

Abstract
Polymeric bicontinuous nanospheres (BCNs) that are analogous to lipid cubosomes possess high internal surface area and porosity that can accommodate the loading of a wide range of hydrophobic and hydrophilic molecules for diverse applications. Self-assembly of BCNs has been reported using complex amphi-philic polymeric structures, with co-solvent dispersion being the only documented method of formation. Here, we report a simple amphiphilic diblock copolymer, poly(ethylene glycol)17-block-poly(propylene sulfide)75 (PEG17-bl-PPS75), to form BCNs using the rapid and scalable technique of flash nanoprecipitation (FNP). Dynamic light scattering (DLS) and cryogenic transmission electron microscopy (cryoTEM) verified low polydispersity and the formation of bicontinuous structures with internal aqueous channels, respectively. Small-angle X-ray scattering (SAXS) confirmed a primitive cubic (Im3m) internal organization for BCNs assembled by FNP. Both hydrophobic and hydrophilic molecules were effectively loaded into BCNs via FNP, and encapsulated payloads were found to release in controlled manner in aqueous solutions. Due to the oxidation-sensitivity of PPS, biologically relevant concentrations of reactive oxygen species could trigger payload release on demand. BCNs were found to be non-toxic and endocytosed by phagocytic cells. Furthermore, an in vitro functional assay showed BCNs co-loaded with antigen oval-bumin and adjuvant monophosphoryl lipid A (MPL) to promote peptide/MHCI surface presentation by dendritic cells, a critical step for vaccine formulations during immunization. In conclusion, FNP supports the facile and scalable assembly and loading of PEG-bl-PPS BCNs, making them an attractive nanoscale delivery vehicle for both hydrophilic and hydrophobic molecules.
Introduction
Amphiphiles can be engineered to serve as molecular building blocks for self-assembled nanostructures possessing physicochemical properties that are advantageous for a wide range of applications.1,2 Assemblies presenting ordered liquid crystal-line architectures uniquely remain thermodynamically stable under a variety of conditions that otherwise promote dissociation of less structured self-assembled systems, like micelles. Bicontinuous nanospheres (BCNs) are one example of such a lyotropic soft nanoarchitecture and have attracted much attention for their large surface area, ability to solubilize payloads of diverse polarity and potential to function as slow release nanomatrices.3,4 Cryogenic transmission electron microscopy (cryoTEM) and small-angle X-ray scattering (SAXS) have revealed diverse internal geometries for BCNs,5,6 which present as continuous self-assembled bilayers perforated with separate non-intersecting water channels of varying degrees of organization7 (Fig. 1). BCNs with inverse bicontinuous cubic organization, referred to as cubosomes, have been categorized into gyroid (Ia3d), diamond (Pn3m) and primitive (Im3m) phases depending on their topology.8,9 These porous phases of mixed hydrophilic and hydrophobic domains make these structures particularly useful for application as controlled delivery vehicles, nanotemplates, photovoltaics and catalytic nanoreactors.10–13
Fig. 1.
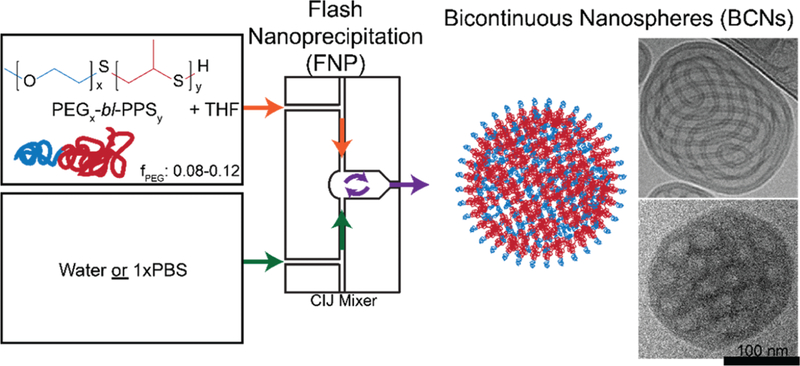
Schematic of the formation of bicontinuous nanospheres (BCNs) using flash nanoprecipitation. PEG17-bl-PPS75 copolymer was dissolved in THF and impinged against water or PBS in a CIJ mixer to form BCNs (illustration and cryoTEM image).
The internal structure of BCNs is dependent on the solvent concentration and temperature during assembly and both the geometry and glass transition temperature of the amphi-phiIe.4,6,7,14 Few amphiphiles are capable of forming stable inverted mesophases, and the majority of BCNs are assembled from unsaturated mono- and diglycerides, usually lipid mixtures of monoolein or oleic acid.8 Despite their stability, lipid-based BCNs are susceptible to aggregation due to their extensive exposed hydrophobic surfaces, resulting in the need for the incorporation of stabilizing surfactants in lipid BCN formulations. Furthermore, these fatty acids are highly susceptible to hydrolysis by esterases, which limits there in vivo application.15 As a result, a need exists for alternative amphiphile chemistries capable of forming stable lyotropic mesophases with BCN internal geometry.
The stability of self-assembled nanoarchitectures can be enhanced through the rational design of block copolymer amphiphiles, but polymeric BCNs usually require complex structures like triblock copolymers,16 comb-like block copolymers,17 semi-crystalline block copolymers,12 dendrimers,18 and dendritic linear block copolymers.6 Formation of BCNs using simpler amphiphilic block copolymers has only rarely been achieved.4,19 For example, poly(ethylene oxide)-block-poly (octadecyl methacrylate) (PEO-bl-PODMA) has been reported to form BCNs with varying fractions of PEO under specific solvent conditions.4 Thin-film hydration and co-solvent dispersion are the most commonly used methods for the self-assembly of nanostructures using amphiphilic block copolymers.20 However, scalability, repeatability, control of particle size, efficient loading of diverse actives and fabrication time remain major concerns with these methods. To date, co-solvent dispersion is the only reported method in the literature to form polymeric BCNs.4,6 Various parameters including the type and volume of organic solvent, rate of addition and salt concentration of the aqueous solvent and stirring speed limit the usage of this method to form BCNs.
We recently demonstrated that flash nanoprecipitation (FNP) can be used to scalably assemble poly(ethylene glycol)-b1ock-poly(propylene sulfide) (PEG-bl-PPS), an oxidation responsive diblock copolymer that assembles into lyotropic mesophases,21 into a wide range of soft nanoarchitectures as a function of the PEG fraction (fPEG).22,23 FNP employs muIti-stream mixers for the turbulent mixing of organic and aqueous phases at millisecond timescales.24,25 The resulting supersaturated conditions induces precipitation and nucleation of hydrophobic payloads and coprecipitation of block copolymers for stabilization of monodisperse nanoparticles. Although FNP had only been used previously to form such solid core nanoparticles,25,26 we hypothesized that the use of a copolymer with the appropriate chain flexibility and glass transition temperature for aggregate shape transformations at short time scales may allow assembly of complex nanoarchitectures using FNP. We found the timescale of PEG-bl-PPS transitions between metastable intermediate morphologies closely matches the mixing time of FNP to allow kinetic entrapment of diverse nanostructures.22 Here, we present FNP as a scalable and reproducible method of assembling BCNs with primitive Im3m cubic phases from PEG17-bl-PPS75 (fPEG = 0.118) diblock copolymers.
