

Abstract
Medulloblastoma (MB) is the most common malignant brain tumor of childhood. Although outcomes have improved in recent decades, new treatments are still needed to improve survival and reduce treatment-related complications. The MB subtypes groups 3 and 4 represent a particular challenge due to their intragroup heterogeneity, which limits the options for “rational” targeted therapies. Here, we report a systems biology approach to drug repositioning that integrates a nonparametric, bootstrapping-based simulated annealing algorithm and a 3D drug functional network to characterize dysregulated driver signaling networks, thereby identifying potential drug candidates. From more than 1300 drug candidates studied, we identified five members of the cardiac glycoside family as potentially inhibiting the growth of groups 3 and 4 MB and subsequently confirmed this in vitro. Systemic in vivo treatment of orthotopic patient-derived xenograft (PDX) models of groups 3 and 4 MB with digoxin, a member of the cardiac glycoside family approved for the treatment of heart failure, prolonged animal survival at plasma concentrations known to be tolerated in humans. These results demonstrate the power of a systematic drug repositioning method in identifying a potential treatment for MB. Our strategy could potentially be used to accelerate the repositioning of treatments for other human cancers that lack clearly defined rational targets.
INTRODUCTION
Medulloblastoma (MB) is the most common malignant brain tumor in children and accounts for 20 to 25% of all pediatric brain tumors (1). With the current standard therapy, which includes maximal safe resection, craniospinal radiation, and multiagent chemotherapy, the average 5-year overall survival for children with newly diagnosed MB is greater than 75% (2). The need for new MB treatments remains substantial, however, because survivors of this disease often face long-term treatment-related neurocognitive and endocrine sequelae. Furthermore, there are no curative second-line therapies currently available for those patients who fail to respond to upfront therapy or suffer a relapse of their disease.
MB is a heterogeneous group of tumors that can be divided into four subtypes based on distinct genomic signatures (3). Two of these subgroups are defined by single dysfunctional signaling pathways, Wingless/integrase-1 (WNT) and sonic hedgehog (SHH), which have raised the prospect of taking a rational target-based approach for the development of new therapies. Conversely, the other two subtypes, groups 3 and 4, which comprise 60 to 65% of all MB cases, contain complex genetic, epigenetic, and genomic abnormalities and display considerable intragroup heterogeneity, making them less amenable to rationally targeted therapies. Group 3 tumors, moreover, are associated with the worst prognosis of all the subgroups and are frequently metastatic at presentation (3), making the need for effective treatments for these tumors particularly acute.
The repositioning of known drugs for new medical indications is a promising strategy to accelerate the discovery of new treatments. Because of the readily available toxicity and pharmacokinetic information for drugs already in use (4, 5), compounds identified through repositioning have the potential for rapid clinical translation. There is also a wealth of publicly available data systematically exploring both the effects of drugs on intracellular signaling pathways and the relationship between genetic diversity and treatment efficacy. These resources include the Connectivity Map (CMAP) and Library of Integrated Network-Based Cellular Signatures (LINCS) projects, which generated gene expression signatures in a large number of cell lines in response to different perturbations by small molecules (6); the Cancer Cell Line Encyclopedia (CCLE) project, which systemically generated genomic profiles, including gene expression, copy number changes, and sequencing profiles for a set of human cancer cell lines after treatment with various anti-cancer drugs (7); and the Catalogue of Somatic Mutations in Cancer (COSMIC), which focuses on the association between somatic mutations and drug sensitivity (8). To date, a number of computational methods have been proposed to make use of this wide array of available pharmacogenomics data for drug repositioning (4, 5, 9, 10). Although comprehensive transcriptomic data provide valuable information about drug responses, integration of other equally important and relevant genomic and epigenetic data, such as DNA mutations, copy number changes, and methylation patterns, may help identify clinically meaningful new treatments, particularly when applied to heterogeneous and genomically complex cancers like groups 3 and 4 MB.
With this in mind, we developed a systems biology approach for driver signaling network identification (DSNI)– and drug functional network (DFN)–based drug repositioning. This approach integrates multiple types of genomic profiles from patients with groups 3 and 4 MB (whole-genome/exome sequencing, DNA copy number changes, DNA methylation, and mRNA expression) with data from human cancer signaling pathway resources and the gene expression profiles of 1309 drugs in CMAP (6). Using the DSNI-DFN method, we identified five members of the cardiac glycoside family, a group of Na+/K− pump inhibitors best known for their role in the treatment of heart failure as potential inhibitors of cancer driver signaling in groups 3 and 4 MB. We validated the therapeutic potential of these compounds in groups 3 and 4 MB in vitro in cell-based assays, and of one member of this family, digoxin, in vivo, in orthotopic patient-derived xenograft (PDX) models. Our findings show both a potential therapeutic strategy for groups 3 and 4 MB and a systematic drug repositioning approach that could potentially be applied to other genomically complex tumors.
RESULTS
Overview of systematic drug repositioning using DSNI-DFNs
The goal of the DSNI-DFN method is to identify existing drugs that can interfere with cancer-specific driver signaling networks. It is composed of two analytical components: identifying signaling networks that drive tumor progression or proliferation (Fig. 1A) and evaluating the effects of drugs on those networks using reconstructed DFNs (Fig. 1B). We used this method as follows. First, we identified potential driver genes for group 3 (or group 4) MB from multiple omics datasets, including DNA sequencing, gene copy number, gene expression, and DNA methylation data. We then used these genes to derive driver gene signaling networks using a nonparametric, bootstrapping-based simulated annealing algorithm (NPBSA; Fig. 1A). NPBSA explores subnetworks from individual driver (seed) genes. It grows the subnetwork by adding genes neighboring the current network genes to increase a predefined network score representing differential gene expression, until an optimum score for the current subnetwork is achieved. By applying NPBSA to RNA sequencing (RNA-seq) (or methylation) data for patients with group 3 (or group 4) MB and to cancer pathway information, we generated transcriptome (or methylation-)– based driver net-works. We defined a network score for those driver networks and calculated a driver network score for each patient. We then clustered the patients with group 3 (or group 4) MB into different groups on the basis of these scores and tested whether these groups correlated with patient survival. We next reconstructed three drug-drug functional similarity networks by independently computing the two-dimensional chemical structure similarity, transcriptional response similarity, or in vitro drug test similarity between the 1309 drugs and then integrating them into one uniform network using a non-linear network fusion technique (11). We then clustered this integrated DFN into drug functional modules using a Bayesian non-negative matrix factorization (NMF) with the β-divergence (BNMFβD) method and applied a network diffusion algorithm to each module to predict the off-target effects of each drug (12). Last, by evaluating drug-induced effects on the subtype-specific MB driver signaling networks, we prioritized the drugs for each siibtvpe of MB
Fig. 1. Workflow of the systems biology–driven drug repositioning strategy.
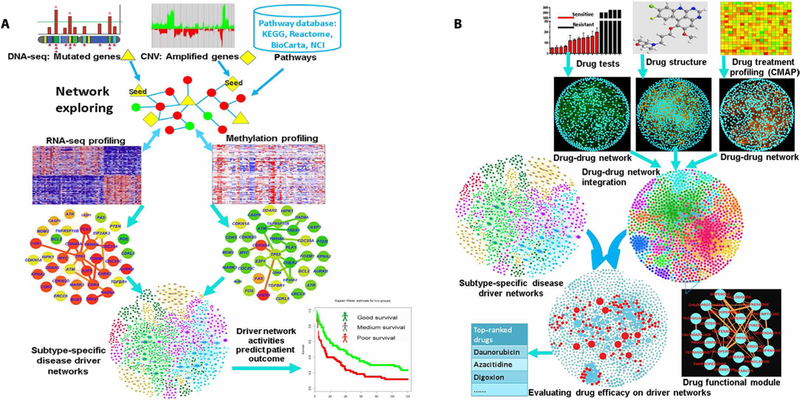
The systematic drug repositioning strategy included two major components: (A) uncovering driver signaling networks by integrating multiple genomic profiles through a NPBSA algorithm and (B) an integrative analysis of DFNs and driver signaling networks for systematic drug repositioning. DNA-seq, DNA sequencing; KEGG, Kyoto Encyclopedia of Genes and Genomes; CNV, copy number variation; NCI-PID, NCI Pathway Interaction Database.
