

Abstract
Pd(II)-catalyzed enantioselective C(sp3)−H cross-coupling of free carboxylic acids with organoborons has been realized using either mono-protected amino acid (MPAA) ligands or mono-protected aminoethyl amine (MPAAM) ligands. A diverse range of aryl- and vinyl-boron reagents can be used as coupling partners to provide chiral carboxylic acids. This reaction provides an alternative approach to the enantioselective synthesis of cyclopropanecarboxylic acids and cyclobutanecarboxylic acids containing α-chiral tertiary and quaternary stereocenters. The utility of this reaction was further demonstrated by converting the carboxylic acid into cyclopropyl amine without loss of optical activity.
Keywords: arylation, vinylation, C–H activation, palladium
Graphical Abstract
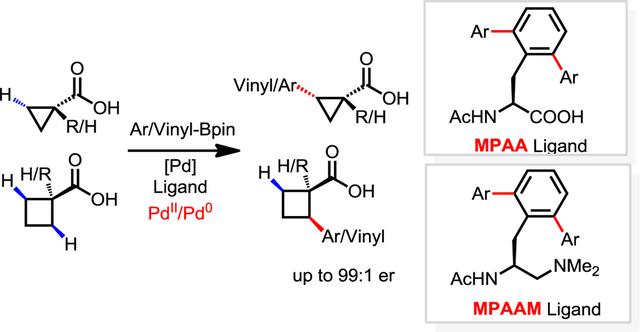
Enantioselective alkyl C(sp3)−H bond activation is a longstanding goal in organic synthesis.1 Recently, Pd-catalyzed asymmetric C−H activation reactions using chiral ligands have been demonstrated. The combination of monodentate coordinating substrates with chiral bidentate mono-N-protected amino acid ligand (MPAA) has led to the development of a wide range of Pd(II)-catalyzed enantioselective intermolecular C−H activation reactions.2–5 Recently, chiral bidentate quinoline ligands and oxazoline ligands were developed to realize the enantioselective functionalization of methylene C−H bonds and methyl C−H bonds in carboxylic acid derivatives, respectively.3c–3d However, substrates in these reactions require preinstalled directing groups that need to be removed after C−H functionalization.3 Therefore, the direct C−H functionalization of free carboxylic acids would afford superior atom and step economies in the context of protecting free synthesis.6 To date, there exists a single example of enantioselective intermolecular C(sp3)–H activation of carboxylic acids without using exogenous directing groups (Scheme 1).7 Key to the development of this method was the design of a novel mono-protected aminoethyl amine (MPAAM) chiral ligand to match the weakly coordinating carboxylic acid for stereocontrol. However, only arylation via a Pd(II)/Pd(IV) catalytic cycle is compatible with this chiral catalyst. In addition, this method was not compatible with cyclobutane substrates, an important class of cycloalkane in synthesis. Herein, we report the first chiral catalyst for the enantioselective C(sp3)–H cross-coupling of both cyclopropanecarboxylic acids and cyclobutanecarboxylic acids with both aryl and vinyl boron reagents. Both chiral ligands (L25, L36) were conveniently synthesized by C−H activation reactions developed in our laboratory. Enantioselective C−H coupling of free carboxylic acids via Pd(II)/Pd(0) catalytic cycle offers a new avenue to broaden the diversity of transformations.
Scheme 1.

Pd-Catalyzed Enantioselective C–H Functionalization of Free Carboxylic Acids.
