

Abstract
For over 100 years, clinicians and scientists have been unraveling the consequences of the A to T substitution in the beta globin gene that produces hemoglobin S, which leads to the systemic manifestations of sickle cell disease (SCD), including vaso-occlusion, anemia, hemolysis, organ injury and pain. However, despite growing understanding of the mechanisms of hemoglobin S polymerization and its effects on red blood cells, only two therapies for SCD — hydroxyurea and L-glutamine — are approved by the US Food and Drug Administration. Moreover, these treatment options do not fully address the manifestations of SCD. The pathophysiology of SCD suggests a complex network of interdependent processes, and in this article, we review efforts to develop new drugs targeting these processes, including agents that reactivate fetal hemoglobin, anti-sickling agents, anti-adhesive agents, modulators of inflammation, ischemia/reperfusion and oxidative stress, anti-thrombotics, anti-platelet agents and agents that counteract free hemoglobin/heme. We also discuss gene therapy, which holds promise of a cure, although its widespread application is currently limited by technical challenges and the expense of treatment. We thus propose that developing systems-oriented multi-agent strategies based on SCD pathophysiology is needed to improve to improve the quality of life and survival of people with SCD.
INTRODUCTION
Sickle cell disease (SCD) is a common monogenic disorder affecting over 100,000 people in the United States alone, and millions more worldwide.1,2 This often devastating disease is characterized by red blood cell (RBC) sickling; chronic hemolytic anemia; episodic vaso-occlusion associated with severe pain and inflammation; acute and cumulative organ damage that manifests as stroke, acute chest syndrome, sickle lung disease, pulmonary hypertension, nephropathy and end-stage renal disease; and other chronic morbidities.3 Lives of patients with SCD are characterized by frequent episodes of severe pain (vaso-occlusive events or “crises”); acute organ dysfunction, including a pneumonia-like syndrome termed acute chest syndrome, and strokes starting in childhood; and progressive multi-organ damage. Not surprisingly, patients with SCD have very high health care utilization (over $1 billion/year in healthcare costs in the United States alone4), and a median life-expectancy of only ~45–58 years, compared to the life expectancy of 78.2 years overall in the United States.3,5
The pathophysiology of sickle cell disease arises from a single amino acid alteration in adult hemoglobin, whose expression is primarily limited to RBCs. Nonetheless, the effects of the causative mutation are far reaching, mediated by the interacting processes of hemolysis and aberrant RBC behavior in the circulation. In this review, we first highlight the complex and multifaceted pathophysiological networks in SCD. We then review progress so far on the various strategies that have been used to intervene therapeutically in these networks, including agents that induce hemoglobin F (HbF), anti-sickling agents, modulators of ischemia–reperfusion injury and oxidative stress, anti-thrombotic therapies, anti-platelet therapies, anti-inflammatory agents, therapies to counteract free hemoglobin/heme and anti-adhesion agents. Here, we focus on agents that are currently either in clinical evaluation or have strong translational potential, while also noting lessons learned from failures of agents that are no longer being actively investigated. We also summarize emerging gene therapy approaches, including therapeutic gene transfer with lentiviral vectors and gene editing, which have the potential to be curative. Nevertheless, such therapies are still at an early stage of development, and their likely costs could limit access in many countries in which SCD is most prevalent. We therefore suggest that systems-oriented strategies based on the use of multiple agents with different targets could have a key role in improving the treatment of SCD, and we discuss challenges in the development of such strategies. Hematopoietic stem cell (HSC) transplantation from a normal donor is an established curative therapy for SCD, but is limited to 10–20% of SCD patients with an appropriately matched donor and not the focus of this review (see refs 6–11 for recent reviews).
[H1] PATHOPHYSIOLOGY OF SICKLE CELL DISEASE
The pathological single amino acid substitution (Glu to Val) at the sixth position of the β chain of hemoglobin S (HbS) results in a loss of negative charge and gain in hydrophobicity that alters hemoglobin dimer–tetramer assembly (Box 1), resulting in hemoglobin-S instability and HbS polymerization.12 Following deoxygenation of hemoglobin-S, deoxy-HbS aggregates densely pack into polymers, and the RBC changes shape (“sickles”) due to this polymer-induced distortion (FIG. 1a), giving the disease its name. This is the fundamental basis for the hemolytic anemia, vaso-occlusion associated with painful events, organ dysfunction and shortened life span in people with SCD. However, this simple Hb defect leads to a plethora of downstream effects, each of which sets in motion a particular pathophysiologic pathway, described below.
Box 1 |. Hemoglobin composition and function.
Human hemoglobin (Hb) is a metalloprotein that possesses a quaternary
structure. The quaternary structure is derived from four globular protein subunits.
The most common combination of globular protein subunits is called hemoglobin A
(HbA) and consists of two α-globin subunits encoded on chromosome 16 and two
β-globin subunits encoded on chromosome 11. Each monomer is attracted
electrostatically to unlike monomers to form α –β dimers and
ultimately assemble as an α2β2 tetramer to
optimally bind oxygen, forming HbA (α2β2). HbA
is the dominant Hb variant in healthy adults and accounts for roughly 97% of all
Hb.
The presence of hemoglobin S (HbS; α2βS2), a variant form of Hb that results from a mutation in the β-globin gene (HBB) at position 6 (GAG to GTG), creates a β6 Glu→Val substitution, which underlies sickle cell disease. Upon deoxygenation, HbS polymerizes into a semisolid crystalline-like polymer structure that is reversible with oxygenation. This property of HbS is responsible for the familiar shape change of the RBC to sickle or hook shapes, due to the sickle polymer-induced distortion.
Variations in the subunit composition of Hb also occur during development. The main form of human Hb during fetal development until approximately six months of age contains two α-globin subunits and two γ-globin subunits and is known as fetal hemoglobin (HbF; α2γ2 ). In early life, expression of the gene coding for the γ-globin subunit is repressed and HbA becomes the dominant form in healthy individuals, whereas HbS is produced in patients with sickle cell disease. As discussed further in the text, however, reactivation of HbF can be achieved pharmacologically, and this is useful in patients with sickle cell disease becausethe stronger negative charge of γ-globin allows it to outcompete βS globin for α globin, leading to the formation of HbF, rather than HbS.

Figure 1 |. Pathophysiology of sickle cell disease.

a | Sickle RBC oxidant and membrane changes. Upon deoxygenation of hemoglobin-S (Hb S), deoxygenated Hb S aggregates densely into polymers, and the red cell changes shape (“sickles”) due to this polymer-induced distortion. Spontaneous auto-oxidation of Hb S Fe2+ leads to the formation of Fe3+-methemoglobin and release of heme from the globin. O2 is reduced to superoxide and superoxide mutase (SOD) reacts to form H2O219. A catalytic cycle between ferric Hb-Fe3+ and the ferryl HbFe4+ can be initiated by H2O2222, which is subsequently eliminated in a pseudoperoxidase-like manner. Ferryl Fe4+ Hb S promotes βCys93 oxidation, Hb dimerization and hemichrome formation. Upregulated NADPH-oxidase19 contributes to the oxidative environment, as do down-regulated oxidative defense pathways.223 Sickle red cells also have retained mitochondria,224 providing another source of oxidants that lead to membrane damage. There is a decrease in glutathione, as well as reduced activity of glutathione peroxidase, vitamin E, catalase, and peroxiredoxin. Oxidative membrane protein and lipid changes, including phosphatidyl serine (PS) expression on the membrane, promote erythrophagocytosis, adhesion, complement activation and prothombinase assembly. Excess oxidants lead band 3 sulfhydryl oxidation, enhanced erythrophagocytosis, altered Na+,Ca2+, and K+ homeostasis, dehydration, mechanosensitivity and deformability, increased fragility and vesiculation of microvesicles (that contain Hb S and heme), increased activity of adhesion receptors on the red blood cell membrane, and hemolysis.b | Hemolysis results in several intravascular events promoting vaso-occlusion in SCD. The abnormal sickle RBC membrane changes utimately lead to hemolysis within the vasculature and the release into the plasma of hemoglobin, reactive oxygen species (ROS) and arginase225, which can accentuate nitric oxide (NO) depletion in the plasma. Free hemoglobin can react with NO and can readily oxidize to methemoglobin (MetHb); ROS can activate the endothelium of the vessel wall to promote adhesion molecule expression and the adherence of SRBCs, white cells (WBCs), and platelets. Within the vessel, free methemoglobin readily gives up its heme46, which can interact with inflammatory cells and the endothelium. Excess plasma hemoglobin and heme depletes the heme scavengers haptoglobin and hemopexin.226 The adhesion of SRBCs, WBCs and platelets slow the flow in postcapillary venules, leading to further SRBC deoxygenation, sickling, and vaso-occlusion.227
Figure 1A outlines how oxidative biochemistry of HbS and the sickle RBC can lead to a variety of cellular and membrane abnormalities that contribute to the pathophysiology. Figure 1 B proposes that these oxidative changes in the SRBC promotes hemolysis, depletion of nitric oxide, excess reactive oxygen species (ROS), and endothelial activation. The adhesion of SRBCs along with WBCs and platelets leads to vaso-occlusion in the post capillary venules, causing hypoxia, worsening deoxygenation and sickling and ultimately organ damage. The depletion of scavengers of free hemoglobin and heme, haptoglobin and hemopexin, enhance the pro-inflammatory, pro-adhesive and pro-coagulant environment within the vessel wall. Ultimately flow is restored and ischemia-reperfusion physiology with activation of xanthine dehydrogenase to xanthine oxidase can occur to promote more ROS production.13,14
[H2] Hemolysis.
It is estimated that about 1/3 of hemolysis in SCD is intravascular, due to mechanical destruction of deformed and inelastic SRBCs, while 2/3 is extravascular, resulting from removal of abnormal SRBCs by the reticuloendothelial system.15 The consequences of hemolysis include the immediate effects of RBC loss, such as anemia and expansion of erythropoiesis with increased reticulocytosis. Hemolysis also releases hemoglobin from RBCs or RBC microparticles into the plasma, leading to increased levels of toxic heme. Increased heme levels associated with chronic hemolysis cause depletion of plasma haptoglobin and hemopexin, which tightly bind heme. Hemolysis, membrane alterations, and their downstream effects also appear to promote decreased NO bioavailability, hemostatic system abnormalities, production and release of vasoactive agents, increased erythrocyte, leukocyte and platelet adhesion and activation, increased endothelial cell activation and injury, iron overload, vaso-occlusion, inflammation, and ischemia-reperfusion injury.16
[H2] Adhesion and coagulation.
SRBCs have increased expression and activation of various signal-transducing proteins, including ones belonging to the β2-adrenergic-Gαi-protein-adenylyl cyclase-protein kinase A and MAPK (MEK-ERK) pathways.17,18 Both of these pathways activate the normally inactive erythroid adhesion molecules BCAM/Lu, ICAM-4, and CD44, as well as NADPH-oxidase (NOX) pathways,19 which increase oxidant stress. Thus, SRBCs not only adhere to the endothelium but also interact with circulating and stationary leukocytes and platelets.20–26 Activation of circulating leukocytes and platelets further exacerbates the propensity to develop vaso-occlusion, and leukocytes that adhere to the endothelium readily “capture” circulating SRBCs. Adhesion of SRBCs also promotes expression of pro-adhesive and procoagulant proteins on the surface of endothelial cells.27 Damage to RBC membranes and alteration of cytoskeletal proteins, likely through the processes of reversible HbS polymerization, oxidant injury, and aggregation of proteins along the inner membrane leaflet, also result in poorly deformable RBCs that can be trapped in vessels, best seen in the spleen in children, independently of polymerized HbS.28
The coagulation system is hyperactivated in SCD. While any process that increases adhesion of SRBCs or leukocytes to endothelial cells can cause microvascular occlusion and promote stasis-induced microthrombi due to coagulation activation, SRBC intrinsic and extrinsic abnormalities themselves can activate the coagulation system and platelets. Phosphatidylserine (PS) externalization is commonly seen in SRBCs and in sickle reticulocytes29 and occurs secondary to increased oxidative stress,30 increased intracellular calcium induced by sickling, and cellular dehydration.31 More recently, it was shown that PS exposure may be a normal developmental process in immature reticulocytes undergoing autophagy, and the autophagic vesicles are removed by the spleen. However, due to trapping of abnormal RBCs in the spleen, patients with SCD have hypofunctional and eventually autoinfarcted spleens. Hyposplenia and eventual functional asplenia amplify the risk of infection by encapsulated bacteria and also result in increased levels of circulating mature reticulocytes expressing inside-out PS-exposed autophagic vesicles.32 All of these contribute to a hypercoagulable state primarily induced by PS-expressing sickle reticulocytes and SRBCs that increase thrombogenesis and promote adhesion.32
Patients with SCD show high plasma levels of coagulation factors involved in thrombin generation.33–36 In support of these clinical findings, a sickle mouse with genetically imposed reduction in thrombin showed significantly improved organ pathology.36 In addition, chronic platelet activation and aggregation is observed in SCD patients, which is evident by the expression of platelet activation markers.21,33 Platelet agonists such as thrombin, adenosine diphosphate (ADP), and epinephrine are increased in the plasma of patients with SCD and contribute to platelet activation. Activated platelets release thrombospondin that mediates adhesion of SRBCs to endothelial cell via both CD47 as well as CD36, cell surface molecules present on both erythroid and endothelial cells.34 CD40 and soluble CD40L, expressed on and shed by activated platelets in SCD, respectively, mediate increased coagulation. Ligation of CD40 on endothelial cells by CD40L increases tissue factor and ICAM-1 expression.34,37 A significant increase in platelet-monocyte and platelet-neutrophil aggregates are also observed in patients and mouse models of SCD, which is postulated to be mediated via a gpIIb/IIIa–fibrinogen interaction,38 although other lines of evidence point to a role for P-selectin in aggregate formation as well.39,40, 41,47,
[H2] Oxidation and inflammation.
Heme derived from SRBCs can act as a damage-associated molecular pattern (DAMP) that activates toll-like receptor 4 (TLR4) of the innate immune system, leading to oxidant production, inflammation, vaso-occlusion, ischemia, and tissue injury, including pulmonary injury.42,43 TLR4 ligation on the endothelium triggers exocytosis of Weibel-Palade bodies, which contributes to endothelial recruitment of leukocytes by increasing the expression of P selectin, and promotes coagulation through release of von Willebrand factor.44 TLR4 signaling also activates the inflammasome in monocytes and macrophages via NF-kB. Scavenging hemoglobin or heme with haptoglobin or hemopexin in sickle mice inhibits TLR4 activation and consequent vaso-occlusion. Excess hemoglobin/heme is degraded by heme oxygenase-1 (HO-1) with production of carbon monoxide. Ferritin—especially ferritin heavy chain (FHC) with its ferroxidase activity, is insufficiently upregulated to handle the heme burden in SCD patients, as evident in patients who have longer GT repeats and more frequent acute chest syndrome and who would presumably make less heme oxygenase in response to heme.45 Increasing HO-1 to degrade heme inhibits oxidative stress, inflammation, vaso-occlusion, and organ pathology in SCD mice.46
[H2] Interconnection in SCD pathophysiology.
As described above, SCD pathophysiology involves multiple processes, primarily affecting RBCs but also other blood cells, the vasculature, and multiple biological pathways.14 Vaso-occlusion and hemolysis are interrelated, amplifying each other and promoting the production of a myriad of biological mediators. Slow microvascular transit resulting from adhesive interactions and reduced RBC deformability enhances deoxygenation, polymerization, sickling, adhesion and vaso-occlusion.12 Ischemia-reperfusion injury, caused by vaso-occlusion followed by reperfusion, likely occurs in every organ of the SCD patient.13 In addition, SRBCs and monocytes directly activate adhesivity of both leukocytes and endothelial cells.47
These constant stimuli of inflammation and underlying systemic endothelial dysfunction represent a vicious circle, with increased RBC, WBC, platelet and endothelial cell adhesion promoting slower flow, and thus further deoxygenation, HbS polymerization, sickling, stasis, hemolysis and release of hemoglobin/free heme, and more vascular occlusion, followed by reperfusion, and more inflammation. This pro-inflammatory, pro-oxidative milieu is a critical contributor to the leukocytosis, WBC activation, acute phase reactants, cytokine elevation, excess oxidants, increased expression and activation of adhesion molecules by all blood cells, endothelial activation, expression of adhesion receptors, and dysfunction, aberrant vaso-regulation, decreased NO bioavailability, and coagulation activation, all of which ultimately result in multi-organ injury and pain in patients. These interconnected pathophysiologies are illustrated in Figure 2.
Figure 2 |. Sickle cell disease pathophysiological pathways and opportunities for targeted therapy.

