares fitting of the experimental data to eq 1 were (1.4 ± 0.2) × 10–2 M–1 s–1, (1.3 ± 0.1) × 10–4 s–1, and (7.1 ± 0.7) × 103 M–1 s–1, respectively.
As previously demonstrated for the hydrolysis of simple H-phosphonate diesters,40,41 all of the partial reactions in all likelihood proceed by a nucleophilic attack on phosphorus (rather than carbon). Accordingly, the most probable explanation for the observed hydronium ion catalysis is facilitation of the nucleophilic attack of a water molecule by preequilibrium protonation of the H-phosphonate. The pH-independence, in turn, is open to two kinetically equivalent interpretations, viz., the attack of a water molecule on neutral H-phosphonate or the attack of a hydroxide ion on a protonated monocationic H-phosphonate. Hydroxide ion catalysis becomes the predominant pathway already at pH 5, suggesting that hydroxide ion is a better nucleophile than water by at least 7 orders of magnitude (pKW under the experimental conditions = 12.8). Similarly, protonation of the H-phosphonate diester makes it more electrophilic, but the magnitude of this activation cannot be estimated as acidity constants of monoprotonated H-phosphonate diesters are not known. Finally, the hydroxide ion catalysis is undoubtedly attributable to the attack of a hydroxide ion on a neutral H-phosphonate.
Sulfurization of 1h was base-catalyzed over the entire pH range studied, but the dependence on hydroxide ion concentration appears to be less than first order. The observed rate constant may be expressed by eq 2
| 2 |
where k2OH is the rate constant for the hydroxide-ion-catalyzed sulfurization, KW is the ion product of water, and n is the reaction order with respect to hydroxide ion. The values obtained for k2 and n by nonlinear least-squares fitting of the experimental data to eq 2 were 1.9 ± 0.8 M–n s–1 and 0.53 ± 0.02, respectively.
Bases play a dual role in the sulfurization of H-phosphonate diesters.42 First, a sufficiently strong base is required to abstract the P–H proton, thereby converting the H-phosphonate into a more reactive phosphite anion.43 Second, bases can activate sulfur by breaking its eight-membered ring structure.44,45 The fractional order dependence on hydroxide ion concentration probably stems from this dual role as well as the heterogeneity of the sulfurization reaction. It can be speculated that the reaction order n is exactly 1/2, but the data at hand do not allow a firm conclusion to be drawn.
Conclusions
Sulfurization of H-phosphonate diesters by elemental sulfur is able to compete with hydrolysis despite the extremely low solubility of elemental sulfur in water. The reaction is base-catalyzed over a wide pH range, with the relative rate of sulfurization to hydrolysis reaching a maximum around pH 5. In other words, once formed, H-phosphonate diester linkages could be stabilized by sulfurization to the respective phosphorothioate diester linkages even in aqueous solutions. One could, for example, envision cycles of de- and rehydration driving nucleic acid polymerization in a volcanic environment enriched in phosphite and sulfur.
Experimental Section
General Methods
NMR spectra were recorded on a Bruker Avance 500 NMR spectrometer, and the chemical shifts are given in ppm. Mass spectra were recorded on a Bruker micrOTOF-Q ESI mass spectrometer. Synthesis of 2′,3′-O-methyleneadenosine (2) has been described in the literature.36 The other chemicals were commercial products and were used as received.
Kinetic Measurements
The reactions were carried out in sealed tubes in a dry bath thermostated to 60.0 ± 0.1 °C. The pH of the reaction solutions was adjusted with hydrogen chloride and formic, acetic, and cacodylic acid buffers and the ionic strength (1.0 M) with sodium chloride. Sublimed elemental sulfur was added to the reaction mixtures and dispersed by sonication before the start of each kinetic run. The initial concentration of the starting material 1h in the kinetic runs was 10.0 μM. The composition of aliquots drawn from the reaction mixtures at appropriate time intervals was determined by RP-HPLC on a Hypersil-Keystone Aquasil C18 column (150 × 4 mm and 5 μm), eluting with a linear gradient (8–55% over 15 min) of acetonitrile in a 60 mM acetic acid buffer (pH = 4.3). The flow rate was 1.0 mL min–1, and the detection wavelength was 260 nm. The observed retention times were as follows: 4.6 min (3h), 7.4 min (2), 7.7 min (1s), and 9.9 min (1h). The identity of these components was verified by HPLC-mass spectometry (MS) (with 1s) or spiking with authentic samples (with 1h, 2, and 3h). Molar absorptivities of the dimeric components (1h and 1s) were assumed to be twice as high as those of the monomeric components (2 and 3h).