Identification of dysregulated driver networks in groups 3 and 4 MB
Dysregulated driver networks that either drive tumor cell proliferation or unlock barriers against tumor progression represent a small fraction of the genomic alterations present in a tumor (13). Key driver events can be identified by their increased frequency in a given set of tumors. Most often, changes in driver genes lead to the activation of multiple tumorigenic signaling pathways. To identify driver alterations for groups 3 and 4 MB, we collected comprehensive data from 144 patients with group 3 MB and 326 patients with group 4 MB including DNA mutation (3), DNA methylation (14), DNA copy number, and gene expression data (table S1) (14). From these data, we identified 660 potential driver genes for group 3 MB and 635 potential driver genes for group 4 tumors. Specifically, we identified 145 mutated genes in group 3 samples and 123 mutated genes in group 4 samples from DNA sequencing data. Using gene copy number data, we also identified 179 amplified genes and 144 deleted genes in group 3 samples and 165 amplified genes and 108 deleted genes in group 4. Last, by comparing gene expression data from these patients to data from WNT (70 patients) and SHH (223 patients) tumors, we identified subgroup-specific changes in gene expression with 194 and 239 differentially expressed genes for groups 3 and 4, respectively [|fold change| ≥ 2, P < 0.05, empirical Bayes test (15)].
We then applied the NPBSA algorithm to search for alternations in signaling networks due to changes in the activity of these potential driver genes. We separately combined mRNA expression profiling or methylation profiling data with our integrated human cancer signaling network to generate two sets of potential driver networks for each tumor type. Using this approach, 318 mRNA-based and 341 methylation-based signaling networks were identified as altered in group 3 tumors. From these networks, genes capable of forming a subnetwork were designated as final likely driver genes based on the rationale that genes involved in highly activated networks are the most likely genes to represent true drivers of oncogenesis. In this way, we identified 403 driver genes for group 3 tumors (table S2). We then applied the same approach to data from group 4, resulting in the identification of 331 mRNA-based and 330 methylation-based driver signaling networks and 378 driver genes (table S3). Our approach successfully identified candidate driver genes that have been previously published for both group 3 (MYC, OTX2, NPR3, SMARCA4, GABRA5, and HLX) (16) and group 4 (CDK6, MYCN, SNCAIP, GRM1, GRM8, KCNA1, KCNA5, LMX1A, and KDM6A) MB (16).
We merged the methylation data-based signaling networks with those derived from mRNA expression data by selecting the genes with high joint confidence scores in both of them. We clustered the dysregulated driver networks using an affinity propagation (AP) algorithm (17), resulting in 48 dysregulated driver networks for group 3 MB and 45 dysregulated driver networks for group 4 MB (tables S4 and S5). As with driver genes, our approach identified previously validated driver signaling networks, specifically transforming growth factor-β (TGFβ) (18) and MYC-p53 signaling in group 3 and the LMX1A regulatory network in group 4 (16). We computed a network score (see Materials and Methods) for each driver signaling network, which represented the differential gene expression activities of that network (termed network activity). Using these scores, we generated a specific network activity profile for each patient.
Driver network-derived clusters correlate with differences in patient outcomes
To compare the strength of our approach to analysis based on mRNA expression only, we performed NMF analysis (19) on both the driver network activity profiles and the driver gene profiles of patients with group 3 MB for whom survival data were available (114 patients in total). Our unsupervised analysis of driver network activity profiles identified two distinct clusters representing patients with significant differences in overall survival (log-rank test, P = 0.0232; Fig. 2A). Applying NMF clustering to driver network gene profiles also identified two clusters with differences in overall survival (log-rank test, P = 0.0349; Fig. 2B). Conversely, patient clusters identified using only genome-wide gene expression profiles did not demonstrate significant differences in outcomes (log-rank test, P = 0.290; Fig. 2C). Similarly, when clusters were generated for group 4 tumors (264 patients in total) in an analogous fashion, driver network activity and driver gene profiles demonstrated differences in patient outcomes (log-rank test, P = 0.00664 and P = 0.0077; Fig. 2, D and E, respectively), whereas clusters identified using gene expression profiles did not (log-rank test, P = 0.441; Fig. 2F). Because clusters based on driver network activities appeared to be associated with improved overall survival in patients with group 3 or 4 MB, we used driver networks as the basis for further analysis and identification of potential drugs.
Fig. 2. Correlation of driver network activity with patient with groups 3 and 4 MB overall survival.
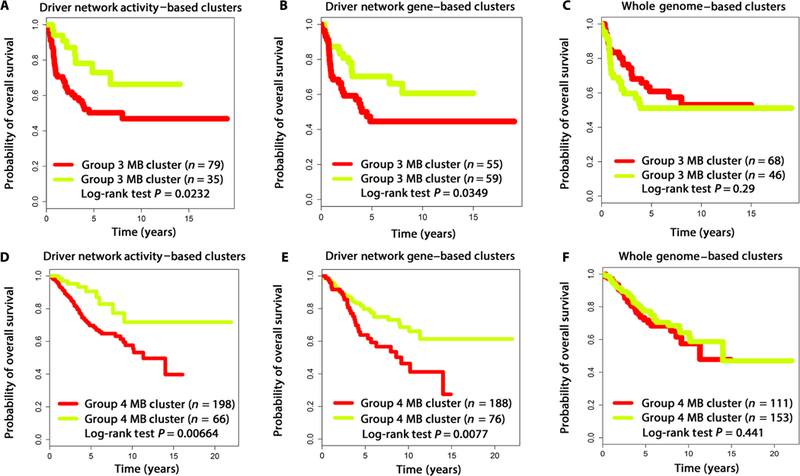
(A) Driver network activity– and (B) driver network gene–based clusters for group 3 MB correlated with differences in patient survival (P = 0.0232 and P = 0.0349, respectively). (C) Genome-wide mRNA expression–based clusters for group 3 MB were not associated with significant differences in patient survival (P = 0.290). (D) Driver network activity– and (E) driver network gene–based clusters for group 4 MB correlated with differences in patient survival (P = 0.00664 and P = 0.0077, respectively). (F) Genome-wide gene expression–based clusters for group 4 MB were not associated with significant differences in survival (P = 0.441).
DSNI-DFN-based drug repositioning accurately predicts in vitro drug effects
Using our DSNI-DFN method, we overlaid the DFNs onto the dysregulated driver networks and looked for clusters of compounds predicted to modulate pathways that were highly active in groups 3 and 4 MB. This allowed us to rank 1309 drugs based on their predicted ability to inhibit tumor growth.
We sought to compare the performance of our DSNI-DFN method with that of a previously reported CMAP-gene set enrichment analysis (GSEA)–based drug repositioning method (6). To this end, the 100 drugs predicted to be the most likely and the 100 drugs predicted to be the least likely to inhibit tumor growth were evaluated for their effect on MED8A cells, a MB cell line reported as belonging to group 3 (20). Cells were plated and incubated with 10 μM of each drug, with viability assessed at 72 hours. Compared to only 3% of the top 100 drugs (see table S6) identified using the CMAP-GSEA approach (Fig. 3A), about 12% of the top 100 drugs (see table S6) identified using the DSNI-DFN approach demonstrated >60% nonviable cells at 72 hours. In addition, 85% of the 100 lowest-ranked drugs predicted by DSNI-DFN had <20% cell death at 72 hours, whereas more than 10% of the 100 lowest-ranked drugs identified using CMAP-GSEA had greater than 50% inhibition of cell viability. We have listed out the full ranking order of drugs in DSNI-DFN and CMAP-GSEA for group 3 MB in tables S7 and S8, respectively. These results indicate that the DSNI-DFN method improved both the reliability and sensitivity of identifying effective drugs.
Fig. 3. Cardiac glycosides as candidate drugs for groups 3 and 4 MB.
