

Abstract
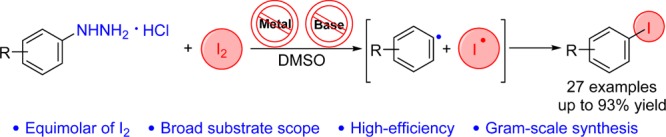
A metal- and base-free method is developed for the synthesis of aryl iodides from arylhydrazine hydrochlorides and iodine. A wide variety of aryl iodides can be conveniently synthesized by an equimolar reaction of arylhydrazine hydrochlorides and I2 in dimethyl sulfoxide at 60 °C for 6 h. In the iodination step, arylhydrazines are oxidized by iodine to form arenediazonium salts, which undergo single-electron transfer from iodide anion to give aryl and iodine radicals; subsequent combination of them affords the corresponding aryl iodides.
Introduction
Aryl iodides are important synthetic building blocks in organic chemistry that are mainly used for cross-coupling and related reactions,1 such as the Mizoroki–Heck reaction,2 Sonogashira coupling,3 Suzuki–Miyaura cross-coupling,4 and Ullmann condensation.5 Besides, aryl iodides are employed in metal–iodine exchange using organometallic reagents such as Grignard reagents6 and in halogen exchange via the halogenation of diaryliodonium salts with cuprous halides.7 In addition, hypervalent iodine compounds, which are important oxidizing agents, are usually synthesized from aryl iodides.8 Furthermore, arenes bearing radioactive iodine isotopes (123I, 124I, 125I, and 131I) play an important role in labeling biomacromolecules, such as proteins, nucleic acids, and cell surfaces in nuclear medicine and radiotherapy science.9
Over the past several decades, many methodologies for aromatic iodination have been developed: (1) direct aromatic iodination with I2 or other iodination reagents such as N-iodosuccinimide;10 (2) aromatic iodination by using activated precursors such as aryl boronic acid,11 aryl triflates,12 phenylazocarboxylates,13 aryl carboxylic acids,14 potassium aryltrifluoroborates,15 and aryl diazonium salts;16 (3) halogen exchange via an aromatic Finkelstein reaction.17 Among these, the Sandmeyer reaction is a classical reaction that proceeds via the diazotization of aromatic amines, followed by iodination with iodides (Scheme 1, eq 1). This method does not require transition metals; however, nitrous acid, which is highly corrosive, and a strongly acidic medium, such as sulfuric acid, hydrochloric acid, or tetrafluoroboric acid are inevitable for the diazotization step.18 Therefore, developing a facile approach to generate diazonium salts without transition metals, corrosive acids, and harsh oxidants or reductants is highly desirable.
Scheme 1. Aromatic Iodination.

Recently, we used commercially available arylhydrazine hydrochlorides as the source of aryl radicals and successfully achieved the arylation of aminoheterocycles and aromatic diamines, as well as the synthesis of unsymmetrical diaryl sulfides and selenides (Scheme 1, eq 2).19 Inspired by the results, we explored the reaction of arylhydrazines with iodine to synthesize aryl iodides (Scheme 1, eq 3). Joshi’s group reported a similar method (Scheme 1, eq 4), but it is applicable only to nitrophenylhydrazines and afforded the corresponding products in moderate yields (54–68%).20 By contrast, our method requires mild conditions, has a much broader substrate scope, and tolerates a wide range of functional groups; further, the desired aryl iodides are formed in good to excellent yields (74–95%). Besides, compared to the Sandmeyer reaction, our protocol is safer, easier to execute, involves simpler work-up, and has higher efficiency.
Results and Discussion
Initially, the cesium carbonate (Cs2CO3)/dimethyl sulfoxide (DMSO) system, which was used for the generation of aryl radicals in our previous work,19 was employed for the present iodination reaction. 4-Chlorophenylhydrazine hydrochloride (1a, 0.5 mmol) was chosen as the model substrate for reaction with I2 (1.0 equiv) in the presence of Cs2CO3 (1.0 equiv) as the base and DMSO as the solvent in the air, and 1-chloro-4-iodobenzene 2a was obtained in 63% yield (Table 1, entry 1). A lower or higher reaction temperature (40 or 80 °C) gave 2a in reduced yield (Table 1, entry 1, footnotes c and d). Screening of the solvent effects demonstrated that DMSO was the best solvent for this iodination (Table 1, entries 2–8). Increasing the amount of I2 (2.0 equiv) failed to improve the yield of 2a (Table 1, entry 9). A higher concentration of reactants led to the slightly improved yield of 2a (Table 1, entry 10), but the exorbitant reaction concentration with the same equivalent of Cs2CO3 was sluggish for the generation of 2a (Table 1, entry 11). Interestingly, a greater amount of Cs2CO3 hindered the formation of 2a (Table 1, entry 12), and conversely, the reaction without the base afforded 2a in 54% yield (Table 1, entry 13). Encouraged by this result, we hypothesized that no additional base was required for the generation of aryl radicals in this reaction, as opposed to our previous work.19 Hence, we further optimized the iodination conditions in the absence of the base. As expected, the use of 2.0 equiv of I2 gave 2a in 84% yield (Table 1, entry 14). A shorter reaction time furnished 2a in a similar yield (Table 1, entry 14, footnote e), whereas a longer reaction time lowered the yield of 2a slightly (Table 1, entry 14, footnote f). Moreover, upon reducing the amount of DMSO to 0.1 mL, the desired product 2a was furnished in 92% yield (Table 1, entry 15). With this substrate concentration, the use of the same equivalent of iodine gave 2a in 92% yield (Table 1, entry 16), which was chosen as the optimized condition for this iodination reaction. Under the optimized condition, a clear dark red solution was formed and gas bubbles appeared. After the reaction was finished, the color of iodine does not disappear and the resulting mixture caused a foul odor, suggests the formation of dimethyl sulfide. Further decreasing the concentration (equivalents) of iodine gave 2a in a slightly reduced yield (Table 1, entry 17).
