

Abstract
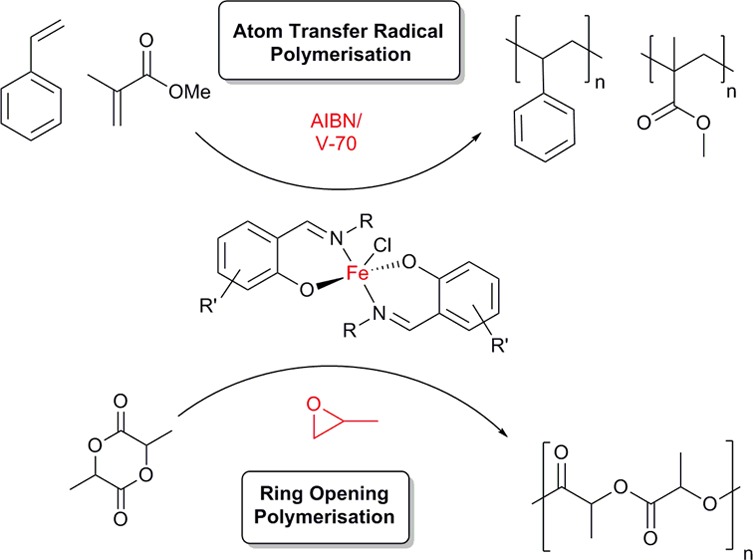
A series of monometallic pentacoordinate FeIII chloride complexes have been prepared and characterized by high-resolution mass spectrometry and elemental analysis. X-ray diffraction analysis showed that the bis-chelated FeIII complexes bear distorted trigonal bipyramidal geometries. The air- and moisture-stable FeIII complexes were screened as mediators in the reverse atom transfer radical polymerization (ATRP) of styrene and methyl methacrylate. Moderate to excellent control was achieved with dispersities as low as 1.1 for both poly(methyl methacrylate) and polystyrene. Kinetic studies showed living characteristics, and end group analysis revealed the presence of olefin-terminated polymer chains, suggesting catalytic chain transfer as a competing polymerization mechanism. Although the catalysts are not the fastest Fe ATRP mediators, they are robust and flexible. Using propylene oxide as an initiator, the complexes were active catalysts for the ring-opening polymerization of rac-lactide with moderate control. While the addition of propylene oxide has been reported as an efficient method of converting a metal–halide bond to a metal–alkoxide bond in situ, we show herein that this initiation mechanism can limit polymerization reproducibility and introduce an induction period.
Introduction
Transition-metal complexes play an important role in the catalysis of polymerization reactions, from providing better control over the microstructure to tuning the properties of the obtained polymers.1 Non-toxic iron is an ideal candidate to be used for this purpose as the removal of catalyst residues from polymer products is still a major challenge for food or biomedical applications.2−4 Moreover, as Fe is the most abundant transition metal, it can maintain low costs even on industrial scales.5,6 With multiple active oxidation states, most commonly FeII and FeIII, to exploit in different catalytic processes, research in Fe-based catalysts has become an area of focus in polymer chemistry; complexes supported with a range of ligands have been characterized and applied in various types of polymerizations.1,7−9
Atom transfer radical polymerization (ATRP) is based on a dynamic equilibrium between the growing and the dormant polymer chains using a metal-mediated halogen-exchange process (Scheme 1). As an alternative to the typical Cu-based systems,10 the first Fe catalysts for ATRP were pioneered by the groups of Matyjaszewski and Sawamoto, originally using FeIIX2(PR3)2 (X = Cl and Br)-type complexes.11,12 Since then, Fe salts and complexes with a variety of ligands (including phosphines, imines, and amines) have been explored in the ATRP of vinyl monomers,7,13,14 but interestingly the use of the ubiquitous salen ligand framework gave only limited control over the polymerization.15 Catalysts with Fe in its more stable oxidation state can have the additional benefit of air and moisture stability that enables simple, user-friendly synthesis and application. These systems traditionally feature an azo initiator (such as AIBN) and proceed via a reverse ATRP (R-ATRP) mechanism (Scheme 1). The first examples of Fe-mediated R-ATRP were reported by Teyssié et al.,16 whereas amine bisphenolate complexes of both FeII and FeIII showed good control in the RP of styrene and methyl methacrylate (MMA),17−21 through joint ATRP and organometallic-mediated radical polymerization mechanisms.22,23
Scheme 1. Mechanism of Fe-Mediated ATRP and R-ATRP.

The ring-opening polymerization (ROP) of cyclic esters, in particular the renewable cyclic diester lactide, is a burgeoning component of sustainable polymer development.24 Industrially, poly(lactic acid) (PLA) is formed via the ROP of lactide catalyzed by Sn carboxylates. These catalysts require harsh reaction conditions, show limited control over the polymerization, and can cause toxicity problems in biomedical applications. An intense effort to develop well-defined, single site catalysts to achieve a better control over the dispersity and tacticity has led to a plethora of new catalysts for this reaction, including limited reports with some Cu complexes with relevance to ATRP.25−28 Iron catalysts are attractive alternatives because of their low cost and biocompatibility, but surprisingly few papers report efficient Fe ROP catalyst systems.29−36 Byers and co-workers developed a range of bis(imino)pyridine Fe alkoxide catalysts and showed that the polymerization was sensitive to both oxidation state and electron density of Fe.9,37 Salen complexes of FeII aalso catalyzed the ROP of l-LA.38 Following the recent trend of using propylene oxide (PO)-triggered initiations,39−42 Duan et al. reported on the in situ generation of salen-based FeIII–alkoxide catalysts for the ROP of lactide and caprolactone.43 The proposed mechanism suggests the insertion of PO into the Fe–Cl bond to form the catalytically active Fe–alkoxide (Scheme 2). Similar to Al-based systems,39 the addition of a massive excess (2000 equivalents) of the carcinogenic PO was required to achieve good conversions, hence greatly reducing the atom efficiency and biocompatibility of this system.
Scheme 2. Coordination–Insertion Mechanism of in Situ Fe–Alkoxide-Catalyzed Lactide Polymerization.

Complimentary to the well-studied tetradentate salen systems, tridentate derivatives of Schiff bases were also applied to support FeIII as a polymerization catalyst.44 Considering the paucity of studies exploring Fe complexes with bidentate phenoxyimine ligands,45−47 we recently developed a novel family of air-stable half salen FeIII chloride catalysts that showed high activity in CO2/epoxide couplings.48 In these reactions, the conformational flexibility of the complexes proved to be an advantage over the standard salen scaffolds. The presence of the Fe–Cl bond in these catalysts opened up the possibility of their applications in R-ATRP (Scheme 1) and ROP (Scheme 2) reactions. Half salen ligands offer the advantage of simple and modular synthesis to achieve stereoelectronically diverse properties (Figure 1). Aluminum complexes of these ligands have shown promising results in the ROP of cyclic esters,49 including the application of monometallic Al–phenoxyimines in the ROP of γ-butyrolactone50 and the stereocontrolled ROP of lactide.51 Moreover, dimeric, oxygen-bridged Li, Na, and K phenoxyimine complexes were extremely active in the ROP of lactide; however, these systems exerted a lower control over molecular weights and tacticity.52
Figure 1.

General structure of a bis-chelated half salen complex.
This work explores the synthesis of five bidentate phenoxyimine ligands, the corresponding novel FeIII chloride complexes, and their application in polymerization catalysis. Combined with the previously reported complexes, all eight Fe–phenoxyimines were screened as mediators in the R-ATRP of styrene and MMA. Moreover, we explored the PO-triggered ROP of rac-lactide, including understanding the limitations of this emerging initiation strategy.
