

Disentangling the mechanisms shaping bacterioplankton communities during a marine ecological event is a core concern for ecologists. Harmful algal bloom (HAB) is a typical ecological disaster, and its formation is significantly influenced by alga-bacterium interactions. Microbial community shifts during the HAB process are relatively well known. However, the assembly processes of microbial communities in an HAB are not fully understood, especially the relative influences of deterministic and stochastic processes. We therefore analyzed the relative contributions of deterministic and stochastic processes during an HAB event. Both free-living and attached bacterial groups had a dramatic response to the HAB, and the relative importance of determinism versus stochasticity varied between the two bacterial groups at various bloom stages. Environmental factors and biotic interactions were the main drivers impacting the microbial shift process. Our results strengthen the understanding of the ecological mechanisms controlling microbial community patterns during the HAB process.
KEYWORDS: microbial community, assembly profile, coregulated, deterministic selection, dinoflagellate bloom, random processes
ABSTRACT
Given the ecological significance of microorganisms in algal blooming events, it is critical to understand the mechanisms regarding their distribution under different conditions. We tested the hypothesis that microbial community succession is strongly associated with algal bloom stages, and that the assembly mechanisms are cocontrolled by deterministic and stochastic processes. Community structures and underlying ecological processes of microbial populations (attached and free-living bacteria) at three algal bloom stages (pre-, during, and postbloom) over a complete dinoflagellate Scrippsiella trochoidea bloom were investigated. Both attached and free-living taxa had a strong response to the bloom event, and the latter was more sensitive than the former. The contribution of environmental parameters to microbial variability was 40.2%. Interaction analysis showed that complex positive or negative correlation networks exist in phycosphere microbes. These relationships were the potential drivers of mutualist and competitive interactions that impacted bacterial succession. Null model analysis showed that the attached bacterial community primarily exhibited deterministic processes at pre- and during-bloom stages, while dispersal-related processes contributed to a greater extent at the postbloom stage. In the free-living bacterial community, homogeneous selection and dispersal limitation dominated in the initial phase, which gave way to more deterministic processes at the two later stages. Relative contribution analyses further demonstrated that the community turnover of attached bacteria was mainly driven by environmental selection, while stochastic factors had partial effects on the assembly of free-living bacteria. Taken together, these data demonstrated that a robust link exists between bacterioplankton community structure and bloom progression, and phycosphere microbial succession trajectories are cogoverned by both deterministic and random processes.
IMPORTANCE Disentangling the mechanisms shaping bacterioplankton communities during a marine ecological event is a core concern for ecologists. Harmful algal bloom (HAB) is a typical ecological disaster, and its formation is significantly influenced by alga-bacterium interactions. Microbial community shifts during the HAB process are relatively well known. However, the assembly processes of microbial communities in an HAB are not fully understood, especially the relative influences of deterministic and stochastic processes. We therefore analyzed the relative contributions of deterministic and stochastic processes during an HAB event. Both free-living and attached bacterial groups had a dramatic response to the HAB, and the relative importance of determinism versus stochasticity varied between the two bacterial groups at various bloom stages. Environmental factors and biotic interactions were the main drivers impacting the microbial shift process. Our results strengthen the understanding of the ecological mechanisms controlling microbial community patterns during the HAB process.
INTRODUCTION
The concept of ecological succession is essential in the field of microbial ecology. The mechanisms and trajectories that control ecological succession are critical in order to accurately predict the responses of ecosystems to environmental change and project their future states (1). However, the mechanisms of community assembly and related critical factors (such as physicochemical parameters, habitat type, and biotic interactions) in shaping species composition and structure remain controversial (2), particularly in microbial ecology. The traditional niche-based theory postulates that specific variables, such as life history traits, species interactions (such as competition, predation, mutualisms, and trade-offs), and abiotic conditions (such as pH, temperature, salt, and moisture) determine how communities are organized (3). This differs from the neutral theory, which describes community structures as being independent of species traits; it assumes that they are dictated by random processes such as births, deaths, immigration, extinction, and speciation (4). The simultaneous occurrence of niche-based and neutral theory processes in local community assemblages is now widely accepted (5). However, what role these processes and factors play in overall community structure, succession, and ecological results is still debated (3, 6).
Deterministic and stochastic processes are the principal representatives for niche-based and neutral theory, respectively (2, 7, 8). The former comprises environmental filtering (including factors such as temperature, pH, and illumination), dispersal-related factors, and biotic interactions. The latter includes ecological drift and random birth and/or death events (9). Deterministic processes involve ecological selection established by both abiotic and biotic factors. This influences organismal fitness and ultimately determines both the composition and relative abundance of species (10). Meanwhile, stochastic processes, including random immigration, emigration, and dispersal events, are events that alter species composition patterns that are tantamount to a result of random chance (4). It is thought that both deterministic and stochastic processes work concomitantly to regulate how ecological communities are structured (2, 11). The balance between these two processes has grown into a dominant theme in microbial ecology.
Interestingly, the relative effects of deterministic and stochastic processes can be modified by environmental and biological interactions (12). With the development of sequencing techniques and null-model approaches, an increasing number of studies have explored microbial biodiversity and distribution patterns using a phylogenetic framework (13, 14). These studies have examined the phylogenetic structure of microbial communities in various habitats, including grassland, forest, desert, reservoir, and marine environments, and these studies have revealed the distribution profiles and underlying ecological mechanisms of environmental microorganisms (1, 7, 8, 14, 15).
In the marine environment, harmful algal bloom (HAB) is a typical ecological event and has been closely linked with microbial ecological structure and function (such as metabolism dependency) (16). Factors that are responsible for this include microbes that mediate biogeochemical cycling, microfood web structure, matter transformation, and alga-bacterium signaling regulation (such as quorum sensing) (17–19). Bacterial community structure during bloom events is complex, changes throughout the duration of the bloom, and is contingent on the algal species, physiological status, environmental conditions, biotic interactions, and bloom stage (20). Many studies have reported bacterial community structuring during natural or mesocosm phytoplankton blooms (21–23), and certain dominant bacterial clusters (such as Roseobacter and Flavobacterium) are commonly associated with blooms (24, 25). The structural and metabolic properties of the associated bacteria alter their ecological functions, including nutrient provision and release of organic compounds. They can also act as a competitor; in certain ecological niches, they take up the same space as some algae, resulting in interspecific competition (26). Such behaviors create a distinct regulatory network that acts across the phases of bloom formation, duration, and collapse (27, 28).
Current advances in next-generation sequencing and theoretical ecological analysis tools offer the opportunity to introduce and test mechanistic concepts in microbial ecology (29). Among these methods, beta-nearest taxon index (β-NTI) has been applied to find the relative role of deterministic and stochastic processes (30). The β-NTI calculates the mean nearest phylogenetic neighbor among the individuals between two communities. β-NTI values that are positive or negative indicate more or less than expected phylogenetic turnover, respectively. Values between −2 and +2 indicate the expectation under neutral community assembly. Additionally, values below −2 or above +2 indicate significantly less than or greater than expected phylogenetic turnover, respectively (see reference 7 for details). The null model randomizes the species labels on the phylogeny and recalculates β-NTI values, randomizing the phylogenetic relationships among species while preserving the compositional β-diversity and species richness of the samples. Numerous studies have shown that deterministic processes are the main ecological force driving the turnover of bacteria in environmental microbiomes, such as soil, aquatic, marine, and fluidic ecosystems (3, 12, 31). However, studies on the assembly mechanisms of bacteria during HAB events remain limited (16). Previously, phycosphere (a region surrounding individual phytoplankton cells) microbial assembly was found to be determined mainly by substance or environmental conditions (16, 21), but there is some evidence that random processes also participate in it (1). The relative importance of deterministic and stochastic processes in structuring distribution patterns of bacteria in algal blooming stages remains unknown, especially the contribution of fundamental ecological processes, such as selection, dispersal, diversification, and drift. In algal ecology, gaining an understanding of the mechanisms that regulate microbial distribution patterns is necessary, since microorganisms regulate the algal-bacterial relationship dynamics and play a key role in microalgal blooming (32, 33).
This study was conducted on a natural marine dinoflagellate bloom (Scrippsiella trochoidea) to examine the microbial communities for their community structure. The roles of deterministic and stochastic mechanisms in regulating their successional patterns were also investigated. We hypothesized that (i) determinism and stochasticity simultaneously control the assembly of microbial communities and (ii) environmental selection, ecological drift, and dispersal have different contributions among the different bloom stages in free-living and attached bacteria. The aims of the study were to identify the microbial structure in an HAB cycle and the main driving factors, as well as understanding how deterministic versus stochastic processes change microbial assemblages under HAB perturbations.
RESULTS
Bloom characteristics.