Bis(2′,3′-O-methyleneadenosin-5′-yl)-H-phosphonate (1h)
2′,3′-O-Methyleneadenosine (2, 0.290 g, and 1.04 mmol) was coevaporated from anhydrous pyridine, and the residue was redissolved in anhydrous pyridine (10 mL). Diphenyl phosphite (95.0 μL and 0.361 mmol) was added, and the resulting mixture was stirred at room temperature for 16 h, during which a solid precipitated. This material was isolated by filtration and found to be the desired product 1h in sufficient purity for the kinetic experiments. The low yield (18.8 mg and 6%) thus obtained suggests that most of the products remained in the pyridine solution, but no attempts were made to recover it. 1H NMR (500 MHz, d6-DMSO): δ 8.32 (s, 1H, H2), 8.32 (s, 1H, H2), 8.17 (s, 1H, H8), 8.17 (s, 1H, H8), 7.37 (m, 4H, NH2), 6.73 (d, J = 718.3 Hz, 1H, PH), 6.19 (d, J = 2.3 Hz, 1H, H1′), 6.19 (d, J = 2.3 Hz, 1H, H1′), 5.36 (dd, J1 = 2.9 Hz, J2 = 6.6 Hz, 1H, H2′), 5.34 (dd, J1 = 2.9 Hz, J2 = 6.5 Hz, 1H, H2′), 5.16 (s, 4H, OCH2O), 4.96 (m, 2H, H3′), 4.27 (m, 2H, H4′), 4.22 (m, 2H, H5′), 4.12 (m, 2H, H5″). 13C NMR (125 MHz, d6-DMSO): δ 156.7 (C6), 153.3 (C8), 150.0 (C4), 149.3 (C4), 140.4 (C2), 140.3 (C2), 119.5 (C5), 95.6 (OCH2O), 88.4 (C1′), 88.3 (C1′), 82.8 (C2′), 82.7 (C4′), 80.4 (C3′), 80.3 (C3′), 65.0 (C5′). 31P NMR (202 MHz, d6-DMSO): δ 9.7. HRMS (ESI+) m/z: calcd for C22H26N10O9P, 605.1616; found, 605.1610 [M + H]+.
2′,3′-O-Methyleneadenosine-5′-H-phosphonate (3h)
2′,3′-O-Methyleneadenosine (2, 61.3 mg, and 0.220 mmol) was coevaporated from anhydrous pyridine, and the residue was redissolved in anhydrous pyridine (1200 μL). Diphenyl phosphite (100 μL and 0.380 mmol) in anhydrous pyridine (500 μL) was added, and the resulting mixture was stirred at room temperature for 23 h. Et3N (150 μL) and H2O (50 μL) were added, and the reaction mixture was stirred for 1 h, after which it was evaporated to dryness. The residue was purified on a silica gel column, eluting with a mixture of Et3N, MeOH, and CH2Cl2 (1:19:80, v/v). The product was obtained as the triethylammonium salt in 69% yield (67.4 mg). 1H NMR (500 MHz, CDCl3): δ 8.44 (s, 1H, H2), 8.28 (s, 1H, H8), 7.17 (br, 2H, NH2), 6.84 (d, J = 620.2 Hz, 1H, PH), 6.24 (d, J = 2.7 Hz, 1H, H1′), 5.23 (s, 1H, OCH2O), 5.16 (dd, J1 = 2.8 Hz, J2 = 6.2 Hz, 1H, H2′), 5.10 (s, 1H, OCH2O), 5.02 (dd, J1 = 2.5 Hz, J2 = 6.2 Hz, 1H, H3′), 4.47 (m, 1H, H4′), 4.06 (m, 2H, H5′, H5″), 2.99 (q, J = 7.3 Hz, 6H, NCH2), 1.21 (t, J = 7.3 Hz, 9H, NCH2CH3). 13C NMR (125 MHz, CDCl3): δ 156.0 (C6), 153.3 (C8), 149.4 (C4), 139.3 (C2), 119.2 (C5), 96.3 (OCH2O), 89.4 (C1′), 84.7 (d, J = 8.2 Hz, C4′), 84.6 (C2′), 81.6 (C3′), 63.7 (d, J = 4.2 Hz, C5′), 45.5 (NCH2), 8.6 (NCH2CH3). 31P NMR (202 MHz, CDCl3): δ 4.1. HRMS (ESI+) m/z: calcd for C11H13N5O6P, 342.0609; found, 342.0597 [M – H]−.
