

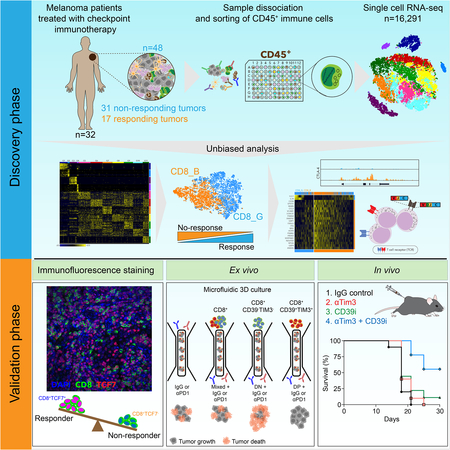
SUMMARY
Treatment of cancer has been revolutionized by immune checkpoint blockade therapies. Despite the high rate of response in advanced melanoma, the majority of patients succumb to disease. To identify factors associated with success or failure of checkpoint therapy, we profiled transcriptomes of 16,291 individual immune cells from 48 tumor samples of melanoma patients treated with checkpoint inhibitors. Two distinct states of CD8+ T cells were defined by clustering, and associated with patient tumor regression or progression. A single transcription factor, TCF7, was visualized within CD8+ T cells in fixed tumor samples and predicted positive clinical outcome in an independent cohort of checkpoint-treated patients. We delineated the epigenetic landscape and clonality of these T cell states, and demonstrated enhanced anti-tumor immunity by targeting novel combinations of factors in exhausted cells. Our study of immune cell transcriptomes from tumors demonstrates a strategy for identifying predictors, mechanisms and targets for enhancing checkpoint immunotherapy.
Graphical Abstract
IN BRIEF
Single cell analysis of immune cells from melanoma uncovers a TCF7+ memory-like state in the cytotoxic T cell population, and demonstrates its association with a positive outcome
INTRODUCTION
Antibodies that block immune checkpoint proteins, including CTLA4, PD-1 and PD-L1 (Callahan et al., 2016), are FDA approved for treating a wide variety of cancers, including melanoma and non-small-cell lung carcinoma. In melanoma, despite the high response rate (Larkin et al., 2015; Robert et al., 2015), most patients are refractory to therapy or acquire resistance. Identification of components that drive or prevent effective responses to checkpoint therapy thus remains an urgent need for understanding and expanding the use of immunotherapy in patients.
Checkpoint therapies were developed to overcome the dysfunction or exhaustion of T cells (Speiser et al., 2016; Wherry et al., 2007) resulting from chronic antigen exposure and suppression by the tumor or cells in its microenvironment. However, it remains unclear why some patients respond to checkpoint therapy while others do not. One factor associated with outcome is the number of infiltrating CD8+ T cells detected before (Tumeh et al., 2014) or during early treatment (Chen et al., 2016). In addition, several studies have found a correlation with signatures of T cell states, including signatures of IFNγ responses, as well as those of T cell activation, exhaustion and cytotoxicity (Ayers et al., 2017; Prat et al., 2017; Riaz et al., 2017), abundance of partially exhausted CD8+ T cells in responding tumors (Daud et al., 2016) and magnitude of T cell reinvigoration in relation to pretreatment tumor burden in blood (Huang et al., 2017). Additional studies have implicated non-T cell factors, including PD-L1 protein expression (Larkin et al., 2015), load of tumor neoantigens (Rizvi et al., 2015; Snyder et al., 2014), defects in antigen presentation and IFNγ pathways (Gao et al., 2016; McGranahan et al., 2017; Sade-Feldman et al., 2017; Zaretsky et al., 2016), and signatures of mesenchymal transition, wound healing and angiogenesis (Hugo et al., 2016).
One limitation of many of these studies is that the exact states of individual cells were not determined, either because bulk tumor biopsies or a limited set of pre-defined markers were used, thus limiting our ability to interpret the cellular basis for response to checkpoint inhibitors. To address these limitations, we applied high-dimensional single-cell RNA sequencing (scRNA-seq) to determine the states of immune cells from dissociated tumor biopsies of patients treated with checkpoint therapy. We focused our analysis on two unique states of CD8+ T cells that predicted the success or failure of checkpoint immunotherapy, and analyzed the immunological programs and functional properties of these T cells in the context of tumor immunity.
RESULTS
Single cell profiling of immune cells in tumors of patients treated with checkpoint inhibitors
To analyze immune cells associated with efficacy of checkpoint therapies, we performed scRNA-seq on immune cells isolated from 48 tumor biopsies taken from 32 metastatic melanoma patients treated with checkpoint therapy (with 35 anti-PD-1; 11 anti-CTLA4+PD-1; and 2 anti-CTLA4 samples), including 11 patients with longitudinal biopsies and 20 patients with one biopsy (or 2 for one patient), taken either at baseline or during treatment (Figure 1A and Table S1). We used the following patient response categories defined by RECIST criteria: complete response (CR) and partial response (PR) for responders, or stable disease (SD) and progressive disease (PD) for nonresponders (Eisenhauer et al., 2009). However, to relate molecular and cellular variables with responses of individual lesions to therapy, we classified each of the 48 tumor samples based on radiologic assessments into progression/non-responder (NR, n=31, including SD/PD samples) or regression/responder (R, n=17, including CR/PR samples) (Table S1). Of 19,392 sorted and sequenced CD45+ cells (using an optimized full length Smart-seq2 protocol (Villani et al., 2017) with a median of ~1.4 million paired-end reads per cell), 16,291 cells passed quality control, with a median of 2,588 genes detected per cell (Methods). Whole exome sequencing (WES) available for 20 patient tumor and normal pairs, identified 4 tumors with somatic mutations in B2M, JAK1, STAT1 and IFNGR1 (Table S1), recently associated with primary or acquired resistance to checkpoint therapy in melanoma (Gao et al., 2016; Sade-Feldman et al., 2017; Zaretsky et al., 2016).
Figure 1. The immune landscape of tumors from patients with melanoma treated with checkpoint therapy.

A. Schematic of cohort. B. tSNE (t-distributed stochastic neighbor embedding) plot of all CD45+ cells that passed QC. Cells are colored based on 11 clusters defined by k-means clustering. C. Heatmap displaying scaled expression values of discriminative gene sets per cluster as defined in (B). A list of representative genes is shown per cluster. D. Box plots showing the % of cells (of all CD45+ cells) per sample for clusters that had a significant difference in frequency between responder and non-responder lesions. Each point represents a single lesion. E-G. Box plots comparing % of cells between responder and non-responder lesions with exhausted or activated signatures for CD45+CD3+ cells (E), B cells and myeloid cells (F) and memory CD8+ and CD4+ T cells (G) based on known markers (Table S1). Each symbol represents a single lesion. H. Heatmap displaying scaled expression values of genes that best discriminate between responder and non-responder lesions for all CD45+ cells. Best marker genes are sorted by fold-change (Table S1). Colored circles on left show the cluster in which the gene is enriched. Data are represented as mean±SEM.
The immune cell composition of melanoma tumors and their association with response to checkpoint therapy
To define the immune landscape and its association with outcome in an unbiased manner, unsupervised clustering of 16,291 CD45+ cells was used to identify a robust 11 cluster solution (Methods), with 2 B cell clusters (G1- B cells; G2- plasma cells), 2 myeloid clusters (G3- monocytes and/or macrophages; G4- dendritic cells) and 7 clusters enriched for T/NK/NKT cells (G5–11) (Figure 1B,C and Table S1). While each patient showed changes in cluster frequencies between baseline and post-treatment samples (Table S1), there were no consistent changes when aggregating all samples (Table S1). When we consider regression or progression of each lesion, 2 clusters (G1, p=0.003; G10- T cells, p=0.03; 2-sided Wilcoxon test) were more frequent in responder lesions while 4 clusters (G3, p=0.003; G4, p=0.015; G6- T cells, p=0.005; G11- T/NK cells, p=1.3×10−5) were more frequent in non-responder lesions (Figure 1D and Table S1). G6 and G11 were both enriched for genes linked to T cell exhaustion (LAG3, PDCD1, HAVCR2, TIGIT, CD38, ENTPD1), and G11 for cell cycle genes (CDK1, CCNB1, MKI67, CDK4, RB1, TP53) (Table S1). Consistent with the unsupervised analysis, known signatures of exhausted and activated T cells (Fuertes Marraco et al., 2015; Wherry et al., 2007) (Table S1) were enriched in non-responder (p=0.002) and responder lesions (p=2×10−4), respectively (Figure 1E), with no significant change between baseline and post-therapy samples (Figure S1 and Table S1); B cells, CD8+ and CD4+ memory T cells were enriched in responder (p=0.004, 0.001, 0.03, respectively; Figure 1F,G) and myeloid cells in non-responder lesions (p=0.002; Figure 1F). We also identified individual markers based on the fraction of cells expressing a marker in responder vs. non-responder lesions (Figure 1H and Table S1), including PLAC8, LTB, LY9, SELL, TCF7, IGKC, CCR7 in responder and CCL3, CD38, HAVCR2, ENTPD1, WARS in non-responder lesions. Our analysis thus identified specific cell types, states and markers associated with regression or progression of individual tumors in response to checkpoint therapy.
Unbiased definition of CD8+ T cell states and their association with therapy response
Based on the high frequency of CD8+ T cells and the association of T cells states with clinical responses, as well as the established role of CD8+ T cells in recognition of tumor antigens and control of tumors (Pardoll, 2012; Tumeh et al., 2014), we focused our analysis on CD8+ T cells. Clustering all CD8+ T cells (n=6,350) revealed 2 major cell states: CD8_G with increased expression of genes linked to memory, activation and cell survival (e.g. IL7R, TCF7, REL, FOXP1, FOSL2 and STAT4) (Hurton et al., 2016) and reduced expression of co-inhibitory molecules; and CD8_B enriched for genes linked to cell exhaustion (e.g. CD38, HAVCR2, ENTPD1, PDCD1, BATF, LAG3, CTLA4 and PTPN6) (Figure 2A,B and Table S2). CD8_G cells mapped primarily to G10, G5 and G8 clusters, while CD8_B cells to G11 and G9 (Figure S2A), and both clusters were found in a published scRNA-seq dataset of melanoma (Tirosh et al., 2016) (Figure S2H,I and Table S2). CD8_G cells were enriched in responding lesions (2-sided Wilcoxon p=1.4×10−6) while CD8_B cells were enriched in non-responding lesions (p=0.005; Figure 2C) with both clusters coexisting in all lesions. Most responders had a cell number ratio of CD8_G/CD8_B>1, and most non-responders a ratio<1 for both baseline and post-treatment samples (Figure 2D and Figure S2B,C). For the 9 non-responding lesions with a ratio>1, we hypothesized that these patients had productive immunity that selected for tumors with de novo resistance to checkpoint therapy. Based on WES, immunohistochemistry and flow cytometry, 6 of 9 samples (no DNA or slides were available for the other 3) showed complete loss of B2M or HLA-A,B,C, as previously reported (Sade-Feldman et al., 2017; Zaretsky et al., 2016) (Table S1 and Figure S2D,E). We classified lesions as responders or non-responders based on CD8_G/CD8 ratios with high predictive power (AUC of ROC=0.87; one-sided Wilcoxon p=1.1×10−5). However, when excluding 6 samples known to lack B2M or HLA-A,B,C, the predictive power was boosted (AUC of ROC=0.95; p=3.8×10−7; Figure S2F). We also identified individual CD8+ T cell markers (based on fraction of cells expressing a marker) (Figure 2E, S2G and Table S2) associated with response (TCF7, IL7R) and lack of response (e.g., CCL3, CD38, CLTA, ENTPD1, EPSTI1, FABP5, HAVCR2, NDUFB3, PDCD1, PRDX3, SIRPG, SNAP47, SNRPD1, UBE2F and WARS). These analyses revealed exhausted-like and memory-like CD8+ T cell states and markers that associate with lesion-level response to checkpoint therapy.
Figure 2. Identification of CD8+ T cell states associated with clinical outcome.

A. tSNE plot of all CD8+ T cells collected in this study, with cells colored based on 2 clusters found by k-means clustering. B. Heatmap showing scaled expression values of discriminating genes for same 2 clusters as in (A). Numbers on right margin indicate number of genes shown in heatmap of the total differential per cluster. C. % cells in CD8_G or CD8_B clusters (of all CD8+ T cells) per sample, in responder and non-responder lesions. D. log10 ratio of number of cells in CD8_G compared to CD8_B per sample for responder and non-responder lesions. Circles outlined in white represent samples with defects in antigen presentation or IFNγ pathways. E. Heatmap displaying scaled expression values of discriminative gene sets from all CD8+ T cells between responder and non-responder lesions. Top marker genes are shown for each group (Table S2). Top bar shows mapping of each cell to CD8_G and CD8_B. Data are represented as mean±SEM.
Elevated frequencies of TCF7+CD8+ T cells in fixed tumor specimens predict positive outcome in an independent anti-PD-1-treated cohort
To test our findings in an independent melanoma cohort (33 patients treated with anti-PD-1, n= 43 samples) with a different method, we stained fixed sections for CD8 and TCF7, a top marker associated with responding lesions and expressed frequently in CD8_G cells (Table S3). TCF7 is part of the Wnt/b-catenin signaling pathway and is crucial for differentiation, self-renewal, and persistence of memory CD8+ T cells (Zhou et al., 2010) and reinvigoration and effective immunity of CD8+ T cells against chronic lymphocytic choriomeningitis mouse virus (LCMV) infection upon anti-PD-1 treatment (Im et al., 2016; Utzschneider et al., 2016). Based on automated image analysis with CellProfiler (Carpenter et al., 2006) (Figures 3A and 3B; File S1), we found more TCF7+CD8+ cells in responding (two sided Wilcoxon p=3.9×10−6) and more TCF7CD8+ cells in non-responding (p = 1.1 x 10-8; Figures 3C and 3D) samples. Most responders had a ratio of TCF7+CD8+ to TCF7CD8+ cell number >1, while non-responders had a ratio <1, in all (n=43; one-sided Wilcoxon p=2.4×10−6), baseline (n=24; p=0.001), or post-treatment (n=19; p=1.7×10−4; Figure 3E) samples. In contrast, the frequency of tumor-associated CD8+ T cells was not different between responder and non-responder patients (Figure 3F), as we found in the single-cell analysis (Table S1). The power to classify responses based on immunofluorescence analysis was high for all (AUC of ROC = 0.91), baseline (AUC of ROC= 0.88), or post-treatment samples (AUC of ROC= 0.98; Figure 3G). Finally, patients with a ratio>1 survived longer than those with a ratio <1 (Kaplan-Meier [KM] log rank p= 0.03, Figure 3H). Thus, staining of the TCF7 protein in CD8+ T cells may serve as a useful and practical marker of clinical outcome in patients treated with anti-PD-1 therapy.
Figure 3. Immunofluorescence staining and automated image analysis for the quantification of CD8+ T cells expressing TCF7.

A. Schematic illustration of the immunofluorescence (IF) analysis pipeline. B. Representative images from the multiplex IF of tissue stained for nuclei using DAPI (blue), CD8 (green) and TCF7 (red) from a responder and non-responder patient prior to therapy with anti-PD-1. Original magnification X400. C. % of CD8+TCF7+ and CD8+TCF7− cells showing each sample. D. % TCF7+ and TCF7− cells, out of all CD8+ T cells, per sample, with clinical status above bars. E. TCF7+CD8+/TCF7−CD8+ cell number ratio. F. % of CD8+cells out of all nuclei. ns-non-significant. G. Receiver operating characteristic (ROC) analysis was constructed to evaluate the prognostic power of the TCF7+CD8+/TCF7−CD8+ ratio. The area under the ROC curve (AUC) was used to quantify response prediction. H. Kaplan-Meier survival curve for 33 patients treated with anti-PD-1 therapy. Patients were divided into two groups based on TCF7+CD8+/TCF7−CD8+ ratio (n=16 >1; n=17 <1) from IF. Data are represented as mean±SEM.