Three phases were documented over the course of the marine dinoflagellate S. trochoidea bloom cycle: pre-, during-, and postbloom stages. During the sampling period, the temperature, salinity, and pH ranged from 22.1 to 26.0°C, 27.9 to 29.5‰, and 8.05 to 8.09, respectively (Table 1). Table 1 also reveals the nutrient concentrations observed at each time point. Nitrogen source (NH4+ + NO3− + NO2−) and phosphorous source (PO43−) concentrations ranged from 0.147 to 0.493 mg liter−1 and 0.0015 to 0.012 mg liter−1, respectively. The highest and lowest concentrations of both NO3− and NO2− were detected at pre- and during-bloom stages, respectively (P < 0.05). A similar phenomenon was observed for PO43−, which had a relatively higher concentration (0.01 ± 0.002 mg liter−1) at the prebloom stage, with the value declining to 20% of the original in the two later stages (0.002 ± 0.0005 mg liter−1) (P < 0.05). A different trend was observed with NH4+ variables. Concentrations followed a shallow U-type curve, in which relatively large amounts of NH4+ were measured during the prebloom (0.14 ± 0.0013 mg liter−1) and postbloom (0.205 ± 0.016 mg liter−1) stages, while a lower concentration (0.091 ± 0.004 mg liter−1) occurred in the onset/during-bloom stage (P < 0.01). The approximate inorganic N/inorganic P (IN/IP) ratio in surface waters ranged from 31.39 ± 6.87 to 57.82 ± 7.69.
TABLE 1.
Environmental and biological parameters observed during the bloom of S. trochoidea
| Environmental parameter | Value by bloom stage of Scrippsiella trochoideaa |
||
|---|---|---|---|
| Before | During | After | |
| Temp (°C) | 25.9 ± 0.1a | 25.3 ± 0.1a | 22.3 ± 0.2a |
| Salinity (‰) | 29.4 ± 0.1a | 28.9 ± 0.1a | 28.1 ± 0.2a |
| pH | 8.07 ± 0.01a | 8.06 ± 0.01a | 8.07 ± 0.02a |
| Nitrate (NO3−) (mg liter−1) | 0.251 ± 0.03a | 0.056 ± 0.01b | 0.084 ± 0.015b |
| Nitrite (NO2−) (mg liter−1) | 0.016 ± 0.002a | 0.01 ± 0.005a | 0.007 ± 0.002b |
| Ammonium (NH4+) (mg liter−1) | 0.14 ± 0.0013a | 0.091 ± 0.004b | 0.205 ± 0.016c |
| Phosphate (PO43−) (mg liter−1) | 0.01 ± 0.002a | 0.002 ± 0.0005b | 0.002 ± 0.0007b |
| IN/IP ratio | 40.71 ± 6.93a | 31.39 ± 6.87a | 57.82 ± 7.69b |
| Biomass of algae (cells ml−1) | (5.5 ± 0.5) × 102a | (9.6 ± 0.3) × 103 b | (8.3 ± 0.2) × 102a |
| Chlorophyll a (μg liter−1) | 15.6 ± 2.3a | 170.59 ± 16.58c | 26.34 ± 4.29a |
| Fv/Fm value | 0.65 ± 0.05a | 0.62 ± 0.05a | 0.47 ± 0.04b |
Data represent the means ± standard deviations from triplicate technical measurements for the 12 collected samples. Different lowercase letters (a, b, and c) indicate statistically significant differences at P < 0.05 (a versus b) or P < 0.01 (a versus c and b versus c).
For biological parameters, during the bloom, S. trochoidea cell (see Fig. S2 in the supplemental material) densities ranged from 5.5 × 102 ± 0.5 × 102 to 9.6 × 103 ± 0.3 × 103 cells ml−1, with the highest biomass appearing at the during-bloom stage. Total chlorophyll a (Chl a) ranged from 13.3 to 187.17 μg liter−1, and the highest value (170.59 ± 16.58 μg liter−1) (P < 0.01) was recorded after the peak of the dinoflagellate bloom. The photosynthetic efficiency (Fv/Fm) value ranged from 0.43 to 0.70, with the highest value appearing at the prebloom stage.
Changes in biodiversity composition according to bloom stages.
In total, 5,640,302 16S rRNA gene sequences were obtained by sequencing. After removal of low-quality sequences, a mean of 78,337 reads were subsampled from each sample. Sequence statistics can be found in Table S2.
Using a 97% similarity cutoff, 3,625 attached bacterial operational taxonomic units (OTUs) and 6,177 free-living bacterial OTUs were obtained from the field samples (Fig. 1A). Different trends in α-diversity changes were observed between the attached and free-living bacteria. The Chao1 index obtained for the attached bacterial group steadily increased throughout the S. trochoidea bloom, with a peak value of 3,050 emerging at the postbloom stage (Fig. 1B). The Shannon index revealed a similar trend, amplifying over the course of the bloom (Fig. 1C). There were no remarkable changes in the Simpson and dominance indices for the attached bacterial group during the whole bloom process (Fig. 1D and E). In the free-living group, fluctuations were detected throughout the HAB period. The prebloom stage had the highest Chao1 diversity index value (4,420), and the onset/during-bloom stage had the lowest value (3,800) (Fig. 1B). The Shannon index value decreased during the bloom, whereas Simpson index values increased from this time point (Fig. 1C and D). The dominance index displayed a trend contrary to that of the Simpson index, with the lowest value (0.045) appearing at the onset/during-bloom stage (Fig. 1E). After Spearman’s correlation analysis, the richness and Chao1 diversity showed strong positive correlations with temperature, PO43− concentration, and IN/IP ratio (P < 0.05) but obvious negative correlations with salinity and Chl a levels (P < 0.05) (Table S3). There were no significant correlations between environmental variables and Shannon/Simpson indices. Taken together, our data demonstrate that both Chao1 diversity and richness are susceptible indices during algal bloom processes.
FIG 1.

α-Diversity indices of microbial communities sampled from the S. trochoidea bloom stages. (A) Richness; (B) Chao1 index; (C) Shannon index; (D) Simpson index; (E) dominance index.
The UniFrac distance between (or within) attached and free-living bacteria of each sample are shown in Fig. 2i and ii. There were significant differences among the three bloom stages in attached bacteria (R = 0.238, P = 0.001) and free-living bacteria (R = 0.401, P = 0.001). Some obvious changes were also observed after further comparisons between the attached taxa and free-living taxa at each stage. At the prebloom stage, there was a significant difference between the attached group and the free-living group (Fig. 2iii) (R = 0.487, P = 0.001). Furthermore, during bloom onset, the UniFrac average distance for the free-living bacterial group was 180 ± 70, considerably higher than that of the attached bacterial group (40 ± 15) (R = 0.356, P < 0.001) (Fig. 2iv). When the bloom entered the postbloom stage, there was greater variation in taxa of the free-living bacterial group than those of the attached bacterial group (R = 0.755, P < 0.001) (Fig. 2v). The Venn diagram and LEfSe (linear discriminant analysis effect size) picture supply further evidence of this. Specifically, there are different unique and overlapped OTUs in attached and free-living bacteria at different bloom stages (Fig. S3). In addition, in the attached bacterial group, despite some intervariability, Alpha- and Gammaproteobacteria and Actinobacteria showed significantly higher abundance in the onset/during-bloom stage samples than the pre- and postbloom stage samples, while Bacteroidetes and Epsilonproteobacteria were significantly more abundant in the attached microbiota after the HAB enters the postbloom stage (Fig. S4). A similar phenomenon was also observed in the free-living bacterial group; Flavobacterium and Verrucomicrobia were the dominant taxa in the prebloom and onset/during-bloom stages, whereas multiple species codominated in the postbloom stage.
FIG 2.

Statistical difference analysis between (or within) attached bacteria and free-living bacteria. The compared results among the different S. trochoidea bloom stages in attached (i) or free-living (ii) taxa are shown. The compared results with the attached and free-living taxa at each bloom stage (prebloom [iii], during bloom [iv], and postbloom [v]) are shown. Different lower case letters (a, b, and c) indicate statistically significant differences at P = 0.005 (a versus b) or P = 0.001 (a versus c and b versus c). Pre.AT, preattached; Pre.FL, pre-free living.
Microbial community dynamics.
At the phylum level, Proteobacteria and Bacteroidetes were the predominant taxa, occupying 72.5% ± 5.3% and 16.2% ± 5.8% of the total OTU reads, respectively; the other phyla shared the remaining portion (11.3% ± 11.1%) (Fig. S5).