The presence of sickle red blood cells (SRBCs) that contain hemoglobin S (HbS), along with SRBC-derived microparticles, are central to the pathophysiology of SCD and have a plethora of effects, ultimately leading to SRBC sickling, hemolysis, adhesive cell-cell interactions, inflammation, activation of coagulation, and endothelial dysfunction, as shown here. Cellular dehydration and deoxygenation along with hemoglobin S are at the center, leading to RBC sickling, hemolysis and release of free hemoglobin. Shown in the bottom left, free hemoglobin has several effects, including causing oxidant injury and endothelial activation. Abnormal membrane characteristics of SRBCs also activate coagulation processes that can then further activate endothelial cells, at bottom center, and inflammation, at bottom right. The SRBCs also adhere abnormally to leukocytes, other SRBCs, and the endothelial (shown on right). Leukocyte activate serves to proposgate the vaso-occlusive event, as well as causing ischemia/reperfusion injury. In each instance, investigators are working to develop agents to counteract these pathways (indicated in red).
[H1] DEVELOPING NEW SCD THERAPIES
Given the complexity of SCD pathophysiology, multiple therapeutic strategies have been pursued. To date, at least some progress has been made on most of the key fronts noted in the introduction of the article, which we review below and summarize in Tables 1–6. We also examine progress of of potentially curative gene therapy approaches, some of which are already in clinical trials while others are in clinical development. However, these approaches will not be at the forefront of treatment options for some time, especially as their application in countries with the highest numbers of people with SCD may be limited by their technical demands and cost. Consequently, we propose that a systems-oriented multi-agent approach targeting several aspects of the pathophysiological process in SCD should be pursued, and we also discuss the challenges for this strategy. General challenges for drug development for SCD are highlighted in Box 2.
Table 1 |.
Hemoglobin F inducers and anti-sickling agents
| Therapeutic agent |
Properties | Comments |
|---|---|---|
| Hemoglobin F inducers | ||
| Decitabine | DNA methyltransferase 1 inhibitor | Raised HbF 4–9% when given with tetrahydrouridine in Phase 1 trial (NCT01685515)56 |
| Suberoylanilide hydroxaminc acid (Vorinostat) | HDAC inhibitor | Phase 1/2 trial (NCT01000155) closed due to insufficient enrolment |
| LBH589 (Panibostat) | HDACinhibitor | Increased HbF in three patients with Hodgkin’s lymphoma)60; completion of phase 1 trial (NCT01245179) in SCD anticipated in 2019 |
| Sodium 2,2 dimethylbutyrate (HQK-1001) | HDAC inhibitor | 0.2 g/dl increase in HbF, mean increase in total Hb of 0.83 g/dl in Phase 2 study [Au:OK?] (NCT01322269)58; showed minimal HbF response in Phase 2 study (NCT01601340)59 |
| Pomalidomide | Reprograms erythropoiesis to fetal pattern, lowering expression of Bcl11a and Sox6 | Increases HbF in myeloma patients228; Phase 1 study in SCD (NCT0522547) completed but no results reported |
| Metformin | Induction of FOX03, but mechanism of induction of HbF in human erythroid cells is unknown | A pilot study (NCT02981329) in SCD patients is being conducted |
| Anti-sickling agents | ||
| Mg pidolate, in combination with hydroxyurea | Improvement in cell hydration (by Mg pidolate), along with ribonuclease reductase inhibition (by hydroxyurea) | A phase 1 study of this combination (NCT00143572) has been completed but results are not available |
| CO(MP4CO, PEG-bHb-CO) | Forms Hb-CO, prevents sickling | MP4CO withdrawn from development during phase 2 trial; Phase 2 trial of PEG-bHb-CO underway (NCT02672540)71,72 |
| 5 hydroxymethylfurfural (Aes-103) | Forms a high-affinity Schiff-base adduct with HbS, shifting oxygen-dissociation curve to the left and reducing sickling | Drug development has ceased despite success reported in early phase and animal studies. 229 |
| SCD-101, Niprisan | Prevents sickling by unknown mechanism74 | Phase 1B study ongoing in US (NCT02380079); niprisan safe and effective in animals230 and in phase 2 in Africa 75 |
| GBT 440(Voxelotor) | Covalently binds to N-terminus of Hb α chain, stabilizing HbS in the oxy-Hb conformation | Increased Hb level in early study76; Phase 3 multicenter study ongoing (NCT03036813) |
| Vanillin derivatives | Vanillin reacts in a transient covalent manner withHbS, resulting in allosteric modulation to a high-affinityHbSmolecule and by stereospecific inhibition of T stateHbSpolymerization. | In vitro studies of chemistry, crystallographic structure bound toHb,and anti-sickling effects 231–233 |
| VZH-039 | Binds to Hb in a transiently covalently manner, increasingHbaffinity for oxygen, with concomitant inhibition of polymerization of deoxygenatedHbS | In vitro studies show inhibition of sickling in hypoxic conditions234 |
| Triazole sulfide | Allosteric effector of Hb O2 affinity, stabilizing Hb in the relaxed state (R3-state) | Decreases RBC sickling in vitro 235 |
| RN1 | Inhibition of lysine-specific demethylase 1 (LSD1), resulting in increased Hgb F | Decreases inflammation and necrotic lesions in liver and spleen in sickle mice 63 |
| Sphingosine kinase 1 inhibitor (PF-543) | Inhibits sphingosine kinase-1-mediated elevation of sphingosine-1-phosphate, whichcontributes to sickling | Reduces sickling, hemolysis, and inflammation in sicklemice236 |
| Clotrimazole | Gardoschannel inhibitor, leads to more cell hydration | Abolishes hypoxia-induced SS RBC dehydration in SCD mice237; improved RBC hydration andHblevels in SCD patients238 |
| ICA-17043 | Gardoschannel inhibitor, leads to more cell hydration | ImprovedHblevel in phase 2, but Phase 3 study resulted in no reduction in VOC episodes239 |
Box 2 |. Challenges in drug development for sickle cell disease.
Preclinical drug development for SCD has benefited from the creation of two murine models of SCD218,219. Although neither faithfully mimics all aspects of SCD, many processes, such as vaso-occlusion and end-organ damage, can be studied reasonably well in these mice.
Once a candidate drug is ready to enter clinical trials, however, there are several challenges. One is the many presentations of SCD, including acute pain (vaso-occlusive crises (VOCs), chronic pain, fatigue, stroke and cognitive impairment, acute chest syndrome, pulmonary hypertension, cardiac dysfunction, nephropathy, leg ulcers, among others. The most common and devastating of these—acute pain episodes, stroke and acute chest syndrome—may arise from different processes, which are incompletely understood. In addition, there is no “gold standard” or biomarker for identifying a VOC, and diagnosis of an event as well as definition of its resolution therefore rely on patient-reported symptoms.
Thus choosing both a therapeutic endpoint (e.g. prevention or amelioration of acute vaso-occlusion) and then deciding how to define the endpoint and what change in the endpoint is clinically meaningful can be difficult. Some of the endpoints considered in recent clinical trials designed to achieve prophylaxis endpoints have been reduction in rate of hospitalization, rate of VOC, frequency of acute chest syndrome, degree of chronic pain, analgesic usage, rate of RBC transfusion due to SCD, reduction in school absence, and incidence of stroke. Drugs designed to improve acute pain episodes have generally defined endpoints as length of pain crises, time to resolution from start of therapy, or length of hospitalization for VOC. Most of these endpoints, however, are made more difficult to analyze by the extreme heterogeneity that characterizes SCD. Not only are there different types of SCD (e.g. hemozygosity for HbS or compound heterozygosity for HbS and HbC), but even within specific genetic types, measures such as frequency and length of painful episodes are quite variable.
In addition, without well established evidence-based guideines, there is abundant institutional heterogeneity in day-to-day and acute event management of patients. Although heterogeneity can be overcome by enrollment of large number of subjects, that is also hard to achieve in SCD. With <100,000 SCD patients in the US, <15,000 in the UK, but millions in Africa and India, the resources and skills needed to conduct clinical trials are geographically mismatched with the potential subjects. And even in the US, many centers with hundreds of SCD patients have few resources available to conduct clinical trials well.
Overall, these factors make conduct and successful completion of clinical trials in SCD difficult, although a number of phase 2 clinical studies have recently met their targeted enrollment and been successfully completed.149,150,220 Most of these have used frequency and/or length of acute painful episodes as their primary endpoint for judging efficacy, much as did the Multicenter Study of Hydroxyurea two decades ago. 221
[H2] Pharmacological reactivation of HbF.
HbF has long been known to retard polymerization of HbS48. Indeed, infants with SCD are not symptomatic until their HbF production declines, and SCD patients with hereditary persistence of HbF have a mild disease phenotype, characterized by fewer VOC, less cumulative organ damage and longer survival (reviewed in 3,49). Indeed, clinical studies have shown that a threshold of 10% HbF reduces major organ damage, while 20% HbF results in amelioration of VOC and pulmonary complications in SCD.50,51 Consequently, reactivation of HbF has been a major focus of both drug development and gene therapies (discussed separately in a later section).
Chemotherapeutic/cytotoxic agents that impose regenerative hematopoietic stress increase HbF production in erythrocytes. One of the first DNA hypomethylating drugs (a DNA methyl transferase [DNMT] inhibitor) used to reactivate HbF was the chemotherapeutic drug, 5-azacytidine.52–54 However, it was quickly abandoned due to its toxicity and carcinogenicity. Next, hydroxyurea, a chemotherapeutic drug that is a ribonucleotide reductase inhibitor without DNA hypomethylating activity and has a known long term-safety profile in myeloproliferative diseases, was shown to reactivate HbF production. Its mechanism of action is still unclear but thought to be from cytotoxicity-induced stress erythropoiesis. The expression of HbF with hydroxyurea is generally heterocellular. In 1998, hydroxyurea became the first drug to be licensed by the FDA for adults with SCD, and its use has led to a reduction of complications of SCD in adults, children, and even infants, as well as prolonged survival (reviewed in 55).
However, since hydroxyurea does not improve HbF production in all patients, requires regular monitoring of blood counts, and in some patients has intolerable side effects, many alternative HbF inducing agents have been explored. Molecular studies of globin gene regulation have identified the γ-globin repressor protein complex: TR2 and TR4 non-steroidal nuclear receptor proteins specifically bind γ-globin regulator elements and subsequently recruit a multi-protein co-repressor complex that includes DNA methyltransferase 1 (DNMT1), lysine specific demethylase 1 (LSD1), and histone deacetylases (HDACs), Bcl11a, and Sox6 to repress gene expression. Hence, drugs targeting repressive complex proteins have been explored (summarized in Table 1).
Decitabine was found to be a potent inhibitor of DNMT1, in addition to having a more favorable safety profile than 5‐azacytidine. The oral formulation of decitabine, however, is rapidly deaminated by cytosine deaminase and inactivated; when given with a cytosine deaminase inhibitor, tetrahydrouridine, this limitation is overcome. This combined therapy was the subject of a phase I trial (NCT01375608) which showed HbF increase by 4%−9% and doubling of HbF-enriched RBCs (F-cells) up to approximately 80% of total RBCs. Importantly, this agent does not induce cytoreduction but increases platelet counts, and while this was not dose limiting in this early phase study, it may become problematic in SCD. An active trial is ongoing (NCT01685515) and results are awaited. 56
Another category of HbF-inducing agents that have been studied are histone deacetylase (HDAC) inhibitors. Arginine butyrate, a short chain fatty acid, was shown to increase HbF in pulsed parenteral dosing,57 although its oral formulations had only modest effects. Several different isoforms of HDAC are present, and it is not surprising that global HDAC inhibition with butyrates was associated with toxicities, and a pulsed or intermittent dosing regimen was necessary to avoid cytotoxicity. Another oral short chain fatty acid derivative, sodium 2,2 dimethylbutyrate (HQK‐1001), with a more favorable toxicity profile and strong preclinical data on HbF induction, went through early phase clinical trials and showed a mean absolute increase in HbF of 0.2 g/dl and a mean increase in total hemoglobin (Hgb) of 0.83 g/dl above baseline in SCD (NCT01322269 58). However, the double blind phase 2 trial showed minimal-modest increases in HbF in 24% of patients (NCT01601340).59 Suberoylanilide hydroxaminc acid (Vorinostat), an orally available HDAC inhibitor approved for treatment of cutaneous T-cell lymphoma, showed efficient HbF induction in vitro and was tested in a phase 1/2 trial in SCD (NCT01000155). However, the trial was closed due to insufficient enrollment. In general, clinical and investigational HDAC inhibitors were limited by non-class-specific HDAC inhibition-associated toxicities. Using small hairpin RNA-based knockdown of HDAC1–9 showed that knockdown of HDAC1/2 improved HbF without global histone acetylation. A small molecule (LBH589, Panabinostat) that inhibits these HDAC isoforms at low concentrations was shown to improve HbF in three patients treated for Hodgkin’s lymphoma.60 Following this, a trial of panabinostat was inititated in SCD (NCT01245179) and is anticipated to be completed in 2019. Overall, studies on HDAC inhibition have shown that the toxicities of global HDAC inhibitors, especially with life-long use, undermined their translation into effective HbF-inducing therapies; specific HDAC-1/2 inhibitors with no effect on global acetylation may have potential.
Thalidomide derivatives have also been tried (shown in Table 1). Other agents that inhibit the γ-globin repressor protein complex, specifically lysine specific demethylase 1 (LSD1), show great promise. Tranylcypromine (TCP), an LSD1 inhibitor, was shown to induce HbF in vitro and in mice.61 Since TCP has known side effects that preclude its use in SCD, a more potent and specific LSD1 inhibitor, RN-1 was investigated and found to increase HbF in sickle mice.62 It has been also tested in baboons, where it showed strong HbF induction,63 although one concerning effect was neutropenia and thrombocytopenia. The LSD1 inhibitor INCB059872 is in a phase 1 trial (NCT03132324).
Finally, metformin, a FOXO3 inducer, has been shown to increase HbF production in erythroid cells in vitro, and is now being tested in patients with SCD (NCT02981329). Other promising HbF reactivating agents currently in preclinical studies include recombinant SIRT164, a hydroxyurea-thalidomide hybrid,65 resveratrol,66 and dimethyl fumarate.67
Overall, with better understanding of the molecular regulation of globin gene switching, key repressors such as Bcl11a, Sox6, Myb, Klf1, TR2/TR4, HDAC1/2, LSD-1 have been identified, with Bcl11a identified as a major repressor of HbF. Compounds that target these with specificity hold the potential to translate to more potent and less toxic HbF inducers.
[H2] Anti-sickling agents.
Hemoglobin polymerization and subsequent cell “sickling” are promoted by HbS deoxygenation and higher Hb concentrations. Therefore, agents that shift the oxygen dissociation curve or otherwise push the conformation of HbS to the R (oxygenated) state will reduce Hb polymerization and cell sickling, resulting in a reduction of hemolysis and improvement in anemia (Figure 1A). Other less well characterized chemicals that influence Hb conformation may also affect Hb polymerization and cell sickling (Table 1).
One anti-sickling approach that has been tested is to deliver carbon monoxide (CO) to HbS. CO in low concentrations may act to prevent sickling, hemolysis, and vaso-occlusion through multiple mechanisms, including a direct effect on Hb conformation, inhibition of dehydration through the Ca2+-permeable cation conductance channel, anti-oxidant effects, and anti-inflammatory effects through heme oxygenase-1 and Nrf2.68 In sickle mice, an infusion of pegylated carboxyHb (MP4CO) improved vascular stasis in response to hypoxia/reperfusion through a heme oxygenase-1-dependent mechanism. MP4CO also induced Nrf2, a transcriptional regulator of several anti-oxidant enzymes, and inhibited NF-kB activation.69 MP4CO successfully completed a phase 1 study in SCD patients in steady state without major safety concerns,70 but development of this compound has since been discontinued for undisclosed reasons.
A somewhat similar product, pegylated bovine carboxyhemoglobin (PEG-bHb-CO), has been proposed to both release carbon monoxide and transfer oxygen to hemoglobin. In animal models of various types of ischemia, infusion of PEG-bHb-CO was able to reduce tissue damage and promote recovery.71 In early phase human studies, this agent has been judged safe in both healthy and steady-state SCD subjects.72 A phase 2 randomized, single-blind study of the drug in SCD patients experiencing vaso-occlusive episodes is now underway (NCT02672540). PEG-bHb-CO is also being studied in other conditions involving severe anemia and tissue damage.