Acknowledgments
Financial support from the Academy of Finland (decision #286478) is gratefully recognized.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.7b00970.
1H, 13C, and 31P NMR spectra of compounds 1h and 3h and the observed rate constants for the hydrolysis and sulfurization of compound 1h (PDF)
The author declares no competing financial interest.
Supplementary Material
References
- Higgs P. G.; Lehman N. The RNA World: molecular cooperation at the origins of life. Nat. Rev. Genet. 2015, 16, 7–17. 10.1038/nrg3841. [DOI] [PubMed] [Google Scholar]
- Bernhardt H. S. The RNA world hypothesis: the worst theory of the early evolution of life (except for all the others). Biol. Direct 2012, 7, 23. 10.1186/1745-6150-7-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. H. C. The origin of the genetic code. J. Mol. Biol. 1968, 38, 367–379. 10.1016/0022-2836(68)90392-6. [DOI] [PubMed] [Google Scholar]
- Orgel L. E. Evolution of the genetic apparatus. J. Mol. Biol. 1968, 38, 381–393. 10.1016/0022-2836(68)90393-8. [DOI] [PubMed] [Google Scholar]
- Neveu M.; Kim H.-J.; Benner S. A. The “Strong” RNA World Hypothesis: Fifty Years Old. Astrobiology 2013, 13, 391–403. 10.1089/ast.2012.0868. [DOI] [PubMed] [Google Scholar]
- Sankaran N. The RNA World at Thirty: A Look Back with its Author. J. Mol. Evol. 2016, 83, 169–175. 10.1007/s00239-016-9767-3. [DOI] [PubMed] [Google Scholar]
- Gull M. Prebiotic Phosphorylation Reactions on the Early Earth. Challenges 2014, 5, 193–212. 10.3390/challe5020193. [DOI] [Google Scholar]
- Schwartz A. W. Phosphorus in prebiotic chemistry. Philos. Trans. R. Soc., B 2006, 361, 1743–1749. 10.1098/rstb.2006.1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gulick A. Phosphorus as a Factor in the Origin of Life. Am. Sci. 1955, 43, 479–489. [Google Scholar]
- Jauker M.; Griesser H.; Richert C. Copying of RNA Sequences without Pre-Activation. Angew. Chem., Int. Ed. 2015, 54, 14559–14563. 10.1002/anie.201506592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzo G.; Pino S.; Ciciriello F.; Di Mauro E. Generation of long RNA chains in water. J. Biol. Chem. 2009, 284, 33206–33216. 10.1074/jbc.m109.041905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohidi M.; Orgel L. E. Polymerization of the cyclic pyrophosphates of nucleosides and their analogues. J. Mol. Evol. 1990, 30, 97–103. 10.1007/bf02099935. [DOI] [PubMed] [Google Scholar]
- Blain J. C.; Ricardo A.; Szostak J. W. Synthesis and Nonenzymatic Template-Directed Polymerization of 2′-Amino-2′-deoxythreose Nucleotides. J. Am. Chem. Soc. 2014, 136, 2033–2039. 10.1021/ja411950n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W.; Ferris J. P. Synthesis of 35–40 mers of RNA oligomers from unblocked monomers. A simple approach to the RNA world. Chem. Commun. 2003, 1458–1459. 10.1039/b303134a. [DOI] [PubMed] [Google Scholar]
- Stawinski J.; Kraszewski A. How To Get the Most Out of Two Phosphorus Chemistries. Studies on H-Phosphonates. Acc. Chem. Res. 2002, 35, 952–960. 10.1021/ar010049p. [DOI] [PubMed] [Google Scholar]
- Glindemann D.; de Graaf R. M.; Schwartz A. W. Chemical Reduction of Phosphate on the Primitive Earth. Origins Life Evol. Biospheres 1999, 29, 555–561. 10.1023/a:1006622900660. [DOI] [PubMed] [Google Scholar]
- Pasek M.; Herschy B.; Kee T. P. Phosphorus: a Case for Mineral-Organic Reactions in Prebiotic Chemistry. Origins Life Evol. Biospheres 2015, 45, 207–218. 10.1007/s11084-015-9420-y. [DOI] [PubMed] [Google Scholar]
