

Abstract
A new family of iodoplumbates based on phosphonium cations have been synthesized and characterized via X-ray crystallography. Thermogravimetric analysis demonstrates that these materials have a remarkably high thermal stability and show potential for applications as organic–inorganic hybrid semiconductors. We also present the synthesis of three novel phosphonium salts and the crystallographic elucidation of these compounds.
Introduction
In the past two decades, lead(II) iodide–organic hybrids (iodoplumbates) have emerged as promising materials for a myriad of applications ranging from photovoltaics and nonlinear optics to semiconductors and dielectric materials.1 This great diversity of applications is a consequence of their variable and interesting optical and electronic properties. The topology of the anionic lead(II) iodide component (which largely determines the electronic and optical properties) is controlled by the packing of the cationic organic components.1,2 Typically, small cations like methylammonium and formamidinium yield three-dimensional (3D) perovskites, which make useful light-harvesting materials.1,3 Larger polycationic organic components give rise to two-dimensional (2D) and one-dimensional (1D) iodoplumbate networks,4,5 which can exhibit interesting optical properties (e.g., efficient photoluminescence,6 nonlinear optics) or magnetic properties,7 respectively. Nitrogen-centered organic cations are components in most examples of iodoplumbate materials, with there being only a few examples containing phosphorus-centered organic cations.8 Thus, we present a few novel examples of phosphonium-templated iodoplumbates that contain an extended network of PbI3– fragments with the potential of being semiconducting materials.
Monophosphonium-Templated Iodoplumbates
Group 14 iodometalates have demonstrated the ability to form perovskitic structures of the formula ABI3, where “A” is a monocation and “BI3” is a monoanion.3 Historically, the A cation site is occupied by a larger alkali metal such as Rb or Cs2,9,10 or an alkylammonium.11−14 These cations render 3D perovskite structures because of their smaller size. When a larger cation is used, a 2D- or 1D- perovskitic structure is observed with possible formulae of the general form ABI3, A2BI4, and A3BI5.13−17 Given that different-sized alkylammonium cations produce different dimensionalities of anion connectivity, and in light of the interesting structures obtained with trimethyl sulfonium cations,17 we sought to elucidate the structures obtained using alkyl- and arylphosphonium cations. Tetramethylphosphonium, tri(n-butyl)phosphonium, and methyltriphenylphosphonium iodide salts were used to react with lead(II) iodide in the hope of obtaining distorted perovskitic structures.
Two of the monophosphonium iodide salts used were synthesized specifically for these reactions. Tetramethylphosphonium iodide has been prepared before;18 however, it was not crystallographically characterized. We isolated colorless crystals of tetramethylphosphonium iodide and performed single-crystal X-ray diffraction (Figure 1). The P–C bond lengths in tetramethylphosphonium iodide of 1.761(7) Å are comparable to those in the analogous tetramethylphosphonium fluoride salt,19 which has P–C bond lengths averaging at 1.778(13) Å. The bond angles in the cation at 109.5(3)° are similar to those previously reported,19 indicating that the methyl groups are tetrahedrally arranged in the phosphonium cation. Furthermore, we report a cleaner, more facile, and higher-yielding synthesis for this salt, which consists of adding iodomethane to trimethylphosphine in toluene at room temperature and stirring the mixture for 24 h. The reported method to produce this salt is considerably less convenient:18 calcium phosphide and iodomethane are mixed in a methanol–water solution at 0 °C for 3 h, and then, the mixture is heated to boiling for 48 h. The volatiles are then removed in vacuo and the resulting residue is mixed with ethanol and heated to boiling, hot-filtered, exposed again to reduced pressure, and then the residue is recrystallized from anhydrous ethanol. Not only is the reported synthesis lengthy, NMR spectroscopy of the resultant material identifies the presence of three other byproduct phosphonium cations,18 whereas the NMR spectrum of our synthesis contains only the target phosphonium. Lastly, our synthesis has a 78% yield, whereas the other method18 reports a 53% yield.
Figure 1.
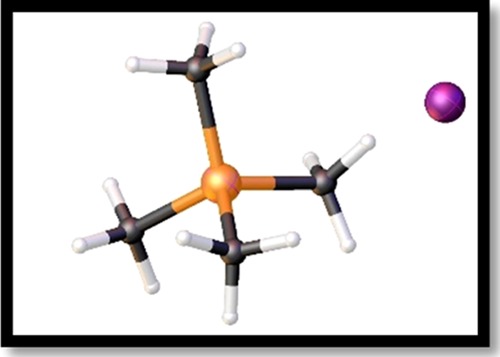
Tetramethylphosphonium iodide ([PMe4][PbI3]).
