

Abstract

Despite the low bond strength of the oxygen–oxygen bond, organic peroxides are often surprisingly resistant to cleavage by nucleophiles and reductants. As a result, achieving decomposition under mild conditions can be challenging. Herein, we explore the reactivity of a selection of peroxides toward thiolates, phenyl selenide, Fe(II) salts, and iron thiolates. Peroxides activated by conjugation, strain, or stereoelectronics are rapidly cleaved at room temperature by thiolate anions, phenylselenide, or Fe(II) salts. Under the same conditions, unhindered dialkyl peroxides are only marginally reactive; hindered peroxides, including triacetone triperoxide and diacetone diperoxide (DADP), are inert. In contrast, all but the most hindered of peroxides are rapidly (<1 min at concentrations down to ∼40 mM) cleaved by mixtures of thiols and iron salts. Our observations suggest the possible intermediacy of strongly reducing complexes that are readily regenerated in the presence of stoichiometric thiolate or hydride. In the case of DADP, an easily prepared explosive of significant societal concern, catalytic amounts of iron and thiol are capable of promoting rapid and complete disproportionation. The availability of inexpensive and readily available catalysts for the mild reductive degradation of all but the most hindered of peroxides could have significant applications for controlled remediation of explosives or unwanted radical initiators.
Introduction
In the course of investigations into reaction of carbanions with organic peroxides, we became intrigued by the often low reactivity of the thermodynamically weak O–O bond,1 an incongruity previously observed in electrochemical and chemical reductions.2,3 The high barriers associated with peroxide cleavage can make it difficult to achieve controlled decomposition under mild conditions during decontamination or reaction work-up procedures. In an effort to better understand the factors controlling reductive and/or nucleophilic attack on an O–O bond, we investigated the reactivity of several classes of peroxides toward thiolates, selenides, and/or iron salts. Individually, the reagents cleave only activated peroxides. However mixtures of Fe(II) or Fe(III) salts and thiols, possibly in the form of Fe/thiol complexes, catalyze reductive cleavage of all but the most hindered of peroxides.
Reductive cleavage of hydrogen peroxide and alkyl hydroperoxides by thiolates and selenides is well known.4,5 The same is true of peroxides activated by conjugation, stereoelectronics, or strain.6 For example, ozonides (1,2,4-trioxolanes) are reduced by glutathione,7 and we have demonstrated rapid inactivation of the enzyme papain by an ozonide-containing dipeptide analog.8 Selenoperoxidases are able to reduce strained cyclic peroxides.9,10 In contrast, unstrained dialkyl peroxides are inert toward thiols,11 and reported reductions by thiolates employ high temperatures12 or involve substrates susceptible to base-promoted fragmentation.13 Similar trends are seen with ferrous iron, which rapidly cleaves hydrogen peroxide (Fenton reaction),14 alkyl hydroperoxides,15,16 and activated or unhindered dialkyl peroxides.17−19 Fe(II)-promoted cleavage of hindered dialkyl peroxides typically requires reaction temperatures at which radical thermolysis of the peroxide is expected to be significant;20,21 however, a hindered 1,2-dioxolane that induces iron-dependent regulated cell death (ferroptosis) has been shown to undergo room-temperature cleavage by soluble Fe(II).22 The peroxyacetal core of the antimalarial artemisinin, which is reactive toward Fe(II) but not thiols, is reductively cleaved by cysteine in the presence of catalytic amounts of iron; follow-up studies observed similar reactivity with other peroxide substrates.23
Results and Discussion
Preparation of peroxide substrates is illustrated in Scheme 1. Peroxyacetal 1 and dialkyl peroxides 2 and 3 were prepared by alkylation of a hydroperoxyacetal,24 hydrogen peroxide (two-fold reaction), or t-butyl hydroperoxide, respectively, with sulfonates derived from 3-phenyl-propanol.1t-Butyl peroxybenzoate (4) is commercially available but also easily prepared through oxidative peresterification.25 Triacetone triperoxide (TATP, 5) and diacetone diperoxide (DADP, 6) were prepared through acid-promoted condensations of acetone with hydrogen peroxide.26Caution: TATP and DADP are shock-, friction-, heat-, and spark-sensitive high explosives and can decompose violently in strong acid.26−29 Peroxide 7 was prepared from the corresponding tertiary bromide following a reported procedure;2b peroxide 8 was available through methylation of the known tertiary hydroperoxide.30
Scheme 1. Peroxide Substrates.

The reactivity of the peroxides toward thiolates and selenides is summarized in Table 1. Three approaches were used for nucleophile generation: (1) stoichiometric deprotonation of thiophenol with potassium tert-butoxide;31 (2) deprotonation with a much weaker base (triethylamine); (3) reductive cleavage of diphenyl disulfide or diphenyl diselenide.32,33 Reactions were monitored by thin-layer chromatography (TLC) using both conventional and peroxide-sensitive indicators.34 With the exception of peroxyacetal 1, which rapidly undergoes base-promoted E1CB (Kornblum) cleavage,35 little or no reactivity was observed with base (rows 1, 2), hydride (row 3) or thiophenol (row 4). Peroxyacetal 1 and perester 4 were rapidly decomposed by phenyl thiolate, regardless of how they were generated. However, the dialkyl peroxides (2 and 3), as well as the two acetone peroxides (5 and 6), was proved inert. A similar reactivity was observed with phenyl selenide, although traces of reduction products were detected from 2 and 3 after prolonged reaction.
Table 1. Reactivity of Peroxides toward Phenyl Thiolate and Phenylselenide.

| peroxide |
|||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | ||
| row | reagent(s) | time for substrate consumption (min)a | |||||
| 1 | KOtBu | <15 | >60 | >60 | >60 | nrb | nr |
| 2 | Et3N | >60 | nr | nr | nr | nr | nr |
| 3 | LiBH4c | >60 | nr | nr | nr | nr | nr |
| 4 | PhSH | nr | nr | nr | nr | nr | nr |
| 5 | PhSH, KOtBu | <15 | nr | nr | <15 | nr | nr |
| 6 | PhSH, Et3N | <15 | nr | nr | <15 | nr | nr |
| 7 | (PhS)2, LiBH4c | <15 | nr | nr | <15 | nr | nr |
| 8 | (PhSe)2, LiBH4c | <15 | >120 | >120 | <15 | nr | nr |
See the Experimental Procedures section.
No detectable change after 2 h.
Similar results obtained in ethanol.
Table 2 illustrates reactions incorporating ferrous iron. Although monoperoxyacetal 1 and perester 4 undergo slow decomposition at room temperature in the presence of FeBr2, the dialkyl peroxides (2 and 3) and the acetone peroxides (5 and 6) are inert (Table 2, row 1). A dramatic enhancement in reactivity was observed from the combination of iron and thiols (Table 2, rows 2–8). Addition of thiophenol to the orange-brown solutions containing FeBr2 and peroxides resulted in a dark suspension, which clarified, often within less than a minute, to a light green solution (Figure S1). Monitoring of reactions by TLC revealed that consumption of the peroxide approximately paralleled the clearing of the suspension.34 Reactions could be conducted in tetrahydrofuran (THF) or CD3CN and were unaffected by addition of small amounts (1–2 v/v %) of water or Et3N (not shown).
Table 2. Reactivity of Peroxides with Fe(II) and/or PhSH.

| peroxide |
|||||||
|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 6 | |||
| equiv of Fe(II) | equiv of PhSH | time for disappearance (min)a | |||||
| 1 | 1.0 | 0 | <15 | nr | nr | <15 | nr |
| 2 | 0.2 | 2 | <1 | <1 | <1 | <1 | <1 |
| 4 | 0.1 | 1 | <1b | <1b | |||
| 5 | 0.1 | 2 | <1b | <1b | <1b | ||
| 6 | 10–2 | 2 | <1 | ||||
| 7 | 10–2 | 1 | <1 | <1 | |||
| 8 | 10–4 | 2 | <1 | <1 | |||
Monitored by TLC (all) or 1H NMR (rxns in CD3CN); “nr” indicates no change in 60 min.
Reaction in CD3CN.
Isolated yields for selected reactions of peroxides 2, 3, and 4 in CD3CN are illustrated in Table 3. In contrast to the 2:1 stoichiometry expected for reaction of alkyl thiols with hydrogen peroxide or alkyl hydroperoxides,36 complete consumption of peroxides 1–4 could be achieved using stoichiometric thiol; the use of 0.5 equiv of thiol resulted in a reduced yield of alcohol and recovery of half of the peroxide substrate (Table 3, row 2). In contrast, the reduction of TATP (5) in the presence of catalytic Fe(II) and excess thiol was extremely slow (eq 1). The use of a more electron-rich thiol had no discernible impact. The stoichiometry of DADP decomposition is discussed below.
 |
1 |
Table 3. Yields from Reactions of Peroxides 2–4.

| equiv Fe | equiv RSH | yield Ph(CH2)3OH (%) | yield PhCO2H (%) | |
|---|---|---|---|---|
| 2 | 0.1 | 2.0 | 87 | na |
| 3 | 0.1 | 0.5 | 46 | na |
| 0.1 | 1.0 | 75a | na | |
| 0.1 | 2.0 | 81 | na | |
| 4 | 0.1 | 1.0 | na | 71 |
| 0.1 | 2.0 | na | 77 |
PhSSPh (70%) isolated.