Fine clustering of CD8+ T cells
While the 2 CD8+ T cell clusters were sufficient to separate responders from nonresponders, unsupervised clustering further defined 6 sub-clusters, with 3 that are mostly contained within CD8_G and 3 within CD8_B (Methods, Figure 4A,B,C). CD8_1 cells expressed markers of exhaustion and cell cycle (Table S4; similar to G11; Table S1), similar to terminally exhausted CD39+ (ENTPD1) CD8+ T cells from chronic infection with hepatitis C virus (Gupta et al., 2015). CD8_2 cells expressed many of the same exhaustion markers along with heat shock proteins (HSPB1, HSPA1A and HSPA4) and additional inhibitory receptors (ENTPD1 and KIR2DL4). CD8_3 cells expressed the known exhaustion markers (HAVCR2, CD38, PDCD1 and PTPN6) but lacked heat shock and cell-cycle genes. CD8_4 (CCR7, IL7R, TCF7, TNF and S100A10), and CD8_6 (SELL, TCF7, LTB, IL7R, FLT3LG, IL16) cells were consistent with a memory and/or effector-like phenotype, while CD8_5 cells had the phenotypes of memory and early activated cells (IL6ST, CXCL13, IL7R and CTLA4) with higher HAVCR2 and PDCD1 compared to CD8_4, 6, but lower than CD8_1,2,3 (Figure S3A). Interestingly, GZMA, GZMB and PRF1 had much higher expression in CD8_1,2,3 (Table S4), resembling programs previously reported in melanoma (Tirosh et al., 2016), in a mouse model of chronic LCMV infection (Wherry et al., 2007), and in resident memory T cells (Mami-Chouaib et al., 2018). CD8_1 and CD8_3 were enriched in non-responder (1-sided Wilcoxon p=6.7×10−5, p=0.001, respectively) while CD8_4 and CD8_5 were enriched in responder lesions when excluding samples deficient for B2M or HLA-A,B,C (p=0.01 and p=0.02, respectively; Figure 4D). CD8_5 was found mostly in post-therapy samples (80%, Figure S3B,C) and appeared to have fewer TCF7-expressing cells relative to CD8_4 and CD8_6 (Figure S3A). However, it split into 2 sub-clusters (Figure 4E): CD8_5.1 (TCF7+GZMB−) and CD8_5.2 (TCF7−GZMB+) cells, both enriched in responder lesions (1-sided Wilcoxon p=0.01, 0.04, respectively). These findings are consistent with a recent study showing loss of TCF7 expression is associated with acquisition of effector phenotype in response to anti-PD-1 treatment in a mouse model of LCMV infection (Im et al., 2016). Since T cells transition to new states in cancer or chronic infections (Im et al., 2016; Speiser et al., 2016), we used trajectory analysis (Qiu et al., 2017) to identify a main trajectory branch, and 2 side branches (Figure 4F), reflecting a possible path for differentiation (CD8_4, CD8_6, CD8_5 followed by CD8_3, CD8_2 and 1, with some overlap of clusters and with no information on the directionality) (Figure 4F and Figure S3D,E). The finding of transitional cells and the proximity between clusters suggests states that may arise or give rise to others. Our analysis of finer T cell states are consistent with our findings of the 2 CD8+ T cell clusters, but provide better resolution of cell states and suggest testable paths for differentiation.
Figure 4. CD8+ T cell state heterogeneity and its association with clinical response.

A. tSNE plot of all CD8+ T cells collected in this study, with cells colored based on 6 clusters found by k-means clustering. B. Heatmap showing scaled expression values of discriminating genes for same 6 clusters as in (A). Numbers on right margin indicate number of genes shown in heatmap of the total differential genes per cluster. Bottom bar depicts mapping of each cells to CD8_G and CD8_B, respectively. C. Hierarchical tree structure for 6 clusters, with each split showing genes up-regulated in the corresponding cluster relative to the rest of the cells found in the last common ancestor. D. % of cells in CD8_1 to 6 clusters (out of all CD8+ T cells). E. tSNE plot of CD8+ T cells with coloring of CD8_5 according to TCF7 expression upper panel and TCF7 and GZMB expression, lower panel. F. Trajectory analysis for the 6 CD8+ T cells clusters. Cell expression profiles in a two-dimensional independent space. Solid black line indicates the main diameter path of the minimum spanning tree (MST) and provides the backbone of Monocle’s pseudotime ordering of the cells. Each dot represents an individual cell colored by cluster (left plot) or by pseudotime (right plot). Data are represented as mean±SEM.
TIM3 and ENTPD1 mark the exhausted-like state of CD8+ T cells
To isolate cells with the different CD8+ T cell states using flow sorting, we used the cell surface markers, CD39 (ENTPD1) and TIM3 (HAVCR2), which both had low expression in clusters associated with response and high expression in those associated with no response (Figures S3A and S4A; Tables S2 and S4). We used scRNA-seq to profile freshly sorted CD39+TIM3+ (DP, double positive) and CD39TIM3 (DN, double negative) CD8+ T cells from four melanoma patients (Figure S4B; Table S5) and found that the profiles recapitulated the original unsorted clusters (Figures 5A and 5B), DN with CD8_4 and 6, and DP mostly with CD8_2 cells. Since CD39 is an ectonucleotidase in the adenosine pathway that modulates immunity (Young et al., 2014), a marker for terminally exhausted CD8+ T cells in patients with chronic hepatitis C virus (HCV) and HIV infections (Gupta et al., 2015), and a marker of exhaustion in tumor-infiltrating CD8+ T cells in melanoma and breast cancer (Canale et al., 2018), we analyzed the properties of CD8+CD39+ from 12 melanoma patients treated with checkpoint blockade therapy (Table S5). We found that while CD8+CD39+ and CD8+CD39 T cells had equal expression of PD-1, CD39 turned out to be a key marker that separates all TIM3+ from TIM3 cells (Figure S4C), the latter being reported as a marker of T cell dysfunction in cancer and chronic infections (Anderson et al., 2016). We prepared single-cell suspensions from the 12 patients and assessed their ability to produce cytokines in response to T cell receptor (TCR) (anti-CD3/CD28) stimulation. While CD39 and CD39+ cells contained equivalent percentages of IL-2-producing cells, CD39+ cells had a significant reduction in the number of TNF⍺- (unpaired- Student’s t test p= 0.0016) and IFNγ-producing cells (p=5×10−4; Figure 5C).
Figure 5. Discriminating exhausted from memory cells using TIM3 and CD39.

A. Heatmap showing scaled expression values of discriminative gene sets between CD8_2 (exhaustion-like) and CD8_4+6 (memory/effector-like) using original unsorted, and sorted (CD39+TIM3+ and CD39−TIM3−) cells. B. Heatmap of scaled expression values of discriminative gene sets between sorted CD39+TIM3+CD8+ and CD39−TIM3−CD8+ T cells. Colored bars above heatmap show the CD8+ cluster (as in Figure 4A) in which the gene is enriched C. Representative flow cytometry plots for intracellular staining of IL-2, IFNγ and TNFα in CD39− and CD39+ cells, with quantification below for 12 patients. Data were combined from 2 replicate experiments. D. Quantification of live/dead cells based on staining of CT26GFP+ MDOTS on day 5 of ex vivo culture. One of two independent experiments is shown, with n=3 replicates per group per experiment. 2-way ANOVA, Tukey’s multiple comparisons test. E. A schematic summary of the therapy regimen used in the transplantable B16-F10 mouse model. F. Tumor volumes for all 4 groups. G. Survival of B16-F10 tumor-bearing mice treated with CD39i in combination with anti-TIM3. H. Tumor volumes for untreated, anti-PD-1, CD39i and anti-PD-1+CD39i treated groups. I. Survival of B16-F10 tumor-bearing mice treated with CD39i + anti-PD-1. J. Tumor volumes for untreated, anti-PD-1/CTLA4, CD39i, anti-PD-1/CTLA4+CD39i. K. Survival of B16-F10 tumor-bearing mice treated with CD39i and anti-PD-1/CTLA4. Data are represented as mean±SEM. For in vivo mouse tumor models one of two independent experiments is shown.
To address which of the two cell states (DN or DP) is important for tumor eradication upon anti-PD-1 therapy, we used a CT26GFP+ mouse tumor cell-line that exhibits modest regression when treated with anti-PD-1 in an ex-vivo 3-D microfluidic culture system for growing murine organotypic tumor spheroids (MDOTS) (Jenkins et al., 2018). We found anti-PD-1 mediated killing using this system was dependent on both CD8+ T cells and IFNγ (Figure 5D). Next, we isolated CD8+CD39−TIM3− (DN) and CD8+CD39+TIM3+ (DP) cells from CT26GFP+ tumors and immediately incubated each population or a mixed one (DN/DP, 1:1 ratio) in the device with the MDOTS for five days with anti-PD-1 (or anti-IgG control antibodies), followed by live/dead staining and fluorescence microscopy to evaluate the viability of the CT26GFP+ MDOTS (Figure S4D). Addition of DN cells (which were also TCF7+) induced the most cell death with ~50% of GFP+ tumor cells eradicated after five days with anti-PD-1 antibodies, while DP cells reduced killing to control (IgG) levels (Figure 5D). We conclude that expression of CD39 and TIM3 discriminated exhausted from memory and/or effector cells, with DN cells supporting antitumor activity of checkpoint blockade ex vivo.
Dual inhibition of TIM3 and CD39 reduces tumor growth and improves survival
Because cells expressing CD39 and TIM3 were associated with non-responding lesions, and their expression was highly correlated with each other (relative to all pairwise correlations between top CD8_B markers, Table S5), we examined the combined effect of CD39 and TIM3 blockade. Mice transplanted with B16-F10 melanoma were treated with a small molecule, POM-1, that inhibits CD39 activity (Sun et al., 2010) and/or anti-TIM3 blocking antibodies (Figure 5E). While either monotherapy transiently reduced tumor growth through day 14, the combination strongly reduced tumor size and increased survival at day 40 to 20% vs. 0% for monotherapy (Figure 5F,G and Figure S4E,F). Combined treatment of POM-1 with anti-PD-1 or anti-PD-1/CTLA4 also reduced tumor growth and increased survival, with POM1/PD-1/CTLA4 therapy having a strong synergistic boost of survival at day 40 to 60% (Figure 5H–K and Figure S4G–J). The effects of POM-1 were dependent on CD8+ but not CD4+ T cells (Figure S5A–E), and led to higher frequencies of IFNγ- but not GZMB/PRF1-producing CD8+ T cells (unpaired-student’s t-test p=0.04), and higher T cell proliferation in response to TCR stimulation (Figure S5F,G,H,I). While surface CD39 levels were not altered by POM-1 in CD8+ and B16-F10GFP+ cells (Figure S5J), ATP levels increased in whole tumor (p=0.02) or B16-F10GFP+ cells (p=0.02; Figure S5K), suggesting that CD39 ATPase is active in B16-F10 tumors. These results are consistent with prior studies in which inhibition of CD39 enzymatic activity enhances proliferation of T cells (Bastid et al., 2015). Altogether, we observed enhanced tumor control when targeting CD39 in combination with TIM3 or other checkpoints, providing new and effective therapeutic combinations.
Chromatin states of exhausted-like and memory-like CD8+ T cells
To better understand the transcriptional regulation that explains the signatures observed in the exhausted-like and memory-like CD8+ T cells, DP and DN cells were isolated from five metastatic melanoma patients (Table S5), and open chromatin was quantified by assay for transposase-accessible chromatin using sequencing (ATAC-seq) and transcript levels by scRNA-seq (Figure 6A). Of the differentially expressed transcription factors (TFs) by scRNA-seq, DN cells expressed higher levels of TCF7, STAT4, FOXP1, and FOSB transcripts, as observed for stem cell memory CD8+ T cells (Hurton et al., 2016), and DP cells expressed higher BATF, PRDM1, TOX, HMGB2, and IRF2, as described for exhausted CD8+ T cells (Waugh et al., 2016; Wherry et al., 2007) (Figure 6B). These TFs were also detected in single cells sorted computationally based on expression of CD39 and TIM3 mRNAs as well as in the original CD8_G and CD8_B clusters (Figures S6A and S6B). ATAC-seq identified differentially accessible regions in genes related to exhaustion and memory (Figures 6C, 6D, and S6C), with a smaller number in DN cells (424; Benjamini-Hochberg false discovery rate [FDR] < 0.01; Table S5) when compared to DP cells (858; FDR < 0.01; Table S5), consistent with a previous study showing increased open regions as cells differentiate in response to chronic LCMV infection (Sen et al., 2016). Next, we searched for TF motifs enriched in open chromatin peaks and found BATF (and other TFs) motifs enriched in DP peaks and TCF7 and FOXP1 motifs in DN peaks (Figure 6E). EOMES was highly expressed in DP cells consistent with previous studies (Paley et al., 2012; Wherry et al., 2007); however, differential peak motif enrichment was found in DN cells, suggesting that TF activity and expression are not coupled. Since BATF and TCF7 had the highest peak motif enrichment and the highest expression in DP and DN, respectively (Figures 6E and S6D), we compared whether differentially expressed genes near significant (FDR < 0.01) open chromatin regions (OCR) in DP or DN cells (as defined by GREAT [McLean et al., 2010]) contain enhancers with BATF or TCF7 motifs. We identified 95 genes in DP (16%, including CXCL13, ENTPD1, CD38, CTLA4, and HAVCR2) and 6 genes in DN cells (20%, including IL7R, PLAC8, and SELL), out of the total differentially expressed genes (584 for DP and 30 for DN), that meet these criteria (Figure 6F), suggesting that both BATF and TCF7 control the expression of key genes unique to each cell state. Furthermore, BATF was associated with nonresponder lesions when looking at all cells (Fisher’s exact test p= 7.2×10−49; Table S1) or CD8+ cells (p= 8.1×10−19; Table S2), and TCF7 with responder lesions (all CD45+, p= 8.03×10−50; CD8+, p= 3.02×10−20). Finally, we compared open chromatin in DP and DN cells to those of recently reported dysfunctional PD-1highCD8+ cells and central memory cells (CD45RA−CD45RO +CD62Llo) (Philip et al., 2017). We found an overlap of DP with PD-1highCD8+ and DN with central memory cells but with some unique peaks in each (Benjamini-Hochberg FDR < 0.01; Figure S6E; Table S5), showing that they share some of their programs. Our analysis reveals key regulatory elements and TFs that regulate the exhaustion-like and memory-like programs in CD8+ T cells found in human melanoma.
Figure 6. Differential chromatin accessibility in CD39+TIM3+ and CD39−TIM3− cells.

A. Schematic of ATAC-seq analysis performed on sorted CD39+TIM3+ and CD39−TIM3-cells. B. Heatmap describing averaged scaled expression values of differentially expressed transcription factors for sorted CD39+TIM3+ and CD39−TIM3− cells. C. Heatmap describing patient specific (n=5) differentially accessible regions (FDR<0.01) in CD39+TIM3+ and CD39−TIM3− sorted populations. D. ATAC-seq traces for open chromatin regions near selected genes in CD39+TIM3+ (orange) and CD39−TIM3− (blue) cells. E. Graph depicting enrichment of TF motifs based on open chromatin specific to CD39−TIM3− (blue) vs. CD39+TIM3+ (orange) cells (x-axis), and differential expression of TFs (y-axis). F. Left, enhancer binding sites for BATF and TCF7 near the listed genes. Significant genes, red; non-significant, white. The same genes are also differentially expressed between CD39+TIM3+ cells and CD39−TIM3− cells. Right, the number of genes that are differentially expressed with a corresponding differential peak containing BATF or TCF7 is shown.