At class or order level, the attached bacteria changed greatly during the bloom process (Fig. 3A and B). Rhodobacterales, Vibrionales, and Flavobacteriales were the most prevalent during the first sampling period, and their relative abundance increased substantially (nearly 70%) during the bloom stage (Fig. 3B). Gammaproteobacteria (Vibrionales, Oceanospirillales, Alteromonadales, Thiotrichales, and Legionellales) and Epsilonproteobacteria (Pseudomonadales and Enterobacteriales) all were present during bloom decline, and the proportional abundances of these taxa increased during the postbloom stage. Together, these taxa made up 30.5% of the total bacterial community (Fig. 3B). The Bacteroidia (especially the Bacteroidales) gradually increased from the prebloom stage (<1%) to during-bloom stage (1.3%) and reached maximum abundance (7.2%) in the postbloom stage. A heat map of differences between taxa at family and genus levels is depicted in Fig. S6.
FIG 3.

Bacterial community composition of samples. Relative abundances of the dominant groups at class (A) and order (B) levels. In panel A, the numbers 1, 2, and 3 represent Proteobacteria, Bacteroidetes, and Actinobacteria, respectively. In panel B, the numbers 1, 2, 3, and 4 represent Alphaproteobacteria, Gammaproteobacteria, Bacteroidia, and Acidimicrobiia, respectively.
Remarkable differences were also observed in the composition of the free-living bacterial community during the HAB event. Rhodobacterales, Flavobacteriales, Bacteroidales, and Oceanospirillales comprised the majority of the free-living bacterial community (Fig. 3B). The relative abundance of Rhodobacterales significantly decreased (from 53.3% to 22.6% abundance) concurrent with termination of the bloom. Oceanospirillales gradually increased during development of the bloom, reached maximum (from 1.2% to 3.5% abundance) during the onset stage, and then declined (2.2%) along with the bloom. Similar to that for Oceanospirillales, levels of Flavobacteriales increased significantly during the bloom process, reached the highest proportional abundance (16.3%) at the onset/during-bloom stage, and then subsequently decreased at the postbloom phase. The order Pseudomonadales was also proportionally abundant, showing an approximately 13.7-fold increase (from 2.3% to 31.6%) as the bloom progressed.
Cluster analysis, correlation of microbial communities with environmental factors, and network analysis.
Bacterial samples were separated into two clusters depending on their taxon type (attached or free living) during the three bloom stages (pre-, during, and postbloom) (Fig. S7A). After assemblage analysis, in the attached group, postbloom stage samples were clustered together and generally divided from samples collected during the two earlier stages. A similar profile was also observed in the free-living groups, with a few overlaps at some stages (Fig. S7A).
Correspondence canonical analysis (CCA) was used to establish whether correlations in microbial structure were associated with environmental parameters. With respect to biochemical factors, PO43− and NO3− made the most significant contributions to the variance in free-living bacterial communities. NH4+ and NO2− were the controlling factors associated with population variation in the attached bacterial community (Fig. S7B). Moreover, the IN/IP ratio was the common factor determining community structure in both taxon types. With respect to physical parameters, temperature and salinity affected the composition of free-living and attached bacteria. We found no obvious negative or positive correlations between population dynamics and pH value. Additional data regarding the relationships between genera and environmental factors are shown in Fig. S8.
Variation partitioning analysis was performed to partition the relative contributions of tested environmental factors and to further assess the influence of environmental parameters on bacterial distribution. Temperature (T), nutrients (N; including NO3−, NO2−, NH4+, PO43−, and IN/IP ratio), and salinity plus pH (SP) accounted for 40.2% of microbial community variation observed in the OTU data (Fig. 4), indicating that they are major factors in determining microbial composition. Independently, T, N, and SP can account for 17.9% (P = 0.01), 14.8% (P = 0.05), and 7.5% (P = 0.05), respectively, of the total variation observed. Interactions between T and N, T and SP, and N and SP explained 9.1%, 4.9%, and 5.6%, respectively, of the variation. Approximately 59.8% of the microbial community variation in OTU data was not explained by these environmental parameters.
FIG 4.
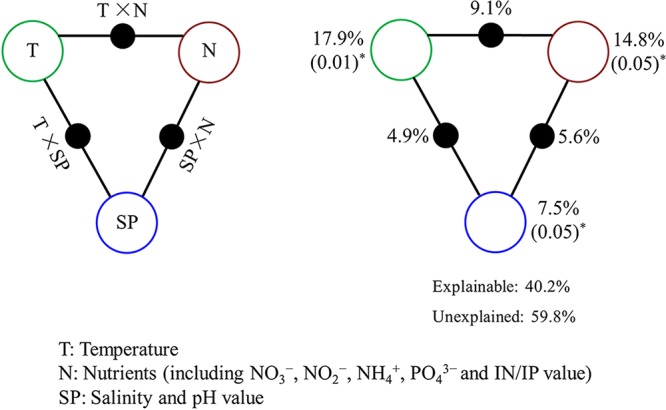
Variation partitioning analysis of microbial distribution explained by environmental factors. (A) General outline. (B) Environmental parameters tested, including temperature (T), nutrients (N), and salinity-plus-pH value (SP). Each diagram represents the variation partitioned into the relative effects of each factor or combination of factors, in which area is proportional to the respective percentages of explained variation. The points of the triangles represent the variation explained by each factor alone. The sides of the triangles represent interactions of any two.
To identify possible biotic factors that contribute to the bacterial community composition, an interaction network based on community correlations was generated (Fig. 5). Among the top 15 (relative abundance) classes of microorganisms, there were 12,152 associations, of which 53.9% were positive and 46.1% were negative. These associations represented inter- and intraspecies interactions in the cooccurring subcommunities, including 7,029 pairs, 3,628 triplets, and 1,495 quadruplets. Among the positive relationships, Alphaproteobacteria, Gammaproteobacteria, Flavobacteria, Actinobacteria, and dinoflagellates were found to be keystone taxa. In pairwise analyses, these five groups cooccurred most frequently, particularly with dinoflagellates and Alphaproteobacteria (Fig. 5A) (P < 0.01). Active interplay was also observed in the negative relationships, which occurred at the highest degree in the Alphaproteobacteria. This class was highly negatively correlated (P < 0.01) with several microorganisms, including Gammaproteobacteria, Flavobacteria, BD1-5, and the target dinoflagellate (such as S. trochoidea) (Fig. 5B). Seven of the 15 most abundant OTUs showed negative nonlinear correlations with S. trochoidea.
FIG 5.

Taxonomic patterns identified within the cooccurrence network. The top 15 interacting taxon groups are depicted as colored segments in a chord diagram, in which ribbons connecting two segments indicate copresence (A) and exclusion (B) links, respectively. The size of the ribbon is proportional to the number of links (copresence and exclusion) between the OTUs assigned to the respective segments, and the color is the segment (of the two involved) with the greater number of total links. Links are dominated by Alphaproteobacteria, Gammaproteobacteria, Flavobacteriia, and Actinobacteria. The chord diagram was generated using the circlized package in R based on the adjacency matrix for the taxon of interest. Beta-Pro, Betaproteobacteria; Epsilon-Pro, Epsilonproteobacteria; Delta-Pro, Deltaproteobacteria.
The assembly processes and relative contribution of fundamental ecological elements.
β-NTI values for most of the attached samples were observed to be beyond the scope of −2 to +2 (P < 0.05) (Fig. 6A) at prebloom and onset/during-bloom stages, indicating that deterministic processes govern community assembly. In contrast, for the samples from the postbloom stage, β-NTI values were less than 2 (P < 0.05) (Fig. 6A). This suggested that stochastic processes shape the assembly of these communities. In the free-living taxa, pairwise comparisons of β-NTI values within each successional bloom phase revealed various patterns in different data sets (Fig. 6B). The β-NTI distributions gradually shifted over the successional stages, from stochastic community assembly (−2 < β-NTI < +2) to homogeneous selection (β-NTI < −2). However, the trend in the β-NTI distribution over successional stages was a little different in the postbloom stage, where the selection was partly weakened (Fig. 6B).
FIG 6.
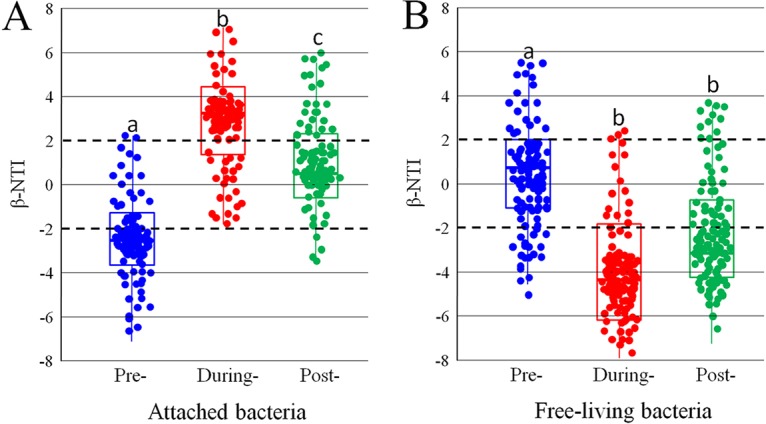
Patterns of β-NTI across HAB stages in attached taxa (A) and free-living taxa (B). Horizontal lines indicate upper and lower significance thresholds at β-NTI of +2 and −2, respectively.