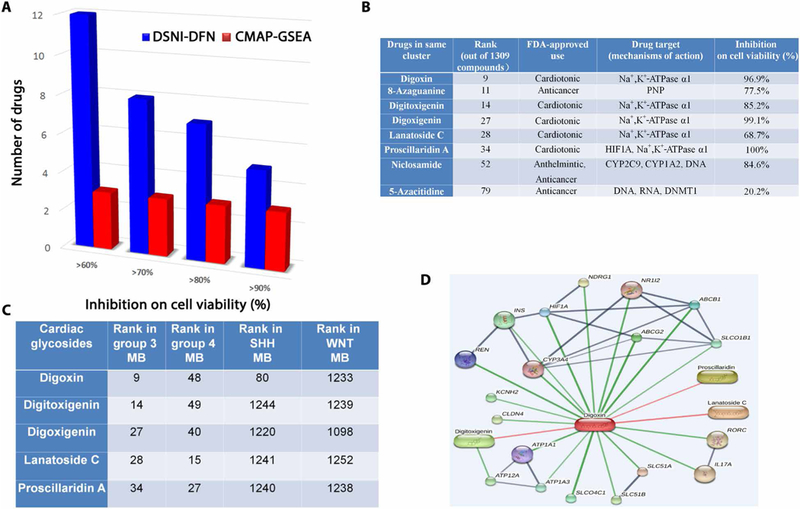
(A) Comparison of in vitro inhibition of cell viability predicted by DSNI-DFN versus CMAP-GSEA. MED8A cells were incubated for 72 hours at 10 μM with predicted drugs, after which cell viability was assessed. (B) Details of a drug cluster predicted to strongly inhibit cell viability in groups 3 and 4 MB. This cluster contains known anticancer compounds and multiple members of the cardiac glycoside family. (C) Ranked order of cardiac glycosides predicted by DSNI-DFN in groups 3 and 4 SHH and WNT MB. FDA, U.S. Food and Drug Administration. (D) Schematic of drug-drug (red) and drug-target (dark blue) interactions for the cardiac glycoside digoxin derived from the STITCH database.
Cardiac glycosides are candidate drugs for groups 3 and 4 MB
Among the 100 drugs predicted to be the most effective by the DSNI-DFN method was a cluster of compounds present in both groups 3 and 4. This cluster contained both a number of known chemotherapeutic agents currently in clinical use (daunorubicin, mitoxantrone, and etoposide) (21) and five members of the cardiac glycoside family: the ionotropic drugs digoxin, proscillaridin A, and lanatoside C, known primarily for their use in the treatment of heart failure (22), and the related compounds digoxigenin and digitoxigenin (Fig. 3B). The cardiac glycosides ranked similarly in groups 3 and 4 MB. In contrast, only digoxin was predicted to show efficacy in SHH MB, with all the other glycosides bottom-ranked in both WNT and SHH MBs (Fig. 3C). For group 3, this prediction was based on the ability of these compounds to modulate the dysregulated driver networks associated with 22 genes (HIF1A, ABCG2, ABCB1, CYP3A4, ATP1A1, ATP1A3, NPPA, REN, FXYD2, RYR2, TOP2B, CACNA2D1, CALM1, DNAH8, TUBA4A, TUBA1A, ABCC1, TUBB, SDC1, TOP2A, KRIT1, and MAPK8), which are frequently altered in group 3 tumors. In addition, the first 11 genes listed above are reported to be direct targets of digoxin in the STITCH database (Fig. 3D) (23). For group 4, the cardiac glycosides are predicted to exert an effect on networks associated with 14 genes (HIF1A, ATP1A1, ATP1A3, ABCB1, SDC1, PDE1B, CACNA1A, CALM1, CYP11A1, ABCC1, PRL, KCNA5, KRIT1, and CFD) frequently altered in this subgroup, with the first five genes listed above being reported as the direct targets of digoxin in STITCH database (23).
Cardiac glycosides inhibit MB cell growth in vitro
To further evaluate the predicted antitumor effects of the cardiac glycosides on MB, we performed in vitro cell viability assays using two established MB cell lines, MED8A and D283. The MED8A line has been reported to belong to group 3, whereas D283 cells have been described in the literature as belonging to both groups 3 and 4 (20). An adult brain tumor (glioblastoma) cell line, U87, and a triple-negative breast cancer cell line, MDA-MB231-Br, were included in the assays as controls for nonspecific cytotoxicity. Cells were plated and incubated with five members of the cardiac glycoside family (proscillaridin A, digoxin, lanatoside C, digi-toxigenin, and digoxigenin) at escalating doses, and cell viability was assessed after 72 hours. Both MED8A and D283 showed substantial decreases in viability, with escalating doses of all cardiac glycoside family members resulting in IC50s (median inhibitory concentrations) ranging from the low nanomolar to low micromolar range depending on the compound (Fig. 4, A and B). By comparison, when these compounds were tested in U87 cells, proscillaridin A and digoxin showed IC50s >5 μM, whereas other cardiac glycosides tested failed to reach 50% decreases in cell viability despite drug concentrations up to 100 mM (Fig. 4A). Similarly, MDA-MB231-Br cells failed to show growth inhibition even at drug concentrations >100 μM (fig. S1). Our findings both demonstrate the in vitro antitumor activities of this class of drugs and highlight the selectivity of the cardiac glycosides for MB cells.
Fig. 4. Cardiac glycosides inhibit MB cell growth in vitro and prolong survival in vivo.
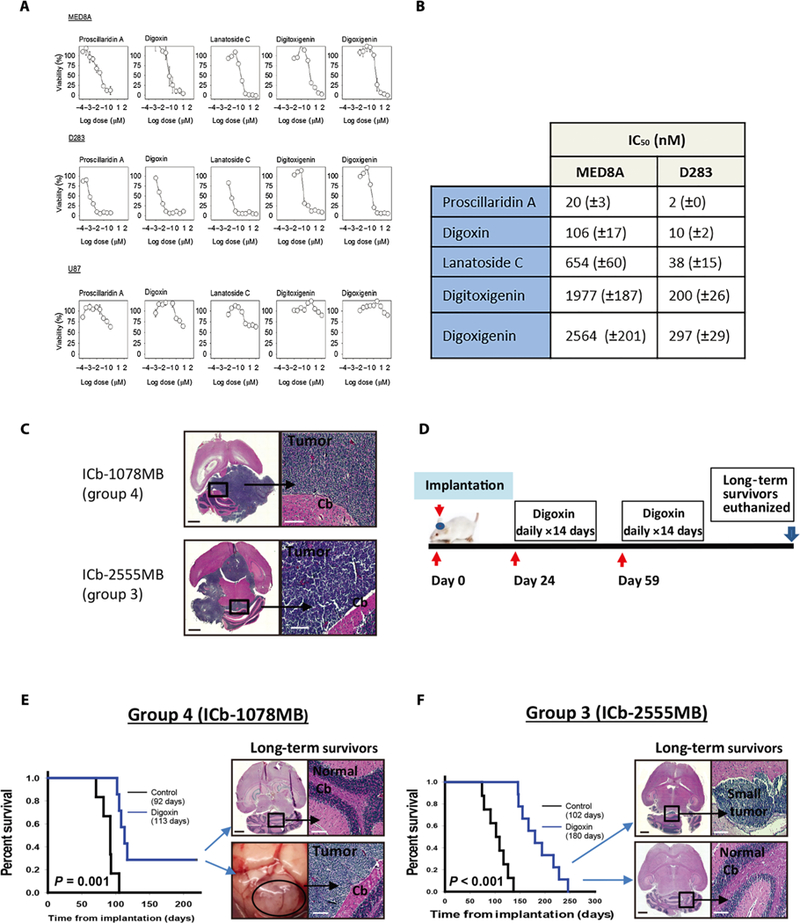
(A) Dose-response curve for the cardiac glycosides (proscillaridin A, digoxin, lanatoside C, digitoxigenin, and digoxigenin) on MB-derived cell lines (MED8A and D283) and the control glioblastoma line (U87). Cell viability was assessed at 72 hours. (B) IC50 values for the dose titration experiment. (C) Schematic of orthotopic PDX model treatment with digoxin. Twenty-four days after tumor cell implantation, mice received digoxin (2 mg/kg ip daily) for 14 days, followed by 21 days without treatment, and followed by another 14 days of treatment. (D) Hematoxylin and eosin (H&E) staining examples of untreated tumors from orthotopic PDX models of group 3 (ICb-2555MB) and group 4 (ICb-1078MB) MB. (E and F) Kaplan-Meier curves showing digoxin-treated mice compared to untreated controls. (E) Digoxin treatment prolonged survival in an orthotopic PDX model of group 4 MB (ICb-1078MB) to 113 days (n = 7) versus 92 days (n = 6) for untreated controls (log-rank test, P = 0.001). Two long-surviving mice were euthanized at 219 days while asymptomatic. Gross-scale image of one long-surviving mouse with large tumor and H&E staining demonstrating no evidence of tumor in the two long-surviving mice, one with a tumor and one without is shown. (F) Digoxin treatment prolonged survival in an orthotopic PDX model of group 3 MB (ICb-2555MB) to 180 days (n = 10) versus 102 days (n = 8) for untreated controls (log-rank test, P < 0.001). H&E comparison of two long-term surviving mice, one with a microscopic tumor and one without is shown. Scale bars, 1 mm (black) and 0.1 mm (white).