With our previously developed β-C−H arylation of free cyclopropyl carboxylic acid with aryl iodides in mind,7 we began our study by investigating the reactivity of cyclopropanecarboxylic acid under Pd(II)/Pd(0) catalysis.8–9 Initially we focused on the C−H cross-coupling reactions with phenylboronic acid pinacol ester using various types of bidentate chiral ligands (Table 1). While 11% yield was obtained in the absence of ligand, we were pleased to observe that mono-protected aminoethyl amine (MPAAM) ligand (L1) provided the desired arylated product in 38% yield with 96:4 er. Other ligand screening showed that our previous two classes of chiral ligands (L2 and L3) gave poor yields or enantioselectivities. Further optimization using other ligands did not afford significant improvement (see supporting information). We then turned our attention to mono-protected amino acid (MPAA) ligands. Pleasingly, N-acetyl-L-phenylalanine (L4) afforded a 58% yield with 97:3 er. Notably, replacing acetyl with other protecting groups (L5–L9) led to either low yields or no reaction. Ligands with different side chains were also examined. The yield decreased when the benzyl group was replaced with other alkyl groups (L10–L14). On the other hand, only 13% yield and 88:12 er was obtained when we replaced benzyl side chain with phenyl substituent (L15) in this reaction, suggesting the importance of the benzyl group. We then focused on the modification of the benzyl group. Though para-OMe substituted N-acetyl-L-phenylalanine (L17) slightly improved the yield to 60%, other groups, such as tert-butyl, naphthalenyl and fluoro (L16, L18–L19), introduced to the para or ortho positions gave lower yields. Gratifyingly, N-acetyl-L-(2,6-di-phenyl)phenylalanine (L20) gave a better yield and high enantioselectivity (64% yield. 94:6 er). Since this type of ligand can be synthesized by our sulfonamide-directed C−H cross-coupling reaction10, we prepared a series of 2,6-di-substituted phenylalanine-derived ligands (L20–L28) for optimization. Among those 2,6-di-substituted ligands, the [2,6-di-(4-tert-butylphenyl)] phenylalanine ligand (L25) gave the best yield and enantioselectivity (70% yield, 97:3 er).
Table1.
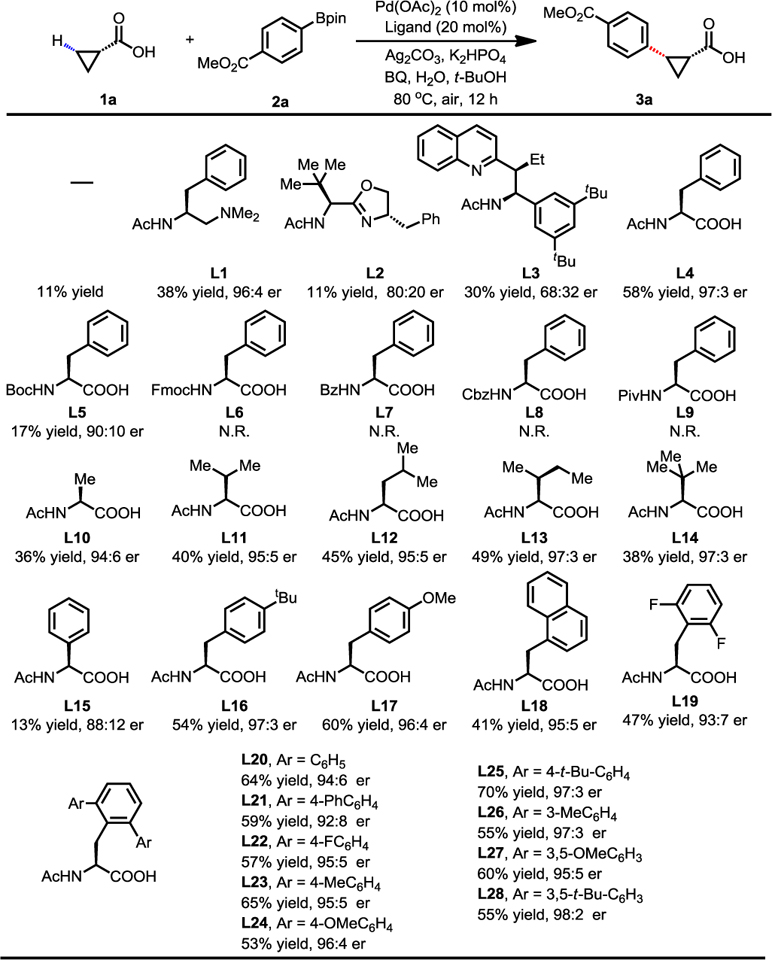
|
Conditions: 1a (0.1 mmol), 2a (1.5 equiv), Pd(OAc)2 (10 mol%), ligand (20 mol%), Ag2CO3 (1.5 equiv), K2HPO4 (1.5 equiv), BQ (0.5 equiv), H2O (10.0 equiv), t-BuOH (1.0 mL), 80 °C, air, 12 h.
1H NMR yields, using CH2Br2 as an internal standard. BQ = benzoquinone.