To estimate the relative contributions of each assembly process over HAB stages, weighted β-NTI was used in combination with Bray-Curtis-based Raup-Crick (RCbray) and null analysis to quantify the ecological processes that influence bacterioplankton composition (Fig. 7 and Table S4). In the attached group (Fig. 7A), the trend in the fraction of homogeneous selection was a decrease with time (from 62.5% at prebloom phase to 27.5% at postbloom phase). However, the contribution of heterogeneous selection gradually increased; values at pre-, during-, and postbloom stages were 7.5%, 17.7%, and 22.1%, respectively. Furthermore, in these data sets dispersal limitation and homogenizing dispersal showed significant influences in phycosphere bacteria; these influences increased in the postbloom phase of the HAB process. For the ecological drift fraction, the percentage was relatively stable (between 14.4 and 18.5%). Compared with that for the attached taxa, a different view was observed in the free-living bacteria (Fig. 7B). The fraction of deterministic selection processes (homogeneous selection and homogenizing dispersal) was higher in prebloom and onset/during-bloom stages, while the fraction of stochastic processes (dispersal limitation and drift) increased in the postbloom stage. Interestingly, the fraction of ecological drift increased from 11.2% (prebloom) to 13.5% (during bloom), up to 24.7% (postbloom). Compared with the initial phase, the ratio occupied by drift in the postbloom period increased 2.2-fold.
FIG 7.

Relative contributions of ecological processes that determine community assembly across sample types. The percentage of turnover in bacterial community assembly governed by various deterministic factors (homogeneous/heterogeneous selection and variable homogenizing dispersal), as well as the random fraction (dispersal limitation, drift, and undominated process), is shown.
DISCUSSION
Phycosphere bacteria demonstrate strong responses to an HAB event (Fig. 3, 6, and 7), and the results confirmed our hypothesis that microbial community succession is interconnected to the stages of the HAB and that the assembly mechanisms are coinfluenced by stochastic-neutral processes. In line with our results, previous work confirmed that during the assembly of prokaryotic communities, both environmental selection as well as neutral processes can play roles (11). Similarly, Lindh et al. (34) found that different taxonomic groups of bacterioplankton in the Baltic Sea were structured by environmental filtering, mass effects, patch dynamics, or the neutral model. For HAB events, only limited work was performed in diatoms. That work demonstrated three main occurrences in the assembly of microbial communities: a shift from strong heterogeneous selection during the first stage of the bloom to stochasticity during the middle stage, followed by strong homogeneous selection in the last stage (16). To the best of our best knowledge, this is the first study using the ecological model theory to evaluate the assembly mechanism in dinoflagellates, which display some similar or special structuring patterns depending on the host type. In this work, we focused on the bacterial community dynamics and assembly profile during the trajectories of an S. trochoidea bloom.
The predominant bacteria associated with S. trochoidea included Rhodobacterales, Bacteroidetes, Rhodospirillales, Flavobacteriales, and Pseudomonadales, which were coexisting in both free-living and attached taxon groups. Of these, Rhodobacterales (22.7 to 56.2%) were the most abundant (Fig. 3). Composition analysis showed a significant difference in community structure between (or within) the free-living and attached bacteria (Fig. 2). Moreover, Bray-Curtis similarity-based dendrogram analysis revealed that free-living and attached bacteria were distinctly clustered (see Fig. S7A in the supplemental material). Algae may provide a distinct microhabitat for attached bacteria that require different activities (e.g., enzymes and functions) than free-living bacteria (35), which may lead to the observed differences. Besides the habitat type (free-living or attached), bacterial community composition also can be affected by the HAB phase (35). Similarly, in the present study, diversity indices indicated that the composition of both free-living and attached bacterial communities was likely to vary depending on S. trochoidea life cycle stages (Fig. 1). During HAB processes, algae release many low-molecular-weight (LMW) molecules, including carbohydrates, organic acids, amino acids, and sugar alcohols, in the earliest stage of blooms (36). However, as the blooms move to the declining phase, algae start to release higher-molecular-weight (HMW) macromolecules, including proteins, nucleic acids, polysaccharides, and lipids. This is predominantly due to cell lysis (37). Therefore, the different stages may have changed the concentration and ratio of LMW to HMW, and this variation can affect free-living and attached bacterial composition. In addition, compared with the attached bacteria, the change in α-diversity indices in the free-living community was relatively greater among the different phases (Fig. 1). A possible reason for this is that attached bacteria benefit more from the substrates provided by algal hosts, which may lead to a reduction in diversity indices (38). In addition, some other unpredictable factors seem to play a role in the assembly of phycosphere communities (39).
To evaluate the relative contribution of environmental parameters to microbial communities, physicochemical data were analyzed to determine which factors (temperature, nutrients, pH, and salinity) drive community variability. These factors independently explained 7.5 to 17.9% of the total observed variations in OTU data (Fig. 4), partly supporting the principle of “everything is everywhere, but the environment selects” (40). However, only 40.2% of microbial community variation in the OTU data in this study was accounted for by the parameters mentioned above, indicating that other abiotic factors not measured in this study (such as dissolved organic carbon, sinicate, chemical oxygen demand, and trace elements) or biotic parameters (such as community interactions) also are significant contributors to community structure. Indeed, diverse network relationships at the class level were observed among the phycosphere microbes (Fig. 5), indicating that biotic factors (cooccurrence or exclusion) possess roles in influencing the distribution of microbial communities during algal events. Although the precise contribution of interactions between community members with community variations is still unknown, the dynamics of biotic interactions might be a factor that shapes the succession of microbial communities. Consistent with previous studies (41–44), we found that environmental factors can partially explain microbial community structure. Further analysis of multiple factors, such as physicochemical parameters, competitive-cooperative relationships, and predator-prey interactions, along with their network relevance, are required to better understand how conditional factors influence changes in community variation.
In terms of the assembly profile, for free-living taxa, our data indicate that in the prebloom stage of an HAB, stochastic processes (more than 30%) participate in shaping the community assembly. However, the relative importance of deterministic factors was greatly enhanced at the peak-bloom stage. Based on the remarkable change in abiotic/biotic parameters (Table 1), we speculated that the peak phase (i.e., during-bloom stage) in the HAB imposed strong environmental filtering on microbial communities. Our observation of β-NTI values ranging from null to negative to weakly negative supports the fact that the influence of deterministic processes in phylogenetic turnover varied across stages (Fig. 6B). When the HAB enters the postbloom stage, in spite of the dominance of environmental selection, partial contributions of stochastic assembly were observed, highlighting the importance of the two processes during the decline phase.
A different pattern was observed in the assembly profile for attached microbes. The β-NTI distributions gradually shifted over time, from homogeneous selection (β-NTI < −2) to variable selection (β-NTI > 2), up to stochastic community assembly (−2 < β-NTI < 2) (Fig. 6A). As expected, the results indicated that at pre- and during-bloom stages of an HAB, the attached microbial communities are controlled mainly by niche-based processes. However, when the HAB enters the postbloom stage, a different picture is apparent, i.e., stochastic processes dominate in this period. This may be due to the disturbance (algal bloom event), which resulted in competition based on limited nutrients or space (45). This is further supported by literature demonstrating that competition contributed to the random process of attached bacterial distribution (46). With respect to the framework of neutral and deterministic processes, when a community suffers from a disturbance, the relative influence of stochastic processes may grow larger due to the subsequent loss of selective forces (12). In our study, the stochastic processes appear in the postbloom phase, indicating that bacteria undergo rearrangements driven by the local conditions. Our findings indicate that the relative effects of stochastic and deterministic community assembly processes vary with successional time.
Based on the published framework, we calculated the relative contributions of variable selection, homogeneous selection, dispersal limitation, homogenizing dispersal, and drift (47). Of these, we showed that homogeneous selection was largely responsible for community assembly in the first stage of the HAB, while drift and dispersal limitation were the dominant contributors in the postbloom stage (Fig. 7). For ecological drift, the impact was greatest at the postbloom stage (especially in free-living taxa), which may cause bacterial abundances to fluctuate, and largely resulted from stochastic changes in population size, including microbial movements and immigration-emigration events (48). Combining previous studies with the results observed here indicates that phycosphere microbial trajectory is governed by stochastic-deterministic balance.
