

Abstract
A wide range of noncanonical amino acids (ncAAs) can be incorporated into proteins in living cells by using engineered aminoacyl-tRNA synthetase/tRNA pairs. However, most engineered tRNA synthetases are polyspecific; that is, they can recognize multiple rather than one ncAA. Polyspecificity of engineered tRNA synthetases imposes a limit to the use of genetic code expansion because it prevents specific incorporation of a desired ncAA when multiple ncAAs are present in the growth media. In this study, we employed directed evolution to improve substrate selectivity of polyspecific tRNA synthetases by developing substrate-selective readouts for flow-cytometry-based screening with the simultaneous presence of multiple ncAAs. We applied this method to improve the selectivity of two commonly used tRNA synthetases, p-cyano-l-phenylalanyl aminoacyl-tRNA synthetase (pCNFRS) and Nε-acetyl-lysyl aminoacyl-tRNA synthetase (AcKRS), with broad specificity. Evolved pCNFRS and AcKRS variants exhibit significantly improved selectivity for ncAAs p-azido-l-phenylalanine (pAzF) and m-iodo-l-phenylalanine (mIF), respectively. To demonstrate the utility of our approach, we used the newly evolved tRNA synthetase variant to produce highly pure proteins containing the ncAA mIF, in the presence of multiple ncAAs present in the growth media. In summary, our new approach opens up a new avenue for engineering the next generation of tRNA synthetases with improved selectivity toward a desired ncAA.
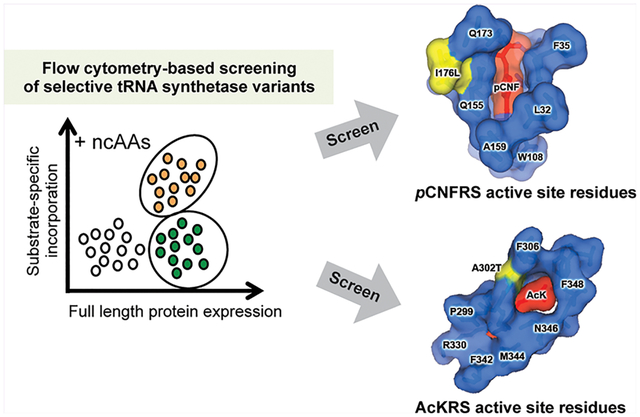
Graphical Abstract
Genetic code expansion (GCE) is a powerful technology to study and manipulate protein function. GCE enables site-specific incorporation of ncAAs into a protein of intere living cells. Some examples of useful functional groups that can be introduced via ncAAs include post-translational modifications, bioorthogonal functional groups for attachment of small affinity tags or fluorophores, photocrosslinking amino acids for capturing transient protein—protein interactions, photocaged amino acids for controlling protein function, and fluorescent groups for live imaging.1–5
Genetic encoding of ncAAs in cells requires orthogonal aminoacyl-tRNA synthetases (aaRSs), which are engineered to acylate ncAAs to an engineered orthogonal tRNA. Extensive engineering efforts of the orthogonal aaRSs over the past two decades have enabled genetic encoding of more than 200 ncAAs.6,7
However, most engineered aaRSs have one significant drawback; they lack high substrate specificity toward a particular ncAA and instead recognize a num ber of ncAAs.8–10 For example, the engineered aaRS pCNFRS derived from Methanocaldococcus jannaschii (Mj) tyrosyl-tRNA synthetase can recognize at least 18 ncAAs, including p-azido-l-phenylalanine (pAzF), p-cyano-l-phenylalanine (pCNF), and other para substituted phenylalanine analogues.9 Hence, when multiple ncAAs are present in the growth media, selective they may react with the same engineered aaRS, which prevents incorporation of the desired ncAA at the specified site of a protein of interest. For this reason, simultaneous incorporation of distinct ncAAs so far has been limited to structurally distinct substrates that can be selectively charged by different aaRS/tRNA pairs.11,12 Thus, polyspecificity of engineered aaRSs remains one major obstacle for genetic encoding of multiple ncAAs, especially when ncAAs represent chemical derivatives of the same amino acid.
Here, we have addressed the problem of polyspecificity by developing a new directed evolution strategy to evolve aaRSs with markedly improved selectivity toward a particular ncAA. This method is based on random mutagenesis of aaRSs, followed by flow cytometry-based screening using substrate- selective readouts tailored to discriminate between two structurally similar ncAAs. Unlike conventional methods of directed evolution of aaRSs, which are based on stop codon readthrough in a reporter gene,13 our approach is based on specific recognition of a desired ncAA in a pool of structurally similar ncAAs. Using our strategy, we evolved aaRSs with improved selectivity, including a pAzF-selective variant of pCNFRS and an mIF-selective variant of AcKRS. We finally demonstrated that this is a facile method for engineering substrate-selective aaRSs that can allow selective incorporation of a desired ncAA into proteins despite the presence of multiple ncAAs in the growth media. Overall, our work provides a general approach to evolve engineered aaRSs with markedly improved substrate selectivity.
To improve the substrate selectivity of aaRSs, we proposed a new directed evolution strategy using two consecutive rounds of positive selection and screening in the simultaneous presence of multiple ncAAs in the experimental system. The first round involves chloramphenicol selection to isolate active tRNA synthetase variants using Escherichia coli cells containing the chloramphenicol acetyltransferase (CAT) gene with a stop codon at a permissive site (CAT-112TAG) and cognate tRNACUA. This is followed by a second round of positive screening to isolate ncAA-selective tRNA synthetase variants using E. coli cells containing superfolder GFP (sfGFP) with a stop codon at a permissive site (sfGFP-2TAG) and cognate tRNACUA. In order to do so, the assay readout is tailored to discriminate between two ncAAs. At least 20 ncAAs bearing strained alkenes or alkynes have been genetically encoded into proteins to facilitate chemoselective modifications via click chemistry.7 Proteins containing pAzF can be chemoselectively modified with an alkyne-probe conjugate via Strain Promoted Azide-Alkyne Click (SPAAC) reaction,14 whereas proteins containing non-click-reactive ncAAs such as pCNF will remain unmodified (Figure 1A and B).
Figure 1.