The formation of BCNs using either FNP or PEG-bl-PPS has not been previously explored. We therefore compared PEG-bl-PPS BCN formation by FNP with both the co-solvent dispersion and thin-film hydration methods. The internal cubic structure of the BCNs was confirmed using cryoTEM and SAXS. BCNs were then loaded with small and macromolecules using FNP and their release kinetics, toxicity and cellular uptake were assessed in vitro. The ability of BCNs to stably load and transport combinations of molecules with diverse solubilities has attracted much attention for the development of vaccine formulations, and thus we performed an in vitro functional assay on immune cells activated by PEG-bl-PPS BCNs dual loaded with antigen and adjuvant. Our results validate FNP for the facile fabrication of monodisperse block copolymer BCNs, potentially allowing wider usage of this typically difficult to assemble, yet highly versatile soft nanoarchitecture.
Materials and methods
Chemicals
The following were purchased from Sigma Aldrich: polyethylene glycol) methyl ether MW 750, methanesulfonyl chloride, triethylamine, potassium carbonate, thioacetic acid, 0.5 M sodium methoxide solution, propylene sulfide, acetic acid, calcein, ethyl eosin, dichloromethane, Celite filter cel, activated charcoal powder, anhydrous tetrahydrofuran, tetrahydrofuran, ethanol, methanol, diethyl ether, deuterated chloroform, and sepharose CL-6B. The following reagents were purchased from ThermoFisher Scientific: tetramethylrhodamine-dextran 70 kDa, Texas red-dextran 10 kDa, DiD, Texas red conjugated ovalbumin, Lysotracker green, DAPI, toluene, hexanes, and phosphate buffered saline (1× PBS) tablets (product BP2944). Monophosphoryl lipid A (MPL) from Salmonella minnesota R595 was purchased from Invivogen. All antibodies and flow cytometry reagents were purchased from BioLegend.
Synthesis of polymer
Poly(ethylene glycol)17-block-poly(propylene sulfide)75 (PEG17-bl-PPS75) copolymer was synthesized as described elsewhere.22 Initially, methyl ether PEG 750 was end-functionalized with a mesylate leaving group using methanesulfonyl chloride, which was then converted to PEG-thioacetate by reaction with thioa-cetatic acid. The thioacetate was then activated with base to form a thiolate anion for initiating ring opening living polymerization of propylene sulfide. Following 1 h of polymerization, the reaction was quenched by protonation of remaining thiolate anions with acetic acid and stored under argon at −20 °C. 1H NMR was used to confirm the degree of polymerization (3H methyl ether (3.36 singlet); 4H PEG −CH2−CH2−(3.60–3.64); 1H CH2−CH−CH3 (2.56−2.65); 2H −CH−CH2−CH−CH3 (2.82−2.95), 3H −CH2−CH3 (1.30−1.38)) and gel permeation chromatography verified polymer purity (Fig. S1†).
Preparation of bicontinuous nanospheres by FNP
Bicontinuous nanospheres (BCNs) were produced using a confined impingement jets (CIJ) mixer as previously described in Allen et al.22 Briefly, PEG-bl-PPS copolymer (5, 10 or 20 mg) was weighed and dissolved in 500 μL of tetrahydrofuran (THF) and loaded into a 1 mL plastic disposable syringe. A second syringe was loaded with 500 μL of water or 1× PBS (pH 7.4). The organic and aqueous phases were then impinged within the CIJ mixer at a rate of approximately 1 mL s−1. The super-saturated solution formed in the mixer was collected into a 2 mL reservoir of water or 1× PBS. The loading of hydrophobic or hydrophilic constituents into BCNs was performed by adding hydrophobic actives to the THF phase whereas hydro-philic actives were added to the aqueous phase. Free form unloaded molecules and THF were then removed using a sepharose CL-6B size exclusion column and 1× PBS mobile phase. The loading efficiency was assessed using a spectrofluo-rometer by measuring sample fluorescence before and after column purification. For lyophilization studies, inulin was added to 10 mg ml−1 of BCNs to create a 2% w/v inulin solution, placed in a freezer at −80 °C for 3 h and then dried over-night in a lyophilizer. Dynamic light scattering (DLS) measurements of BCNs were determined using a Zetasizer Nano-ZS (Malvern Instruments, UK). All samples were diluted 1 in 1000 using PBS and then analyzed for DLS measurements.
Loading of hydrophobic and hydrophilic molecules via FNP
Hydrophilic molecules calcein (4 mM, 50 μL), Texas Red-dextran (10 kDa) (10 mg ml−1, 20 μL), tetramethylrhodamine dextran (70 kDa) (5 mg ml−1, 20 μL) and Texas Red-ovalbumin (5 mg ml−1, 25 μL) were separately loaded inside BCNs. Hydrophobic molecules ethyl eosin (5 mg ml−1, 10 μL), and DiD (20 mg ml−1, 5 μL) were loaded inside BCNs. For each individual loading experiment, hydrophilic or hydrophobic molecules were added to water or THF, respectively to attain a final volume of 500 μL before impingement. For co-loading of ovalbumin and MPL, the aqueous phase containing ovalbumin (5 mg ml−1, 25 μL) and the THF phase containing MPL (1 mg ml−1, 20 μL) were impinged against each other to form BCNs. For control experiments, PEG17-bl-PPS32 was utilized to form polymersomes using FNP and loaded with Ovalbumin and MPL as described above. The loading efficiency was determined by fluorescence measurements using the following wavelengths: (excitation/emission, filter used): calcein: 470/509, 495 filter, ethyl eosin: 525/560, 550 filter, tetramethyl-rhodamine: 555/580, 570 filter, DiD: 644/670, 665 filter, and Texas Red-ovalbumin: 594/615, 610 filter.
BCN formation via co-solvent dispersion and thin film rehydration
The standard methods of co-solvent dispersion and thin film hydration were used to assemble BCNs from PEG17-bl-PPS75 copolymer for comparison with the FNP protocol. BCNs were prepared by co-solvent dispersion with a slight modification to the method reported elsewhere.6 Briefly, 10 mg of polymer was dissolved in 1 ml of THF in a 5 mL scintillation vial. 1.5 ml of water or 1× PBS was then slowly added at a speed of 1 mL h−1 with continuous stirring. After addition of water, THF was removed overnight under vacuum. For the thin film hydration method, 10 mg of polymer was dissolved in 1 ml of dichloro-methane and desiccated overnight under vacuum to form a lamellar polymer film. The thin film formed in the vial was hydrated using 2 mL of water or 1× PBS and then vortexed at 1600 rpm for 12 h prior to analysis.