To intuitively understand the assembly mechanism of phycosphere microbes in an HAB event, a conceptual model was constructed (Fig. 8) based on the null deviation value (12). We demonstrated that the HAB caused a quantitative change in succession and how communities were assembled (the relative importance of niche versus neutral processes), and that this shifted with bloom stage. For free-living bacteria, phase 1 (prebloom stage) is characterized by a brief upsurge in the relative role of neutral processes in community assembly, most likely because random processes are strongly influencing community structure (49). During phase 2 (during-bloom stage), algae start to grow and bloom. Niche-based processes in the disturbance act as strong filters on microbial community composition in this phase. This can be depicted by increases in β-diversity if the disturbance is heterogeneous or decreases in β-diversity if the disturbance is more homogenous (50). The importance of niche-based processes following bloom events has been reported in other experimental systems (15). Weak environmental filtering from increased levels of nutrients relaxes communities partially back toward the neutral state in the postbloom stage (homogeneous selection, phase 3). Distinct trajectories and multiple equilibria are likely initiated at this stage. To some degree, this supports other work that suggests limit-random processes participate in community assembly within successional stages (51, 52). Unlike the free-living members, a different phenomenon is observed in attached taxa (Fig. 8). Weak variable selection is observed at the prebloom stage (phase 1). Strong environmental filtering from the bloom host (homogeneous and heterogeneous selection, phase 2) then decreases community diversity at the onset stage of the press disturbance. Finally, as the bloom enters the poststage (phase 3), the niche-filter role is relatively weaker in shaping community structure; it may be that the homogenizing dispersal provides a buffer against environmental filters (53). It should be noted that Fig. 8 is applicable only to one kind of event in S. trochoidea. In the future, we would aim to broaden the scope of this potential model by studying more microbial profiles in different types of algae.
FIG 8.

Conceptual model depicting two different possible scenarios in bacterial community assembly processes during the HAB event. Close to 0, neutral processes are more important; larger (+2) or negative (−2) niche-based processes are more important.
Conclusions.
Microbial communities exhibited clear successional patterns during the HAB process, and environmental parameters and biotic interactions are both responsible for the structural changes. Two main theories (niche theory and neutral theory) were examined to ascertain relative contributions toward explaining the composition of microbial communities in an HAB event. In attached bacteria, community assembly is mainly governed by environmental filtering in the pre- and onset/during-bloom stages, while natural selection participates in the postbloom stage. However, a different picture was observed in the free-living group. In this group, bacterial community assembly in the early successional phase is affected in part by stochastic processes. Moreover, the relative importance of deterministic processes progressively increases in later successional phases. Our study increases our understanding of the specific ecological processes that influence microbial community assembly in the phycosphere microecosystem over the course of an HAB. To obtain a more comprehensive understanding about the assembly profile of bacterioplankton in HAB events, identification of additional factors (host type, hydrological parameters, substrate status, and longer time series) that alter the balance between stochastic and deterministic assembly processes are critical factors to investigate moving forward.
MATERIALS AND METHODS
Site description and sample collection.
A high-precision global positioning system (GPS) recorded the locations of the sampling sites. Samples were collected from the coast of Shenzhen, China (117°0.03′24 E, 23°0.44′0.19 N), where a Scrippsiella trochoidea bloom regularly occurs. To obtain a complete algal blooming sample series, the phytoplankton biomass was monitored twice a week by track sampling. An algal bloom occurred from 15 to 28 August 2017. Surface seawater samples (0.5 to 1.0 m) were collected during the 2-week bloom period.
Three phases (each of 3 to 5 days) were sampled in the bloom cycle, prebloom, during bloom, and postbloom (see Fig. S1 in the supplemental material), with 12 parallel samples collected at each phase. Each sample was comprised of 2 liters of seawater collected with a Niskin bottle. Water samples were transferred to a polyethylene container and kept under ice bath conditions during delivery to the laboratory. Samples were successively filtered through a 300-mesh plankton net to remove large zooplankton and impurity particles. Attached and free-living microbes were then obtained according to the methods of Tan et al. (28). Briefly, water samples were filtered through a 10-μm-pore-size fiber membrane (diameter, 47 mm; Millipore) to collect the first group of microorganisms (i.e., the attached taxa, part 1). The filtrate was then passed through a 0.22-μm-pore-size fiber membrane (diameter, 47 mm; Millipore) to obtain the second group of microorganisms (i.e., the free-living taxa, part 2). The filters (parts 1 and 2) were immediately stored at −80°C before DNA extraction. In addition, part of each water sample was preserved with 1% Lugol’s iodine solution for phytoplankton identification and enumeration. S. trochoidea organisms were observed and counted under an optical microscope (×100 to ×400 magnification; Zeiss, Germany).
Environmental parameters.
At each sampling time, the physicochemical factors (temperature, salinity, and pH) of the seawater were recorded using a multiparameter water quality Sonde 6600 V2 (YSI, USA). Chlorophyll a (Chl a) concentration and photosynthetic activity (Fv/Fm) were measured using a chlorophyll fluorescence system (PHYTO-PAM; Walz, Germany). Other environmental factors, including nitrate nitrogen (NO3−), nitrite nitrogen (NO2−), ammonium nitrogen (NH4+), and phosphate phosphorus (PO43−), were measured on a discrete chemistry analyzer (CleverChem; Anna, Germany) according to the manufacturer’s operating instructions. The complete protocols are available online at http://cmore.soest.hawaii.edu. The approximate inorganic nitrogen/inorganic phosphate (IN/IP) value was measured according to the methods of Murphy and Riley (54) and Greenberg et al. (55). Standards for NO3−, NO2−, and PO43− were obtained from Shanghai Macklin Biochemical Co., Ltd. (China), and the purity was >99%. Statistical analyses of the tested environmental variables (NH4+, NO2−, NO3−, PO43−, pH, salinity, and temperature) were performed using SPSS 10.0 software (SPSS, Chicago, IL), and P values of <0.05 or <0.01 were considered statistically significant.
DNA extraction, PCR amplification, and pyrosequencing.
Total DNA was extracted from filters using a FastDNA spin kit (mBio, USA) according to the manufacturer’s instructions. The extracted DNA was dissolved in 100 μl Tris-EDTA (TE) buffer, quantified based on optical density (OD) using a NanoDrop 2000 spectrophotometer (OD260/OD230 of >1.8), and stored at −20°C until further use.
Prokaryote 16S rRNA genes were amplified by following the procedure described by Fierer et al. (56) using the primer pair F515 (5′-GTGCCAGCMGCCGCGG-3′) and R907 (5′-CCGTCAATTCMTTTRAGTTT-3′). This targeted gene region (392 bp) can provide sufficient resolution for accurate taxonomic classification of microbial sequences (57, 58). The forward primer includes an 8-bp barcode, indicated by “M” in the above-described sequence, for multiplexing of samples during sequencing. The barcode sequence for each sample is listed in Table S1. Samples were amplified in triplicate as described previously (56). Briefly, reactions were carried out in a 50-μl volume using a Bio-Rad PCR cycler with predenaturation at 95°C for 3 min; 28 cycles of denaturation at 95°C for 30 s, annealing at 55°C for 30 s, and elongation at 72°C for 45 s; and a final extension for 10 min at 72°C. Replicate PCRs for each sample were pooled and purified using a QIAquick G01 extraction kit (Qiagen, CA, USA). A single composite sample was prepared for pyrosequencing by combining approximately equimolar amounts of PCR products from each sample. Sequencing was performed at MeiGe Biotech, Co., Ltd. (Guangzhou, China), using a MiSeq platform (Illumina) in a paired-end 250-bp sequence read run.
Processing sequencing data.
Raw sequences were processed and checked using mothur and QIIMe tools and were then trimmed and filtered according to previously described methods (59). Briefly, sequencing primers and barcodes were removed from the raw sequence reads by allowing 1.5 mismatches to the barcode and 2 mismatches to the primer sequence. Sequences were removed if they had homopolymeric regions of more than 6 nucleotides (nt), were smaller than 200 nt, had quality scores lower than 25, or were identified as being chimeric. After removal of low-quality reads, representative sequences were annotated using an alignment tool (BLAST) by searching against the Ribosomal Database Project and the Silva database augmented with sequences from major marine bacterial taxa (for 16S rRNA gene sequences) (60). Operational taxonomic units (OTUs) were clustered at 97% identity using the UPARSE (version 7.1; http://drive5.com/uparse/) pipeline (61). To prevent artificial diversity inflation, singletons were removed as putative sequencing errors or PCR amplification artifacts (62). Taxonomic assignments were achieved by UCLUST with the SILVA 128 reference database. OTUs affiliated with chloroplast, mitochondrion, unassigned, and unclassified sequences and singletons were removed from the data set before downstream analysis. The final number of sequences in each sample ranged from 52,907 to 124,666 reads, yielding a total of 144,133 OTUs (Table S2).