Results and Discussion
Synthesis and Characterization
A series of FeIII complexes were synthesized from half salen ligands, incorporating substituents with varying steric bulk [methyl or 2,6-diisopropylphenyl (DIPP)] at the imine position and peripheral Cl or tBu substituents on the phenol moiety.52,53 Initially, the pro-ligands were deprotonated by NaH to form the corresponding Na–phenolates. Subsequent transmetalation with anhydrous FeCl3 (0.5 equiv) in tetrahydrofuran (THF) solvent at ambient temperature gave an immediate color change from yellow to dark purple, which indicated the formation of the desired L2FeCl complexes (C1–C8). The resulting Fe complexes were isolated in moderate to high yields (45–89%) as dark purple-brown solids. As the paramagnetically shifted 1H NMR spectra of the complexes provided limited information, the stoichiometry was confirmed with high-resolution mass spectrometry and elemental analysis. All eight complexes were oxygen and moisture stable, and single crystals of C5, C6, and C8 were obtained under air via the slow evaporation of the dichloromethane (DCM) solvent (Scheme 3). X-ray diffraction studies revealed that all three complexes display a mononuclear structure with distorted trigonal bipyramidal geometries (τ = 0.94, 0.87, and 0.66 for C5, C6, and C8, respectively) around the pentacoordinate FeIII center, using the τ factor developed by Addison et al. (where τ = 0 for perfect square pyramidal and τ = 1 for perfect trigonal bipyramidal geometries).54 In line with the previously reported molecular structures of C1, C3 and C7,48C5 and C8, both feature a N–Fe–N axis which is almost linear [C5, N(1)–Fe(1)–N(1′), 177.44(5)°; C8, N(1)–Fe(1)–N(2), 169.98(6)°], with one Cl and two phenoxy-O centers occupying the equatorial positions (Figures 2 and 3). In contrast, C6 displays an O–Fe–O axis [O(1)–Fe(1)–O(2), 173.7(2)°]. This switch in axial and equatorial occupants may arise from the significant increase in steric bulk of the DIPP imine substituents. For C5, C6, and C8 (Figure 4), the bond metrics are comparable to previously reported Fe half salen complexes,55−59 with Fe–N bond lengths ranging from 2.101(1) to 2.111(1) Å and Fe–O bond lengths of 1.8757(9)–1.911 (1) Å. For all six structurally characterized complexes, the ancillary phenoxyimine ligands are arranged such that both nitrogen and oxygen atoms are in mutually trans positions. These geometries differ significantly from those observed with FeIII salen and salalen complexes, which typically possess a square pyramidal geometry with mutually cis N and O ligands.41,43 Upon the introduction of electronegative chloro substituents to the ligand scaffold, the Fe–Cl bond length is significantly shortened and strengthened, from 2.2713(5) Å for unsubstituted C1,48 to 2.2517(6) Å for monochloro-substituted C7 and to 2.2271(5) Å for ortho, para-dichloro-substituted C8. The magnetic moments of all eight complexes were determined via the Evans method and displayed μeff values of 4.6–5.5 B. M., which suggests that high spin FeIII complexes predominate.60
Scheme 3. Synthesis of FeIII complexes C1–C8.

Figure 2.
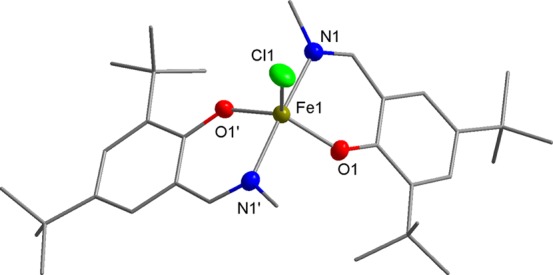
Molecular structure of C5 with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): Fe1–Cl1 2.2713(5), Fe1–O1 1.8758(9), and Fe1–N1 2.111(1). Selected bond angles (deg): O1–Fe1–Cl1 119.50(3), O1′–Fe1–O1 121.00(6), O1–Fe1–N1′ 88.46(4), O1–Fe1–N1 90.28(4), N1–Fe1–Cl1 91.28(3), and N1–Fe1–N1′ 177.44(5).
Figure 3.
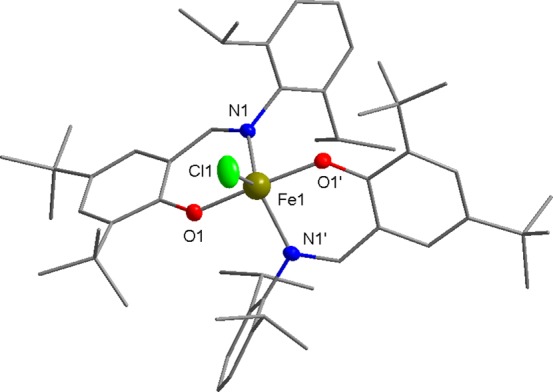
Molecular structure of C6 with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): Fe1–Cl1 2.215(2), Fe1–O1 1.921(3), and Fe1–N1 2.080(3). Selected bond angles (deg): O1–Fe1–Cl1 93.2(1), O1–Fe1–O1′ 173.7(2), O1–Fe1–N1 86.2(1), O1′–Fe1–N1 90.7(1), N1–Fe1–Cl1 119.4(1), and N1–Fe1–N1′ 121.1(2).
Figure 4.
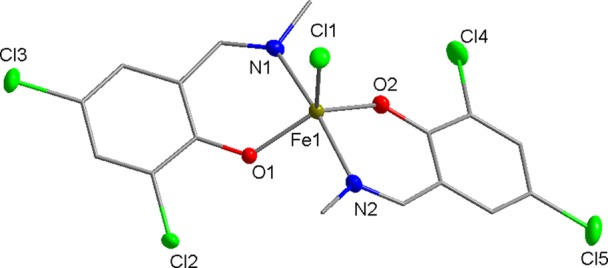
Molecular structure of C8 with ellipsoids set at the 50% probability level. Hydrogen atoms have been omitted for clarity. Selected bond lengths (Å): Fe1–Cl1 2.2271(5), Fe1–O1 1.881(1), Fe1–O2 1.911(1), Fe1–N1 2.101(1), and Fe1–N2 2.104(2). Selected bond angles (deg): O1–Fe1–Cl1 114.06(4), O1–Fe1–O2 130.30(5), O1–Fe1–N1 87.91(5), O1–Fe1–N2 90.12(5), O2–Fe1–Cl1 115.64(4), O2–Fe1–N1 87.02(5), O2–Fe1–N2 86.78(5), N1–Fe1–Cl1 96.19(4), N1–Fe1–N2 169.98(6), and N2–Fe1–Cl1 93.58(4).
Atom Transfer Radial Polymerization
Complexes C1–C8 were screened as mediators in the R-ATRP of styrene using the standard AIBN initiator at 120 °C or an alternative azo-initiator (V-70) which ‘degrades quickly to radicals at 75 °C (Table 1). As expected, the lower temperature of reaction used with V-70 leads to lower conversions and slightly narrower molecular weight distributions. Following the Fe–Cl bond strengths, the conversion increased with the Lewis acidity of the Fe center using AIBN and C1, C7, and C8 as mediators (entries 1, 13, and 15). Surprisingly, along with providing one of the highest conversions (56%), C8 also displayed good control over the dispersities, with D̵ values (1.2, entries 15 and 16) that are comparable with the best reported for FeIII R-ATRP systems.19,61 Of particular interest, catalyst C6 was an excellent mediator using V-70, affording polystyrene with very narrow dispersity (D̵ = 1.05, entry 12) with matching theoretical and experimental molecular weights. This may suggest that the bulky tBu substituents can sterically stabilize the reduced FeII derivative during the R-ATRP process. The presence of these electron-donating groups may also decrease the Lewis acidity of the metal center and therefore limit the concentration of active chains. On the contrary, when L6 was investigated by Gibson et al. in a mono-chelated FeLCl2-type complex, poor control was achieved, attributed to the relative instability of the reduced tricoordinate FeII species formed during polymerization.44 Interestingly, C5 with identical tBu substituents on the phenol moiety displayed significantly lower control, which can be linked to the lack of steric hindrance and thus protection of the coordination sphere by the DIPP group on the imine. Complexes C3 and C4 provided a moderate degree of control (D̵ = 1.4–1.5), whereas C1, C2, C5, and C7 were relatively poor mediators providing broad dispersities (D̵ > 1.5). These findings suggest that overall, the steric bulk at the imine position (Me or DIPP substituents) does not have a significant effect upon the degree of control. In some cases (e.g., entries 3–4 and 9–10), the number average molecular weights (Mn) were higher than the theoretical values (Mn,th), suggesting inefficient initiation.