Table 1. Optimization of Reaction Conditions for Iodination of Arylhydrazines with Iodinea.

| entry | I2 (mmol) | additive (mmol) | solvent (mL) | yieldb (%) |
|---|---|---|---|---|
| 1g | 0.5 | Cs2CO3 (0.5) | DMSO (1.5) | 63, 43c, 57d |
| 2 | 0.5 | Cs2CO3 (0.5) | DMF (1.5) | 50 |
| 3 | 0.5 | Cs2CO3 (0.5) | DMA (1.5) | 38 |
| 4 | 0.5 | Cs2CO3 (0.5) | CH3CN (1.5) | 35 |
| 5 | 0.5 | Cs2CO3 (0.5) | acetone (1.5) | 29 |
| 6 | 0.5 | Cs2CO3 (0.5) | MeOH (1.5) | 28 |
| 7 | 0.5 | Cs2CO3 (0.5) | CHCl3 (1.5) | 13 |
| 8 | 0.5 | Cs2CO3 (0.5) | toluene (1.5) | 14 |
| 9 | 1.0 | Cs2CO3 (0.5) | DMSO (1.5) | 61 |
| 10 | 0.5 | Cs2CO3 (0.5) | DMSO (0.5) | 68 |
| 11 | 0.5 | Cs2CO3 (0.5) | DMSO (0.25) | 58 |
| 12 | 0.5 | Cs2CO3 (0.75) | DMSO (0.5) | 36 |
| 13 | 0.5 | none | DMSO (0.5) | 54 |
| 14h | 1.0 | none | DMSO (0.5) | 84, 83e, 78f |
| 15 | 1.0 | none | DMSO (0.1) | 92 |
| 16 | 0.5 | none | DMSO (0.1) | 92 (87) |
| 17 | 0.3 | none | DMSO (0.1) | 82 |
Conditions: 1a, I2, additive, and solvent were stirred at 60 °C for 6 h in the air.
Determined by 1H NMR using an internal standard 1,3,5-trioxane (isolated yield).
Reaction temperature was 40 °C.
Reaction temperature was 80 °C.
Reaction time was 4 h.
Reaction time was 8 h.
In this reaction, azide and aniline, besides aryl iodide, were generated as byproducts. After the reaction, arylhydrazine was already consumed but I2 was not consumed. Most probably, air acted as an oxidant for this iodination.
Iodine not only acts as iodination reagent but also oxidizes phenylhydrazines to diazonium salt, and therefore excessive iodine is more beneficial to the iodination.
With the optimized reaction conditions in hand (Table 1, entry 16), we next investigated a series of phenylhydrazine hydrochlorides to synthesize aryl iodides (Table 2). First, substrates with a chloro-substituent at the para-, ortho-, and meta-positions were examined and the corresponding aryl iodides were isolated in high yields (Table 2, 2a–2c). Substrates with other halo-substituents (iodo, bromo, and fluoro) at the para-position were also tested under these reaction conditions; 2d and 2e were formed in high yields, but the volatile product 2f was generated in somewhat low isolated yield. A similar scenario was encountered during the purification of iodobenzene, which decreased the yield of the isolated product 2g (70% yield). Substrates with a methyl-substituent at the ortho-, meta-, and para-position, and an ethyl-substituent at the ortho-position gave the corresponding aryl iodides 2h–2k in good to excellent yields. The reaction of 4-methoxyphenylhydrazine hydrochloride gave the product 2l in 90% yield. Besides, a gram-scale iodination of 4-methoxyphenylhydrazine hydrochloride was performed under the standard conditions (Table 2, 2lb) and 1.44 g (6.15 mmol, 88% isolated yield) of 2l was formed from 7 mmol of the starting compound (1.22 g). 3-Methoxy- and 2-methoxy substituents were also tolerated under these reaction conditions (Table 2, 2m and 2n). Substrates with electron-withdrawing groups such as the nitro group at the ortho-, meta-, and para-positions (1o–1q), as well as 4-trifluoromethyl (1s), 4-cyano (1t), and 4-carboxyl (1v) moieties could be iodinated to afford the corresponding products in good to excellent yields. 4-Isopropylphenylhydrazine hydrochloride also afforded 2r in excellent yield. Furthermore, 2-iodonaphthalene 2u was generated in good yield under these iodination conditions but the formation of 2-iodopyridine 2w did not proceed well probably due to the presence of the basic pyridyl group. Disubstituted substrates, i.e., substrates with 3,5-dichloro-, 2,4-dichloro-, 3,4-dichloro-, and 2,4-dinitro substituents, could be employed under the standard reaction conditions to afford the desired products in good to excellent yields (Table 2, 2x–2a′). Unfortunately, when an aliphatic hydrazine such as tert-butylhydrazine hydrochloride was employed as substrate, 2b′ was not formed under the standard condition.
Table 2. Substrate Scopea.

Yield of isolated product is based on 1 (1H NMR yield using an internal standard 1,3,5-trioxane).
Gram scale.
DMSO (0.2 mL) was used.