Digoxin treatment prolongs survival in orthotopic PDX models of groups 3 and 4 MB
Next, to evaluate whether the growth inhibition that we observed in vitro translated into a clinically relevant prolongation of survival in vivo, we turned to two patient-derived orthotopic PDX models, ICb-2555MB and ICb-1078MB, representing groups 3 and 4 disease, respectively. Orthotopic PDX models have a number of advantages in survival studies in that they replicate the location of the patient’s original disease and also preserve both inter- and intratumoral heterogeneity, thereby more closely mirroring patient tumors (24,25). Digoxin was selected for our survival studies because of its long history of clinical use and known toxicity profile.
ICb-1078MB, as we have previously reported (24), belongs to group 4 MB and harbors an N-MYC amplification. After tumor cell implantation, we allowed 24 days for tumor growth before starting treatment. The treatment group subsequently received two cycles of digoxin [2 mg/kg ip (intraperitoneally) daily (26) for 14 days] 3 weeks apart (Fig. 4C). Digoxin-treated and untreated control mice were monitored for signs of tumor progression, and brains were evaluated after euthanasia for the presence of a tumor.
There were no long-surviving control mice, and all died with grossly visible tumors (Fig. 4D). Digoxin treatment significantly prolonged survival, in ICb-1078MB showing a median survival of 113 days (n = 7) compared to 92 days (n = 6) for untreated controls (P = 0.001; Fig. 4E). Furthermore, the digoxin-treated group included two long-surviving mice that still appeared well at twice the median survival age of the control group. These mice were euthanized on postimplantation day 219 while asymptomatic. One long-surviving mouse had a cerebellar tumor histologically consistent with MB, whereas the other showed no evidence of disease (Fig. 4E).
To evaluate the efficacy of digoxin treatment in group 3 disease, we treated a newly established orthotopic PDX model of group 3 MB (ICb-2555MB) as described above (Fig. 4D). ICb-2555MB was initially derived from a tumor harboring a C-MYC amplification, which is associated with aggressive disease and poor patient outcomes (3). ICb-2555MB was validated as a model through multiple in vivo passages (fig. S2A) and was identified as belonging to group 3 via quantitative polymerase chain reaction (fig. S2B).
Digoxin treatment also significantly prolonged survival in mice implanted with ICb-2555MB. Digoxin-treated mice (n = 10) displayed a median survival of 180 days compared to 102 days for untreated controls (n = 8) (P < 0.001; Fig. 4F). Furthermore, whereas all control mice had grossly visible tumors, the final three mice in the digoxin treatment group showed no evidence of intracranial tumors at the time of euthanasia, and only a small tumor was detectable after microscopic evaluation (Fig. 4F and fig. S3). This suggests that these mice may have been cured of their tumors, and instead, the physical decline that led to their being euthanized was related to other etiologies that occurred as they approached the end of their life span. Together, our data suggest that digoxin has an antitumor effect in vivo against preformed orthotopic PDX tumors from both groups 3 and 4 MB.
Digoxin-mediated prolongation of survival occurs at clinically relevant plasma levels
Our results, combined with the fact that digoxin is known to cross the blood-brain barrier in humans (27), suggested that digoxin may be a potential treatment for patients with groups 3 and 4 MB. The known narrow therapeutic window of digoxin, however, is a major concern in translating these findings to a clinical setting. Safe therapeutic concentrations are well established in humans (22), and plasma trough concentrations of digoxin can be used in patients to monitor dosing and predict potential toxicities.
To examine whether our dosing schedule resulted in clinically tolerable plasma troughs, we treated six mice implanted with ICb-1078MB with digoxin (2 mg/kg ip daily for 16 days; fig. S4A). Twenty-four hours after the final digoxin dose (a standard time point in clinical drug dose monitoring), the mice were euthanized, and plasma digoxin concentrations were analyzed (table S9). Average plasma trough concentrations in digoxin-treated mice were 2.4 ± 0.2 ng/ml (fig. S4B), which was only slightly above the recommended goal troughs of 0.8 to 2 ng/ml targeted in human patients and well below the concentrations of >10 ng/ml described as being “highly toxic” (22). These data suggest that clinical benefits for human patients may be achievable at safe digoxin dosing concentrations.
Comparison of single-agent digoxin therapy relative to and in combination with ionizing radiation
Because radiation therapy is a key component of the standard therapy for MB (2), we sought to compare the efficacy of single-agent digoxin treatment with that of radiation and to evaluate the therapeutic efficacy of the two as a combination therapy. Mice were implanted with ICb-2555MB, and radiation treatments were initiated 17 days after implantation. The radiation treatment schedule was based on current clinical protocols in which patients receive cranio-spinal irradiation followed by additional boost to the tumor bed. Mice in the radiation and combination therapy groups received 2 gray (Gy) daily to the craniospinal axis for 5 days, followed by 2 Gy daily for 5 days to the region of tumor implantation (Fig. 5, A and B). In mice receiving combination therapy, digoxin was initiated on day 24 (Fig. 5B).
Fig. 5. Comparison of the effects of digoxin and ionizing radiation alone and in combination on survival in an orthotopic PDX model of group 3 MB.
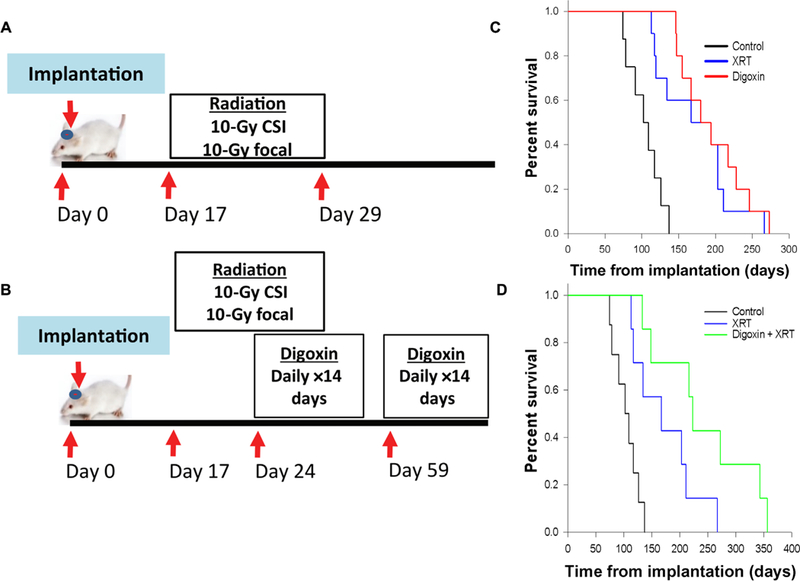
(A) Treatment schematic for radiation alone. Seventeen days after implantation, mice received fractionated radiation to the craniospinal axis (2 Gy/day) for 5 days, followed by 2 days of recovery, and then 5 days focal radiation to the cerebellum (2 Gy/day). CSI, craniospinal irradiation. (B) Treatment schematic of combination therapy. Radiation therapy (XRT) was initiated on day 17 described in (A), with initiation of digoxin treatments 24 days after implantation, mice received digoxin (2 mg/kg ip daily) for 14 days, followed by 21 days with no treatment, and then an additional 14 days of treatment. (C) Kaplan-Meier curve comparing the median survival of ICb-2555MB tumor-bearing mice: untreated (n = 8, 102 days), digoxin-treated (n = 10, 180 days), and radiation alone (n = 10, 167 days). Both digoxin single-agent therapy (log-rank test, P = 0.007) and radiation alone (log-rank test, P < 0.001) showed significant prolongation of survival relative untreated controls but were comparable when compared to one another (log-rank test, P = 0.91) (D) Kaplan-Meier curve comparing the median survival of untreated (n = 8, 102 days), radiation only (n = 10, 167 days), and combination therapy-treated (n = 10, 219 days) mice. Combination therapy showed a significant prolongation of survival compared to both radiation alone (log-rank test, P = 0.04) and untreated controls (log-rank test, P < 0.001).