Table 1. Screening of Catalysts C1–C8 in Styrene R-ATRPa.

| entry | complex | init. | conv. (%) | Mn,th (Da) | Mn (Da) | D̵ |
|---|---|---|---|---|---|---|
| 1 | C1 | AIBN | 29 | 3000 | 3800 | 1.7 |
| 2 | V-70b | 18 | 1900 | 3600 | 1.4 | |
| 3 | C2 | AIBN | 61 | 6400 | 11 600 | 1.7 |
| 4 | V-70 | 36 | 3800 | 6800 | 1.5 | |
| 5 | C3 | AIBN | 34 | 3500 | 4000 | 1.5 |
| 6 | V-70 | 24 | 2700 | 4000 | 1.5 | |
| 7 | C4 | AIBN | 40 | 4200 | 4200 | 1.4 |
| 8 | V-70 | 31 | 3200 | 3300 | 1.5 | |
| 9 | C5 | AIBN | 79 | 8200 | 15 500 | 2.2 |
| 10 | V-70 | 33 | 6800 | 13 800 | 1.8 | |
| 11 | C6 | AIBN | 23 | 2400 | 4200 | 1.4 |
| 12 | V-70 | 25 | 2600 | 2600 | 1.1 | |
| 13 | C7 | AIBN | 45 | 4700 | 14 200 | 1.6 |
| 14 | V-70 | 23 | 2400 | 6000 | 1.4 | |
| 15 | C8 | AIBN | 56 | 5800 | 10 900 | 1.2 |
| 16 | V-70b | 14 | 1500 | 5700 | 1.2 |
Conditions: [styrene]/[FeIII]/[I] = 100:1:0.6, styrene/toluene = 1:1 (v/v), 120 °C for AIBN or 75 °C for V-70, 2 h. Conversion was determined using 1H NMR spectra of crude reaction mixtures. Mn,th [Fe] = [styrene]0/[Fe] × M(styrene) × conversion.
Insufficient polymer was precipitated after 2 h to allow analysis; therefore, a sample precipitated after 4 h reaction time was used.
Kinetic experiments were performed using complex C8, which exerted the highest control over dispersity using AIBN as the initiator. The molecular weights increased linearly with conversion, suggesting a controlled polymerization. The reaction kinetics deviated from the expected first-order dependence on monomer concentration, which may be due to the presence of competing side reactions (vide infra) (Figure 5).
Figure 5.

Kinetic plots for styrene polymerization: ln([M]0/[M]t) vs time (on left) and molecular weight vs conversion (right).
To provide insight into the nature of the polymer end groups, the purified polystyrene samples were investigated by 1H NMR spectroscopy. Besides the expected halide capping group (ClCH(Ph)CH2 at 4.4 ppm), the presence of olefin-terminated polymer chains was also observed, as indicated by diagnostic doublets at 5.27 and 5.78 ppm (Figure 6). These findings suggest that catalytic chain-transfer (CCT) side reactions occur, which compete with the ATRP mechanism.18 However, molecular weights increased linearly with the conversion, as observed previously for intermediate spin α-diimine Fe complexes.62,63
Figure 6.
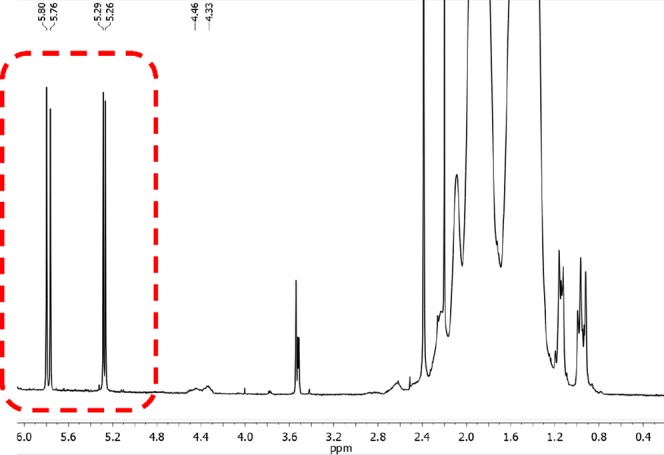
1H NMR spectrum (CDCl3, 20 °C) showing doublet peaks of olefin-terminated polymer chains formed via CCT.
The R-ATRP of MMA using C1–C8 as mediators afforded polymers with Mn significantly in excess of the theoretical values (Mn,th), which is a common phenomenon using this monomer with azo initiators (AIBN and V-70).17 Similarly to the styrene polymerizations, using C1, C3, and C5 as mediators, the conversion increased with the presence of sterically bulky tBu substituents on the half salen ligand scaffold (Table 2, entries 1, 5 and 9). The complexes were found to be moderate to poor mediators achieving relatively broad dispersities (D̵ = 1.4–2.3). The exception was the unsubstituted complex C1, which achieved moderate activities (28–30% conversion) while exerting good control (D̵ = 1.1 and 1.2 using AIBN and V-70, respectively). This observation is consistent with the general principle in normal ATRP, where the mediator must be fine-tuned so that the polymerization rate is sufficiently rapid, but not so active that radical termination dominates the reaction.64
Table 2. Screening of Catalysts C1–C8 in MMA R-ATRPa.

| entry | complex | init. | conv. (%) | Mn,th (Da) | Mn (Da) | D̵ |
|---|---|---|---|---|---|---|
| 1 | C1 | AIBN | 28 | 2800 | 8400 | 1.1 |
| 2 | V-70 | 30 | 3000 | 13 000 | 1.2 | |
| 3 | C2 | AIBN | 76 | 7600 | 17 000 | 1.8 |
| 4 | V-70 | 66 | 6600 | 18 900 | 2.3 | |
| 5 | C3 | AIBN | 47 | 4700 | 12 700 | 1.6 |
| 6 | V-70 | 24 | 2400 | 18 800 | 1.5 | |
| 7 | C4 | AIBN | 85 | 8500 | 18 400 | 1.4 |
| 8 | V-70 | 68 | 6800 | 15 700 | 1.4 | |
| 9 | C5 | AIBN | 61 | 6100 | 16 700 | 2.2 |
| 10 | V-70 | 54 | 5400 | 16 500 | 1.9 | |
| 11 | C6 | AIBN | 65 | 6500 | 22 800 | 1.4 |
| 12 | V-70 | 62 | 6200 | 14 900 | 1.4 | |
| 13 | C7 | AIBN | 68 | 6800 | 18 900 | 1.7 |
| 14 | V-70 | 62 | 6200 | 19 400 | 1.6 | |
| 15 | C8 | AIBN | 64 | 6400 | 16 100 | 1.8 |
| 16 | V70 | 63 | 6300 | 16 400 | 1.4 |
Conditions: [MMA]/[FeIII]/[I] = 100:1:0.6, MMA/toluene = 1:1 (v/v), 120 °C for AIBN or 75 °C for V-70, 2 h. Conversion was determined using 1H NMR spectra of crude reaction mixtures. Mn,th [Fe] = [MMA]0/[Fe] × M(MMA) × conversion.
Kinetic experiments using C1 showed that the polymerization was well controlled; the molecular weight increased linearly with conversion. However, the reaction slowed down considerably, reaching a plateau at moderate conversions (Figure 7), possibly because of bimolecular termination side reactions.7 Similarly to the polystyrene samples, 1H NMR analysis revealed the presence of olefin-terminated polymer chains (doublets at 5.50 and 6.23 ppm, Figure S14). However, the integrals of these peaks were relatively low compared to the polymer peaks, which suggested significantly less CCT than that observed for styrene.
Figure 7.

Kinetic plots for MMA polymerization with C1: ln([M]0/[M]t) vs time (left) and molecular weight vs conversion (right).