Cryogenic transmission electron microscopy (cryoTEM)
For cryoTEM studies, a pretreated holey carbon 400 mesh TEM grid was applied with 4 μL of sample (3 mg ml−1) and plunge-frozen with a Gatan Cryoplunge freezer. These specimens were imaged using a JEOL 3200FS transmission electron micro-scope operating at 300 keV at 4000× nominal magnification. All the images were collected in vitreous ice using a total dose of ~10 e− Å−2 and a nominal defocus range of 2.0–5.0 μm. Micrographs were acquired as 20-frame movies during a 5 s exposure using a Gatan 3.710 × 3.838 pixel K2 Summit direct electron detector operating in counting mode. A Digital Micrograph software (Gatan) was utilized to align individual frames of each micrograph and compensate for stage and beam-induced drift. These aligned images were summed up and used for image processing. For electron tomography studies, the data was collected using SerialEM and using a defocus of about 5 μm and a total dose of about 30 e− A−2. A tilt range of −40° to +40° at 2° increments was utilized and individual images per angle were taken. The data obtained was processed and analyzed using the IMOD 4.9 package.
Small angle X-ray scattering (SAXS)
Small angle X-ray scattering (SAXS) studies were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) beamline at Argonne National Laboratory’s Advanced Photon Source (Argonne, IL, USA) with 10 keV (wave-length λ = 1.24 Å) collimated X-rays. All the samples were analyzed in the q-range (0.001 to 0.5 Å−1), with a sample-to-detector distance of approximately 7.5 m and an exposure time of 1 s. The diffraction patterns of silver behenate was utilized to calibrate the q-range. The momentum transfer vector q is defined as q = 4π sinθ/λ, where θ is the scattering angle. The data collected was then analyzed using PRIMUS 2.8.2 software, where the solvent buffer scattering was subtracted and the final scattering curve was obtained.
In vitro release studies
In vitro release studies of BCNs manufactured through FNP were performed in 1× PBS with, in some cases, added hydrogen peroxide, using a dialysis membrane. Briefly, 1 ml (5 mg ml−1) of BCNs loaded with calcein, DiD, Texas Red-ovalbumin (45 kDa) or tetramethylrhodamine dextran (75 kDa) were transferred into the dialysis unit (Spectra/Por Float-A-Lyzer G2, MWCO 100 kDa) from Spectrum® Laboratories. The dialyzer was then introduced into a 250 mL glass beaker containing release media (1× PBS or 100 μM hydrogen peroxide), which was stirred at 400 rpm in the dark. At pre-determined time intervals, 10 μL of sample was collected from the dialyzer and then replaced with 1× PBS. The samples were analyzed for fluorescence using a spectrofluorometer and then assessed for amount released at each time point.
In vitro degradation studies
In vitro degradation studies of BCNs were performed in 100 μM hydrogen peroxide. 200 μL of BCNs (5 mg ml−1) were transferred to a centrifuge tube with 200 μL of 200 μM hydrogen peroxide. This mixture was then placed on a vortex mixer and at predetermined intervals samples were analyzed using DLS and cryoTEM as described above.
Cell culture
RAW 264.7 macrophages were obtained from the American Type Culture Collection (ATCC, Rockville, MD, USA). These cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% fetal bovine serum (FBS), penicillin (100 IU mL−1) and streptomycin (100 μg mL−1) at 37 °C and 5% CO2. Bone marrow derived dendritic cells (BMDCs) were obtained after processing of bone marrow from the tibias and femurs of C57BL/6 mice as reported elsewhere.27 Briefly, tibias and femurs collected from the mice were flushed with 10% FBS in HBSS. Cells were collected in a petri dish, and red blood cells were lysed with ACK lysis buffer, followed by centrifugation and suspension of a cell pellet in RPMI medium supplemented with 10% fetal bovine serum (FBS), penicillin (100 IU mL−1) and streptomycin (100 μg mL−1). These cells were then plated in 100 mm petri dishes with 20 ng mL−1 of GM-CSF and 10 ng mL−1 of IL-4, refreshed every three days. After 8 days, matured dendritic cells were collected and utilized for the assay.
MTT assay
RAW 264.7 macrophages (2 × 105 cells per ml, 100 μL) were seeded in each well of a 96-well plate. 10 μL of BCNs at different concentrations (0, 1, 2.5, 5, 10, 25 and 50 mg ml−1 in PBS) was added to the wells in quadruplicate and incubated for 24 and 48 h. MTT (5 mg ml−1 in PBS, 10 μL) was then added to each well and incubated for 4 h. Media was removed from each well and replaced with 200 μL of dimethyl sulfoxide to dissolve formazan crystals deposited on the plate. The absorbance of the plate was measured using a microplate reader at 570 nm. The percentage cell viability was then calculated using the formula:
% cell viability = (OD of BCNs treated sample/OD of BCNs untreated sample) × 100.
Cellular uptake
RAW 264.7 macrophages (2 × 105 cells per ml, 500 μL) were seeded in each well of a 24-well plate and cultured overnight. DiD loaded BCNs (5 mg ml−1 in PBS, 20 μL) were added to each well and incubated for 2, 4, 8 and 24 h. After incubation, cells were washed thrice with PBS and harvested in 0.5 mL PBS. Samples were then stained with Zombie Aqua™ (live/dead stain) and analyzed using a LSRII flow cytometer (BD Biosciences), and data were analyzed by Cytobank online soft-ware (https://www.cytobank.org/).28
Confocal microscopy
RAW 264.7 macrophages (2 × 105 cells per ml, 150 μL) were plated in each well of an 8-well chamber slide and cultured overnight. Texas Red dextran-loaded BCNs (5 mg ml−1 in PBS, 10 μL) were added to each well and incubated for 2, 4 and 8 h. After incubation, the cells were washed with DMEM, and stained with LysoTracker green (lysosome stain) and DAPI (nuclear stain). The cells were then fixed and imaged using a 63× oil-immersion objective on a SP5 Leica confocal microscope.
Functional BMDC antigen presentation assay
A functional assay to demonstrate in vitro delivery of antigen (ovalbumin) and adjuvant (MPL) to BMDCs was performed using BCNs. In this study, BMDCs were plated into a non-tissue culture treated 48-well plate at 1 × 105 cells per well. After 6 hours of acclimation, cells were incubated with 20 μL of OVA-MPL-loaded BCNs or PSs (5 mg mL−1 polymer, 20 μg mL−1 OVA, 10 μg mL−1 MPL), OVA-loaded BCNs or PSs (5 mg mL−1 polymer, 20 μg mL−1 OVA), Blank BCNs or PSs (5 mg mL−1 polymer), free OVA (20 μg mL−1) with or without free MPL (10 μg mL−1), or 1× PBS. After 14 h of incubation, BMDCs were blocked with anti-mouse CD16/32 antibody and stained with: BV421 anti-mouse CD11c, PE/Cy7 anti-mouse MHCI-SIINFEKL, APC anti-mouse CD80, APC/Cy7 anti-mouse CD86, PerCP/Cy5.5 anti-mouse CD40, and Zombie Aqua viability dye. Cells were analyzed using a LSRII flow cytometer (BD Biosciences) and data was analyzed by Cytobank online software.28 Results were analyzed for statistical significance with GraphPad Prism 7 using two-way ANOVA followed by Tukey’s multiple comparison tests.