Tri-(n-butyl)phosphonium iodide was also synthesized for the first time via addition of hydroiodic acid to a solution of tri(n-butyl)phosphine in toluene. Although no crystals suitable for X-ray diffraction were isolated, NMR and elemental analysis are consistent with the synthesis of this new phosphonium salt. The 1H NMR chemical shift that arises from the proton directly attached to the phosphorus atom occurs at 7.40 ppm as a doublet of septets and is clearly indicative of the protonation of tri(n-butyl)phosphine.
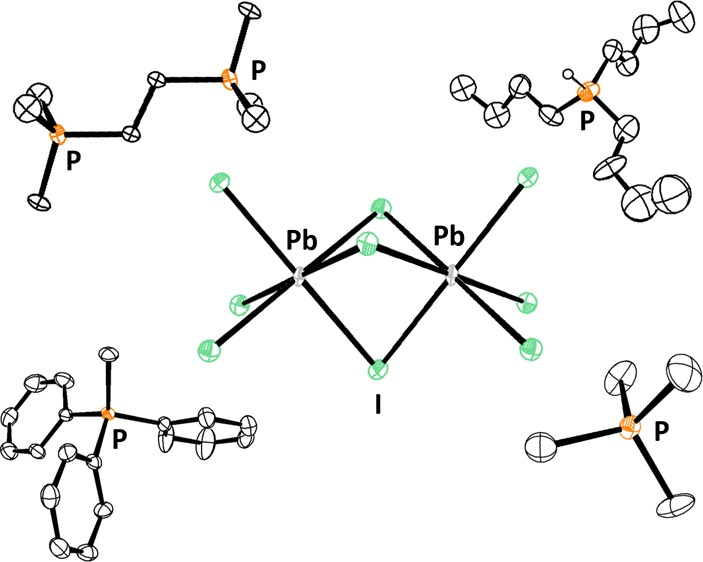
Crystals of the reactions of the three monophosphonium ions of varying substituent size with lead(II) iodide were obtained using two different methods. For tetramethylphosphonium, the starting materials were combined in dimethyl sulfoxide (DMSO) and heated at 120 °C to remove the solvent. After 2 days, mustard-yellow crystals suitable for X-ray diffraction were collected ([PMe4][PbI3]). Alternatively, crystals for the reaction of lead(II) iodide with tri(n-butyl)phosphonium ([PH(n-butyl)3][PbI3]) and methyltriphenylphosphonium iodide ([PPh3Me][PbI3]) were obtained by slow evaporation from acetonitrile (Figure 2).
Figure 2.

Monophosphoniums reacted with lead(II) iodide. From left: tetramethyphosphonium iodide [PMe4][I], tri-(n-butyl)phosphonium iodide [PH(n-butyl)3][I], and methyltriphenylphosphonium iodide [PPh3Me][I].
NMR data revealed that the phosphonium cations persisted in the product mixture. Interestingly, the single-crystal X-ray diffraction data of the monophosphonium-lead iodide reactions did not yield traditional perovskites of the form ABI3, but provided instead an extended 1D network of PbI3– fragments in which each lead atom has an octahedral geometry and the resultant octahedra are linked to each other in a face-sharing fashion. A Cambridge structural database (CSD) search indicated that the P–C bond lengths and angles in the cations are typical of those reported in other phosphoniums, ranging from 1.781 to 1.793 Å and 108.97(10) to 109.5(2)°, respectively. The bond lengths of the PbI3– fragments in the 1D network range from 3.1379(4) to 3.398(3) Å, which is similar to the iodoplumbate networks that have been previously reported.4,5,11,14,16,17,20 The bond angles in this network deviate from those observed in a perfectly octahedral network. The trans I–Pb–I bond angles in this 1D network average 175.37(4)°, with the range being 165.87(11) to 180.00(12)°. Consequently, the bond angles of cis iodine atoms in the molecule have an average of 90.96(3)° with the minimum at 81.93(3)° and the maximum at 96.90(2)°. It is not surprising that the bond angles deviate from the ideal, since [PbI3–]∞ networks that have previously been reported feature trans-molecule and cis bond angles of less than 180° and greater or less than 90°, respectively (Figures 3, 4, and 5).4,5,14,17,20−22
Figure 3.

[PbI3]∞ chain of [PMe4][PbI3]. The iodine atoms labeled with a prime symbol indicate the symmetry-generated iodine atoms.
Figure 4.

Diagrams of the monophosphonium-templated iodoplumbates highlighting the face-sharing octahedra of the [PbI3]∞ chains.
Figure 5.

Crystal packing of the monophosphonium-templated iodoplumbates.