Ring-Opening Polymerization of rac-Lactide
Iron catalysts remain underexplored within ring-opening polymerization reactions, yet can offer benefits in terms of low cost, biocompatibility, and air stability. Complexes C1–C8 were therefore tested as catalysts for the ROP of lactide (LA). Most catalysts for this reaction include a metal–alkoxide initiating group, which is typically formed via the in situ conversion of a metal–alkyl group through deprotonation of an alcohol.25 The in situ generation of metal–alkoxide catalysts through the addition of an epoxide to metal–chlorides is currently gaining momentum as an alternative initiation method (Scheme 2),41,43 thus we tested our Fe–Cl complexes for the ROP of rac-LA in the presence of PO as an activator. On previously reported procedures,41,43 we aimed to avoid the use of a large excess of PO, which would compromise the benefits of the Fe/LA system because of the toxicity of this solvent. Initial optimization of the [Fe]/PO ratio with C8 showed that excellent conversions can be achieved with as low as 50 equivalents of PO when the reaction is carried out at 120 °C for 24 h in the toluene solvent (Table S1, entries 1–4). This modification enabled the polymerizations to be performed in airtight vials instead of autoclaves and significantly improved the overall atom efficiency. Control reactions without an Fe complex and without the addition of PO showed that both components are essential for the initiation (Table 3, entries 1 and 2). Under the optimized conditions, C1–C8 all achieved almost complete conversion of LA (>89%), although inconsistent molecular weights and broad dispersities indicated uncontrolled polymerizations (Table 3, entries 3–10). This was attributed to detrimental side reactions, such as intramolecular and intermolecular transesterifications, which are a commonly observed in ROP at high conversions.25 The tacticity was investigated via homonuclear decoupled 1H NMR experiments of the purified polymers, which revealed a lack of control over the tacticity with Pi values around 0.52 indicating atactic enchainments (Figure S15). To reduce the rate of undesired side reactions, the polymerization was tested under milder conditions (85 °C), affording only traces of PLA (Table S1, entry 5). Attempts to reduce the reaction time from 24 to 2 h using C1–C8 led to variable conversions (Table 3, entries 11–18) with significant errors in reproducibility (Table S1, entries 6–9), suggesting that the rate-limiting step is the in situ formation of the active Fe–alkoxide catalyst. It may also suggest the presence of an induction period for the epoxide ring opening by the metal–halide, a phenomenon that was previously often observed in epoxide/CO2 copolymerization reactions,65−67 but may have been overlooked in the in situ formation of metal–alkoxide catalysts for ROP. The higher average molecular weights observed after 2 h of reaction time (compared to 24 h reactions) suggest the presence of extensive transesterification reactions that cleave longer polymer chains.24 Efforts to minimize the variability through the use of a PO stock solution or a higher boiling activator (butylene oxide, bp 63 °C) were unsuccessful (Table S1, entries 6–12). The inefficient initiation observed after 2 h of reaction time using C2, C5, C7, and C8 (Table 3, entries 12, 15, 17, and 18, respectively) may arise from the strength of the Fe–Cl bond. For example, the molecular structures of C1, C7, and C8 suggest that the presence of electronegative chloro-substituents shortens and strengthens the Fe–Cl bond (vide supra). This falls in line with the conversions obtained with C1 (78%, entry 11), C7 (49%, entry 17), and C8 (1%, entry 18). These results suggest that in situ initiation can lead to greater experimental variation in comparison to the use of isolated metal–alkoxide complexes. Indeed, preforming the active Fe–alkoxide species by stirring the FeIII–Cl species C8 with excess PO for 16 h, prior to the addition of LA, drastically increased the conversion from 1% (entry 18) to 90% (entry 19). Moreover, using the preformed catalyst, the reaction temperature could be lowered to 85 °C while maintaining high activity (89%, Table 3, entry 20). In accordance with initiation via epoxide opening, the matrix-assisted laser desorption ionization time-of-flight (MALDI-ToF) spectrum of the purified polymer showed a series of chains terminated by −OCH(Me)CH2Cl and −OH groups (Figures S19 and S20). Additional polymer series bearing −OCH(Me)CH2OH end groups were also observed, suggesting that initiation also occurs through the opening of epoxide due to the presence of trace water. In addition to the expected lactide repeat units (144 Da), the spectra also showed equally intense repeating units corresponding to a half lactide unit (72 Da), which further corroborates the prevalence of transesterification reactions. It is worth noting that poly(propylene oxide) was absent from both the MALDI-ToF and the NMR spectra of the polymer products, suggesting that no polymerization of the epoxide occurred.
Table 3. ROP of rac-Lactide Catalyzed by Complexes C1–C8 and Propylene Oxidea.

| entry | complex | time (h) | conv. (%) | Mn,th (Da) | Mn (Da) | D̵ |
|---|---|---|---|---|---|---|
| 1 | 24 | 0 | ||||
| 2b | C8 | 24 | 0 | |||
| 3 | C1 | 24 | 95 | 13 700 | 8800 | 1.4 |
| 4 | C2 | 24 | 95 | 13 700 | 1500 | 1.7 |
| 5 | C3 | 24 | 95 | 13 700 | 3300 | 1.4 |
| 6 | C4 | 24 | 95 | 13 700 | 9000 | 1.6 |
| 7 | C5 | 24 | 93 | 13 400 | 40 600 | 1.6 |
| 8 | C6 | 24 | 95 | 13 700 | 2100 | 4.0 |
| 9 | C7 | 24 | 93 | 13 400 | 12 600 | 1.8 |
| 10 | C8 | 24 | 89 | 12 800 | 17 200 | 1.5 |
| 11c | C1 | 2 | 78 | |||
| 12 | C2 | 2 | 2 | |||
| 13 | C3 | 2 | 89 | 12 900 | 5500 | 1.1 |
| 14 | C4 | 2 | 90 | 13 000 | 45 900 | 1.7 |
| 15 | C5 | 2 | 49 | 7100 | 50 500 | 1.3 |
| 16 | C6 | 2 | 91 | 13 100 | 42 200 | 1.5 |
| 17 | C7 | 2 | 49 | 7100 | 22 000 | 1.4 |
| 18 | C8 | 2 | 1 | |||
| 19d | C8 | 2 | 90 | 13 000 | 15 900 | 2.1 |
| 20d,e | C8 | 2 | 89 | 12 800 | 17 200 | 1.5 |
Conditions: [lactide]/[Fe]/[PO] = 100:1:50, lactide concentration in toluene = 1 M, 120 °C. Conversion was determined using 1H NMR spectra of crude reaction mixtures. Mn,th [Fe] = [lactide]0/[Fe] × M(lactide) × conversion.
Reaction was carried out without the addition of PO.
Efforts to recover polymer were unsuccessful, indicating the formation of shorter polymer chains.
The Fe complex was stirred for 16 h in excess PO at room temperature prior to the addition of lactide.
The reaction was performed at 85 °C.
Conclusions
In summary, five new monometallic pentacoordinate FeIII chloride complexes were prepared based on a half salen ligand scaffold. The complexes were characterized through a combination of elemental analysis, high-resolution mass spectrometry, and in three cases, X-ray crystallography. Combined with three previously reported analogues, this series of eight complexes bearing a range of ligand substituents has been tested as mediators in the R-ATRP of styrene and MMA, achieving good to moderate control over the polymerization. The complexes have also been applied in the ROP of rac-lactide, generally giving >95% conversion after 24 h and showing that the in situ formation of FeIII–alkoxide via epoxide opening is necessary for efficient initiation. Although this method of initiation is currently gaining momentum within ROP, some of our FeIII half salen catalysts give little or no conversion after 2 h, suggesting that epoxide opening by a metal–halide may require an extreme excess of toxic epoxide (i.e., as a solvent) to achieve control. In general, these results demonstrate the versatility of simple, air-stable FeIII half salen complexes and show that this abundant, inexpensive, and non-toxic metal can be a relevant alternative in polymerization catalysis.