For a better understanding of this iodination reaction, several control experiments were conducted, as shown in Scheme 2 (eqs 5–12). During the optimization of the reaction conditions, we found that 1-azido-4-chlorobenzene 3 and 4-chloroaniline 4 were generated as byproducts in the presence of a base (Scheme 2, eq 5). Under the standard reaction conditions, when the amount of I2 was decreased to 0.1 mmol, aryl iodide 2a, azide 3, aniline 4, and dimethyl sulfide 5 were detected by 1H NMR analysis of the crude mixture. Besides, the formation of azide 3 and aniline 4 could be further confirmed by gas chromatography–mass spectrometry (Scheme 2, eq 6). As well known, DMSO can oxidize hydrogen iodide to iodine, whereas dimethyl sulfide is generated as the reduction product.21 Therefore, we speculated that hydrogen iodide may be generated during this iodination, which further confirms that acidic conditions favor this reaction. To clarify the role of DMSO in the iodination reaction, dimethylformamide was used as the solvent in combination with 0.5 mmol DMSO (Scheme 2, eq 7); 86% of 2a was formed along with 0.17 mmol of dimethyl sulfide 5 (which was detected by the crude 1H NMR spectra), whereas DMSO was not detected after the iodination reaction was finished. The reaction with 0.5 mmol of tert-butyl hydroperoxide (tBuOOH) as an oxidant instead of DMSO also afforded 2a in 80% yield (Scheme 2, eq 8). In contrast, the iodination in the absence of DMSO only yielded 26% of 2a (Scheme 2, eq 9). These results suggest that DMSO acted as not only solvent but also co-oxidant during the iodination reaction: comparing the results of eqs 7, 9, and 11, DMSO seems to be the major oxidant in this iodination reaction. In addition, when the radical scavenger (2,2,6,6-tetramethylpiperidin-1-yl)oxyl was added to the reaction mixture under the standard conditions (Scheme 2, eq 10), 2a was formed in a low yield with 10% of 6, which implies that the iodination proceeds through a free-radical pathway. Besides, this reaction can proceed well under argon protection (Scheme 2, eq 11), suggesting that the oxidation of arylhydrazine to aryl radical can proceed not only with molecular oxygen but also iodine itself. Free 4-chlorophenylhydrazine 7 was also employed as a substrate for the iodination reaction but only to generate 42% of 2a, which implied that acidic condition is essential for stabilizing arylhydrazine under this iodination reaction (Scheme 2, eq 12).
Scheme 2. Control Experiments.

On the basis of these control experiments, a mechanism for the iodination is proposed, as shown in Scheme 3. Specifically, the starting material 1a produces the free 4-chlorophenylhydrazine 7, which reacts with iodine to afford intermediate 8. Dehydroiodination of 8 leads to 9, which further reacts with iodine to form 10. Charge transfer of 10 affords diazonium salt 11, which upon single-electron transfer (SET) and nitrogen release, generates phenyl radical 12 and an iodine radical. The combination of the phenyl and iodine radicals leads to the formation of aryl iodide 2a. Excess amount of 7 (free form) may react with diazonium salt 11 to generate byproducts 3 and 4.22 During this iodination, hydrogen iodide can be oxidized to iodine by DMSO with the release of dimethyl sulfide.21
Scheme 3. Possible Pathway for the Synthesis of Aryl Iodides.

Conclusions
In summary, we have developed a facile and efficient method to synthesize aryl iodides from arylhydrazine hydrochlorides and iodine in the absence of any metal catalysts and additives. During this iodination, iodine plays a dual role: (1) an oxidant for converting arylhydrazines to arenediazonium salts, which subsequently undergo SET to form aryl radicals; (2) an iodination reagent to afford aryl iodides. Arylhydrazine hydrochlorides with a diverse range of functional groups are tolerated under these iodination conditions, and the corresponding aryl iodides are obtained in good to excellent yields. Further studies on halogenation of the arylhydrazine hydrochlorides using other halogenation reagents are underway, and the results will be reported in due course.
Experimental Section
General Remarks
Unless otherwise stated, all starting materials and solvents were purchased from commercial sources and used without further purification. 1H NMR spectra were recorded on JEOL JNM-ECS400 (400 MHz) FT NMR system or JEOL JNM-ECX400 (400 MHz) FT NMR system in CDCl3 and DMSO-d6 with Me4Si as an internal standard. 13C NMR spectra were recorded on JEOL JNM-ECX400 (100 MHz) FT NMR or JEOL JNM-ECS400 (100 MHz) FT NMR in CDCl3 and DMSO-d6.
General Procedure for the Synthesis of Aryl Iodides (2)
The desired arylhydrazine hydrochloride derivatives 1 (0.5 mmol), I2 (126.9 mg, 0.5 mmol), and DMSO (0.1 mL) were added to a round-bottomed flask, and the reaction mixture was stirred at 60 °C for 6 h under air. The resulting mixture was cooled to room temperature and then sat. Na2S2O8 (aq, 5 mL) and water (10 mL) were added. The mixture was extracted with CHCl3 (4 × 5 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. Finally, the residue was purified by silica gel chromatography (eluent: hexane/ethyl acetate) to give 2 (eluent for 2v: chloroform/methanol).
1-Chloro-4-iodobenzene (2a)
[CAS: 637-87-6].23 White solid, 103.7 mg, 87% (isolated yield), mp 52–53 °C; 1H NMR (400 MHz, CDCl3): δ 7.60 (d, J = 8.8 Hz, 2H), 7.08 (d, J = 8.8 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 138.9, 134.6, 130.7, 91.3; MS (EI) [M]+m/z = 238.
1-Chloro-2-iodobenzene (2b)
[CAS: 615-41-8].23 Light yellow oil, 101.6 mg, 85% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.86 (d, J = 8.0 Hz, 1H), 7.45 (d, J = 8.0 Hz, 1H), 7.29 (t, J = 8.4 Hz, 1H), 6.96 (t, J = 8.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 140.4, 138.6, 129.5, 128.0, 98.2; MS (EI) [M]+m/z = 238.
1-Chloro-3-iodobenzene (2c)
[CAS: 625-99-0].23 Light yellow oil, 102.0 mg, 85% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.72 (t, J = 1.8 Hz, 1H), 7.59 (dq, J = 7.6, 0.9 Hz, 1H), 7.32 (dq, J = 8.4, 1.1 Hz, 1H), 7.03 (t, J = 8.2 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 137.3, 135.8, 135.2, 131.1, 128.1, 94.1; MS (EI) [M]+m/z = 238.
1,4-Diiodobenzene (2d)
[CAS: 624-38-4].12 White solid, 145.3 mg, 85% (isolated yield), mp 126–127 °C; 1H NMR (400 MHz, CDCl3): δ 7.41 (s, 4H); 13C{1H} NMR (100 MHz, CDCl3): δ 139.5, 93.5; MS (EI) [M]+m/z = 330.