Labeling of pAzF-containing sfGFP reporter protein allows selection of tRNA synthetase variants with improved selectivity toward pAzF. (A) Scheme for discrimination between incorporation of pAzF and pCNF in sfGFP in live E. coli cells by flow cytometry. pCNFRS incorporates either pAzF or pCNF into sfGFP. pAzF-containing sfGFP chemoselectively reacts with DBCO-derivatized Cy5 via Strain Promoted Azide-Alkyne Click (SPAAC) reaction. Thus, cells containing pAzF-incorporated sfGFP will be positive for Cy5 and GFP, whereas cells containing pCNF-incorporated sfGFP will be positive for GFP only on flow cytometry. (B) Scheme for directed evolution of substrate-selective tRNA synthetase variants from the parental enzyme pCNFRS. tRNA synthetase variant library was prepared with error-prone PCR (epPCR). First, the variant library was transformed into DH10B cells expressing chloramphenicol acetyltransferase (CAT) for positive selection of functional variants in the presence of pAzF (1 mM) and pCNF (1 mM). Then, the enriched library was transformed into the BL21 (DE3) strain of E. coli cells expressing sfGFP-2TAG for screening substrate-selective variants in the presence of pAzF (1 mM) and pCNF (1 mM). Cells were treated with DBCO-derivatized Cy5 (1 μM) and FACS sorted for Cy5+/GFP+ (potentially enriched in pAzF-selective variants) and Cy5−/GFP+ (potentially enriched in pCNF- selective variants). (C) In-gel fluorescence reveals specific labeling of pAzF-containing sfGFP. In-gel fluorescence image of Cy5 labeling (top), Western blotting with anti-His6 tag for full-length sfGFP (middle), Coomassie blue staining of gel (bottom). Cells expressing pCNFRS/tRNACUATyr and sfGFP-2TAG were grown in LB media containing the indicated ncAAs (1 mM). Cell lysates were incubated with DBCO-Cy5 (5 μM) and resolved on SDS-PAGE. (D) Flow cytometry reveals specific labeling of cells that express pAzF-containing sfGFP. Cells expressing pCNFRS/tRNACUATyr and sfGFP-2TAG were grown in LB media containing pAzF (1 mM) or pCNF (5 mM) and labeled with DBCO-Cy5 (1μM). Representative fluorescence density plots showing live E. coli cells expressing sfGFP with no ncAA (left), pCNF (middle), or pAzF (right) incorporation. At least 2 × 104 cells were analyzed and presented on each FACS plot.
To test the validity of this strategy, we endeavored to improve substrate selectivity of MjTyrRS-derived pCNFRS.9 We chose pCNFRS because this enzyme is highly polyspecific and can recognize at least 18 ncAAs, including pAzF, pCNF, and other para substituted phenylalanine analogues.9
We first asked if we can observe pCNFRS polyspecificity in our experimental system based on a stop codon readthrough assay with sfGFP as a reporter protein. In our assay, the sfGFP gene carries an amber mutation at Ser2 (sfGFP-2TAG). We selected this mutation because previous studies showed that Ser2 replacement by other amino acids does not affect sfGFP stability and fluorescence.15 To measure ncAAs incorporation efficiency, we used E. coli cells [BL21(DE3)] containing pCNFRS, its cognate tRNACUA, and the reporter gene sfGFP-2TAG-His6. Consistent with previous studies,9 we observed that pCNFRS allowed efficient incorporation of multiple phenylalanine analogues including pAzF, pCNF, l −4,4′-biphenylalanine (bipA), p-acetyl-l-phenylalanine (pAcF), and p-nitro-l-phenylalanine (pNO2F), as indicated by expression of full-length sfGFP (Figure 1C, Supporting Information Figure 1). We also observed production of low but detectable levels of full-length sfGFP in the absence of ncAAs, suggesting the mischarging of tRNACUATyr with canonical amino acids (Figure 1C). This observation is consistent with previous findings that polyspecific MjTyrRS-derived variants recognize canonical amino acids such as phenylalanine and tyrosine in the absence of ncAAs.10,16,17 Thus, we could observe that pCNFRS acts as a polyspecific enzyme in our experimental system.
We next asked if we can use click chemistry to discriminate between sfGFP containing pAzF (a click-reactive amino acid) and sfGFP containing non-click-reactive ncAAs. For this purpose, we labeled sfGFP with an alkyne-conjugated fluorophore via SPAAC reaction14 (Figure 1C). We prepared E. coli lysates containing sfGFP that is produced in the presence of various ncAAs and then treated these lysates with SPAAC reagent dibenzocyclooctyl (DBCO)-derivatized Cy5. We found that incorporation of pAzF, but not the other ncAAs, including pCNF, bipA, pAcF, and pNO2F, permitted chemical modification of sfGFP with DBCO-Cy5 (Figure 1C). Thus, we confirmed that pAzF incorporation in the reporter protein could be discriminated from incorporation of non-click-reactive ncAAs.
We next tested if SPAAC labeling of pAzF-containing sfGFP can be performed directly in live E. coli cells (Figure 1D). By treating E. coli cells with DBCO-Cy5, we found that E. coli grown in pAzF-containing media displayed increased fluorescence in both GFP and Cy5, whereas E. coli grown in p CNF-containing media displayed increased fluorescence in GFP only (Figure 1D). These data showed that discrimination between incorporation of click-reactive pAzF and non-click-reactive pCNF is possible in live cells.
Having established a scheme to detect pAzF incorporation into sfGFP by SPAAC labeling in live cells, we then sought to use this scheme to create new pCNFRS variants with improved substrate selectivity. For this reason, we generated novel pCNFRS variant libraries by using error-prone PCR (epPCR) to introduce random mutations in the pCNFRS-encoding sequence (Figure 1B). We chose epPCR over the more conventional mutagenesis of active site residues because recent studies indicate that substrate selectivity of aaRSs could be altered not only by mutations in the catalytic site but also by mutations elsewhere in the protein structure.10,17 By using epPCR, we generated variants with an average of five base mutations per sequence of theoretical library size of 4.6 × 107(data not shown).18
We anticipated that some of the pCNFRS variants generated by random mutagenesis could be enzymatically impaired. Therefore, we first wanted to eliminate the nonfunctional pCNFRS variants from our library in order to reduce the library and simplify subsequent screening. For this purpose, the pCNFRS variant library was first transformed into E. coli cells (DH10B) containing CAT-112TAG and cognate tRNACUA. The transformed E. coli cells were grown in LB media containing pAzF and pCNF. To eliminate nonfunctional pCNFRS variants, positive selection was performed with chloramphenicol, and pCNFRS-encoding plasmids were isolated from the surviving clones (Figure 1B). This allowed us to reduce the library of pCNFRS variants and potentially enrich this library with only functional pCNFRS variants, as suggested by stop codon readthrough in the CAT gene.