Experimental Section
Materials and Methods
All air- and/or moisture-sensitive experiments were performed under argon atmosphere using an MBRAUN LABmaster glovebox and standard Schlenk techniques. Solvents were obtained from a solvent purification system (Innovative Technologies) consisting of columns of alumina and copper catalyst and were further degassed by freeze–pump–thaw cycles prior to use. Styrene and MMA were dried by stirring over calcium hydride, vacuum transferred, and stored at −35 °C. rac-Lactide was sublimed three times and stored at −35 °C. 2,2′-Azobis(4-methoxy-2,4-dimethylvaleronitrile) (V-70) was washed with methanol, dried under vacuum, and stored at −35 °C. 2,2′-Azobis(2-methylpropionitrile) (AIBN) was recrystallized from methanol, dried under vacuum, and stored at −35 °C. 2,6-Di-isopropylaniline was distilled under vacuum prior to use. N-Methylamine (40 m/m % in H2O), salicylaldehyde, 3,5-dichlorosalicylaldehyde, and 3,5-di-tert-butylsalicylaldehyde were used as received. Gel permeation chromatography (GPC) was carried out in the HPLC grade THF solvent at a flow rate of 1 mL/min at 35 °C on a Malvern Instruments Viscotek 270 GPC Max triple detection system with 2× mixed bed styrene/DVB columns (300 × 7.5 mm). Absolute molar masses were obtained using the refractive index increment (dn/dc values) for this sample/solvent combination of 0.185 mL/g for poly(styrene),68 0.088 mL/g for poly(methyl methacrylate),69 and 0.05 mL/g for PLA.70 NMR spectra were obtained on a 500 MHz Bruker AVANCE III spectrometer. Solution magnetic moments were determined via NMR spectroscopy using Evans’ method.60 EI mass spectra were obtained on a Bruker DALTONICS micro TOF instrument operating in the positive ion electrospray mode. MALDI-ToF mass spectrometry was performed on a Bruker ultrafleXtreme MALDI-ToF spectrometer; samples were prepared with dithranol as matrix and sodium or potassium iodide as the ionization source. Elemental analyses were performed by Stephen Boyer at London Metropolitan University.
Synthetic Procedures
Pro-ligands L1–L8 and complexes C1, C3, and C7 were prepared according to previously reported literature methods.48,53,71−73 Characterization data for L1,48L2,52L3,48L4,73L5,74L6,52 and L7(48) were in agreement with reported values in the literature.
Data for L8
(2.90 g, 91%) 1H NMR (500 MHz CDCl3): δ 14.49 (s, 1H, OH), 8.23 (s, 1H, HC=N), 7.39 (d, J = 2.5 Hz, 1H, ArH), 7.12 (d, J = 2.5 Hz, 1H, ArH), 3.51 (d, J = 1.5 Hz, 3H, CH3); 13C NMR (126 MHz, CDCl3): δ 164.9 (C=N), 157.8 (C–OH), 132.4, 128.9, 123.3, 122.2, 119.4 (Ar-C), 44.9 (CH3). HRMS (EI): m/z [M]+ 202.9906; calcd [M]+ 202.9905. Elemental Anal. Calcd for C8H7Cl2NO: C, 47.1; H, 3.5; N, 6.9. Found: C, 47.3; H, 3.35; N, 7.0.
General Procedure for Complexes C2, C4–C6, and C8
To a solution of pro-ligand (L1–L8) in THF solvent, 1.1 equiv of NaH was gradually added and the mixture was stirred at ambient temperature for 1 h. A solution of anhydrous FeCl3 (0.5 equiv) in THF was added dropwise to afford a dark colored mixture that was stirred for a further 16 h at room temperature. Solvents were evaporated in vacuo, and the crude product was taken up in toluene solvent. The NaCl by-product was removed by filtration through celite, and the filtrate was dried in vacuo. The crude products were washed three times with hexane to afford dark purple-brown powders. Single crystals suitable for X-ray diffraction analysis were obtained via slow evaporation of DCM under air. Structures of C5, C6, and C8 can be found under CCDC numbers 1868412–1868414.
Data for C2
(1.1 g, 52%) HRMS (EI): m/z [M]+ 651.2441; calcd [M]+ 651.2441. Elemental Anal. Calcd for C38H44ClFeN2O2: C, 70.0; H, 6.8; N, 4.3. Found: C, 70.2; H, 6.8; N, 4.4.
Data for C4
(1.1 g, 49%) HRMS (EI): m/z [M]+ 763.3718; calcd [M]+ 763.3693. Elemental Anal. Calcd for C46H60ClFeN2O2: C, 72.3; H, 7.9; N, 3.7. Found: C, 70.1; H, 7.8; N, 4.0.
Data for C5
(1.4 g, 89%) HRMS (EI): m/z [M]+ 583.2790; calcd [M]+ 583.2754. Elemental Anal. Calcd for C32H48ClFeN2O2: C, 65.8; H, 8.3; N, 4.8. Found: C, 65.9; H, 8.4; N, 4.9.
Data for C6
(1.0 g, 49%) HRMS (EI): m/z [M]+ 875.5019; calcd [M]+ 875.4945. Elemental Anal. Calcd for C54H76ClFeN2O2: C, 74.0; H, 8.7; N, 3.2. Found: C, 74.1; H, 8.8; N, 3.2.
Data for C8
(0.6 g, 45%) HRMS (EI): m/z [M]+ 494.8716; calcd [M]+ 494.8691. Elemental Anal. Calcd for C16H12Cl5FeN2O2: C, 38.6; H, 2.4; N, 5.6. Found: C, 38.8; H, 2.5; N, 5.6.
General Procedure for the R-ATRP of Styrene and MMA
In a glovebox, the complexes (C1–C8) (24.0 μmol), monomer (100 equiv, 2.40 mmol), toluene (toluene/monomer, 1:1, v/v), and initiator (AIBN or V-70) (0.6 equiv, 14.4 μmol) were sequentially added to an airtight screwcap vial equipped with a magnetic stirrer bar. The vial was removed from the glovebox and heated to 75 or 120 °C for 2 h. The vial was subsequently cooled to room temperature, and an aliquot of the crude mixture was analyzed via 1H NMR spectroscopy to determine the monomer conversion. The remainder of the reaction mixture was quenched with chloroform (approximately 1 mL), and the polymer was purified by precipitation in acidified methanol (MeOH/HCl(aq), approximately 75:1 mL). The polymer was isolated by filtration and was subsequently dried in vacuo until constant weight was achieved. Samples for GPC analysis were prepared in HPLC grade THF and filtered through a 0.22 μm pore-sized syringe filter.
General Procedure for the ROP of rac-Lactide
In a glovebox, complexes (C1–C8) (10.0 μmol), PO (50 equiv, 0.5 mmol), rac-lactide (100 equiv, 1.0 mmol), and toluene (1 mL) were added to an airtight screwcap vial equipped with a magnetic stirrer bar. The vial was transferred to the bench and heated to 120 °C in an oil bath for 24 h. The vial was subsequently cooled to room temperature, the reaction mixture was quenched with hexane, and an aliquot of the crude mixture was analyzed via 1H NMR spectroscopy to determine monomer conversion. The remainder of the reaction mixture was dissolved in chloroform (approximately 1 mL), and the polymer was precipitated by the addition of the solution to cold (0 °C) acidified methanol (MeOH/HCl(aq), 75:1 mL). The polymer was then isolated by filtration and dried in vacuo until constant weight was achieved. Samples for GPC analysis were prepared in HPLC grade THF solvent and filtered through a 0.20 μm pore sized syringe filter prior to analysis.
Crystallography
Single-crystal X-ray diffraction data were collected on an Rigaku Oxford diffraction SuperNova diffractometer fitted with an Atlas CCD detector with Mo Kα radiation (λ = 0.7107 Å) or Cu Kα radiation (λ = 1.5418 Å). Crystals were mounted under Paratone on MiTeGen loops. The structures were solved by direct methods using SHELXS or SHELXT and refined by full-matrix least-squares on F2 using SHELXL interfaced through Olex2.75,76 Molecular graphics for all structures were generated using Diamond.