1-Bromo-4-iodobenzene (2e)
[CAS: 589-87-7].24 White solid, 117.2 mg, 83% (isolated yield), mp 87–88 °C; 1H NMR (400 MHz, CDCl3): δ 7.55 (dt, J = 8.8, 2.4 Hz, 2H), 7.23 (dt, J = 8.8, 2.4 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 139.2, 133.6, 122.3, 92.2; MS (EI) [M]+m/z = 282.
1-Fluoro-4-iodobenzene (2f)
[CAS: 352-34-1].11 Colorless oil, 71.2 mg, 64% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.60–7.65 (m, 2H), 6.84 (tt, J = 8.8, 2.6 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 162.8 (d, J = 249.1 Hz), 139.1 (d, J = 7.6 Hz), 117.9 (d, J = 23.0 Hz), 87.1 (d, J = 2.9 Hz); MS (EI) [M]+m/z = 222.
Iodobenzene (2g)
[CAS: 591-50-4].12 Colorless oil, 71.5 mg, 70% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.70 (d, J = 7.6 Hz, 2H), 7.33 (t, J = 7.4 Hz, 1H), 7.11 (t, J = 7.8 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 137.6, 130.4, 127.6, 94.5; MS (EI) [M]+m/z = 204.
1-Iodo-2-methylbenzene (2h)
[CAS: 615-37-2].23 Colorless oil, 78.6 mg, 72% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.80 (d, J = 7.6 Hz, 1H), 7.25–7.22 (m, 2H), 6.84–6.88 (m, 1H), 2.43 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 141.5, 139.1, 129.9, 128.3, 127.5, 101.6, 28.3; MS (EI) [M]+m/z = 218.
1-Iodo-3-methylbenzene (2i)
[CAS: 625-95-6].23 Colorless oil, 80.9 mg, 75% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.55 (s, 1H), 7.49 (d, J = 7.6 Hz, 1H), 7.12 (d, J = 7.6 Hz, 1H), 6.98 (t, J = 8.0 Hz, 1H), 2.29 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 140.4, 138.2, 134.6, 130.1, 128.5, 94.5, 21.1; MS (EI) [M]+m/z = 218.
1-Iodo-4-methylbenzene (2j)
[CAS: 624-31-7].12 White solid, 90.1 mg, 82% (isolated yield), mp 31–32 °C; 1H NMR (400 MHz, CDCl3): δ 7.56 (d, J = 8.0 Hz, 2H), 6.92 (d, J = 8.4 Hz, 2H), 2.29 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 137.6, 137.4, 131.3, 90.3, 21.2; MS (EI) [M]+m/z = 218.
1-Ethyl-2-iodobenzene (2k)
[CAS: 18282-40-1].25 Colorless oil, 82.1 mg, 71% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.81 (dd, J = 8.3, 1.6 Hz, 1H), 7.21–7.30 (m, 2H), 6.87 (td, J = 8.3, 1.6 Hz, 1H), 2.73 (q, J = 7.5 Hz, 2H), 1.21 (t, J = 7.5 Hz, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 146.6, 139.5, 128.7, 128.5, 100.6, 34.3, 14.7; MS (EI) [M]+m/z = 232.
1-Iodo-4-methoxybenzene (2l)
[CAS: 696-62-8].12 White solid, 106.5 mg, 90% (isolated yield), mp 48–49 °C; 1H NMR (400 MHz, CDCl3): δ 7.55 (d, J = 9.2 Hz, 2H), 6.68 (d, J = 9.2 Hz, 2H), 3.77 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 159.6, 138.3, 116.5, 82.8, 55.4; MS (EI) [M]+m/z = 234.
1-Iodo-3-methoxybenzene (2m)
[CAS: 766-85-8].23 Light yellow oil, 78.3 mg, 67% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.25–7.29 (m, 2H), 7.00 (t, J = 8.1 Hz, 1H), 6.87 (dd, J = 8.3, 1.6 Hz, 1H), 3.78 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 160.3, 130.9, 129.9, 123.1, 113.9, 94.5, 55.5; MS (EI) [M]+m/z = 234.
1-Iodo-2-methoxybenzene (2n)
[CAS: 529-28-2].23 Light yellow oil, 71.5 mg, 61% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.77 (d, J = 7.7 Hz, 1H), 7.29–7.33 (m, 1H), 6.83 (d, J = 8.2 Hz, 1H), 6.71 (t, J = 7.6 Hz, 1H), 3.88 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 158.2, 139.6, 129.7, 122.6, 111.1, 86.1, 56.4; MS (EI) [M]+m/z = 234.
1-Iodo-2-nitrobenzene (2o)
[CAS: 609-73-4].26 Yellow solid, 108.6 mg, 87% (isolated yield), mp 48–49 °C; 1H NMR (400 MHz, CDCl3): δ 8.05 (d, J = 8.3 Hz, 1H), 7.87 (d, J = 8.3 Hz, 1H), 7.50 (t, J = 7.7 Hz, 1H), 7.25–7.29 (m, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 153.1, 142.0, 133.5, 129.2, 125.6, 86.3; MS (EI) [M]+m/z = 249.
1-Iodo-3-nitrobenzene (2p)
[CAS: 645-00-1].27 Colorless oil, 106.5 mg, 84% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 8.57 (t, J = 2.0 Hz, 1H), 8.21 (dq, J = 8.8, 1.1 Hz, 1H), 8.03 (dt, J = 7.6, 1.1 Hz, 1H), 7.30 (t, J = 8.2 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 148.7, 143.6, 132.6, 130.8, 122.9, 93.6; MS (EI) [M]+m/z = 249.
1-Iodo-4-nitrobenzene (2q)
[CAS: 636-98-6].28 Yellow solid, 92.4 mg, 74% (isolated yield), mp 171–172 °C; 1H NMR (400 MHz, CDCl3): δ 7.95 (d, J = 9.6 Hz, 2H), 7.91 (d, J = 9.6 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 147.9, 138.8, 125.0, 102.8; MS (EI) [M]+m/z = 249.