We then used the reduced library of the pCNFRS library to screen for pCNFRS variants with improved substrate selectivity. For this purpose, the library was transformed into E. coli cells [BL21(DE3)] expressing sfGFP-2TAG and cognate tRNACUA. The transformed cells were grown in LB media containing pAzF and pCNF and treated with SPAAC reagent (DBCO-Cy5) to detect incorporation of pAzF in the reporter protein. SPAAC-treated cells were sorted for Cy5+/GFP+ cells (potentially enriched in pAzF-selective tRNA synthetase variants) and Cy5−/GFP+ cells (potentially enriched in pCNF-selective tRNA synthetase variants) using a flow cytometer (Figure 1B). The sorted cells were plated out on LB agar plates to isolate single clones and were assessed for their substrate selectivity toward pAzF and pCNF by using an sfGFP readthrough assay.
In total, we characterized 10 variants from the Cy5+/GFP+ fraction and 20 variants from the Cy5−/G FP+ fraction by using an sfGFP readthrough assay with individual ncAAs. Of these, we identified five variants that exhibited significant improvements in selectivity for pAzF (Supporting Information Figure 2). Unexpectedly, none of the variants showed preference toward pCNF, suggesting that our current conditions were not optimal for selection of pCNF-selective variants. Multiple rounds of mutagenesis and selection are typically needed to accumulate enough beneficial mutations for generating enzyme variants with desirable activities.19,20 It is possible that a single round of mutagenesis and screening was not sufficient to improve selectivity for p CNF. We further analyzed the most pAzF-selective variant (which we named pAzFRS-1) and found that this variant showed 2- to 3-fold less incorporation for pCNF compared to parental enzyme pCNFRS (Figure 2A). Also, pAzFRS-1 showed a 2-fold reduction in misincorporation in the absence of any ncAAs, as quantified in the sfGFP readthrough assay (Figure 2A). Western blotting, which is a more sensitive assay for detection of the full-length sfGFP protein, confirmed selective use of pAzF by pAzFRS-1 with no detectable misincorporation in the absence of ncAAs (Figure 2B). Thus, our approach enabled evolution of a highly selective tRNA synthetase variant from a polyspecific tRNA synthetase.
Figure 2.

Directed evolution of pAzF-selective aminoacyl tRNA synthetase. (A) The variant pAzFRS-1 has reduced misincorporation levels of canonical amino acids and is more selective for pAzF than parental enzyme pCNFRS. Cells expressing pCNFRS or pAzFRS-1 with its cognate tRNACUATyr and sfGFP-2TAG were grown in LB media containing varying concentrations of indicated ncAAs. Incorporation efficiencies were evaluated by monitoring sfGFP fluorescence. Normalized fluorescence intensities were calculated from fluorescence readings at 24 h time points divided by absorbance at 600 nm. Mean values and standard deviations were calculated from three replicates. (B) Western blotting with anti-His6 tag for full-length sfGFP (top), Ponceau S staining of membrane (bottom). Cells expressing pCNFRS or pAzFRS-1 with its cognate tRNACUATyr and sfGFP-2TAG were grown in LB media containing the indicated ncAAs (1 mM). (C) Four amino acid substitutions (K90Q, I176L, R257W, E272V) were found in pAzFRS-1. The mutated residues are indicated in yellow in the pCNFRS/pCNF complex crystal structure (left). Shown is the close-up view of the active site residues with the mutation I176L (yellow) and pCNF substrate (red) (right). PDB ID: 3QE4. (D) The mutation I176L is largely responsible for pAzF selectivity in pAzFRS-1. Cells expressing pAzFRS-1, parental enzyme pCNFRS, or pCNFRS with single mutations K90Q/I176L/R257W/E272V, its cognate tRNACUATyr, and sfGFP-2TAG were grown in LB media containing pAzF (1 mM) or pCNF (1 mM). Incorporation efficiencies were evaluated by monitoring sfGFP fluorescence. Normalized fluorescence intensities were calculated from fluorescence readings at 24 h time points divided by absorbance at 600 nm. Mean values and standard deviations were calculated from three replicates.
The parental enzyme pCNFRS is highly polyspecific.9 To determine the activity of evolved enzyme pAzFRS-1 for ncAAs that were originally substrates for pCNFRS, we tested pAzFRS-1 against seven ncAAs, which are not used during the selection. pAzFRS-1 showed reduced incorporation for most ncAAs, including pAcF (2.5-fold), pNO2F (2.5-fold), mIF (2.6-fold), bip A (1.7-fold), and pMeF (1.3-fold), while retaining activity for pIF (0.9-fold) and pClF (1.1-fold; Supporting Information Figures 1 and 3). As pAzFRS-1 exhibited a modest reduction in pAzF incorporation and a modest increase in pIF incorporation, we further tested the selectivity of pAzFRS-1 by expressing sfGFP-2TAG in LB media containing pAzF and pIF simultaneously. Intact mass spectrometry of the His6-purified sfGFP revealed that both pAzFRS-1 and pCNFRS produced sfGFP with predominantly pIF incorporation (Supporting Information Figure 4A and B). We were unable to detect the expected mass which corresponds to incorporation of pAzF. It is known that pAzF can be readily reduced to p-amino-l - phenylalanine (pAmF) during sample preparation.21,22 Con-sistent with previous reports, we observed the mass that corresponds to pAmF when pAzFRS-1 is used to express sfGFP-2TAG in LB media containing pAzF (data not shown). These data indicate that both pCNFRS and pAzFRS-1 retain high activity for pIF.