Results and discussion
Flash nanoprecipitation allows rapid assembly of uniform monodisperse bicontinuous nanospheres
We have previously demonstrated the ability to form nano-structures resembling BCNs from PEG17-bl-PPS75 using FNP.22 However, these BCNs were not fully characterized and, to-date, no study has demonstrated the formation of BCNs from PEG-bl-PPS using traditional techniques, such as thin film rehydration or co-solvent dispersion. To fully characterize these nano-structures and confirm their identity, we assembled BCNs from PEG17-bl-PPS75 using our previously published FNP protocol (Fig. 1). In order to assess differences in nanostructure morphology or yield between different formation techniques, we also fabricated formulations utilizing the same copolymer by co-solvent dispersion and thin film rehydration, using either water or 100 mM phosphate buffered saline (1× PBS).
BCNs were analyzed by cryoTEM and SAXS to elucidate differences in the aggregate morphology and homogeneity. While FNP successfully formed BCNs in aqueous solvent conditions with or without salt, co-solvent dispersion only produced BCNs in pure water (Fig. 2a–c). Thin film rehydration failed to produce BCNs in either aqueous solvent, and macroscopically formed unstable precipitates similar to those seen in the 1× PBS co-solvent dispersion sample (Fig. S2†). BCNs formed by FNP were the predominant structure in solution, formed aqueous channels visible by cryoTEM tomography and displayed uniformity in structure across the sample population (Fig. S3, Video 1†). BCNs formed by FNP in water were smaller than BCNs formed in 1× PBS and by co-solvent dispersion, and had similarly low poly-dispersity (<0.2) to BCNs formed by co-solvent dispersion, though BCNs formed by FNP in 1× PBS had a higher PDI (Table 1). While the aggregate morphology of water co-solvent dispersion formulations was predominantly BCNs, heterogeneity across the sample population was observed in the form of vesicles and nonuniformity in internal structure (Fig. 2c).
Fig. 2.
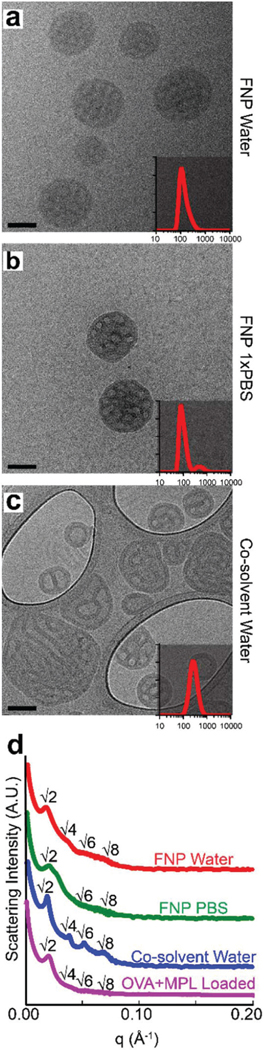
PEG17-bl-PPS75 BCNs displayed primitive Im3m cubic phases. CryoTEM images and overlaid DLS size distributions of BCNs formed by flash nanoprecipitation using water (a) or 1× PBS (b) as the aqueous solvent. BCNs formed using co-solvent dispersion (c) with water as the aqueous solvent is also shown. (d) SAXS data with labeled Bragg peaks for BCN formulations shown in images from (a)–(c) along with BCNs loaded with ovalbumin and MPL demonstrating primitive Im3m cubic internal organization. In (a)–(c), scale bars represent 100 nm.
Table 1.
DLS size and polydispersity index (PDI) for BCNs shown in Fig. 2, before and after lyophilization and reconstitution in the presence of a 2% w/v inulin cryoprotectant. Data represents the averages from three independent samples
| Pre-lyophilization |
Post-lyophilization |
||||
|---|---|---|---|---|---|
| Formation method |
Aqueous solvent |
Diameter (nm) |
PDI | Diameter (nm) |
PDI |
| FNP | Water | 210.5 | 0.166 | 186 | 0.285 |
| FNP | 1× PBS | 392.8 | 0.287 | 429 | 0.51 |
| Co-solvent | Water | 319.8 | 0.169 | — | — |
SAXS analysis confirmed the internal primitive cubic organization of BCNs formed by FNP and co-solvent dispersion. Bragg reflections for BCNs formed by FNP in water and PBS showed relative spacing ratios at , , and , and , , , respectively (Fig. 2d). These Bragg spacing ratios indicate the presence of primitive cubic (Im3m) lattice with lattice parameters aIm3m = 32.5 nm (FNP in water) and aIm3m = 28.5 nm (FNP in PBS). Interestingly, BCNs formed by co-solvent method in water also showed Bragg reflections at relative spacing ratios, , and, indicating a primitive cubic lattice (Im3m) with a lattice parameter of 31.4 nm (Fig. 2d). This result agrees with cryoTEM analysis showing predominant formation of BCN structures (Fig. 2c).
Since polymer concentration may influence nanostructure formation, we fabricated BCNs via FNP using lower (5 mg mL−1) and higher (20 mg mL−1) concentrations of PEG-bl-PPS. The 5 mg mL−1 sample formed BCN structures in both water and 1× PBS, however DLS analysis showed high poly-dispersity for these formulations (Fig. S4†), while 20 mg mL−1 of polymer generated small precipitates in the reservoir and making these samples unsuitable for DLS analysis. Cryo-TEM analysis of these samples revealed phases with spongy-type morphology and no observable BCNs (Fig. S4†). We therefore selected a 10 mg mL−1 polymer concentration for further analysis of BCN formation by FNP.
Polymeric nanospheres and polymersomes have been reported to preserve their structure after lyophilization in the presence of a cryoprotectant.29,30 Taking this into consideration, we have performed a simple lyophilization study on BCNs in the presence of the cryoprotectant inulin. BCNs formed via FNP in water and PBS were added with 2% w/v inulin as a cryoprotectant and then lyophilized. DLS analysis before and after lyophilization demonstrate that BCNs formed in water showed a smaller increase in PDI after lyophilization and reconstitution compared to BCNs formed in PBS (Table 1). These results demonstrate that BCNs formed in water may be lyophilized for practical storage and reconstitution, a desirable capability for translational applications.
FNP can load hydrophilic and hydrophobic actives into BCNs for controlled delivery and release
The presence of both ample hydrophobic volume and internal aqueous channels makes BCNs an attractive potential nano-carrier for both lipophilic and hydrophilic molecules.7 To investigate their capacity to load diverse molecules, BCNs were loaded with molecules ranging in molecular weight and hydro-phobicity: calcein, ethyl eosin, lipophilic dye DiD, dextran (10 and 70 kDa), and ovalbumin. SAXS profile of BCNs loaded with hydrophobic and hydrophilic molecules showed relative spacing ratios at , , and , which indicates presence of primitive cubic (Im3m) lattice like the blank cubosomes (Fig. 2d). Loading efficiency of larger hydrophilic macro-molecules ovalbumin and 70 kDa dextran (33%, each) were higher than that for smaller hydrophilic molecules calcein and 10 kDa dextran (5.3% and 9.46%, respectively) (Fig. 3a). The lower loading efficiency of small molecules may be due to their higher diffusion rate into the aqueous reservoir prior to completion of nanostructure assembly. After assembly, larger molecules may also be more easily entrapped within the aqueous channels of the BCNs. Hydrophobic molecules ethyl eosin and DiD demonstrated loading efficiencies >70% (Fig. 3a), likely due to partition into the large hydrophobic domains. These results demonstrate the ability of FNP to efficiently load diverse payloads into BCNs. Hydrophilic mole-cules smaller than 10 kDa may have reduced loading into BCNs due to the presence of larger aqueous channels. However, payload-specific customization, such as the inclusion of appropriate charge moieties into BCNs, may enhance the accommodation of these molecules inside the aqueous channels.