Peroxide 7, developed as a clock for electrochemically generated alkoxy radicals,2 proved unreactive (Scheme 2). However, a less hindered analog (8) underwent rapid reduction (minutes), to furnish a low yield of 2-methyl-1-phenyl-2-propanol and larger amounts of acetone, toluene, and phenyl benzyl sulfide.
Scheme 2. Reactions with Radical “Clock” Precursors.

Relative yields (NMR); isolated yield in parentheses.
Nature of the Thiol and the Iron Source
As illustrated in eq 2, a variety of thiols, including a dithiol, could be successfully applied to the reduction; for reasons that remain unclear, no reduction was observed in the presence of dithiothreitol (not shown). No reaction was observed when diphenyl disulfide was used in place of thiophenol (eq 3). The dependence upon iron source was investigated via the reduction of DADP (6) in the presence of stoichiometric thiophenol (Table 4). Rapid and complete decomposition of the peroxide was observed in the presence of catalytic amounts of multiple Fe(II) salts as well as iron(III) chloride; in each case, analysis of the crude reaction mixtures by NMR revealed acetone as the only detectable product (Figure S2). Reaction in the presence of iron tetraphenylporphyrin was quite slow and no reaction was observed in the presence of ferrocene or iron oxide (Fe2O3).
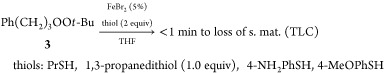 |
2 |
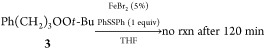 |
3 |
Table 4. Influence of Iron Source.

| Fe source | conversion (time)a |
|---|---|
| FeBr2, FeCl2, or FeI2 | >90% (<1 min) |
| FeCl3 (dry or wet) | >90% (<1 min) |
| FeTPP | traces (>120 min) |
| ferrocene | nr (>120 min) |
| ferrocene, NaCl | nr (>120 min) |
| Fe2O3 | nr (>120 min) |
1H NMR (see Experimental Procedures section).
Stoichiometry
The amount of reductant required was probed using dialkyl peroxide 3 and acetone peroxide 6 (Table 5). Complete consumption of 3 was observed using 1% of FeBr2 and either stoichiometric or excess thiophenol. Reductions in CD3CN generated nearly pure samples of the phenylpropanol product (TLC, NMR; Figure S2); reactions in THF generated the same major product but in addition displayed evidence [TLC, NMR, gas chromatography/mass spectrometry (GC/MS)] of a number of byproducts, each present at a low level. Use of substoichiometric thiol resulted in incomplete reaction. DADP (6) was completely decomposed by the combination of catalytic Fe(II) and either stoichiometric or substoichiometric thiol to generate acetone as the only detectable product; rapid reaction was observed with as little as 0.1% FeBr2 (Figure S2).
Table 5. Investigation of Reductant Stoichiometry.

| subst | Fe (equiv) | PhSH (equiv) | t (min) |
|---|---|---|---|
| 3a | 0.01 | 3 | <1 |
| 3a | 0.01 | 1 | <1 |
| 3a | 0.01 | 0.5 | <1c |
| 6b | 0.01 | 3 | <1 |
| 6b | 0.01 | 1 | <1 |
| 6b | 0.001 | 0.5 | <1 |
| 6b | 0.001 | 0.15 | <1 |
Rxn in THF; monitored by TLC.
Rxn in CD3CN; monitored by NMR.
Reaction incomplete.
Calorimetry
Reduction of dialkyl peroxide 3 in the presence of catalytic Fe(II) and stoichiometric thiol was exothermic by >30 kcal/mol when conducted in CH3CN and by a somewhat smaller amount when conducted in THF (Scheme 3). As will be discussed later, neither value approaches that calculated for a catalyzed reduction of a peroxide bond at the expense of two molecules of thiol. The differing heats of reaction measured in the two solvents, along with our qualitative observations of a number of minor byproducts from reactions in THF, may suggest a more complicated mechanism (vide infra). The decomposition of diperoxide 6 to acetone, a transformation which can be accomplished in the presence of catalytic amounts of Fe and thiol (see Table 4) was even less exothermic.
Scheme 3. Reaction Thermochemistry.

See the Experimental Procedures section for details.
Reduction could also be accomplished with stoichiometric hydride (Table 6). Dialkyl peroxide 3 is inert toward iBu2AlH (DIBAL-H) or BH3·THF, but is rapidly cleaved to 3-phenyl-1-propanol by either reagent in the presence of catalytic amounts of FeBr2 and thiophenol. No reaction was observed with less reactive hydride sources.
Table 6. Reductants Other Than Thiols.

| FeBr2 (equiv) | PhSH (equiv) | reductant (1.0 equiv) | loss of 3a |
|---|---|---|---|
| 0 | 0 | iBu2AlH | nr |
| 0.05 | 0.1 | iBu2AlH | <1 min |
| 0.05 | 0.1 | BH3·THF or BH3·Me2S | <1 min |
| 0.05 | 0.1 | NaCNBH3 | nr |
| 0.05 | 0.1 | Et3SiH | nr |
| 0.05 | 0.1 | HCO2H | nr |
| 0.05 | 0.1 | ascorbic acid | nr |
Monitored by TLC (ref (34)).
Discussion
Reduction of activated peroxides, for example, peroxyacetals, ozonides, or peresters, been accomplished using a variety of reagents.37 However, the reduction of unactivated dialkyl peroxides is more challenging.3,37 Reduction of DADP and TATP, of particular interest due to their ready availability as well as their use in acts of terrorism,26−29 has been achieved with SnCl2 in boiling ethanol,28 with Cr3+ or Co2+ species,29a,29b or with alloyed bimetals or emulsified metals.38 Although Fe(II)-promoted activation of hindered dialkyl peroxides has been described, the reported reaction temperatures are often commensurate with peroxide thermolysis.20 Reported Fe(III)-promoted decompositions of di-t-butyl peroxide and a ketone peroxide are likely to involve Lewis-acid-promoted release of an intermediate ferric peroxide.39 However, we note that low-temperature activation of di-t-alkyl peroxides has been achieved with electron-rich copper complexes.40
The greatly enhanced reactivity observed from the combination of thiols and iron salts suggests the possible intermediacy of Fe/thiol complexes or similar intermediates possessing greater reducing potential relative to either individual reductant (eq 4).41 The combination of Fe(II) and cysteine has been reported to achieve a rapid reduction of artemisinin and related peroxides.23 However, whereas the peroxyacetal of artemisinin is sufficiently activated to react with Fe(II) in the absence of thiol,18,23 we observed reduction of several substrates inert toward either Fe(II) or thiophenolate.
| 4 |
The Fe/thiol-promoted reductions display a number of interesting features: (1) the active species can be regenerated by thiolate or hydride; (2) decomposition of a dialkyl peroxide requires only 1 equiv of thiol or hydride; and (3) the reductions of dialkyl peroxides are considerably less exothermic than what would be predicted by bond energies for a two-proton, two-electron reduction (eq 5).42
 |
5 |
The intermediacy of alkoxy radicals is strongly supported by the products derived from cleavage of probe molecule 8 (Scheme 4).2b,43 The much greater yield of acetone (87%) compared with 2-methyl-1-phenyl-2-propanol (14%) indicates that unimolecular scission of the alkoxide radical is much more rapid than bimolecular trapping by thiol.44 The preferential capture of the resulting benzyl radical to form thioether versus toluene suggests that the released carbon radical reacts more rapidly with catalyst than with thiol, paralleling observations made by Wu.23
Scheme 4. Evidence for an Alkoxy Radical Intermediate.

Relative yields in parentheses.
A possible mechanism, illustrated in Scheme 5, begins with Fenton-type cleavage of the peroxide by the presumed Fe/thiol complex to generate an oxidized complex, an alkoxide and an alkoxy radical. Selective oxidation at Fe has been observed in Fe–S complexes.45 Although we do not yet have good kinetic data, we note that under some of our most commonly employed conditions, decomposition of peroxide and thiol (each 0.34 M) in the presence of 3.4–17 mM Fe(II) is complete in less than a minute. Several species are likely capable of reduction of the oxidized intermediate: thiol or hydride, one or the other of which is present in stoichiometric amounts in most of the reductions described here; thiophenyl radical,46 available from the thiol from oxidation or via hydrogen abstraction by the alkoxy radical intermediate;47 or carbon radicals, whether derived from scission of the initially formed alkoxy radical or via attack of the alkoxy radical on a hydrogen atom donor (e.g., THF).48 Diphenyl disulfide, observed as a major product in every reaction employing stoichiometric thiol, was isolated in 70% yield from a preparative trial. However, the amounts of thiol consumed and disulfide produced are insufficient to explain reaction stoichiometry. Similarly, while we cannot rule out the oxidative dimerization or polymerization of thiyl radicals,46 we do not observe byproducts associated with these pathways. Scission of alkoxy radicals, including tert-butoxy radical, is well established in reactions of Fe(II) with hydroperoxides or peroxides, and carbon radicals could play a role here in the reduction of the putative Fe(III) intermediate.15a,49,50 Interestingly, the Fe(II)/cysteine-promoted decomposition of artemisinin was found to result in formation of thioethers derived by trapping of an intermediate carbon radical.22 We did not observed thiylated co-products, for example, 2-phenylthio tetrahydrofuran, from our model reductions; however, we did observe a significant amount of thioether upon reductive cleavage of alkoxy radical probe 8 (see Scheme 4).
Scheme 5. Proposed Mechanism for Fe/SR-Promoted Reductions.