Mice that received radiation therapy alone (n = 10) had a median survival of 167 days versus 102 days for untreated controls (n = 8) and 180 days for single-agent digoxin therapy (n = 10). These results demonstrate that in this model, single-agent digoxin has efficacy comparable to 20 Gy of radiation (Fig. 5C). Furthermore, when the digoxin treatment regimen was modified to prolonged continuous treatment of 60 days (n = 10), the observed survival benefit was superior to that of radiation, with a median survival of 235 days (P < 0.01; fig. S5, A to C). The treatment of mice with a combination of radiation therapy and digoxin (n = 10) resulted in a median survival of 219 days (Fig. 5B). This represents a statistically significant improvement in survival over the radiation-only group (P = 0.04; Fig. 5D) but did not meet statistical significance when compared to single-agent digoxin therapy (219 days versus 180 days, P = 0.33; fig. S6). These results highlight both the potency of digoxin antitumor activity in vivo and the need for further study regarding digoxin’s interaction with radiation to determine the optimal approach to integrating it into future clinical contexts.
Digoxin treatment leads to increased apoptosis and changes in gene expression consistent with modulation of ERK/AKT signaling and induction of mitochondrial dysfunction
To gain insight into the mechanisms by which digoxin inhibited tumor growth and prolonged survival, we evaluated changes in cell morphology and gene expression after digoxin treatment. Tumor cells harvested from mice undergoing digoxin treatment showed an increase in apoptosis compared to untreated tumors or those that progressed after completion of treatment (Fig. 6A). We subsequently performed deep RNA-seq (GSE115542) on group 3 (ICb-2555MB) and group 4 (ICb-1078MB) tumors harvested before and immediately after completion of digoxin treatment to evaluate changes in gene expression (Fig. 6 and tables S10 and S11). Lists of genes showing the largest changes in expression were then subjected to Ingenuity Pathway Analysis (IPA) (28) to identify the signaling pathways most altered in response to digoxin treatment. Consistent with the observed increase in apoptosis (Fig. 6, A and B), the most enriched pathways in both group 3 [sirtuin signaling pathway, nuclear factor, erythroid 2-like 2 (NFE2L2)-mediated oxidative stress response, regulation of eIF4, and p70S6K signaling; table S12] and group 4 (oxidative phosphorylation, mitochondrial dysfunction, and sirtuin signaling pathway; table S13) were associated with apoptosis and cellular stress responses. Group 3 (ICb-2555MB) tumors, in particular, showed changes in mitochondrially encoded genes, consistent with previous studies in melanoma that found that cardiac glycosides impaired cellular function, in part, by altering mitochondrial function (29).
Fig. 6. Cellular responses to digoxin in orthotopic PDX models of MB.
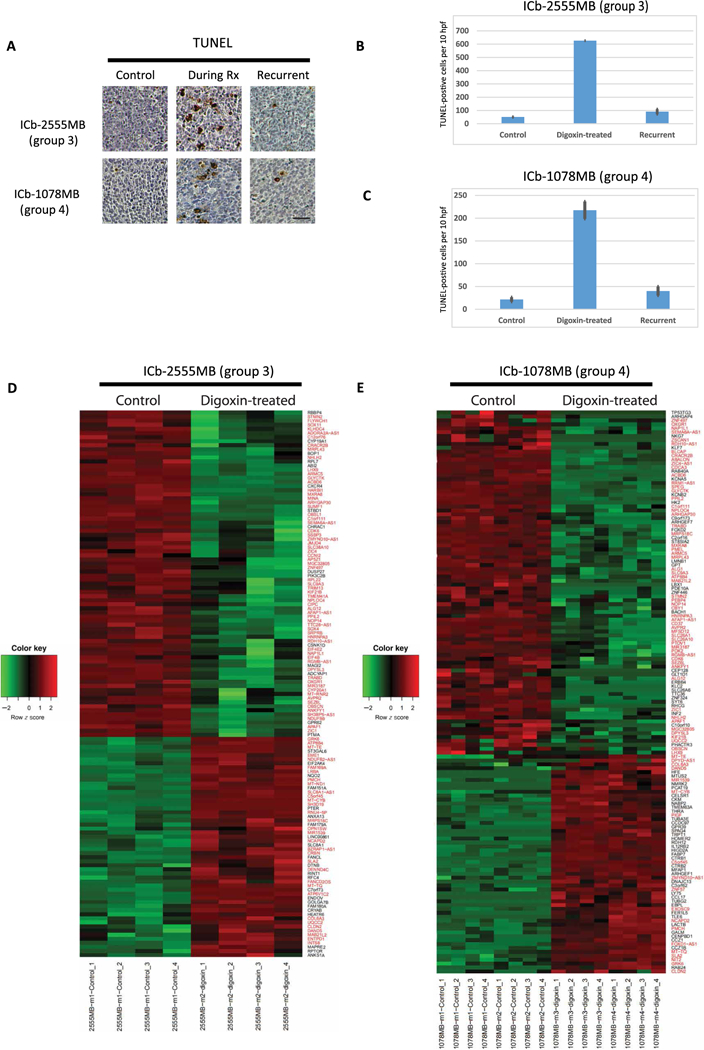
(A) TUNEL (terminal deoxynucleotidyl transferase–mediated deoxyuridine triphosphate nick end labeling) staining of tumors from group 3 (ICb-2555MB) and group 4 (ICb-1078MB) models before digoxin treatment, during treatment, or at time of recurrence. Scale bar, 25 mM. (B and C) Quantitation of TUNEL staining from group 3 (ICb-2555MB) (B) and group 4 (ICb-1078MB) (C). hpf, high-power field. (D and E) Heatmaps depicting changes gene expression in group 3 tumor (D) (ICb-2555 MB) and group 4 (E) (ICb-1078MB) tumors before and after digoxin treatment. Red text denotes genes that are differentially expressed in both groups 3 and 4 MB. For the heatmap, red indicates up-regulation, and green indicates down-regulation.
Because genes that have been less extensively characterized or demonstrate tissue-restricted expression are less likely to be represented in pathways identified by IPA, we also individually examined the 15 most up-regulated and 15 most down-regulated genes in each model (Fig. 6, C and D, and tables S10 and S11). We found that many of these genes have been reported to either play a role in or be a target of extracellular signal–regulated kinase (ERK) or AKT signaling. Specifically, in group 3 tumors (ICb-2555MB) treated with digoxin, 7 of the 15 most down-regulated genes (ADCYAP1, DPYSL3, STBD1, CXCR4, NDUFB9, STMN2, and CYP19A1) (30–32) and 5 of the 15 most up-regulated genes (NQO2, SLC81A, RPTOR, ST3GAL6, and CRYAB) (33–35) have been linked to these pathways. In the group 4 model (ICb-1078MB), 6 of the 15 most down-regulated genes (KCNA5, FOXD2, HK2, BACH1, ST8SIA2, and ZNF446) (36–38) and 1 of the 15 most up-regulated genes (FABP7) (39) have been associated with AKT or ERK signaling. These findings are consistent with a report that the cardiac glycoside, ouabain, is capable of inhibiting epidermal growth factor receptor (EGFR)–mediated activation of ERK and AKT (40). Furthermore, mitochondrial AKT signaling has recently been reported to play a key role in altering cellular metabolism to inhibit apoptosis in tumor cells undergoing metabolic stress (41). Together, therefore, these findings suggest a potential model in which digoxin modulation of AKT and ERK signaling sensitizes MB cells to metabolic stress resulting in widespread cell death in these aggressive and rapidly dividing tumors.