Diphosphonium-Templated Iodoplumbates
Given that the monophosphonium cations generated an interesting 1D face-sharing octahedral structure, we synthesized a diphosphonium cation to probe the effect of a dication in the reaction with lead iodide. A novel diphosphonium iodide salt, bis(trimethylphosphonio)ethane iodide, was synthesized by adding iodomethane to bis(dimethylphosphino)ethane (Dmpe) in toluene. Bis(trimethylphosphonio)ethane iodide was then dissolved in DMSO with lead iodide, which produced a yellow solution, and evaporation of the DMSO yielded pale yellow crystals ([P2C8H22][Pb2I6]). NMR spectroscopy revealed that the phosphonium ions persisted in the product and single-crystal X-ray diffraction revealed an extended 1D face-sharing network of octahedral PbI3– fragments charge-balanced with the diphosphonium in the same manner as the monophosphonium-templated iodoplumbates (Figure 6).
Figure 6.

Bis(trimethylphosphonio)ethane iodide.
The average P–C bond length is 1.792(12)Å, which is typical of other P–C bond lengths in phosphonium ions in the CSD. Furthermore, the C–P–C bond angles average at 109.6(9)°, indicating a tetrahedral coordination environment around the phosphorus atoms. As for the 1D iodoplumbate network, the average Pb–I bond length is 3.219(4) Å, which is in agreement with our monophosphonium iodoplumbate networks and with those reported previously.4,5,14,17,20−24 The I–Pb–I bond angles are close to that of the ideal for an octahedral network, with the angle between cis iodide ions averaging at 90.0(10)°, the minimum angle being 83.644(12)°, and the maximum angle being 96.356(12)°. The angles between trans iodide ions were all 180.0(0)° (Figure 7).
Figure 7.

(a) Face-sharing octahedra of the [PbI3]∞ chain in [P2C8H22][Pb2I6]. (b) Crystal packing of [P2C8H22][Pb2I6].
Thermogravimetric Analysis (TGA) and Differential Scanning Calorimetry (DSC)
Thermogravimetric analysis (TGA) was performed to assess the thermal stability of the phosphonium iodoplumbates and the results are outlined in Figure 8. We observed that the mono- and diphosphonium with methyl substituents afforded the best stability at higher temperatures, with [PMe4][PbI3] having no significant mass loss up to 400 °C and [P2C8H22][Pb2I6] having lost approximately 8% of its original mass. Conversely, the phosphoniums with longer alkyl and aryl substituents showed decomposition and significant mass loss at much lower temperatures. The TGA curves for [PH(n-butyl)3][PbI3] and [PPh3Me][PbI3] display two events, with the first occurring at approximately 129 °C, which can be attributed to evaporation of the residual toluene used to prepare the starting material. The second event at approximately 300 °C for [PH(n-butyl)3][PbI3] and 325 °C for [PPh3Me][PbI3] represents a significant mass loss due to decomposition of the product, with [PH(n-butyl)3][PbI3] losing approximately 25% of its original mass and [PPh3Me][PbI3] losing 34% of its original mass.
Figure 8.
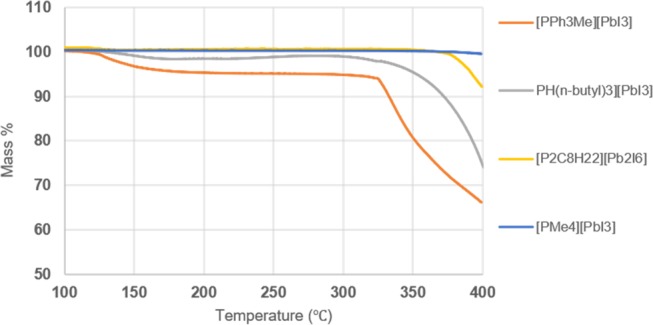
TGA curves of phosphonium-templated iodoplumbates.
Importantly, the TGA curves of our phosphonium-templated iodoplumbates show that they have a much greater thermal stability in comparison with the previously reported amide-templated iodoplumbates. Conversely, the transition metal-templated iodoplumbates decompose at approximately 270 °C,7 which is just below the decomposition temperature of our compounds. Most notably, the phosphonium-templated iodoplumbates’ decomposition pattern resembles that of the methylammonium-PbI3– perovskite, which initiates decomposition after 300 °C.3 Both the phosphonium-templated iodoplumbates and the methylammonium-PbI3– perovskite display a one-step decomposition, which likely corresponds to the loss of the organic cation and iodide content. These similarities between perovskites and our compounds with respect to thermal stability point toward a potential for making functional organic–inorganic hybrid materials from phosphonium-templated iodoplumbates.