1-Iodo-4-isopropylbenzene (2r)
[CAS: 17356-09-1].12 Light yellow oil, 111.0 mg, 90% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.60 (d, J = 7.9 Hz, 2H), 6.98 (d, J = 8.7 Hz, 2H), 2.80–2.90 (m, 1H), 1.23 (s, 3H), 1.21 (s, 3H); 13C{1H} NMR (100 MHz, CDCl3): δ 148.6, 137.4, 128.8, 90.8, 33.9, 24.0; MS (EI) [M]+m/z = 246.
1-Iodo-4-(trifluoromethyl)benzene (2s)
[CAS: 455-13-0].29 Colorless oil, 99.0 mg, 73% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 7.7 Hz, 2H), 7.35 (d, J = 8.2 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 138.2, 130.3 (q, J = 33.0 Hz), 127.0 (q, J = 3.8 Hz), 124.1 (q, J = 272.2 Hz), 98.7; MS (EI) [M]+m/z = 272.
4-Iodobenzonitrile (2t)
[CAS: 3058-39-7].28 White solid, 103.4 mg, 90% (isolated yield), mp 124–125 °C; 1H NMR (400 MHz, CDCl3): δ 7.85 (d, J = 7.9 Hz, 2H), 7.37 (d, J = 7.9 Hz, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 138.6, 133.3, 118.3, 111.9, 100.4; MS (EI) [M]+m/z = 229.
2-Iodonaphthalene (2u)
[CAS: 612-55-5].28 Light yellow solid, 110.7 mg, 86% (isolated yield), mp 49–50 °C; 1H NMR (400 MHz, CDCl3): δ 8.24 (s, 1H), 7.80 (q, J = 3.2 Hz, 1H), 7.72 (dt, J = 9.6, 1.6 Hz, 2H), 7.58 (d, J = 8.8 Hz, 1H), 7.51–7.47 (m, 2H); 13C{1H} NMR (100 MHz, CDCl3): δ 136.7, 135.1, 134.5, 132.2, 129.6, 128.0, 126.9, 126.8, 126.6, 91.6; MS (EI) [M]+m/z = 254.
4-Iodobenzoic Acid (2v)
[CAS: 619-58-9].23 White solid, 89.1 mg, 72% (isolated yield), mp 269–270 °C; 1H NMR (400 MHz, DMSO-d6): δ 13.04 (br, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.69 (d, J = 8.7 Hz, 2H); 13C{1H} NMR (100 MHz, DMSO-d6): δ 166.9, 137.6, 131.1, 130.3, 101.2; MS (EI) [M]+m/z = 248.
1,3-Dichloro-5-iodobenzene (2x)
[CAS: 3032-81-3].27 White solid, 119 mg, 87% (isolated yield), mp 49–50 °C; 1H NMR (400 MHz, CDCl3): δ 7.61 (d, J = 1.6 Hz, 2H), 7.34 (t, J = 2.0 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 135.7, 128.5, 93.8; MS (EI) [M]+m/z = 272.
2,4-Dichloro-1-iodobenzene (2y)
[CAS: 29898-32-6].30 Colorless oil, 125.1 mg, 92% (isolated yield); 1H NMR (400 MHz, CDCl3): δ 7.76 (d, J = 8.2 Hz, 1H), 7.46 (d, J = 2.3 Hz, 1H), 6.96 (dd, J = 8.7, 2.3 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 140.9, 139.6, 135.2, 129.4, 128.5, 95.6; MS (EI) [M]+m/z = 272.
1,2-Dichloro-4-iodobenzene (2z)
[CAS: 20555-91-3].27 White solid, 126.6 mg, 93% (isolated yield), mp 30–31 °C; 1H NMR (400 MHz, CDCl3): δ 7.79 (d, J = 1.8 Hz, 1H), 7.51 (dd, J = 8.5, 2.1 Hz, 1H), 7.16 (d, J = 8.7 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 138.9, 136.9, 133.8, 132.8, 131.9, 91.1; MS (EI) [M]+m/z = 272.
1-Iodo-2,4-dinitrobenzene (2a′)
[CAS: 709-49-9].24 Yellow solid, 114.4 mg, 78% (isolated yield), mp 83–84 °C; 1H NMR (400 MHz, CDCl3): δ 8.68 (d, J = 2.4 Hz, 1H), 8.31 (d, J = 8.8 Hz, 1H), 8.11 (dd, J = 8.8, 2.4 Hz, 1H); 13C{1H} NMR (100 MHz, CDCl3): δ 153.3, 148.1, 143.5, 127.1, 120.5, 94.9; MS (EI) [M]+m/z = 294.
General Procedure for the Gram-Scale Synthesis of 1-Iodo-4-methoxybenzene (2l)
4-Methoxyphenylhydrazine hydrochloride 1l (1.22 g, 7 mmol), I2 (1.80 g, 7 mmol), and DMSO (1.4 mL) were added to a round-bottomed flask, and the reaction mixture was stirred at 60 °C for 6 h under air. The resulting mixture was cooled to room temperature, and then sat. Na2S2O8 (aq, 25 mL) and water (100 mL) were added. The mixture was extracted with CHCl3 (4 × 20 mL), dried over anhydrous Na2SO4, and concentrated in vacuo. Finally, the residue was purified by silica gel chromatography (eluent: hexane/ethyl acetate) to give 2l (1.44 g).
Acknowledgments
C.-p.D. acknowledges the Ministry of Education, Culture, Sports, Science and Technology (MEXT) Scholarship Program. This work is also supported by Grant-in-Aid for Scientific Research on Scientific Research (B, 16H04138) and Exploratory Research (16K14049) from The Japan Society for the Promotion of Science (JSPS) and also supported by the Nanotechnology Platform Program of the Nara Institute of Science and Technology (NAIST).