With the optimized reaction conditions in hand, we next explored the substrate scope of this method. The reaction was compatible with a wide range of aryl-boronic acid pinacol esters (Table 2). The coupling of 1a with electron-withdrawing arylboronic acid pinacol esters afforded the arylated products in good yields with excellent enantioselectivities (3a–3g). On the other hand, arylboronic acid pinacol esters containing electron-rich groups tended to give the desired arylated products in slightly lower yields with similar enantioselectivities (3h–3i). In addition, both meta-substituted and ortho-substituted arylboron reagents were also viable coupling partners (3j–3l). We were also pleased to find that heteroaryl boronic ester can also participate in the reaction, albeit in a lower yield (3o). Furthermore, the reaction worked well with various 1-substituted 1-cyclopropanecarboxylic acids (3p–3u) to give similar enantioselectivity despite the drastic change of sterics. 1-Aryl-1-cyclopropanecarboxylic acids (3p–3q) were arylated to give the desired products in good yields with good enantioselectivities. 1-Butyl (3r), phenylpropyl (3s), chloropentyl (3t) and benzyl-protected 1-hydroxymethyl (3u) cyclopropanecarboxylic acids were also smoothly arylated in moderate to good yields with good enantioselectivities.
Table 2.
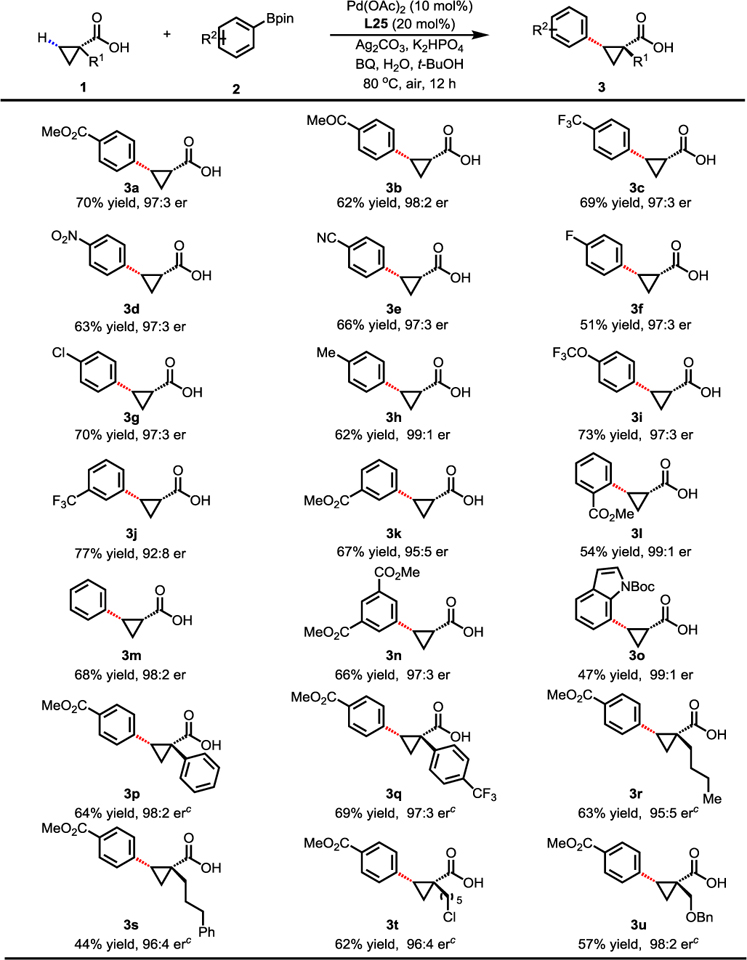
|
Conditions: 1 (0.1 mmol), 2 (1.5 equiv), Pd(OAc)2 (10 mol%), L25 (20 mol%), Ag2CO3 (1.5 equiv), K2HPO4 (1.5 equiv), BQ (0.5 equiv), H2O (10.0 equiv), t-BuOH (1.0 mL), 80 °C, air, 12 h.
Isolated yields.
Using L36 instead of L25.
We next investigated the hitherto unreported coupling of cyclobutanecarboxylic acids with arylboronic acid pinacol esters. Asymmetric synthesis of diverse chiral cyclobutanes remains a significant challenge.11 Previous attempts to enantioselectively functionalize cyclobutyl C−H bonds require the installation of a directing group.12 Through ligand screening, we found that the mono-protected aminoethyl amine (MPAAM) ligand (L1) was promising for enantioselective C−H activation of cyclobutane carboxylic acid, affording a moderate yield and enantioselectivity. Though MPAA ligands worked well for cyclopropanecarboxylic acids, poor enantioselectivity was obtained with cyclobutanecarboxylic acid (L4 and L25) in this reaction. We thus decided to focus on the modification of MPAAM ligands to further optimize this transformation (L29–L39). Changing the benzyl group to other alkyl groups (L30–L33) led to lower yields and moderate er. Remarkably, 2,6-di-aryl substituents on the phenyl ring (L36–L39) dramatically improved both the yield and er. Of the ligands prepared, the 2,6-phenyl substituted MPAAM ligand (L36) gave the best yield and enantioselectivity of 62% and 94:6 er, respectively.