Fig. 3.
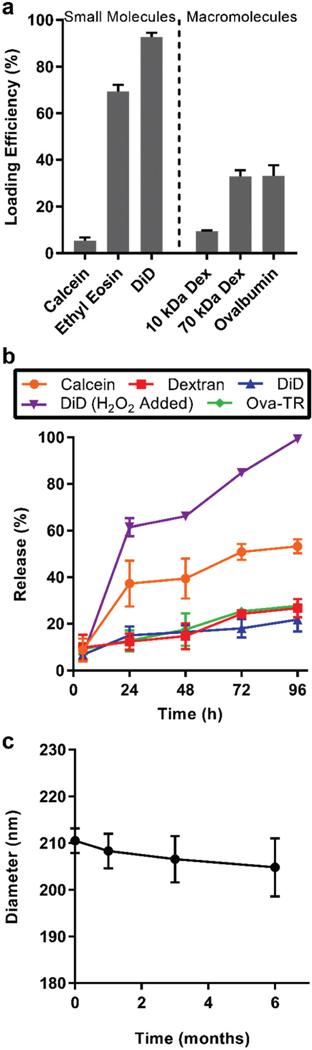
Loading, release, and stability characteristics of BCNs. (a) Loading efficiency of small and macromolecules into BCNs formed by FNP. (b) Release of hydrophilic and hydrophobic molecules from BCNs over 96 hours. (c) Diameter of BCN samples measured over the course of 6 months by DLS. In all cases, error bars represent SD, n = 3.
The release of various payloads was quantified in vitro using a dialysis membrane in PBS for 4 days. Within the first 4 h, 8–9% of hydrophilic payloads (calcein, 70 kDa dextran, and ovalbumin) and 6% of hydrophobic DiD was released from BCNs, indicative of an initial burst release from BCN nanostructures (Fig. 3b). By the end of 4 days, 55% of calcein and 27–30% of dextran and ovalbumin were released from BCNs. A greater release of calcein was observed compared to the larger molecules, which may be due to faster diffusion of calcein from the aqueous channels. DiD showed slower release than hydrophilic payloads, with 21% being released from BCNs after 4 days, suggesting that hydrophobic payload can be retained in the structures for longer periods of time (Fig. 3b). Both hydrophilic and hydrophobic payloads showed controlled release behavior for greater than 7 days from BCNs (Fig. S5†). This slow release kinetics of hydrophilic and hydrophobic pay-loads from BCNs could make them unique platforms for controlled release applications.
PEG-bl-PPS copolymers show oxidation responsive behavior due to the susceptibility of the hydrophobic PPS chain to oxidation and resulting conversion to more hydrophilic sulfoxide and sulfone derivatives.31 To assess the oxidation responsiveness of PEG-bl-PPS BCNs and the impact on controlled release of payloads, we performed degradation studies in the presence of biologically relevant concentration of hydrogen peroxide (100 μM)32 using DiD-Ioaded BCNs. In the presence of hydrogen peroxide, 6% of DiD was released in the first 4 h, 61% by 24 h, and 100% by the end of the study (Fig. 3b). This indicates no effect of oxidation on the reIease of DiD from BCNs during the first 4 h. To further confirm these resuIts, cryoTEM analysis was performed at different time points after incubation of BCNs in hydrogen peroxide. After 2 days of incubation, cryoTEM images revealed the majority of BCNs to have reassembled into micellar structures, which is consistent with an increase in the hydrophilic fraction of the copolymer and previously published results for oxidized PEG-bl-PPS nano-structures (Fig. S6†).33 A complete loss of turbidity was observed after 4 days of incubation with hydrogen peroxide (Fig. S6a†), correlating well with the observed complete release of DiD from BCNs. CryoTEM images after 4 days of incubation showed the presence of micelles and small polymer aggregates, and a rare BCN (Fig. S6c†). These results indicate that PEG-bl-PPS BCNs undergo triggered payload release in the presence of biologically relevant levels of oxidation.
Stability plays a vital role in the development and translation of nanoparticulate formulations. We therefore performed stability studies on BCNs over a period of 6 months at room temperature and analyzed the formulations for changes in size using DLS. DLS data showed a progressive increase in size distribution over time, however, no statistically significant changes in size were observed after 1, 3 and 6 months when compared to the initial DLS measurement at the first time point (Fig. 3c). This result verifies the stability of PEG-bl-PPS BCNs formed via FNP, which, unlike lipid-based BCNs, demonstrate no aggregation in the absence of stabilizing surfactants.
BCNs are non-toxic and enter macrophage endolysosomes
Interactions of PEG17-bl-PPS75 BCNs with cells were assessed in vitro using a phagocytic macrophage cell line. Other PEG-bl-PPS based nanostructures have been extensively utilized for cell based assays and in vivo applications, as they are non-toxic at therapeutically relevant concentrations and promote intra-cellular delivery of payloads.34–36 The viability of RAW 264.7 macrophages in the presence of BCNs was determined at various polymer concentrations (0.25–5 mg ml−1) for 24 and 48 h. While the highest concentration tested (5 mg mL−1) decreased cell viability to 80%, no significant changes in viability were observed for concentrations at or below 2.5 mg mL−1 (Fig. 4a). This verified the non-toxic nature of PEG17-bl-PPS75 BCNs to be consistent with previously published PEG-bl-PPS nanocarrier formulations.23,37–39
Fig. 4.