DISCUSSION
Our informatics-driven drug repositioning strategy identified consistent (robust) dysfunctional signaling networks with high statistical confidence by jointly analyzing multigenomics profiling data of group-specific MB patients, thereby overcoming the deficiencies associated with analyzing data from only a specific genomic space. Further-more, because MB is a pediatric brain tumor with low incidence rate, this bootstrapping strategy provided an added advantage by estimating the robustness of the identified driver networks given the limited sample size. Unlike “on-target repositioning,” which applies a drug’s known pharmacologic mechanism for a different therapeutic indication, our approach used a DFN to incorporate predicted off-target effects, an important element given that the antitumor mechanism of many compounds is likely multifactorial. This approach allowed us to identify a potential treatment for two difficult-to-target subgroups of MB, which, if borne out in future clinical studies, has the potential to both improve patient survival and reduce treatment-related complications by allowing for de-escalation of other treatment modalities.
Although in these studies our systems biology-driven predictions were validated by the experimental results from the orthotopic PDX models, our ability to generalize this approach remains limited by the available data. Specifically, the cellular responses to perturbation reported in the CMAP database were derived from a specific set of cancer cell lines (breast and prostate), and it remains to be seen how consistently the effects observed can be extrapolated to other tumors. In addition, this database represents a limited number of compounds (1309), limiting the pool of potential agents for repurposing. Moving forward, the ability to gather additional drug-related information will be an important step to maximizing the impact of this approach, thereby allowing for the construction of large-scale DFNs and enabling large-scale drug repositioning.
In addition, we used subtype-specific genomics data from MB patients to identify drug candidates. However, cellular heterogeneity exists even within a given tumor, resulting in different drug responses and the potential for the development of drug-resistant subpopulations of cells. The increasing availability of single-cell RNA-seq data raises the potential for applying the DSNI-DFN approach data from individual tumors to discover personalized driver signaling mechanisms, thereby deriving personalized treatment plans for individual cancer patients that could further demonstrate the power of integrative pharmacogenomics.
We believe that the activity of cardiac glycosides against groups 3 and 4 MBs is not an off-target effect because otherwise it would be hard to explain why all five members of this class of drugs are ranked highly in our systems approach analysis. In recent years, cardiac glycosides have been found to have antitumor effects in multiple types of cancers (42), with increasing amount of evidence suggesting the signal transduction activity of Na+- and K+-dependent adenosine triphosphatases (ATPases) as a possible mechanism in addition to its more conventional role as a sodium pump. Most notable is the observation that inhibition of Na+,K+-ATPases by ouabain induces both apoptosis and necrosis and can enhance the cytotoxicity of chemotherapy in human cancer cells that have up-regulated Na+,K+-ATPases, such as glioblastoma cells (43). Other mechanisms that have been suggested for the effects of cardiac glycosides on various classes of tumor cells include the inhibition of hypoxia signaling (26, 44), global suppression of protein synthesis (45), modulation of EGFR/ERK/AKT signaling (40), and disruption of mitochondrial function via calcium-based signaling (29, 46). Although the results presented here are very encouraging, we acknowledge that our studies do not provide a definitive mechanism for the marked effect that digoxin exerts on groups 3 and 4 tumors and would need further experiments to fully characterize the nature of cell death and delineate the detail dose-response relationship between the inhibition of Na+,K+-ATPases and other potential downstream effects. However, these data do suggest a potential model that can serve as the basis of future studies, one in which digoxin inhibition of AKT-mediated signaling sensitizes MB cells to metabolic stress. How exactly digoxin influences AKT-mediated signaling, and whether this represents the primary or one of several ways in which digoxin induces apoptosis, remain to be elucidated. However, our results suggest an exciting potential role for the use of digoxin in the treatment of these devastating tumors.
MATERIALS AND METHODS
Study design
To identify repurposable drugs for the treatment of MB, we developed a systems biology approach, which identifies candidate drugs targeting driver signaling networks of groups 3 and 4. It consists of two components: identification of the driver signaling networks using DNA sequencing, gene copy number, DNA methylation, and RNA-seq data of patients with groups 3 and 4 MB. These driver signaling networks were used to evaluate the targeting effects of drugs or bioactive compounds. We hypothesized that drugs that can target and reverse the gene expression pattern of driver signaling networks might be useful therapeutics. In vitro and in vivo experiments were designed to validate the antitumor activity of the cardiac glycosides against groups 3 and 4 MB predicted by the systems biology model. In vitro assays were conducted using a single–time point assay (72 hours of drug exposure) with escalating doses of five cardiac glycosides, with the end point being the cell viability in two MB-derived cell lines (MED8A and D283) as well as two control cell lines to control for nonspecific cytotoxicity. In vivo survival studies were carried out in NOD.129S7 (B6)-Rag1tm1Mom/J severe combined immunodeficient (SCID) mice (the Jackson Laboratory) because of their ability to tolerate radiation compared to other strains of SCID mice. To detect the difference in means of 0.01 or greater at the SD of 0.06, the power of detection at 0.90, and α (the risk of a false positive conclusion) at 0.05000, we chose 10 mice per group (digoxin-treated and controls). Treatment start times after implantation with tumor cells were determined on the basis of prior experience with serial sectioning of mouse brains to target tumors of about 2 to 3 mm in size when treatment was initiated. Fractionated radiation was administered at 2 Gy/day via with an RS-2000 Biological irradiator (Rad Source Technologies), and digoxin was given 2 mg/kg ip daily. Start times between digoxin (21 days) and radiation (17 days) were staggered to limit any potential acute toxicity in the mice receiving combination therapy. End points were evidence of neurologic compromise, loss of >20% of body weight, substantially diminished physical activity, or respiratory distress. Mice were screened for evidence of B and T cell populations, and evidence of reversion to immunocompetence was an exclusion criterion.
Statistical analysis
The Kaplan-Meier estimator was used to estimate the survival curve from lifetime data of patients with MB as well as the mice survival times for the in vivo study. The log-rank test was used to estimate the P values for the survival analysis. Differential expression analysis for comparing the digoxin-treated gene expression data with the untreated control was based on the read counts using DESeq2 (47). In DESeq2, we modeled the raw read counts data to follow a negative binomial distribution. A generalized linear model was used to fit the raw read counts data of each gene. A Wald test was applied on the fitting coefficients for differential expression. The Benjamin and Hochberg procedure was used for multiple testing on P values from the Wald test to estimate the false discovery rate (FDR). Differentially expressed genes were selected by two concurrent criteria, absolute fold change (digoxin-treated versus controls) larger than 2 and adjusted P values (FDR) less than 0.05.
Supplementary Material
Acknowledgments:
We would like to thank members of the Cancer Systems Biology Laboratory in the Department of Systems Medicine and Bioengineering at Houston Methodist Research Institute, members of the X.-N.L. Laboratory at Texas Children’s Hospital for helpful discussions, and R. Danforth of the Department of Systems Medicine and Bioengineering for proofreading.
Funding: This work is supported by NIH U54CA149196 (to S.T.C.W.), NIH U01CA188388 (to S.T.C.W.), CPRIT RP110532 (to S.T.C.W.), Ting Tsung and Wei Fong Chao Foundation (to S.T.C.W.), John S. Dunn Research Foundation (to S.T.C.W.), NIH RO1 CA185402 (to X.-N.L.), CPRIT RP-170169 (to X.-N.L.), Cancer Fighters of Houston (to S.T.C.W., X.-N.L., and C.C.L.), and Virginia and L.E. Simmons Foundation (to T.-K.M., C.C.L., and S.T.C.W.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability: All data associated with this study can be found in the paper or the Supplementary Materials. RNA-seq data for group 3 (ICb-2555MB) and group 4 (ICb-1078MB) MB are available at the NCBI Gene Expression Omnibus under accession no. GSE115542.