TCR analysis identifies patterns of expansion associated with cell states and clinical outcome
We reconstructed T cell receptor (TCR) sequences for all identified CD8+ T cells and defined 4 patterns of TCR clonality based on shared CDR3 sequences in both α and β chains (Figure 7A, Methods): persistent TCRs detected in pre- and post-therapy samples from the same patient; enriched TCRs detected in more than one T cell in a single sample; singlet TCRs found in only one T cell at one time point; common TCRs shared across two or more patients. Since the overall number of persistent TCRs was very low, especially in responders, we could not make many conclusions about their relationships to clinical response; still, we detected a significant enrichment for persistent TCRs in non-responders in clusters CD8_3 (2-sided Wilcoxon p=0.01) and CD8_6 (p=0.006) (Figure 7B), and when aggregating exhaustion clusters (CD8_1–3, p=0.02) but not the memory/effector ones (Figure 7C). Interestingly, very few persistent TCRs were detected in the CD8_5 cluster (which was present predominantly in post-therapy samples) when looking at all patient CD8+ cells (Figure 7D), suggesting that these T cell clones did not exist prior to therapy. Enriched TCRs were more common in exhausted clusters and singlet TCRs in effector/memory clusters (Figure 7G,J), but both were enriched in non-responders for CD8_1–3 and in responders for CD8_4–6 (Figure 7F,I). We hypothesize that enriched TCRs are likely to have been exposed to persistent tumor antigen stimulation, explaining their higher proportions in the exhausted than effector/memory clusters, while singlet TCRs are not as expanded because they are more recently generated against tumor antigens (or are bystanders) and have fewer exhaustion markers. Although common TCRs were predominantly present in clusters CD8_2 and 3, no significant association was found with clinical outcome (Figure 7K,L,M). Collectively, this analysis allowed us to connect the transcriptional phenotype of cells and therapeutic outcomes with TCR clonality, and could aid in investigating T cell dynamics and cell state plasticity. Indeed, when looking at the transitions of T cell states (CD8_1–6) within a specific clone (based on identical TCR sequence) across longitudinal samples in the same patient (Table S6), we discovered bilateral transitions between exhausted and memory/effector states.
Figure 7. TCR analysis and its relationship with cell state and clinical outcome.

A. Schematic illustration of the TCR analysis pipeline. B, E, H, K. tSNE plot delineating 6 CD8+ T cell clusters and persistent (B), enriched (E), singlet (H) and common (K) TCRs. Bar plot summarizes fraction of TCRs per patient across the different clusters between responder (R) and non-responder (NR) lesions. C, F, I, L. Fraction of persistent (C), enriched (F), singlet (I) and common (L) TCRs per patient, aggregated for CD8_1,2,3 and CD8_4,5,6 clusters for R and NR lesions. D, G, J, M. Fraction of persistent (D), enriched (G), singlet (J) and common (M) TCRs in each cluster, out of total persistent, enriched, singlet and common TCRs. Data are represented as mean±SEM.
DISCUSSION
Although immune checkpoint blockade leads to durable responses in patients with metastatic melanoma, refractory disease and progression after initial response remain major causes of mortality (Larkin et al., 2015; Robert et al., 2015). By profiling single immune cells in baseline and on or post-therapy samples in melanoma patients treated with checkpoint therapy, we identified and characterized several CD8+ T cell states associated with lesion growth, and studied their properties using a series of molecular and functional experiments.
A central finding from our study is that the presence of TCF7 protein in CD8+ T cells can predict clinical response to checkpoint therapy, suggesting that the state of T cells, in addition to the number of T cells and spatial distribution (Galon et al., 2006; Mahmoud et al., 2011; Sharma et al., 2007), found in a patient’s tumor is critical for induction of effective tumor immunity. Consistent with our findings, TCF7 (TCF1 in mice) is required for reinvigorating CD8+ T cells in response to PD-1 blockade to resolve chronic LCMV infection (Im et al., 2016; Utzschneider et al., 2016), and for the expansion of CXCR5+TIM3−CD8+ T cells (but not TIM3+cells) for control of virus in mice (Im et al., 2016), although we did not observe CXCR5 expression in TCF7+ cells in human melanoma. Finally, our results also agree with recent studies showing a reduction in open chromatin regions at TCF7 sites in non-programmable, dysfunctional PD-1hi T cells (Philip et al., 2017), and with the importance of TCF7 role for WNT signaling in stem cell-like memory cells (Gattinoni et al., 2009).
In contrast to TCF7+CD8+ T cells, we found cells expressing exhausted or dysfunctional signatures associated with lack of response to checkpoint therapy. Our finding of CD39 (an enzyme in the adenosine pathway) as a marker of exhausted CD8+ T cells is consistent with recent observations in melanoma (Canale et al., 2018), and HCV and HIV infected patients (Gupta et al., 2015). The ability of CD39 inhibitors together with TIM3, PD-1 or PD-1/CTLA4 blockade to reduce tumor growth and increase survival of mice with B16-F10 tumors is consistent with studies using checkpoint blockade with inhibitors of CD73 (a downstream component of the adenosine pathway) (Allard et al., 2013), and suggests new therapeutic combinations for treating melanoma and other cancers.
Some studies used bulk tumor expression data to identify several signatures (IFNg, exhaustion, cytotoxicity, antigen presentation and others) that associate with outcome (Ayers et al., 2017; Prat et al., 2017; Riaz et al., 2017). However, since many distinct signatures are upregulated at the same time in responders, one interpretation of these findings is that the bulk transcriptome change (which mixes many immune and non-immune cells together) likely reflects an overall increase in the T cell infiltrate, and does not discriminate specific T cell states. Consistent with this notion, we find that signatures of CD8_G and CD8_B, as well as known markers of T cell states (exhausted, effector, memory), are equally correlated with CD3 transcript number in samples from these cohorts (Figure S7).
We also show evidence that T cells can transition between states based on identical TCRs in T cells from exhausted-like and memory-like states, but we do not know the order or the exact transitions between states. Surprisingly we found that CD 8_5 T cells, which were predominantly found in post-therapy tumor samples, hardly share TCRs with the baseline sample, suggesting that members of the CD8_5 cluster are generated outside of the tumor and subsequently migrate to the tumor, consistent with observations from a recent study (Spitzer et al., 2017) demonstrating that lymphoid-organ derived T cells are required for anti-PD-1 potency.
Future studies will need to purify cells in each of the different T cell states based on surface protein markers (which may not always correlate with transcripts), validate their purity by scRNA-seq, and study their properties (as we did for some of the states here). Indeed, we identified a T cell state associated with response and showed that CD39-TIM3−CD8+ T cells (which are TCF7+) contribute to the antitumor activity of anti-PD-1 therapy in culture, but we have yet not determined which cells kill the tumors, the role of PD-1 in this process or the factors that induce or attract TCF7+ T cells.
Our finding that specific memory-like signatures are associated with response, together with recent studies showing that chimeric antigen receptor (CAR) T cell activity is enhanced by generating more memory-like cells through IL-7 or IL-15 pathways, leading to better outcome in preclinical models (Hurton et al., 2016; Shum et al., 2017) —suggests that methods to increase the ratio of CD8_G to CD8_B would enhance immunotherapies. Indeed, we were able to increase ex vivo tumor killing by removal of CD39+TIM3+CD8+ T cells (from tumor infiltrates) prior to treatment with anti-PD-1.
Building on the results and datasets presented here, one can envision designing trials that select patients for anti-PD-1 therapy based on T cell states and markers, and then testing whether this strategy increases the rate of durable responses. In addition, it may be possible to use the change in the ratio of T cell states as an assay to prioritize therapeutic approaches prior to anti-PD-1 therapy. Future studies will also need to test whether our predictive markers of response are relevant to other types of malignancies and therapies.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Nir Hacohen (NHACOHEN@mgh.harvard.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Patient samples.
Metastatic melanoma patients treated with checkpoint blockade therapy at Massachusetts General Hospital (Boston, MA) and University of Texas MD Anderson Cancer Center (Houston, TX) provided written informed consent for the collection of tissue and blood samples for research and genomic profiling, as approved by the Dana-Farber/Harvard Cancer Center Institutional Review Board (DF/HCC Protocol 11–181) and UT MD Anderson Cancer Center (IRB LAB00–063 and 2012–0846). For the single cell RNAseq analysis 48 tumor samples at baseline and/or after checkpoint treatment were collected from 32 patients, with 20 patients having matched normal blood samples for whole exome sequencing.
Mice.
Female C57BL/6 or BALB/cJ mice, age of 8–9 weeks were purchased from Jackson Laboratory and were housed at Massachusetts General Hospital under SPF conditions. All experiments followed protocols approved by the Massachusetts General Hospital Institutional Animal Care and use Committee (IACUC).
In vivo tumor transplant experiments.
B16-F10 and CT26 cells were generously provided by Mikael Pittet and Umar Mahamood respectively. B16-F10 or B16-F10GFP+ cells (0.5×106) were intradermally injected into the right flank using a 30g needle and tumors were measured every 4 days in two dimensions using a digital caliper. Tumor volume (mm3) was calculated using the following formula V= (L * W2)/2 (V=volume, L= tumor length, tumor width). For MDOTS experiments, CT26GFP+cells (2×106) were subcutaneously injected into the right flank using a 27g needle and tumors were harvested 14 days post transplantation. All blocking treatments started on day 4 post transplantation after 100% of tumors were visible. Invivo plus rat IgG2a isotype control (BioXCell; 2A3; BE0089) 100μg/dose was intraperitoneally (i.p.) injected to the control (untreated) group every 3 days. Invivo plus anti-mouse TIM3 (BioXCell; RMT3–23; BE0115) 100μg/dose, Invivo plus anti-mouse PD-1 (BioXCell; 29F.1A12; BE0273) 200μg/dose and Invivo plus anti-mouse CTLA4 (BioXCell; 9D9; BE0164) 100μg/dose were i.p. injected every 3 days. POM-1 (polyoxometalate-1) 5mg/kg/day (Santa Cruz Biotechnology; sc-203205), a CD39 inhibitor, was i.p. injected on a daily basis, starting on day 4 post transplantation.
METHOD DETAILS
Sample dissociation.
Fresh isolated tumor samples were collected immediately after surgery and were dissociated within 1 hour using the human tumor dissociation kit (Miltenyi Biotec; 130–095-929) with the following modifications. Tissue was minced into small pieces using a scalpel and put into a 1.5ml eppendorf tube containing 100μl of enzyme H, 50μl of enzyme R, 12.5μl of enzyme A (all provided in the kit), and 837.5μl of RPMI, followed by a 20 minute incubation in a thermomixer (Eppendorf; F1.5) at 37°C, 600 rpm. After incubation, debris were removed by filtering through a 70μm cell strainer, followed by mincing of the remaining tissue left on the strainer with a plunger in order to increase cell yield. Dissociated cells were subsequently washed with cold 1X PBS containing 1.5% heat inactivated FCS, spun down at 1300 rpm, 4°C for 5 minutes, resuspended, and counted for yield and viability with trypan blue using a Countess automated cell counter (Invitrogen).
Flow cytometry and cell sorting.
For both flow cytometry and cell sorting, Human or mouse TrueStain FcX (Biolegend, 422302 or 101320) was used for blocking Fc receptors before labeling cells. To discriminate live from dead cells, we used Zombie Violet Dye (Biolegend, 423114) or Zombie Green (Biolegend, 423111) for 15 min at 4°C, followed by surface labelling of cells for 30 min at 4°C, using standard protocols. The antibodies used for cell surface labelling were PE anti-human CD45 (Biolegend, 304008), APC anti-human CD3 (Biolegend, 300412), FITC anti-human HLA-A,B,C (Biolegend, 311404), APC/Cy7 anti-human CD235a (Biolegend, 349116), PE/Cy5 anti-human CD3 (Biolegend, 300309), BV421 anti-human PD-1 (Biolegend, 329919), PE/Cy7 anti-human TIM3 (Biolegend, 345013), APC/Cy7 anti-human CD39 (Biolegend, 328226), AF700 anti-human CD4 (Biolegend, 317425) and BV650 anti-human CD8 (biolegend, 301041). Antibodies used for cell surface of mouse cells were AF647 anti-mouse CD39 (Biolegend, 143808), BV605 or Pacific blue anti-mouse CD3 (Biolegend, 100351 or 100334), PE/Cy5 or FITC anti-mouse CD8a (Biolegend, 100710 or 100705), APC/Cy7 anti-mouse CD4 (Biolegend, 100414), BV650 or FITC anti-mouse CD45.2 (Biolegend, 109836 or 109806) and APC or Pacific Blue anti-mouse Thy1.2 (Biolegend, 140312 or 105324). The antibodies used for intracellular staining were FITC anti-human IFNγ (Miltenyi Biotec, 130–097-936), PE anti-human IL2 (Miltenyi Biotec, 130–099-391), APC anti-human TNFα (Miltenyi Biotec, 130–099-197), APC anti-mouse TNFα (Biolegend, 506307), PE anti-mouse IL2 (Biolegend, 503807), FITC anti-mouse IFNγ (Biolegend, 505805), PE anti-mouse Perforin-1 (Biolegend, 154305) and APC anti-mouse Granzyme-B (Biolegend, 372203). Intracellular cellular labelling for granzyme-B and perforin-1 was performed following surface staining, fixation and permeabilization using the BD Transcription Factor Buffer Set (BD, 562574) according to the manufacturer’s instructions. Sorting of single cells was performed on a BD Fusion instrument using the following antibody panel: Zombie dye, CD45, CD235a and HLAA,B,C. CD45+ cells from dissociated samples were sorted into 96-well plates (Eppendorf, 951020401) containing 10μl of lysis buffer (TCL buffer, Qiagen 1031576, supplemented with 1% β-mercaptoethanol), sealed, vortexed, spun down at 2500 rpm for 30 seconds, immediately placed on dry ice, and then stored at −80°C until processing with the Smart-Seq2 protocol. Sorting of human and mouse CD8+CD39+TIM+(DP) and CD8+CD39−TIM3−(DN) was performed using the following antibody panel: Zombie dye, CD45, CD3, CD8, CD39, and TIM3. For flow cytometry, we used the Beckman Coulter CytoFLEX instrument and analyzed the data with FlowJo 10.4.2 software.
Single cell RNA sequencing procedure.