With the optimized reaction conditions in hand, the scope of substrates and coupling partners was evaluated (Table 4). Electron-withdrawing groups (5a–5g), such as methoxycarbonyl, acetyl, nitro, cyano and phenyl groups provided the arylated products in moderate yields and with good enantioselectivities. On the other hand, the coupling of 4a with electron-rich arylboronic acid pinacol esters (5h and 5i) afforded the desired products in slightly lower yields and with similar enantioselectivities. Pleasingly, 1- and 2-naphthylboronic acid pinacol esters (5l and 5m) were also successful coupling partners, albeit in moderate yield and with good enantioselectivities. The reaction also worked well with other 1-substituted 1-cyclobutanecarboxylic acids (5p–5t). 1-Aryl-1-cyclobutanecarboxylic acid and 1-alkyl cyclobutanecarboxylic acid (5p–5r) were arylated to give the desired products in moderate yields and enantioselectivities. Substrates containing either an ether (5s) or a protected amine group (5t) were also compatible with this reaction conditions. Surprisingly, α-hydrogen cyclobutanecarboxylic acid (5u) gave poor enantioselectivity with this MPAAM ligand (57% yield, 64:36 er). Interestingly, the use of MPAA ligand L25 afforded the desired product 5u in 61% yield with 92:8 er. The absolute configuration of the product 5u was confirmed by X-ray crystallographic analysis (see Supporting Information).
Table 4.
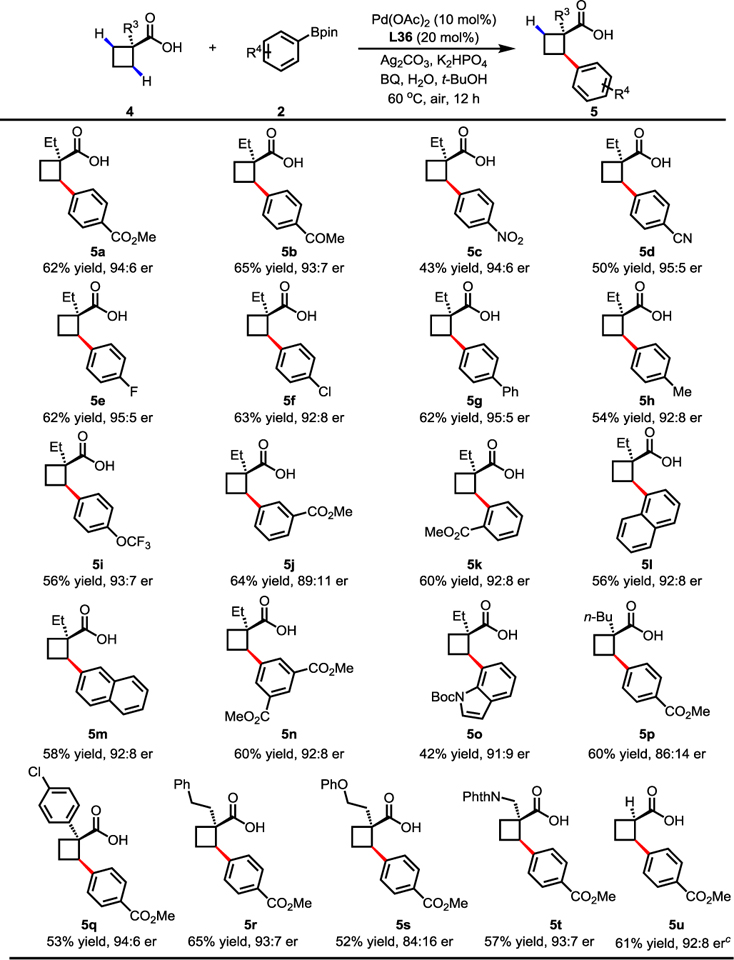
|
Conditions: 4 (0.1 mmol), 2 (1.5 equiv), Pd(OAc)2 (10 mol%), L36 (20 mol%), Ag2CO3 (2.0 equiv), K2HPO4 (1.5 equiv), BQ (0.5 equiv), H2O (5.0 equiv), t-BuOH (1.0 mL), 60 °C, air, 12 h.