Acknowledgments
We would like to thank Dr Benjamin Lake and Daniel Coward for useful discussions about controlled radical polymerizations. We gratefully acknowledge the University of Edinburgh and the Ramsay Memorial Trust (J.A.G.) for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02432.
GPC and HRMS data, 1H NMR and MALDI spectra, and single-crystal X-ray diffraction data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Chikkali S.; Ambade A. V.; de Bruin B.; Shaver M.. Metal-Catalyzed Polymerization: Fundamentals to Applications, 1st ed.; CRC Press: Boca Raton, 2017. [Google Scholar]
- Platel R.; Hodgson L.; Williams C. Biocompatible initiators for lactide polymerization. Polym. Rev. 2008, 48, 11–63. 10.1080/15583720701834166. [DOI] [Google Scholar]
- Tsarevsky N. V.; Matyjaszewski K. “Green” Atom Transfer Radical Polymerization: From Process Design to Preparation of Well-Defined Environmentally Friendly Polymeric Materials. Chem. Rev. 2007, 107, 2270–2299. 10.1021/cr050947p. [DOI] [PubMed] [Google Scholar]
- Hoppe J. O.; Agnew Marcelli M. G.; Tainter M. L. A review of the toxicity of iron compounds. Am. J. Med. Sci. 1955, 230, 558–571. 10.1097/00000441-195523050-00012. [DOI] [PubMed] [Google Scholar]
- Gopalaiah K. Chiral iron catalysts for asymmetric synthesis. Chem. Rev. 2013, 113, 3248–3296. 10.1021/cr300236r. [DOI] [PubMed] [Google Scholar]
- Jordan T. H. Structural geology of the Earth’s interior. Proc. Natl. Acad. Sci. U.S.A. 1979, 76, 4192–4200. 10.1073/pnas.76.9.4192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poli R.; Allan L. E. N.; Shaver M. P. Iron-mediated reversible deactivation controlled radical polymerization. Prog. Polym. Sci. 2014, 39, 1827–1845. 10.1016/j.progpolymsci.2014.06.003. [DOI] [Google Scholar]
- Raynaud J.; Wu J. Y.; Ritter T. Iron-catalyzed polymerization of isoprene and other 1,3-dienes. Angew. Chem. 2012, 124, 11975–11978. 10.1002/ange.201205152. [DOI] [PubMed] [Google Scholar]
- Biernesser A. B.; Li B.; Byers J. A. Redox-controlled polymerization of lactide catalyzed by bis(imino)pyridine iron bis(alkoxide) complexes. J. Am. Chem. Soc. 2013, 135, 16553–16560. 10.1021/ja407920d. [DOI] [PubMed] [Google Scholar]
- Matyjaszewski K.; Xia J. Atom transfer radical polymerization. Chem. Rev. 2001, 101, 2921–2990. 10.1021/cr940534g. [DOI] [PubMed] [Google Scholar]
- Ando T.; Kamigaito M.; Sawamoto M. Iron(II) Chloride Complex for Living Radical Polymerization of Methyl Methacrylate. Macromolecules 1997, 30, 4507–4510. 10.1021/ma961478j. [DOI] [Google Scholar]
- Matyjaszewski K.; Wei M.; Xia J.; McDermott N. E. Controlled/“Living” Radical Polymerization of Styrene and Methyl Methacrylate Catalyzed by Iron Complexes. Macromolecules 1997, 30, 8161–8164. 10.1021/ma971010w. [DOI] [Google Scholar]
- Ding M.; Jiang X.; Peng J.; Zhang L.; Cheng Z.; Zhu X. An atom transfer radical polymerization system: catalyzed by an iron catalyst in PEG-400. Green Chem. 2015, 17, 271–278. 10.1039/c4gc01169d. [DOI] [Google Scholar]
- Zhang B.; Jiang X.; Zhang L.; Cheng Z.; Zhu X. Fe(III)-mediated ICAR ATRP in a p-xylene/PEG-200 biphasic system: facile and highly efficient separation and recycling of an iron catalyst. Polym. Chem. 2015, 6, 6616–6622. 10.1039/c5py01045d. [DOI] [Google Scholar]
- Claverie J. Controlled living radical polymerization of styrene with iron complexes. Res. Discl. 1998, 1595–1604. [Google Scholar]
- Moineau G.; Dubois P.; Jérôme R.; Senninger T.; Teyssié P. Alternative Atom Transfer Radical Polymerization for MMA Using FeCl3 and AIBN in the Presence of Triphenylphosphine: An Easy Way to Well-Controlled PMMA. Macromolecules 1998, 31, 545–547. 10.1021/ma971132o. [DOI] [Google Scholar]
- Lake B. R. M.; Shaver M. P. Iron(ii) β-ketiminate complexes as mediators of controlled radical polymerisation. Dalton Trans. 2016, 45, 15840–15849. 10.1039/c6dt01208f. [DOI] [PubMed] [Google Scholar]
- Coward D. L.; Lake B. R. M.; Shaver M. P. Understanding Organometallic-Mediated Radical Polymerization with an Iron(II) Amine-Bis(phenolate). Organometallics 2017, 36, 3322–3328. 10.1021/acs.organomet.7b00473. [DOI] [Google Scholar]
- Allan L. E. N.; MacDonald J. P.; Reckling A. M.; Kozak C. M.; Shaver M. P. Controlled Radical Polymerization Mediated by Amine-Bis(phenolate) Iron(III) Complexes. Macromol. Rapid Commun. 2012, 33, 414–418. 10.1002/marc.201100872. [DOI] [PubMed] [Google Scholar]
- Schroeder H.; Buback M.; Shaver M. P. Kinetics of Amine-Bis(phenolate) Iron-Mediated ATRP Up to High Pressure. Macromolecules 2015, 48, 6114–6120. 10.1021/acs.macromol.5b01390. [DOI] [Google Scholar]
- Schroeder H.; Lake B. R. M.; Demeshko S.; Shaver M. P.; Buback M. A Synthetic and Multispectroscopic Speciation Analysis of Controlled Radical Polymerization Mediated by Amine-Bis(phenolate)iron Complexes. Macromolecules 2015, 48, 4329–4338. 10.1021/acs.macromol.5b01175. [DOI] [Google Scholar]
- Lake B. R. M.; Shaver M. P.. Controlled Radical Polymerization: Mechanisms; ACS, 2015; Vol. 1187, pp 311–326. [Google Scholar]
- Poli R.; Shaver M. P. Atom transfer radical polymerization (ATRP) and organometallic mediated radical polymerization (OMRP) of styrene mediated by diaminobis(phenolato)iron(II) complexes: A DFT study. Inorg. Chem. 2014, 53, 7580–7590. 10.1021/ic5009347. [DOI] [PubMed] [Google Scholar]
- Dechy-Cabaret O.; Martin-Vaca B.; Bourissou D. Controlled ring-opening polymerization of lactide and glycolide. Chem. Rev. 2004, 104, 6147–6176. 10.1021/cr040002s. [DOI] [PubMed] [Google Scholar]
- Stanford M. J.; Dove A. P. Stereocontrolled ring-opening polymerisation of lactide. Chem. Soc. Rev. 2010, 39, 486–494. 10.1039/b815104k. [DOI] [PubMed] [Google Scholar]
- MacDonald J. P.; Shaver M. P.. Green Polymer Chemistry: Biobased Materials and Biocatalysis; ACS, 2015; Vol. 1192, pp 147–167. [Google Scholar]
- Mandal M.; Oppelt K.; List M.; Teasdale I.; Chakraborty D.; Monkowius U. Copper(II) complexes with imino phenoxide ligands: synthesis, characterization, and their application as catalysts for the ring-opening polymerization of rac-lactide. Chem. Mon. 2016, 147, 1883–1892. 10.1007/s00706-016-1830-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routaray A.; Nath N.; Maharana T.; Sutar A. k. Synthesis and Immortal ROP of L-Lactide Using Copper Complex. J. Macromol. Sci., Part A: Pure Appl.Chem. 2015, 52, 444–453. 10.1080/10601325.2015.1029370. [DOI] [Google Scholar]