The Fe/thiol catalyzed disproportionation of DADP to acetone presumably also begins with a Fenton-type cleavage (eq 6). The modest exothermicity of the overall process, as well as the requirement for only catalytic amounts of Fe and thiol, suggests the formation of a downstream species capable of reducing the oxidized catalyst. We suggest that the alkoxide/alkoxy radical derived from the Fenton reaction undergoes loss of acetone to generate an α-metallodioxy alkoxy radical which can regenerate the Fe(II)-catalyst via formation of acetone and oxygen. Our measured heat of reaction agrees closely with values reported for a thermal decomposition of DADP.51
 |
6 |
Conclusion
The limited reactivity of many peroxides toward reductive or nucleophilic cleavage can be useful in terms of storage, shipping, and the serum stability of peroxide-based antimalarial agents.52 However, this kinetic stability can create a challenge to decomposing hindered ozonides or peroxides,53 including peroxide explosives.27−29 The ability of Fe/thiol complexes to catalyze the rapid reduction or disproportionation of all but the most hindered of peroxides under mild conditions could have significant applications for controlled remediation of peroxide-based explosives or unwanted peroxides28−30 or as an approach for activation of peroxides in biological settings.22
Experimental Procedures
General Methods
All reactions were conducted in flame-dried glassware under an atmosphere of N2 except where indicated. Reagents and solvents were used as supplied commercially, except for THF, which was distilled from Na/benzophenone and CH2Cl2, acetonitrile, and pyridine, which were distilled from CaH2. Extracted organic layers were dried using sodium sulfate and filtered through a cotton plug. 1H and 13C spectra were acquired in CDCl3, d8-THF, or CD3CN at 400 MHz (1H) or 100 MHz (13C) unless noted otherwise. Chemical shifts are reported relative to residual chloroform (7.26 ppm for 1H and 77.0 ppm for 13C) and residual acetonitrile (1.96 ppm for 1H). IR spectra were obtained on neat films (ZnSe, attenuated total reflection mode); selected absorbances are reported in wavenumbers (cm–1). Flash column chromatography was performed on 230–400 μM silica gel. TLC was performed on 0.25 mm hard-layer silica G plates containing a fluorescent indicator; developed TLC plates were visualized with a hand-held UV lamp, or by heating after staining with either: a peroxide-sensitive solution prepared from 1.2 g N,N′-dimethyl-p-phenylenediamine monohydrochloride, 1 mL acetic acid, 20 mL of water, and 100 mL of methanol;34 or a general purpose dip composed of vanillin and sulfuric acid in ethanol. Abbreviations throughout: EA = ethyl acetate; Hex = hexane; DCM = dichloromethane. THF = tetrahydrofuran.
3-Phenylpropyl Trifluoromethanesulfonate1
![]() To a 0 °C solution of 3-phenyl-propanol
(2.51 g, 18.4
mmol) in 50 mL of dichloromethane (DCM) was added 5.21 g (19 mmol)
of trifluoromethanesulfonic (triflic) anhydride. Pyridine (1.48 g,
19.0 mmol) was then added over a period of 5 min, and the reaction
was allowed to stir for 30 min. The reaction was then quenched by
addition of 10 mL of cold hexane and 10 mL of cold 0.1 M aq KHSO4. The mixture was extracted with cold Hex (2 × 30 mL).
The combined organic extracts were dried and then concentrated under
reduced pressure, with the bath temperature held below 10 °C
during concentration. The crude product, a light to medium pink oil,
was used immediately and without further purification for peroxide
synthesis. The reagent can be held for a few hours at −20 °C
without excessive decomposition but should be used as soon as possible.
To a 0 °C solution of 3-phenyl-propanol
(2.51 g, 18.4
mmol) in 50 mL of dichloromethane (DCM) was added 5.21 g (19 mmol)
of trifluoromethanesulfonic (triflic) anhydride. Pyridine (1.48 g,
19.0 mmol) was then added over a period of 5 min, and the reaction
was allowed to stir for 30 min. The reaction was then quenched by
addition of 10 mL of cold hexane and 10 mL of cold 0.1 M aq KHSO4. The mixture was extracted with cold Hex (2 × 30 mL).
The combined organic extracts were dried and then concentrated under
reduced pressure, with the bath temperature held below 10 °C
during concentration. The crude product, a light to medium pink oil,
was used immediately and without further purification for peroxide
synthesis. The reagent can be held for a few hours at −20 °C
without excessive decomposition but should be used as soon as possible.
3-Phenylpropyl Methanesulfonate
![]() The methanesulfonate was prepared using a modification of
a reported procedure.54 To a room temperature
solution of 3-phenyl-propanol (2.00 g, 14.71 mmol) in 20 mL of DCM
was added triethylamine (2.97 g, 29.42 mmol) followed by 1.84 g (16.18
mmol) of methanesulfonyl chloride slowly over 2 min. The reaction
was allowed to stir for 2 h. The reaction was then quenched by addition
of aq 6 M HCl (10 mL). The mixture was extracted with ether (3 ×
30 mL). The combined organic extracts were dried and then concentrated
under reduced pressure. The crude product, a light to medium yellow
brown oil, was used immediately and without further purification for
peroxide synthesis. If further purification is needed, the oil can
be dissolved in a 70/30 mixture of ether/ethyl acetate (EA) and run
through a silica plug. Spectra matched those reported previously.55
The methanesulfonate was prepared using a modification of
a reported procedure.54 To a room temperature
solution of 3-phenyl-propanol (2.00 g, 14.71 mmol) in 20 mL of DCM
was added triethylamine (2.97 g, 29.42 mmol) followed by 1.84 g (16.18
mmol) of methanesulfonyl chloride slowly over 2 min. The reaction
was allowed to stir for 2 h. The reaction was then quenched by addition
of aq 6 M HCl (10 mL). The mixture was extracted with ether (3 ×
30 mL). The combined organic extracts were dried and then concentrated
under reduced pressure. The crude product, a light to medium yellow
brown oil, was used immediately and without further purification for
peroxide synthesis. If further purification is needed, the oil can
be dissolved in a 70/30 mixture of ether/ethyl acetate (EA) and run
through a silica plug. Spectra matched those reported previously.55
2-Hydroperoxytetrahydro-2H-pyran
![]() This hydroperoxyacetal was synthesized
using a known procedure.1 To a 0 °C
solution of hydrogen peroxide
(35% in H2O, 20 mL, ∼200 mmol) was added concentrated
H2SO4 (0.1 mL) slowly. After the solution had
been stirred for 10 min, 2.00 g of 3,4-dihydro-2H-pyran (25.88 mmol) was added slowly over 5 min. The reaction was
stirred for 4 h at 0 °C and then quenched with 10 mL of saturated
NH4Cl in H2O. The resulting mixture was extracted
using ether (3 × 20 mL). The combined organic extracts were washed
with H2O (20 mL), dried over a small amount of Na2SO4, and then concentrated under reduced pressure. The
residue was then purified by chromatography (5% EA/Hex) to yield 1.92
g (16.3 mmol, 63%) of a colorless oil. The spectra matched those previously
reported.1
This hydroperoxyacetal was synthesized
using a known procedure.1 To a 0 °C
solution of hydrogen peroxide
(35% in H2O, 20 mL, ∼200 mmol) was added concentrated
H2SO4 (0.1 mL) slowly. After the solution had
been stirred for 10 min, 2.00 g of 3,4-dihydro-2H-pyran (25.88 mmol) was added slowly over 5 min. The reaction was
stirred for 4 h at 0 °C and then quenched with 10 mL of saturated
NH4Cl in H2O. The resulting mixture was extracted
using ether (3 × 20 mL). The combined organic extracts were washed
with H2O (20 mL), dried over a small amount of Na2SO4, and then concentrated under reduced pressure. The
residue was then purified by chromatography (5% EA/Hex) to yield 1.92
g (16.3 mmol, 63%) of a colorless oil. The spectra matched those previously
reported.1
2-((3-Phenylpropyl)peroxy)tetrahydro-2H-pyran (1)
![]() Peroxyacetal 2 was
synthesized using a known
procedure.1 To a 0 °C solution of
KOtBu (2.24 g, 20.0 mmol) in 50 mL of THF was added
2.4 g (20.3 mmol) of the 2-hydroperoxytetrahydropyran (above), followed,
over 5 min, by addition of previously prepared 3-phenylpropyl trifluoromethanesulfonate
(∼18.4 mmol, added neat). The reaction was stirred for 4 h
and then quenched with 30 mL of water. The mixture was extracted with
EA (3 × 40 mL). The combined organic extracts were dried and
then concentrated under reduced pressure. The residue was then purified
by chromatography (20% EA/Hex) to yield 2.21 g (9.38 mmol, 51%) of
peroxyacetal 1. The 1H NMR spectra closely
matched those in a previous report:1Rf: 0.70 (10% EA/Hex); 1H NMR: δ
1.57–1.61 (overlapping peaks, 4H), 1.76 (m, 2H), 1.99 (m, 2H),
2.74 (t, 2H, J = 7.6), 4.02 (t, 1H, J = 11.6), 3.83 (t, 1H, J = 11.6), 4.14 (t, 2H, J = 6.4), 5.18 (t, 1H, J = 3.6), 7.21–7.30
(overlapping peaks, 5H).