Differential scanning calorimetry (DSC) analyses were performed on each of the samples over the temperature range of 25–225 °C. The experiments revealed no unanticipated phase transitions for any of the materials: de-solvation events were observed for systems containing solvents of crystallization, and melting and crystallization points were observed only for [PPh3Me][PbI3].
Conclusions
This work has demonstrated that phosphonium-templated iodoplumbates can be synthesized via direct addition of a phosphonium salt to lead(II) iodide in solution. The materials have been crystallographically characterized, revealing an extended network of anionic lead iodide fragments charge-balanced with phosphonium cations. Thermogravimetric analysis indicates that these compounds exhibit remarkable thermal stability. Study of these materials as potential high-temperature semiconductors is currently underway.
Experimental Section
General Remarks
All manipulations were carried out using standard inert atmosphere techniques. All chemicals and reagents were purchased from Sigma-Aldrich, except for tri(n-butyl)phosphine, which was purchased from Strem Chemicals, and used without further purification. Deuterated solvents were dried according to literature procedures when necessary, and all other solvents were dried over a series of Grubbs’-type columns and degassed prior to use. NMR spectra were recorded at room temperature on a Bruker Avance III 500 MHz, Bruker Avance Ultrashield 300 MHz, or Bruker Avance DPX 300 MHz spectrometer. Chemical shifts are reported in ppm relative to internal standards for 1H and 13C (the given deuterated solvent) and the external standard for 31P (85% H3PO4). Coupling constants |J| are given in Hertz. Elemental analysis was performed by the University of Windsor Mass Spectrometry Service Laboratory using a PerkinElmer 2400 combustion CHN analyzer. UV–vis absorption spectra were recorded on a Varian Cary 50 Conc UV–vis spectrophotometer. All samples were dissolved in DMSO and run in a quartz cuvette with a path length of 1 cm. Thermogravimetric analysis was conducted on a Mettler Toledo TGA SDTA 851e. Helium (99.99%) was used to purge the system at a flow rate of 60 mL/min. Samples were held at 25 °C for 30 min before being heated to 400 °C at a rate of 5 °C/min. All samples were run in aluminum crucibles. Calorimetric studies were performed on Mettler Toledo DSC 822e.
X-ray Crystallography
Crystals for investigation were covered in Paratone, mounted into a goniometer head, and then rapidly cooled under a stream of cold N2 of the low-temperature apparatus (Oxford Cryostream) attached to the diffractometer. Data were then collected using the APEXII software suite25 on a Bruker Photon 100 CMOS diffractometer using a graphite monochromator with Mo Kα (λ = 0.71073 Å) or Cu Kα (λ = 1.54178 Å) radiation. For each sample, data were collected at low temperature. The APEXII software was used for data reductions and SADABS26 was used for absorption corrections (multiscan; semiempirical from equivalents). XPREP was used to determine the space group, and the structures were solved and refined using the SHELX27 software suite as implemented in the WinGX28 or OLEX229 program suites. Validation of the structures was conducted using PLATON.30
Synthesis of Tetramethylphosphonium Iodide
To a 100 mL Schlenk flask, trimethylphosphine (0.75 mL, 7.5 mmol) was added and dissolved in ca. 30 mL of toluene. Iodomethane (0.47 mL, 7.50 mmol) was added to the Schlenk flask in a dropwise fashion with stirring under a nitrogen atmosphere. Upon addition of MeI, a white precipitate formed. After approximately 24 h of stirring, the toluene was pumped off and a white solid was collected. Some of this solid was dissolved in acetonitrile and left for slow evaporation, bestowing crystals suitable for X-ray diffraction. Yield: 78.0% (1.275 g, 5.84 mmol). 31P{1H} NMR (D2O) δ: 23.65 (s). 13C{1H} NMR (CD3CN) δ: 9.98 (d, PCH3, 1JCP = 56.3 Hz). 1H NMR (CD3CN) δ: 1.84 (d, 12H, PCH3, 2JHP = 15 Hz). Elemental analysis: calculated for C4H12PI·0.33 tol: C, 30.51; H, 5.94; N, 0.00; found: C, 30.42; H, 5.73; N, 0.21.