- O’Keefe B. J.; Monnier S. M.; Hillmyer M. A.; Tolman W. B. Rapid and controlled polymerization of lactide by structurally characterized ferric alkoxides. J. Am. Chem. Soc. 2001, 123, 339–340. 10.1021/ja003537l. [DOI] [PubMed] [Google Scholar]
- O’Keefe B. J.; Breyfogle L. E.; Hillmyer M. A.; Tolman W. B. Mechanistic Comparison of Cyclic Ester Polymerizations by Novel Iron(III)–Alkoxide Complexes: Single vs Multiple Site Catalysis. J. Am. Chem. Soc. 2002, 124, 4384–4393. 10.1021/ja012689t. [DOI] [PubMed] [Google Scholar]
- Gibson V. C.; Marshall E. L.; Navarro-Llobet D.; White A. J. P.; Williams D. J. A well-defined iron(ii) alkoxide initiator for the controlled polymerisation of lactide. Dalton Trans. 2002, 4321–4322. 10.1039/b209703f. [DOI] [Google Scholar]
- Wang X.; Liao K.; Quan D.; Wu Q. Bulk ring-opening polymerization of lactides initiated by ferric alkoxides. Macromolecules 2005, 38, 4611–4617. 10.1021/ma047545o. [DOI] [Google Scholar]
- Kricheldorf H. R.; Damrau D.-O. Polylactones, 38. Polymerization of L-lactide with Fe(II) lactate and other resorbable Fe(II) salts. Macromol. Chem. Phys. 1997, 198, 1767–1774. 10.1002/macp.1997.021980606. [DOI] [Google Scholar]
- Stolt M.; Södergård A. Use of Monocarboxylic Iron Derivatives in the Ring-Opening Polymerization of L-Lactide. Macromolecules 1999, 32, 6412–6417. 10.1021/ma9902753. [DOI] [Google Scholar]
- McGuinness D. S.; Marshall E. L.; Gibson V. C.; Steed J. W. Anionic iron(II) alkoxides as initiators for the controlled ring-opening polymerization of lactide. J. Polym. Sci., Part A: Polym. Chem. 2003, 41, 3798–3803. 10.1002/pola.10946. [DOI] [Google Scholar]
- Kricheldorf H. R.; Boettcher C. Polylactones 21. Polymerization of L,L-lactide and rac-D,L-lactide with hematin and related porphyrin complexes. Makromol. Chem. 1993, 194, 463–473. 10.1002/macp.1993.021940209. [DOI] [Google Scholar]
- Delle Chiaie K. R.; Biernesser A. B.; Ortuño M. A.; Dereli B.; Iovan D. A.; Wilding M. J. T.; Li B.; Cramer C. J.; Byers J. A. The role of ligand redox non-innocence in ring-opening polymerization reactions catalysed by bis(imino)pyridine iron alkoxide complexes. Dalton Trans. 2017, 46, 12971–12980. 10.1039/c7dt03067c. [DOI] [PubMed] [Google Scholar]
- Idage B. B.; Idage S. B.; Kasegaonkar A. S.; Jadhav R. V. Ring opening polymerization of dilactide using salen complex as catalyst. Mater. Sci. Eng., B 2010, 168, 193–198. 10.1016/j.mseb.2009.10.037. [DOI] [Google Scholar]
- Robert C.; Schmid T. E.; Richard V.; Haquette P.; Raman S. K.; Rager M.-N.; Gauvin R. M.; Morin Y.; Trivelli X.; Guérineau V.; del Rosal I.; Maron L.; Thomas C. M. Mechanistic aspects of the polymerization of lactide using a highly efficient aluminum(III) catalytic system. J. Am. Chem. Soc. 2017, 139, 6217–6225. 10.1021/jacs.7b01749. [DOI] [PubMed] [Google Scholar]
- Anker M.; Balasanthiran C.; Balasanthiran V.; Chisholm M. H.; Jayaraj S.; Mathieu K.; Piromjitpong P.; Praban S.; Raya B.; Simonsick W. J. A new route for the preparation of enriched iso-polylactide from rac-lactide via a Lewis acid catalyzed ring-opening of an epoxide. Dalton Trans. 2017, 46, 5938–5945. 10.1039/c6dt03198f. [DOI] [PubMed] [Google Scholar]
- Cozzolino M.; Leo V.; Tedesco C.; Mazzeo M.; Lamberti M. Salen, salan and salalen iron(iii) complexes as catalysts for CO2/epoxide reactions and ROP of cyclic esters. Dalton Trans. 2018, 47, 13229. 10.1039/c8dt03169j. [DOI] [PubMed] [Google Scholar]
- Doherty S.; Errington R. J.; Housley N.; Clegg W. Dimeric Aluminum Chloride Complexes of N-Alkoxyalkyl-β-ketoimines: Activation with Propylene Oxide To Form Efficient Lactide Polymerization Catalysts. Organometallics 2004, 23, 2382–2388. 10.1021/om0343770. [DOI] [Google Scholar]
- Duan R.; Hu C.; Li X.; Pang X.; Sun Z.; Chen X.; Wang X. Air-Stable Salen-Iron Complexes: Stereoselective Catalysts for Lactide and ε-Caprolactone Polymerization through in Situ Initiation. Macromolecules 2017, 50, 9188–9195. 10.1021/acs.macromol.7b01766. [DOI] [Google Scholar]
- O’Reilly R. K.; Gibson V. C.; White A. J. P.; Williams D. J. Design of Highly Active Iron-Based Catalysts for Atom Transfer Radical Polymerization: Tridentate Salicylaldiminato Ligands Affording near Ideal Nernstian Behavior. J. Am. Chem. Soc. 2003, 125, 8450–8451. 10.1021/ja035458q. [DOI] [PubMed] [Google Scholar]
- Silvino A. C.; Rodrigues A. L. C.; Resende J. A. L. C. Synthesis, structure and application to l-lactide polymerization of a new phenoxy-imine iron(III) complex. Inorg. Chem. Commun. 2015, 55, 39–42. 10.1016/j.inoche.2015.03.006. [DOI] [Google Scholar]
- Al-Qaisi F.; Genjang N.; Nieger M.; Repo T. Synthesis, structure and catalytic activity of bis(phenoxyiminato)iron(III) complexes in coupling reaction of CO2 and epoxides. Inorg. Chim. Acta 2016, 442, 81–85. 10.1016/j.ica.2015.11.023. [DOI] [Google Scholar]
- van der Bergen A.; Murray K. S.; O’Connor M. J.; Rehak N.; West B. O. N-Substituted salicylaldimine complexes of iron(III). I. Synthesis, infrared and mass spectra. Aust. J. Chem. 1968, 21, 1505–1515. 10.1071/ch9681505. [DOI] [Google Scholar]
- Fazekas E.; Nichol G. S.; Shaver M. P.; Garden J. A. Stable Fe(III) phenoxyimines as selective and robust CO2/epoxide coupling catalysts. Dalton Trans. 2018, 47, 13106–13112. 10.1039/c8dt02919a. [DOI] [PubMed] [Google Scholar]
- Jianming R.; Anguo X.; Hongwei W.; Hailin Y. Review - recent development of ring-opening polymerization of cyclic esters using aluminum complexes. Des. Monomers Polym. 2013, 17, 345–355. 10.1080/15685551.2013.840509. [DOI] [Google Scholar]
- García-Valle F. M.; Tabernero V.; Cuenca T.; Mosquera M. E. G.; Cano J.; Milione S. Biodegradable PHB from rac-β-butyrolactone: highly controlled ROP mediated by a pentacoordinated aluminum complex. Organometallics 2018, 37, 837. 10.1021/acs.organomet.7b00843. [DOI] [Google Scholar]
- Normand M.; Dorcet V.; Kirillov E.; Carpentier J.-F. {Phenoxy-imine}aluminum versus -indium Complexes for the Immortal ROP of Lactide: Different Stereocontrol, Different Mechanisms. Organometallics 2013, 32, 1694–1709. 10.1021/om301155m. [DOI] [Google Scholar]