Isolated yields.
Using L25 instead of L36.
Subsequently, we explored the Pd(II)-catalyzed enantioselective C(sp3)−H vinylation of free carboxylic acids (Table 5) as this transformation will provide access to a broader range of chiral carboxylic acids. Through extensive experimentation, we established the optimal reaction conditions that consisted of 2 equiv. of vinyl boronic ester with 20% ligand L28 as the chiral ligand (8a). A number of disubstituted and trisubstituted (Z)-vinylboron reagents (8a–8e) were effective coupling partners for this reaction. Moreover, 1-cyclohexenyl-Bpin (8f) was compatible with this reaction, affording a lower yield with good er. We were also pleased to see that the heterocyclohexenyl boronic ester (8g) could also be subjected in the vinylation, giving the desired product in moderate yield with excellent enantioselectivity. Furthermore, this protocol was also successfully extended to the vinylation of cyclobutanecarboxylic acid, affording the desired product in 61% yield with 88:12 er (8h). Such olefin-containing chiral acids could provide valuable scaffold for organic synthesis and medicinal chemistry.
Table 5.
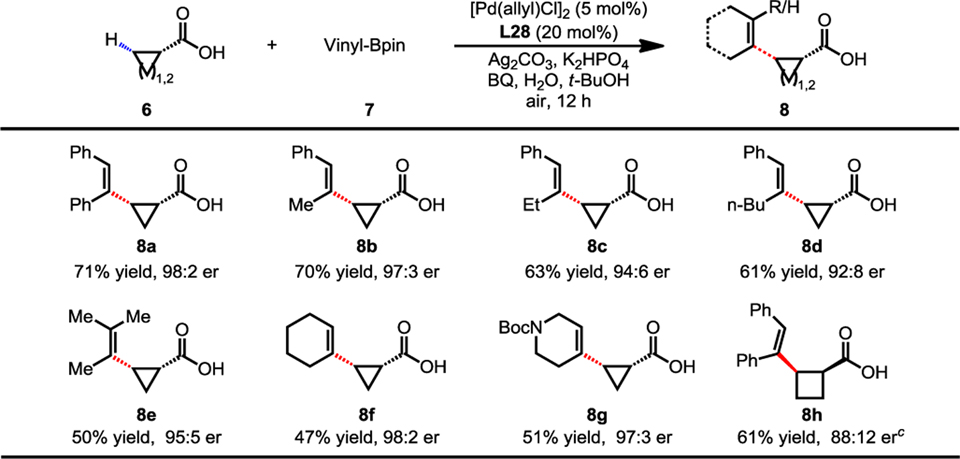
|
Conditions: 6 (0.1 mmol), 7 (2.0 equiv), [Pd(allyl)Cl]2 (5 mol%), L28 (20 mol%), Ag2CO3 (1.5 equiv), K2HPO4 (1.5 equiv), BQ (0.5 equiv), H2O (2.0 equiv), t-BuOH (1.0 mL), 80 °C, air, 12 h.
Isolated yields.
60 °C.
To demonstrate the synthetic utility of these C(sp3)−H arylation products, the cross-coupling product 3f was further transformed into chiral amine 9 without loss of optical activity (Scheme 2).13 It should be noted that 2-substituted cyclopropanamines and 2-substituted cyclopropanecarboxylic acids are important structural motifs in biologically active molecules and natural products.
Scheme 2.

Synthesis of Cis-Chiral Amine from Carboxylic Acid
In summary, we have developed an efficient Pd(II)-catalyzed enantioselective C(sp3)−H cross-coupling reaction of free carboxylic acids. The key to the success of this method was the use of the bidentate MPAA ligands or MPAAM ligands. This reaction is also compatible with aryl and vinyl coupling partners and affords aryl and olefin-containing chiral acids in high enantioselectivities. The synthetic utility of this reaction was also demonstrated by converting the chiral carboxylic acid into the chiral cyclopropyl amine without loss of optical activity.
Supplementary Material
Table 3.