Synthesis of Tri(n-butyl)phosphonium Iodide
Tri-n-butylphosphine (0.75 mL, 3.0 mmol) was added to a 100 mL Schlenk flask with ca. 30 mL of toluene. Under stirring, hydroiodic acid (0.41 mL, 3.0 mmol) was added dropwise to the flask under a nitrogen atmosphere. After 24 h, the toluene was pumped off and an orange oily substance was left in the flask. The orange substance was placed under vacuum, and after 6h, a pale pink solid was collected. Yield: 96.0% (1.566 g, 4.74 mmol). 31P{1H} NMR (CD3CN) δ: 12.98 (s). 13C{1H} NMR (CD3CN) δ: 12.70 (s, PCCCCH3), 16.10 (d, PCH2CCC, 1JCP = 47.2 Hz), 23.36 (d, PCCH2CC, 2JPC = 15.5 Hz), 24.20 (d, PCCCH2C, 3JPC = 4.7 Hz). 1H NMR (CD3CN) δ: 0.95 (t, 9H, PCCCCH3, 3JHH = 7.0 Hz), 1.49 (m, 6H, PCCCH2C, unresolved coupling), 1.62 (m, 6H, PCCH2CC, unresolved coupling), 2.35 (m, 6H, PCH2CCC, unresolved coupling), 7.400 (dsept, 1H, PH, 1JHP = 489 Hz, 3JHH = 5.5 Hz). Elemental analysis: calculated for PC12H28I: C, 43.65; H, 8.55; N, 0.00; found: C, 43.36; H, 8.54; N, 0.02.
Synthesis of Bis(trimethyl)phosphonioethane Iodide
Dmpe (0.25 mL, 1.53 mmol) was dissolved in ca. 10 mL of toluene in a Schlenk flask. Two equivalents of MeI (0.19 mL, 3.06 mmol) were added to the flask in a dropwise fashion under stirring. A white precipitate was observed at the surface of the solution upon addition of the first drops of MeI. The reaction mixture was stirred for 18 h and the solvent was pumped off. A white powder was collected. Yield: 77.6% (0.515 g, 1.19 mmol). 31P{1H} NMR (DMSO-d6) δ: 32.49 (s). 13C{1H} NMR (D2O) δ: 6.92 (t, P(CH3)3, 1JCP = 16.2 Hz), 16.00 (t, P(μ-CH2), 1JPC = 15.2 Hz). 1H NMR (D2O) δ: 1.99 (d, 18H, P(CH3)3, 2JPH = 13.8 Hz), 2.65 (d, 4H, P(μ-CH2)2, 3JPH = 6 Hz). Elemental analysis: calculated for P2C8H22I2·tol: C, 34.24; H, 5.75; N, 0.00; found: C, 33.93; H, 5.48; N, −0.03.
Synthesis of [PMe4][PbI3]
A 20 mL scintillation vial was charged with tetramethylphosphonium iodide (50 mg, 0.23 mmol) and PbI2 (106 mg, 0.23 mmol) and then dissolved in DMSO (ca. 5 mL). The vial was placed in an oil bath set at 120 °C and the solvent was left to evaporate from the vial with the cap off. After 48 h, mustard-yellow crystals suitable for X-ray diffraction formed at the bottom of the vial. Yield: 76% (118 mg, 0.17 mmol). 31P{1H} NMR (DMSO-d6, 121.5 MHz) δ: 22.88 (s). 1H NMR (DMSO-d6, 300 MHz); δ: 1.82 (PCH3, d, 12H, 2JPH = 15.3 Hz). 13C{1H} NMR (DMSO-d6, 75.5 MHz); δ: 9.70 (PCH3, d, 1JPC = 55 Hz). Activation energy (EA) calculated for C4H12PPbI3: C, 7.08; H, 1.78; N, 0.00%; found: C, 7.08; H, 1.19; N, −0.05%. UV–vis absorption peak: 365 nm.
Synthesis of [HP(n-bu)3][PbI3]
To 50 mg (0.151 mmol) of tri-n-butylphosphonium iodide, 70 mg of PbI2 was added in a 20 mL scintillation vial with approximately 10 mL of acetonitrile, creating a yellow solution. The mixture was stirred for 24 h, after which a yellow solution with a pale-yellow precipitate was observed. The yellow solution was separated and left for recrystallization by slow evaporation. Yellow crystals suitable for X-ray diffraction were collected. Yield: 55% (33 mg, 41.7 mmol). 31P{1H} NMR (CD3CN, 202.5 MHz); δ: 12.72 (s). 1H NMR (CD3CN, 500 MHz); δ: 0.95 (PCCCCH3, t, 9H, 3JHH = 7 Hz), 1.48 (PCCCH2C, m, 6H, unresolved coupling), 1.62 (PCCH2CC, m, 6H, unresolved coupling), 2.23 (PCH2CCC, m, 6H, unresolved coupling). 13C{1H} NMR (CD3CN, 125.8 MHz); δ: 12.65 (PCCCCH3, s), δ: 16.10 (PCH2CCC, d, 1JPC = 47 Hz), δ: 12.65 (PCCH2CC, d, 2JPC = 16 Hz), δ: 12.65 (PCCCH2C, d, 3JPC = 4.5 Hz). EA calculated for [HP(n-bu)3]+[PbI3]−·0.5 toluene: C, 22.23; H, 3.85; N, 0.00%; found: C, 21.99; H, 3.88; N, −0.06%. UV–vis absorption peak: 365 nm.