SCD-101 is an oral botanically derived drug that has an anti-sickling effect in vitro, albeit by an unknown mechanism.73 A similar preparation, NIX-0699 (Niprisan; Xechem International), was developed from a combination of African plants as an herbal medicine and has been shown to significantly prolong the delay time prior to polymerization (i.e. the length of the delay until polymer forms due to the conversion of HbS from R (oxy) to T (deoxy ) states),12 although it has only a minor effect on the oxygen dissociation curve.74 NIX-0699 was reported as both safe and effective in reducing the frequency of vaso-occlusive symptoms in a phase 2 study in Africa.75 A phase 1B two-part study of SCD-101 (NCT02380079) comprising a non-randomized dose-escalation phase followed by a randomized, placebo-controlled, crossover study, is currently ongoing in the U.S.
GBT-440 is another oral anti-sickling drug under development and currently has both U.S. Food and Drug Administration (FDA) Fast Track and Orphan Drug designations. This compound forms a reversible covalent bond to the N-terminus of the Hb α chain, thereby stabilizing HbS in the oxy-Hb conformation. As reported in abstract form, GBT-440 treatment of 41 SCD patients for up to 6 months resulted in hematologic responses, including increased Hb and decreased reticulocytes and/or bilirubin.76 Slightly less than half of treated patients experienced a clinically meaningful Hb increase of > 1 g/dL. A phase 3, double-blind, randomized, placebo-controlled, multicenter study of GBT-440 in SCD (NCT03036813) is now underway in the U.S. Nonetheless, there remain at least some concerns that treatments that shift hemoglobin oxygen affinity may bring significant risks to cerebrovascular oxygenation.77 GBT-440 and other agents with similar effects on hemoglobin decrease the oxygen-carrying capacity of hemoglobin and thus have been theorized to have potential negative effects on tissues with especially high oxygen demand, such as the brain and kidney.78 However, data presented to date have not shown evidence of adverse effects due to decreased oxygen availability to such organs.
[H2] Modulators of ischemia–reperfusion injury and oxidative stress.
In SCD, ischemia–reperfusion physiology — caused by the transient vaso-occlusion followed by the opening of vessels to re-establish flow — seems to lead to enhanced oxidant production (Figure 1B). The conversion of xanthine dehydrogenase to xanthine oxidase in tissues results in the production of superoxide anion and hydrogen peroxide. These ROS activate NF-kB, stimulating the production of inflammatory cytokines such as tumor necrosis α (TNFα), upregulating adhesion molecule expression, and promoting leukocyte, platelet and pro-coagulant activation (see Table 2).
Table 2 |.
Modulators of ischemia/reperfusion, oxidative stress, and inflammation
| Therapeutic agent | Properties | Comments |
|---|---|---|
| Modulators of ischemia reperfusion and oxidative stress | ||
| Allopurinol | Inhibits xanthine oxidase | No effect on sickle blood cell adhesion after hypoxia/reoxygenation (H/R) 84 |
| N-Acetyl cysteine | Enhances sulfhydryls, anti-oxidant | Reduced oxidative stress in SCD patients86 |
| Superoxide dismutase | Dismutates superoxide | Blocks blood cell adhesion in sickle mice84 |
| Suberoylanilidehydroxamic acid (SAHA) | Iron chelator, induces hemoglobin F, HDAC inhibitor | Decreased VOC, decreased tissue factor in SCD mice240 |
| Trimidox | Iron chelator | Decreased oxidants, inflammation and VO in SCD mice241 |
| PolynitroxylAlbumin | Scavenges superoxide | Decreased H/R induced VOC in SCD mice242,243 |
| Alpha lipoic acid | Antioxidant | Increased catalase in sickle RBCs244 |
| L carnitine | Mitochondrial ATP | Pulmonary hypertension improved245; no improvement in leg ulcers246 |
| Glutamine | Increase NAD | Decreased SRBC adhesion81 |
| NADPH oxidase inhibitors | Blocks superoxide production | Improved endothelial dysfunction in SCD mice247 |
| Sirolimus | mTOR inhibitor | Decreased ROS in SRBCs and decreased mitochondrial retention224 |
| PF-04447943 | Phosphodiesterase-9 inhibitor, increases cellular cGMO | Safe when tested in a phase 1 study (NCT02114203) in SCD |
| Riociguat | Activates soluble guanylyl cyclase | In phase 2 study for severe SCD |
| Anti-inflammatory agents | ||
| Regadenoson | Modulates invariant NKT cells | Reduced inflammation in SCD patients248 |
| Omega 3 fatty acids | Alters membrane fatty acids | Protected against vasculopathy in SCD mice 249,250 |
| Etanercept | Blocks TNF alpha | Improved abnormal endothelial activation, vaso-occlusion, and pulmonary hypertension in SCD mice93 |
| TAK-242 | TLR4 antagonist | Blocked P selectin expression and VOC in SCD mouse43,44 |
| Carbon monoxide | Blocks polymerization, anti-inflammatory | Decreases inflammation and VOC in SCD mice68,251,252 |
| Statins | Down regulate inflammatory mediators, decrease adhesion molecule expression increase NO | Decreased inflammatory markers in SCD patients101 |
| Dexamethasone | Anti-inflammatory | Decreased acute chest duration hospitalization in SCD patients172 |
| AKT 2 inhibitor | Regulate platelet P selectin and Beta 2 integrins | Decreased VOC and neutrophil heterotypic aggregates in mice140,141 |
| Zileuton | 5-lipoxygenase inhibitor, anti-inflammatory | Increased HbF in sickle erythroid precursors, decreases airway response in SCD mice253,254 |
| Leukotriene inhibitors | Block leukotrienes | Increased leukotrienes in acute chest in SCD 255 |
| Intravenous gamma globulin (IVIg) | Stabilizes neutrophil Mac-1 activation during vaso-occlusion, thus decreasing neutrophil adhesion to endothelium and RBC-neutrophil interactions | Decreased Mac-1 activation in SCD patients experiencing vaso-occlusion108,109 |
| Endothelin receptor blockade | Blocks endothelin receptor on endothelium | Reduced vaso-constriction and inhibits neutrophil adhesion in SCD mice105 |
| 2F-Fucose | Block fucosylation, Interferes with PMN/selectins | Blocked VOC in SCD mice decreased WBC rolling and adhesion256 |
| Antibiotics | Alters microbiome | Decreased aged neutrophils and inflammation in mice257 |
| Complement inhibitors | Inhibits C5a | Inhibited hypoxia /re-oxygenation vaso-occlusion in mice258 |
| Curcumin | Decrease inflammatory cytokines, increase antioxidants, | Decreased inflammation and pain responses in SCD mice259 |
Glutamine and glutathione levels have been found to be decreased in SRBCs. Moreover, RBC glutathione depletion in SCD has been linked to hemolysis and oxidative stress.79 Glutamine taken up by RBCs can be converted to glutamate by deamination and used as a precursor of glutathione.80 L-glutamine therapy also reduces the endothelial adhesion of SRBCs to human endothelial cells.81 Following a phase 3 randomized trial in SCD, in which oral L-glutamine therapy demonstrated clinical benefit, with reduced frequency of VOC and hospitalizations over 6 months,82 as well as decreased incidence of acute chest syndrome, L-glutamine became only the second agent to be approved by the FDA for SCD in 2017.83
A direct approach to reducing oxidative stress could also be to inhibit the production of ROS. Allopurinol is a xanthine oxidase inhibitor that is approved for the treatment of high uric acid levels derived from purine metabolism. One study of this drug in sickle mice showed no benefit in blunting blood cell/endothelial cell adhesion responses, while another suggested benefit with improved blood flow and decreased leukocyte recruitment.84,85 However, during ischemia purine triphosphates are released from cells, stoking these purine pathways. A trial in human SCD seems warranted, possibly performed in combination with other agents.
N-acetyl cysteine (NAC) has been used to treat a variety of pro-oxidative injury states, such as acetaminophen toxicity. In SCD, providing enhanced sulfhydryl thiols using agents such as NAC to increase levels of amino-thiol glutathione (GSH) in RBCs and thereby decrease oxidative stress seems a promising strategy. An open label pilot study of NAC treatment in SCD patients for 6 weeks decreased RBC phosphatidylserine exposure, increased GSH levels and decreased the plasma concentration of free hemoglobin.86 A follow-up study focused on NAC’s ability to reduce von Willebrand factor (VWF) activity suggested improvements in RBC parameters, decreased platelet adhesion, reduced VWF adhesive activity and protection against oxidative stress.87 However a randomized controlled Phase 3 trial of NAC in SCD patients did not provide any clinical benefit, although this was possibly due to poor patient compliance.88 Further studies are therefore warranted.
[H2] Anti-inflammatory agents.
Inflammation is a key factor in propagating and exacerbating the tissue injury caused by sickle cell disease. Essentially all cellular components of the immune system, including neutrophils, lymphocytes, monocytes, and platelets, contribute to this process, and many pro-inflammatory cytokines are either chronically elevated or become acutely elevated in sickle cell disease. Therefore, various anti-inflammatory treatment approaches have been investigated.
Invariant natural killer cells (iNKT) contribute to inflammation that promotes vaso-occlusion and pulmonary injury in sickle mice89 via release of cytokines and cytolytic mediators.90 However, while infusion of regadenoson to adults with SCD decreased the activation of iNKT cells91, it was not sufficient to produce statistically significant changes in iNKT activation or in measures of clinical efficacy.92
Production of tumor necrosis factor α (TNFα) by monocytes and macrophages is an early response in ischemia reperfusion injury. The myriad of inflammatory responses mediated by TNFα mirrors the sickle milieu, leading to vascular dysfunction and vaso-occlusion. Etanercept, a TNFα receptor blocker, given long term to sickle mice, decreased vaso-occlusion, responses to painful agonists, monocyte activation, biomarkers of inflammation, liver injury and surrogate markers of pulmonary hypertension.93 No human trials have been initiated to date, although a report of two cases of concomitant rheumatoid arthritis and SCD suggest that the drug can be safely used in SCD.94
Intravascular hemolysis is a hallmark of SCD. Infusion of heme into SCD mice causes inflammation, microvascular stasis, pulmonary injury and death,43,44 and these adverse events can all be prevented by an antagonist of TLR4, TAK-242. Like TLR4, the complement system also contributes to SCD pathogenesis in part due to ischemia-reperfusion physiology.95 The alternative complement pathway is abnormally activated in SCD and is amplified by phosphatidylserine (PS) on the outer leaflet of SRBCs and microparticles(MPs).96 PS on the surface of SRBCs and activated platelets accelerates the assembly of prothrombinase complexes, leading to thrombin generation, which can generate biologically active complement fragments.97,98 Recent studies suggest a possible role for antibodies to C5 that block microvascular stasis in sickle mice.99 C5a receptor antagonists are also under development.
Statins probably reduce cardiovascular events not only through cholesterol lowering but also through their anti-inflammatory effects. Simvastatin given to SCD patients decreased expression of CRP, sICAM-1, soluble E-selectin, VEGF and frequency of pain.100,101 These preliminary data support a larger trial in SCD.
Endothelin-1 (ET-1) levels are increased in SCD.102 ET-1 is a potent vasoconstrictor that plays a potent pro-inflammatory and pro-oxidative role in SCD. ET-1 receptor antagonists in SCD mice alter RBC membranes, cell adhesion, pain, and hypoxia-induced death.103–105 Endothelin B receptor blockade may thus be a potent anti-inflammatory approach in SCD by blocking inflammatory PMNs and lymphocytes and decreasing endothelial cell activation.106
Intravenous immunoglobulin has been shown to reverse vaso-occlusion in sickle mice.107 IgG can modulate neutrophil activation and vascular injury through FcγRIII and SHP-1.108 A single dose of intravenous gammaglobulin can stabilize neutrophil Mac-1 activation during sickle pain.109 A Phase 1–2 clinical trial of intravenous IgG is now underway for treating VOC (NCT01757418).
Phosphodiesterase-9 (PDE9) inhibitors block the degradation of cGMP and increase cellular cGMP levels. Increased cGMP is anti-inflammatory and has been shown to reduce the adhesion of leukocytes to vascular endothelium.110 In conjunction with hydroxyurea, the PDE9 inhibitor BAY73–6691 in sickle mice improved leukocyte rolling and decreased heterotypic RBC-leukocyte interactions, which was coupled with prolonged animal survival. In 35 SCD patients, BAY 73–6691 decreased levels of the inflammatory markers TNF and myeloperoxidase and increased levels of NO-containing species and and the antioxidant enzyme superoxide dismutase.111 The PDE9 inhibitor IMR-687 increased cGMP levels in K562 cells, and in sickle mice IMR-687 decreased the percentage of sickled RBCs, increased HbF positive cells, and decreased microvascular stasis in response to hypoxia/reoxygenation.112 And the PDE9 inhibitor PF-04447943 was also safe when tested in a phase 1 study (NCT02114203) in sickle cell patients. Investigations of PDE9 inhibitors, as well as activators of soluble guanylate cyclase, such as riociguat (NCT02633397), continue to be enthusiastically pursued in order to increase cGMP levels, as does exploration of additional anti-inflammatory approaches to sickle cell disease.
[H2] Counteracting free hemoglobin/heme.
Hemolysis of SRBCs has several negative effects, including decreased nitric oxide (NO) bioavailability, increased oxidative stress, arginine depletion due to excess arginase, and inflammation.
NO can vasodilate, inhibit platelets and alter inflammation, and it also plays a role in pulmonary hypertension. Various strategies to increase levels of NO have been explored in clinical trials in SCD (Table 4), and some have shown promising effects. For example, arginine supplementation has been shown to have efficacy in leg ulcers and pain in SCD.113 In an early clinical trial there were no adverse effects from arginine, and narcotic use was decreased by >50%.114 A current study (NCT02447874) is examining the role of supplemental arginine in the treatment of VOC in children. Hydroxyurea’s beneficial effects in SCD in countering hemolysis-induced inflammation may also be mediated by NO through peroxidase-mediated formation of NO from hydroxyurea, hydrolysis of hydroxyurea to hydroxylamine,and/or the NO-producing structure-activity relationships of hydroxyurea.115,116
Table 4 |.
Agents that counteract free hemoglobin, heme and iron
| Therapeutic agent | Properties | Comments |
|---|---|---|
| Nitric oxide | Vasodilates, anti-adhesive, replaces bioavailable NO | Inhaled NO did not improve SCD patients hospitalized with VOC266; NO helped priapism in SCD mice 267 |
| Arginine | Substrate for NO synthesis | Efficacy in leg ulcers and pain in children114 |
| Tetrahydrobiopterin | Co-factor for nitric oxide synthase | Improved endothelial dysfunction in several clinical settings268 |
| Sildenafil | Blocksphosophodiesterase, increases cGMP | Increased hospitalizations in SCD patients269 |
| Nitrite | Vasodilates, supplies NO | Enhanced healing of leg ulcers in SCD270 |
| Hemeoxygenase inducers | Increases CO, biliverdin/bilirubin, ferritinand CO | Decreased VOC and inflammation in SCD mice42 |
| Dimethyl fumarate | Increases Nrf2 | Increased HO1 and decreased VOC in SCD mice271 |
| Nrf2 inducers | Increases anti-oxidant, anti-inflammatory response | Decreased inflammation in SCD mice272,273 |
| Haptoglobin | Clears free hemoglobin via274 CD163 | Decreased inflammation in SCD mice44,275 |
| Hemopexin | Clearshemevia CD91 | Decreased TLR4 signaling in SCD mice43,44,118,276 |
| Iron chelators | Bind and clear excess iron, decrease oxidative stress | Decreased inflammation and VOC in SCD mice44,241 |
In SCD, chronic hemolysis also depletes plasma haptoglobin and hemopexin. Studies in SCD mice44,117–119 suggest beneficial effects of supplementation, as co-infusion of human haptoglobin or hemopexin with hemoglobin/haptoglobin or hemoglobin/hemopexin reduced vascular stasis, with these effects involving activation of Nrf2 and HO-1. Importantly, haptoglobin or hemopexin supplementation also inhibited VOC in unchallenged sickle mice, demonstrating the ongoing nature of hemolysis-driven VOC in SCD.44,118,119 Although not currently available therapeutically as purified proteins, both haptoglobin and hemopexin are potentiallyextractable from human plasma for therapeutic interventions. In all, approaches to reducing inflammation and oxidative injury remain among the most exciting new areas in drug development for sickle cell disease.
[H2] Anti-thrombotic therapies.
In SCD, where vascular occlusion, increased blood viscosity, activation of the coagulation system and endothelial injury/dysfunction promote thrombosis, anticoagulation has been long considered a potential therapeutic avenue. Coagulation activation occurs via multiple mechanisms in SCD: via tissue factor (TF) and phosphatidylserine (PS) expressing microparticles and abnormal exposure of PS on SRBCs and reticulocytes, erythrocyte-platelet and neutrophil-platelet aggregates in the circulation, endothelial cell activation, and hemolysis itself.120,121,35,122 Increased thrombin (Factor IIa) generation occurs in SCD, both by activated contact pathway123–128 and the TF pathway, due to aberrant and increased TF expression by monocytes and endothelial cells, which together with its ligand Factor VIIa, activates Factor X (FX).129,130 Activated Factor X (FXa) results in generation of thrombin (Factor IIa) from prothrombin (Factor II) and increased fibrin generation.