Libraries from single cell lysates were generated with the Smart-Seq2 protocol (Picelli et al., 2013) with some modifications in the reverse transcription step as recently described (Villani et al., 2017). 96-well plates containing cell lysates were thawed on ice, spun down at 1500 rpm for 30 seconds, and mixed with Agencourt RNAClean XP SPRI beads (Beckman Coulter) for RNA purification. Purified RNA was resuspended in 4μl of Mix-1, denatured at 72°C for 3 min and placed immediately on ice for 1 min before 7μl of Mix-2 was added (Table S6). Reverse transcription was carried out at 50°C for 90 min, followed by 5 min incubation at 85°C. 14μl of Mix-3 was added in each well and the whole-transcriptome amplification step was performed at 98°C for 3 min, followed by 21 cycles at (98°C for 15 sec, 67°C for 20 sec and 72°C for 6 min), and final extension at 72°C for 5min. cDNA was then purified with Agencourt AMPureXP SPRI beads (Beckman Coulter) as described (Villani et al., 2017), to remove all primer dimers residues. Quality control steps were performed on samples before library construction and included the following steps: (1) concentration measurements, using the Qubit dsDNA high sensitivity assay kit on the Synergy H1 Hybrid Microplate Reader (BioTek); (2) cDNA size distribution using the High-Sensitivity DNA Bioanalyzer Kit (Table S6). Libraries were generated using the Nextera XT Library Prep kit (Illumina) with custom indexing adapters (Villani et al., 2017) in a 384-well PCR plate, followed by a cleanup step to remove residual primer dimers. Combined libraries from 384 cells were then sequenced on a NextSeq 500 sequencer (Illumina), using paired-end 38-base reads.
Immunofluorescence assay and analysis.
Multiplex staining was performed on 4μm formalin-fixed paraffin-embedded sections using the Opal multiplex IHC system (PerkinElmer; NEL800001KT) according to the manufacturer’s instructions. Briefly, slides were baked for 1 hour at 65C followed by deparaffinization with xylene and a graded series of ethanol dilutions (100%, 95% and 70%), fixation with 10% neutral buffered formalin for 30 minutes, microwave antigen retrieval using the AR9 buffer (PerkinElmer; AR900250ML), and blocking. Primary antibodies used for staining were: CD8a (Biolegend; C8/144B; 372902; 1:100) detected with OPAL520 (1:100; Cy2); TCF7 (Cell Signaling; #2203; 1:100) detected with OPAL690 (1:100; Cy5.5). Counterstain was done using DAPI (1:1000) and subsequently mounted using Vectashield (Vectra; H-1000) fluorescence media. Slides were imaged using the Olympus IX83 confocal microscope by scanning 10 random fields on each sample at 40X magnification, and analyzed with CellProfiler 2.2.0 (Carpenter et al., 2006) to detect the total number of nuclei, CD8+, TCF7+, and CD8+TCF7+ cells. Due to cellular heterogeneity between different slides/patients, in each sample the percentage of CD8+TCF7− or CD8+TCF7+ was calculated out of the total nuclei detected. For the analysis, a new pipeline was made for detection of cells positive for CD8 and TCF7 (File S1).
Immunohistochemistry.
Procedures were done on the automated Ventana Discovery Ultra staining system, using 4μm formalin-fixed paraffin-embedded sections. Sections were deparaffinized in xylene and graded alcohols, followed by antigen retrieval (EDTA), blocking with Discovery inhibitor (Ventana; 760–4840), incubation with primary antibodies for 16 minutes, washing and incubation with a secondary antibody conjugated with horseradish peroxidase (HRP). Sections were developed with discovery purple chromogen kit (Ventana; 760–229) and were then counterstained with hematoxylin. Primary antibodies used were: B2M (Abcam; ab27588; 1:1000); anti melanoma triple cocktail (Ventana; 790–4677; 1:100) containing antibodies against melanosome (HMB45), Mart-1/melan A (A103), tyrosinase (T311). The melanoma triple cocktail was used to separate tumor from normal cells enabling detection of B2M in the cancerous cell fraction.
Intracellular cytokine detection.
For intracellular cytokine analysis of human CD8+ T cells, 5×105 cells from dissociated samples (n=12) were cultured in the presence of soluble LEAF purified anti-CD3 (Biolegend, 317303, 2μg/ml), anti-CD28 (Biolegend, 302913, 1μg/ml) and GolgiPlug (BD, 555029) for 6 hours at 37°C. Intracellular cytokine labelling was performed following surface staining, fixation and permeabilization using the BD Cytofix/Cytoperm Plus kit (BD, 555028) according to the manufacturer’s instructions.
Generation of B16-F10GFP+ and CT26GFP+ cells.
B16-F10 and CT26 cell lines were transduced with the lenti-GFP+virus FUGW (a gift from David Baltimore; Addgene, 14883) (Lois et al., 2002), and GFPhigh-positive cells were sorted on day +5 to generate B16-F10GFP+ and CT26GFP+ cells. Before each experiment the percentage of GFP+ cells was evaluated by flow-cytometry. For lentivirus production 293T cells were transfected with psPAX2 (a gift from Didier Trono; Addgene, 12260), FUGW and pMD2.G (a gift from Didier Trono; Addgene, 12259) at a 10:10:1 ratio using TransIT-LT1 reagent (MIRUS, MIR2300) according to the manufacturer’s guidelines.
In vivo depletion of CD4+ and CD8+ T cells.
For depletion of CD4+ and CD8+ T cells 400μg/dose of Invivo MAb anti-mouse CD8a (BioXCell; 2.43; BE0061), 400μg/dose Invivo MAb anti-mouse CD4 (BioXCell; GK1.5; BE0003–1) or 400μg/dose rat IgG2b isotype control (BioXCell; LTF-2; BE0090), were i.p injected every 3 days, starting from day +7 post tumor transplantation until day +21. Depletion efficacy was evaluated on day +14 by flow cytometry analysis.
Ex vivo culture and live/dead imaging of Murine-derived Organotypic Tumor Spheroids (MDOTS).
CT26GFP+ tumors from untreated BALB/cJ mice were harvested on day +14 following implantation. MDOTS (S2 fraction; 40–100μm) isolation was performed as previously described (Jenkins et al., 2018). Following isolation, MDOTS were resuspended in type I rat tail collagen and the spheroid-collagen mixture was injected into the center gel region of the 3D microfluidic culture device (10μl per AIM device). After incubation (30 min, 37°C in sterile humidity chambers), the collagen-MDOTS mixture was hydrated with media (10% FBS in RPMI) with the indicated therapeutic monoclonal antibodies: isotype control IgG2a (10μg/ml, BioXCell, BE0089), anti-PD-1 (10μg/ml, BioXCell, BE0146), anti-IFNγ (10μg/ml, BioXCell, BE0054), and anti-CD8a (10μg/ml, BioXcell, BE0004–1). For TIM3/CD39 subpopulation studies, sorted CD8+ double-positive derived from the S3 (<40μm) MDOTS fraction (DP; CD39+TIM3+), double-negative (DN; CD39−TIM3−) and mixed (1:1 mixture of DP and DN cells) populations were pelleted and resuspended in MDOTS/collagen mixture at an estimated effector:target (E:T) ratio of ~1:3–1:4 (based on estimated ~10,000 cells per device). MDOTS were cultured in the DAX-1 3-D cell culture chip from AIM Biotech, as described (Jenkins et al., 2018). On day +5 live/dead fluorescence staining was performed as previously described (Jenkins et al., 2018) with the following modifications. After incubation with Hoechst/PI (40 min, 37oC, 5% CO2) images were obtained. Image-capture and analysis were performed using a Nikon Eclipse 80i fluorescence microscope equipped with Z-stack (Prior), motorized stage (ProScan) and ZYLA5.5 sCMOS camera (Andor) and NIS-Elements AR software package. Live/dead cell quantification was performed by measuring total cell area of each dye in GFP-positive CT26 cells.
CFSE labeling and ex vivo proliferation assay.
Single cell suspensions (10×106) of digested B16-F10GFP+ tumors isolated on day 14 post implantation (from POM-1 treated and untreated mice) were incubated with PBS (without Ca2+ and Mg2+) containing 5μM CFSE (Biolegend, 423801) for 10 min at 37°C. Heat inactivated fetal calf serum (FCS) was then added for 1 min, and cells were washed three times in RPMI + 10% FCS. CFSE-labeled cells (3×105) were seeded in triplicates in a 96-flat bottom plate with or without the presence of 1μg/ml anti-CD3/CD28 antibodies (Biolegend, 100314 and 102112) for 72h. The number of cell divisions of Thy1.2+CD8+ cells was determined by flow-cytometry analysis.
Intra-tumoral ATP level measurements.
Intra-ATP levels were measured immediately either in total single cell suspensions of digested B16-F10GFP+ tumors or sorted B16-F10GFP+ cells (1×105), isolated from POM-1 treated and untreated mice on day 14, using the CellTiter-Glo Luminescent Cell Viability Assay (Promega, G7571) according to the manufacturer’s guidelines. ATP levels were measured (in triplicates) using the Synergy H1M plate reader (BioTek), and its concentration was calculated using an ATP standard curve.
ATAC-seq tagmentation.
Methods for tagmentation are as previously reported (Corces et al., 2016). Briefly, 5,000–10,000 cells were cell sorted into RPMI containing 10% FBS, 1% Pen/Strep, 1% L-Glutamine, and 1% HEPES. The cells were then centrifuged at 500xg at 4°C for 10 minutes, the supernatant aspirated, and resuspended in tagmentation mixture (25 μl tagmentation buffer (Illumina, FC-121–1031), 2.5 μl TBE (Illumina, FC-121–1031), 0.5 μl 1% digitonin (Promega, G9441), and 22 μl H2O). The cells were then incubated at 37°C in a thermomixer, mixing at 300 RPM for 30 min. Following tagmentation, the sample was immediately purified via minElute PCR cleanup column (QIAGEN, 28006), and eluted in 10 μl. The tagmented DNA was then PCR’ed using Nextera indexing primers with sequencing adapters for 5 cycles in a 50 μl reaction. 5 μl of the reaction was then used for qPCR to determine the remaining number of PCR cycles required (as determined by the cycle number of each sample when it reaches 1/3 the fluorescence threshold), followed by PCR of each individual sample according to this cycle number. The samples were purified using 1.5X Agencourt AMPure XP beads (A63880), followed by two 70% EtOH washes, and elution of DNA in 15 μl buffer EB (QIAGEN, 19086). Each sample was quantified by Qubit, and measured for fragment lengths on a Tape Station. The samples were pooled and sequenced on an Illumina Nextseq 500 using 75 bp PE reads to a sequencing depth of 30 million reads per sample.
Whole exome sequencing (WES).
WES of DNA from tumor and matched normal blood samples was done as previously described (Sade-Feldman et al., 2017). Briefly, 250–500ng of extracted DNA, using Qiagen AllPrep DNA/RNA Mini Kit (cat# 80204), was used as input for library preparation. Sample were barcoded using unique 8 base molecular barcodes followed by a library enrichment process, and all libraries above 40ng/μl were considered acceptable for solution-phase hybrid selection and sequencing. Libraries preparation was carried out using the SureSelect Target Enrichment System Sequencing Platform Library Prep v2 (Agilent Technologies, G3360–90000), according to manufacturer’s specifications, followed by quantification and normalization using PicoGreen to ensure equal concentration. Libraries were then quantified using qPCR (KAPA Biosystems, KK4832), denturated with 0.2M NaOH and diluted to 20pM using hybridization buffer (Illumina). Cluster amplification was performed according to the manufacturer’s protocol (Illumina), HiSeq 2500 v4 cluster chemistry and flowcells, as well as Illumina’s Multiplexing Sequencing Primer Kit. Libraries were sequenced using the HiSeq 2500 v4 Sequencing-by-Synthesis method (paired end 76bp reads) followed by analysis with RTA v.1.12.4.2. The minimum depth of coverage was 150X and 80X for tumor and normal samples respectively. All procedures were done at the Genomics Platform of the Broad Institute of Harvard and MIT.
QUANTIFICATION AND STATISTICAL ANALYSIS
Single cell RNA-seq data generation and processing.
FASTQ files were aligned to the NCBI Human Reference Genome Build GRCh37 (hg19) using STAR (Dobin et al., 2013). Expression levels were quantified as Transcripts Per Million (TPM) and were computed by the RSEM tool (Li and Dewey, 2011). For each cell, we used three quality control (QC) measures. We excluded: (1) cells with a zero expression of both CD45 and CD3E; (2) cells expressing less than 1000 genes; (3) cells with an average expression of housekeeping genes (Table S6), log2(TPM+1) < 2.5. For downstream analysis, we focused on protein coding genes (Table S6), out of which, we used the set of genes with expression levels log2(TPM+1) > 4.5 in at least 10 cells per sample or genes with a particularly high expression level (log2(TPM+1) > 12) in one or more cells, per sample.
Supervised classification of single cells to cell types.
To classify each single cell that passed QC to a pre-defined cell type, we performed a supervised analysis based on a list of known marker genes (Table S1). This was done by defining a set of genes per cell type which must or must not be expressed. On average, this approach led to the unambiguous classification of 80% of the cells. The remaining cells were then annotated using a manual review process. Following this step, we validated that no cell had an ambiguous classification (e.g., a T cell and a B cells).
Dimensionality reduction
The t-Distributed Stochastic Neighbor Embedding (t-SNE) method (Maaten and Hinton, 2008) was used for dimensionality reduction with the default perplexity parameter of 30 and initial dimension parameter of 10. Of note, t-SNE was used only for visualization and not for clustering, as defined below.
Unsupervised clustering of immune cells.
To cluster all cells that passed QC we applied the k-means algorithm with a correlation distance metric, testing k = 3 … 15. The algorithm was applied using all genes with variance >6, yielding ~4000 genes. This value was selected based on the relation between the variance and the fraction of cells expressing each gene (Figure S8A). To determine the optimal number of clusters we applied the following steps: (1) We first examined how much of the complexity each cluster captures by applying the elbow method. This was done by computing the Pearson correlation matrix R and the distance matrix as D (1 − R). We then computed the sum of pair-wise distances between all cells in different clusters and the total distance Dist= ∑i,j D(i,j). The ratio between these two measures V = Disb/Dist was used to estimate the variance explained by a given solution (Figure S8B), such that in the extreme case where all cells are clustered together or the case where each cell is a single cluster, this ratio would be 0 and 1, respectively. Exploring this ratio, we then select the solutions that are near plateau (k = 10, … ,15). (2) We then performed differential expression analysis (see below) to search for gene markers that are significantly more highly expressed in a specific cluster as compared to all other clusters. Then, in order avoid complex solutions, we excluded solutions with clusters that have too few marker genes (<20) distinguishing between them and the rest of the cells. (3) Finally, we performed a robustness analysis and selected the clustering solution with the highest median robustness score. Specifically, to determine the robustness of each clustering solution, we performed 100 iterations in which we randomly removed 10% of the cells, and re-ran the k-means algorithm and checked the stability of the clustering solution. We quantified the agreement of a given solution with the original one as the number of pairs of cells that were either clustered together, or not clustered together, in both solutions, divided by the total number pairs shared between the runs. This process yielded a median robustness measure of 0.96 for the selected = 11 (Figure S8C).
To examine if there is a significant difference between responders and non-responder lesions for a given cluster, we computed the fraction of cells in each lesion assigned to cluster, and applied the Wilcoxon rank-sum test to the corresponding values of responder and non-responder lesions. P-values were corrected using the Benjamini-Hochberg False Discovery Rate (FDR) procedure and were considered significant if the FDR q-value was 0.1.
Unsupervised clustering of CD8 T cells.
To identify different CD8+ T cell clusters we first extracted all single-cells classified as CD8+ in our supervised analysis. Our clustering process for CD8+ T cells followed the exact steps described above, testing possible clustering solutions for k = 2, … ,13 (Figure S8D). We then further explored the solutions with the highest variance explained (k = 6, … ,13), and identified = 6 as the optimal number of clusters, with a median robustness value of 0.93 (Figure S8E). In addition, we note for k = 2, … ,6 solutions had a hierarchical pattern in which whenever we increased k, a single cluster was split into two sub-clusters (Figure S8F,G). Similar to the analysis of all cells, to examine if there is a significant difference between responder and non-responder lesions for a given cluster, we computed the fraction of CD8+ cells in each lesion assigned to cluster, and applied the Wilcoxon rank-sum test to the corresponding values of responders and non-responder lesions. P-values were corrected using the Benjamini-Hochberg False Discovery Rate (FDR) procedure and were considered significant if the FDR q-value was ≤ 0.1.