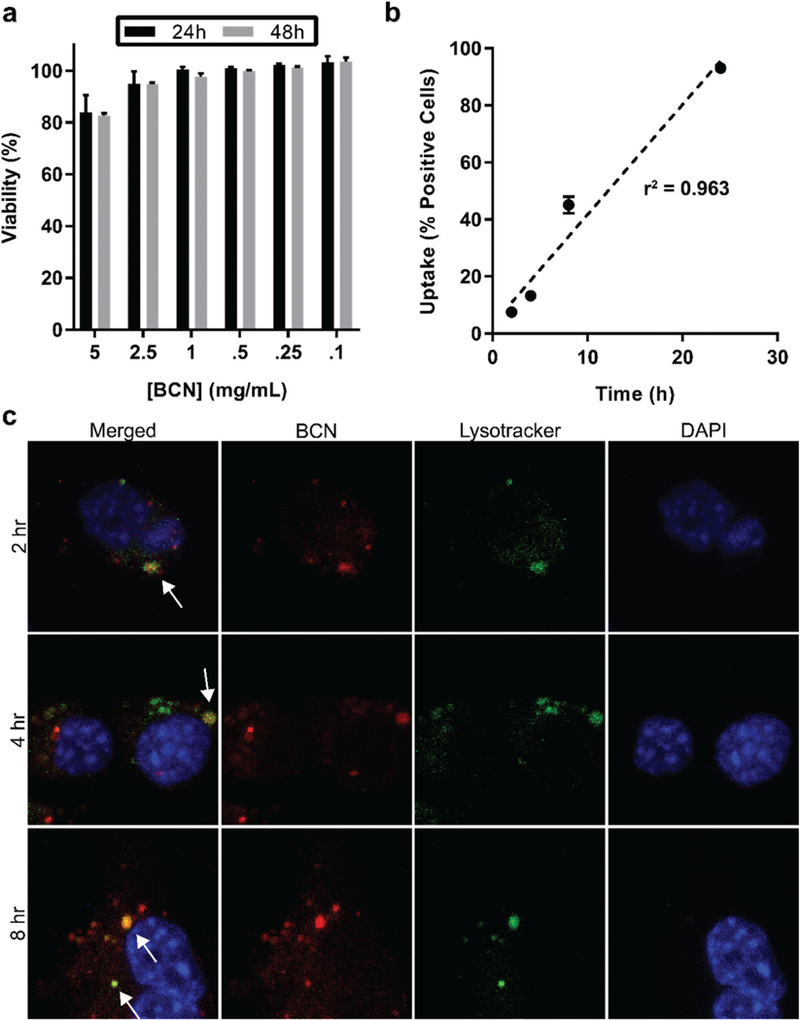
Uptake of BCNs and effect on cell viability. (a) MTT assay of RAW 264.7 macrophages incubated with BCNs for 24 or 48 hours at different concentrations. Error bars represent SD, n = 3. (b) Flow cytometric analysis of BCN uptake by RAW macrophages at different timepoints of incubation, error bars represent SD, n = 3. (c) Confocal images of macrophages stained with lysosomal dye Lysotracker (green) and DNA dye DAPI (blue) following incubation with Texas Red labeled BCNs (red) for 2, 4, or 8 h. White arrows in the merged image point to examples of colocalization of Lysotracker and Texas Red signals, demonstrating endolysosomal uptake of BCNs.
Next, we assessed uptake of BCNs by cells using flow cytometry and confocal microscopy. BCNs loaded with DiD were incubated with RAW 264.7 macrophages over a period of 24 h and the percentages of cells positive for BCNs was measured by flow cytometry. BCNs demonstrated time-dependent uptake upon incubation with RAW 264.7 macrophages. After 4, 8 and 24 h of incubation, 13%, 45% and 93% of cells were positive for BCNs, respectively (Fig. 4b). For confocal microscopy, BCNs loaded with hydrophilic Texas Red-dextran (10 kDa) were incubated with RAW 264.7 macrophages and cells were further stained with LysoTracker green and DAPI to respectively track cell lysosomaI compartments and nuclei. After 2, 4 and 8 h of incubation, punctae of Texas Red dye was observed within cells to colocalize with lysosomal signals. This demonstrated uptake of BCNs by the cells into endolysosomes, which would promote antigen processing for peptide complexation with MHC molecules38–41 (Fig. 4c).
BCNs formed via FNP demonstrate intracellular delivery of hydrophilic and hydrophobic payloads simultaneously
Due to their non-toxic nature and propensity for uptake into phagocytic cells, BCNs show significant potential for use in a number of biomedical applications. As an example, we applied BCNs as a model nanoparticle vaccine formulation for the activation of bone marrow derived dendritic cells (BMDCs), which are phagocytes capable of initiating immune responses against antigenic molecular components of pathogens and thus key cellular targets during immunization. Activation of BMDCs requires simultaneous cytoplasmic delivery of a typically hydrophilic antigen and immunostimulation of pattern recognition receptors (PRRs) by pathogen associated molecular patterns (PAMPs).42–44 While protein and peptide antigens can be loaded within the aqueous channels of BCNs, common PAMPs, such as lipophilic toll-like receptor (TLR) agonists from bacterial cell walls, are well suited for stable retention within the lyotropic BCN hydrophobic phase.
To demonstrate the dual delivery of hydrophilic and lipophilic payloads from PEG-bl-PPS BCNs, an in vitro BMDC activation and antigen presentation assay was performed. In this study, BCNs were co-loaded with the model protein antigen ovalbumin (Ova) and TLR4 agonist MPL as an adjuvant using FNP and incubated with BMDCs (Fig. 5a). PEG-bl-PPS polymer-somes loaded with Ova and MPL was included in the study as a positive control. Cell activation was quantified via the expression level of cell surface markers CD80, CD86, and CD40 that are upregulated following TLR stimulation. BMDCs were additionally analyzed for the surface presentation of the immunodominant antigenic peptide component of Ova, SIINFEKL, which forms a complex with MHCI receptors. SIINFEKL is a peptide fragment generated by the proteolytic cleavage of Ova by the proteasome within the cell cytosol. While BMDC activation is possible with just the delivery of adjuvant, the presentation of SIINFEKL occurs when Ova is successfully delivered into the cytosol for processing. This process is enhanced by nanocarriers that promote escape of payloads from endosomal compartments, a step that is also critical for gene delivery and can be cytotoxic without moderation.
Fig. 5.