Peroxyacetal 2 was
synthesized using a known
procedure.1 To a 0 °C solution of
KOtBu (2.24 g, 20.0 mmol) in 50 mL of THF was added
2.4 g (20.3 mmol) of the 2-hydroperoxytetrahydropyran (above), followed,
over 5 min, by addition of previously prepared 3-phenylpropyl trifluoromethanesulfonate
(∼18.4 mmol, added neat). The reaction was stirred for 4 h
and then quenched with 30 mL of water. The mixture was extracted with
EA (3 × 40 mL). The combined organic extracts were dried and
then concentrated under reduced pressure. The residue was then purified
by chromatography (20% EA/Hex) to yield 2.21 g (9.38 mmol, 51%) of
peroxyacetal 1. The 1H NMR spectra closely
matched those in a previous report:1Rf: 0.70 (10% EA/Hex); 1H NMR: δ
1.57–1.61 (overlapping peaks, 4H), 1.76 (m, 2H), 1.99 (m, 2H),
2.74 (t, 2H, J = 7.6), 4.02 (t, 1H, J = 11.6), 3.83 (t, 1H, J = 11.6), 4.14 (t, 2H, J = 6.4), 5.18 (t, 1H, J = 3.6), 7.21–7.30
(overlapping peaks, 5H).
(Peroxybis(propane-3,1-diyl))dibenzene (2)
![]() Dialkyl peroxide 2 was prepared
by a modification
of a reported procedure.54 To a 0 °C
solution of 3-phenylpropyl methanesulfonate (1.00 g, 4.07 mmol)1 in 50 mL of MeOH was added 1.65 g (18.68 mmol)
of aq H2O2 (35%, used as received), followed,
over 5 min, by 7.43 g (13.52 mmol) of KOH dissolved in water as a
aq 50% solution. The reaction was allowed to warm to room temperature
and then stirred for 4 h prior to quenching by addition of 20 mL of
3 M HCl. The crude reaction mixture was extracted with Hex (3 ×
40 mL). The combined organic extracts were dried and then concentrated
under reduced pressure. The residue was then purified by chromatography
(10% EA/Hex) to yield 0.20 g (0.74 mmol, 20%) of dialkyl peroxide 2 accompanied by varying amounts of the phenylpropyl hydroperoxide.
If desired, the latter can be reacted with additional methanesulfonate
to generate additional quantities of 2. Rf: 0.73 (10% EA/Hex); 1H NMR (400 MHz): δ
2.02 (m, 4H), 2.73 (t, 4H, J = 7.2), 4.07 (t, 4H, J = 6.4), 7.22–7.30 (overlapping peaks, 10H); 13C NMR: δ 29.28 (CH2), 32.15 (CH2), 76.36 (CH2), 126.10 (CH), 128.55 (CH), 141.60 (C);
HRMS (ESI): calcd for C18H22O2Na
[M + Na]+, 293.1517; found, 293.1516.
Dialkyl peroxide 2 was prepared
by a modification
of a reported procedure.54 To a 0 °C
solution of 3-phenylpropyl methanesulfonate (1.00 g, 4.07 mmol)1 in 50 mL of MeOH was added 1.65 g (18.68 mmol)
of aq H2O2 (35%, used as received), followed,
over 5 min, by 7.43 g (13.52 mmol) of KOH dissolved in water as a
aq 50% solution. The reaction was allowed to warm to room temperature
and then stirred for 4 h prior to quenching by addition of 20 mL of
3 M HCl. The crude reaction mixture was extracted with Hex (3 ×
40 mL). The combined organic extracts were dried and then concentrated
under reduced pressure. The residue was then purified by chromatography
(10% EA/Hex) to yield 0.20 g (0.74 mmol, 20%) of dialkyl peroxide 2 accompanied by varying amounts of the phenylpropyl hydroperoxide.
If desired, the latter can be reacted with additional methanesulfonate
to generate additional quantities of 2. Rf: 0.73 (10% EA/Hex); 1H NMR (400 MHz): δ
2.02 (m, 4H), 2.73 (t, 4H, J = 7.2), 4.07 (t, 4H, J = 6.4), 7.22–7.30 (overlapping peaks, 10H); 13C NMR: δ 29.28 (CH2), 32.15 (CH2), 76.36 (CH2), 126.10 (CH), 128.55 (CH), 141.60 (C);
HRMS (ESI): calcd for C18H22O2Na
[M + Na]+, 293.1517; found, 293.1516.
(3-(tert-Butylperoxy)propyl)benzene (3)
![]() To a 0 °C solution of KOtBu (2.24
g, 20.0 mmol) in 50 mL of THF was added 3.64 mL of t-butyl hydroperoxide as a solution in decane (nominally 5.5 M, 20.3
mmol). The previously prepared 3-phenylpropyl trifluoromethanesulfonate
(∼18.4 mmol)1 was added over a period
of 5 min and the reaction was allowed to stir for 4 h before being
quenched with 30 mL of water. The resulting mixture was extracted
using EA (3 × 40 mL). The combined organic extracts were dried
and then concentrated under reduced pressure. The residue was then
purified by chromatography (20% EA/Hex) to yield 1.88 g (9.02 mmol,
49%) of dialkyl peroxide 3. Rf: 0.71 (10% EA/Hex); 1H NMR: δ 1.29 (s, 9H), 1.98
(t, 2H, J = 7.4), 2.74 (t, 2H, J = 7.8), 4.00 (t, 2H, J = 6.4), 7.22–7.289
(overlapping peaks, 5H); 13C NMR: δ 26.5 (CH3), 29.7 (CH2), 32.53 (CH2), 74.33 (CH2), 80.19 (C), 125.96 (CH), 128.47 (CH), 128.53 (CH), 141.92
(C); IR: 2977, 2928, 1361. Spectra matched those in a previous report.1
To a 0 °C solution of KOtBu (2.24
g, 20.0 mmol) in 50 mL of THF was added 3.64 mL of t-butyl hydroperoxide as a solution in decane (nominally 5.5 M, 20.3
mmol). The previously prepared 3-phenylpropyl trifluoromethanesulfonate
(∼18.4 mmol)1 was added over a period
of 5 min and the reaction was allowed to stir for 4 h before being
quenched with 30 mL of water. The resulting mixture was extracted
using EA (3 × 40 mL). The combined organic extracts were dried
and then concentrated under reduced pressure. The residue was then
purified by chromatography (20% EA/Hex) to yield 1.88 g (9.02 mmol,
49%) of dialkyl peroxide 3. Rf: 0.71 (10% EA/Hex); 1H NMR: δ 1.29 (s, 9H), 1.98
(t, 2H, J = 7.4), 2.74 (t, 2H, J = 7.8), 4.00 (t, 2H, J = 6.4), 7.22–7.289
(overlapping peaks, 5H); 13C NMR: δ 26.5 (CH3), 29.7 (CH2), 32.53 (CH2), 74.33 (CH2), 80.19 (C), 125.96 (CH), 128.47 (CH), 128.53 (CH), 141.92
(C); IR: 2977, 2928, 1361. Spectra matched those in a previous report.1
tert-Butyl Benzoperoxoate (4)
![]() Perester 5 was
prepared by a known procedure.25 To a room
temperature solution of tetrabutyl
ammonium iodide (1.7 g, 4.6 mmol) in 40 mL of H2O under
a balloon of oxygen was added 1.00 g (9.2 mmol) of benzyl alcohol,
followed by 6.7 mL of t-butyl hydroperoxide solution
(nominally 5.5 M in decane, 37.0 mmol, all at once). The reaction
was allowed to stir for 16 h and then diluted with 30 mL of ether.
The separated aqueous layer was extracted with additional ether (2
× 30 mL) and the combined organic extracts were dried over Na2SO4. The filtrate was concentrated under reduced
pressure and the residue purified by chromatography (10% EA/Hex) to
yield 1.44 g (7.45 mmol, 81%) of perester 4. Rf: 0.63 (10% EA/Hex); 1H NMR: δ
1.44 (s, 9H), 1.98 (t, 2H, J = 7.4), 7.48 (t, 2H, J = 8), 7.61 (t, 1H, J = 8), 7.98 (d, 2H, J = 8); 13C NMR: δ 26.37 (CH3), 84.1 (C), 127.86 (CH), 128.74 (CH), 129.25 (CH), 133.46 (C); IR:
2981, 2936, 1754, 1189.
Perester 5 was
prepared by a known procedure.25 To a room
temperature solution of tetrabutyl
ammonium iodide (1.7 g, 4.6 mmol) in 40 mL of H2O under
a balloon of oxygen was added 1.00 g (9.2 mmol) of benzyl alcohol,
followed by 6.7 mL of t-butyl hydroperoxide solution
(nominally 5.5 M in decane, 37.0 mmol, all at once). The reaction
was allowed to stir for 16 h and then diluted with 30 mL of ether.
The separated aqueous layer was extracted with additional ether (2
× 30 mL) and the combined organic extracts were dried over Na2SO4. The filtrate was concentrated under reduced
pressure and the residue purified by chromatography (10% EA/Hex) to
yield 1.44 g (7.45 mmol, 81%) of perester 4. Rf: 0.63 (10% EA/Hex); 1H NMR: δ
1.44 (s, 9H), 1.98 (t, 2H, J = 7.4), 7.48 (t, 2H, J = 8), 7.61 (t, 1H, J = 8), 7.98 (d, 2H, J = 8); 13C NMR: δ 26.37 (CH3), 84.1 (C), 127.86 (CH), 128.74 (CH), 129.25 (CH), 133.46 (C); IR:
2981, 2936, 1754, 1189.