Differential expression analysis.
In all cases, differential expression analysis was applied to all genes that had an average expression level log2(TPM+1) > 2 in either tested groups, % and %&. Then, for each gene, we count the number of cells in % and %& that express it with an expression level log2(TPM+1) > 2 or ≤ 2. We then apply Fisher’s Exact test for the corresponding 2×2 table. To identify significant differences we considered genes with a Bonferroni-corrected q-value ≤ 0.05 and log2(fold-change) > 0.5.
Trajectory analysis of CD8+ T cells.
To analyze the trajectory of CD8+ T cells based on single-cell RNA-seq expression data, we used Monocle v. 2.5.4 (Qiu et al., 2017). As input to Monocle’s Reversed Graph Embedding algorithm, we selected a set of 426 genes that was the union of the top 100 differentially expressed genes ordered by ascending q-value (as described above) for each of the six CD8+ T cell clusters (or all such genes for two clusters that had fewer than 100 significant genes).
T cell Receptor (TCR) reconstruction.
We applied the MixCr tool for reconstructing TCRs from all identified T cells (Bolotin et al., 2015). We defined persistent TCRs as TCRs having an identical CDR3 sequence in both chains and were detected in baseline (pre-therapy) and post-therapy samples from the same patient. Enriched TCRs were defined as TCRs having an identical CDR3 sequence in both chains and detected in the same patient at a single time point, or in two parallel time points (e.g., multiple biopsies collected at the same time point). Singlet TCRs found in only one T cell at one time point. Lastly, common TCRs were defined as those having an identical CDR3 sequence in both chains and detected in different patients. P-values were corrected using the Benjamini-Hochberg False Discovery Rate (FDR) procedure and were considered significant if the FDR q-value was 0.1.
ATAC-seq analysis.
Sequencing reads for each sample were aligned to hg19 using Bowtie 2.2.1 (Langmead and Salzberg, 2012) with a max insert size of 2000 bp. SAM files were converted to BAM files and sorted using Samtools 1.3 (Li et al., 2009). Duplicate (as defined by http://broadinstitute.github.io/picard) and mitochondrial reads were removed, and peaks were called, initially by making tag directories according to chromosome and then by finding peaks (areas with more sequencing reads than expected by chance) for each sample, using the “DNase” peak finding style (‘makeTagDirectory –format sam’ and ‘findPeaks -style dnase’, Homer version 4.9) (Heinz et al., 2010). Overlapping peaks were then merged. The number of Tn5 transposition events (5’ ends of reads) lying within each peak were quantified for each sample, yielding a matrix of peaks by samples containing ATAC read counts. EdgeR 3.14.0 was used to call CD39+TIM3+(DP)/CD39−TIM3− (DN)-specific peaks, first by grouping the samples by cell type (DP and DN) and pairing the samples from each patient, and then using EdgeR (Robinson et al., 2010) to estimate the tagwise dispersion using generalized linear models (estimateGLMTagwiseDisp function). We then performed a likelihood ratio test to identify differential accessibility between paired samples from each patient (glmFit, glmLRT). We obtained the top differential peaks (topTags), sorting peaks by their FDR q-value. Differential peaks between DP and DN were called significant if their FDR q-value was 0.01. Similar analysis was performed on the data set (GSE89308) from the Schietinger group (Philip et al., 2017), for the identification of unique peaks in PD-1high and central memory (CM) cells.
Motif analysis.
To identify TF motifs that distinguish DP- and DN-specific peaks from non-specific (background) peaks, each peak was scanned with the human motifs from the CIS-BP database (Weirauch et al., 2014), using the GOMER approach (Granek and Clarke, 2005), yielding a binding score for each peak for each TF motif. The minimum hypergeometric (minHG) test was then used to gauge how well motif scores enrich DP-or DN-specific peaks (FDR q < 0.01) compared to background peaks, considering the top N (1 up to 3000) highest scoring peaks. Here, background peaks included those whose ATAC DP-vs-DN FDR was over 0.1 (i.e. not significantly DP- or DN-specific) and had an average count per million (CPM) greater than the minimum CPM of DP/DN-specific peaks (i.e. enough reads that a difference could have been detected). MinHG P-values were corrected by Benjamini-Hochberg FDR, counting each minHG test as independent (resulting in more conservative FDR q-values).
Survival analysis.
We used the TCF7+CD8+/TCF7−CD8+ ratio generated from our immunofluorescence analysis to split samples into two groups (ratio>1 and <1). A standard Kaplan-Meier survival analysis was then used to determine the association of these groups with survival rate. In case where two or more samples for the same patient exist, we selected the baseline sample for this analysis.
Mutation calling pipeline.
WES BAM files were aligned to the NCBI Human Reference Genome Build GRCh37 (hg19) and were checked for contamination by DNA originating from a different individual using ContEst (Cibulskis et al., 2011). Somatic single nucleotide variations (sSNVs) were then detected using MuTect (Cibulskis et al., 2013). Following this standard procedure, we filtered sSNVs by: (1) removing potential DNA oxidation artifacts (Costello et al., 2013); (2) realigning identified sSNVs with NovoAlign (www.novocraft.com) and performing an additional iteration of MuTect with the newly aligned BAM files; (3) removing technology- and site-specific artifacts using a panel of ~7000 TCGA normal samples (PoN filtering)(Ellrott et al., 2018). Finally, sSNVs were annotated using Oncotator (Ramos et al., 2015).
Whole transcriptome analysis of bulk tumor samples.
We used bulk RNA-seq data from the Van Allen et al. (Van Allen et al., 2015) (n=37) and Riaz et al. (Riaz et al., 2017) (n=51) datasets. To make our comparison consistent with our single cell dataset, we aligned the RNA-seq reads with the protocol described above and used log2(TPM+1) values for quantification of expression levels. To compute the bad and good signature scores we computed the average expression of each set of marker genes (CD8_G=34 genes, CD8_B=1114 genes, Table S2).
DATA AND SOFTWARE AVAILABILITY
Data availability.
Raw sequencing data (single cell RNAseq, WES and ATACseq) from this study have been deposited in dbGAP database (https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs001680.v1.p1) under accession code phs001680.v1.p1. Processed Single cell RNAseq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE120575 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120575). The Van Allen (Van Allen et al., 2015) bulk RNA dataset used in this study is available in dbGAP database under accession number phs000452.v2.p1. Data from Riaz (Riaz et al., 2017) and Tirosh (Tirosh et al., 2016) studies used in this paper are available in GEO with the accession number GSE91061 and GSE72056 for the Riaz and Tirosh datasets respectively. ATAC-seq data from the Schietinger group (Philip et al., 2017), used for the identification of unique peaks in PD-1high and central memory (CM) cells is available in GEO with the accession number GSE89308.
Supplementary Material
Figure S1. Comparing the composition of known cell types to clinical outcomes and checkpoint therapy, Related to Figure 1. A. tSNE plot of all CD45+ cells collected in this study. Cells are colored by cell type on the basis of pre-defined markers (Table S1). Pie chart summarizes the corresponding percentage of known cell types across all CD45+ cells collected in this cohort. B. A comparison between the supervised classification of single cells to cell types (right color code) to the unsupervised clustering of immune cells identified by k-means clustering (left color code). For each one of the 11 unsupervised clusters identified, the percentage of cell types as defined by the supervised analysis is shown.
Figure S4. Characterization of CD39+CD8+ cells in melanoma patients and combined inhibition of CD39 with anti-PD-1 or anti-PD-1/CTLA4 improves survival and reduces tumor growth, Related to Figure 5. A. Gene expression level distribution (log2(TPM+1)) of CD39 (ENTPD1, left) and TIM3 (HAVCR2, right) in the six CD8+ T cell clusters is shown. Each dot represents an individual cell. 1-sided Wilcoxon rank-sum p-value is shown. B. Gating strategy that was used to isolate CD39+TIM3+ and CD39−TIM3−, CD8+ T cells from melanoma patients. C. Flow-cytometry quantification of PD-1 and TIM3 in CD39+, CD39− (CD8+ T cells), and CD39 expression in PD-1+, PD-1−, TIM3+ and TIM3− (CD8+ T cells) in metastatic melanoma patients is shown. Data were combined from 2 replicate experiments. Unpaired-student’s t-test is shown; ns - not significant. D. Schematic representation for the preparation, isolation and live/dead analysis of CT26GFP+ MDOTS. E. Individual tumor volumes of B16-F10 implants in the untreated (IgG control), anti-TIM3, CD39i, and anti-TIM3+CD39i groups is shown. F. Box plots showing the kinetics of tumor growth between the different groups of mice on days +4, +7, +11, +14 and +18 post tumor transplantation. P-value was determined by unpaired-student’s t-test. G. A schematic summary of the therapy regimen used in the transplantable B16-F10 mouse model. H-I. Box plots showing the kinetics of tumor growth between the different groups of mice on days +4, +7, +11, +14, +18 and +21 post tumor transplantation for the CD39i+PD-1 (H) and CD39i+PD-1/CTLA4 (I) experiments. P-value was determined by unpaired-student’s t-test. J. Individual tumor volumes of B16-F10 implants in the untreated (IgG control), CD39i, anti-PD-1, anti-PD-1/CTLA4, anti-PD-1+CD39i and anti-PD-1/CTLA4+CD39i groups is shown. To simplify the presentation, we split the two different arms of therapy and showed them separately. Data are represented as mean±SEM. For in vivo mouse tumor models one of two independent experiments is shown.
Figure S5. CD8+ T cells are required for the success of in vivo CD39 inhibition which modulates CD8+ TILs and tumor cells, Related to Figure 5. A. A schematic summary of the depletion experiments used in the transplantable B16-F10 mouse model (n=5 per group). B. Representative flow cytometric plots of surface staining for CD4+CD3+ (left) and CD8+CD3+ (right) in depleted and non-depleted groups on day +14 are shown. C. Box plots showing the kinetics of tumor growth between the different groups of mice on days +4, +7, +11, +14, +18 and +21 post tumor transplantation. D. Survival at day 35 of tumor-bearing mice for all 4 groups. Log-rank P-value is shown. E. Individual tumor volumes of B16-F10 implants in the untreated (IgG control), CD39i, CD39i+anti-CD4 and CD39i+anti-CD8 groups is shown. F. A schematic summary for the in-vivo POM-1 experiments used in the transplantable B16-F10GFP+ mouse model (n=5 per group). Representative flow cytometry plot on the left shows the gating strategy used to characterize and isolate GFP+ B16-F10 tumor cells. G. The percentage of CD45+CD3+CD8+ cells expressing granzyme-B (left) and perforin-1 (right) in POM-1 treated and untreated groups. H. Representative flow cytometric plots (left) of intracellular staining for IL-2, IFNγ and TNFα in CD45+CD3+CD8+cells. Flow cytometry quantification of cytokine-producing cells obtained from the POM-1 treated and untreated groups is shown (right). I. Representative histograms (left) showing the CFSE profiles of Thy1.2+CD8+ gated cells after 72h stimulation with anti-CD3/CD28 antibodies from POM-1 treated and untreated animals. Bar plot (right) summarizes the percentage of proliferating cells analyzed by flow cytometry in each group. J. The percentage of CD45+CD3+CD8+ cells (left) or GFP+B16-F10 tumor cells (right) expressing CD39 in POM-1 treated and untreated groups. K. Intra-ATP levels in mixed tumor cell suspensions containing CD45+ and CD45− cells (left) or isolated B16-F10GFP+ cells (right) from POM-1 treated and untreated mice, with n=3 replicates for each cell suspension per experiment that were measured on day +14. An equivalent number of cells from each mouse (1×105) was added to each well prior to ATP measurement. Data are represented as mean±SEM. For A-D one of two independent experiments is shown. For G-K combined data from 2 replicates is shown. P-value was determined by unpaired-student’s t-test, ns - not significant.
Figure S6. CD39+TIM3+ and CD39−TIM3− cells have a distinctive epigenetic landscape, Related to Figure 6. A. Heatmap of averaged scaled expression values of discriminative transcription factors for non-sorted CD39+TIM3+ and CD39−TIM3− cells as defined by single cell RNA expression. B. Heatmap of averaged scaled expression values of discriminative transcription factors for CD8_B and CD8_G cells as in Figure 2. C. ATAC-seq traces for open chromatin regions (OCRs) near selected genes in CD39-TIM3− cells (blue) and CD39+TIM3+ cells (orange) in all 5 patients is shown. D. Transcription factor (TF) enrichment graphs for BATF and TCF7 in CD39+TIM3+ and CD39−TIM3− sorted cells are shown. Each graph shows the enrichment peaks relative to background (x-axis). Black bars indicate CD39+TIM3+ (top) or CD39−TIM3− (bottom) peaks, while white bars indicate background peaks. Motif enrichment was calculated using the minimum hypergeometric (minHG) test. E. Venn diagrams showing the distribution of ATAC-seq OCRs in DP (red), PD-1highCD8+ (blue), DN (green) and CM (orange).
Figure S7. Correlation of T cell markers in bulk RNA-seq data, Related to STAR Methods. A,E. Pairwise Spearman correlation between different immune markers in the Van Allen (A) and Riaz (E) cohorts. B,F. Spearman correlation between the expression levels of the different immune markers shown in the table and CD3 in the Van Allen (B) and Riaz (F) cohorts. C,G. Scatter plot showing the correlation between the good signatures based on CD8_G marker genes and CD3 expression levels in the Van Allen (C) and Riaz (G) cohorts. D,H. Scatter plot showing the correlation between the bad signatures based on CD8_B marker genes and CD3 expression levels in the Van Allen (D) and Riaz (H) cohorts.
Figure S8. Summary of variance and clustering robustness analysis, Related to STAR Methods. A. Variance of each gene vs. the fraction of cells expressing each gene (log2(TPM+1)>0). Left panel: genes expressed in more than 10% of the cells and less than 90% are colored in red. Right panel: genes with variance 6 are colored in red. As the set of genes expressed in less than 10% of the cells are of less interest for clustering analysis, we set as a minimal threshold the maximal variance observed in this group of genes, as indicated by the black arrow. B. Variance explained by each k-means solution ranging from k=3,…,15, when applied to all analyzed single cells. C. A box plot summarizing the distribution of robustness scores for each k=3, …,15. D. Variance explained by each k-means solution ranging from k=2, …,13, when applied to all analyzed CD8+ T cells. E. A box plot summarizing the distribution of robustness scores for each k=2, …,13. F,G. Clustering of CD8_B and CD8_G, separately, into two (F) or three (G) clusters. Hierarchical structure is shown where CD8_B and CD8_G are each split into 2 and 3 clusters, respectively.
File S1. Analysis pipeline for CellProfiler, Related to Figure 3.
Table S1. A summary of data related to all single cells analysis, Related to Figure 1.
Table S2. A summary of data related to CD8+ T cells analysis and 2 clusters solution, Related to Figures 2 and S2.
Table S3. A summary of data related to IF analysis, Related to Figure 3.
Table S4. A summary of data related to CD8+ T cells analysis and 6 clusters solution, Related to Figure 4.
Table S5. A summary of data related to sorted (CD39+TIM3+ and CD39−TIM3−) cells and ATAC-seq analysis, Related to Figures 5,6 and S7.
Table S6. A summary of identified TCRs and Methods material, Related to Figure 7 and STAR Methods.