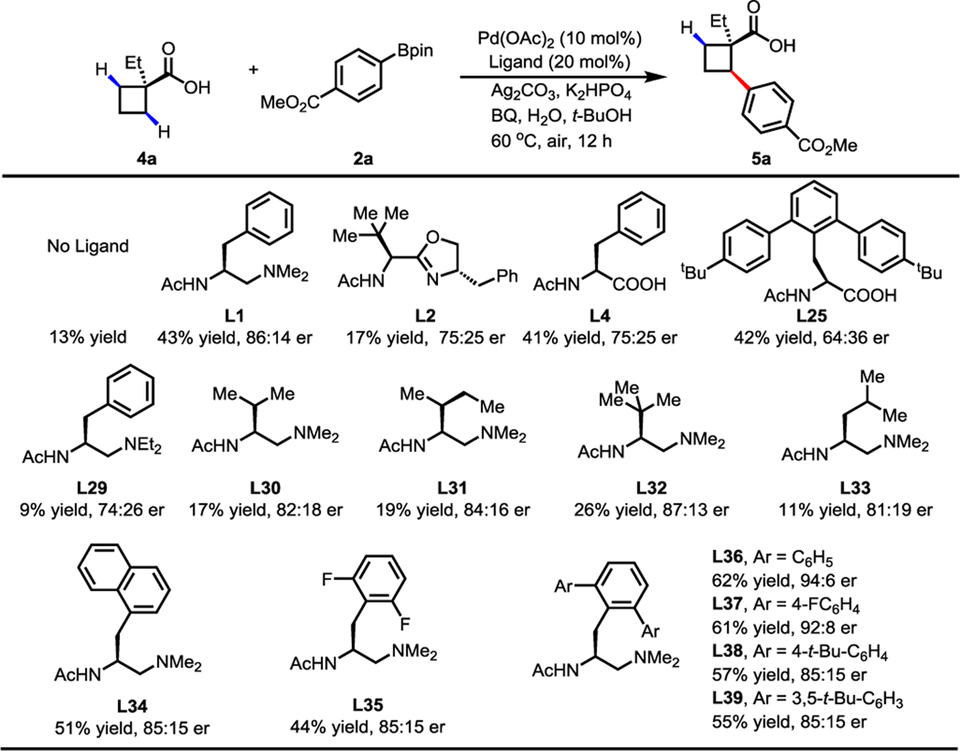
|
Conditions: 4a (0.1 mmol), 2a (1.5 equiv), Pd(OAc)2 (10 mol%), ligand (20 mol%), Ag2CO3 (2.0 equiv), K2HPO4 (1.5 equiv), BQ (0.5 equiv), H2O (5.0 equiv), t-BuOH (1.0 mL), 60 °C, air, 12 h.
1H NMR yields, using CH2Br2 as an internal standard.
Acknowledgement
We gratefully acknowledge The Scripps Research Institute, the NIH (NIGMS 2R01 GM084019) and Shanghai RAAS Blood Products Co. Ltd. for financial support. We thank Dr. Jason Chen from Automated Synthesis Facility, The Scripps Research Institute for his assistance with 2D HPLC/SFC analysis. We also thank China Scholarship Council (fellowship to L.H., Hunan University, China).
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].For selected reviews of enantioselective C-H Functionalization:Giri R, Shi B-F, Engle KM, Maugel N, Yu J-Q. Chem. Soc. Rev 2009, 38, 3242;Newton CG, Wang S-G, Oliveira CC, Cramer N. Chem. Rev 2017, 117, 8908;Saint-Denis TG, Zhu R-Y, Chen G, Wu Q-F, Yu J-Q. Science 2018, 359, eaao4798.
- [2].For the early observation of enantioselective C(sp3)−H functionalization using mono-N-protected amino acid ligands (MPAA) and pyridine directing group, see: Shi B-F, Maugel N, Zhang Y-H, Yu J-Q. Angew. Chem. Int. Ed 2008, 47, 4882; Angew. Chem. 2008, 120, 4960. [DOI] [PubMed] [Google Scholar]
- [3].Wasa M, Engle KM, Lin DW, Yoo EJ, Yu J-Q. 2011, 133, 19598;Chan KSL; Fu H-Y; Q J. J. Am. Chem. Soc. 2015, 137, 2042;Chen G, Gong W, Zhuang Z, Andra MS, Chen Y-Q, Hong X, Yang Y-F, Liu T, Houk KN, Yu J-Q. Science 2016, 353, 1023;Wu Q-F, Shen P-X, He J, Wang X-B, Zhang F, Shao Q, Zhu R-Y, Mapelli C, Qiao JX, Poss MA, Yu J-Q. Science 2017, 355, 499;Shao Q, Wu Q-F, He J, Yu J-Q. J. Am. Chem. Soc. 2018, 140, 5322.