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01559.
1H NMR and 13C NMR spectra (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- a Yanagisawa S.; Ueda K.; Taniguchi T.; Itami K. Potassium t-butoxide alone can promote the biaryl coupling of electron-deficient nitrogen heterocycles and haloarenes. Org. Lett. 2008, 10, 4673–4676. 10.1021/ol8019764. [DOI] [PubMed] [Google Scholar]; b Taniguchi N.; Onami T. Magnesium-induced copper-catalyzed synthesis of unsymmetrical diaryl chalcogenide compounds from aryl iodide via cleavage of the Se–Se or S–S bond. J. Org. Chem. 2004, 69, 915–920. 10.1021/jo030300+. [DOI] [PubMed] [Google Scholar]; c Turner G. L.; Morris J. A.; Greaney M. F. Direct arylation of thiazoles on water. Angew. Chem., Int. Ed. 2007, 46, 7996–8000. 10.1002/anie.200702141. [DOI] [PubMed] [Google Scholar]; d Yanagisawa S.; Sudo T.; Noyori R.; Itami K. Direct C–H arylation of (hetero)arenes with aryl iodides via rhodium catalysis. J. Am. Chem. Soc. 2006, 128, 11748–11749. 10.1021/ja064500p. [DOI] [PubMed] [Google Scholar]; e Antilla J. C.; Baskin J. M.; Barder T. E.; Buchwald S. L. Copper–diamine-catalyzed N-arylation of pyrroles, pyrazoles, indazoles, imidazoles, and triazoles. J. Org. Chem. 2004, 69, 5578–5587. 10.1021/jo049658b. [DOI] [PubMed] [Google Scholar]; f Singh D.; Deobald A. M.; Camargo L. R.; Tabarelli G.; Rodrigues O. E.; Braga A. L. An efficient one-pot synthesis of symmetrical diselenides or ditellurides from halides with CuO nanopowder/Se0 or Te0/base. Org. Lett. 2010, 12, 3288–3291. 10.1021/ol100558b. [DOI] [PubMed] [Google Scholar]
- Pavia C.; Giacalone F.; Bivona L. A.; Salvo A. M. P.; Petrucci C.; Strappaveccia G.; Vaccaro L.; Aprile C.; Gruttadauria M. Evidences of release and catch mechanism in the Heck reaction catalyzed by palladium immobilized on highly cross-linked-supported imidazolium salts. J. Mol. Catal. A: Chem. 2014, 387, 57–62. 10.1016/j.molcata.2014.02.025. [DOI] [Google Scholar]
- a Tang B. X.; Wang F.; Li J. H.; Xie Y. X.; Zhang M. B. Reusable Cu2O/PPh3/TBAB system for the cross-couplings of aryl halides and heteroaryl halides with terminal alkynes. J. Org. Chem. 2007, 72, 6294–6297. 10.1021/jo070538o. [DOI] [PubMed] [Google Scholar]; b Xu W.; Yu B.; Sun H.; Zhang G.; Zhang W.; Gao Z. Copper-catalyzed C(sp2)–C(sp) Sonogashira-type cross-coupling reactions accelerated by polycyclic aromatic hydrocarbons. Appl. Organomet. Chem. 2015, 29, 353–356. 10.1002/aoc.3298. [DOI] [Google Scholar]
- Senra J. D.; Malta L. F. B.; da Costa M. E.; Michel R. C.; Aguiar L.; Simas A. B.; Antunes O. E. E. Hydroxypropyl-α-cyclodextrin-capped palladium nanoparticles: Active scaffolds for efficient carbon-carbon bond forming cross-couplings in water. Adv. Synth. Catal. 2009, 351, 2411–2422. 10.1002/adsc.200900348. [DOI] [Google Scholar]
- Hosseini-Sarvari M.; Razmi Z. Highly active recyclable heterogeneous Pd/ZnO nanoparticle catalyst: Sustainable developments for the C–O and C–N bond cross-coupling reactions of aryl halides under ligand-free conditions. RSC Adv. 2014, 4, 44105–44116. 10.1039/C4RA06486K. [DOI] [Google Scholar]
- a Zhao Y.; Wang Y.; Sun H.; Li L.; Zhang H. Ullmann reaction in tetraethyl orthosilicate: A novel synthesis of triarylamines and diaryl ethers. Chem. Commun. 2007, 3186–3188. 10.1039/b706449g. [DOI] [PubMed] [Google Scholar]; b Knochel P.; Dohle W.; Gommermann N.; Kneisel F. F.; Kopp F.; Korn T.; Sapountzis I.; Vu V. A. Highly functionalized organomagnesium reagents prepared through halogen–metal exchange. Angew. Chem., Int. Ed. 2003, 42, 4302–4320. 10.1002/anie.200300579. [DOI] [PubMed] [Google Scholar]
- Li J.; Liu L.; Ding D.; Sun J. T. Halogen exchange via a halogenation of diaryliodonium salts with cuprous halide. Lett. Org. Chem. 2013, 10, 541–548. 10.2174/15701786113109990011. [DOI] [Google Scholar]
- a Wirth T. Hypervalent iodine chemistry in synthesis: Scope and new directions. Angew. Chem., Int. Ed. 2005, 44, 3656–3665. 10.1002/anie.200500115. [DOI] [PubMed] [Google Scholar]; b Hossain M. D.; Kitamura T. Unexpected, drastic effect of triflic acid on oxidative diacetoxylation of iodoarenes by sodium perborate. A facile and efficient one-pot synthesis of (diacetoxyiodo)arenes. J. Org. Chem. 2005, 70, 6984–6986. 10.1021/jo050927n. [DOI] [PubMed] [Google Scholar]; c Ueno M.; Nabana T.; Togo H. Novel oxidative α-tosyloxylation of alcohols with iodosylbenzene and p-toluenesulfonic acid and its synthetic use for direct preparation of heteroaromatics. J. Org. Chem. 2003, 68, 6424–6426. 10.1021/jo030045t. [DOI] [PubMed] [Google Scholar]; d Kiyokawa K.; Watanabe T.; Fra L.; Kojima T.; Minakata S. Hypervalent iodine(III)-mediated decarboxylative Ritter-type amination leading to the production of α-tertiary amine derivatives. J. Org. Chem. 2017, 82, 11711–11720. 10.1021/acs.joc.7b01202. [DOI] [PubMed] [Google Scholar]