Figure S2. Annotating CD8_G and CD8_B clusters to the CD45+ immune cell population clusters, Related to Figure 2. A. tSNE plot of all CD45+ clusters (n=11) collected in this study (left) is shown. Cells are colored based on 11 clusters identified by k-means clustering analysis. Right tSNE plot shows the distribution of CD8+ T cells that are classified as CD8_G (blue) and CD8_B (orange) in relation to all CD45+ cells analyzed in this study. B-C. For each sample, the percentage of cells found in CD8_G and CD8_B (out of all CD8+ T cells) in responder lesions (B) and non-responder lesions (C) is shown. * symbol marks samples with defects in antigen presentation and the IFNγ pathway as inferred from WES, IHC and flow-cytometry analysis. P# indicates patient number as described in Table S1. D. Representative immunohistochemistry staining (1 out of 3) of sections from patient #3 with homozygous mutations in B2M (as inferred from WES). Sections were stained with an antibody cocktail for melanoma cells (mel.cocktail) using anti-melanosome (HMB45), anti-MART-1/melan A and anti-Tyrosinase, to discern melanoma cells from normal cells; or with an antibody specific for B2M. Original Magnification X100. E. Flow-cytometry plot (left) and histogram (right), showing the expression of HLA-A,B,C in immune (blue) and tumor cells (orange) in patient #15. F. Receiver operating characteristic (ROC) analysis was constructed to evaluate the prognostic power of the ratio between CD8_G/CD8_B as shown in Figure 2D between responder and non-responder lesions. The area under the ROC curve (AUC) was used to quantify response prediction, and one-sided Wilcoxon test was used to assess significance of the AUC results. G. Heatmap describing scaled expression values of discriminative gene sets out of top 34 markers in CD8_B and CD8_G (Table S2), between responder and non-responder lesions in CD8+ T cells. Significant marker genes are shown for each group (Table S2). Top blue and orange bar depicts a cell-level association to CD8_G and CD8_B, respectively. H. tSNE plot of all CD8+ T cells found in the Tirosh et al. data, before (left panel) and after (right panel) filtering, as identified by k-means clustering, and correspond to CD8_G and CD8_B identified in this study (Table S2). Clustering of all CD8+ T-cells in this cohort (n=1450) identified 3 clusters, 2 confirming the presence of our CD8_G and CD8_B cellular states and a third one that was contaminated with melanoma markers (left). After removal of all CD8+ T-cells from this cohort, having either a melanoma (MITF, S100B, TYR, PMEL, and MLANA) NK\NKT cells (NCAM1 and NCR1), regulatory T-cells (FOXP3), and B cells (MS4A1 and CD79a) markers, analysis was performed on the remaining cells (n=260, right). I. Heatmap showing scaled expression values of discriminating genes for the clusters defined in (H). A list of representative genes is shown for each cluster next to the right margin color bars.
Figure S3. The distribution of exhaustion and memory markers across the six clusters and summary of trajectory analysis, Related to Figure 4. A. tSNE plot colored such that single-cells with a high expression level (log2(TPM+1)>2) of ENTPD1, HAVCR2, PDCD1, LAG3, TIGIT, TCF7, TNF, and IL7R are colored in red, and those with a low expression level (log2(TPM+1)<2) are colored in gray. Bar plots (to the right) summarize the corresponding percentages of each gene in the 6 identified clusters. B. tSNE plot of all CD8+ T cells profiled with coloring of CD8_5 cells associated with baseline (blue, left) or post-treatment lesions (green, right). C. tSNE plot of all CD8+ T cells profiled with coloring of CD8_5 cells associated with responder (blue, left) or non-responder lesions (green, right). D. Violin plots showing organization of cells corresponding to the six CD8 clusters by pseudotime as inferred by Monocle. E. Cell expression profiles in a two-dimensional independent space for each cluster (CD8_1 to 6) is shown. Solid black line indicates the main diameter path of the minimum spanning tree (MST) and provides the backbone of Monocle’s pseudotime ordering of the cells. Each dot represents an individual cell colored by cluster.
KEY RESOURCES TABLE:
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| InVivoMab rat IgG2a isotype control (clone 2A3) | BioXCell | Cat# BE0089, RRID:AB_1107769 |
| InVivoMab anti-mouse TIM3 (clone RMT3-23) | BioXCell | Cat# BE0115, RRID:AB_10949464 |
| InVivoMab anti-mouse PD-1 (clone 29F.1A12) | BioXCell | Cat# BE0273, RRID:AB_2687796 |
| InVivoMab anti-mouse CTLA-4 (clone 9D9) | BioXCell | Cat# BE0164, RRID:AB_10949609 |
| InVivoMab anti-mouse CD8a (clone 2.43) | BioXCell | Cat# BE0061, RRID:AB_1125541 |
| InVivoMab anti-mouse CD4 (clone GK1.5) | BioXCell | Cat# BE0003–1, RRID:AB_1107636 |
| InVivoMab rat IgG2b isotype control (clone LTF-2) | BioXCell | Cat# BE0090, RRID:AB_1107780 |
| InVivoMab anti-mouse PD-1 (clone RMP1-14) | BioXCell | Cat# BE0146, RRID:AB_10949053 |
| InVivoMab anti-mouse IFNγ (clone R4–6A2) | BioXCell | Cat# BE0054, RRID:AB_1107692 |
| InVivoMab anti-mouse CD8a (clone 53–6.7) | BioXCell | Cat# BE0004–1, RRID:AB_1107671 |
| Human TruStain FcX | Biolegend | Cat# 422302 |
| TruStain FcX anti-mouse CD16/32 (clone 93) | Biolegend | Cat# 101320, RRID:AB_1574975 |
| PE anti-human CD45 (clone HI30) | Biolegend | Cat# 304008, RRID:AB_314396 |
| APC anti-human CD3 (clone UCHT1) | Biolegend | Cat# 300412, RRID:AB_314066 |
| FITC anti-human HLA-A,B,C (clone W6/32) | Biolegend | Cat# 311404, RRID:AB_314873 |
| APC/Cy7 anti-human CD235a (clone HI264) | Biolegend | Cat# 349116, RRID:AB_2650978 |
| PE/Cy5 anti-human CD3 (clone HIT3a) | Biolegend | Cat# 300309, RRID:AB_314045 |
| Brilliant Violet 421 anti-human CD279 (PD-1; clone EH12.2H7) | Biolegend | Cat# 329919, RRID:AB_10900818 |
| PE/Cy7 anti-human CD366 (TIM3; clone F38–2E2) | Biolegend | Cat# 345013, RRID:AB_2561719 |
| APC/Cy7 anti-human CD39 (clone A1) | Biolegend | Cat# 328226, RRID:AB_2571981 |
| Alexa Fluor 700 anti-human CD4 (clone OKT4) | Biolegend | Cat# 317425, RRID:AB_571942 |
| Brilliant Violet 650 anti-human CD8a (clone RPA-T8) | Biolegend | Cat# 301041, RRID:AB_11125174 |
| Alexa Fluor 647 anti-mouse CD39 (clone Duha59) | Biolegend | Cat# 143808, RRID:AB_2563978 |
| Brilliant Violet 605 anti-mouse CD3 (clone 145–2C11) | Biolegend | Cat# 100351, RRID:AB_2565842 |
| Pacific Blue anti-mouse CD3 (clone 145-2C11) | Biolegend | Cat# 100334, RRID:AB_2028475 |
| PE/Cy5 anti-mouse CD8a (clone 53–6.7) | Biolegend | Cat# 100710, RRID:AB_312749 |
| FITC anti-mouse CD8a (clone 53–6.7) | Biolegend | Cat# 100705, RRID:AB_312744 |
| APC/Cy7 anti-mouse CD4 (clone GK1.5) | Biolegend | Cat# 100414, RRID:AB_312699 |
| Brilliant Violet 650 anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109836, RRID:AB_2563065 |
| FITC anti-mouse CD45.2 (clone 104) | Biolegend | Cat# 109806, RRID:AB_313443 |
| APC anti-mouse CD90.2 (Thy1.2; clone 53–2.1) | Biolegend | Cat# 140312, RRID:AB_10640728 |
| Pacific Blue anti-mouse CD90.2 (thy1.2; clone 30-H12) | Biolegend | Cat# 105324, RRID:AB_2201291 |
| APC anti-mouse TNFα (clone MP6-XT22) | Biolegend | Cat# 506307, RRID:AB_315428 |
| PE anti-mouse IL-2 (clone JES6–5H4) | Biolegend | Cat# 503807, RRID:AB_315301 |
| FITC anti-mouse IFNγ (clone XMG1.2) | Biolegend | Cat# 505805, RRID:AB_315399 |
| PE anti-mouse Perforin (clone S16009A) | Biolegend | Cat# 154305, RRID:AB_2721638 |
| APC anti-human/mouse Granzyme B (clone QA16A02) | Biolegend | Cat# 372203, RRID:AB_2687027 |
| Anti-IFNγ FITC, human antibody (clone 45–15) | Miltenyi Biotec | Cat# 130-097-936, RRID:AB_2661059 |
| Anti-IL-2-PE, human antibody (clone N7.48 A) | Miltenyi Biotec | Cat# 130-099-391, RRID:AB_2661066 |
| Anti-TNFα-APC, human antibody (clone cA2) | Miltenyi Biotec | Cat# 130-099-197, RRID:AB_2661079 |
| LEAF Purified anti-human CD3 (clone OKT3) | Biolegend | Cat# 317303, RRID:AB_571924 |
| LEAF Purified anti-human CD28 (clone CD28.2) | Biolegend | Cat# 302913, RRID:AB_314315 |
| LEAF Purified anti-mouse CD3 (clone 145-2C11) | Biolegend | Cat# 100314, RRID:AB_312679 |
| LEAF Purified anti-mouse CD28 (clone 37.51) | Biolegend | Cat# 102112, RRID:AB_312877 |
| Purified anti-human CD8a (clone C8/144B) | Biolegend | Cat# 372902, RRID:AB_2650657 |
| TCF1 (C63D9) Rabbit mAb | Cell Signaling Technology | Cat# 2203, RRID:AB_2199302 |
| Beta 2 Microglobulin antibody | Abcam | Cat# ab27588, RRID:AB_471058 |
| Melanoma Triple Cocktail Primary Antibody (clone HMB45+A103+T311) | Ventana (Roche) | Cat# 790–4677 |
| Biological Samples | ||
| Patient samples used in this study are detailed in tables S1, S3 and S5 | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| POM-1 (polyoxometalate-1) 5mg/kg/day (ChemCruz; sc-203205) | Santa Cruz Biotechnology | Cat# sc-203205 |
| Zombie green fixable viability dye | Biolegend | Cat# 423111 |
| Zombie violet fixable viability dye | Biolegend | Cat# 423114 |
| CFSE cell division tracker kit | Biolegend | Cat# 423801 |
| Critical Commercial Assays | ||
| Tumor Dissociation Kit, Human | Miltenyi Biotec | Cat# 130-095-929 |
| Transcription Factor Buffer Set | BD Biosciences | Cat# 562574 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat# 555028 |
| Qubit dsDNA HS assay kit | Invitrogen | Cat# Q32854 |
| High sensitivity DNA kit | Agilent | Cat# 5067–4626 |
| Nextera XT Library Prep kit | illumina | Cat# FC-131–1096 |
| OPAL 4-color multiplex manual IHC kit | PerkinElmer | Cat# NEL800001KT |
| 3D culture chips | AIM Biotech | Cat# DAX-1 |
| CellTiter-Glo Luminescent Cell Viability Assay | Promega | Cat# G7571 |
| Nextera DNA Library prep kit | illumina | Cat# FC-121–1031 |
| Allprep DNA/RNA Mini kit | Qiagen | Cat# 80204 |
| Deposited Data | ||
| Raw data for single cell RNA sequencing and Whole Exome sequencing (this paper) | dbGAP | phs001680.v1.p1 https://www.ncbi.nlm.nih.gov/proiects/gap/cgi-bin/study.cgi?studyid=phs001680.v1.p1 |
| Processed data for single cell RNA | GEO | GSE120575 |
| sequencing (this paper) | http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120575 | |
| Bulk RNA expression data Van Allen cohort | (Van Allen et al., 2015) | dbGap accession number: phs000452.v2.p1 |
| Bulk RNA expression data Riaz cohort | (Riaz et al., 2017) | GEO accession number: GSE91061 |
| Single cell sequencing data Tirosh cohort | (Tirosh et al., 2016) | GEO accession number: GSE72056 |
| Experimental Models: Cell Lines | ||
| B16-F10 | Mikael Pittet | N/A |
| CT26 | Umar Mahamood | N/A |
| Experimental Models: Organisms/Strains | ||
| C57BL/6J | Jackson Laboratory | Cat# JAX:000664; RRID:IMSR_JAX:000664 |
| BALB/cJ | Jackson Laboratory | Cat# JAX:000651; RRID:IMSR_JAX:000651 |
| Oligonucleotides | ||
| RT primer (DNA oligo) | IDT | 5’-AAGCAGTGGTATCAACGCAGAGTACT30VN-3’ |
| TSO primer (RNA oligo with LNA) | Exigon | 5’-AAGCAGTGGTATCAACGCAGAGTACATrGrG+G-3’ |
| ISPCR primer (DNA oligo) | IDT | 5’-AAGCAGTGGTATCAACGCAGAGT-3’ |
| Recombinant DNA | ||
| FUGW | David Baltimore | Cat# 14883; addgene |
| psPAX2 | Didier Trono | Cat# 12260; addgene |
| pMD2.G | Didier Trono | Cat# 12259; addgene |
| Software and Algorithms | ||
| STAR | (Dobin et al., 2013) | https://github.com/alexdobin/STAR |
| RSEM | (Li and Dewey, 2011) | https://github.com/deweylab/RSEM |
| t-SNE | (Maaten and Hinton, 2008) | https://lvdmaaten.github.io/tsne/ |
| Matlab | MATLAB and Statistics Toolbox Release 2016b, The MathWorks, Inc., Natick, Massachusetts, United States. | https://www.mathworks.com/products/matlab.html |
| Monocle v. 2.5.4 | (Qiu et al., 2017) | http://cole-trapnell-lab.github.io/monocle-release/ |
| MixCr | (Bolotin et al., 2015) | https://github.com/milaboratory/mixcr |
| Bowtie 2.2.1 | (Langmead and Salzberg, 2012) | http://bowtie-bio.sourceforge.net/index.shtml |
| Samtools 1.3 | (Li et al., 2009) | http://samtools.sourceforge.net/ |
| ContEst | (Cibulskis et al., 2011) | https://www.broadinstitute.org/node/358411 |
| MuTect | (Cibulskis et al., 2013) | https://www.broadinstitute.org/node/358411 |
| oxoG filtering | (Costello et al., 2013) | |
| NovoAlign | Novocraft | www.novocraft.com |
| Oncotator | (Ramos et al., 2015) | https://portals.broadinstitute.org/oncotator/ |
| GOMER | (Granek and Clarke, 2005) | http://people.duke.edu/~josh/biophysicsweb/GOMER/index.html |
| CIS-BP database | (Weirauch et al., 2014) | http://cisbp.ccbr.utoronto.ca/ |
| HOMER version 4.9 | (Heinz et al., 2010) | http://homer.ucsd.edu/homer/download.html |
| edgeR 3.14.0 | (Robinson et al., 2010) | https://bioconductor.org/packages/release/bioc/html/edgeR.html |
| FlowJo 10.4.2 | FlowJo | https://www.flowjo.com/solutions/flowjo/downloads/previous-versions |
| CellProfiler 2.2.0 | (Carpenter et al., 2006) | http://cellprofiler.org/previous_releases/ |
| GraphPad PRISM 7.0c | GraphPad Software | https://www.graphpad.com/scientific-software/prism/ |
HIGHLIGHTS.