To gain insights into a possible mechanism of the improved substrate selectivity of pAzFRS-1, we sequenced the pAzFRS-1 gene. We found four nonsynonymous mutations (K90Q, I176L, R257W, and E272V) in pAzFRS-1 compared to l-phenyllactic acid.23 Also, crystal structures showed that I176 is located in the direct vicinity of the MjTyrRS substrate-binding pocket, suggesting that I176L improves pCNFRS selectivity by changing the substrate-binding pocket to discriminate other ncAAs better.9
To demonstrate that our method can be extended to improve selectivity of other aaRSs for different ncAAs, we performed directed evolution with our new strategy on another polyspecific synthetase, Methanosarcina mazei (Mm) PylRS-derived AcKRS1.24 AcKRS1 recognizes Nε-acetyl-l-lysine (AcK), a widely found protein post-translational modification (PTM ) that is important for various cellular processes.25 Besides AcK, AcKRS1 can recognize other meta-substituted phenylalanine analogues including m-iodo-l-phenylalanine (mIF), m-bromo-l-phenylalanine (mBrF), m-trifluoro-l-phenylalanine (mCF3F), and m-m ethoxy-l-phenylalanine (mM eOF).8
We tested if we can discriminate the incorporation between AcK and other ncAAs in a reporter protein using an anti-acetyl-lysine antibody. We employed surface display technology by using a well-established display system, the Lpp-OmpA scaffold, to present a reporter peptide on the bacterial cell surface because antibodies are unable to reach intracellular proteins in live cells.26–29 The Lpp-OmpA scaffold includes a signal peptide (29 amino acids) from the E. coli lipoprotein (Lpp) fused to a transmembrane domain (114 amino acids) from the E. coli outer membrane protein A (OmpA).26 We appended His6-tagged histone 3 reporter peptide (H3-His6) (amino acid sequence: 1ARTKQTARKSTGGKAPRHHH-HHH23) onto the C-terminus of the Lpp-OmpA scaffold to enable its presentation on the cell surface (Figure 3A). The corresponding residue for histone 3 lysine 9 (ARTKQTARK-STGGKAPR) is mutated to a stop codon (Lpp-OmpA-H3–9TAG) for ncAA incorporation. To measure ncAA incorporation efficiency, we used E. coli cells [BL21(DE3)] containing AcKRS, its cognate tRNACUA, and the reporter gene Lpp-OmpA-H3–9TAG-His6. We observed that AcKRS allowed incorporation of AcK and multiple meta-substituted phenylalanine analogues including mBrF, mIF, mCF3F, and mMeOF, as indicated by expression of a full-length Lpp-OmpA-H3 protein (Figure 3B, Supporting Information Figure 1). We found that the anti-acetyl H3K9 antibody could specifically recognize the AcK-containing reporter protein (Figure 3B). These data indicate AcK incorporation can be discriminated from incorporation of meta-substituted phenylalanine analogues in the reporter protein.
Figure 3.

Specific labeling of AcK-containing Lpp-OmpA-H3 reporter protein allowing selection of engineered tRNA synthetase variants with improved selectivity toward AcK. (A) Presentation of His6-tagged histone 3 (H3) peptide on the surface of E. coli cells by the Lpp-OmpA display scaffold (top). Full-length Lpp-OmpA-H3 can be detected by anti-His6 antibody, and AcK incorporation can be detected by anti-acetyl-lysine antibody in live E. coli cells. Schematic representation of Lpp-OmpA-H3 fusion gene structure (bottom). (B) Anti-acetyl lysine H3K9 antibody specifically detects AcK-incorporated Lpp-OmpA-H3. Western blotting with anti-acetyl-lysine H3K9 (top) for AcK incorporation and anti-His6 for full-length Lpp-OmpA-H3 (bottom). Cells expressing AcKRS1/tRNACUAPyl and Lpp-OmpA-H3 (9TAG) were grown in LB media containing AcK (1 mM or 10 mM), mBrF (1 mM), mIF (1 mM), mCF3F (1 mM), or mMeOF (1 mM). (C) Flow cytometry reveals specific labeling of cells expressing AcK-incorporated Lpp-OmpA-H3. Cells expressing AcKRS1/tRNACUAPyl and Lpp-OmpA-H3 (9TAG) were grown in LB media containing AcK (10 mM) or mIF (1 mM) and labeled with anti-acetyl-lysine H3K9 (indicated by PE stain) and anti-His6 (indicated by FITC stain) antibodies. Representative fluorescence density plots showing live E. coli cells expressing Lpp-OmpA-H3 with no ncAA (left), AcK (middle), or mIF (right) incorporation. At least 2 × 104 cells were analyzed and presented on each FACS plot.
To verify the proper display of H3 peptide on the E. coli surface, we carried out immunofluorescence staining with fluorescein (FITC)-conjugated anti-His6 antibody on live E. coli cells expressing Lpp-OmpA-H3. The expression of Lpp-OmpA-H3 is driven by isopropyl β-d −1-thiogalactopyranoside (IPTG)-inducible T7 promoter. We observed cell surface-localized fluorescence signals, indicating the expression of H3 peptide in IPTG-induced cells. No fluorescence was detected in the noninduced cells (Supporting Information Figure 5A). Flow cytometry analysis revealed that 25.3% of cells were displaying the H3 peptide (Supporting Information Figure 6). We next probed for AcK incorporation by performing immunofluorescence staining with primary anti-acetyl H3K9 and phycoerythrin (PE)-conjugated secondary anti-rabbit antibodies. The antibodies were applied on live cells expressing AcKRS, its cognate tRNACUA, and the reporter gene Lpp-Om pA-H3–9TAG-His6 in the presence of AcK. We observed cell surface-localized fluorescence signals, indicating the expression of AcK-containing H3 peptide in IPTG-induced cells. No fluorescence was detected in the noninduced cells (Supporting Information Figure 5B). Thus, we confirmed that the H3 peptide reporter is properly expressed on the cell surface for antibody recognition.
Next, we examined if we can discriminate between incorporation of AcK and mIF in live cells with the above established reporter protein and antibody staining conditions by using flow cytometry. By costaining cells with anti-His6 (indicated by FITC) and anti-acetyl H3K9 (indicated by PE) antibodies, we found that E. coli grown with AcK displayed increased FITC and PE fluorescence, whereas E. coli grown with mIF displayed increased FITC fluorescence only (Figure 3C). These data indicate that discrimination between incorporation of AcK and mIF is possible in live cells.