SUPPLEMENTARY MATERIALS
www.sciencetranslationalmedicine.org/cgi/content/full/10/464/eaat0150/DC1
REFERENCES AND NOTES
- 1.Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A,Scheithauer BW, Kleihues P, The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 114, 97–109 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gottardo NG, Gajjar A, Current therapy for medulloblastoma. Curr. Treat. Options. Neurol. 8, 319–334 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Kool M, Korshunov A, Remke M, Jones DT, Schlanstein M, Northcott PA, Cho YJ, Koster J, Schouten-van Meeteren A, van Vuurden D, Clifford SC, Pietsch T, von Bueren AO, Rutkowski S, McCabe M, Collins VP, Backlund ML, Haberler C, Bourdeaut F, Delattre O, Doz F, Ellison DW, Gilbertson RJ, Pomeroy SL, Taylor MD, Lichter P, Pfister SM, Molecular subgroups of medulloblastoma: An international meta-analysis of transcriptome, genetic aberrations, and clinical data of WNT, SHH, Group 3, and Group 4 medulloblastomas. Acta Neuropathol. 123, 473–484 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ, Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci. Transl. Med 3, 96ra76 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.lorio F, Bosotti R, Scacheri E, Belcastro V, Mithbaokar P, Ferriero R, Murino L, Tagliaferri R, Brunetti-Pierri N, Isacchi A, di Bernardo D, Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc. Natl. Acad. Sci. U.SA. 107, 14621–14626 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN, Reich M, Hieronymus H, Wei G, Armstrong SA, Haggarty SJ, Clemons PA, Wei R, Carr SA, Lander ES, Golub TR, The Connectivity Map: Using gene-expression signatures to connect small molecules, genes, and disease. Science 313, 1929–1935 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehar J, Kryukov GV, Sonkin D, Reddy A, Liu M, Murray L, Berger MF, Monahan JE, Morais P, Meltzer J, Korejwa A, Jané-Valbuena J, Mapa FA, Thibault J, Bric-Furlong E, Raman P, Shipway A, Engels IH, Cheng J, Yu GK, Yu J, Aspesi P Jr., de Silva M, Jagtap K, Jones MD, Wang L, Hatton C, Palescandolo E, Gupta S, Mahan S, Sougnez C, Onofrio RC, Liefeld T, MacConaill L, Winckler W, Reich M, Li N, Mesirov JP, Gabriel SB, Getz G, Ardlie K, Chan V, Myer VE, Weber BL, Porter J, Warmuth M, Finan P, Harris JL, Meyerson M, Golub TR, Morrissey MP, Sellers WR, Schlegel R, Garraway LA , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, Liu Q, lorio F, Surdez D, Chen L, Milano RJ, Bignell GR, Tam AT, Davies H, Stevenson JA, Barthorpe S, Lutz SR,Kogera F, Lawrence K, McLaren-Douglas A, Mitropoulos X, Mironenko T, Thi H, Richardson L, Zhou W, Jewitt F, Zhang T, O’Brien P, Boisvert JL, Price S, Hur W, Yang W, Deng X, Butler A, Choi HG, Chang JW, Baselga J, Stamenkovic I, Engelman JA, Sharma SV, Delattre O, Saez-Rodriguez J, Gray NS, Settleman J, Futreal PA, Haber DA, Stratton MR, Ramaswamy S, McDermott U, Benes CH, Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature 483, 570–575 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Jin G, Fu C, Zhao H, Cui K, Chang J, Wong STC, A novel method of transcriptional response analysis to facilitate drug repositioning for cancer therapy. Cancer Res. 72, 33–44 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, Sage J, Butte AJ, Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci. Transl. Med. 3, 96ra77 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang B, Mezlini AM, Demir F, Fiume M, Tu Z, Brudno M, Haibe-Kains B, Goldenberg A, Similarity network fusion for aggregating data types on a genomic scale. Nat. Methods 11, 333–337 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Huang L, Li F, Sheng J, Xia X, Ma J, Zhan M, Wong STC, DrugComboRanker: Drug combination discovery based on target network analysis. Bioinformatics 30, i228–i236 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bozic I, Antal T, Ohtsuki H, Carter H, Kim D, Chen S, Karchin R, Kinzler KW, Vogelstein B, Nowak MA, Accumulation of driver and passenger mutations during tumor progression. Proc. Natl. Acad. Sci. U.S.A. 107, 18545–18550 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cavalli FMG, Remke M, Rampasek L, Peacock J, Shih DJH, Luu B, Garzia L, Torchia J, Nor C, Morrissy AS, Agnihotri S, Thompson YY, Kuzan-Fischer CM, Farooq H, Isaev K, Daniels C, Cho BK, Kim SK, Wang KC, Lee JY, Grajkowska WA, Perek-Polnik M, Vasiljevic A, Faure-Conter C, Jouvet A, Giannini C, Nageswara Rao A. A., Li KKW, Ng HK, Eberhart CG, Pollack IF, Hamilton RL, Gillespie GY, Olson JM, Leary S, Weiss WA, Lach B, Chambless LB, Thompson RC, Cooper MK, Vibhakar R, Hauser P, van Veelen MC, Kros JM, French PJ, Ra YS, Kumabe T, López-Aguilar E, Zitterbart K, Sterba J, Finocchiaro G, Massimino M, Van Meir EG, Osuka S, Shofuda T, Klekner A, Zollo M, Leonard JR, Rubin JB, Jabado N, Albrecht S, Mora J, Van Meter TE, Jung S, Moore AS, Hallahan AR, Chan JA, Tirapelli DPC, Carlotti G, Fouladi M, Pimentel J, Faria CC, Saad AG, Massimi L, Liau LM, Wheeler H, Nakamura H, Elbabaa SK, Perezpeña-Diazconti M, de León Chico Ponce F., Robinson S, Zapotocky M, Lassaletta A, Huang A, Hawkins CE, Tabori U, Bouffet E, Bartels U, Dirks PB, Rutka JT, Bader GD, Reimand J, Goldenberg A, Ramaswamy V, Taylor MD , Intertumoral heterogeneity within medulloblastoma subgroups. Cancer Cell 31, 737–754. e6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smyth GK, Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3 (2004). [DOI] [PubMed] [Google Scholar]
- 16.Lin CY, Erkek S, Tong Y, Yin L, Federation AJ, Zapatka M, Haldipur P, Kawauchi D, Risch T, Warnatz H-J, Worst BC, Ju B, Orr BA, Zeid R, Polaski DR, Segura-Wang M, Waszak SM, Jones DTW, Kool M, Hovestadt V, Buchhalter I, Sieber L, Johann P, Chavez L, Gröschel S, Ryzhova M, Korshunov A, Chen W, Chizhikov VV, Millen KJ, Amstislavskiy V, Lehrach H, Yaspo M-L, Eils R, Lichter P, Korbel JO, Pfister SM, Bradner JE, Northcott PA, Active medulloblastoma enhancers reveal subgroup-specific cellular origins. Nature 530, 57–62 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frey BJ, Dueck D, Clustering by passing messages between data points. Science 315, 972–976 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Northcott PA, Jones DT, Kool M, Robinson GW, Gilbertson RJ, Cho YJ, Pomeroy SL, Korshunov A, Lichter P, Taylor MD, Pfister SM, Medulloblastomics: The end of the beginning. Nat. Rev. Cancer 12, 818–834 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaujoux R, Seoighe C, A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ivanov DP, Coyle B, Walker DA, Grabowska AM, In vitro models of medulloblastoma: Choosing the right tool for the job. J. Biotechnol. 236, 10–25 (2016). [DOI] [PubMed] [Google Scholar]
- 21.Pizzo PA, Poplack DG, Principles and Practice of Pediatric Oncology (Wolters Kluwer/Lippincott Williams & Wilkins Health, Philadelphia, PA, ed. 6th, 2011), pp. xx, 1531 p. [Google Scholar]
- 22.Ehle M, Patel C, Giugliano RP, Digoxin: Clinical highlights: A review of digoxin and its use in contemporary medicine. Crit. Pathw. Cardiol. 10, 93–98 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Kuhn M, Szklarczyk D, Franceschini A, von Mering C, Jensen LJ, Bork P, STITCH 3: Zooming in on protein-chemical interactions. Nucleic Acids Res. 40, D876–D880 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shu Q, Wong KK, Su JM, Adesina AM, Yu LT, Tsang YTM, Antalffy BC, Baxter P, Perlaky L, Yang J, Dauser RC, Chintagumpala M, Blaney SM, Lau CC, Li X-N, Direct orthotopic transplantation of fresh surgical specimen preserves CD133+ tumor cells in clinically relevant mouse models of medulloblastoma and glioma. Stem Cells 26, 1414–1424 (2008). [DOI] [PubMed] [Google Scholar]