Synthesis of [PMePh3][PbI3]
Methyltriphenylphosphonium iodide (50 mg, 0.12 mmol) and lead(II) iodide (57 mg, 0.12 mmol) were combined in a 20 mL scintillation vial and dissolved in ca. 5 mL of DMSO, forming a yellow solution. The vial was placed in an oil bath and the solvent was left to evaporate. After 24 h, a pale yellow solid was found at the bottom of the vial and collected. The solid product was dissolved in acetonitrile and yellow crystals suitable for X-ray diffraction were collected upon evaporation of the MeCN. Yield: 70% (74 mg, 0.085 mmol). 31P{1H} NMR (DMSO-d6, 121.5 MHz); δ: 22.590 (s). 1H NMR (DMSO-d6, 300 MHz) δ: 3.12 (PCH3, d, 3H, 2JPH = 14.4 Hz), 7.71–7.86 (aromatic protons, m, 15H). 13C{1H} NMR (DMSO-d6, 75.5 MHz) δ: 7.89 (PCH3, d, 1C, 1JPC = 58 Hz), 120.43 (Cipso, d, 3C, 1JPC = 88.3 Hz), 133.77 (Cortho, d, 6C, 2JPC = 10.8 Hz), 130.66 (Cmeta, d, 6C, 3JPC = 12.7 Hz), 135.34(Cpara, s, 3C). EA calculated for [PMePh3]+[PbI3]−·MeCN: C, 25.95; H, 2.06; N, 1.59%; found: C, 25.94; H, 1.99; N, 1.58%. UV–vis absorption peak: 365 nm.
Synthesis of [C8H22P2 ][Pb2I6]
To a 20 mL scintillation vial, 50 mg (0.12 mmol) of bis-(trimethylphosphonio)ethane iodide and 2 equiv of lead(II) iodide (106 mg, 0.23 mmol) were added in ca. 5 mL of DMSO, which generated a pale-yellow solution. The vial was placed in an oil bath and the solvent was allowed to evaporate. After 24 h, pale-yellow crystals were found at the bottom of the vial and collected for X-ray diffraction. Yield: 86.5% (135 mg, 85 μmol). 31P{1H} NMR (DMSO-d6, 121.5 MHz) δ: 32.86 (s). 1H NMR (DMSO-d6, 500 MHz); δ: 1.89 (P(CH3)3, d, 18H, 2JPH = 15 Hz), 2.55 (P(CH2)2, d, 4H, 2JPH = 6.5 Hz). 13C{1H} NMR (DMSO-d6, 125.8 MHz); δ: 7.13 (PCH3, t, 1JPC = 26.7 Hz), 15.42 (PCH2, t, 1JPC = 25.0 Hz). EA calculated for C8H22P2Pb2I6: C, 7.09; H, 1.64; N, 0.00%; found: C, 6.78; H, 1.13; N, −0.06%. UV–vis absorption peak: 365 nm.
Acknowledgments
We acknowledge the Natural Sciences and Engineering Research Council (Canada) and the Canada Foundation for Innovation for funding, a scholarship (JFB), and equipment.
Glossary
Abbreviations
- NMR
nuclear magnetic resonance
- DMSO
dimethyl sulfoxide
- CSD
Cambridge structural database
- 1D
one-dimensional
- 3D
three-dimensional
- dmpe
bis(dimethylphosphino)ethane
- UV–vis
ultraviolet–visible
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02240.
DSC results; 1,2-bis(trimethylphosphonio)ethane triiodoplumbate(II), [P2C8H22][Pb2I6]; tributylphosphonium triiodoplumbate(II), [PHBu3][PbI3]; crystal data and structure refinement for [PMe4][I] (Table S1); crystal data and structure refinement for [PMe4][PbI3] (Table S2); crystal data and structure refinement for [P2C8H22][Pb2I6] (Table S3) (PDF)
Author Present Address
§ Department of Chemistry, Carlton University, 1125 Colonel By Drive, Ottawa, Ontario K1S 5B6, Canada (C.L.B.M.).