- [4].For selected Pd(0)-catalyzed intramolecular enantioselective C-H functionalization, see:Nakanishi M, Katayev D, Besnard C, Kündig EP. Angew. Chem. Int. Ed 2011, 50, 7438; Angew. Chem. 2011, 123, 7576.Anas S, Cordi A, Kagan HB. Chem. Commun 2011, 47, 11483;Saget T, Cramer N. Angew. Chem. Int. Ed. 2012, 51, 12842; Angew. Chem. 2012, 124, 13014.Martin N, Pierre C, Davi M, Jazzar R, Baudoin O, Chem. Eur. J. 2012, 18, 4480;Pedroni J, Cramer N. Angew. Chem. Int. Ed. 2015, 54, 11826; Angew. Chem. 2015, 127, 11992.Ladd CL, Charette AB. Org. Lett. 2016, 18, 6046.
- [5].For Pd(II)-catalyzed enantioselective C-H functionalization using phosphoric acids/amides ligands, see:Yan S-B, Zhang S, Duan W-L. Org. Lett. 2015, 17, 2458;Wang H, Tong H-R, He G, Chen G. Angew. Chem. Int. Ed. 2016, 55, 15387; Angew. Chem. 2016, 128, 15613.Jain P, Verma P, Xia G, Q J. Yu. Nat. Chem. 2017, 9, 140.
- [6].Baran PS, Maimone TJ. J. M. Richter. Nature 2007, 446, 404. [DOI] [PubMed] [Google Scholar]
- [7].Shen P-X, Hu L, Shao Q, Hong K, Yu J-Q. J. Am. Chem. Soc. 2018, 140, 6545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].For selected reviews and reactions of asymmetric cyclopropanation, see:Doyle MP, McKervey MA, Ye T. Modern Catalytic Methods for Organic Synthesis with Diazo Compounds: From Cyclopropanes to Ylides; Wiley: New York, 1998;Doyle MP, Protopopova MN. Tetrahedron 1998, 54, 7919;Denmark SE, Beutner G. In Cycloaddition Reactions in Organic Synthesis; Kobayashi S, Jørgensen KA, Eds.; Wiley-VCH: Weinheim, Germany, 2002; pp 85ff;Lebel H, Marcoux J-F, Molinaro C, Charette AB. Chem. Rev 2003, 103, 977;Lou Y, Horikawa M, Kloster RA, Hawryluk NA, Corey EJ. J. Am. Chem. Soc. 2004, 126, 8916;Goudreau SR, Charette AB. J. Am. Chem. Soc. 2009, 131, 15633;Zhu S, Cui X, Zhang XP. Eur. J. Inorg. Chem. 2012, 2012, 430;Wang Y, Wen X, Cui X, Wojtas L, Zhang XP. J. Am. Chem. Soc. 2017, 139, 1049.
- [9].For Rh(I)-catalyzed intramolecular enantioselective C?H functionalization of cyclopropanes, see:For Rh(I)-catalyzed intramolecular enantioselective C−H functionalization of cyclopropanes, see: Lee T, Hartwig JF. Angew. Chem. Int. Ed. 2016, 55, 8723; Angew. Chem. 2016, 128, 8865.
- [10].Xiao K-J, Lin DW, Miura M, Zhu R-Y, Gong W, Wasa M, Yu J-Q. J. Am. Chem. Soc. 2014, 136, 8138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].For reviews of the synthesis of enantiopure cyclobutane derivatives, see:Lee-Ruff E, Mladenova G. Chem. Rev. 2003, 103, 1449;Secci F, Frongia A, Piras PP. Molecules 2013, 18, 15541.
- [12].For selected examples of Pd-catalyzed enantioselective C-H functionalization of cyclobutanes, see:He J, Shao Q, Wu Q-F, Yu J-Q. J. Am. Chem. Soc. 2017, 139, 3344;Wu Q-F, Wang X-B, Shen P-X, Yu J-Q, ACS Catal. 2018, 8, 2577.
- [13].Shaw MH, McCreanor NG, Whittingham WG, Bower JF. J. Am. Chem. Soc. 2015, 137, 463. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.