- García-Valle F. M.; Estivill R.; Gallegos C.; Cuenca T.; Mosquera M. E. G.; Tabernero V.; Cano J. Metal and ligand-substituent effects in the immortal polymerization of rac-lactide with Li, Na, and K phenoxo-imine complexes. Organometallics 2015, 34, 477–487. 10.1021/om501000b. [DOI] [Google Scholar]
- Gotthardt H.; Hoffmann N. Synthese und neue [4 + 2]-Cycloadditionen von Cumarin-Abkömmlingen. Liebigs Ann. Chem. 1985, 1985, 901–912. 10.1002/jlac.198519850504. [DOI] [Google Scholar]
- Addison A. W.; Rao T. N.; Reedijk J.; van Rijn J.; Verschoor G. C. Synthesis, structure, and spectroscopic properties of copper(II) compounds containing nitrogen-sulphur donor ligands; the crystal and molecular structure of aqua[1,7-bis(N-methylbenzimidazol-2′-yl)-2,6-dithiaheptane]copper(II) perchlorate. Dalton Trans. 1984, 1349–1356. 10.1039/dt9840001349. [DOI] [Google Scholar]
- Becker J. M.; Barker J.; Clarkson G. J.; van Gorkum R.; Johal G. K.; Walton R. I.; Scott P. Chirality and diastereoselection in the μ-oxo diiron complexes L2Fe-O-FeL2 (L = bidentate salicylaldiminato). Dalton Trans. 2010, 39, 2309–2326. 10.1039/b905706d. [DOI] [PubMed] [Google Scholar]
- Amini M.; Arab A.; Derakhshandeh P. G.; Bagherzadeh M.; Ellern A.; Woo L. K. A novel iron complex containing an N,O-type bidentate oxazoline ligand: Synthesis, X-ray studies, DFT calculations and catalytic activity. Spectrochim. Acta, Part A 2014, 133, 432–438. 10.1016/j.saa.2014.06.012. [DOI] [PubMed] [Google Scholar]
- Davies J. E.; Gatehouse B. M. The crystal and molecular structure of chlorobis-(N-n-propylsalicylaldiminato)iron(III). Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1972, 28, 3641–3645. 10.1107/s0567740872008490. [DOI] [Google Scholar]
- Magurany C. J.; Strouse C. E. Structure and magnetism of monomeric chlorobis(N-(2-phenylethyl)salicylideniminato)iron(III), Fe(SANE)2Cl. Inorg. Chem. 1982, 21, 2348–2350. 10.1021/ic00136a045. [DOI] [Google Scholar]
- Elerman Y.; Paulus H. A five-coordinate bis(p-bromophenylsalicylaldimino)chloroiron(III) complex. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 1996, 52, 1971–1973. 10.1107/s0108270196005422. [DOI] [Google Scholar]
- Evans D. F. 400. The determination of the paramagnetic susceptibility of substances in solution by nuclear magnetic resonance. Chem. Commun. 1959, 2003–2005. 10.1039/jr9590002003. [DOI] [Google Scholar]
- Allan L. E. N.; MacDonald J. P.; Nichol G. S.; Shaver M. P. Single component iron catalysts for atom transfer and organometallic mediated radical polymerizations: mechanistic studies and reaction scope. Macromolecules 2014, 47, 1249–1257. 10.1021/ma402381x. [DOI] [Google Scholar]
- Allan L. E. N.; Shaver M. P.; White A. J. P.; Gibson V. C. Correlation of Metal Spin-State in α-Diimine Iron Catalysts with Polymerization Mechanism. Inorg. Chem. 2007, 46, 8963–8970. 10.1021/ic701500y. [DOI] [PubMed] [Google Scholar]
- Shaver M. P.; Allan L. E. N.; Gibson V. C. Organometallic Intermediates in the Controlled Radical Polymerization of Styrene by α-Diimine Iron Catalysts. Organometallics 2007, 26, 4725–4730. 10.1021/om700395h. [DOI] [Google Scholar]
- Konkolewicz D.; Matyjaszewski K.. Controlled Radical Polymerization: Mechanisms; American Chemical Society, 2015; Vol. 1187, pp 87–103. [Google Scholar]
- Garden J. A.; Saini P. K.; Williams C. K. Greater than the sum of its parts: A heterodinuclear polymerization catalyst. J. Am. Chem. Soc. 2015, 137, 15078–15081. 10.1021/jacs.5b09913. [DOI] [PubMed] [Google Scholar]
- Darensbourg D. J.; Moncada A. I. Mechanistic Insight into the Initiation Step of the Coupling Reaction of Oxetane or Epoxides and CO2 Catalyzed by (salen)CrX Complexes. Inorg. Chem. 2008, 47, 10000–10008. 10.1021/ic801231p. [DOI] [PubMed] [Google Scholar]
- Lu X.-B.; Shi L.; Wang Y.-M.; Zhang R.; Zhang Y.-J.; Peng X.-J.; Zhang Z.-C.; Li B. Design of Highly Active Binary Catalyst Systems for CO2/Epoxide Copolymerization: Polymer Selectivity, Enantioselectivity, and Stereochemistry Control. J. Am. Chem. Soc. 2006, 128, 1664–1674. 10.1021/ja056383o. [DOI] [PubMed] [Google Scholar]
- Grcev S.; Schoenmakers P.; Iedema P. Determination of molecular weight and size distribution and branching characteristics of PVAc by means of size exclusion chromatography/multi-angle laser light scattering (SEC/MALLS). Polymer 2004, 45, 39–48. 10.1016/j.polymer.2003.10.077. [DOI] [Google Scholar]
- Min K.; Gao H.; Yoon J. A.; Wu W.; Kowalewski T.; Matyjaszewski K. One-pot synthesis of hairy nanoparticles by emulsion ATRP. Macromolecules 2009, 42, 1597–1603. 10.1021/ma8026244. [DOI] [Google Scholar]
- Cairns S. A.; Schultheiss A.; Shaver M. P. A broad scope of aliphatic polyesters prepared by elimination of small molecules from sustainable 1,3-dioxolan-4-ones. Polym. Chem. 2017, 8, 2990–2996. 10.1039/c7py00254h. [DOI] [Google Scholar]
- Huang J.; Lian B.; Qian Y.; Zhou W.; Chen W.; Zheng G. Syntheses of titanium(IV) complexes with mono-Cp and Schiff base ligands and their catalytic activities for ethylene polymerization and ethylene/1-hexene copolymerization. Macromolecules 2002, 35, 4871–4874. 10.1021/ma012064f. [DOI] [Google Scholar]
- Cameron P. A.; Gibson V. C.; Redshaw C.; Segal J. A.; Solan G. A.; White A. J. P.; Williams D. J. Synthesis and characterisation of neutral dialkylaluminium complexes stabilised by salicylaldiminato ligands, and their conversion to monoalkylaluminium cations. Dalton Trans. 2001, 1472–1476. 10.1039/b100743m. [DOI] [Google Scholar]
- Opstal T.; Couchez K.; Verpoort F. Easily accessible ring opening metathesis and atom transfer radical polymerization catalysts based on arene, norbornadiene and cyclooctadiene ruthenium complexes bearing Schiff base ligands. Adv. Synth. Catal. 2003, 345, 393–401. 10.1002/adsc.200390045. [DOI] [Google Scholar]
- Safaei E.; Kabir M. M.; Wojtczak A.; Jagličić Z.; Kozakiewicz A.; Lee Y.-I. Synthesis, crystal structure, magnetic and redox properties of copper(II) complexes of N-alkyl(aryl) tBu-salicylaldimines. Inorg. Chim. Acta 2011, 366, 275–282. 10.1016/j.ica.2010.11.017. [DOI] [Google Scholar]
- Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Cryst. Struct. Commun. 2015, 71, 3–8. 10.1107/s2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolomanov O. V.; Bourhis L. J.; Gildea R. J.; Howard J. A. K.; Puschmann H. OLEX2: a complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. 10.1107/s0021889808042726. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.