- Kee T. P.; Bryant D. E.; Herschy B.; Marriott K. E. R.; Cosgrove N. E.; Pasek M. A.; Atlas Z. D.; Cousins C. R. Phosphate activation via reduced oxidation state phosphorus (P). Mild routes to condensed-P energy currency molecules. Life 2013, 3, 386–402. 10.3390/life3030386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant D. E.; Marriott K. E. R.; MacGregor S. A.; Kilner C.; Pasek M. A.; Kee T. P. On the prebiotic potential of reduced oxidation state phosphorus: The H-phosphinate–pyruvate system. Chem. Commun. 2010, 46, 3726–3728. 10.1039/c002689a. [DOI] [PubMed] [Google Scholar]
- Pasek M. A.; Kee T. P.; Bryant D. E.; Pavlov A. A.; Lunine J. I. Production of potentially prebiotic condensed phosphates by phosphorus redox chemistry. Angew. Chem., Int. Ed. 2008, 47, 7918–7920. 10.1002/anie.200802145. [DOI] [PubMed] [Google Scholar]
- de Graaf R. M.; Schwartz A. W. Reduction and Activation of Phosphate on the Primitive Earth. Origins Life Evol. Biospheres 2000, 30, 405–410. 10.1023/a:1006700512902. [DOI] [PubMed] [Google Scholar]
- Pasek M. A.; Lauretta D. S. Aqueous Corrosion of Phosphide Minerals from Iron Meteorites: A Highly Reactive Source of Prebiotic Phosphorus on the Surface of the Early Earth. Astrobiology 2005, 5, 515–535. 10.1089/ast.2005.5.515. [DOI] [PubMed] [Google Scholar]
- Bryant D. E.; Greenfield D.; Walshaw R. D.; Johnson B. R. G.; Herschy B.; Smith C.; Pasek M. A.; Telford R.; Scowen I.; Munshi T.; Edwards H. G. M.; Cousins C. R.; Crawford I. A.; Kee T. P. Hydrothermal modification of the Sikhote-Alin iron meteorite under low pH geothermal environments. A plausibly prebiotic route to activated phosphorus on the early Earth. Geochim. Cosmochim. Acta 2013, 109, 90–112. 10.1016/j.gca.2012.12.043. [DOI] [Google Scholar]
- Bryant D. E.; Kee T. P. Direct evidence for the availability of reactive, water soluble phosphorus on the early Earth. H-Phosphinic acid from the Nantan meteorite. Chem. Commun. 2006, 2344–2346. 10.1039/b602651f. [DOI] [PubMed] [Google Scholar]
- Gorrell I. B.; Wang L.; Marks A. J.; Bryant D. E.; Bouillot F.; Goddard A.; Heard D. E.; Kee T. P. On the origin of the Murchison meteorite phosphonates. Implications for pre-biotic chemistry. Chem. Commun. 2006, 1643–1645. 10.1039/b517497j. [DOI] [PubMed] [Google Scholar]
- Pasek M. A.; Harnmeijer J. P.; Buick R.; Gull M.; Atlas Z. Evidence for reactive reduced phosphorus species in the early Archean ocean. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 10089–10094. 10.1073/pnas.1303904110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasek M. A.; Sampson J. M.; Atlas Z. Redox chemistry in the phosphorus biogeochemical cycle. Proc. Natl. Acad. Sci. U.S.A. 2014, 111, 15468–15473. 10.1073/pnas.1408134111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Graaf R. M.; Schwartz A. W. Thermal Synthesis of Nucleoside H-Phosphonates Under Mild Conditions. Origins Life Evol. Biospheres 2005, 35, 1–10. 10.1007/s11084-005-0093-9. [DOI] [PubMed] [Google Scholar]
- Lönnberg T. Nucleic acids through condensation of nucleosides and phosphorous acid in the presence of sulfur. Beilstein J. Org. Chem. 2016, 12, 670–673. 10.3762/bjoc.12.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyser J. R.; Ferris J. P. The Rates of Hydrolysis of Thymidyl-3′, 5′-Thymidine-H-Phosphonate: The Possible Role of Nucleic Acids Linked by Diesters of Phosphorous Acid in the Origins of Life. Origins Life Evol. Biospheres 2001, 31, 363–380. 10.1023/a:1011871726600. [DOI] [PubMed] [Google Scholar]