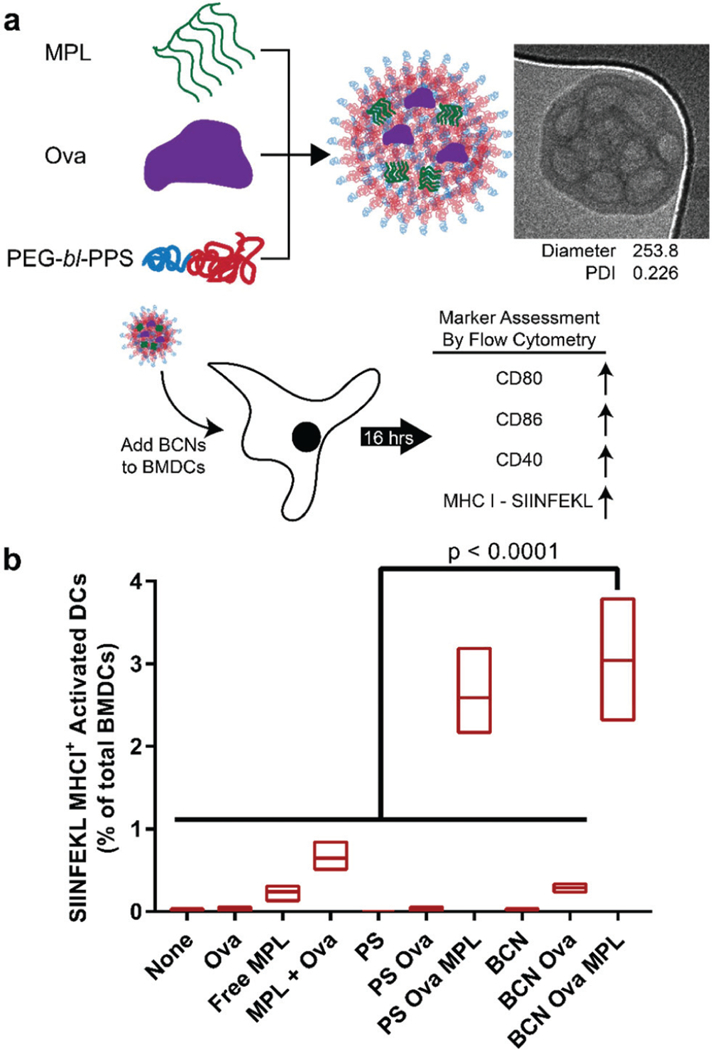
In vitro delivery of antigen and adjuvant to bone marrow derived dendritic cells (BMDCs) using BCNs. (a) Schematic of the experimental process. TLR4 agonist MPL was loaded into BCNs with model protein antigen ovalbumin (Ova) by FNP. BCNs were incubated with BMDCs for 14 hours, which were then assessed for activation and antigen presentation by flow cytometry. (b) Activation of BMDCs by BCN formulations was determined by upregulation of cell surface markers CD80, CD86 and CD40. Antigen processing and presentation were determined using a fluorescent antibody specific for the SIINFEKL/MHCI complex. For comparison, polymersome (PS) formulations loaded with Ova and MPL were also tested. Floating bars represent min, mean, and max values, n = 3. Significance determined by 1-way ANOVA and Tukey’s multiple comparisons test.
Interestingly, BCNs loaded with Ova and MPL showed significantly greater SIINFEKL/MHCI surface presentation on BMDCs compared to all other control groups (p < 0.0001), outperforming free antigen and adjuvant added into the cell culture medium (Fig. 5b). There was no statistical difference observed between Ova-MPL loaded BCNs and the positive control (Fig. 5b). BCNs loaded with only Ova and no MPL neither activated cells nor resulted in antigen presentation, which agrees with literature signifying the requirement of adjuvant for improved activation of BMDCs. This also indicates that BCNs themselves are non-immunogenic and non-inflammatory in vitro on a cellular level unless loaded with an immunostimulant. Furthermore, the enhanced SIINFEKL/MHCI expression demonstrates that BCNs can safely promote endosomal escape of encapsulated payloads, consistent with previous applications of PEG-bl-PPS nanocarriers.34 These results indicate FNP can be used to rapidly assemble BCNs and load them with antigen and adjuvant that in turn can be efficiently delivered to the cytosol of BMDCs. The presence of high internal surface area, extensive hydrophobic domains and aqueous internal channels allow BCNs to load and deliver molecules without the need for chemical conjugation that can modulate or decreases their activity.
Conclusion
Here, we have demonstrated that PEG17-bl-PPS75 block copolymers can be assembled into BCNs that possess a cubic lattice internal organization by FNP. Monodisperse uniform BCN populations could be fabricated by FNP in either water or PBS, while co-solvent dispersion, the only previous method shown to form stable polymeric BCNs, could only assemble BCNs in water and with high heterogeneity. Concentrations of 5 mg mL−1 and 10 mg mL−1 supported BCN formation using a simple hand-driven CIJ mixer, with 10 mg mL−1 formulations demonstrating lower PDI. Further systematic testing with additional relevant block copolymer systems and solvent conditions will be required to determine the key properties of PEG-bl-PPS that support BCN formation by FNP. BCNs were able to be lyophilized and reconstituted successfully using standard inulin-based cryoprotectant protocols. Unlike lipid-based formulations, PEG-bl-PPS BCNs displayed long-term stability in aqueous solutions without the aid of surfactants, with no observable aggregation for 6 months, and potentially longer. FNP allowed facile co-loading of diverse molecular pay-loads that could be retained within either the extensive hydrophobic phase volume or aqueous channels, which were observed by cryoTEM and electron tomography. Water soluble payloads demonstrated loading efficiencies and sustained release rates that were dependent on molecular size. Oxidation-sensitive reassembly of BCNs into micelles allowed triggered payload release at biologically relevant concentrations of reactive oxygen species. These BCNs were non-toxic and were taken up by both macrophages and BMDCs into their endolysosomal pathway for cytoplasmic delivery of cargo. This set of characteristics and facile scalable assembly by FNP make PEG-bl-PPS BCNs a very attractive nanoscale delivery vehicle for both hydrophilic and hydrophobic molecules in vitro and potentially in vivo.
Supplementary Material
Acknowledgements
We wouId Iike to thank Devang Amin and PhiIip Messersmith for their generous aid in the initiaI stages of procuring a CIJ mixer. We wouId Iike to thank J. Remis for CryoTEM assistance and the foIIowing faciIities at Northwestern University: Robert H. Lurie Comprehensive Cancer Center FIow Cytometry Core; BioIogicaI imaging faciIity and Keck-II faciIity of Northwestern University’s NUANCE Center. SAXS experiments were performed at the DuPont-Northwestern-Dow Collaborative Access Team (DND-CAT) Iocated at Sector 5 of the Advanced Photon Source (APS). DND-CAT is supported by Northwestern University, E.I. DuPont de Nemours & Co., and The Dow ChemicaI Company. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User FaciIity operated for the DOE Office of Science by Argonne NationaI Laboratory under Contract No. DE-AC02–06CH11357. This research was supported by the NationaI Science Foundation CAREER Award (grant no. 1453576), the NationaI Institutes of HeaIth Director’s New Innovator Award (grant no. 1DP2HL132390–01), and the Louis A. Simpson & KimberIy K. Querrey Center for Regenerative Nanomedicine Regenerative Nanomedicine CataIyst Award.
Footnotes
Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr06779h
Conflicts of interest
There are no conflicts to declare.