Having established the system to specifically detect AcK incorporation, we sought to use this system to generate AcKRS variants with improved substrate selectivity. The C-terminal region of AcKRS1 comprises of the catalytic core of the enzyme.30 By using epPCR to introduce random mutations within the C-terminal region (amino acids 184–454), we created AcKRS variants with an average of 6.2 base mutations per sequence of theoretical library size of 4.9 × 107 (data not shown).18 We applied our new directed evolution strategy on AcKRS1 in the simultaneous presence of AcK and mIF (Supporting Information Figure 7). Following staining with anti-His6 (FITC) and anti-acetyl H3K9 (PE) antibodies, cells were sorted for FITC+/PE + (potentially enriched in AcK- selective variants) and FITC+/PE− cells (potentially enriched in mIF-selective variants) by using a flow cytometer. The sorted cells were plated out on LB agar plates to isolate single clones and were assessed for their substrate selectivity toward AcK and mIF by using an sfGFP readthrough assay.
In total, we characterized six variants from the FITC+/PE + fraction and 10 variants from the FITC+/PE− fraction by using an sfGFP readthrough assay with individual ncAAs. We identified two mIF-selective clones and three AcK-selective variants (Supporting Information Figure 8). While the AcK- selective variants are promising, significant levels of mIF incorporation remained detectable. Additional rounds of evolutions will be needed to further improve AcK selectivity. For this reason, we chose to only further analyze the most mIF-selective variant, which we named mIFRS-1. mIFRS-1 showed remarkable selectivity toward mIF (1 .4–7.4-fold) compared to parental enzyme AcKRS1 despite a slight reduction in activity (1 .3–1.6-fold; Figure 4A). Also, we assessed the activity of mIFRS-1 for three other ncAAs, which were originally substrates for the parental enzyme AcKRS and were not used during the selection. mIFRS-1 showed reduced incorporation of mMeOF (2.1-fold; Supplementary Figure 9). Together, these results dem onstrated the general applicability of our approach to evolve highly selective tRNA synthetase variants from polyspecific tRNA synthetases.
Figure 4.

Directed evolution of mIF-selective aminoacyl tRNA synthetase. (A) The variant mIFRS-1 is more selective for mIF compared to parental enzyme AcKRS1. Cells expressing mIFRS-1 or AcKRS1 with its cognate tRNACUAPyl and sfGFP-2TAG were grown in LB media containing varying concentrations of indicated ncAAs. Incorporation efficiencies were evaluated by monitoring sfGFP fluorescence. Normalized fluorescence intensities were calculated from fluorescence readings at 24 h time points divided by absorbance at 600 nm. Mean values and standard deviations were calculated from three replicates. (B) A single amino acid substitution (A302T) was found in mIFRS-1. The mutated residue is indicated in yellow in the AcKRS1/AcK complex crystal structure (left). Shown on the right is the close-up view of the active site residues with the mutation A302T (yellow) and AcK substrate (red). PDB ID: 4Q6G. (C) mIFRS-1 produces highly pure mIF-incorporated sfGFP proteins in the presence of AcK (10 mM) and mIF (1 mM). ESI-MS spectrum of purified full-length intact sfGFP. (D) AcKRS1 produces a mixture of mIF-incorporated and AcK-incorporated sfGFP proteins in the presence of AcK (10 mM) and mIF (1 mM). ESI-MS spectrum of purified full-length intact sfGFP.
To identify the mutation(s) that is(are) responsible for improved substrate specificity of mIFRS-1, we sequenced the mIFRS-1-encoding gene and found a single nonsynonymous mutation (A302T) compared to the parental enzyme AcKRS1 (Figure 4B). A302 is located within the substrate-binding pocket of MmPylRS and has been previously reported to be a key determinant for recognition of phenylalanine analogues.30,31 MmPylRS-derived variants charging O-methyl-l-tyrosine, l −3-(2-naphthyl)alanine, and p-benzoyl-l-phenylalanine were reported to contain A302T mutation.31,32 Our results are consistent with previous findings on the role of A302 for determination of substrate selectivity.
Highly pure proteins with incorporation of the desired ncAA are crucial for protein studies as well as industrial applications and obviate the need for further purification from proteins containing undesired amino acids at the target residue(s). We further tested the selectivity of mIFRS-1 by expressing sfGFP-2TAG in LB media containing mIF and AcK simultaneously. Intact mass spectrometry of the His6-purified sfGFP revealed the desired mass corresponding to incorporation of mIF with only trace incorporation of AcK (Figure 4C). On the contrary, AcKRS1 produced a mixture of sfGFP with AcK and mIF incorporation (Figure 4D). These results indicated that highly selective aaRSs would be particularly useful for producing highly pure proteins.
In summary, we have developed a novel scheme for generating substrate-selective aaRSs by carrying out directed evolution with multiple ncAAs simultaneously. To this end, we have significantly improved the substrate selectivity of two polyspecific aaRSs—pCNFRS and AcKRS— and generated variants that are highly selective for pAzF and mIF, respectively.
A post-translational proofreading-based method has been recently developed for engineering bipA aminoacyl-tRNA synthetase selectivity.10 Discrimination of ncAAs, which is dependent on the N-recognin binding pocket, is largely based on the size and polarity of the amino acids. In comparison, our method is customized to distinguish specific desired ncAAs and provides an attractive complementary approach for engineering substrate-selective tRNA synthetase variants.
Our approach reveals critical residues for substrate selectivity in polyspecific aaRSs. Since the mutations that influence substrate selectivity are located within the catalytic pocket, enzymatic properties are likely to be affected. An important consideration during directed evolution of tRNA synthetases is to achieve an optimal tradeoff between the activity and selectivity. Uncovering these key residues may advance our understanding on the molecular basis of substrate selectivity and guide future efforts in more intensive rational engineering of aaRSs. Our strategy can be further extended to evolve additional substrate-selective aaRS variants. Our future goal is to evolve aaRSs for selective incorporation of post-translational modifications (PTMs) using PTM-specific antibodies, as well as trans-cyclooctene-or norbornene-containing amino acids using tetrazine-derivatized probes.2,33–35 We envision that our scheme will facilitate the production of additional substrate-selective aaRS variants with enhanced utility for genetic code expansion.
MATERIALS A ND METHODS
Reagents and Strains.
All cloning and plasmid propagation was conducted using Stellar (Clontech) cells. Selection and screening experiments were conducted using DH10B and BL21 (DE3) strains of E. coli cells. All ncAAs used in this study were purchased from Chem-Impex International. Cy5-DBCO was from Click Chemistry Tools. IPTG and antibiotics were from AmericanBio. Antibiotics were used in the following concentrations: 100 μg/mL of ampicillin, 50 μg/mL of spectinomycin, 34 μg/mL of chloramphenicol, and 10 μg/mL of tetracycline. Oligonucleotide synthesis and DNA sequencing were performed by the Keck Foundation Biotechnology Resource Laboratory at Yale University.