TATP (5)
Caution: TATP and
DADP (following entry) are shock-, friction-, heat-, and spark-sensitive
high explosives.26,28−30![]()
To acetone (1.2 g, 20 mmol) cooled to 0 °C was added 1.97 g (20 mmol) of 35% hydrogen peroxide solution. Then, 0.1 mL of concentrated HCl was added to the solution. The reaction was allowed to warm to room temperature and allowed to stir for 24 h, resulting in the formation of a white crystal. The filtered solid (vacuum filtration) was washed with cold H2O (3 × 10 mL) and then dried by pulling air through the product mass for 1 h. The resulting solid was allowed to air dry for 24 h before use. No further purification is needed. Yields: 0.49 g (2.2 mmol, 11%) of TATP 7. Rf: 0.74 (10% EA/Hex); 1H NMR (400 MHz): δ 1.49 (s, 18H); 13C NMR (100 MHz): δ 21.44 (CH3), 107.62 (C); IR: 2997, 2945, 1177. The spectra matched those in a previous report.26b
DADP (6)
Caution: See safety
note above.![]()
DADP was prepared by a modification of a reported procedure.26b To a stirred sample of acetone (1.2 g, 20 mmol) cooled to 0 °C was added 1.97 g (20 mmol) of 35% aq hydrogen peroxide used as received. The solution was diluted with water (8 mL) and concentrated H2SO4 (4 mL) was then added slowly (over 2 min interval). The reaction was allowed to warm up to room temperature and stirred for 48 h, resulting in the formation of a white crystalline solid. The vacuum filtered solid was washed with cold H2O (3 × 10 mL) and then tried by drawing air through the mass for 1 h. The solid was allowed to sit and dry in open air for 24 h, and was then used without further purification. Yield: 0.68 g (4.6 mmol, 23%) of DADP 6. Rf: 0.74 (10% EA/Hex); 1H NMR: δ 1.38 (s, 6H), 1.82 (s, 6H); 13C NMR: δ 20.65 (CH3), 22.55 (CH3), 107.67 (C); IR: 3000, 2295, 1196. The spectra matched a previous report.26b
t-Butyl 1-Phenyl-2-methyl-2-propyl peroxide (7)
t-Butyl 1-phenyl-2-methyl-2-propyl peroxide (7) was prepared using a reported procedure.2b Spectra matched those previously reported.
Methyl 1-Phenyl-2-methyl-2-propyl peroxide (8)
Methyl 1-phenyl-2-methyl-2-propyl peroxide (8) was
prepared using a variation on reported procedures.30,43
1-Phenyl-2-methyl-2-propyl hydroperoxide was prepared through an adaptation of reported procedures:30,43 1-phenyl-2-methyl-2-propanol (3.1 mL, 20 mmol, 1 equiv), hydrogen peroxide (50 v/v % H2O, 2.3 mL, 40 mmol, 2 equiv), and phosphoric acid (85%, 5.8 mL, 100 mmol, 5 equiv) were combined in a round-bottom flask backfilled with nitrogen. The reaction was heated to 45 °C for 5 h at which point the reaction was deemed complete by disappearance of starting material (TLC). The reaction mixture was cooled to room temperature and diluted with Hex (150 mL). The separated organic layer was washed with deionized water (2 × 10 mL) and brine (1 × 10 mL), dried over sodium sulfate, and concentrated in vacuo. The resulting thick and dark oil was purified by silica column chromatography (1.9 × 30.5 cm) with 5% EA in Hex to furnish, following concentration in vacuo, 2.192 g (66%) of the hydroperoxide as a light yellow oil. Rf: 0.40 (10% EA/Hex); IR (thin film): 3399 (br), 2981 (m), 1453 (s) cm–1; 1H NMR: δ 1.22 (s, 6H), 2.90 (s, 2H), 7.24 (br m, 5H); 13C NMR: δ 24.0, 44.6, 83.2, 126.4, 128.1, 130.6, 137.7.
The tertiary hydroperoxide (0.3329 g, 2.0 mmol, 1 equiv), methyl iodide (0.37 mLl, 6 mmol, 3 equiv), and THF (10 mL, 0.2 M) were combined in a flame-dried round-bottom flask backfilled with nitrogen. Potassium tert-butoxide (0.2470 g, 2.2 mmol, 1.1 equiv) was added to the clear solution, and the resulting reaction was stirred at room temperature until the starting material could no longer be observed (TLC, 18 h). The reaction was quenched with saturated NH4Cl (50 mL) and extracted with EA (3 × 10 mL). The combined organic layers were washed with brine (1 × 10 mL), dried over sodium sulfate, and concentrated in vacuo to give an oily residue. The mixture was purified by column chromatography (1.9 × 20 cm) with 1% EA in Hex to furnish, following concentration in vacuo, 0.2389 g (66%) of the dialkyl peroxide as a clear oil which smelled lightly of apples/pears. Rf: 0.78 (10% EA/Hex); IR (thin film): 2980 (s), 2929 (s), 2891 (s), 1464 (s) cm–1; 1H NMR: δ 1.19 (s, 6H), 2.87 (s, 2H), 3.85 (s, 3H), 7.23 (br m, 5H); 13C NMR: δ 24.3, 45.0, 63.1, 82.6, 126.3, 128.0, 130.7, 137.9; HRMS-EI: calcd for C11H16O2 (M)+, 180.1150; found, 180.1156.
Reduction Procedure Using Thiolate Generated via Deprotonation
This example describes the reduction of peroxide 3; the same procedure was applied to other substrates: into 10 mL of THF is dissolved 0.118 g (1.06 mmol) of KOtBu, or, alternatively, 0.107 g (1.06 mmol) of Et3N. To this solution 0.116 g (1.06 mmol) of phenyl thiol is added, followed by 0.201 g (0.964 mmol) of peroxide 3. The reaction was monitored by TLC for disappearance of peroxide using N,N′-dimethyl-p-phenylenediamine as a redox-active indicator;34 for most substrates, progress of reactions could also be followed using more conventional oxidizing TLC indicators.
Control Reactions with Base
Control reactions with base employed the same procedure outlined above for reactions with thiolate and base but omitted the thiol. Reactions were monitored by TLC and/or NMR.
Reductive Generation of Thiolate
This example describes the reduction of peroxide 3; the same procedure was applied to other substrates: in 10 mL of EtOH, 0.230 g (1.056 mmol) of diphenyl sulfide is dissolved. Then, 0.72 mL (1.44 mmol) of a solution of LiBH4 in THF (nominally 2 M) is added slowly. Once bubbling ceased, 0.201 g (0.964 mmol) of dialkyl peroxide 3 was added slowly. Disappearance of peroxide was monitored by TLC as described previously.34
Control Reaction Using Only Peroxide and LiBH4
Control reaction using only peroxide and LiBH4 was accomplished as in the preceding example except that the disulfide was omitted.
General Thiol/Fe Reduction Procedure
The procedure is exemplified for the reduction of peroxide 3.
To a solution of 0.0051 g (0.024 mmol) of FeBr2 in 5 mL of THF is added 0.10 g (0.48 mmol) dialkyl peroxide 3. At this point, the solution is typically clear with a light orange to light red coloration. Addition of 0.052 mL (0.48 mmol) of thiophenol results in the immediate formation of a black colloidal suspension. The reaction can be followed by disappearance of peroxide substrate (TLC) as described previously;34 in most cases, the reactions are complete in less than 60 s. The solutions from reactions employing stoichiometric thiol, even if remaining dark and opaque upon disappearance of substrate (TLC), will clear to a light-to-dark green solution within a few minutes.
Reactions in CD3CN were conducted in an identical manner but were monitored by both TLC and by 1H NMR of crude aliquots.
Thiol/Fe Reduction of TATP
To a solution of 0.0100 g (0.047 mmol) of FeBr2 in 3 mL of CD3CN was added 0.110 g (0.47 mmol) of TATP (5), followed by addition of 0.160 g (1.41 mmol) of phenyl thiol. The black suspension was allowed to stir for 3 days after which 0.043 g (0.470 mmol) of toluene is added as an internal standard. An aliquot drawn from the solution was then analyzed by NMR.
Control Reaction Using Only Peroxide and Iron
A control reaction of FeBr2 and peroxide was conduced as for the Fe/thiol procedures described above except that thiol was omitted. Reactions were monitored by TLC, as described previously, and/or NMR.
General Procedure for Fe/Thiol-Promoted Reduction of DADP
To a solution of 0.0072 g (0.034 mmol) of FeBr2 in 2 mL of CD3CN is added 0.10 g (0.68 mmol) of DADP (6), followed by addition of 0.129 g (1.35 mmol) of thiophenol. The reaction is allowed to sit for 3 min, and then, 0.062 g (0.6755 mmol) of toluene is added. The entire solution is then analyzed by 1H NMR, comparing the integration of the signals for toluene (2.33 ppm) versus acetone (2.08 ppm).
Radical Clock Experiment (See Also Scheme 4)
The general thiol/Fe reduction procedure was applied to a solution of peroxide 8 (0.100 g, 0.562 mmol) in CD3CN (3.0 mL). 1H NMR was recorded on the crude reaction mixture. Four major products were observed (Figure S4). As a first approximation, we assume that all products are derived from the alkoxy radical, either through hydrogen atom transfer or reduction (to form the alcohol) or fragmentation to afford acetone, plus either toluene or the benzyl phenyl sulfane (see Scheme 4). The tertiary alcohol was quantified by isolation; we note that the isolated yield and the relative yield calculated from the NMR assay are very similar. With the exception of acetone, the products could also be detected by GC/MS (not shown).