References
- 1.Blanazs A, Armes SP and Ryan AJ, Macromol. Rapid Commun, 2009, 30, 267–277. [DOI] [PubMed] [Google Scholar]
- 2.Ramanathan M, Shrestha LK, Mori T, Ji Q, Hill JP and Ariga K, Phys. Chem. Chem. Phys, 2013, 15, 10580–10611. [DOI] [PubMed] [Google Scholar]
- 3.Azhari H, Strauss M, Hook S, Boyd BJ and Rizwan SB, Eur.J. Pharm. Biopharm, 2016, 104, 148–155. [DOI] [PubMed] [Google Scholar]
- 4.McKenzie BE, Friedrich H, Wirix MJ, de Visser JF, Monaghan OR, Bomans PH, Nudelman F, Holder SJ and Sommerdijk NA, Angew. Chem., Int. Ed, 2015, 54, 2457–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Angelov B, Angelova A, Drechsler M, Garamus VM, Mutafchieva R and Lesieur S, Soft Matter, 2015, 11, 3686–3692. [DOI] [PubMed] [Google Scholar]
- 6.La Y, Park C, Shin TJ, Joo SH, Kang S and Kim KT, Nat. Chem, 2014, 6, 534–541. [DOI] [PubMed] [Google Scholar]
- 7.McKenzie BE, De Visser J, Portale G, Hermida-Merino D, Friedrich H, Bomans P, Bras W, Monaghan OR, Holder SJ and Sommerdijk NA, Soft Matter, 2016, 12, 4113–4122. [DOI] [PubMed] [Google Scholar]
- 8.Rizwan SB and Boyd BJ, in Subunit Vaccine Delivery, Springer, 2015, pp. 125–140. [Google Scholar]
- 9.Oka T, Langmuir, 2015, 3–1, 3180–3185.25050712 [Google Scholar]
- 10.Kimber RG, Walker AB, Schröder-Turk GE and Cleaver DJ, Phys. Chem. Chem. Phys, 2010, 12, 844–851. [DOI] [PubMed] [Google Scholar]
- 11.Angelova A, Angelov B, Papahadjopoulos-Sternberg B, Ollivon M and Bourgaux C, Langmuir, 2005, 21, 4138–4143. [DOI] [PubMed] [Google Scholar]
- 12.Holder SJ, Woodward G, McKenzie B and Sommerdijk NA, RSCAdv, 2014, 4, 26354–26358. [Google Scholar]
- 13.Gao Y, Wang Y, Jiang M and Chen D, ACS Macro Lett, 2012, 1, 1312–1316. [DOI] [PubMed] [Google Scholar]
- 14.McKenzie BE, Holder SJ and Sommerdijk NA, Curr. Opin. Colloid Interface Sci, 2012, 17, 343–349. [Google Scholar]
- 15.Chen Y, Ma P and Gui S, BioMed Res. Int, 2014, 2014, 815981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hales K, Chen Z, Wooley KL and Pochan DJ, Nano Lett, 2008, 8, 2023–2026. [DOI] [PubMed] [Google Scholar]
- 17.McKenzie BE, Nudelman F, Bomans PH, Holder SJ and Sommerdijk NA, J. Am. Chem. Soc, 2010,132, 10256–10259. [DOI] [PubMed] [Google Scholar]
- 18.Percec V, Wilson DA, Leowanawat P, Wilson CJ, Hughes AD, Kaucher MS, Hammer DA, Levine DH, Kim AJ and Bates FS, Science, 2010, 328, 1009–1014. [DOI] [PubMed] [Google Scholar]
- 19.McKenzie BE, de Visser J. l. F., Friedrich H, Wirix MJ, Bomans PH, de With G, Holder SJ and Sommerdijk NA, Macromolecules, 2013, 46, 9845–9848. [Google Scholar]
- 20.Napoli A, Sebök D, Senti A and Meier W, in Block Copolymers in Nanoscience, Wiley-VCH Verlag GmbH & Co. KGaA, 2008, pp. 39–71, DOI: 10.1002/9783527610570.ch3. [DOI] [Google Scholar]
- 21.Napoli A, Tirelli N, Wehrli E and Hubbell JA, Langmuir, 2002, 18, 8324–8329. [Google Scholar]
- 22.Allen S, Osorio O, Liu Y-G and Scott E, J. Controlled Release, 2017, 262, 91–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yi S, Allen SD, Liu YG, Ouyang BZ, Li X, Augsornworawat P, Thorp EB and Scott EA, ACS Nano, 2016, 10, 11290–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han J, Zhu Z, Qian H, Wohl AR, Beaman CJ, Hoye TR and Macosko CW, J. Pharm. Sci, 2012, 101, 4018–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Saad WS and Prud’homme RK, Nano Today, 2016, 11, 212–227. [Google Scholar]
- 26.Tang C, Amin D, Messersmith PB, Anthony JE and Prud’homme RK, Langmuir, 2015, 31, 3612–3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lutz MB, Kukutsch N, Ogilvie AL, Rößner S, Koch F, Romani N and Schuler G, J. Immunol. Methods, 1999, 223, 77–92. [DOI] [PubMed] [Google Scholar]
- 28.Kotecha N, Krutzik PO and Irish JM, Curr. Protoc. Cytom, 2010, 10.17.11–10.17.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ayen WY and Kumar N, Eur. J. Pharm. Sci, 2012, 46, 405–414. [DOI] [PubMed] [Google Scholar]
- 30.Goyal R, Macri L and Kohn J, Sci. Rep, 2015, 5, 13065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rehor A, Hubbell JA and Tirelli N, Langmuir, 2005, 21, 411–417. [DOI] [PubMed] [Google Scholar]
- 32.de Gracia Lux C, Joshi-Barr S, Nguyen T, Mahmoud E, Schopf E, Fomina N and Almutairi A, J. Am. Chem. Soc, 2012, 134, 15758–15764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vasdekis AE, Scott EA, O’Neil CP, Psaltis D and Hubbell JA, ACS Nano, 2012, 6, 7850–7857. [DOI] [PubMed] [Google Scholar]
- 34.Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA and Hubbell JA, Biomaterials, 2012, 33, 6211–6219. [DOI] [PubMed] [Google Scholar]
- 35.Yi S, Allen SD, Liu Y-G, Ouyang BZ, Li X, Augsornworawat P, Thorp EB and Scott EA, ACS Nano, 2016, 10, 11290–11303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rincon-Restrepo M, Mayer A, Hauert S, Bonner DK, Phelps EA, Hubbell JA, Swartz MA and Hirosue S, Biomaterials, 2017, 132, 48–58. [DOI] [PubMed] [Google Scholar]
- 37.Du F, Liu Y-G and Scott EA, Cell. Mol. Bioeng, 2017, 10, 357–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dowling DJ, Scott EA, Scheid A, Bergelson I, Joshi S, Pietrasanta C, Brightman S, Sanchez-Schmitz G, Van Haren SD, Ninkovic J, Kats D, Guiducci C, de Titta A, Bonner DK, Hirosue S, Swartz MA, Hubbell JA and Levy O, J. Allergy Clin. Immunol, 2017, 140, 1339–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stano A, Scott EA, Dane KY, Swartz MA and Hubbell JA, Biomaterials, 2013, 34, 4339–4346. [DOI] [PubMed] [Google Scholar]
- 40.Scott EA, Stano A, Gillard M, Maio-Liu AC, Swartz MA and Hubbell JA, Biomaterials, 2012, 33, 6211–6219. [DOI] [PubMed] [Google Scholar]
- 41.Vasdekis AE, Scott EA, O’Neil CP, Psaltis D and Hubbell JA, ACS Nano, 2012, 6, 7850–7857. [DOI] [PubMed] [Google Scholar]
- 42.S. R. Bonam, C. D. Partidos, S. K. M. Haimuthur and S. Muller, Trends Pharmacol. Sci, 2017, 38(9), 771–793. [DOI] [PubMed] [Google Scholar]
- 43.Sahdev P, Ochyl LJ and Moon JJ, Pharm. Res, 2014, 31, 2563–2582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bobbaia S and Hook S, Pharm. Res, 2016, 33, 2078–2097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.