Constructions of Plasmids.
PCR and site-directed mutagenesis were performed with Herculase II Fusion DNA Polymerase (Agilent Technologies). For construction of pET-Lpp-OmpA-H3-His6, a gene fragment encoding Lpp and OmpA was synthesized by Quintara Biosciences. H3 peptide was amplified from pET-H3 to make Lpp-OmpA-H3.36 The gene fragments were inserted into the pET-His6 vector using an In-Fusion cloning kit (Clonetech) to make pET-LPP-OmpA-H3-His6. pULTRA-CNF was obtained from Addgene (#48215).
Lpp-OmpA-H3-His6 protein sequence: MKATKLVLGAVILGSTLLAGCSSNAKIDQNNNGPTHENQLGAGAFGGYQyNPYVGFEMGYDWLGRMPYKGSVENGAYKAQGVQLTAKLGYPITDDLDIYTRLGGMVWRADTKSNVYGKNHDTGVSPVFAGGVEYAITPEIATRARTKQTARKSTGGKAPRHHHHHH
Protein Expression.
To express sfGFP, pULTRA-pCNFRS or pCDF-AcKRS1 or variant aaRSs and pET-sfGFP-2TAG were cotransformed into the BL21 (DE3) strain of E. coli cells. To express Lpp-OmpA-H3, pCDF-AcKRS1 or variant aaRSs and pET-Lpp-OmpA-H3–9TAG were cotransformed into the BL21 (DE3) strain of E. coli cells. Overnight growth culture from a single colony was used to inoculate Lysogeny Broth (LB) media supplemented with appropriate antibiotics. When cell OD600 reached 0.6, the following chemicals were added to the indicated final concentrations: IPTG (0.5 mM; induction of sfGFP, Lpp-OmpA-H3, and pCNFRS proteins expression), ncAAs (1 mM, except AcK is 10 mM unless otherwise stated), and nicotinamide (Thermo Fisher Scientific; 20 mM; to prevent deacetylation of AcK). To improve protein expression and ncAA incorporation, cells were grown at 30 °C. For sfGFP, cells were harvested after 24 h of incubation at 30 °C with shaking (250 rpm). For Lpp-OmpA-H3, cells were harvested after 24 h of incubation at 30 °C with shaking for protein expression or 1 h of incubation at 37 °C with shaking for screening experiments.
Protein Purification.
sfGFP was purified by Ni-NTA affinity chromatography (Qiagen). A cell pellet from a 25 mL culture of sfGFP expression was resuspended in 10 mL of lysis buffer containing 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, 10 mM imidazole, and protease inhibitor cocktail tablets (cOmplete, Roche), followed by sonication. Lysed cells were centrifuged for 10 min at 4 °C at 10 000g and passed through a 0.45 μm membrane filter. The filtrate was applied onto an equilibrated Ni-NTA column (Qiagen), washed twice with a wash buffer containing 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, and 20 mM imidazole, and sfGFP was eluted with elution buffer containing 50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, and 150 mM imidazole. The eluate was concentrated using Amicon centrifugal filter devices (Millipore), buffer exchanged with 1 × PBS to remove imidazole, and analyzed by intact mass spectrometry.
Western Blotting and in-Gel Fluorescence.
Cells were lysed with Bugbuster (Novagen) containing 25 units/mL of benzonase nuclease (Millipore) and protease inhibitor cocktail tablets (cOmplete, Roche). The lysates were incubated at RT for 20 min with constant rotation and centrifuged at 13 000g at 4 °C for 30 min. The supernatant was collected, and protein quantification was performed with Bradford assay. For in-gel fluorescence detection of pAzF- containing proteins, lysates were incubated with Cy5-DBCO (5 μM) at 37 °C for 1 h. The samples were separated by 15% SDS-PAGE and transferred to a 0.2 μm nitrocellulose membrane. The membrane was blocked with blocking buffer containing 5% (w/v) nonfat dry milk and 0.1% Tween-20 in PBS for 1 h at RT. The primary antibodies used were anti-His6 tag (1:5000 dilution, Cell Signaling Technology (CST) catalog #2366) and anti-histone 3 H3 (acetyl-K9) (1:1000 dilution, Abcam catalog #ab4441), incubated at 4 °C overnight. The membrane was washed thrice with PBS with 0.1% Tween-20 (PBS-T) and incubated with antimouse IgG HRP (1:5000, CST catalog #7076) or anti-rabbit IgG (1:5000, CST catalog #7074) secondary antibodies for 1 h at RT. The membrane was washed with PBS-T and visualized using Western Lightning Plus-ECL and Enhanced Chemiluminescence Substrate (PerkinElmer) on a ChemiDoc MP Imaging System (Biorad Laboratories).
Directed Evolution of pCNFRS and AcKRSI.
Variant libraries for pCNFRS and AcKRS1 were generated via epPCR using GeneMorph II Random Mutagenesis kit (Stratagene), following the manufacturer’s instructions. A total of 100 ng of target DNA was used as a template for 50 μL-EP-PCR reaction. The reaction mixture was amplified for 30 cycles. Four separate 50 μL EP-PCR reactions were performed for each tRNA synthetase and pooled. The plasmid backbone was amplified by PCR using Herculase II Fusion DNA Polymerase. Both PCR products were resolved on 1% agarose by gel electrophoresis, purified, DpnI digested, and ligated by Gibson assembly (New England Biolabs). Assemblies were purified by ethanol precipitation, and 200 ng of library was electroporated into DH10B cells.
Positive chloramphenicol selection was performed as previously described to isolate functional tRNA synthetase variants.24 The selection requires readthrough of the TAG codon at position 112 in chloramphenicol acetyltransferase (CAT) in the presence of ncAAs. Cells were selected on 50 μg/mL or 100 μg/mL chloramphenicol- containing LB agar plates. Chloramphenicol-resistant colonies were pooled, grown for 4 h at 37 °C, mini-prepped for plasmid DNA, and retransformed into the BL21 (DE3) strain of E. coli cells with pET-Lpp-OmpA-H3–9TAG-His6. For directed evolution of pAzF/pCNF-selective variants, cells were incubated with DBCO-Cy5. For directed evolution of AcK/mIF-selective variants, cells were incubated with anti-acetyl lysine (H3K9) and anti-His6 antibodies. Then, cells were sorted using FACSAria III (BD Biosciences) and plated on LB agar plates containing appropriate antibiotics to isolate single clones for downstream sequencing and analysis. Cells grown in the absence of ncAAs were used as controls for setting sorting gates.