-
(1)
2-Methyl-1-phenylpropan-2-ol (14%), which has been previously characterized,56 was quantified by the 1H NMR singlets at 2.74 (2H) and 1.15 ppm (6H). This product was also isolated (extraction, chromatography) in 13% yield.
-
(2)
Acetone (87%) was quantified by the 1H NMR singlet (6H) at 2.10 ppm.
-
(3)
Toluene (24%) was quantified by the 1H NMR singlet (3H) at 2.35 ppm.
-
(4)
The benzyl (phenyl) sulfane (62%), which has been previously characterized,57 was quantified by the integration of the 1H NMR singlet (2H), at 4.19 ppm.
Thiol/Fe Catalyzed Reduction Using Added Hydride
To a solution of 0.005 g (0.024 mmol) of FeBr2 in 10 mL of THF is added 0.050 g (0.24 mmol) of dialkyl peroxide 3 (transferred from preweighed vial in minimum amount of THF) followed by 0.48 mL (0.024 mmol) of phenyl thiol in THF stock solution (nominally 0.05 M) and last 0.24 mL (0.24 mmol) of a solute of DIBAL in THF (nominally 1.0 M). The resulting light red/light green reaction was allowed to sir for 5 min, during which time the solution became translucent with very light red/green color. TLC analysis confirmed the disappearance of the peroxide and the formation of 3-phenyl-1-propanol.
Reaction Calorimetry for Fe/SPh-Catalyzed Decomposition of DADP (6)
Caution: see notes above related to dangers of the acetone peroxides.
In a glass vial with a septum cap is placed 0.021 g (0.10 mmol) of FeBr2 and 0.150 g (1.01 mmol) of DADP 6. The vial is placed under an atmosphere of N2 and then wrapped with several layers of a PIG cloth mat. THF (4.0 mL) is added to the vial. A metal temperature probe (Fisher Scientific, Traceable, uncalibrated) is inserted through the septum into the solution. After the solution temperature has stabilized (22.1 °C), a solution of 0.011 g (0.101 mmol) of phenyl thiol in 1 mL THF is added to the solution all at once, resulting in an observed temperature rise to 27.2 °C. This value corresponded to a heat of reaction of 15.4 kcal/mol, which was rounded to 15 kcal/mol given experimental uncertainties.
Reaction Calorimetry for Fe/SR Cleavage of a Dialkyl Peroxide in THF
Calorimetry was conducted as in the previous experiment except that dialkyl peroxide 3 (0.208 g, 1.00 mmol) was used as the peroxide substrate and the reaction vial was wrapped with a 150 mm thickness of foam covering every part of the vial except for the septum. A temperature rise from 24.5 to 32.7 °C was observed over a period of 3.0 min.
Reaction Calorimetry of Fe/SR Cleavage of a Dialkyl Peroxide in CH3CN
Calorimetry was conducted as previously using dialkyl peroxide 4 (0.200 g, 0.961 mmol) and acetonitrile as solvent. A temperature rise from 22.9 to 33.6 °C was observed over a 3.0 min period.
Acknowledgments
This work was supported by NSF (CHE 1464914) and carried out in facilities remodeled with support from NIH (RR016544). We thank the following individuals for technical assistance and/or useful discussions: B. Cheung, A. Diepenbrock, M. Morton, J. Redepenning, and J. Takacs (Chemistry, University of Nebraska–Lincoln); J. Vennerstrom (Pharmaceutical Sciences, Univ. of Nebraska-Medical Center).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01977.
Reaction progress monitoring (visual and NMR); 1H and 13C NMR spectra of radical probe 8 and the precursor hydroperoxide; 1H NMR of crude reaction mixture from reduction of radical probe; and 1H NMR spectra of reaction products from reduction of substrates 1–4 (PDF)
The authors declare no competing financial interest.
Notes
Note on Safety: Although no safety issues were encountered in the course of this work, any preparative work with peroxides, particularly known explosives such as the acetone peroxides, should be conducted with an awareness of the potential for spontaneous and exothermic decompositions.27−30,58 The reader is directed to a digital collection related to peroxide safety: https://digitalcommons.unl.edu/chemistryperoxides/.
Supplementary Material
References
- a Kyasa S.; Meier R. N.; Pardini R. A.; Truttmann T. K.; Kuwata K. T.; Dussault P. H. Synthesis of Ethers via Reaction of Carbanions and Monoperoxyacetals. J. Org. Chem. 2015, 80, 12100–12114. 10.1021/acs.joc.5b02043. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Willand-Charnley R.; Puffer B. W.; Dussault P. H. Oxacycle Synthesis via Intramolecular Reaction of Carbanions and Peroxides. J. Am. Chem. Soc. 2014, 136, 5821–5823. 10.1021/ja5026276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Donkers R. L.; Maran F.; Wayner D. D. M.; Workentin M. S. Kinetics of the Reduction of Dialkyl Peroxides. New Insights into the Dynamics of Dissociative Electron Transfer. J. Am. Chem. Soc. 1999, 121, 7239–7248. 10.1021/ja9906148. [DOI] [Google Scholar]; b Magri D. C.; Workentin M. S. Model dialkyl peroxides of the Fenton mechanistic probe 2-methyl-1-phenyl-2-propyl hydroperoxide (MPPH): kinetic probes for dissociative electron transfer. Org. Biomol. Chem. 2003, 1, 3418–3429. 10.1039/b305348b. [DOI] [PubMed] [Google Scholar]
- a Dai P.; Dussault P. H.; Trullinger T. K. Magnesium/methanol: An effective reducing agent for peroxides. J. Org. Chem. 2004, 69, 2851–2852. and references within 10.1021/jo035191d. [DOI] [PubMed] [Google Scholar]; b Jin H.-X.; Liu H.-H.; Zhang Q.; Wu Y. On the Susceptibility of Organic Peroxy Bonds to Hydride Reduction. J. Org. Chem. 2005, 70, 4240–4247. 10.1021/jo050139y. [DOI] [PubMed] [Google Scholar]
- Winterbourn C. C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- Trujillo M.; Alvarez B.; Radi R. One- and two-electron oxidation of thiols: mechanisms, kinetics and biological fates. Free Radical Res. 2016, 50, 150–171. 10.3109/10715762.2015.1089988. [DOI] [PubMed] [Google Scholar]
- a Murray R. W. Chemistry of dioxiranes. 12. Dioxiranes. Chem. Rev. 1989, 89, 1187–1201. 10.1021/cr00095a013. [DOI] [Google Scholar]; b Shi L.; Zhao R.; Chang D. Recent Advances in Cyclic Diacyl Peroxides: Reactivity and Selectivity Enhancement Brought by the Cyclic Structure. Synthesis 2017, 49, 3357–3365. 10.1055/s-0036-1588458. [DOI] [Google Scholar]
- Rietjens I. M. C. M.; Lemmink H. H.; Alink G. M.; Van Bladeren P. J. The role of glutathione and glutathione S-transferases in fatty acid ozonide detoxification. Chem. Biol. Interact. 1987, 62, 3–14. 10.1016/0009-2797(87)90075-5. [DOI] [PubMed] [Google Scholar]
- Dussault P. H.; Denise George A.; Trullinger T. K. Peroxides as oxidative enzyme inhibitors: mechanism-based inhibition of a cysteine protease by an amino acid ozonide. Bioorg. Med. Chem. Lett. 1999, 9, 3255–3258. 10.1016/s0960-894x(99)00563-6. [DOI] [PubMed] [Google Scholar]
- Yang Y.; Cheng J.-Z.; Singhal S. S.; Saini M.; Pandya U.; Awasthi S.; Awasthi Y. C. Role of Glutathione S-Transferases in Protection against Lipid Peroxidation. J. Biol. Chem. 2001, 276, 19220–19230. 10.1074/jbc.m100551200. [DOI] [PubMed] [Google Scholar]
- Perkins A.; Nelson K. J.; Parsonage D.; Poole L. B.; Karplus P. A. Peroxiredoxins: guardians against oxidative stress and modulators of peroxide signaling. Trends Biochem. Sci. 2015, 40, 435–445. 10.1016/j.tibs.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas P.; Fleury E.; Estur J.-F.; Lapinte V.; Robin J.-J. Peroxide-Grafted PDMS: Hydrosilylation Reaction and Thiol-Ene Chemistry as an Alternative Pathway. Macromol. Chem. Phys. 2009, 210, 1933–1941. 10.1002/macp.200900310. [DOI] [Google Scholar]
- Griesbaum K.; Oswald A. A.; Naegele W. Polymeric Peroxide of 2,5-Dimethyl-2,4-hexadiene and a New Selective Reduction of Its Peroxide Linkage. J. Org. Chem. 1964, 29, 1887–1892. 10.1021/jo01030a054. [DOI] [Google Scholar]
- Mayo F. R.; Miller A. A. Oxidation of Unsaturated Compounds. II. Reactions of Styrene Peroxide. J. Am. Chem. Soc. 1956, 78, 1023–1034. 10.1021/ja01586a043. [DOI] [Google Scholar]
- Winterbourn C. C. Toxicity of iron and hydrogen peroxide: the Fenton reaction. Toxicol. Lett. 1995, 82–83, 969–974. 10.1016/0378-4274(95)03532-x. [DOI] [PubMed] [Google Scholar]