Hence lowering thrombin generation has long been a therapeutic target in SCD (Table 3). Studies with warfarin and heparins beginning nearly 70 years ago have provided some initial indications of therapeutic activity131,132,133,134, but warfarin is limited by its bleeding risk and heparins by the need for parenteral administration. With the advent of oral direct thrombin inhibitors (dabigatran) or factor Xa inhibitors (such as apixaban and rivaroxaban), these disadvantages of warfarin and heparin can be overcome. Indeed, apixaban and rivaroxaban are now in initial short-term clinical trials, the results of which await publication. Long-term use of these anticoagulant strategies may have the potential to ameliorate chronic organ damage based on animal studies where such an effect has been shown.36
Table 3 |.
Anti-platelet agents and anticoagulants
| Therapeutic agent | Properties | Comments |
|---|---|---|
| Anti-platelet therapies | ||
| Prasugrel | An oral P2Y12 adenosine diphosphate (ADP) receptor antagonist that inhibits ADP-mediated platelet activation and aggregation | After early promising results, a large multicenter randomized placebo-controlled trial showed modest platelet inhibition but no significant difference in rate of VOC events or other clinical endpoints220 |
| Ticlopidine | An oral P2Y12 adenosine diphosphate (ADP) receptor antagonist that inhibits ADP-mediated platelet activation and aggregation; discontinued due to serious side effects and newer P2Y12 inhibitors | Despite some mildly promising results of reduction in painful events260,261, no further clinical trials have been pursued |
| Aspirin | NSAID; prostaglandin synthase inhibitor that irreversibly inhibits platelets by decreasing conversion of arachidonic acid to prostaglandin | Four trials of aspirin in SCD in the 1980s showed no consistent effect on frequency of VOC or RBC indices |
| Piroxicam | NSAID, inhibits prostaglandin synthesis to inhibit platelet aggregation | Peroxicam but not aspirin showed a reduction in mean pain scores and mean mobility scores, and was not associated with side effects262 |
| Eptifibatide | Cyclic heptapeptide and reversible gp IIb/IIIa inhibitor | After small proof-of-principle study showed decreased platelet activation and reduction in CD40L shedding, a small randomized pilot study showed safety but no improvement in median times to discharge/crisis resolution or total opioid use263 |
| Anti-thrombotic therapies | ||
| Warfarin, icumarol, acenocoumarol | Oral vitamin K antagonists that inhibit production of procoagulant factors II, VII, IX and X as well as anticoagulant factors (protein C and protein S) | Evidence limited to small studies; long-term use of dicumarol resulted in healing of leg ulcers but no benefit in VOC132; warfarin showed modest reduction in VOC from 1.3 VOC/year to 0.9 VOC/year in one small study131; acenocoumarol reduced markers of coagulation activation but did not affect frequency VOC264 |
| Heparin | Anti-thrombin | Mini-dose heparin for 2–6 years showed ~70% reduction in VOC hospitalizations/ED visits in one small study133 |
| Low-molecular weight Heparins | Anti-thrombin | One large phase 2 study showed reduction in VOC duration134; a smaller randomized study of dalteparin (NCT01419977) showed reduction in D-dimer and thrombin generation, as well as pain scores265 |
| Rivaroxiban, apixaban | Factor Xa inhibitors | Randomized placebo-controlled trials underway (NCT02072668, NCT02179177) |
[H2] Anti-platelet therapies.
Platelet counts are higher in SCD patients under steady-state conditions and further increase during VOC.21,33 Chronic platelet activation is observed in SCD21,33,135 and is conceivably from increased circulating platelet agonists, such as thrombin, adenosine diphosphate (ADP) and epinephrine, as well as increased platelet-monocyte and platelet-neutrophil aggregates. Activated platelets promote adhesion of SRBCs to vascular endothelium and contribute to micro-thrombosis and pulmonary hypertension in SCD.33 Thrombin activates platelets through proteolytic cleavage of two major protease-activated receptors, PAR1 and PAR4.136,137 Cell-free hemoglobin, increased secondary to hemolysis, can also trigger platelet activation, and the bioavailability of NO, a key inhibitor of platelet activation, is reduced in SCD.33,138 Hence a plethora of factors mediate platelet activation in SCD, and it has therefore been targeted to reduce acute events.
Preclinical studies with antiplatelet agents such as the P2Y12 antagonist clopidogrel supported the possibility that blocking platelet activation and aggregation may be beneficial in SCD39,139,140.141. However, clinical trials of antiplatelet therapies in SCD have yielded modest to disappointing results so far (summarized in Table 3). For example, several recent studies with the third-generation P2Y12 antagonist, prasugrel, have been disappointing. While no major bleeding complications were seen with prasugrel in four trials,142–146 including one large randomized placebo controlled multicenter trial,143 these showed a non-significant decrease in VOC despite significant platelet inhibition.
Overall, the studies summarized in Table 3 suggest that platelet activation may not play a major role in sickle cell acute VOC. Indeed, these studies are reminiscent of results observed in studies with anticoagulants, where no clear benefit in acute events was evident. However, reducing thrombin generation chronically in SCD mice led to a remarkable improvement in multi-organ pathologies and improved survival.36 An early clinical report showed no difference in acute VOC with prolonged use of the anticoagulant dicumarol, but healing of leg ulcers was observed, suggesting that chronic anticoagulation or anti-platelet therapy may remain worthy of investigation with organ damage as the endpoint of study rather than VOC.132 None of these anti-platelet studies have addressed the potential effects of chronically reducing platelet activation and the implications for long-term organ damage, which remains a major cause of death in SCD despite improvements in preventive care. Also, antiplatelet therapy is usually best at preventing arterial complications, and in SCD, these conditions (stroke, pulmonary hypertension or priapism) have not been studied in SCD trials with antiplatelet agents.
[H2] Anti-adhesion agents.
Considerable progress in has been with anti-adhesion agents for SCD, and drugs that interfere with selectin-mediated adhesion currently appear the most promising in this area. Selectins are expressed by endothelial cells as well as all circulating blood cells except erythrocytes. Receptors for selectins are present on all blood cells, including erythrocytes. In general, selectins mediate fast-on–fast-off cell–cell interactions and therefore facilitate “rolling,” a phenomenon in which cells have intermittent or even continuous contact with endothelial cells as they traverse the circulation. Furthermore, these interactions lead to firmer attachment through integrin-mediated interactions, resulting in stable attachment and, in the case of leukocytes, migration of the cells through the endothelial layer into tissues in the presence of appropriate chemokine signals. However, it remains unclear whether E- or P-selectin is most important to the pathophysiological mechanism of vaso-occlusion. Animal studies to date are somewhat conflicting, with at least some suggesting that both are important.24,147,148
Table 5 summarizes the anti-adhesion agents that have been tested clinically. Thus far, three pharmacological inhibitors of selectin-mediated adhesion have been or are in human clinical trials. Rivipansel (formerly known as GMI-1070) is a small-molecule pan-selectin inhibitor that requires intravenous infusion and has strongest activity against E-selectin. After completion of a successful phase 2 study (NCT01119833), in which the drug markedly reduced opioid requirements during VOC,149 a phase 3 randomized placebo-controlled multi-center study of the drug for vaso-occlusive episodes (NCT02187003) was undertaken and is now well underway.
Table 5 |.
Anti-adhesive agents
| Therapeutic agent | Properties | Comments |
|---|---|---|
| Rivipansel | Small molecule pan-selectin inhibitor with strongest activity against E selectin | Successful phase 2 (NCT01119833)149; currently in phase 3 trial (NCT02187003) |
| Crizanlizumab | Humanized monoclonal antibody that inhibits P selectin. | Successful phase 2 study showing safety and efficacy (NCT01895361)150 |
| Sevuparin | Heparinoid that blocks P selectin and other adhesive ligands | Inhibited RBC and leukocyte adhesion to endothelial cells in vitro and in SCD mice151 |
| Nonsulfated heparins | Blocks P-selectin and possibly other adhesive ligands | Blocked RBC adhesion in vitro and pretreatment of both erythrocytes and decreases ICAM-1 and sP-SEL in SCD mice153 |
| Anti-selectin aptamers | Blocks P selectin-mediated adhesion | Blocks adhesion in vitro and in animal studies 277,278 |
| αvβ3 inhibition | Blocks adhesion of RBC via ICAM-4 to endothelial cell αvβ3 | Blocked RBC and WBC adhesion to endothelial cells in vitro and in animal models 17,154,155 |
| MAST-188 (Poloxamer 188) | Non-specific inhibition of cell adhesion | Unsuccessful in repeated phase 3 studies (NCT00004408, NCT01737814) |
| Beta blockers | Prevents activation of RBC adhesion receptors ICAM-4, BCAM/Lu, and CD44 | Prevented vaso-occlusion in SCD mice; tolerated in phase 1 and 2 human studies, with decrease in biomarkers of vasculopathy.157,158 |
|
Phosphodiesterase-9 inhibitors (BAY73–6691, PF-04447943, IMR-687) |
Vasodilates, anti-oxidant, anti- inflammatory | Decreased TNF and MPO, and increased SOD and NOX in sickle polymorphonuclear leukocytes279; decreased microvascular stasis in response to hypoxia/ reoxygenation in SCD mice111, 112. Phase 1 completed (NCT02114203), no results available. |
| MEK inhibitors | Prevents activation of RBC and leukocyte adhesion molecules | Showed efficacy in animal models160 |
Crizanlizumab is a humanized monoclonal antibody that also requires intravenous infusion and solely inhibits P-selectin. A phase 2 study of monthly infusions of this drug for the prevention of vaso-occlusion (NCT01895361) was recently concluded and showed, at the highest dose used, a significant reduction in VOC requiring hospitalization, although without a significant effect on quality of life.150
There is also an ongoing phase 2 trial of another P-selectin blocker, sevuparin, which is a heparinoid derivative that retains P-selectin binding ability but has almost no anticoagulant effect. This compound was effective in reducing vaso-occlusion after inflammatory stimuli in a mouse model,151 and it has also been safely tested in a phase 2 study of malaria.152 Tinzaparin was used successfully for VOC in a phase 2 study,134 although whether its effect was attributable to its anti-coagulant activity or its P-selectin-blocking activity is not clear. A sulfated non-anticoagulant version of tinzaparin has also been shown to reduce SRBC adhesion to endothelial cells.153
As noted above, while selectins mediate early adhesive events, they do not cause firm adhesion, which is the province of integrins. SRBCs bind firmly to endothelial cells largely through binding to endothelial αvβ3 integrin,154 whose erythroid ligand is ICAM-4 (LW blood group antigen protein).17,155 However, although at least one FDA-approved drug (abciximab) blocks both this interaction as well as SRBC interactions with platelet αIIbβ3, it remains to be studied in SCD patients, perhaps due to the profound platelet dysfunction associated with this drug.
Interestingly, however, the erythroid ICAM-4 protein, as well as several other erythroid adhesion receptors (BCAM/Lu – the RBC laminin receptor, and CD44 – the RBC hyaluronan and α4β1 integrin receptor) are activatable via erythroid signaling pathways involving β2-adrenergic receptors17,47,155,156 and the MAPK pathway.18 Indeed, propranolol, which effectively blocks both epinephrine and TNF-induced vaso-occlusion in vivo in mice, has been studied in both phase 1 and phase 2 human trials.157,158 However, to date, no investigator or pharmaceutical company has proposed testing of propranolol in a randomized, placebo-controlled phase 3 study. Likewise, MEK inhibitors, some of which are FDA-approved drugs, can also prevent vaso-occlusion in murine models by down-regulating both SRBC and leukocyte adhesion,159,160 but human trials in SCD have thus far not been undertaken.
[H2] Agents that address specific end-organ damage.
Given the complex pathophysiology of SCD, there are many potential pathways by which individual organ processes might be addressed (Table 6). However, mortality in SCD is closely tied to three acute and chronic organ damage syndromes: acute chest syndrome, pulmonary hypertension, and sickle cell nephropathy.5,161–166
Table 6 |.
Organ-specific therapies
| Therapeutic agent | Properties | Comments |
|---|---|---|
| Varespladib | Inhibits secretary phospholipase A2 | Studied in 30 patients at risk for acute chest syndrome (NCT00434473), results not published |
| Bosentan | Inhibits endothelin | Prevented hypoxia-induced mortality and morbidity in SCD mice;175 Modest improvement in some SCD patients with pulmonary hypertension176 |
| ACE inhibitors (e.g. lisinopril, enalapril, captopril) | Angiotensin converting enzyme inhibition reduces albuminuria and slows progression of other nephropathies | Mixed results regarding ability to reduce albuminuria in SCD 180,181,280,281 |
| Losartan | Inhibits angiotensin receptor, expected to reduce albuminuria | A phase 2 trial showed efficacy in reducing albuminuria;183 no phase 3 randomized clinical trials conducted |
Acute chest syndrome (ACS) remains the most common acute event leading to mortality in both children and adults with SCD. ACS typically presents with hypoxemia and pulmonary infiltrates consistent with pneumonia on chest X-ray. This syndrome can have a rapidly downhill course, which may result from the effect hypoxia has on pulmonary endothelium, including upregulation of adhesion molecules and down-regulation of protective mechanisms, such as NO.167 Recently, in its approval of L-glutamine, the FDA cited not only its ability to reduce the frequency of VOC, but also its efficacy in reducing the incidence of acute chest syndrome. 168
Levels of secretory phospholipase A2 (sPLA2) often rise during the period preceding onset of ACS during a vaso-occlusive event.169 Varespladib (A-001), an inhibitor of sPLA2, is theorized to act as an anti-inflammatory agent by disrupting the first step of the arachidonic acid pathway of inflammation. However, studies of varespladib to treat ACS have not proved successful, including a phase 3 trial that was terminated due to lack of efficacy. Other approaches to treatment of ACS have included inhaled NO,170 which appeared to have benefitted patients with hypoxemia in a post-hoc analysis, and, in an ongoing study, L-citrulline (NCT02697240).171 Steroids have also been studied as a treatment for or to prevent recurrence of ACS,172–174 although concern about rebound pain and other side effects has prevented wide adoption of this approach.
Pulmonary artery hypertension is a chronic condition also associated with premature mortality in SCD. In adults with Hb SS and Sβ0, very mild elevations of tricuspid regurgitant jet velocity (> 2.5 m/sec) that reflect only modestly increased pulmonary artery pressures that would be clinically unimportant in other settings are strongly linked to increased mortality. SCD is associated with markedly increased plasma levels of both endothelin-1 and endothelin-3. Inhibitors of endothelin receptors, such as bosentan, have been proposed to have therapeutic value in this setting and are sometimes used to treat pulmonary hypertension in SCD. In an animal model of SCD, bosentan prevented vascular congestion, systemic inflammation, and infiltration of activated neutrophils in both the lungs and kidneys after a hypoxia/reoxygenation challenge.175 A small nonrandomized prospective study of bosentan and ambrisentan has shown some promise in severely affected SCD patients with pulmonary hypertension.176 Unfortunately, randomized, double-blinded studies of bosentan failed to accrue adequately to judge efficacy.177
Sickle cell nephropathy affects nearly 30% of adults with Hb SS/Sβ0 thalassemia.5,178–180 Angiotensin-converting enzyme inhibitors have been proposed and studied for treatment of proteinuria in SCD, as in diabetic and other nephropathies involving proteinuria. In many centers, such therapy is common practice, although strong evidence for efficacy of this approach is lacking.181 More recently, angiotensin receptor blockers, such as such as losartan, have also been considered for sickle cell nephropathy, and a prospective phase 2 trial has shown it reduces micro- and macro-albuminuria without adverse events.182,183
Overall, most efforts to develop new treatments for SCD are focusing on the systemic pathophysiologic processes responsible for both pain and end-organ damage, such as SRBC sickling, oxidative damage, cell adhesion, inflammation, and coagulation. It is hoped that ameliorating these widespread phenomena will be effective in preventing much of the end-organ damage. Nonetheless, developing drugs that can specifically address injurious organ-specific processes remains an important avenue of drug development given that these cause the vast majority of SCD-related deaths in resource-rich settings such as Western Europe and the United States.
[H2] Gene therapy.
Gene therapy to correct autologous HSCs could also be an attractive curative option, as it would overcome the obstacle of finding a matched donor needed for HSC transplantation and be devoid of immune complications associated with an allogeneic transplant, such as graft versus host disease or graft rejection.