Flow Cytometry.
Analysis of live cells by flow cytometry was carried out on Attune NxT flow cytometer (Invitrogen). For detection of pAzF incorporation, cells are incubated with DBCO-Cy5 (1 μM) overnight at 4 °C. For detection of AcK incorporation, cells were stained with the following primary and secondary antibodies at 4 °C: anti-His6 tag FITC conjugate (1:11 dilution; 15 min; Miltenyi Biotec; catalog #130–098-808), anti-histone 3 H3 (acetyl-K9) (1:1000 dilution, overnight; Abcam catalog # ab4441), and anti-rabbit IgG PE conjugate (1:100 dilution; 15 min; Biolegend catalog #406421). After staining, cells were washed three times with PBS. At least 20 000 events were recorded in each experiment. FlowJo v10 software was used to analyze the flow cytometry data.
sfGFP Fluorescence Readthrough Assay. Overnight growth culture from a single colony was used to inoculate LB media supplemented with appropriate antibiotics. Cells were grown to an OD600 of 0.6, and the following chemicals were added to the indicated final concentrations: IPTG (0.5 mM; induction of sfGFP and pCNFRS proteins expression) and ncAAs (1 mM; except AcK is 10 mM unless otherwise stated). Cells were transferred to a 96-well clear-bottom plate and incubated at 30 °C for 24 h in a shaking plate incubator. GFP fluorescence was measured on a Biotek Synergy HT spectrophotometric plate reader using excitation and emission wavelengths of 485 and 525 nm. Fluorescence was normalized via the OD600 reading to obtain FL/OD.
LC-MS of Intact Purified Proteins.
Purified protein samples were diluted in PBS to analyze using Waters XEVO Q-TOF mass spectrometer equipped with Acquity UPLC BEH C18 1.7 μM column. The mass spectra obtained were processed with Maximum Entropy program (MaxEnt) to deconvolute multiple charged ESI data to give a measurement of average molecular weight. The average molecular weights of the proteins were predicted using ExPASy Compute pI/Mw tool (http://web.expasy.org/compute_pi/) and adjusted for chromophore maturation in sfGFP and ncAA substitutions.
Supplementary Material
ACKNOWLEDGMENTS
We thank all members of the Söll laboratory for insightful discussions and critical feedback and L. Qin for critical reading of the manuscript and valuable suggestions. We thank O. Ad and L. Qin for mass spectrometry analysis. We gratefully acknowledge the laboratory of A. Schepartz for use of the ESI mass spectrometer and flow cytometer and the Yale Flow Cytometry Core Facility (K. Nelson) and Keck Mass Spectrometry and Proteomics Resource (R. W ilson) for technical assistance. This work was supported by National Institutes of Health Grant R35GM122560 (to D.S.).
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.9b00088.
Chemical structures of ncAAs (Supporting Figure 1); assessment of pAzF-selective variants (Supporting Figure 2); assessment of substrate range of pAzFRS-1 (Supporting Figure 3); assessment of pAzFRS-1 and pCNFRS activity for pIF and pAzF incorporation (Supporting Figure 4); confirmation of surface expression of Lpp-OmpA-H3 (Supporting Figure 5); assessment of Lpp-OmpA-H3 display efficiency (Supporting Figure 6); scheme for directed evolution of AcKRS1 (Supporting Figure 7); assessment of mIF-selective and AcK-selective variants (Supporting Figure 8); assessment of substrate range of mFRS-1 (Supporting Figure 9) (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Park HS, Hohn MJ, Umehara T, Guo LT, Osborne EM, Benner J, Noren CJ, Rinehart J, and Söll D (2011) Expanding the genetic code of Escherichia coli with phosphoserine. Science 333, 1151–1154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Lang K, Davis L, Wallace S, Mahesh M, Cox DJ, Blackman ML, Fox JM, and Chin JW (2012) Genetic Encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels-Alder reactions. J. Am. Chem. Soc 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Chin JW, Martin AB, King DS, Wang L, and Schultz PG (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichiacoli. Proc. Natl. Acad. Sci. U. S. A 99, 11020–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Lemke EA, Summerer D, Geierstanger BH, Brittain SM, and Schultz PG (2007) Control of protein phosphorylation with a genetically encoded photocaged amino acid. Nat. Chem. Biol 3, 769–772. [DOI] [PubMed] [Google Scholar]
- (5).Chatterjee A, Guo J, Lee HS, and Schultz PG (2013) A genetically encoded fluorescent probe in mammalian cells. J. Am. Chem. Soc 135, 12540–12543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mukai T, Lajoie MJ, Englert M, and Söll D (2017) Rewriting the Genetic Code. Annu. Rev. Microbiol 71, 557–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wan W, Tharp JM, and Liu WR (2014) Pyrrolysyl-tRNA synthetase: an ordinary enzyme but an outstanding genetic code expansion tool. Biochim. Biophys. Ada, Proteins Proteomics 1844, 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Guo LT, Wang YS, Nakamura A, Eiler D, Kavran JM, Wong M, Kiessling LL, Steitz TA, O’Donoghue P, and Söll D (2014) Polyspecific pyrrolysyl-tRNA synthetases from directed evolution. Proc. Natl. Acad. Sci. U. S. A 111, 16724–16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Young DD, Young TS, Jahnz M, Ahmad I, Spraggon G, and Schultz PG (2011) An evolved aminoacyl-tRNA synthetase with atypical polysubstrate specificity. Biochemistry 50, 1894–1900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kunjapur AM, Stork DA, Kuru E, Vargas-Rodriguez O, Landon M, Söll D, and Church GM (2018) Engineering posttranslational proofreading to discriminate nonstandard amino acids. Proc. Natl. Acad. Sci. U. S. A 115, 619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Zheng Y, Addy PS, Mukherjee R, and Chatterjee A (2017) Defining the current scope and limitations of dual noncanonical amino acid mutagenesis in mammalian cells. Chem Sci 8, 7211–7217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Neumann H, Wang K, Davis L, Garcia-Alai M, and Chin JW (2010) Encoding multiple unnatural amino acids via evolution of a quadruplet-decoding ribosome. Nature 464, 441–444. [DOI] [PubMed] [Google Scholar]