- a Kochi J. K. Chemistry of alkoxy radicals: Cleavage reactions. J. Am. Chem. Soc. 1962, 84, 1193–1197. 10.1021/ja00866a026. [DOI] [Google Scholar]; b Ivanov S. K.; Kropf H.. Metal-Catalyzed and Induced Reactions. Houben Weyl Methoden Der Organischen Chemie, 4th ed.; Georg Thieme Verlag: Stuttgart, 1988; Vol. E13, pp 1029–1104 (German). [Google Scholar]
- Labeque R.; Marnett L. J. 10-Hydroperoxy-8,12-octadecadienoic acid. A diagnostic probe of alkoxyl radical generation in metal-hydroperoxide reactions. J. Am. Chem. Soc. 1987, 109, 2828–2829. 10.1021/ja00243a048. [DOI] [Google Scholar]
- a Moane S.; Raftery D. P.; Smyth M. R.; Leonard R. G. Decomposition of peroxides by transition metal ions in anaerobic adhesive cure chemistry. Int. J. Adhes. Adhes. 1999, 19, 49–57. 10.1016/s0143-7496(98)00056-6. [DOI] [Google Scholar]
- O’Neill P. M.; Chadwick J.; Rawe S. L.. Biomimetic Fe(II) chemistry and synthetic studies on antimalarial and antitumour endoperoxides. In The Chemistry of Peroxides; Rappoport Z., Ed.; The Chemistry of Functional Groups; John Wiley & Sons: Chichester, 2006; Vol. 2, pp 1279–1346. [Google Scholar]
- a Wieland H.; Chrometzka F. Die katalytische Zersetzung von Diäthylperoxyd durch Eisen. Ber. Dtsch. Chem. Ges. B 1930, 63, 1028–1032. 10.1002/cber.19300630505. [DOI] [Google Scholar]; b Turner J. A.; Herz W. Iron(II)-induced decomposition of unsaturated cyclic peroxides derived from butadienes. A simple procedure for synthesis of 3-alkylfurans. J. Org. Chem. 1977, 42, 1900–1904. 10.1021/jo00431a016. [DOI] [Google Scholar]
- a Zhao J.; Fang H.; Zhou W.; Han J.; Pan Y. Iron-Catalyzed Cross-Dehydrogenative Coupling Esterification of Unactive C(sp3)-H Bonds with Carboxylic Acids for the Synthesis of α-Acyloxy Ethers. J. Org. Chem. 2014, 79, 3847–3855. 10.1021/jo500192h. [DOI] [PubMed] [Google Scholar]; b Niu Y.-N.; Xia X.-F.; Yuan Y. Metal-Free Cascade Methylation/Cyclization of N-Alkyl-N-methacryloylbenzamides with Dicumyl Peroxide. Synlett 2018, 29, 617–620. 10.1055/s-0036-1591522. [DOI] [Google Scholar]
- Myers T. N.Radical Initiators. Kirk-Othmer Encyclopedia of Chemical Technology, 5th ed.; Wiley-Interscience: Hoboken, NJ, 2005; Vol. 14, pp 274–311. [Google Scholar]
- Gaschler M. M.; Andia A. A.; Liu H.; Csuka J. M.; Hurlocker B.; Vaiana C. A.; Heindel D. W.; Zuckerman D. S.; Bos P. H.; Reznik E.; Ye L. F.; Tyurina Y. Y.; Lin A. J.; Shchepinov M. S.; Chan A. Y.; Peguero-Pereira E.; Fomich M. A.; Daniels J. D.; Bekish A. V.; Shmanai V. V.; Kagan V. E.; Mahal L. K.; Woerpel K. A.; Stockwell B. R. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat. Chem. Biol. 2018, 14, 507–515. 10.1038/s41589-018-0031-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Wu Y.; Yue Z.-Y.; Wu Y.-L. Interaction of Qinghaosu (Artemisinin) with Cysteine Sulfhydryl Mediated by Traces of Non-Heme Iron. Angew. Chem., Int. Ed. 1999, 38, 2580–2582. . [DOI] [PubMed] [Google Scholar]; b Liu H.-H.; Wu Y.-K.; Shen X. Transfer of Alkylation of Sulfur Ligand in Cysteinate-Iron Chelates by a 1, 2, 4, 5-Tetraoxane. Chin. J. Chem. 2003, 21, 875–877. 10.1002/cjoc.20030210731. [DOI] [Google Scholar]; c Liu H.-H.; Jin H.-X.; Wu Y.-K. Synthesis and cleavage studies of a 1, 2-dioxolane-type peroxide. Chin. J. Chem. 2010, 22, 1029–1033. 10.1002/cjoc.20040220930. [DOI] [Google Scholar]
- Milas N. A.; Peeler R. L. Jr.; Mageli O. L. Organic Peroxides. XIX. α-Hydroperoxyethers and Related Peroxides. J. Am. Chem. Soc. 1954, 76, 2322–2325. 10.1021/ja01638a012. [DOI] [Google Scholar]
- Zhang H.; Dong D.-Q.; Hao S.-H.; Wang Z.-L. Bu4NI-catalyzed construction of tert-butyl peresters from alcohols. RSC Adv. 2016, 6, 8465–8468. 10.1039/c5ra27500h. [DOI] [Google Scholar]
- a Matyáš R.; Pachman J. Study of TATP: influence of reaction conditions on product composition. Propellants, Explos. Pyrotech. 2010, 35, 31–37. 10.1002/prep.200800044. [DOI] [Google Scholar]; b Oxley J. C.; Smith J. L.; Bowden P. R.; Rettinger R. C. Factors influencing triacetone triperoxide (TATP) and diacetone diperoxide (DADP) formation: Part I. Propellants, Explos. Pyrotech. 2013, 38, 244–254. 10.1002/prep.201200116. [DOI] [Google Scholar]; c Oxley J. C.; Smith J. L.; Steinkamp L.; Zhang G. Factors influencing triacetone triperoxide (TATP) and diacetone diperoxide (DADP) formation: part 2. Propellants, Explos. Pyrotech. 2013, 38, 841–851. 10.1002/prep.201200215. [DOI] [Google Scholar]
- Bellamy A. J. Triacetone triperoxide: its chemical destruction. J. Forensic Sci. 1999, 44, 14517J. 10.1520/jfs14517j. [DOI] [Google Scholar]
- a Oxley J. C.; Smith J. L.; Huang J.; Luo W. Destruction of peroxide explosives. J. Forensic Sci. 2009, 54, 1029–1033. 10.1111/j.1556-4029.2009.01130.x. [DOI] [PubMed] [Google Scholar]; b Oxley J. C.; Smith J. L.; Brady J. E.; Steinkamp L. Factors influencing destruction of triacetone triperoxide (TATP). Propellants, Explos. Pyrotech. 2014, 39, 289–298. 10.1002/prep.201300063. [DOI] [Google Scholar]
- a Pachman J.; Matyáš R. Study of TATP: Stability of TATP Solutions. Forensic Sci. Int. 2011, 207, 212–214. 10.1016/j.forsciint.2010.10.010. [DOI] [PubMed] [Google Scholar]; b Matyáš R.; Šelešovský J.; Musil T. Decreasing the Friction Sensitivity of TATP, DADP, and HMTD. Cent. Eur. J. Energ. Mater. 2013, 10, 263–275. [Google Scholar]; c Oxley J.; Smith J.. Peroxide Explosives. In Detection and Disposal of Improvised Explosives; Schubert H., Kuznetsov A., Eds.; Springer: Netherlands, 2006; pp 113–121. [Google Scholar]
- Kropf H.; Bernert C.-R. Organische Peroxide, VII. Verhalten von Phenyl- und Benzyl-tert.-butylhydroperoxiden bei thermischer Zersetzung, Hock- und Criegee-Umlagerung. Liebigs Ann. Chem. 1971, 751, 109–120. 10.1002/jlac.19717510113. [DOI] [Google Scholar]
- a Poole L. B. The Basics of Thiols and Cysteines in Redox Biology and Chemistry. Free Radical Biol. Med. 2015, 80, 148–157. 10.1016/j.freeradbiomed.2014.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Semenov S. N.; Kraft L. J.; Ainla A.; Zhao M.; Baghbanzadeh M.; Campbell V. E.; Kang K.; Fox J. M.; Whitesides G. M. Autocatalytic, bistable, oscillatory networks of biologically relevant organic reactions. Nature 2016, 537, 656–660. 10.1038/nature19776. [DOI] [PubMed] [Google Scholar]
- Jocelyn P. C. Chemical reduction of disulfides. Methods Enzymol. 1987, 143, 246–256. 10.1016/0076-6879(87)43048-6. [DOI] [PubMed] [Google Scholar]
- Liotta D.; Sunay U.; Santiesteban H.; Markiewicz W. Phenyl selenide anion, a superior reagent for the SN2 cleavage of esters and lactones. J. Org. Chem. 1981, 46, 2605–2610. 10.1021/jo00326a001. [DOI] [Google Scholar]
- Smith L. L.; Hill F. L. Detection of sterol hydroperoxides on thin-layer chromatoplates by means of the Wurster dyes. J. Chromatogr. A 1972, 66, 101–109. 10.1016/s0021-9673(01)82933-2. [DOI] [PubMed] [Google Scholar]
- Yaremenko I. A.; Vil’ V. A.; Demchuk D. V.; Terent’ev A. O. Rearrangements of organic peroxides and related processes. Beilstein J. Org. Chem. 2016, 12, 1647–1748. 10.3762/bjoc.12.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sato R.; Kimura T. Variation 2: Oxidation with Peroxides. Sci. Synth. 2008, 39, 577. 10.1055/sos-SD-039-00665. [DOI] [Google Scholar]; b Crane D.; Häussinger D.; Graf P.; Sies H. Decreased flux through pyruvate dehydrogenase by thiol oxidation during t-butyl hydroperoxide metabolism in perfused rat liver. Hoppe Seyler’s Z. Physiol. Chem. 1983, 364, 977–988. 10.1515/bchm2.1983.364.2.977. [DOI] [PubMed] [Google Scholar]
- Kropf H.durch Reduktion. Houben Weyl Methoden Der Organischen Chemie, 4th ed.; Kropf H., Ed.; Georg Thieme Verlag: Stuttgart, 1988; Vol. E13, pp 1102–1116 (German). [Google Scholar]
- a Fidler Albo R. L.; Legron T.; Gittings M. J.; Elie M. R.; Holland Saitta E.; Sigman M. E.; Geiger C. L.; Clausen C. Degradation of triacetone triperoxide (TATP) using mechanically alloyed Mg/Pd. Propellants, Explos. Pyrotech. 2009, 35, 100–104. 10.1002/prep.200900011. [DOI] [Google Scholar]; b Clausen C. III; Geiger C. L.; Sigman M.; Fidler R.. Safe, in situ methodologies for the destruction of triacetone triperoxide and other explosive peroxides. U.S. Patent 8,062,442 B1, Nov 22, 2011.