Gene therapies based on gene addition using viral vectors to carry therapeutic genes into HSCs are currently in clinical trials, while gene editing is in preclinical development. Preclinical studies and anecdotal clinical reports on patients following allogeneic transplants have shown that if 10–20 percent of normal or gene-corrected HSCs are present, the selective advantage of RBCs with normal or non-sickling hemoglobin is sufficient to replace the majority of SRBCs with normal RBCs or with those that cannot sickle.184,185 Hence, genetic modification of a fraction of HSCs may suffice as a one-time treatment for SCD. Based on the potent anti-sickling properties of HbF noted above, gene therapy strategies are being pursued either to express γ-globin in postnatal RBCs or reactivate endogenous γ-globin expression. Preclinical studies of additive gene therapy have shown that even <20% gene-modified HSC chimerism is beneficial in improving organ pathology and prolonging life significantly,185 an effect akin to the heterocellular expression of HbF with hydroxyurea.
Gene addition strategies that have reached clinical trials utilize lentivirus vectors to deliver an erythroid-specific anti-sickling gene. Lentiviral vectors can readily transfer therapeutic genes into the relatively quiescent HSCs at very high efficiency186,187 and have shown clinical efficacy and safety in a number of genetic disorders188–194. The downside of this approach is that the gene, along with appropiate promoter and regulatory elements, have to be delivered via the vector, which integrates semi-randomly into the genome. There is therefore a potential of genotoxicity due to inadvertent dysregulation of a cellular gene. Importantly, however, a decade-long clinical trial experience has not shown genotoxicity of lentiviral vectors in patients.
While anti-sickling globins have the potential to greatly ameliorate disease, patients still make HbS, and hence, even if “cured” of SCD symptoms and organ damage, they are not genetically cured of SCD. Nevertheless, preclinical studies with antisickling globins have shown complete correction of the SCD phenotype, due to a large selective advantage of HbF-containing sickle erythrocytes over uncorrected ones, and a 20–40% gene modified HSC chimerism results in a near pancellular F-cell production. Three versions of nonsickling or antisickling genes are being used in erythroid-specific lentivirus vectors in three clinical trials for SCD: NCT02140554, utilizing a β-globin-derived gene [βT87Q] that prevents sickle hemoglobin polymerization;192,195 NCT02247843, utilizing a β-globin gene (βAS3) with three point mutations that confer antisickling properties;196–198 and NCT02186418, utilizing a γ-globin coding sequence embedded in a β-globin gene).185 Thus far, one phenotypic cure has been reported from the use of βT87Q lentivirus vector192, and other results are being awaited.
Using recent advances in gene editing, it is now also possible to develop therapies that achieve precise genetic modifications in a patient’s HSCs ex vivo199,200. The major advantage of this approach is that it is a highly targeted approach to the gene of interest. And, if the endogenous gene can be fixed, it would be regulated naturally by the endogenous promoters and enhancers. There would be no random integration of vector DNA. The key disadvantage is that gene editing strategies typically make a targeted DNA double-strand break (DSB) but rely on the cell to repair this break. By far, the most common mode of DSB repair is non-homologous end-joining, often resulting in gene disruption/knockout. In the presence of a homologous donor template, a fraction of cells repair the DSB via homologous recombination, a precise repair mechanism and the more clinically desirable outcome201 Intensive efforts to improve homologous recombination frequency in hematopoietic stem cells via gene editing are currently underway.202–206 Second, site-directed nucleases can have off-target activity creating DSB at unintended sites.207,208 These hurdles are currently being tackled by researchers in order to be able to translate gene editing to the clinic. Thus far, two major approaches are being actively investigated. The first is de-repression of the γ-globin gene by either knocking out its repressor, Bcl11a,209,210 or putative binding sites for γ−globin repressors211. The second is genetic correction of the βS mutation, by providing a homology template with the correct sequence at the sixth codon.196,199,212 Correction of the βS mutation, if at sufficient levels, would indeed be a “cure” of SCD.
However, while specific gene knockouts using these approaches can be done quite efficiently in a large proportion of hematopoietic stem and progenitor cells, precise gene correction in long term repopulating cells is still inefficient 199,203,212,213. Moreover, gene editing results in significant loss of the HSC long term repopulating potential.196,199,212 Hence, gene correction technologies are at a preclinical stage for the treatment of SCD.
One hurdle that is rate limiting for genetic therapies is difficulty in obtaining adequate numbers of HSCs from SCD patients. SCD patients cannot tolerate mobilization of HSCs via the conventional GCSF-mediated mobilization. Thus, bone marrow harvest is the predominant way of obtaining HSCs in SCD, and limited numbers of HSCs can be obtained via this route. Trials of the use of plerixafor to mobilize HSCs are underway (NCT02989701, NCT02193191, NCT02212535 and NCT03226691), and preliminary results show this may be safe and feasible.214–216 Another hurdle to successful genetic therapies in general is ensuring that genetically manipulated HSCs retain long-term repopulating potential, as major losses in HSC potential occur with in vitro culture and manipulation. A third hurdle for genetic therapies in SCD is the toxicity/morbidity associated with pre-transplant conditioning. Reduced intensity pre-transplant conditioning is being clinically tested for gene therapy for SCD (NCT02186418). Antibody-mediated non-chemotherapy conditioning is currently being tested for severe combined immune deficiency transplants (NCT02963064) and advancements in this technology may make it useful in SCD in the future. Progress on these three major challenges will go a long way to making gene therapy more easily applicable and transportable, increasing the potential for application, including in resource-poor countries. Finally, use of closed system automated gene transfer systems are now developed to transfer chimeric antigen receptors into T-cells for cancer immunotherapy, which will ease the gene transfer process.
[H1] TIME FOR MULTI-AGENT THERAPY FOR SCD
As described above, a complex network of intertwined and interdependent processes results from the A to T substitution in the beta globin gene that produces hemoglobin S. The optimal therapeutic strategy would eliminate this genetic alteration in the RBCs and solve the downstream consequences of vaso-occlusion, anemia and hemolysis. However, HSC transplantation is the only currently available strategy capable of achieving this goal, and it is expensive, limited to a small group of patients and has substantial safety risks. Gene therapy also holds the potential to be curative, but is at an early stage of development, with substantial technical challenges, and is likely to be very costly, at least initially. This, together with the technical challenges mentioned above, is likely to strongly limit the potential for application in resource-poor countries with high numbers of people affected by SCD, at least in the near future.
So, while the hurdles associated with curative therapies are being overcome, we propose that a multi-agent systems-oriented approach has the potential to greatly improve outcomes for patients with SCD more broadly. As described above, the use of hydroxyurea, which inhibits HbS polymerization by inducing HbF, has been very effective in altering the course of SCD for many patients. However, despite higher HbF levels, patients still suffer debilitating VOC, strokes, organ dysfunction and pain. Moreover, therapies targeted at different pathophysiologies have shown modest effect. We therefore ask: can we take the approach commonly used in cardiology, oncology and HIV therapy and utilize multiple agents from among the types described above to more effectively alter the interconnected processes aand ultimately the course of SCD, thus improving the quality and quantity of life for patients? What would such a cocktail look like? Which processes should be targeted? At what age should this approach begin: infancy, childhood, adulthood, or before organ damage? Can the toxicity of adding multiple drugs be tolerated? Would more complicated regimes and greater “pill burden” hamper compliance with therapy? Will host defenses be dangerously compromised, leading to infections and auto-immunity? Should there be a separate cocktail for acute events versus chronic therapy?
Thus far, three leading drug candidates for SCD in late-stage development — crizanlizumab, rivipansel, and voxelotor (GBT440)149,150,217 — have all been shown to have positive effects in the presence or absence of hydroxyurea. If any were to receive final approval, they—like L-glutamine—woud be probably be used either without or added to hydroxyurea and would be likely to have at least somewhat additive benefits. If approved, these could also become important ‘building blocks’ in the assembly of multiagent treatment regimens, which might include different medications to be used in prophylactic or acute treatment settings (Figure 3).
Figure 3 |. The pipeline of sickle cell disease therapies.
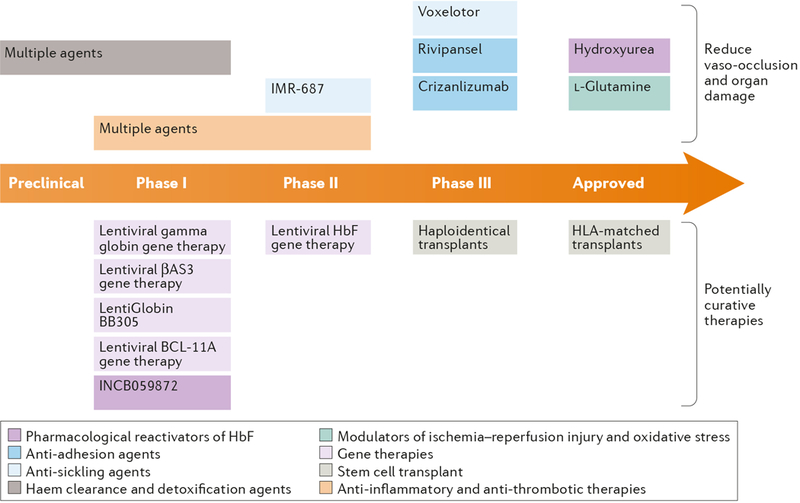
The figure indicates promising therapeutic strategies and specific candidate therapeutics that are being actively pursued, as well as the two approved therapies (hydroxyurea and L-glutamine), classified as in the main text. Agents that are intended to amelioriate the various consequences of abnormal sickle red cells are shown in the upper half of the figure, and agents and approaches that coud potentially cure sickle cell disease are shown in the bottom half of the figure.
Although anticoagulants, anti-platelet agents, and agents with anti-inflammatory effects such as prasugrel and regadenoson have so far not shown clear benefit when used alone or with hydroxyurea, drugs of these classes may nonetheless provide incremental benefit when used with drugs targeting other pathophysiological processes. Strategically the order of addition of each agent to treat a specific patient may be guided by the previous response, with defined endpoints for each, such as HbF content or Hb p50, markers of oxidative stress or inflammation, WBC, RBC and platelet count, reticulocyte count, markers of hemolysis, plasma hemoglobin/haptoglobin/hemopexin, frequency of VOC, duration of pain crises, quality of life measures, etc. For example, combining an anti-sickling agent with a scavenger of heme/hemoglobin to address sickling and hemolysis could be tried. An antithrombotic, antioxidant, anti-adhesive or anti-inflammatory agent could then be added to one or both of these, together or sequentially, and different combinations compared. Clinical trials would need to be developed based on preclinical studies and rigorous planning, in order to best discover additive or synergistic effects, given the limited patient populations available and the costs of such trials. Biomarkers of pathophysiological activation of various pathways may also help steer therapeutic approaches and should be examined for correlation with clinical responses. To date, however, biomarkers of short-term disease activity have been disappointing. Although there are obvious barriers to such an approach, we feel that they can potentially be overcome. The health of millions of SCD patients depends upon the success of such an endeavor.
KEY POINTS.
This review discusses therapies targeted at key pathophysiologies of sickle cell disease, using a systems biology approach.
When sickle hemoglobin polymerizes and produces reactive oxidative species, it causes pleiotropic effects on red blood cells (RBCs): dehydration, membrane damage, increased RBC microparticles and hemolysis, all resulting in enhanced erythropoiesis with increased reticulocytes and young RBC that avidly adhere to leukocytes and endothelium and enhance inflammation. Novel agents are being developed to target sickling, oxidative stress, and cell-cell adhesion to ameliorate these processes and reduce vascular occlusion and ischemia reperfusion injury.
RBC sickling, oxidative stress and membrane damage increase hemolysis, which exhausts the scavengers of heme and hemoglobin, causing further inflammation, oxidative stress, endothelial dysfunction and increased adhesion. Agents are being developed to abrogate the deleterious effects of hemolysis, reducing inflammatory signaling, oxidative stress and endothelial dysfunction.
RBC microparticles, phosphatidylserine exposure on RBCs, inflammation and endothelial dysfunction all promote increased activation of the coagulation system and platelets, further augmenting inflammation and thrombosis. Novel antithrombotic and antiplatelet strategies are being investigated to reverse acute and chronic organ damage.
Genetic modification of hematopoietic stem cells, either to prevent sickling or correct the sickle mutation, has the potential to reverse the pathophysiological processes of SCD, although gene therapy is thus far limited by ease of translation and availability.
Importantly, all the pathophysiologic processes of SCD are linked and perpetuate/augment each other. Thus, a systems biology approach and a multi-pronged attack with new agents is likely necessary to abate the pathophysiology of sickle cell disease.
ACKNOWLEDGMENTS
This work was partially supported by the following grant awards from the National Institutes of Health: R21DK106509 (NIDDK, to MJT), U01-HL117709 and RO1-HL112603 (NHLBI, to PM), and R01HL114567 (NHLBI, to GV). MJT and PM also received support from the Doris Duke Charitable Foundation.