- (13).Wang L, and Schultz PG (2005) Expanding the genetic code. Angew. Chem. Int. Ed 44, 34–66. [DOI] [PubMed] [Google Scholar]
- (14).Agard NJ, Prescher JA, and Bertozzi CR (2004) A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- (15).Englert M, Nakamura A, Wang YS, Eiler D, Söll D, and Guo LT (2015) Probing the active site tryptophan of Staphylococcus aureus thioredoxin with an analog. Nucleic Acids Res. 43, 11061–11067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Amiram M, Haimovich AD, Fan C, Wang YS, Aerni HR, Ntai I, Moonan DW, Ma NJ, Rovner AJ, Hong SH, Kelleher NL, Goodman AL, Jewett MC, Söll D, Rinehart J, and Isaacs FJ (2015) Evolution of translation machinery in recoded bacteria enables multi-site incorporation of nonstandard amino acids. Nat. Biotechnol 33, 1272–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bryson DI, Fan C, Guo LT, Miller C, Söll D, and Liu DR (2017) Continuous directed evolution of aminoacyl-tRNA synthetases. Nat. Chem. Biol 13, 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Firth AE, and Patrick WM (2008) GLUE-IT and PEDEL-AA: new programmes for analyzing protein diversity in randomized libraries. Nucleic Acids Res. 36, W 281–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, Bryson DI, and Liu DR (2017) Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Hammer SC, Kubik G, Watkins E, Huang S, Minges H, and Arnold FH (2017) Anti-Markovnikov alkene oxidation by metal-oxo-mediated enzyme catalysis. Science 358, 215–218. [DOI] [PubMed] [Google Scholar]
- (21).Shao N, Singh NS, Slade SE, Jones AM, and Balasubramanian MK (2015) Site Specific Genetic Incorporation of Azidophenylalanine in Schizosaccharomyces pombe. Sci. Rep 5, 17196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Chin JW, Cropp TA, Anderson JC, Mukherji M, Zhang Z, and Schultz PG (2003) An expanded eukaryotic genetic code. Science 301 , 964–967. [DOI] [PubMed] [Google Scholar]
- (23).Guo J, Wang J, Anderson JC, and Schultz PG (2008) Addition of an alpha-hydroxy acid to the genetic code of bacteria. Angew. Chem. Int. Ed 47, 722–725. [DOI] [PubMed] [Google Scholar]
- (24).Umehara T, Kim J, Lee S, Guo LT, Söll D, and Park HS (2012) N-acetyl lysyl-tRNA synthetases evolved by a CcdB-based selection possess N-acetyl lysine specificity in vitro and in vivo. FEBS Lett. 586, 729–733. [DOI] [PubMed] [Google Scholar]
- (25).Choudhary C, Weinert BT, Nishida Y, Verdin E, and Mann M (2014) The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol 15, 536–550. [DOI] [PubMed] [Google Scholar]
- (26).Daugherty PS, Olsen MJ, Iverson BL, and Georgiou G (1999) Development of an optimized expression system for the screening of antibody libraries displayed on the Escherichia coli surface. Protein Eng. Des. Sel 12, 613–621. [DOI] [PubMed] [Google Scholar]
- (27).Francisco JA, Campbell R, Iverson BL, and Georgiou G (1993) Production and fluorescence-activated cell sorting of Escherichia coli expressing a functional antibody fragment on the external surface. Proc. Natl. Acad. Sci. U. S. A 90, 10444–10448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Georgiou G, Stephens DL, Stathopoulos C, Poetschke HL, Mendenhall J, and Earhart CF (1996) Display of beta-lactamase on the Escherichia coli surface: outer membrane phenotypes conferred by Lpp’-OmpA’-beta-lactamase fusions. Protein Eng. Des. Sel 9, 239–247. [DOI] [PubMed] [Google Scholar]
- (29).Harvey BR, Georgiou G, Hayhurst A, Jeong KJ, Iverson BL, and Rogers GK (2004) Anchored periplasmic expression, a versatile technology for the isolation of high-affinity antibodies from Escherichia coli-expressed libraries. Proc. Natl. Acad. Sci. U. S. A 101, 9193–9198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Kavran JM, Gundllapalli S, O’Donoghue P, Englert M, Söll D, and Steitz TA (2007) Structure of pyrrolysyl-tRNA synthetase, an archaeal enzyme for genetic code innovation. Proc. Natl. Acad. Sci. U. S. A 104, 11268–11273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Lacey VK, Louie GV, Noel JP, and Wang L (2013) Expanding the library and substrate diversity of the pyrrolysyl-tRNA synthetase to incorporate unnatural amino acids containing conjugated rings. ChemBioChem 14, 2100–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Takimoto JK, Dellas N, Noel JP, and Wang L (2011) Stereochemical basis for engineered pyrrolysyl-tRNA synthetase and the efficient in vivo incorporation of structurally divergent non-native amino acids. ACS Chem. Biol 6, 733–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Fan C, Ip K, and Söll D (2016) Expanding the genetic code of Escherichia coli with phosphotyrosine. FEBS Lett. 590, 3040–3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Luo X, Fu G, Wang RE, Zhu X, Zambaldo C, Liu R, Liu T, Lyu X, Du J, Xuan W, Yao A, Reed SA, Kang M, Zhang Y, Guo H, Huang C, Yang PY, Wilson IA, Schultz PG, and Wang F (2017) Genetically encoding phosphotyrosine and its nonhydrolyzable analog in bacteria. Nat. Chem. Biol 13, 845–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Wang ZA, Kurra Y, Wang X, Zeng Y, Lee YJ, Sharma V, Lin H, Dai SY, and Liu WR (2017) A versatile approach for site-specific lysine acylation in proteins. Angew. Chem. Int. Ed 56, 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Xiong H, Reynolds NM, Fan C, Englert M, Hoyer D, Miller SJ, and Söll D (2016) Dual genetic encoding of acetyl-lysine and non-deacetylatable thioacetyl-lysine mediated by flexizyme. Angew. Chem. Int. Ed 55, 4083–4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.