Author Contributions
All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhao Y.; Zhu K. Organic–inorganic Hybrid Lead Halide Perovskites for Optoelectronic and Electronic Applications. Chem. Soc. Rev. 2016, 45, 655–689. 10.1039/c4cs00458b. [DOI] [PubMed] [Google Scholar]
- Li Z.; Yang M.; Park J. S.; Wei S. H.; Berry J. J.; Zhu K. Stabilizing Perovskite Structures by Tuning Tolerance Factor: Formation of Formamidinium and Cesium Lead Iodide Solid-State Alloys. Chem. Mater. 2016, 28, 284–292. 10.1021/acs.chemmater.5b04107. [DOI] [Google Scholar]
- Stoumpos C. C.; Malliakas C. D.; Kanatzidis M. G. Semiconducting Tin and Lead Iodide Perovskites with Organic Cations: Phase Transitions, High Mobilities, and Near-Infrared Photoluminescent Properties. Inorg. Chem. 2013, 52, 9019–9038. 10.1021/ic401215x. [DOI] [PubMed] [Google Scholar]
- Eppel S.; Fridman N.; Frey G. Amide-Templated Iodoplumbates: Extending Lead-Iodide Based Hybrid Semiconductors. Cryst. Growth Des. 2015, 15, 4363–4371. 10.1021/acs.cgd.5b00655. [DOI] [Google Scholar]
- Li H.-H.; Chen Z.-R.; Cheng L.-C.; Wang Y.-J.; Feng M.; Wang M. Hybrid Polymeric Iodoplumbates Constructed from Morpholine and Its Derivatives: Structures and Properties. Dalton Trans. 2010, 39, 11000 10.1039/c0dt00622j. [DOI] [PubMed] [Google Scholar]
- Dohner E. R.; Jaffe A.; Bradshaw L. R.; Karunadasa H. I. Intrinsic White-Light Emission from Layered Hybrid Perovskites. J. Am. Chem. Soc. 2014, 136, 13154–13157. 10.1021/ja507086b. [DOI] [PubMed] [Google Scholar]
- Zhang Z. J.; Xiang S. C.; Zhang Y. F.; Wu A. Q.; Cai L. Z.; Guo G. C.; Huang J. S. A New Type of Hybrid Magnetic Semiconductor Based upon Polymeric Iodoplumbate and Metal-Organic Complexes as Templates. Inorg. Chem. 2006, 45, 1972–1977. 10.1021/ic051350v. [DOI] [PubMed] [Google Scholar]
- Bartlett P. N.; Burt J.; Hasan M. M.; Hector A. L.; Levason W.; Reid G.; Richardson P. W. Haloplumbate Salts as Reagents for the Non-Aqueous Electrodeposition of Lead. RSC Adv. 2016, 6, 73323–73330. 10.1039/C6RA12942K. [DOI] [Google Scholar]
- Tan Z.-K.; Moghaddam R. S.; Lai M. L.; Docampo P.; Higler R.; Deschler F.; Price M.; Sadhanala A.; Pazos L. M.; Credgington D.; et al. Bright Light-Emitting Diodes Based on Organometal Halide Perovskite. Nat. Nanotechnol. 2014, 9, 687–692. 10.1038/nnano.2014.149. [DOI] [PubMed] [Google Scholar]
- Saidaminov M. I.; Mohammed O. F.; Bakr O. M. Low-Dimensional-Networked Metal Halide Perovskites: The Next Big Thing. ACS Energy Lett. 2017, 2, 889–896. 10.1021/acsenergylett.6b00705. [DOI] [Google Scholar]
- Aamir M.; Shah Z. H.; Sher M.; Iqbal A.; Revaprasadu N.; Malik M. A.; Akhtar J. Enhanced Photocatalytic Activity of Water Stable Hydroxyl Ammonium Lead Halide Perovskites. Mater. Sci. Semicond. Process. 2017, 63, 6–11. 10.1016/j.mssp.2017.01.001. [DOI] [Google Scholar]
- Stoumpos C. C.; Mao L.; Malliakas C. D.; Kanatzidis M. G. Structure-Band Gap Relationships in Hexagonal Polytypes and Low-Dimensional Structures of Hybrid Tin Iodide Perovskites. Inorg. Chem. 2017, 56, 56–73. 10.1021/acs.inorgchem.6b02764. [DOI] [PubMed] [Google Scholar]
- Stoumpos C. C.; Frazer L.; Clark D. J.; Kim Y. S.; Rhim S. H.; Freeman A. J.; Ketterson J. B.; Jang J. I.; Kanatzidis M. G. Hybrid Germanium Iodide Perovskite Semiconductors: Active Lone Pairs, Structural Distortions, Direct and Indirect Energy Gaps, and Strong Nonlinear Optical Properties. J. Am. Chem. Soc. 2015, 137, 6804–6819. 10.1021/jacs.5b01025. [DOI] [PubMed] [Google Scholar]