Single cell RNA-seq reveals distinct CD45+ cells associated with clinical outcome
The balance between two CD8+ T cell states is linked with tumor regression
TCF7+CD8+ T-cell frequency in tumor tissue predicts response and better survival
Dual blockade of CD39 with different checkpoint proteins enhances immunity
ACKNOWLEDGEMENTS
We thank the Genomics Platform of the Broad Institute of Harvard and MIT for the whole-exome sequencing performed in this study. All cytometric findings reported here were performed in the MGH Department of Pathology Flow and Image Cytometry Research Core, which obtained support from the NIH Shared Instrumentation program with grants 1S10OD012027-01A1, 1S10OD016372-01, 1S10RR020936-01, and 1S10RR023440-01A1. Additional thanks to Natasha Mathur of the Belfer Center for applied Cancer Sciences and assistance with immunofluorescence staining. The research was supported by grants from Broad Next-10 (N.H.), Cancer Research Institute (Clinic and Laboratory Integration Program, N.H.), Adelson Medical Research Foundation (N.H), Sanofi (N.H.), the David P. Ryan, MD, Chair funded by a gift from Arthur, Sandra, and Sarah Irving (N.H.), the Paul C. Zamecnick, MD, Chair in Oncology at MGH (G.G.), NIH/NHGRI (5U54HG003067; PIs: Stacey Gabriel, Eric S. Lander), NIH/NCI (R01CA208756; PI: N.H.; U54CA224068; PIs: Ryan Corcoran, Keith Flaherty). We also thank the following grants for their support: Institute for Medical Research Israel-Canada (M.S.-F.), Tosteson & Fund for Medical Discovery fellowships (M.S.-F.), the Broad-ISF fellowship (K.Y.) Weizmann award for Women in Science (K.Y.), NIH award number F32AI129249 (J.P.R.), 1T32CA207021-01 (J.H.C.), Robert A. and Renee E. Belfer (R.W.J.), John R. Svenson Fellowship (R.W.J.), NCI-R01 CA190394-01 (D.A.B.), NCI-U01 CA214381 (D.A.B., R.W.J.), and the Expect Miracles Foundation (C.P.P.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
Parts of the study have been submitted as a patent application. N.H. is on the SAB, owns shares in Neon Therapeutics, and consults for IFM Therapeutics. G.G. is receiving research funds from IBM and Pharmacyclics. G.G. is an inventor on multiple patent applications, including one related to MuTect. R.W.J. and D.A.B are inventors on pending patent 15/540,346 and PCT/US2016/012450. R.W.J. is a consultant for Apricity Health, LLC. D.A.B. is a consultant for N of One. C.P.P. is on the SAB of DropWorks and received honoraria from AstraZeneca, Biorad. Z.A.C. is an employee of MedImmune. A.S.-R. consults for Recombinetics and Novartis. J.A.W. is a paid speaker for Imedex, Dava Oncology, Omniprex, Illumina, Gilead, MedImmune, and Bristol Meyers Squibb; is a consultant/SAB member for Roche-Genentech, Novartis, Astra-Zeneca, Glaxo Smith Klein, Bristol Meyers Squibb, and Merck; and receives clinical trial support from Glaxo Smith Klein, Roche-Genentech, Bristol Meyers Squibb, and Novartis. V.G. and J.A.W. are inventors on USPTO patent (PCT/US17/53.717) and report consultancy fees from MicrobiomeDX and ExpertConnect. G.M.B. has a sponsored research agreement with Takeda Oncology. K.T.F. owns equity in Shattuck Labs, Checkmate, X4 Pharmaceuticals; is a consultant/advisory board member for Novartis, Genentech, BMS, Merck, Takeda, Verastem, Checkmate, X4 Pharmaceuticals, Sanofi, Amgen, Incyte, Adaptimmune, Shattuck Labs, Arch Oncology, and Apricity; and receives research support from Novartis, Genentech, Sanofi, and Amgen. R.J.S. is a paid speaker for Array, Novartis, and Genentech; is a paid consultant/advisory board member for Amgen, Array, Novartis, Merck, Compugen, and Syndax; and has received research funding from Amgen and Merck.
REFERENCES
- Allard B, Pommey S, Smyth MJ, and Stagg J (2013). Targeting CD73 enhances the antitumor activity of anti-PD-1 and anti-CTLA-4 mAbs. Clin Cancer Res 19, 5626–5635. [DOI] [PubMed] [Google Scholar]
- Anderson AC, Joller N, and Kuchroo VK (2016). Lag-3, Tim-3, and TIGIT: Co-inhibitory Receptors with Specialized Functions in Immune Regulation. Immunity 44, 989–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, Albright A, Cheng JD, Kang SP, Shankaran V, et al. (2017). IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 127, 2930–2940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastid J, Regairaz A, Bonnefoy N, Dejou C, Giustiniani J, Laheurte C, Cochaud S, Laprevotte E, Funck-Brentano E, Hemon P, et al. (2015). Inhibition of CD39 enzymatic function at the surface of tumor cells alleviates their immunosuppressive activity. Cancer Immunol Res 3, 254–265. [DOI] [PubMed] [Google Scholar]
- Bolotin DA, Poslavsky S, Mitrophanov I, Shugay M, Mamedov IZ, Putintseva EV, and Chudakov DM (2015). MiXCR: software for comprehensive adaptive immunity profiling. Nat Methods 12, 380–381. [DOI] [PubMed] [Google Scholar]
- Callahan MK, Postow MA, and Wolchok JD (2016). Targeting T Cell Co-receptors for Cancer Therapy. Immunity 44, 1069–1078. [DOI] [PubMed] [Google Scholar]
- Canale FP, Ramello MC, Nunez N, Araujo Furlan CL, Bossio SN, Gorosito Serran M, Tosello Boari J, Del Castillo A, Ledesma M, Sedlik C, et al. (2018). CD39 Expression Defines Cell Exhaustion in Tumor-Infiltrating CD8(+) T Cells. Cancer Res 78, 115–128. [DOI] [PubMed] [Google Scholar]
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, et al. (2006). CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PL, Roh W, Reuben A, Cooper ZA, Spencer CN, Prieto PA, Miller JP, Bassett RL, Gopalakrishnan V, Wani K, et al. (2016). Analysis of Immune Signatures in Longitudinal Tumor Samples Yields Insight into Biomarkers of Response and Mechanisms of Resistance to Immune Checkpoint Blockade. Cancer Discov 6, 827–837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, Gabriel S, Meyerson M, Lander ES, and Getz G (2013). Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 31, 213–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cibulskis K, McKenna A, Fennell T, Banks E, DePristo M, and Getz G (2011). ContEst: estimating cross-contamination of human samples in next-generation sequencing data. Bioinformatics 27, 2601–2602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Buenrostro JD, Wu B, Greenside PG, Chan SM, Koenig JL, Snyder MP, Pritchard JK, Kundaje A, Greenleaf WJ, et al. (2016). Lineage-specific and single-cell chromatin accessibility charts human hematopoiesis and leukemia evolution. Nat Genet 48, 1193–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, Fostel JL, Friedrich DC, Perrin D, Dionne D, et al. (2013). Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res 41, e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daud AI, Loo K, Pauli ML, Sanchez-Rodriguez R, Sandoval PM, Taravati K, Tsai K, Nosrati A, Nardo L, Alvarado MD, et al. (2016). Tumor immune profiling predicts response to anti-PD-1 therapy in human melanoma. J Clin Invest 126, 3447–3452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, Dancey J, Arbuck S, Gwyther S, Mooney M, et al. (2009). New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 45, 228–247. [DOI] [PubMed] [Google Scholar]
- Ellrott K, Bailey MH, Saksena G, Covington KR, Kandoth C, Stewart C, Hess J, Ma S, Chiotti KE, McLellan M, et al. (2018). Scalable Open Science Approach for Mutation Calling of Tumor Exomes Using Multiple Genomic Pipelines. Cell Syst 6, 271–281 e277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertes Marraco SA, Neubert NJ, Verdeil G, and Speiser DE (2015). Inhibitory Receptors Beyond T Cell Exhaustion. Front Immunol 6, 310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galon J, Costes A, Sanchez-Cabo F, Kirilovsky A, Mlecnik B, Lagorce-Pages C, Tosolini M, Camus M, Berger A, Wind P, et al. (2006). Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science 313, 1960–1964. [DOI] [PubMed] [Google Scholar]
- Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, Chen T, Roszik J, Bernatchez C, Woodman SE, et al. (2016). Loss of IFN-gamma Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 167, 397–404 e399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Zhong XS, Palmer DC, Ji Y, Hinrichs CS, Yu Z, Wrzesinski C, Boni A, Cassard L, Garvin LM, et al. (2009). Wnt signaling arrests effector T cell differentiation and generates CD8+ memory stem cells. Nat Med 15, 808–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granek JA, and Clarke ND (2005). Explicit equilibrium modeling of transcription-factor binding and gene regulation. Genome Biol 6, R87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta PK, Godec J, Wolski D, Adland E, Yates K, Pauken KE, Cosgrove C, Ledderose C, Junger WG, Robson SC, et al. (2015). CD39 Expression Identifies Terminally Exhausted CD8+ T Cells. PLoS Pathog 11, e1005177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. (2017). T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature 545, 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, Berent-Maoz B, Pang J, Chmielowski B, Cherry G, et al. (2016). Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 165, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurton LV, Singh H, Najjar AM, Switzer KC, Mi T, Maiti S, Olivares S, Rabinovich B, Huls H, Forget MA, et al. (2016). Tethered IL-15 augments antitumor activity and promotes a stem-cell memory subset in tumor-specific T cells. Proc Natl Acad Sci U S A 113, E7788–E7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jenkins RW, Aref AR, Lizotte PH, Ivanova E, Stinson S, Zhou CW, Bowden M, Deng J, Liu H, Miao D, et al. (2018). Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumor Spheroids. Cancer Discov 8, 196–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, Lao CD, Schadendorf D, Dummer R, Smylie M, Rutkowski P, et al. (2015). Combined Nivolumab and Ipilimumab or Monotherapy in Untreated Melanoma. N Engl J Med 373, 23–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, and Genome Project Data Processing, S. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lois C, Hong EJ, Pease S, Brown EJ, and Baltimore D (2002). Germline transmission and tissue-specific expression of transgenes delivered by lentiviral vectors. Science 295, 868–872. [DOI] [PubMed] [Google Scholar]
- Maaten L.v.d., and Hinton G (2008). Visualizing Data using t-SNE. Journal of Machine Learning Research 9 (2008) 9, 2579–2605. [Google Scholar]
- Mahmoud SM, Paish EC, Powe DG, Macmillan RD, Grainge MJ, Lee AH, Ellis IO, and Green AR (2011). Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J Clin Oncol 29, 1949–1955. [DOI] [PubMed] [Google Scholar]
- Mami-Chouaib F, Blanc C, Corgnac S, Hans S, Malenica I, Granier C, Tihy I, and Tartour E (2018). Resident memory T cells, critical components in tumor immunology. J Immunother Cancer 6, 87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. (2017). Allele-Specific HLA Loss and Immune Escape in Lung Cancer Evolution. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, and Bejerano G (2010). GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol 28, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paley MA, Kroy DC, Odorizzi PM, Johnnidis JB, Dolfi DV, Barnett BE, Bikoff EK, Robertson EJ, Lauer GM, Reiner SL, et al. (2012). Progenitor and terminal subsets of CD8+ T cells cooperate to contain chronic viral infection. Science 338, 1220–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardoll DM (2012). The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 12, 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, Scott AC, Viale A, Lauer P, Merghoub T, et al. (2017). Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 545, 452–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Bjorklund AK, Faridani OR, Sagasser S, Winberg G, and Sandberg R (2013). Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10, 1096–1098. [DOI] [PubMed] [Google Scholar]
- Prat A, Navarro A, Pare L, Reguart N, Galvan P, Pascual T, Martinez A, Nuciforo P, Comerma L, Alos L, et al. (2017). Immune-Related Gene Expression Profiling After PD-1 Blockade in Non-Small Cell Lung Carcinoma, Head and Neck Squamous Cell Carcinoma, and Melanoma. Cancer Res 77, 3540–3550. [DOI] [PubMed] [Google Scholar]
- Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, and Trapnell C (2017). Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 14, 979–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos AH, Lichtenstein L, Gupta M, Lawrence MS, Pugh TJ, Saksena G, Meyerson M, and Getz G (2015). Oncotator: cancer variant annotation tool. Hum Mutat 36, E2423–2429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martin-Algarra S, Mandal R, Sharfman WH, et al. (2017). Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 171, 934–949 e915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, et al. (2015). Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science 348, 124–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, Daud A, Carlino MS, McNeil C, Lotem M, et al. (2015). Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 372, 2521–2532. [DOI] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sade-Feldman M, Jiao YJ, Chen JH, Rooney MS, Barzily-Rokni M, Eliane JP, Bjorgaard SL, Hammond MR, Vitzthum H, Blackmon SM, et al. (2017). Resistance to checkpoint blockade therapy through inactivation of antigen presentation. Nat Commun 8, 1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen DR, Kaminski J, Barnitz RA, Kurachi M, Gerdemann U, Yates KB, Tsao HW, Godec J, LaFleur MW, Brown FD, et al. (2016). The epigenetic landscape of T cell exhaustion. Science 354, 1165–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Shen Y, Wen S, Yamada S, Jungbluth AA, Gnjatic S, Bajorin DF, Reuter VE, Herr H, Old LJ, et al. (2007). CD8 tumor-infiltrating lymphocytes are predictive of survival in muscle-invasive urothelial carcinoma. Proc Natl Acad Sci U S A 104, 3967–3972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shum T, Omer B, Tashiro H, Kruse RL, Wagner DL, Parikh K, Yi Z, Sauer T, Liu D, Parihar R, et al. (2017). Constitutive Signaling from an Engineered IL7 Receptor Promotes Durable Tumor Elimination by Tumor-Redirected T Cells. Cancer Discov 7, 1238–1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, Walsh LA, Postow MA, Wong P, Ho TS, et al. (2014). Genetic basis for clinical response to CTLA-4 blockade in melanoma. N Engl J Med 371, 2189–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speiser DE, Ho PC, and Verdeil G (2016). Regulatory circuits of T cell function in cancer. Nat Rev Immunol 16, 599–611. [DOI] [PubMed] [Google Scholar]
- Spitzer MH, Carmi Y, Reticker-Flynn NE, Kwek SS, Madhireddy D, Martins MM, Gherardini PF, Prestwood TR, Chabon J, Bendall SC, et al. (2017). Systemic Immunity Is Required for Effective Cancer Immunotherapy. Cell 168, 487–502 e415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wu Y, Gao W, Enjyoji K, Csizmadia E, Muller CE, Murakami T, and Robson SC (2010). CD39/ENTPD1 expression by CD4+Foxp3+ regulatory T cells promotes hepatic metastatic tumor growth in mice. Gastroenterology 139, 1030–1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. (2016). Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science 352, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al. (2014). PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 515, 568–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T Cell Factor 1-Expressing Memory-like CD8(+) T Cells Sustain the Immune Response to Chronic Viral Infections. Immunity 45, 415–427. [DOI] [PubMed] [Google Scholar]
- Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, Sucker A, Hillen U, Foppen MHG, Goldinger SM, et al. (2015). Genomic correlates of response to CTLA-4 blockade in metastatic melanoma. Science 350, 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villani AC, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, Griesbeck M, Butler A, Zheng S, Lazo S, et al. (2017). Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waugh KA, Leach SM, Moore BL, Bruno TC, Buhrman JD, and Slansky JE (2016). Molecular Profile of Tumor-Specific CD8+ T Cell Hypofunction in a Transplantable Murine Cancer Model. J Immunol 197, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weirauch MT, Yang A, Albu M, Cote AG, Montenegro-Montero A, Drewe P, Najafabadi HS, Lambert SA, Mann I, Cook K, et al. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell 158, 1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wherry EJ, Ha SJ, Kaech SM, Haining WN, Sarkar S, Kalia V, Subramaniam S, Blattman JN, Barber DL, and Ahmed R (2007). Molecular signature of CD8+ T cell exhaustion during chronic viral infection. Immunity 27, 670–684. [DOI] [PubMed] [Google Scholar]
- Young A, Mittal D, Stagg J, and Smyth MJ (2014). Targeting cancer-derived adenosine: new therapeutic approaches. Cancer Discov 4, 879–888. [DOI] [PubMed] [Google Scholar]
- Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, Torrejon DY, Abril-Rodriguez G, Sandoval S, Barthly L, et al. (2016). Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. N Engl J Med 375, 819–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, and Xue HH (2010). Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33, 229–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Comparing the composition of known cell types to clinical outcomes and checkpoint therapy, Related to Figure 1. A. tSNE plot of all CD45+ cells collected in this study. Cells are colored by cell type on the basis of pre-defined markers (Table S1). Pie chart summarizes the corresponding percentage of known cell types across all CD45+ cells collected in this cohort. B. A comparison between the supervised classification of single cells to cell types (right color code) to the unsupervised clustering of immune cells identified by k-means clustering (left color code). For each one of the 11 unsupervised clusters identified, the percentage of cell types as defined by the supervised analysis is shown.