- Garegg P. J.; Regberg T.; Stawinski J.; Strömberg R. Nucleoside phosphonates: part 7. Studies on the oxidation of nucleoside phosphonate esters. J. Chem. Soc., Perkin Trans. 1 1987, 1269–1273. 10.1039/p19870001269. [DOI] [Google Scholar]
- Mitchell A. D. CCL.—The reaction between phosphorous acid and iodine. J. Chem. Soc., Trans. 1923, 123, 2241–2254. 10.1039/ct9232302241. [DOI] [Google Scholar]
- Silver B.; Luz Z. Oxidation of Phosphorous Acid. J. Phys. Chem. 1962, 66, 1356–1359. 10.1021/j100813a503. [DOI] [Google Scholar]
- Blackburn G. M.; Cohen J. S.; Todd L. Studies in phosphorylation. Part XXIX. The synthesis of dialkyl phosphates from monoalkyl phosphonates: direct oxidative esterification. J. Chem. Soc. C 1966, 239–245. 10.1039/j39660000239. [DOI] [Google Scholar]
- Xiao Q.; Ju Y.; Zhao Y. A facile approach to phosphonic acid diesters. Heteroat. Chem. 2003, 14, 208–210. 10.1002/hc.10130. [DOI] [Google Scholar]
- Norman D. G.; Reese C. B.; Serafinowska H. T. 2′,3′-O-Methylene Derivatives Of Ribonucleosides. Synthesis 1985, 751–754. 10.1055/s-1985-31332. [DOI] [Google Scholar]
- Sun Q.; Liu S.; Sun J.; Gong S.; Xiao Q.; Shen L. One-pot synthesis of symmetrical P1,P2-dinucleoside-5′-diphosphates from nucleoside-5′-H-phosphonates: mechanistic insights into reaction path. Tetrahedron Lett. 2013, 54, 3842–3845. 10.1016/j.tetlet.2013.05.040. [DOI] [Google Scholar]
- Boulegue J. Solubility of Elemental Sulfur in Water at 298 K. Phosphorus Sulfur Relat. Elem. 1978, 5, 127–128. 10.1080/03086647808069875. [DOI] [Google Scholar]
- Steudel R.; Holdt G. Solubilization of Elemental Sulfur in Water by Cationic and Anionic Surfactants. Angew. Chem., Int. Ed. Engl. 1988, 27, 1358–1359. 10.1002/anie.198813581. [DOI] [Google Scholar]
- Mitchell M. C.; Taylor R. J.; Kee T. P. On the hydrolysis of dimethyl-H-phosphonate. An 18O-labelling and 31P-NMR study. Polyhedron 1998, 17, 433–442. 10.1016/s0277-5387(97)00369-0. [DOI] [Google Scholar]
- Gerrard W.; Green W. J.; Nutkins R. A.; Sykes A.; Tatlow J. C.; Addison C. C.; Lewis J.; Jones R. L.; Le Fèvre R. J. W.; Northcott J.; Hall R. H.; Stern E. S.; Naylor J. R.; Islam A. M.; Raphael R. A. Dealkylation and Hydrolysis of Alkyl Phosphites, Phosphates, and Phosphonates. J. Chem. Soc. 1952, 4076–4087. 10.1039/jr9520004076. [DOI] [Google Scholar]
- Nguyen T. B. Recent Advances in Organic Reactions Involving Elemental Sulfur. Adv. Synth. Catal. 2017, 359, 1066–1130. 10.1002/adsc.201601329. [DOI] [Google Scholar]
- Wallin R.; Kalek M.; Bartoszewicz A.; Thelin M.; Stawinski J. On the Sulfurization of H-Phosphonate Diesters and Phosphite Triesters Using Elemental Sulfur. Phosphorus, Sulfur Silicon Relat. Elem. 2009, 184, 908–916. 10.1080/10426500802715619. [DOI] [Google Scholar]
- Bartlett P. D.; Lohaus G.; Weis C. D. Reactions of Elemental Sulfur. III.1 A Preliminary Study of the Conversion of Hexatomic to Octatomic Sulfur. J. Am. Chem. Soc. 1958, 80, 5064–5069. 10.1021/ja01552a018. [DOI] [Google Scholar]
- Bartlett P. D.; Cox E. F.; Davis R. E. Reactions of Elemental Sulfur. IV. Catalytic Effects in the Reaction of Sulfur with Triphenylphosphine. J. Am. Chem. Soc. 1961, 83, 103–109. 10.1021/ja01462a020. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.