BIBLIOGRAPHY
- 1.Bunn HF Pathogenesis and treatment of sickle cell disease. The New England journal of medicine 337, 762–769, doi: 10.1056/NEJM199709113371107 (1997). [DOI] [PubMed] [Google Scholar]
- 2.Rees DC, Williams TN & Gladwin MT Sickle-cell disease. Lancet 376, 2018–2031, doi: 10.1016/S0140-6736(10)61029-X (2010). [DOI] [PubMed] [Google Scholar]
- 3.Platt OS et al. Mortality in sickle cell disease. Life expectancy and risk factors for early death [see comments]. New England Journal of Medicine 330, 1639–1644 (1994). [DOI] [PubMed] [Google Scholar]
- 4.Kauf TL, Coates TD, Huazhi L, Mody-Patel N & Hartzema AG The cost of health care for children and adults with sickle cell disease. Am J Hematol 84, 323–327, doi: 10.1002/ajh.21408 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Elmariah H et al. Factors associated with survival in a contemporary adult sickle cell disease cohort. American journal of hematology 89, 530–535, doi: 10.1002/ajh.23683 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnold SD, Bhatia M, Horan J & Krishnamurti L Haematopoietic stem cell transplantation for sickle cell disease - current practice and new approaches. British journal of haematology 174, 515–525, doi: 10.1111/bjh.14167 (2016). [DOI] [PubMed] [Google Scholar]
- 7.Fitzhugh CD, Abraham AA, Tisdale JF & Hsieh MM Hematopoietic stem cell transplantation for patients with sickle cell disease: progress and future directions. Hematology/oncology clinics of North America 28, 1171–1185, doi: 10.1016/j.hoc.2014.08.014 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hulbert ML & Shenoy S Hematopoietic stem cell transplantation for sickle cell disease: Progress and challenges. Pediatric blood & cancer, e27263, doi: 10.1002/pbc.27263 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Kassim AA & Sharma D Hematopoietic stem cell transplantation for sickle cell disease: The changing landscape. Hematol Oncol Stem Cell Ther 10, 259–266, doi: 10.1016/j.hemonc.2017.05.008 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Oringanje C, Nemecek E & Oniyangi O Hematopoietic stem cell transplantation for people with sickle cell disease. The Cochrane database of systematic reviews, CD007001, doi: 10.1002/14651858.CD007001.pub4 (2016). [DOI] [PubMed] [Google Scholar]
- 11.Walters MC Update of hematopoietic cell transplantation for sickle cell disease. Current opinion in hematology 22, 227–233, doi: 10.1097/MOH.0000000000000136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Eaton WA & Bunn HF Treating sickle cell disease by targeting HbS polymerization. Blood 129, 2719–2726, doi: 10.1182/blood-2017-02-765891 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hebbel RP Ischemia-reperfusion injury in sickle cell anemia: relationship to acute chest syndrome, endothelial dysfunction, arterial vasculopathy, and inflammatory pain. Hematology/oncology clinics of North America 28, 181–198, doi: 10.1016/j.hoc.2013.11.005 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Hebbel RP, Vercellotti G & Nath KA A systems biology consideration of the vasculopathy of sickle cell anemia: the need for multi-modality chemo-prophylaxsis. Cardiovascular & hematological disorders drug targets 9, 271–292 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hebbel RP Reconstructing sickle cell disease: a data-based analysis of the “hyperhemolysis paradigm” for pulmonary hypertension from the perspective of evidence-based medicine. American journal of hematology 86, 123–154, doi: 10.1002/ajh.21952 (2011). [DOI] [PubMed] [Google Scholar]
- 16.Hebbel RP, Osarogiagbon R & Kaul D The endothelial biology of sickle cell disease: inflammation and a chronic vasculopathy. Microcirculation 11, 129–151 (2004). [PubMed] [Google Scholar]
- 17.Zennadi R et al. Epinephrine acts through erythroid signaling pathways to activate sickle cell adhesion to endothelium via LW-alphavbeta3 interactions. Blood 104, 3774–3781, doi: 10.1182/blood-2004-01-0042 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Zennadi R et al. Erythrocyte plasma membrane-bound ERK1/2 activation promotes ICAM-4-mediated sickle red cell adhesion to endothelium. Blood 119, 1217–1227, doi: 10.1182/blood-2011-03-344440 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.George A et al. Erythrocyte NADPH oxidase activity modulated by Rac GTPases, PKC, and plasma cytokines contributes to oxidative stress in sickle cell disease. Blood 121, 2099–2107, doi: 10.1182/blood-2012-07-441188 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wun T et al. Platelet-erythrocyte adhesion in sickle cell disease. J Investig Med 47, 121–127 (1999). [PubMed] [Google Scholar]
- 21.Wun T et al. Platelet activation and platelet-erythrocyte aggregates in patients with sickle cell anemia. The Journal of laboratory and clinical medicine 129, 507–516 (1997). [DOI] [PubMed] [Google Scholar]
- 22.Frenette PS Sickle cell vaso-occlusion: multistep and multicellular paradigm. Current opinion in hematology 9, 101–106 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Frenette PS Sickle cell vasoocclusion: heterotypic, multicellular aggregations driven by leukocyte adhesion. Microcirculation 11, 167–177 (2004). [PubMed] [Google Scholar]
- 24.Turhan A, Weiss LA, Mohandas N, Coller BS & Frenette PS Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proceedings of the National Academy of Sciences of the United States of America. 99, 3047–3051 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brittain JE, Knoll CM, Ataga KI, Orringer EP & Parise LV Fibronectin bridges monocytes and reticulocytes via integrin alpha4beta1. British journal of haematology 141, 872–881, doi: 10.1111/j.1365-2141.2008.07056.x (2008). [DOI] [PubMed] [Google Scholar]
- 26.Hidalgo A et al. Heterotypic interactions enabled by polarized neutrophil microdomains mediate thromboinflammatory injury. Nature medicine 15, 384–391, doi: 10.1038/nm.1939 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shiu YT, Udden MM & McIntire LV Perfusion with sickle erythrocytes up-regulates ICAM-1 and VCAM-1 gene expression in cultured human endothelial cells. Blood 95, 3232–3241 (2000). [PubMed] [Google Scholar]
- 28.Li H & Lykotrafitis G Erythrocyte membrane model with explicit description of the lipid bilayer and the spectrin network. Biophysical journal 107, 642–653, doi: 10.1016/j.bpj.2014.06.031 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.de Jong K, Larkin SK, Styles LA, Bookchin RM & Kuypers FA Characterization of the phosphatidylserine-exposing subpopulation of sickle cells. Blood 98, 860–867 (2001). [DOI] [PubMed] [Google Scholar]
- 30.Joiner CH, Jiang M & Franco RS Deoxygenation-induced cation fluxes in sickle cells. IV. Modulation by external calcium. The American journal of physiology 269, C403–409 (1995). [DOI] [PubMed] [Google Scholar]
- 31.Mankelow TJ et al. Autophagic vesicles on mature human reticulocytes explain phosphatidylserine-positive red cells in sickle cell disease. Blood 126, 1831–1834, doi: 10.1182/blood-2015-04-637702 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kuypers FA & de Jong K The role of phosphatidylserine in recognition and removal of erythrocytes. Cell Mol Biol (Noisy-le-grand) 50, 147–158 (2004). [PubMed] [Google Scholar]
- 33.Villagra J et al. Platelet activation in patients with sickle disease, hemolysis-associated pulmonary hypertension, and nitric oxide scavenging by cell-free hemoglobin. Blood 110, 2166–2172, doi: 10.1182/blood-2006-12-061697 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brittain HA, Eckman JR, Swerlick RA, Howard RJ & Wick TM Thrombospondin from activated platelets promotes sickle erythrocyte adherence to human microvascular endothelium under physiologic flow: a potential role for platelet activation in sickle cell vaso-occlusion. Blood 81, 2137–2143 (1993). [PubMed] [Google Scholar]
- 35.Setty BN, Kulkarni S, Rao AK & Stuart MJ Fetal hemoglobin in sickle cell disease: relationship to erythrocyte phosphatidylserine exposure and coagulation activation. Blood 96, 1119–1124 (2000). [PubMed] [Google Scholar]
- 36.Arumugam PI et al. Genetic diminution of circulating prothrombin ameliorates multiorgan pathologies in sickle cell disease mice. Blood 126, 1844–1855, doi: 10.1182/blood-2015-01-625707 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SP, Ataga KI, Orringer EP, Phillips DR & Parise LV Biologically active CD40 ligand is elevated in sickle cell anemia: potential role for platelet-mediated inflammation. Arteriosclerosis, thrombosis, and vascular biology 26, 1626–1631, doi: 10.1161/01.ATV.0000220374.00602.a2 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Lee SP et al. Phase I study of eptifibatide in patients with sickle cell anaemia. British journal of haematology 139, 612–620, doi: 10.1111/j.1365-2141.2007.06787.x (2007). [DOI] [PubMed] [Google Scholar]
- 39.Polanowska-Grabowska R et al. P-selectin-mediated platelet-neutrophil aggregate formation activates neutrophils in mouse and human sickle cell disease. Arteriosclerosis, thrombosis, and vascular biology 30, 2392–2399, doi: 10.1161/ATVBAHA.110.211615 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bennewitz MF et al. Lung vaso-occlusion in sickle cell disease mediated by arteriolar neutrophil-platelet microemboli. JCI Insight 2, e89761, doi: 10.1172/jci.insight.89761 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dominical VM et al. Prominent role of platelets in the formation of circulating neutrophil-red cell heterocellular aggregates in sickle cell anemia. Haematologica 99, e214–217, doi: 10.3324/haematol.2014.108555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Belcher JD et al. Heme oxygenase-1 is a modulator of inflammation and vaso-occlusion in transgenic sickle mice. The Journal of clinical investigation 116, 808–816, doi: 10.1172/JCI26857 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ghosh S et al. Extracellular hemin crisis triggers acute chest syndrome in sickle mice. The Journal of clinical investigation 123, 4809–4820, doi: 10.1172/JCI64578 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Belcher JD et al. Heme triggers TLR4 signaling leading to endothelial cell activation and vaso-occlusion in murine sickle cell disease. Blood 123, 377–390, doi: 10.1182/blood-2013-04-495887 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bean CJ et al. Heme oxygenase-1 gene promoter polymorphism is associated with reduced incidence of acute chest syndrome among children with sickle cell disease. Blood 120, 3822–3828, doi: 10.1182/blood-2011-06-361642 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Belcher JD, Nath KA & Vercellotti GM Vasculotoxic and Proinflammatory Effects of Plasma Heme: Cell Signaling and Cytoprotective Responses. ISRN Oxidative Med 2013, doi: 10.1155/2013/831596 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zennadi R, Chien A, Xu K, Batchvarova M & Telen MJ Sickle red cells induce adhesion of lymphocytes and monocytes to endothelium. Blood 112, 3474–3483, doi: 10.1182/blood-2008-01-134346 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nagel RL et al. Structural bases of the inhibitory effects of hemoglobin F and hemoglobin A2 on the polymerization of hemoglobin S. Proceedings of the National Academy of Sciences of the United States of America 76, 670–672 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Akinsheye I et al. Fetal hemoglobin in sickle cell anemia. Blood 118, 19–27, doi: 10.1182/blood-2011-03-325258 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Estepp JH et al. A clinically meaningful fetal hemoglobin threshold for children with sickle cell anemia during hydroxyurea therapy. American journal of hematology 92, 1333–1339, doi: 10.1002/ajh.24906 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Powars DR, Weiss JN, Chan LS & Schroeder WA Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 63, 921–926 (1984). [PubMed] [Google Scholar]
- 52.Charache S et al. Treatment of sickle cell anemia with 5-azacytidine results in increased fetal hemoglobin production and is associated with nonrandom hypomethylation of DNA around the gamma-delta-beta-globin gene complex. Proceedings of the National Academy of Sciences of the United States of America 80, 4842–4846 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DeSimone J, Heller P, Hall L & Zwiers D 5-Azacytidine stimulates fetal hemoglobin synthesis in anemic baboons. Proceedings of the National Academy of Sciences of the United States of America 79, 4428–4431 (1982). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ley TJ et al. 5-azacytidine selectively increases gamma-globin synthesis in a patient with beta+ thalassemia. The New England journal of medicine 307, 1469–1475, doi: 10.1056/NEJM198212093072401 (1982). [DOI] [PubMed] [Google Scholar]
- 55.McGann PT & Ware RE Hydroxyurea therapy for sickle cell anemia. Expert Opin Drug Saf 14, 1749–1758, doi: 10.1517/14740338.2015.1088827 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Molokie R et al. Oral tetrahydrouridine and decitabine for non-cytotoxic epigenetic gene regulation in sickle cell disease: A randomized phase 1 study. PLoS Med 14, e1002382, doi: 10.1371/journal.pmed.1002382 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Atweh GF et al. Sustained induction of fetal hemoglobin by pulse butyrate therapy in sickle cell disease. Blood 93, 1790–1797 (1999). [PMC free article] [PubMed] [Google Scholar]
- 58.Kutlar A et al. A dose-escalation phase IIa study of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. American journal of hematology 88, E255–260, doi: 10.1002/ajh.23533 (2013). [DOI] [PubMed] [Google Scholar]
- 59.Reid ME et al. A double-blind, placebo-controlled phase II study of the efficacy and safety of 2,2-dimethylbutyrate (HQK-1001), an oral fetal globin inducer, in sickle cell disease. American journal of hematology 89, 709–713, doi: 10.1002/ajh.23725 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Bradner JE et al. Chemical genetic strategy identifies histone deacetylase 1 (HDAC1) and HDAC2 as therapeutic targets in sickle cell disease. Proceedings of the National Academy of Sciences of the United States of America 107, 12617–12622, doi: 10.1073/pnas.1006774107 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shi L, Cui S, Engel JD & Tanabe O Lysine-specific demethylase 1 is a therapeutic target for fetal hemoglobin induction. Nature medicine 19, 291–294, doi: 10.1038/nm.3101 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Rivers A et al. RN-1, a potent and selective lysine-specific demethylase 1 inhibitor, increases gamma-globin expression, F reticulocytes, and F cells in a sickle cell disease mouse model. Experimental hematology 43, 546–553 e541–543, doi: 10.1016/j.exphem.2015.04.005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Cui S et al. The LSD1 inhibitor RN-1 induces fetal hemoglobin synthesis and reduces disease pathology in sickle cell mice. Blood 126, 386–396, doi: 10.1182/blood-2015-02-626259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dai Y, Chen T, Ijaz H, Cho EH & Steinberg MH SIRT1 activates the expression of fetal hemoglobin genes. American journal of hematology 92, 1177–1186, doi: 10.1002/ajh.24879 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Lanaro C et al. A thalidomide-hydroxyurea hybrid increases HbF production in sickle cell mice and reduces the release of proinflammatory cytokines in cultured monocytes. Experimental hematology 58, 35–38, doi: 10.1016/j.exphem.2017.10.003 (2018). [DOI] [PubMed] [Google Scholar]
- 66.Theodorou A et al. The investigation of resveratrol and analogs as potential inducers of fetal hemoglobin. Blood cells, molecules & diseases 58, 6–12, doi: 10.1016/j.bcmd.2015.11.007 (2016). [DOI] [PubMed] [Google Scholar]
- 67.Krishnamoorthy S et al. Dimethyl fumarate increases fetal hemoglobin, provides heme detoxification, and corrects anemia in sickle cell disease. JCI Insight 2, doi: 10.1172/jci.insight.96409 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gomperts E et al. The Role of Carbon Monoxide and Heme Oxygenase in the Prevention of Sickle Cell Disease Vaso-Occlusive Crises. American journal of hematology, doi: 10.1002/ajh.24750 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Belcher JD et al. MP4CO, a pegylated hemoglobin saturated with carbon monoxide, is a modulator of HO-1, inflammation, and vaso-occlusion in transgenic sickle mice. Blood 122, 2757–2764, doi: 10.1182/blood-2013-02-486282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Howard J et al. Safety and tolerability of MP4CO: A dose escalation study In stable patients with sickle cell disease. Blood 122, 2205 (abstract) (2013).23945154 [Google Scholar]
- 71.Misra H, Lickliter J, Kazo F & Abuchowski A PEGylated carboxyhemoglobin bovine (SANGUINATE): results of a phase I clinical trial. Artificial organs 38, 702–707, doi: 10.1111/aor.12341 (2014). [DOI] [PubMed] [Google Scholar]
- 72.Abuchowski A PEGylated Bovine Carboxyhemoglobin (SANGUINATE): Results of Clinical Safety Testing and Use in Patients. Advances in experimental medicine and biology 876, 461–467, doi: 10.1007/978-1-4939-3023-4_58 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Swift R et al. SCD-101: A New Anti-Sickling Drug Reduces Pain and Fatigue and Improves Red Blood Cell Shape in Peripheral Blood of Patients with Sickle Cell Disease. Blood 128, 121–121 (2016). [Google Scholar]
- 74.Iyamu EW, Turner EA & Asakura T In vitro effects of NIPRISAN (Nix-0699): a naturally occurring, potent antisickling agent. British journal of haematology 118, 337–343 (2002). [DOI] [PubMed] [Google Scholar]
- 75.Wambebe C et al. Double-blind, placebo-controlled, randomised cross-over clinical trial of NIPRISAN in patients with Sickle Cell Disorder. Phytomedicine 8, 252–261, doi: 10.1078/0944-7113-00040 (2001). [DOI] [PubMed] [Google Scholar]
- 76.Lehrer-Graiwer J et al. Long-Term Dosing in Sickle Cell Disease Subjects with GBT440, a Novel HbS Polymerization Inhibitor. Blood 128, 2488 (2016). [Google Scholar]
- 77.Hebbel RP & Hedlund BE Sickle hemoglobin oxygen affinity-shifting strategies have unequal cerebrovascular risks. American journal of hematology, doi: 10.1002/ajh.24975 (2017). [DOI] [PubMed] [Google Scholar]