- Liu G.; Liu J.; Sun Z.; Zhang Z.; Chang L.; Wang J.; Tao X.; Zhang Q. Thermally Induced Reversible Double Phase Transitions in an Organic–Inorganic Hybrid Iodoplumbate C 4 H 12 NPbI 3 with Symmetry Breaking. Inorg. Chem. 2016, 55, 8025–8030. 10.1021/acs.inorgchem.6b01143. [DOI] [PubMed] [Google Scholar]
- Cao D. H.; Stoumpos C. C.; Farha O. K.; Hupp J. T.; Kanatzidis M. G. 2D Homologous Perovskites as Light-Absorbing Materials for Solar Cell Applications. J. Am. Chem. Soc. 2015, 137, 7843–7850. 10.1021/jacs.5b03796. [DOI] [PubMed] [Google Scholar]
- Huang Z. J.; Cheng H. J.; Dai M.; Ni C. Y.; Li H. X.; Hou K. P.; Ren Z. G.; Lang J. P. Solvothermal Syntheses and Crystal Structures of One 1D and Two 3D [PbxIy]-Based Coordination Polymers. Inorg. Chem. Commun. 2013, 31, 33–36. 10.1016/j.inoche.2013.02.016. [DOI] [Google Scholar]
- Kaltzoglou A.; Stoumpos C. C.; Kontos A. G.; Manolis G. K.; Papadopoulos K.; Papadokostaki K. G.; Psycharis V.; Tang C. C.; Jung Y.-K.; Walsh A.; et al. Trimethylsulfonium Lead Triiodide: An Air-Stable Hybrid Halide Perovskite. Inorg. Chem. 2017, 56, 6302–6309. 10.1021/acs.inorgchem.7b00395. [DOI] [PubMed] [Google Scholar]
- Herrmann F.; Kuhn N. Ein Einfacher Zugang Zu Tetramethylphosphoniumiodid/A Simple Access to Tetramethylphosphonium Iodide. Z. Naturforsch., B 2012, 67, 853–854. 10.5560/znb.2012-0129. [DOI] [Google Scholar]
- Kornath A.; Neumann F.; Oberhammer H. Tetramethylphosphonium Fluoride: “Naked” Fluoride and Phosphorane. Inorg. Chem. 2003, 42, 2894–2901. 10.1021/ic020663c. [DOI] [PubMed] [Google Scholar]
- Tang Z.; Guloy A. M. A Methylviologen Lead(II) Iodide: Novel [PbI 3 - ] ∞ Chains with Mixed Octahedral and Trigonal Prismatic Coordination. J. Am. Chem. Soc. 1999, 121, 452–453. 10.1021/ja982702i. [DOI] [Google Scholar]
- Liu J.-J.; Guan Y.-F.; Jiao C.; Lin M.-J.; Huang C.-C.; Dai W.-X. A Panchromatic Hybrid Crystal of Iodoplumbate Nanowires and J-Aggregated Naphthalene Diimides with Long-Lived Charge-Separated States. Dalton Trans. 2015, 44, 5957–5960. 10.1039/C4DT03785E. [DOI] [PubMed] [Google Scholar]
- Krautscheid H.; Lode C.; Vielsack F.; Vollmer H. Synthesis and Crystal Structures of Iodoplumbate Chains, Ribbons and Rods with New Structural Types. J. Chem. Soc., Dalton Trans. 2001, 1099–1104. 10.1039/b009488i. [DOI] [Google Scholar]
- Bedlivy D.; Mereiter K. The Structures of Potassium Lead Triiodide Dihydrate and Ammonium Lead Triiodide Dihydrate. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1980, 36, 782–785. 10.1107/S0567740880004529. [DOI] [Google Scholar]
- Vincent B. R.; Robertson K. N.; Cameron T. S.; Knop O. Alkylammonium Lead Halides. Part 1. Isolated PbI64– Ions in (CH3NH3)4PbI6•2H2O. Can. J. Chem. 1987, 65, 1042–1046. 10.1139/v87-176. [DOI] [Google Scholar]
- APEX II; Bruker AXS Inc.: Madison, WI, 2012.
- SADABS; Bruker AXS Inc.: Madison, WI, 2008.
- Sheldrick G. M. A Short History of SHELX. Acta Crystallogr., Sect. A: Found. Crystallogr. 2008, 64, 112–122. 10.1107/S0108767307043930. [DOI] [PubMed] [Google Scholar]
- Farrugia L. J. WinGX Suite for Small-Molecule Single-Crystal Crystallography. J. Appl. Crystallogr. 1999, 32, 837–838. 10.1107/S0021889899006020. [DOI] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: A Complete Structure Solution, Refinement and Analysis Program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/S0021889808042726. [DOI] [Google Scholar]
- Spek A. L. Single-Crystal Structure Validation with the Program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. 10.1107/S0021889802022112. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.