- 25.Zhao X, Liu Z, Yu L, Zhang Y, Baxter P, Voicu H, Gurusiddappa S, Luan J, Su JM, Leung HC, Li XN, Global gene expression profiling confirms the molecular fidelity of primary tumor-based orthotopic xenograft mouse models of medulloblastoma. Neuro Oncol. 14, 574–583 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nigim F, Cavanaugh J, Patel AP, Curry WT Jr., Esaki S, Kasper EM, Chi AS, Louis DN, Martuza RL, Rabkin SD, Wakimoto H, Targeting hypoxia-inducible factor 1α in a New orthotopic model of glioblastoma recapitulating the hypoxic tumor microenvironment. J. Neuropathol. Exp. Neurol. 74, 710–722 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Allonen H, Anderson KE, Iisalo E, Kanto J, Stromblad LG, Wettrell G, Passage of digoxin into cerebrospinal fluid in man. Acta Pharmacol. Toxicol. 41, 193–202 (1977). [DOI] [PubMed] [Google Scholar]
- 28.Krämer A, Green J, Pollard J Jr., S. Tugendreich, Causal analysis approaches in ingenuity pathway analysis. Bioinformatics 30, 523–530 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Eskiocak U, Ramesh V, Gill JG, Zhao Z, Yuan SW, Wang M, Vandergriff T, Shackleton M, Quintana E, Johnson TM, DeBerardinis RJ, Morrison SJ, Synergistic effects of ion transporter and MAP kinase pathway inhibitors in melanoma. Nat. Commun. 7, 12336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chiellini C, Grenningloh G, Cochet O, Scheideler M, Trajanoski Z, Ailhaud G, Dani C, Amri EZ, Stathmin-like 2, a developmentally-associated neuronal marker, is expressed and modulated during osteogenesis of human mesenchymal stem cells. Biochem. Biophys. Res. Commun. 374, 64–68 (2008). [DOI] [PubMed] [Google Scholar]
- 31.Reichelt ME, Mellor KM, Curl CL, Stapleton D, Delbridge LM, Myocardial glycophagy - a specific glycogen handling response to metabolic stress is accentuated in the female heart. J. Mol. Cell. Cardiol. 65, 67–75 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Tan T, Wang L, Wang B, Collagen and prostaglandin E2 regulate aromatase expression through the PI3K/AKT/IKK and the MAP kinase pathways in adipose stromal cells. Mol. Med. Rep. 12, 4766–4772 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Hsieh TC, Lin CY, Bennett DJ, Wu E, Wu JM , Biochemical and cellular evidence demonstrating AKT-1 as a binding partner for resveratrol targeting protein NQO2. PLOS ONE 9, e101070 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Earwaker P, Anderson C, Willenbrock F, Harris AL, Protheroe AS, Macaulay VM, RAPTOR up-regulation contributes to resistance of renal cancer cells to PI3K-mTOR inhibition. PLOS ONE 13, e0191890 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Malin D, Strekalova E, Petrovic V, Rajanala H, Sharma B, Ugolkov A, Gradishar WJ, Cryns VL, ERK-regulated αB-crystallin induction by matrix detachment inhibits anoikis and promotes lung metastasis in vivo. Oncogene 34, 5626–5634 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakanome A, Brydun A, Matsumoto M, Ota K, Funayama R, Nakayama K, Ono M, Shiga K, Kobayashi T, Igarashi K, Bachl is critical for the transformation of mouse embryonic fibroblasts by Ras(Vl2) and maintains ERK signaling. Oncogene 32, 3231–3245 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Liu F, Zhu C, Xiao J, Wang Y, Tang W, Yuan W, Zhao Y, Li Y, Xiang Z, Wu X, Liu M, A novel human KRAB-containing zinc-finger gene ZNF446 inhibits transcriptional activities of SRE and AP-1. Biochem. Biophys. Res. Commun. 333, 5–13 (2005). [DOI] [PubMed] [Google Scholar]
- 38.Johansson CC, Dahle MK, Blomqvist SR, Grønning LM, Aandahl EM, Enerbӓck S, Taskén K, A winged helix forkhead (FOXD2) tunes sensitivity to cAMP in T lymphocytes through regulation of cAMP-dependent protein kinase RIalpha. J. Biol. Chem. 278, 17573–17579 (2003). [DOI] [PubMed] [Google Scholar]
- 39.Tripathi S, Kushwaha R, Mishra J, Gupta MK, Kumar H, Sanyal S, Singh D, Sanyal S, Sahasrabuddhe AA, Kamthan M, Mudiam MK, Bandyopadhyay S, Docosahexaenoic acid up-regulates both PI3K/AKT-dependent FABP7-PPARg interaction and MKP3 that enhance GFAP in developing rat brain astrocytes. J. Neurochem. 140, 96–113 (2017). [DOI] [PubMed] [Google Scholar]
- 40.Wolle D, Lee SJ, Li Z, Litan A, Barwe SP, Langhans SA, Inhibition of epidermal growth factor signaling by the cardiac glycoside ouabain in medulloblastoma. Cancer Med. 3, 1146–1158 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chae YC, Vaira V, Caino MC, Tang HY, Seo JH, Kossenkov AV, Ottobrini L, Martelli C, Lucignani G, Bertolini I, Locatelli M, Bryant KG, Ghosh JC, Lisanti S, Ku B, Bosari S, Languino LR, Speicher DW, Altieri DC, Mitochondrial Akt regulation of hypoxic tumor reprogramming. Cancer Cell 30, 257–272 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kepp O, Menger L, Vacchelli E, Adjemian S, Martins I, Ma Y, Sukkurwala AQ, Michaud M, Galluzzi L, Zitvogel L, Kroemer G, Anticancer activity of cardiac glycosides. Oncoimmunology 1, 1640–1642 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen D, Song M, Mohamad O, Yu SP, Inhibition of Na+/K+-ATPase induces hybrid cell death and enhanced sensitivity to chemotherapy in human glioblastoma cells. BMC Cancer 14, 716 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang H, Qian DZ, Tan YS, Lee K, Gao P, Ren YR, Rey S, Hammers H, Chang D, Pili R, Dang CV, Liu JO, Semenza GL, Digoxin and other cardiac glycosides inhibit HIF-1 a synthesis and block tumor growth. Proc. Natl. Acad. Sci. U.S.A. 105, 19579–19586 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perne A, Muellner MK, Steinrueck M, Craig-Mueller N, Mayerhofer J, Schwarzinger I, Sloane M, Uras IZ, Hoermann G, Nijman SM, Mayerhofer M, Cardiac glycosides induce cell death in human cells by inhibiting general protein synthesis. PLOS ONE 4, e8292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McConkey DJ, Lin Y, Nutt LK, Ozel HZ, Newman RA, Cardiac glycosides stimulate Ca2+ increases and apoptosis in androgen-independent, metastatic human prostate adenocarcinoma cells. Cancer Res. 60, 3807–3812 (2000). [PubMed] [Google Scholar]
- 47.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, Kiezun A, Hammerman PS, McKenna A, Drier Y, Zou L, Ramos AH, Pugh TJ, Stransky N, Helman E, Kim J, Sougnez C, Ambrogio L, Nickerson E, Shefler E, Cortes ML, Auclair D, Saksena G, Voet D, Noble M, DiCara D, Lin P, Lichtenstein L, Heiman DI, Fennell T, Imielinski M, Hernandez B, Hodis E, Baca S, Dulak AM, Lohr J, Landau D-A, Wu CJ, Melendez-Zajgla J, Hidalgo-Miranda A, Koren A, McCarroll SA, Mora J, Lee RS, Crompton B, Onofrio R, Parkin M, Winckler W, Ardlie K, Gabriel SB, Roberts WM, Biegel JA, Stegmaier K, Bass AJ, Garraway LA, Meyerson M, Golub TR, Gordenin DA, Sunyaev S, Lander ES, Getz G, Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.GISTIC 2 Manuscript and Software; ftp.broadinstitute.org/pub/genepattern/modules_public_server_doc/GISTIC2.pdf.
- 50.Chen L, Xuan J, Riggins RB, Wang Y, Clarke R, Identifying protein interaction subnetworks by a bagging Markov random field-based method. Nucleic Acids Res. 41, e42 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Glaab E, Baudot A, Krasnogor N, Schneider R, Valencia A, EnrichNet: Network-based gene set enrichment analysis. Bioinformatics 28, i451–i457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trapnell C, Pachter L, Salzberg SL, TopHat: Discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Anders S, Pyl PT, Huber W, HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.