- Seevers R. H.; Counsell R. E. Radioiodination techniques for small organic molecules. Chem. Rev. 1982, 82, 575–590. 10.1021/cr00052a002. [DOI] [Google Scholar]
- a Mulholland G. K.; Zheng Q. H. A direct iodination method with iodine and silver triflate for the synthesis of spect and pet imaging agent precursors. Synth. Commun. 2001, 31, 3059–3068. 10.1081/SCC-100105877. [DOI] [Google Scholar]; b Bergström M.; Ganji S.; Naidu V. R.; Unelius R. N-Iodosuccinimide (NIS) in direct aromatic iodination. Eur. J. Org. Chem. 2017, 3234–3239. 10.1002/ejoc.201700173. [DOI] [Google Scholar]
- a Yang H.; Li Y.; Jiang M.; Wang J.; Fu H. General copper-catalyzed transformations of functional groups from arylboronic acids in water. Chem. - Eur. J. 2011, 17, 5652–5660. 10.1002/chem.201003711. [DOI] [PubMed] [Google Scholar]; b Zhang P.; Zhuang R.; Guo Z.; Su X.; Chen X.; Zhang X. A highly efficient copper-mediated radioiodination approach using aryl boronic acids. Chem. - Eur. J. 2016, 22, 16783–16786. 10.1002/chem.201604105. [DOI] [PubMed] [Google Scholar]
- Liu W.; Yang X.; Gao Y.; Li C. J. Simple and efficient generation of aryl radicals from aryl triflates: Synthesis of aryl boronates and aryl iodides at room temperature. J. Am. Chem. Soc. 2017, 139, 8621–8627. 10.1021/jacs.7b03538. [DOI] [PubMed] [Google Scholar]
- a Höfling S. B.; Bartuschat A. L.; Heinrich M. R. 4-Substituted tert-butyl phenylazocarboxylates–synthetic equivalents for the para-phenyl radical cation. Angew. Chem., Int. Ed. 2010, 49, 9769–9772. 10.1002/anie.201004508. [DOI] [PubMed] [Google Scholar]; b Jasch H.; Höfling S. B.; Heinrich M. R. Nucleophilic substitutions and radical reactions of phenylazocarboxylates. J. Org. Chem. 2012, 77, 1520–1532. 10.1021/jo202406k. [DOI] [PubMed] [Google Scholar]
- Perry G. J. P.; Quibell J. M.; Panigrahi A.; Larrosa I. Transition-metal-free decarboxylative iodination: New routes for decarboxylative oxidative cross-couplings. J. Am. Chem. Soc. 2017, 139, 11527–11536. 10.1021/jacs.7b05155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabalka G. W.; Mereddy A. R. A facile synthesis of aryl iodides via potassium aryltrifluoroborates. Tetrahedron Lett. 2004, 45, 343–345. 10.1016/j.tetlet.2003.10.149. [DOI] [Google Scholar]
- a Yang H.; Fu L.; Wei L.; Liang J.; Binks B. P. Compartmentalization of incompatible reagents within pickering emulsion droplets for one-pot cascade reactions. J. Am. Chem. Soc. 2015, 137, 1362–1371. 10.1021/ja512337z. [DOI] [PubMed] [Google Scholar]; b Isaad J. Acidic ionic liquid supported on silica-coated magnetite nanoparticles as a green catalyst for one-pot diazotization–halogenation of the aromatic amines. RSC Adv. 2014, 4, 49333–49341. 10.1039/C4RA05705H. [DOI] [Google Scholar]; c Bochare M. D.; Degani M. S. Polyethylene glycol nitrite (PEG-ONO) as a novel diazotizing agent. ACS Sustainable Chem. Eng. 2017, 5, 3716–3720. 10.1021/acssuschemeng.6b03210. [DOI] [Google Scholar]; d Leas D. A.; Dong Y.; Vennerstrom J. L.; Stack D. E. One-pot, metal-free conversion of anilines to aryl bromides and iodides. Org. Lett. 2017, 19, 2518–2521. 10.1021/acs.orglett.7b00771. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kolvari E.; Amoozadeh A.; Koukabi N.; Otokesh S.; Isari M. Aryl diazonium nanomagnetic sulfate and potassium iodide: An iodination process. Tetrahedron Lett. 2014, 55, 3648–3651. 10.1016/j.tetlet.2014.02.073. [DOI] [Google Scholar]
- a Li L.; Liu W.; Zeng H.; Mu X.; Cosa G.; Mi Z.; Li C. J. Photo-induced metal-catalyst-free aromatic Finkelstein reaction. J. Am. Chem. Soc. 2015, 137, 8328–8331. 10.1021/jacs.5b03220. [DOI] [PubMed] [Google Scholar]; b Jin X.; Davies R. P. Copper-catalysed aromatic-Finkelstein reactions with amine-based ligand systems. Catal. Sci. Technol. 2017, 7, 2110–2117. 10.1039/C7CY00538E. [DOI] [Google Scholar]