Diversity, environmental factor correlation, and network analyses.
For estimation of α-diversity, sequences in each sample were normalized at the lowest sequence depth (1/100 depth compared to the largest grouping), and the α-diversity indices for each sample were calculated using mothur (63). To track the successional trajectories of microbial communities, the phylogenetic-based UniFrac distance (64) was used to investigate the dissimilarities of microbial community composition at different stages of the bloom process. Phylogenetic diversity was measured using the picante package in R (v.3.2.5). Statistically significant differences in taxon abundance were identified using Welch’s t test in the STAMP program (65). The structure of the bacterial community in each sample was compared by using 2STAGE analysis in the PRIMER package (PRIMER, version 6; PRIMER-E, Ltd., Lutton, UK) (66). A heat map of microbial communities was created using the PhyloTemp tool (http://www.phylotemp.microeco.org) developed by Polson (67), whereby relative abundance data are clustered based on the Bray-Curtis dissimilarity algorithm. Canonical correspondence analysis (CCA) was used to link variations in microbial communities to environmental properties and was carried out using Canoco 5 software. A partial redundancy analysis (RDA)-based variance partitioning analysis (VPA) was conducted to examine the relative contribution of environmental factors in influencing microbial community structure (68). Significant differences were defined as a P value of <0.05 or <0.01. Environmental variables were standardized before the VPA analysis, which was achieved using the betadisper function in the vegan package (69).
To evaluate the cooccurrence patterns among the phycosphere microbes, interaction network analysis was performed according to the methods of Faust et al. (70) and Yang et al. (71). To reduce network complexity, only the top 15 classes (highest relative abundance) in the microbial community were determined. An affiliation network was generated using the R package igraph, version 1.0.1 (available at http://igraph.org), and is defined as a network where the members are affiliated with one another based on comembership of a group or coparticipation in some type of event. The direct connections among the different genera were extracted from the two-mode affiliation network using igraph’s bipartite.projection function. Nodes in the reconstructed network represented variable taxa, and edges connecting the nodes represented correlations between genera. Topographic features of the network, including centrality and edge weights, were also analyzed using functions available in the igraph package. Information on the target network was further organized in matrices and visualized in chord diagrams using the R package circlize, version 0.3.5 (available at https://cran.r-project.org/web/packages/circlize/index.html).
Quantifying influences of ecological processes.
To investigate the assembly mechanisms of the bacterial communities, the ses.mntd function in the picante package of R (72) was applied to calculate the mean phylogenetic distance to the closest relative of each taxon (the mean nearest taxon distance, or MNTD) and the standardized-effect size (SES) of the MNTD (ses.MNTD) of each sample (73). Permutational multivariate analysis of variance (PERMANOVA) was used to test the significance of the difference of the stage communities between the observed similarity matrices with the null model expectation (74). Negative SES values and low quantiles (P < 0.05) indicate that community members are clustered, i.e., that phylogenetic distances across a phylogenetic tree in a given community, relative to the regional pool of taxa, are more closely related than expected by chance. Conversely, positive SES values and high quantiles (P > 0.95) indicate phylogenetic distance over dispersion, i.e., greater-than-expected phylogenetic distances among cooccurring taxa. Based on the random shuffling of OTU labels across the tips of the phylogeny, the β-nearest taxon index (β-NTI) was computed as the number of standard deviations that observed mean nearest taxon distance (β-MNTD) values deviated from the mean of the null distribution (999 null replicates) (1, 75). The nearest taxon index (NTI) is equal to the inverse of ses.MNTD and is calculated as the difference between the observed MNTD and mean expected MNTD divided by the standard deviation for random expected values (12). β-MNTD and β-NTI were calculated using the R function picante as previously described (7, 50). Briefly, if the NTI values are greater than −2 but less than +2, then community assembly is mainly governed by stochastic or dispersal-related processes for a given pairwise comparison. However, if the NTI values are higher than +2 or less than −2, then deterministic processes play more important roles in structuring microbial phylogenetic turnover (76).
To evaluate the relative influence of ecological processes (variable selection, homogeneous selection, dispersal limitation, and drift) governing the microbial community assembly of each bloom stage, a null-model-based statistical framework developed by Stegen et al. (7, 50) was used. First, β-NTI was quantified for all pairwise community comparisons using an in-house pipeline (http://mem.rcees.ac.cn:8080). β-NTI values of >2 or <−2 indicate the influence of deterministic processes in structuring the community, whereas β-NTI values between −2 and 2 indicate the influence of stochastic processes (9). Second, the combination of β-NTI and Bray-Curtis-based Raup-Crick (RCbray) was further used to infer the relative importance of major ecological processes governing the bacterial communities. RCbray was calculated according to the method of Chase and Meyers (9) using Bray-Curtis dissimilarities. β-NTI values of >+2 or <−2 indicate that community turnover is determined by variable or homogeneous selection (causes community composition to be similar under consistent environmental conditions), respectively (9). β-NTI values of >−2 and <+2, but with an RCbray value of >0.95 or <−0.95, indicates that community turnover is determined by dispersal limitation or homogenizing dispersal, respectively. Furthermore, β-NTI values of >−2 and <+2, with an RCbray of >−0.95 and <0.95, suggests that the community assembly is not dominated by any single process described above (47).
Data availability.
Sequence data reported here have been deposited in the NCBI Sequence Read Archive database under the accession number SRP154568.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NSFC (41476092 and 41741015), the S&T Projects of Shenzhen Science and Technology Innovation Committee (JCYJ20170412171959157, JCYJ20170817160708491, and JCYJ20170412171947159), the Trade and Information Commission of Shenzhen (20180124085935704), and China Ocean Mineral Resources R & D Association (COMRA) Special Foundation (DY135-E2-5-4).
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/AEM.00349-19.
REFERENCES
- 1.Zhou J, Ning D. 2017. Stochastic community assembly: does it matter in microbial ecology? Microbiol Mol Biol Rev 81:e00002-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhou J, Deng Y, Zhang P, Xue K, Liang Y, Van Nostrand JD, Yang Y, He Z, Wu L, Stahl DA, Hazen TC, Tiedje JM, Arkin AP. 2014. Stochasticity, succession, and environmental perturbations in a fluidic ecosystem. Proc Natl Acad Sci U S A 111:E836–E845. doi: 10.1073/pnas.1324044111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chesson P. 2000. Mechanisms of maintenance of species diversity. Annu Rev Ecol Syst 31:343–366. doi: 10.1146/annurev.ecolsys.31.1.343. [DOI] [Google Scholar]
- 4.Chave J. 2004. Neutral theory and community ecology. Ecol Lett 7:241–253. doi: 10.1111/j.1461-0248.2003.00566.x. [DOI] [Google Scholar]
- 5.Ofiteru ID, Lunn M, Curtis TP, Wells GF, Criddle CS, Francis CA, Sloan WT. 2010. Combined niche and neutral effects in a microbial wastewater treatment community. Proc Natl Acad Sci U S A 107:15345–15350. doi: 10.1073/pnas.1000604107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vellend M, Srivastava DS, Anderson KM, Brown CD, Jankowski JE, Kleynhans EJ, Kraft NJB, Letaw AD, Macdonald AAM, Maclean JE, Myers-Smith IH, Norris AR, Xue X. 2014. Assessing the relative importance of neutral stochasticity in ecological communities. Oikos 123:1420–1430. doi: 10.1111/oik.01493. [DOI] [Google Scholar]