- 78.Hebbel RP & Hedlund BE Sickle hemoglobin oxygen affinity-shifting strategies have unequal cerebrovascular risks. American journal of hematology 93, 321–325, doi: 10.1002/ajh.24975 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Morris CR et al. Erythrocyte glutamine depletion, altered redox environment, and pulmonary hypertension in sickle cell disease. Blood 111, 402–410, doi: 10.1182/blood-2007-04-081703 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Whillier S, Garcia B, Chapman BE, Kuchel PW & Raftos JE Glutamine and alpha-ketoglutarate as glutamate sources for glutathione synthesis in human erythrocytes. The FEBS journal 278, 3152–3163, doi: 10.1111/j.1742-4658.2011.08241.x (2011). [DOI] [PubMed] [Google Scholar]
- 81.Niihara Y et al. L-glutamine therapy reduces endothelial adhesion of sickle red blood cells to human umbilical vein endothelial cells. BMC blood disorders 5, 4, doi: 10.1186/1471-2326-5-4 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Niihara Y et al. A Phase 3 Study of L-Glutamine Therapy for Sickle Cell Anemia and Sickle ß0-Thalassemia. Blood 124, 86–86 (2014). [Google Scholar]
- 83.Wilmore DW Food and Drug Administration Approval of Glutamine for Sickle Cell Disease: Success and Precautions in Glutamine Research. JPEN J Parenter Enteral Nutr 41, 912–917, doi: 10.1177/0148607117727271 (2017). [DOI] [PubMed] [Google Scholar]
- 84.Wood KC, Hebbel RP & Granger DN Endothelial cell NADPH oxidase mediates the cerebral microvascular dysfunction in sickle cell transgenic mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 19, 989–991, doi: 10.1096/fj.04-3218fje (2005). [DOI] [PubMed] [Google Scholar]
- 85.Kaul DK et al. Anti-inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. American journal of physiology. Heart and circulatory physiology 287, H293–301, doi: 10.1152/ajpheart.01150.2003 (2004). [DOI] [PubMed] [Google Scholar]
- 86.Nur E et al. N-acetylcysteine reduces oxidative stress in sickle cell patients. Annals of hematology 91, 1097–1105, doi: 10.1007/s00277-011-1404-z (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ozpolat HT et al. A Pilot Study of High-Dose N-Acetylcysteine Infusion in Patients with Sickle Cell Disease. Blood 128, 1299–1299 (2016). [Google Scholar]
- 88.Sins JWR et al. N-Acetylcysteine in Patients with Sickle Cell Disease: A Randomized Controlled Trial. Blood 128, 123–123 (2016). [Google Scholar]
- 89.Wallace KL & Linden J Adenosine A2A receptors induced on iNKT and NK cells reduce pulmonary inflammation and injury in mice with sickle cell disease. Blood 116, 5010–5020, doi: 10.1182/blood-2010-06-290643 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Reilly EC, Wands JR & Brossay L Cytokine dependent and independent iNKT cell activation. Cytokine 51, 227–231, doi: 10.1016/j.cyto.2010.04.016 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Field JJ et al. Sickle cell vaso-occlusion causes activation of iNKT cells that is decreased by the adenosine A2A receptor agonist regadenoson. Blood 121, 3329–3334, doi: 10.1182/blood-2012-11-465963 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Field JJ et al. Randomized phase 2 trial of regadenoson for treatment of acute vaso-occlusive crises in sickle cell disease. Blood Adv 1, 1645–1649, doi: 10.1182/bloodadvances.2017009613 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Solovey A et al. A monocyte-TNF-endothelial activation axis in sickle transgenic mice: Therapeutic benefit from TNF blockade. Am J Hemat. 12292, 1119–1130 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Adelowo O & Edunjobi AS Juvenile idiopathic arthritis coexisting with sickle cell disease: two case reports. BMJ case reports 2011, doi: 10.1136/bcr.10.2011.4889 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gorsuch WB, Chrysanthou E, Schwaeble WJ & Stahl GL The complement system in ischemia-reperfusion injuries. Immunobiology 217, 1026–1033, doi: 10.1016/j.imbio.2012.07.024 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wang RH, Phillips G Jr., Medof ME & Mold C Activation of the alternative complement pathway by exposure of phosphatidylethanolamine and phosphatidylserine on erythrocytes from sickle cell disease patients. The Journal of clinical investigation 92, 1326–1335, doi: 10.1172/jci116706 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zwaal RF & Schroit AJ Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 89, 1121–1132 (1997). [PubMed] [Google Scholar]
- 98.Krisinger MJ et al. Thrombin generates previously unidentified C5 products that support the terminal complement activation pathway. Blood 120, 1717–1725, doi: 10.1182/blood-2012-02-412080 (2012). [DOI] [PubMed] [Google Scholar]
- 99.Schaid TRNJ, Chen C, Abdulla F, Killeen T, Nguyen H, Edmund J, Lindorfer MA, Taylor RP, Dalmasso AP, Belcher JD, Vercellotti GM. Complement Activation in a Murine Model of Sickle Cell Disease: Inhibition of Vaso-Occlusion By Blocking C5 Activation. Blood 128, 158 (2016). [Google Scholar]
- 100.Hoppe C et al. A pilot study of the short-term use of simvastatin in sickle cell disease: effects on markers of vascular dysfunction. British journal of haematology 153, 655–663, doi: 10.1111/j.1365-2141.2010.08480.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hoppe C et al. Simvastatin reduces vaso-occlusive pain in sickle cell anaemia: a pilot efficacy trial. British journal of haematology, doi: 10.1111/bjh.14580 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rybicki AC & Benjamin LJ Increased levels of endothelin-1 in plasma of sickle cell anemia patients. Blood 92, 2594–2596 (1998). [PubMed] [Google Scholar]
- 103.Sabaa N Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. 118, 1924–1933, doi: 10.1172/jci33308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Prado GN, Romero JR & Rivera A Endothelin-1 receptor antagonists regulate cell surface-associated protein disulfide isomerase in sickle cell disease. The FASEB Journal 27, 4619–4629, doi: 10.1096/fj.13-228577 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Koehl B et al. The endothelin B receptor plays a crucial role for the adhesion of neutrophils to the endothelium in sickle cell disease. Haematologica, doi: 10.3324/haematol.2016.156869 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Elisa T et al. Endothelin Receptors Expressed by Immune Cells Are Involved in Modulation of Inflammation and in Fibrosis: Relevance to the Pathogenesis of Systemic Sclerosis. J Immunol Res 2015, 147616, doi: 10.1155/2015/147616 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chang J, Shi PA, Chiang EY & Frenette PS Intravenous immunoglobulins reverse acute vaso-occlusive crises in sickle cell mice through rapid inhibition of neutrophil adhesion. Blood 111, 915–923, doi: 10.1182/blood-2007-04-084061 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jang JE, Hidalgo A & Frenette PS Intravenous immunoglobulins modulate neutrophil activation and vascular injury through FcgammaRIII and SHP-1. Circulation research 110, 1057–1066, doi: 10.1161/CIRCRESAHA.112.266411 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Manwani D et al. Single-dose intravenous gammaglobulin can stabilize neutrophil Mac-1 activation in sickle cell pain crisis. American journal of hematology 90, 381–385, doi: 10.1002/ajh.23956 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Almeida CB et al. Hydroxyurea and a cGMP-amplifying agent have immediate benefits on acute vaso-occlusive events in sickle cell disease mice. Blood 120, 2879–2888, doi: 10.1182/blood-2012-02-409524 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Barbosa MC et al. The Effect of a Selective Inhibitor of Phosphodiesterase-9 on Oxidative Stress, Inflammation and Cytotoxicity in Neutrophils from Patients with Sickle Cell Anaemia. Basic & clinical pharmacology & toxicology 118, 271–278, doi: 10.1111/bcpt.12487 (2016). [DOI] [PubMed] [Google Scholar]
- 112.McArthur JCMT, Chen C, Fricot A,Kobayashi D, Nguyen J, Nguyen P, Parachova A, Abudulla F, Vercellotti GM, Svenstrup Nand Belcher JD. A Novel Highly Potent and Selcetive PDE9 Inhibitor for the Treament of Sickle Cell Disease. Blood 128, 268 (2016). [Google Scholar]
- 113.Morris CR Alterations of the arginine metabolome in sickle cell disease: a growing rationale for arginine therapy. Hematol Oncol Clin North Am 28, 301–321, doi: 10.1016/j.hoc.2013.11.008 (2014). [DOI] [PubMed] [Google Scholar]
- 114.Morris CR et al. A randomized, placebo-controlled trial of arginine therapy for the treatment of children with sickle cell disease hospitalized with vaso-occlusive pain episodes. Haematologica 98, 1375–1382, doi: 10.3324/haematol.2013.086637 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Almeida CB et al. Acute hemolytic vascular inflammatory processes are prevented by nitric oxide replacement or a single dose of hydroxyurea. Blood 126, 711–720, doi: 10.1182/blood-2014-12-616250 (2015). [DOI] [PubMed] [Google Scholar]
- 116.King SB Nitric oxide production from hydroxyurea. Free radical biology & medicine 37, 737–744, doi: 10.1016/j.freeradbiomed.2004.02.073 (2004). [DOI] [PubMed] [Google Scholar]
- 117.Belcher JD et al. Haptoglobin and hemopexin inhibit vaso-occlusion and inflammation in murine sickle cell disease: Role of heme oxygenase-1 induction. PloS one 13, e0196455, doi: 10.1371/journal.pone.0196455 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Vercellotti GM et al. Hepatic Overexpression of Hemopexin Inhibits Inflammation and Vascular Stasis in Murine Models of Sickle Cell Disease. Mol Med 22, doi: 10.2119/molmed.2016.00063 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Belcher JD et al. Haptoglobin and Hemopexin Infusion Efficiently Activates the Nrf2/HO-1 Axis and Inhibits Inflammation and Vaso-Occlusion in Murine Sickle Cell Disease. Blood 128, 2477–2477 (2016).27856474 [Google Scholar]
- 120.Sparkenbaugh E & Pawlinski R Interplay between coagulation and vascular inflammation in sickle cell disease. Br J Haematol 162, 3–14, doi: 10.1111/bjh.12336 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang D, Xu C, Manwani D & Frenette PS Neutrophils, platelets, and inflammatory pathways at the nexus of sickle cell disease pathophysiology. Blood 127, 801–809, doi: 10.1182/blood-2015-09-618538 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Setty BN & Stuart MJ Vascular cell adhesion molecule-1 is involved in mediating hypoxia-induced sickle red blood cell adherence to endothelium: potential role in sickle cell disease. Blood 88, 2311–2320 (1996). [PubMed] [Google Scholar]
- 123.Lim MY, Ataga KI & Key NS Hemostatic abnormalities in sickle cell disease. Current opinion in hematology 20, 472–477, doi: 10.1097/MOH.0b013e328363442f (2013). [DOI] [PubMed] [Google Scholar]
- 124.Noubouossie D, Key NS & Ataga KI Coagulation abnormalities of sickle cell disease: Relationship with clinical outcomes and the effect of disease modifying therapies. Blood Rev 30, 245–256, doi: 10.1016/j.blre.2015.12.003 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Whelihan MF et al. Thrombin generation and cell-dependent hypercoagulability in sickle cell disease. J Thromb Haemost 14, 1941–1952, doi: 10.1111/jth.13416 (2016). [DOI] [PubMed] [Google Scholar]
- 126.Gordon EM et al. Reduction of contact factors in sickle cell disease. The Journal of pediatrics 106, 427–430 (1985). [DOI] [PubMed] [Google Scholar]
- 127.LourenCo D, Sampaio MU, Kerbauy J & Sampaio CA Estimation of plasma kallikrein in sickle-cell anemia, and its relation to the coagulation and fibrinolytic systems. Advances in experimental medicine and biology 247B, 553–557 (1989). [DOI] [PubMed] [Google Scholar]
- 128.Miller RL, Verma PS & Adams RG Studies of the kallikrein-kinin system in patients with sickle cell anemia. Journal of the National Medical Association 75, 551–556 (1983). [PMC free article] [PubMed] [Google Scholar]
- 129.Solovey A, Gui L, Key NS & Hebbel RP Tissue factor expression by endothelial cells in sickle cell anemia. J Clin Invest 101, 1899–1904, doi: 10.1172/JCI1932 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Solovey A et al. Endothelial cell expression of tissue factor in sickle mice is augmented by hypoxia/reoxygenation and inhibited by lovastatin. Blood 104, 840–846, doi: 10.1182/blood-2003-10-3719 (2004). [DOI] [PubMed] [Google Scholar]
- 131.Salvaggio JE, Arnold CA & Banov CH Long-term anti-coagulation in sickle-cell disease. A clinical study. The New England journal of medicine 269, 182–186, doi: 10.1056/nejm196307252690403 (1963). [DOI] [PubMed] [Google Scholar]
- 132.Adelson HT Long-term dicumarol administration as a therapeutic trial in sicklemia; report of a case. The New England journal of medicine 256, 353–354, doi: 10.1056/nejm195702212560807 (1957). [DOI] [PubMed] [Google Scholar]
- 133.Chaplin H Jr. et al. Preliminary trial of minidose heparin prophylaxis for painful sickle cell crises. East African medical journal 66, 574–584 (1989). [PubMed] [Google Scholar]
- 134.Qari MH et al. Reduction of painful vaso-occlusive crisis of sickle cell anaemia by tinzaparin in a double-blind randomized trial. Thrombosis and haemostasis 98, 392–396 (2007). [PubMed] [Google Scholar]
- 135.Wun T et al. Platelet activation in patients with sickle cell disease. Br J Haematol 100, 741–749 (1998). [DOI] [PubMed] [Google Scholar]
- 136.Kataoka H et al. Protease-activated receptors 1 and 4 mediate thrombin signaling in endothelial cells. Blood 102, 3224–3231, doi: 10.1182/blood-2003-04-1130 (2003). [DOI] [PubMed] [Google Scholar]
- 137.Coughlin SR Protease-activated receptors in hemostasis, thrombosis and vascular biology. J Thromb Haemost 3, 1800–1814, doi: 10.1111/j.1538-7836.2005.01377.x (2005). [DOI] [PubMed] [Google Scholar]
- 138.Loscalzo J Nitric oxide insufficiency, platelet activation, and arterial thrombosis. Circ Res 88, 756–762 (2001). [DOI] [PubMed] [Google Scholar]
- 139.Kim K et al. Neutrophil Akt2 Plays a Critical Role In Heterotypic Neutrophil-Platelet Interactions During Vascular Inflammation. Blood 122, 321–321 (2013).23645838 [Google Scholar]
- 140.Li J et al. Neutrophil AKT2 regulates heterotypic cell-cell interactions during vascular inflammation. The Journal of clinical investigation 124, 1483–1496, doi: 10.1172/jci72305 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Barazia A, Li J, Kim K, Shabrani N & Cho J Hydroxyurea with AKT2 inhibition decreases vaso-occlusive events in sickle cell disease mice. Blood 126, 2511–2517, doi: 10.1182/blood-2015-02-626234 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Hoppe CC et al. Design of the DOVE (Determining Effects of Platelet Inhibition on Vaso-Occlusive Events) trial: A global Phase 3 double-blind, randomized, placebo-controlled, multicenter study of the efficacy and safety of prasugrel in pediatric patients with sickle cell anemia utilizing a dose titration strategy. Pediatr Blood Cancer 63, 299–305, doi: 10.1002/pbc.25771 (2016). [DOI] [PubMed] [Google Scholar]
- 143.Heeney MM et al. A Multinational Trial of Prasugrel for Sickle Cell Vaso-Occlusive Events. N Engl J Med 374, 625–635, doi: 10.1056/NEJMoa1512021 (2016). [DOI] [PubMed] [Google Scholar]
- 144.Styles L et al. Prasugrel in children with sickle cell disease: pharmacokinetic and pharmacodynamic data from an open-label, adaptive-design, dose-ranging study. J Pediatr Hematol Oncol 37, 1–9, doi: 10.1097/MPH.0000000000000291 (2015). [DOI] [PubMed] [Google Scholar]
- 145.Jakubowski JA et al. A phase 1 study of prasugrel in patients with sickle cell disease: effects on biomarkers of platelet activation and coagulation. Thromb Res 133, 190–195, doi: 10.1016/j.thromres.2013.12.008 (2014). [DOI] [PubMed] [Google Scholar]
- 146.Wun T et al. A double-blind, randomized, multicenter phase 2 study of prasugrel versus placebo in adult patients with sickle cell disease. Journal of hematology & oncology 6, 17, doi: 10.1186/1756-8722-6-17 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Chang J et al. GMI-1070, a novel pan-selectin antagonist, reverses acute vascular occlusions in sickle cell mice. Blood 116, 1779–1786, doi: 10.1182/blood-2009-12-260513 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Turhan A, Weiss LA, Mohandas N, Coller BS & Frenette PS Primary role for adherent leukocytes in sickle cell vascular occlusion: a new paradigm. Proceedings of the National Academy of Sciences of the United States of America 99, 3047–3051, doi: 10.1073/pnas.052522799 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Telen MJ et al. Randomized phase 2 study of GMI-1070 in SCD: reduction in time to resolution of vaso-occlusive events and decreased opioid use. Blood 125, 2656–2664, doi: 10.1182/blood-2014-06-583351 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ataga KI et al. Crizanlizumab for the Prevention of Pain Crises in Sickle Cell Disease. The New England journal of medicine 376, 429–439, doi: 10.1056/NEJMoa1611770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Telen MJ et al. Sevuparin binds to multiple adhesive ligands and reduces sickle red blood cell-induced vaso-occlusion. British journal of haematology 175, 935–948, doi: 10.1111/bjh.14303 (2016). [DOI] [PubMed] [Google Scholar]
- 152.Leitgeb AM et al. Low anticoagulant heparin disrupts Plasmodium falciparum rosettes in fresh clinical isolates. The American journal of tropical medicine and hygiene 84, 390–396, doi: 10.4269/ajtmh.2011.10-0256 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Alshaiban A, Muralidharan-Chari V, Nepo A & Mousa SA Modulation of Sickle Red Blood Cell Adhesion and its Associated Changes in Biomarkers by Sulfated Nonanticoagulant Heparin Derivative. Clinical and applied thrombosis/hemostasis : official journal of the International Academy of Clinical and Applied Thrombosis/Hemostasis, doi: 10.1177/1076029614565880 (2015). [DOI] [PubMed] [Google Scholar]
- 154.Kaul DK et al. Monoclonal antibodies to alphaVbeta3 (7E3 and LM609) inhibit sickle red blood cell-endothelium interactions induced by platelet-activating factor. Blood 95, 368–374 (2000). [PubMed] [Google Scholar]