- a Zhu N.; Zhao J.; Bao H. Iron catalyzed methylation and ethylation of vinyl arenes. Chem. Sci. 2017, 8, 2081–2085. 10.1039/c6sc04274k. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Uchiyama N.; Shirakawa E.; Nishikawa R.; Hayashi T. Iron-catalyzed oxidative coupling of arylboronic acids with benzene derivatives through homolytic aromatic substitution. Chem. Commun. 2011, 47, 11671–11673. 10.1039/c1cc14694g. [DOI] [PubMed] [Google Scholar]
- Gephart R. T. III; McMullin C. L.; Sapiezynski N. G.; Jang E. S.; Aguila M. J. B.; Cundari T. R.; Warren T. H. Reaction of CuI with Dialkyl Peroxides: CuII-Alkoxides, Alkoxy Radicals, and Catalytic C-H Etherification. J. Am. Chem. Soc. 2012, 134, 17350–17353. 10.1021/ja3053688. [DOI] [PubMed] [Google Scholar]
- a Krebs B.; Henkel G. Transition-Metal Thiolates: From Molecular Fragments of Sulfidic Solids to Models for Active Centers in Biomolecules. Angew. Chem., Int. Ed. 1991, 30, 769–788. 10.1002/anie.199107691. [DOI] [Google Scholar]; b Cahiez G.; Gager O.; Buendia J.; Patinote C. Iron thiolate complexes: efficient catalysts for coupling alkenyl halides with alkyl Grignard reagents. Chem.—Eur. J. 2012, 18, 5860–5863. 10.1002/chem.201200184. [DOI] [PubMed] [Google Scholar]
- Luo Y.-R.Handbook of Bond Dissociation Energies in Organic Compound; CRC Press: Boca Raton, 2002; p 392. [Google Scholar]
- Arends I. W. C. E.; Ingold K. U.; Wayner D. D. M. A Mechanistic Probe for Oxygen Activation by Metal Complexes and Hydroperoxides and Its Application to Alkane Functionalization by [FeIIICl2tris(2-pyridinylmethyl)amine]+ BF4–. J. Am. Chem. Soc. 1995, 117, 4710–4711. 10.1021/ja00121a031. [DOI] [Google Scholar]
- Dénès F.; Pichowicz M.; Povie G.; Renaud P. Thiyl radicals in organic synthesis. Chem. Rev. 2014, 114, 2587–2693. 10.1021/cr400441m. [DOI] [PubMed] [Google Scholar]
- McDonald A. R.; Bukowski M. R.; Farquhar E. R.; Jackson T. A.; Koehntop K. D.; Seo M. S.; De Hont R. F.; Stubna A.; Halfen J. A.; Münck E.; Nam W.; Que L. Jr. Sulfur versus Iron Oxidation in an Iron–Thiolate Model Complex. J. Am. Chem. Soc. 2010, 132, 17118–17129. 10.1021/ja1045428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsuchida E.; Nishide H.; Yamamoto K.; Yoshida S. Electrooxidative polymerization of thiophenol to yield poly(p-phenylene sulfide). Macromolecules 1987, 20, 2315–2316. 10.1021/ma00175a046. [DOI] [Google Scholar]
- Denisov E.; Chatgilialoglu C.; Shestakov A.; Denisova T. Rate constants and transition-state geometry of reactions of alkyl, alkoxyl, and peroxyl radicals with thiols. Int. J. Chem. Kinet. 2009, 41, 284–293. 10.1002/kin.20404. [DOI] [Google Scholar]
- Paul H.; Small R. D. Jr.; Scaiano J. C. Hydrogen abstraction by tert-butoxy radicals. A laser photolysis and electron spin resonance study. J. Am. Chem. Soc. 1978, 100, 4520–4527. 10.1021/ja00482a033. [DOI] [Google Scholar]
- a Guan H.; Sun S.; Mao Y.; Chen L.; Lu R.; Huang J.; Liu L. Iron(II)-Catalyzed Site-Selective Functionalization of Unactivated C(sp3 )–H Bonds Guided by Alkoxyl Radicals. Angew. Chem., Int. Ed. 2018, 57, 11413–11417. 10.1002/anie.201806434. [DOI] [PubMed] [Google Scholar]; b Cekovic Z.; Green M. M. Formation of remote double bonds by ferrous sulfate-cupric acetate promoted decomposition of alkyl hydroperoxides. J. Am. Chem. Soc. 1974, 96, 3000–3002. 10.1021/ja00816a059. [DOI] [Google Scholar]
- Wieland H.; Chrometzka F. Die katalytische Zersetzung von Diäthylperoxyd durch Eisen. Ber. Dtsch. Chem. Ges. 1930, 63, 1028–1032. 10.1002/cber.19300630505. [DOI] [Google Scholar]
- a Sinditskii V. P.; Kolesov V. I.; Egorshev V. Y.; Patrikeev D. I.; Dorofeeva O. V. Thermochemistry of cyclic acetone peroxides. Thermochim. Acta 2014, 585, 10–15. 10.1016/j.tca.2014.03.046. [DOI] [Google Scholar]; b Acetone—NIST Chemistry Webbook, SRD 69. https://webbook.nist.gov/cgi/cbook.cgi?ID=67641# (accessed August 2018).
- O’Neill P. M.; Barton V. E.; Ward S. A. The Molecular Mechanism of Action of Artemisinin—The Debate Continues. Molecules 2010, 15, 1705–1721. 10.3390/molecules15031705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher T. J.; Dussault P. H. Alkene ozonolysis. Tetrahedron 2017, 73, 4233–4258. 10.1016/j.tet.2017.03.039. [DOI] [Google Scholar]
- Welch F.; Williams H. R.; Mosher H. S. Organic Peroxides. IV. Higher Dialkyl Peroxides. J. Am. Chem. Soc. 1955, 77, 551–554. 10.1021/ja01608a011. [DOI] [Google Scholar]; Williams H. R.; Mosher H. S. Organic Peroxides. II. Secondary Alkyl Hydroperoxides. J. Am. Chem. Soc. 1954, 76, 2987–2990. 10.1021/ja01640a037. [DOI] [Google Scholar]
- Defoin A.; Albrecht S.; Tarnus C. Simple preparation of O-substituted hydroxylamines from alcohols. Synthesis 2006, 1635–1638. 10.1055/s-2006-926440. [DOI] [Google Scholar]
- a Li C.-C.; Dai X.-J.; Wang H.; Zhu D.; Gao J.; Li C.-J. Iron-Catalyzed Nucleophilic Addition Reaction of Organic Carbanion Equivalents via Hydrazones. Org. Lett. 2018, 20, 3801–3805. 10.1021/acs.orglett.8b01391. [DOI] [PubMed] [Google Scholar]; b Alonso E.; Guijarro D.; Martínez P.; Ramón D. J.; Yus M. Generation of allylic and benzylic organolithium reagents from the corresponding ester, amide, carbonate, carbamate and urea derivatives. Tetrahedron 1999, 55, 11027–11038. 10.1016/s0040-4020(99)00610-9. [DOI] [Google Scholar]
- a Abbasi M.; Mohammadizadeh M. R.; Moradi Z. Efficient reduction of sulfoxides with NaHSO3 catalyzed by I2. Tetrahedron Lett. 2015, 56, 6610–6613. 10.1016/j.tetlet.2015.10.035. [DOI] [Google Scholar]; b Ding F.; Jiang Y.; Gan S.; Bao R. L.-Y.; Lin K.; Shi L. B(C6 F5 )3 -Catalyzed Deoxygenation of Sulfoxides and Amine N -Oxides with Hydrosilanes. Eur. J. Org. Chem. 2017, 3427–3430. 10.1002/ejoc.201700489. [DOI] [Google Scholar]
- a Clark D. E. Peroxides and Peroxide-forming Compounds. J. Chem. Health Saf. 2001, 8, 12–22. 10.1016/s1074-9098(01)00247-7. [DOI] [Google Scholar]; b Zabicky J.Analytical and Safety Aspects of Organic Peroxides and Related Functional Groups. In The Chemistry of the Peroxide Group; Rappoport Z., Ed.; John Wiley & Sons: Chichester, 2006; pt 2, pp 597–773. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.