- 7.Stegen JC, Lin X, Konopka AE, Fredrickson JK. 2012. Stochastic and deterministic assembly processes in subsurface microbial communities. ISME J 6:1653–1664. doi: 10.1038/ismej.2012.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang J, Shen J, Wu Y, Tu C, Soininen J, Stegen JC, He J, Liu X, Zhang L, Zhang E. 2013. Phylogenetic beta diversity in bacterial assemblages across ecosystems: deterministic versus stochastic processes. ISME J 7:1310–1321. doi: 10.1038/ismej.2013.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chase JM, Myers JA. 2011. Disentangling the importance of ecological niches from stochastic processes across scales. Philos Trans R Soc Lond B Biol Sci 366:2351–2363. doi: 10.1098/rstb.2011.0063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fargione J, Brown CS, Tilman D. 2003. Community assembly and invasion: an experimental test of neutral versus niche processes. Proc Natl Acad Sci U S A 100:8916–8920. doi: 10.1073/pnas.1033107100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Langenheder S, Székely AJ. 2011. Species sorting and neutral processes are both important during the initial assembly of bacterial communities. ISME J 5:1086–1094. doi: 10.1038/ismej.2010.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dini-Andreote F, Stegen JC, van Elsas JD, Salles JF. 2015. Disentangling mechanisms that mediate the balance between stochastic and deterministic processes in microbial succession. Proc Natl Acad Sci U S A 112:E1326–E1332. doi: 10.1073/pnas.1414261112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bryant JA, Lamanna C, Morlon H, Kerkhoff AJ, Enquist BJ, Green JL. 2008. Colloquium paper: microbes on mountainsides: contrasting elevation patterns of bacterial and plant diversity. Proc Natl Acad Sci U S A 105:11505–11511. doi: 10.1073/pnas.0801920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ren L, Jeppesen E, He D, Wang J, Liboriussen L, Xing P, Wu QL. 2015. pH influences the importance of niche-related and neutral processes in lacustrine bacterioplankton assembly. Appl Environ Microbiol 81:3104–3114. doi: 10.1128/AEM.04042-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xue Y, Chen H, Yang JR, Liu M, Huang B, Yang J. 2018. Distinct patterns and processes of abundant and rare eukaryotic plankton communities following a reservoir cyanobacterial bloom. ISME J 12:2263–2277. doi: 10.1038/s41396-018-0159-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang H, Wang K, Shen L, Chen H, Hou F, Zhou X, Zhang D, Zhu X. 2018. Microbial community dynamics and assembly follow trajectories of an early-spring diatom bloom in a semi-enclosed bay. Appl Environ Microbiol 84:e01000-18. doi: 10.1128/AEM.01000-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayali X. 2018. Editorial: metabolic interactions between bacteria and phytoplankton. Front Microbiol 9:727. doi: 10.3389/fmicb.2018.00727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhou J, Lyu YH, Richlen ML, Anderson DM, Cai ZH. 2016. Quorum sensing is a language of chemical signals and plays an ecological role in algal-bacterial interaction. Crit Rev Plant Sci 1:82–105. doi: 10.1080/07352689.2016.1172461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou J, Lin GH, Cai ZH. 2016. The roles of microbes in matter cycles in phycosphere niche. Chin J Appl Ecol 27:2708–2716. [DOI] [PubMed] [Google Scholar]
- 20.Zheng TL. (ed). 2011. The microbial control of harmful algal bloom. The Nanqiang Publisher of Xiamen University, Xiamen, China. [Google Scholar]
- 21.Jones KL, Mikulski CM, Barnhorst A, Doucette GJ. 2010. Comparative analysis of bacterioplankton assemblages from Karenia brevis bloom and nonbloom water on the west Florida shelf (Gulf of Mexico, USA) using 16S rRNA gene clone libraries. FEMS Microbiol Ecol 73:468–485. doi: 10.1111/j.1574-6941.2010.00914.x. [DOI] [PubMed] [Google Scholar]
- 22.Riemann L, Steward GF, Azam F. 2000. Dynamics of bacterial community composition and activity during a mesocosm diatom bloom. Appl Environ Microbiol 66:578–587. doi: 10.1128/aem.66.2.578-587.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Theroux S, Huang Y, Amaral-Zettler L. 2012. Comparative molecular microbial ecology of the spring haptophyte bloom in a Greenland arctic oligosaline lake. Front Microbiol 3:415. doi: 10.3389/fmicb.2012.00415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Buchan A, LeCleir GR, Gulvik CA, González JM. 2014. Master recyclers: features and functions of bacteria associated with phytoplankton blooms. Nat Rev Microbiol 12:686–698. doi: 10.1038/nrmicro3326. [DOI] [PubMed] [Google Scholar]
- 25.Teeling H, Fuchs BM, Becher D, Klockow C, Gardebrecht A, Bennke CM, Kassabgy M, Huang S, Mann AJ, Waldmann J, Weber M, Klindworth A, Otto A, Lange J, Bernhardt J, Reinsch C, Hecker M, Peplies J, Bockelmann FD, Callies U, Gerdts G, Wichels A, Wiltshire KH, Glöckner FO, Schweder T, Amann R. 2012. Substrate-controlled succession of marine bacterioplankton populations induced by a phytoplankton bloom. Science 336:608–611. doi: 10.1126/science.1218344. [DOI] [PubMed] [Google Scholar]
- 26.Amin SA, Hmelo LR, van Tol HM, Durham BP, Carlson LT, Heal KR, Morales RL, Berthiaume CT, Parker MS, Djunaedi B, Ingalls AE, Parsek MR, Moran MA, Armbrust EV. 2015. Interaction and signaling between a cosmopolitan phytoplankton and associated bacteria. Nature 522:98–101. doi: 10.1038/nature14488. [DOI] [PubMed] [Google Scholar]
- 27.Needham DM, Fuhrman JA. 2016. Pronounced daily succession of phytoplankton, archaea and bacteria following a spring bloom. Nat Microbiol 1:1–7. doi: 10.1038/nmicrobiol.2016.5. [DOI] [PubMed] [Google Scholar]
- 28.Tan SJ, Zhou J, Zhu XS, Yu S, Zhan W, Wang B, Cai ZH. 2015. An association network analysis among microeukaryotes and bacterioplankton reveals algal bloom dynamics. J Phycol 51:120–132. doi: 10.1111/jpy.12259. [DOI] [PubMed] [Google Scholar]
- 29.Poisot T, Péquin B, Gravel D. 2013. High-throughput sequencing: a roadmap toward community ecology. Ecol Evol 3:1125–1139. doi: 10.1002/ece3.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mori AS, Fujii S, Kitagawa R, Koide D. 2015. Null model approaches to evaluating the relative role of different assembly processes in shaping ecological communities. Oecologia 178:261–273. doi: 10.1007/s00442-014-3170-9. [DOI] [PubMed] [Google Scholar]
- 31.Zhou J, Song X, Zhang CY, Chen GF, Lao YM, Jin H, Cai ZH. 2018. Distribution patterns of microbial community structure along a 7000-mile latitudinal transect from the Mediterranean Sea across the Atlantic ocean to the Brazilian coastal sea. Microb Ecol 76:592–609. doi: 10.1007/s00248-018-1150-z. [DOI] [PubMed] [Google Scholar]
- 32.Asplund-Samuelsson J, Sundh J, Dupont CL, Allen AE, McCrow JP, Celepli NA, Bergman B, Ininbergs K, Ekman M. 2016. Diversity and expression of bacterial metacaspases in an aquatic ecosystem. Front Microbiol 7:1043. doi: 10.3389/fmicb.2016.01043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilbert JA, Field D, Huang Y, Edwards R, Li W, Gilna P, Joint I. 2008. Detection of large numbers of novel sequences in the metatranscriptomes of complex marine microbial communities. PLoS One 3:e3042. doi: 10.1371/journal.pone.0003042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindh MV, Sjostedt J, Casini M, Andersson A, Legrand C, Pinhassi J. 2016. Local environmental conditions shape generalist but not specialist components of microbial metacommunities in the Baltic Sea. Front Microbiol 7:2078. doi: 10.3389/fmicb.2016.02078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bagatini IL, Eiler A, Bertilsson S, Klaveness D, Tessarolli LP, Vieira A. 2014. Host-specificity and dynamics in bacterial communities associated with bloom-forming freshwater phytoplankton. PLoS One 9:e85950. doi: 10.1371/journal.pone.0085950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myklestad SM. 2000. Dissolved organic carbon from phytoplankton, p 111–148. In Wan-Gersky P. (ed), The handbook of environmental chemistry, vol 5D Springer, New York, NY. [Google Scholar]
- 37.Azam F, Malfatti F. 2007. Microbial structuring of marine ecosystems. Nat Rev Microbiol 10:782–791. doi: 10.1038/nrmicro1747. [DOI] [PubMed] [Google Scholar]
- 38.Park BS, Guo RY, Lim WA, Ki JS. 2018. Importance of free-living and particle-associated bacteria for the growth of the harmful dinoflagellate Prorocentrum minimum: evidence in culture stages. Mar Freshw Res 69:290–299. doi: 10.1071/MF17102. [DOI] [Google Scholar]
- 39.Chase JM. 2007. Drought mediates the importance of stochastic community assembly. Proc Natl Acad Sci U S A 104:17430–17434. doi: 10.1073/pnas.0704350104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.O'Malley MA. 2008. 'Everything is everywhere: but the environment selects': ubiquitous distribution and ecological determinism in microbial biogeography. Stud Hist Philos Biol Biomed Sci 39:314–325. doi: 10.1016/j.shpsc.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 41.Rohwer F, Thurber RV. 2009. Viruses manipulate the marine environment. Nature 459:207–212. doi: 10.1038/nature08060. [DOI] [PubMed] [Google Scholar]