- a Hubbard A.; Okazaki T.; Laali K. K. Halo- and azidodediazoniation of arenediazonium tetrafluoroborates with trimethylsilyl halides and trimethylsilyl azide and Sandmeyer-type bromodediazoniation with Cu(I)Br in [BMIM][PF6] ionic liquid. J. Org. Chem. 2008, 73, 316–319. 10.1021/jo701937e. [DOI] [PubMed] [Google Scholar]; b Eshghi H.; Bakavoli M.; Ghasemzadeh M. Nitrite ionic liquid as a new reagent for in situ synthesis of aryl iodides and azides. Res. Chem. Intermed. 2015, 41, 3999–4007. 10.1007/s11164-013-1505-5. [DOI] [Google Scholar]; c Kolvari E.; Amoozadeh A.; Koukabi N.; Otokesh S.; Isari M. Aryl diazonium nanomagnetic sulfate and potassium iodide: An iodination process. Tetrahedron Lett. 2014, 55, 3648–3651. 10.1016/j.tetlet.2014.02.073. [DOI] [Google Scholar]; d Zarei A.; Hajipour A. R.; Khazdooz L. A one-pot method for the iodination of aryl amines via stable aryl diazonium silica sulfates under solvent-free conditions. Synthesis 2009, 2009, 941–944. 10.1055/s-0028-1087981. [DOI] [Google Scholar]
- a Taniguchi T.; Imoto M.; Takeda M.; Matsumoto F.; Nakai T.; Mihara M.; Mizuno T.; Nomoto A.; Ogawa A. Metal-free C–H arylation of aminoheterocycles with arylhydrazines. Tetrahedron 2016, 72, 4132–4140. 10.1016/j.tet.2016.05.056. [DOI] [Google Scholar]; b Taniguchi T.; Imoto M.; Takeda M.; Nakai T.; Mihara M.; Mizuno T.; Nomoto A.; Ogawa A. Regioselective radical arylation of aromatic diamines with arylhydrazines. Synthesis 2017, 49, 1623–1631. 10.1055/s-0036-1588921. [DOI] [Google Scholar]; c Taniguchi T.; Murata A.; Takeda M.; Mizuno T.; Nomoto A.; Ogawa A. Atom-economical synthesis of unsymmetrical diaryl selenides from arylhydrazines and diaryl diselenides. Eur. J. Org. Chem. 2017, 2017, 4928–4934. 10.1002/ejoc.201700938. [DOI] [Google Scholar]; d Taniguchi T.; Naka T.; Imoto M.; Takeda M.; Nakai T.; Mihara M.; Mizuno T.; Nomoto A.; Ogawa A. Transition-metal-free and oxidant-free cross-coupling of arylhydrazines with disulfides: Base-promoted synthesis of unsymmetrical aryl sulfides. J. Org. Chem. 2017, 82, 6647–6655. 10.1021/acs.joc.7b00767. [DOI] [PubMed] [Google Scholar]
- Joshi S. S.; Deorha D. S. Replacement of the hydrazino group in substituted nitro-phenylhydrazines by bromine or iodine. J. Chem. Soc. 1957, 2414. [Google Scholar]
- Vieira A. A.; Azeredo J. B.; Godoi M.; Santi C.; da Silva Junior E. N.; Braga A. L. Catalytic chalcogenylation under greener conditions: A solvent-free sulfur- and seleno-functionalization of olefins via I2/DMSO oxidant system. J. Org. Chem. 2015, 80, 2120–2127. 10.1021/jo502621a. [DOI] [PubMed] [Google Scholar]
- Pearce L. B.; Feingold M. H.; Cerny K. F.; Anselme J. P. Phenyl azide from the reaction of phenylhydrazine with thionyl chloride. Preparation of N-thionylbenzylhydrazine and N-thionyl-β-phenethylhydrazine. J. Org. Chem. 1979, 44, 1881–1883. 10.1021/jo01325a033. [DOI] [Google Scholar]
- Niu L.; Zhang H.; Yang H.; Fu H. Metal-free iodination of arylboronic acids and the synthesis of biaryl derivatives. Synlett 2014, 25, 995–1000. 10.1055/s-0033-1340871. [DOI] [Google Scholar]
- Sloan N. L.; Luthra S. K.; McRobbie G.; Pimlott S. L.; Sutherland A. Late stage iodination of biologically active agents using a one-pot process from aryl amines. RSC Adv. 2017, 7, 54881–54891. 10.1039/C7RA11860K. [DOI] [Google Scholar]
- a Uemura S.; Toshimitsu A. Aromatic thallation and nitration with thallium(III) nitrate. Chem. Express 1986, 1, 99–102. [Google Scholar]; b Friedrich W.; Helmut W.; Eturo M.; Gerd E. Über den Nachweis von aliphatischen und aromatischen primären Aminen mit o-Diacetyl-benzol. Chem. Ber. 1956, 89, 1994–1999. 10.1002/cber.19560890830. [DOI] [Google Scholar]
- Fu Z.; Li Z.; Song Y.; Yang R.; Liu Y.; Cai H. Decarboxylative halogenation and cyanation of electron-deficient aryl carboxylic acids via Cu mediator as well as electron-rich ones through Pd catalyst under aerobic conditions. J. Org. Chem. 2016, 81, 2794–2803. 10.1021/acs.joc.5b02873. [DOI] [PubMed] [Google Scholar]
- Partridge B. M.; Hartwig J. F. Sterically controlled iodination of arenes via iridium-catalyzed C–H borylation. Org. Lett. 2013, 15, 140–143. 10.1021/ol303164h. [DOI] [PubMed] [Google Scholar]
- Cant A. A.; Bhalla R.; Pimlott S. L.; Sutherland A. Nickel-catalysed aromatic Finkelstein reaction of aryl and heteroaryl bromides. Chem. Commun. 2012, 48, 3993–3995. 10.1039/c2cc30956d. [DOI] [PubMed] [Google Scholar]
- Ye Y.; Künzi S. A.; Sanford M. S. Practical method for the Cu-mediated trifluoromethylation of arylboronic acids with CF3 radicals derived from NaSO2CF3 and tert-butyl hydroperoxide (TBHP). Org. Lett. 2012, 14, 4979–4981. 10.1021/ol3022726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulbitski K.; Nisnevich G.; Gandelman M. Metal-free efficient, general and facile iododecarboxylation method with biodegradable co-products. Adv. Synth. Catal. 2011, 353, 1438–1442. 10.1002/adsc.201100145. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.