Figure S4. Characterization of CD39+CD8+ cells in melanoma patients and combined inhibition of CD39 with anti-PD-1 or anti-PD-1/CTLA4 improves survival and reduces tumor growth, Related to Figure 5. A. Gene expression level distribution (log2(TPM+1)) of CD39 (ENTPD1, left) and TIM3 (HAVCR2, right) in the six CD8+ T cell clusters is shown. Each dot represents an individual cell. 1-sided Wilcoxon rank-sum p-value is shown. B. Gating strategy that was used to isolate CD39+TIM3+ and CD39−TIM3−, CD8+ T cells from melanoma patients. C. Flow-cytometry quantification of PD-1 and TIM3 in CD39+, CD39− (CD8+ T cells), and CD39 expression in PD-1+, PD-1−, TIM3+ and TIM3− (CD8+ T cells) in metastatic melanoma patients is shown. Data were combined from 2 replicate experiments. Unpaired-student’s t-test is shown; ns - not significant. D. Schematic representation for the preparation, isolation and live/dead analysis of CT26GFP+ MDOTS. E. Individual tumor volumes of B16-F10 implants in the untreated (IgG control), anti-TIM3, CD39i, and anti-TIM3+CD39i groups is shown. F. Box plots showing the kinetics of tumor growth between the different groups of mice on days +4, +7, +11, +14 and +18 post tumor transplantation. P-value was determined by unpaired-student’s t-test. G. A schematic summary of the therapy regimen used in the transplantable B16-F10 mouse model. H-I. Box plots showing the kinetics of tumor growth between the different groups of mice on days +4, +7, +11, +14, +18 and +21 post tumor transplantation for the CD39i+PD-1 (H) and CD39i+PD-1/CTLA4 (I) experiments. P-value was determined by unpaired-student’s t-test. J. Individual tumor volumes of B16-F10 implants in the untreated (IgG control), CD39i, anti-PD-1, anti-PD-1/CTLA4, anti-PD-1+CD39i and anti-PD-1/CTLA4+CD39i groups is shown. To simplify the presentation, we split the two different arms of therapy and showed them separately. Data are represented as mean±SEM. For in vivo mouse tumor models one of two independent experiments is shown.
Figure S5. CD8+ T cells are required for the success of in vivo CD39 inhibition which modulates CD8+ TILs and tumor cells, Related to Figure 5. A. A schematic summary of the depletion experiments used in the transplantable B16-F10 mouse model (n=5 per group). B. Representative flow cytometric plots of surface staining for CD4+CD3+ (left) and CD8+CD3+ (right) in depleted and non-depleted groups on day +14 are shown. C. Box plots showing the kinetics of tumor growth between the different groups of mice on days +4, +7, +11, +14, +18 and +21 post tumor transplantation. D. Survival at day 35 of tumor-bearing mice for all 4 groups. Log-rank P-value is shown. E. Individual tumor volumes of B16-F10 implants in the untreated (IgG control), CD39i, CD39i+anti-CD4 and CD39i+anti-CD8 groups is shown. F. A schematic summary for the in-vivo POM-1 experiments used in the transplantable B16-F10GFP+ mouse model (n=5 per group). Representative flow cytometry plot on the left shows the gating strategy used to characterize and isolate GFP+ B16-F10 tumor cells. G. The percentage of CD45+CD3+CD8+ cells expressing granzyme-B (left) and perforin-1 (right) in POM-1 treated and untreated groups. H. Representative flow cytometric plots (left) of intracellular staining for IL-2, IFNγ and TNFα in CD45+CD3+CD8+cells. Flow cytometry quantification of cytokine-producing cells obtained from the POM-1 treated and untreated groups is shown (right). I. Representative histograms (left) showing the CFSE profiles of Thy1.2+CD8+ gated cells after 72h stimulation with anti-CD3/CD28 antibodies from POM-1 treated and untreated animals. Bar plot (right) summarizes the percentage of proliferating cells analyzed by flow cytometry in each group. J. The percentage of CD45+CD3+CD8+ cells (left) or GFP+B16-F10 tumor cells (right) expressing CD39 in POM-1 treated and untreated groups. K. Intra-ATP levels in mixed tumor cell suspensions containing CD45+ and CD45− cells (left) or isolated B16-F10GFP+ cells (right) from POM-1 treated and untreated mice, with n=3 replicates for each cell suspension per experiment that were measured on day +14. An equivalent number of cells from each mouse (1×105) was added to each well prior to ATP measurement. Data are represented as mean±SEM. For A-D one of two independent experiments is shown. For G-K combined data from 2 replicates is shown. P-value was determined by unpaired-student’s t-test, ns - not significant.
Figure S6. CD39+TIM3+ and CD39−TIM3− cells have a distinctive epigenetic landscape, Related to Figure 6. A. Heatmap of averaged scaled expression values of discriminative transcription factors for non-sorted CD39+TIM3+ and CD39−TIM3− cells as defined by single cell RNA expression. B. Heatmap of averaged scaled expression values of discriminative transcription factors for CD8_B and CD8_G cells as in Figure 2. C. ATAC-seq traces for open chromatin regions (OCRs) near selected genes in CD39-TIM3− cells (blue) and CD39+TIM3+ cells (orange) in all 5 patients is shown. D. Transcription factor (TF) enrichment graphs for BATF and TCF7 in CD39+TIM3+ and CD39−TIM3− sorted cells are shown. Each graph shows the enrichment peaks relative to background (x-axis). Black bars indicate CD39+TIM3+ (top) or CD39−TIM3− (bottom) peaks, while white bars indicate background peaks. Motif enrichment was calculated using the minimum hypergeometric (minHG) test. E. Venn diagrams showing the distribution of ATAC-seq OCRs in DP (red), PD-1highCD8+ (blue), DN (green) and CM (orange).
Figure S7. Correlation of T cell markers in bulk RNA-seq data, Related to STAR Methods. A,E. Pairwise Spearman correlation between different immune markers in the Van Allen (A) and Riaz (E) cohorts. B,F. Spearman correlation between the expression levels of the different immune markers shown in the table and CD3 in the Van Allen (B) and Riaz (F) cohorts. C,G. Scatter plot showing the correlation between the good signatures based on CD8_G marker genes and CD3 expression levels in the Van Allen (C) and Riaz (G) cohorts. D,H. Scatter plot showing the correlation between the bad signatures based on CD8_B marker genes and CD3 expression levels in the Van Allen (D) and Riaz (H) cohorts.
Figure S8. Summary of variance and clustering robustness analysis, Related to STAR Methods. A. Variance of each gene vs. the fraction of cells expressing each gene (log2(TPM+1)>0). Left panel: genes expressed in more than 10% of the cells and less than 90% are colored in red. Right panel: genes with variance 6 are colored in red. As the set of genes expressed in less than 10% of the cells are of less interest for clustering analysis, we set as a minimal threshold the maximal variance observed in this group of genes, as indicated by the black arrow. B. Variance explained by each k-means solution ranging from k=3,…,15, when applied to all analyzed single cells. C. A box plot summarizing the distribution of robustness scores for each k=3, …,15. D. Variance explained by each k-means solution ranging from k=2, …,13, when applied to all analyzed CD8+ T cells. E. A box plot summarizing the distribution of robustness scores for each k=2, …,13. F,G. Clustering of CD8_B and CD8_G, separately, into two (F) or three (G) clusters. Hierarchical structure is shown where CD8_B and CD8_G are each split into 2 and 3 clusters, respectively.
File S1. Analysis pipeline for CellProfiler, Related to Figure 3.
Table S1. A summary of data related to all single cells analysis, Related to Figure 1.
Table S2. A summary of data related to CD8+ T cells analysis and 2 clusters solution, Related to Figures 2 and S2.
Table S3. A summary of data related to IF analysis, Related to Figure 3.
Table S4. A summary of data related to CD8+ T cells analysis and 6 clusters solution, Related to Figure 4.
Table S5. A summary of data related to sorted (CD39+TIM3+ and CD39−TIM3−) cells and ATAC-seq analysis, Related to Figures 5,6 and S7.
Table S6. A summary of identified TCRs and Methods material, Related to Figure 7 and STAR Methods.
Figure S2. Annotating CD8_G and CD8_B clusters to the CD45+ immune cell population clusters, Related to Figure 2. A. tSNE plot of all CD45+ clusters (n=11) collected in this study (left) is shown. Cells are colored based on 11 clusters identified by k-means clustering analysis. Right tSNE plot shows the distribution of CD8+ T cells that are classified as CD8_G (blue) and CD8_B (orange) in relation to all CD45+ cells analyzed in this study. B-C. For each sample, the percentage of cells found in CD8_G and CD8_B (out of all CD8+ T cells) in responder lesions (B) and non-responder lesions (C) is shown. * symbol marks samples with defects in antigen presentation and the IFNγ pathway as inferred from WES, IHC and flow-cytometry analysis. P# indicates patient number as described in Table S1. D. Representative immunohistochemistry staining (1 out of 3) of sections from patient #3 with homozygous mutations in B2M (as inferred from WES). Sections were stained with an antibody cocktail for melanoma cells (mel.cocktail) using anti-melanosome (HMB45), anti-MART-1/melan A and anti-Tyrosinase, to discern melanoma cells from normal cells; or with an antibody specific for B2M. Original Magnification X100. E. Flow-cytometry plot (left) and histogram (right), showing the expression of HLA-A,B,C in immune (blue) and tumor cells (orange) in patient #15. F. Receiver operating characteristic (ROC) analysis was constructed to evaluate the prognostic power of the ratio between CD8_G/CD8_B as shown in Figure 2D between responder and non-responder lesions. The area under the ROC curve (AUC) was used to quantify response prediction, and one-sided Wilcoxon test was used to assess significance of the AUC results. G. Heatmap describing scaled expression values of discriminative gene sets out of top 34 markers in CD8_B and CD8_G (Table S2), between responder and non-responder lesions in CD8+ T cells. Significant marker genes are shown for each group (Table S2). Top blue and orange bar depicts a cell-level association to CD8_G and CD8_B, respectively. H. tSNE plot of all CD8+ T cells found in the Tirosh et al. data, before (left panel) and after (right panel) filtering, as identified by k-means clustering, and correspond to CD8_G and CD8_B identified in this study (Table S2). Clustering of all CD8+ T-cells in this cohort (n=1450) identified 3 clusters, 2 confirming the presence of our CD8_G and CD8_B cellular states and a third one that was contaminated with melanoma markers (left). After removal of all CD8+ T-cells from this cohort, having either a melanoma (MITF, S100B, TYR, PMEL, and MLANA) NK\NKT cells (NCAM1 and NCR1), regulatory T-cells (FOXP3), and B cells (MS4A1 and CD79a) markers, analysis was performed on the remaining cells (n=260, right). I. Heatmap showing scaled expression values of discriminating genes for the clusters defined in (H). A list of representative genes is shown for each cluster next to the right margin color bars.
Figure S3. The distribution of exhaustion and memory markers across the six clusters and summary of trajectory analysis, Related to Figure 4. A. tSNE plot colored such that single-cells with a high expression level (log2(TPM+1)>2) of ENTPD1, HAVCR2, PDCD1, LAG3, TIGIT, TCF7, TNF, and IL7R are colored in red, and those with a low expression level (log2(TPM+1)<2) are colored in gray. Bar plots (to the right) summarize the corresponding percentages of each gene in the 6 identified clusters. B. tSNE plot of all CD8+ T cells profiled with coloring of CD8_5 cells associated with baseline (blue, left) or post-treatment lesions (green, right). C. tSNE plot of all CD8+ T cells profiled with coloring of CD8_5 cells associated with responder (blue, left) or non-responder lesions (green, right). D. Violin plots showing organization of cells corresponding to the six CD8 clusters by pseudotime as inferred by Monocle. E. Cell expression profiles in a two-dimensional independent space for each cluster (CD8_1 to 6) is shown. Solid black line indicates the main diameter path of the minimum spanning tree (MST) and provides the backbone of Monocle’s pseudotime ordering of the cells. Each dot represents an individual cell colored by cluster.
Data Availability Statement
Data availability.
Raw sequencing data (single cell RNAseq, WES and ATACseq) from this study have been deposited in dbGAP database (https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs001680.v1.p1) under accession code phs001680.v1.p1. Processed Single cell RNAseq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE120575 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120575). The Van Allen (Van Allen et al., 2015) bulk RNA dataset used in this study is available in dbGAP database under accession number phs000452.v2.p1. Data from Riaz (Riaz et al., 2017) and Tirosh (Tirosh et al., 2016) studies used in this paper are available in GEO with the accession number GSE91061 and GSE72056 for the Riaz and Tirosh datasets respectively. ATAC-seq data from the Schietinger group (Philip et al., 2017), used for the identification of unique peaks in PD-1high and central memory (CM) cells is available in GEO with the accession number GSE89308.
Raw sequencing data (single cell RNAseq, WES and ATACseq) from this study have been deposited in dbGAP database (https://www.ncbi.nlm.nih.gov/projects/gap/cgibin/study.cgi?study_id=phs001680.v1.p1) under accession code phs001680.v1.p1. Processed Single cell RNAseq data discussed in this publication have been deposited in NCBI’s Gene Expression Omnibus and are accessible through GEO Series accession number GSE120575 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE120575). The Van Allen (Van Allen et al., 2015) bulk RNA dataset used in this study is available in dbGAP database under accession number phs000452.v2.p1. Data from Riaz (Riaz et al., 2017) and Tirosh (Tirosh et al., 2016) studies used in this paper are available in GEO with the accession number GSE91061 and GSE72056 for the Riaz and Tirosh datasets respectively. ATAC-seq data from the Schietinger group (Philip et al., 2017), used for the identification of unique peaks in PD-1high and central memory (CM) cells is available in GEO with the accession number GSE89308.