- 42.Smetacek V. 2012. Making sense of ocean biota: how evolution and biodiversity of land organisms differ from that of the plankton. J Biosci 37:589–607. doi: 10.1007/s12038-012-9240-4. [DOI] [PubMed] [Google Scholar]
- 43.Lima-Mendez G, Faust K, Henry N, Decelle J, Colin S, Carcillo F, Chaffron S, Ignacio-Espinosa JC, Roux S, Vincent F, Bittner L, Darzi Y, Wang J, Audic S, Berline L, Bontempi G, Cabello AM, Coppola L, Cornejo-Castillo FM, d'Ovidio F, De Meester L, Ferrera I, Garet-Delmas MJ, Guidi L, Lara E, Pesant S, Royo-Llonch M, Salazar G, Sánchez P, Sebastian M, Souffreau C, Dimier C, Picheral M, Searson S, Kandels-Lewis S, Tara Oceans Coordinators, Gorsky G, Not F, Ogata H, Speich S, Stemmann L, Weissenbach J, Wincker P, Acinas SG, Sunagawa S, Bork P, Sullivan MB, Karsenti E, Bowler C, de Vargas C, Raes J. 2015. Ocean plankton. Determinants of community structure in the global plankton interactome. Science 348:1262073. doi: 10.1126/science.1262073. [DOI] [PubMed] [Google Scholar]
- 44.Worden AZ, Follows MJ, Giovannoni SJ, Wilken S, Zimmerman AE, Keeling PJ. 2015. Rethinking the marine carbon cycle: factoring in the multifarious lifestyles of microbes. Science 347:1257594. doi: 10.1126/science.1257594. [DOI] [PubMed] [Google Scholar]
- 45.Zhou J, Richlen ML, Sehein TR, Kulis DM, Anderson DM, Cai ZH. 2018. Microbial community structure and associations during a marine dinoflagellate bloom. Front Microbiol 9:1201. doi: 10.3389/fmicb.2018.01201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zheng YM, Cao P, Fu B, Hughes JM, He JZ. 2013. Ecological drivers of biogeographic patterns of soil archaeal community. PLoS One 8:e63375. doi: 10.1371/journal.pone.0063375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stegen JC, Lin X, Fredrickson JK, Kratz TK, Wu CH, McMahon KD. 2015. Estimating and mapping ecological processes influencing microbial community assembly. Front Microbiol 6:370. doi: 10.3389/fmicb.2015.00370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ruokolainen L, Ranta E, Kaitala V, Fowler MS. 2009. When can we distinguish between neutral and non-neutral processes in community dynamics under ecological drift? Ecol Lett 12:909–919. doi: 10.1111/j.1461-0248.2009.01346.x. [DOI] [PubMed] [Google Scholar]
- 49.Leibold MA, McPeek MA. 2006. Coexistence of the niche and neutral perspectives in community ecology. Ecology 87:1399–1410. doi: 10.1890/0012-9658(2006)87[1399:COTNAN]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 50.Stegen JC, Lin X, Fredrickson JK, Chen X, Kennedy DW, Murray CJ, Rockhold ML, Konopka A. 2013. Quantifying community assembly processes and identifying features that impose them. ISME J 7:2069–2079. doi: 10.1038/ismej.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ellner SP, Fussmann G. 2003. Effects of successional dynamics on metapopulation persistence. Ecology 84:882–889. doi: 10.1890/0012-9658(2003)084[0882:EOSDOM]2.0.CO;2. [DOI] [Google Scholar]
- 52.Cadotte MW. 2007. Concurrent niche and neutral processes in the competition-colonization model of species coexistence. Proc Biol Sci 274:2739–2744. doi: 10.1098/rspb.2007.0925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Graham EB, Stegen JC. 2017. Dispersal-based microbial community assembly decreases biogeochemical function. Processes 5:65. doi: 10.3390/pr5040065. [DOI] [Google Scholar]
- 54.Murphy J, Riley JP. 1962. A modified single solution method for the determination of phosphate in natural waters. Anal Chim Acta 27:30. [Google Scholar]
- 55.Greenberg AE, Clesceri LS, Eaton AD. 1992. Standard methods for the examination of water and wastewater. American Public Health Association, Washington, DC. [Google Scholar]
- 56.Fierer N, Hamady M, Lauber CL, Knight R. 2008. The influence of sex, handedness, and washing on the diversity of hand surface bacteria. Proc Natl Acad Sci U S A 105:17994–17999. doi: 10.1073/pnas.0807920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bates ST, Berg-Lyons D, Caporaso JG, Walters WA, Knight R, Fierer N. 2011. Examining the global distribution of dominant archaeal populations in soil. ISME J 5:908–917. doi: 10.1038/ismej.2010.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. 2007. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 35:e120. doi: 10.1093/nar/gkm541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Huse SM, Huber JA, Morrison HG, Sogin ML, Welch DM. 2007. Accuracy and quality of massively parallel DNA pyrosequencing. Genome Biol 8:R143. doi: 10.1186/gb-2007-8-7-r143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. doi: 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. 2011. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kunin V, Engelbrektson A, Ochman H, Hugenholtz P. 2010. Wrinkles in the rare biosphere: pyrosequencing errors can lead to artificial inflation of diversity estimates. Environ Microbiol 12:118–123. doi: 10.1111/j.1462-2920.2009.02051.x. [DOI] [PubMed] [Google Scholar]
- 63.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lozupone C, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl Environ Microbiol 71:8228–8235. doi: 10.1128/AEM.71.12.8228-8235.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Parks DH, Tyson GW, Hugenholtz P, Beiko RG. 2014. STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30:3123–3124. doi: 10.1093/bioinformatics/btu494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Clarke K, Gorley R. 2016. PRIMER v6: user manual/tutorial. PRIMER-E, Plymouth, United Kingdom. [Google Scholar]
- 67.Polson SW. 2007. Comparative analysis of microbial community structure associated with acroporid corals during a disease outbreak in the Florida Reef Tract. Ph.D. thesis Molecular and Cellular Biology and Pathobiology Program, Marine Biomedicine and Environmental Sciences, Medical University of South Carolina, Charleston, SC. [Google Scholar]
- 68.Ramette A, Tiedje JM. 2007. Multiscale responses of microbial life to spatial distance and environmental heterogeneity in a patchy ecosystem. Proc Natl Acad Sci U S A 104:2761–2766. doi: 10.1073/pnas.0610671104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tracy CR, Streten-Joyce C, Dalton R, Nussear KE, Gibb KS, Christian KA. 2010. Microclimate and limits of photosynthesis in a diverse community of hypolithic bacteria in northern Australia. Environ Microbiol 12:592–607. doi: 10.1111/j.1462-2920.2009.02098.x. [DOI] [PubMed] [Google Scholar]
- 70.Faust K, Sathirapongsasuti JF, Izard J, Segata N, Gevers D, Raes J, Huttenhower C. 2012. Microbial co-occurrence relationships in the human microbiome. PLoS Comput Biol 8:e1002606. doi: 10.1371/journal.pcbi.1002606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yang CY, Li Y, Zhou YY, Lei X, Zheng W, Tian Y, Zheng TL. 2016. A comprehensive insight into functional profiles of free-living microbial community responses to a toxic Akashiwo sanguinea bloom. Sci Rep 6:34645. doi: 10.1038/srep34645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kembel SW, Cowan PD, Helmus MR, Cornwell WK, Morlon H, Ackerly DD, Blomberg SP, Webb CO. 2010. Picante: R tools for integrating phylogenies and ecology. Bioinformatics 26:1463–1464. doi: 10.1093/bioinformatics/btq166. [DOI] [PubMed] [Google Scholar]
- 73.Moroenyane I, Chimphango SB, Wang J, Kim HK, Adams JM. 2016. Deterministic assembly processes govern bacterial community structure in the Fynbos, South Africa. Microb Ecol 72:313–323. doi: 10.1007/s00248-016-0761-5. [DOI] [PubMed] [Google Scholar]
- 74.Poudel R, Jumpponen A, Kennelly MM, Rivard CL, Gomez-Montano L, Garrett KA. 2019. Rootstocks shape the rhizobiome: rhizosphere and endosphere bacterial communities in the grafted tomato system. Appl Environ Microbiol 85:e01765-18. doi: 10.1128/AEM.01765-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Faust K, Raes J. 2012. Microbial interactions: from networks to models. Nat Rev Microbiol 10:538–550. doi: 10.1038/nrmicro2832. [DOI] [PubMed] [Google Scholar]
- 76.Meyerhof MS, Wilson JM, Dawson MN, Michael Beman J. 2016. Microbial community diversity, structure and assembly across oxygen gradients in meromictic marine lakes, Palau. Environ Microbiol 18:4907–4919. doi: 10.1111/1462-2920.13416. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data reported here have been deposited in the NCBI Sequence Read Archive database under the accession number SRP154568.