- 155.Zennadi R et al. Epinephrine-induced activation of LW-mediated sickle cell adhesion and vaso-occlusion in vivo. Blood 110, 2708–2717, doi: 10.1182/blood-2006-11-056101 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Zennadi R et al. Role and regulation of sickle red cell interactions with other cells: ICAM-4 and other adhesion receptors. Transfusion clinique et biologique : journal de la Societe francaise de transfusion sanguine 15, 23–28, doi: 10.1016/j.tracli.2008.04.009 (2008). [DOI] [PubMed] [Google Scholar]
- 157.De Castro LM, Zennadi R, Jonassaint JC, Batchvarova M & Telen MJ Effect of propranolol as antiadhesive therapy in sickle cell disease. Clinical and translational science 5, 437–444, doi: 10.1111/cts.12005 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.De Castro LM in 9th Annual Sickle Cell Disease Research and Educational Symposium and 38th National Sickle Cell Disease Scientific Meeting. [Google Scholar]
- 159.Soderblom EJ et al. Proteomic analysis of ERK1/2-mediated human sickle red blood cell membrane protein phosphorylation. Clinical proteomics 10, 1, doi: 10.1186/1559-0275-10-1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zennadi R MEK inhibitors, novel anti-adhesive molecules, reduce sickle red blood cell adhesion in vitro and in vivo, and vasoocclusion in vivo. PloS one 9, e110306, doi: 10.1371/journal.pone.0110306 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Allareddy V et al. Outcomes of acute chest syndrome in adult patients with sickle cell disease: predictors of mortality. PloS one 9, e94387, doi: 10.1371/journal.pone.0094387 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Ataga KI et al. Pulmonary hypertension in patients with sickle cell disease: a longitudinal study. British journal of haematology 134, 109–115, doi: 10.1111/j.1365-2141.2006.06110.x (2006). [DOI] [PubMed] [Google Scholar]
- 163.Gladwin MT et al. Risk factors for death in 632 patients with sickle cell disease in the United States and United Kingdom. PloS one 9, e99489, doi: 10.1371/journal.pone.0099489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Lima AR, Ribeiro VS & Nicolau DI Trends in mortality and hospital admissions of sickle cell disease patients before and after the newborn screening program in Maranhao, Brazil. Revista brasileira de hematologia e hemoterapia 37, 12–16, doi: 10.1016/j.bjhh.2014.11.009 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Sabarense AP, Lima GO, Silva LM & Viana MB Characterization of mortality in children with sickle cell disease diagnosed through the Newborn Screening Program. Jornal de pediatria, doi: 10.1016/j.jped.2014.08.006 (2014). [DOI] [PubMed] [Google Scholar]
- 166.Wang Y et al. Mortality of New York children with sickle cell disease identified through newborn screening. Genetics in medicine : official journal of the American College of Medical Genetics, doi: 10.1038/gim.2014.123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Stuart MJ & Setty BN Sickle cell acute chest syndrome: pathogenesis and rationale for treatment. Blood 94, 1555–1560 (1999). [PubMed] [Google Scholar]
- 168.Administration, U. S. F. D. FDA approved L-glutamine powder for the treatment of sickle cell disease. (2017). [Google Scholar]
- 169.Styles L et al. Refining the value of secretory phospholipase A2 as a predictor of acute chest syndrome in sickle cell disease: results of a feasibility study (PROACTIVE). British journal of haematology 157, 627–636, doi: 10.1111/j.1365-2141.2012.09105.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Maitre B et al. Inhaled nitric oxide for acute chest syndrome in adult sickle cell patients: a randomized controlled study. Intensive care medicine 41, 2121–2129, doi: 10.1007/s00134-015-4060-2 (2015). [DOI] [PubMed] [Google Scholar]
- 171.Barr FE et al. Pharmacokinetics and safety of intravenously administered citrulline in children undergoing congenital heart surgery: potential therapy for postoperative pulmonary hypertension. The Journal of thoracic and cardiovascular surgery 134, 319–326, doi: 10.1016/j.jtcvs.2007.02.043 (2007). [DOI] [PubMed] [Google Scholar]
- 172.Quinn CT et al. Tapered oral dexamethasone for the acute chest syndrome of sickle cell disease. British journal of haematology 155, 263–267, doi: 10.1111/j.1365-2141.2011.08827.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Kumar R, Qureshi S, Mohanty P, Rao SP & Miller ST A short course of prednisone in the management of acute chest syndrome of sickle cell disease. Journal of pediatric hematology/oncology 32, e91–94, doi: 10.1097/MPH.0b013e3181c29c52 (2010). [DOI] [PubMed] [Google Scholar]
- 174.Strouse JJ, Takemoto CM, Keefer JR, Kato GJ & Casella JF Corticosteroids and increased risk of readmission after acute chest syndrome in children with sickle cell disease. Pediatric blood & cancer 50, 1006–1012, doi: 10.1002/pbc.21336 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Sabaa N et al. Endothelin receptor antagonism prevents hypoxia-induced mortality and morbidity in a mouse model of sickle-cell disease. The Journal of clinical investigation 118, 1924–1933, doi: 10.1172/jci33308 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Minniti CP et al. Endothelin receptor antagonists for pulmonary hypertension in adult patients with sickle cell disease. British journal of haematology 147, 737–743, doi: 10.1111/j.1365-2141.2009.07906.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Barst RJ et al. Exercise capacity and haemodynamics in patients with sickle cell disease with pulmonary hypertension treated with bosentan: results of the ASSET studies. British journal of haematology 149, 426–435, doi: 10.1111/j.1365-2141.2010.08097.x (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ashley-Koch AE et al. MYH9 and APOL1 are both associated with sickle cell disease nephropathy. British journal of haematology 155, 386–394, doi: 10.1111/j.1365-2141.2011.08832.x (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.De Castro LM, Jonassaint JC, Graham FL, Ashley-Koch A & Telen MJ Pulmonary hypertension associated with sickle cell disease: clinical and laboratory endpoints and disease outcomes. American journal of hematology 83, 19–25, doi: 10.1002/ajh.21058 (2008). [DOI] [PubMed] [Google Scholar]
- 180.Falk RJ et al. Prevalence and pathologic features of sickle cell nephropathy and response to inhibition of angiotensin-converting enzyme. The New England journal of medicine 326, 910–915, doi: 10.1056/NEJM199204023261402 (1992). [DOI] [PubMed] [Google Scholar]
- 181.Sasongko TH, Nagalla S & Ballas SK Angiotensin-converting enzyme (ACE) inhibitors for proteinuria and microalbuminuria in people with sickle cell disease. The Cochrane database of systematic reviews, CD009191, doi: 10.1002/14651858.CD009191.pub3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Ataga KI, Derebail VK & Archer DR The glomerulopathy of sickle cell disease. American journal of hematology 89, 907–914, doi: 10.1002/ajh.23762 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Quinn CT et al. Losartan for the Nephropathy of Sickle Cell Anemia: A Phase-2, Multi-Center Trial. American journal of hematology, doi: 10.1002/ajh.24810 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Walters MC et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biology of blood and marrow transplantation : journal of the American Society for Blood and Marrow Transplantation 7, 665–673 (2001). [DOI] [PubMed] [Google Scholar]
- 185.Perumbeti A et al. A novel human gamma-globin gene vector for genetic correction of sickle cell anemia in a humanized sickle mouse model: critical determinants for successful correction. Blood 114, 1174–1185, doi: 10.1182/blood-2009-01-201863 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Naldini L Lentiviruses as gene transfer agents for delivery to non-dividing cells. 9, 457–463 (1998). [DOI] [PubMed] [Google Scholar]
- 187.Case SS et al. Stable transduction of quiescent CD34(+)CD38(−) human hematopoietic cells by HIV-1-based lentiviral vectors. 96, 2988–2993 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sessa M et al. Lentiviral haemopoietic stem-cell gene therapy in early-onset metachromatic leukodystrophy: an ad-hoc analysis of a non-randomised, open-label, phase 1/2 trial. Lancet 388, 476–487, doi: 10.1016/S0140-6736(16)30374-9 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Aiuti A et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science 341, 1233151, doi: 10.1126/science.1233151 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Eichler F et al. Hematopoietic Stem-Cell Gene Therapy for Cerebral Adrenoleukodystrophy. N Engl J Med 377, 1630–1638, doi: 10.1056/NEJMoa1700554 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Cartier N et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science 326, 818–823 (2009). [DOI] [PubMed] [Google Scholar]
- 192.Ribeil JA et al. Gene Therapy in a Patient with Sickle Cell Disease. The New England journal of medicine 376, 848–855, doi: 10.1056/NEJMoa1609677 (2017). [DOI] [PubMed] [Google Scholar]
- 193.Cavazzana-Calvo M et al. Transfusion independence and HMGA2 activation after gene therapy of human beta-thalassaemia. Nature 467, 318–322, doi: 10.1038/nature09328 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Thompson AA et al. Gene Therapy in Patients with Transfusion-Dependent beta-Thalassemia. N Engl J Med 378, 1479–1493, doi: 10.1056/NEJMoa1705342 (2018). [DOI] [PubMed] [Google Scholar]
- 195.Pawliuk R et al. Correction of sickle cell disease in transgenic mouse models by gene therapy. Science (New York, N.Y.) 294, 2368–2371, doi: 10.1126/science.1065806 (2001). [DOI] [PubMed] [Google Scholar]
- 196.Hoban MD et al. Correction of the sickle cell disease mutation in human hematopoietic stem/progenitor cells. Blood 125, 2597–2604, doi: 10.1182/blood-2014-12-615948 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Levasseur DN, Ryan TM, Pawlik KM & Townes TM Correction of a mouse model of sickle cell disease: lentiviral/antisickling beta-globin gene transduction of unmobilized, purified hematopoietic stem cells. Blood 102, 4312–4319, doi: 10.1182/blood-2003-04-1251 (2003). [DOI] [PubMed] [Google Scholar]
- 198.Levasseur DN et al. A recombinant human hemoglobin with anti-sickling properties greater than fetal hemoglobin. The Journal of biological chemistry 279, 27518–27524, doi: 10.1074/jbc.M402578200 (2004). [DOI] [PubMed] [Google Scholar]
- 199.Dever DP et al. CRISPR/Cas9 beta-globin gene targeting in human haematopoietic stem cells. Nature 539, 384–389, doi: 10.1038/nature20134 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.De Ravin SS et al. CRISPR-Cas9 gene repair of hematopoietic stem cells from patients with X-linked chronic granulomatous disease. Sci Transl Med 9, doi: 10.1126/scitranslmed.aah3480 (2017). [DOI] [PubMed] [Google Scholar]
- 201.Cornu TI, Mussolino C & Cathomen T Refining strategies to translate genome editing to the clinic. Nat Med 23, 415–423, doi: 10.1038/nm.4313 (2017). [DOI] [PubMed] [Google Scholar]
- 202.Bak RO & Porteus MH CRISPR-Mediated Integration of Large Gene Cassettes Using AAV Donor Vectors. Cell Rep 20, 750–756, doi: 10.1016/j.celrep.2017.06.064 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wang J et al. Homology-driven genome editing in hematopoietic stem and progenitor cells using ZFN mRNA and AAV6 donors. Nat Biotechnol 33, 1256–1263, doi: 10.1038/nbt.3408 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Lin S, Staahl BT, Alla RK & Doudna JA Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. Elife 3, e04766, doi: 10.7554/eLife.04766 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Charpentier M et al. CtIP fusion to Cas9 enhances transgene integration by homology-dependent repair. Nat Commun 9, 1133, doi: 10.1038/s41467-018-03475-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Canny MD et al. Inhibition of 53BP1 favors homology-dependent DNA repair and increases CRISPR-Cas9 genome-editing efficiency. Nat Biotechnol 36, 95–102, doi: 10.1038/nbt.4021 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kuscu C, Arslan S, Singh R, Thorpe J & Adli M Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat Biotechnol 32, 677–683, doi: 10.1038/nbt.2916 (2014). [DOI] [PubMed] [Google Scholar]
- 208.Fu Y et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol 31, 822–826, doi: 10.1038/nbt.2623 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Basak A et al. BCL11A deletions result in fetal hemoglobin persistence and neurodevelopmental alterations. The Journal of clinical investigation 125, 2363–2368, doi: 10.1172/JCI81163 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Brendel C et al. Lineage-specific BCL11A knockdown circumvents toxicities and reverses sickle phenotype. The Journal of clinical investigation 126, 3868–3878, doi: 10.1172/JCI87885 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Traxler EA et al. A genome-editing strategy to treat beta-hemoglobinopathies that recapitulates a mutation associated with a benign genetic condition. Nature medicine 22, 987–990, doi: 10.1038/nm.4170 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Hoban MD et al. CRISPR/Cas9-Mediated Correction of the Sickle Mutation in Human CD34+ cells. Mol Ther 24, 1561–1569, doi: 10.1038/mt.2016.148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.DeWitt MA et al. Selection-free genome editing of the sickle mutation in human adult hematopoietic stem/progenitor cells. Sci Transl Med 8, 360ra134, doi: 10.1126/scitranslmed.aaf9336 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Boulad F et al. Safety and efficacy of plerixafor dose escalation for the mobilization of CD34(+) hematopoietic progenitor cells in patients with sickle cell disease: interim results. Haematologica 103, 770–777, doi: 10.3324/haematol.2017.187047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Hsieh MM & Tisdale JF Hematopoietic stem cell mobilization with plerixafor in sickle cell disease. Haematologica 103, 749–750, doi: 10.3324/haematol.2018.190876 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Lagresle-Peyrou C et al. Plerixafor enables safe, rapid, efficient mobilization of hematopoietic stem cells in sickle cell disease patients after exchange transfusion. Haematologica 103, 778–786, doi: 10.3324/haematol.2017.184788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Hoppe CC et al. Initial results from a cohort in a phase 2a study (GBT440–007) evaluating adolescents with sickle cell disease treated with multiple doses of GBT440, a HbS polymerization inhibitor. Blood 689, 689 (2017). [Google Scholar]
- 218.Paszty C et al. Transgenic knockout mice with exclusively human sickle hemoglobin and sickle cell disease. Science (New York, N.Y.) 278, 876–878 (1997). [DOI] [PubMed] [Google Scholar]
- 219.Ryan TM et al. Human sickle hemoglobin in transgenic mice. Science (New York, N.Y.) 247, 566–568 (1990). [DOI] [PubMed] [Google Scholar]
- 220.Heeney MM, Hoppe CC & Rees DC Prasugrel for Sickle Cell Vaso-Occlusive Events. The New England journal of medicine 375, 185–186, doi: 10.1056/NEJMc1603499 (2016). [DOI] [PubMed] [Google Scholar]
- 221.Charache S et al. Hydroxyurea and sickle cell anemia. Clinical utility of a myelosuppressive “switching” agent. The Multicenter Study of Hydroxyurea in Sickle Cell Anemia. Medicine 75, 300–326 (1996). [DOI] [PubMed] [Google Scholar]
- 222.Alayash AI Oxidative pathways in the sickle cell and beyond. Blood cells, molecules & diseases, doi: 10.1016/j.bcmd.2017.05.009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Sangokoya C, Telen MJ & Chi JT microRNA miR-144 modulates oxidative stress tolerance and associates with anemia severity in sickle cell disease. Blood 116, 4338–4348, doi: 10.1182/blood-2009-04-214817 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Jagadeeswaran R et al. Pharmacological inhibition of LSD1 and mTOR reduces mitochondrial retention and associated ROS levels in the red blood cells of sickle cell disease. Experimental hematology, doi: 10.1016/j.exphem.2017.02.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Kato GJ, Steinberg MH & Gladwin MT Intravascular hemolysis and the pathophysiology of sickle cell disease. The Journal of clinical investigation 127, 750–760, doi: 10.1172/JCI89741 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Schaer DJ, Buehler PW, Alayash AI, Belcher JD & Vercellotti GM Hemolysis and free hemoglobin revisited: exploring hemoglobin and hemin scavengers as a novel class of therapeutic proteins. Blood 121, 1276–1284, doi: 10.1182/blood-2012-11-451229 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Manwani D & Frenette PS Vaso-occlusion in sickle cell disease: pathophysiology and novel targeted therapies. Blood 122, 3892–3898, doi: 10.1182/blood-2013-05-498311 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Dulmovits BM et al. Pomalidomide reverses gamma-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood 127, 1481–1492, doi: 10.1182/blood-2015-09-667923 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Abdulmalik O et al. 5-hydroxymethyl-2-furfural modifies intracellular sickle haemoglobin and inhibits sickling of red blood cells. British journal of haematology 128, 552–561, doi: 10.1111/j.1365-2141.2004.05332.x (2005). [DOI] [PubMed] [Google Scholar]
- 230.Iyamu EW, Turner EA & Asakura T Niprisan (Nix-0699) improves the survival rates of transgenic sickle cell mice under acute severe hypoxic conditions. British journal of haematology 122, 1001–1008 (2003). [DOI] [PubMed] [Google Scholar]
- 231.Abdulmalik O et al. Crystallographic analysis of human hemoglobin elucidates the structural basis of the potent and dual antisickling activity of pyridyl derivatives of vanillin. Acta crystallographica. Section D, Biological crystallography 67, 920–928, doi: 10.1107/s0907444911036353 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Abraham DJ et al. Vanillin, a potential agent for the treatment of sickle cell anemia. Blood 77, 1334–1341 (1991). [PubMed] [Google Scholar]
- 233.Ouattara B et al. Antisickling properties of divanilloylquinic acids isolated from Fagara zanthoxyloides Lam. (Rutaceae). Phytomedicine 16, 125–129, doi: 10.1016/j.phymed.2008.10.013 (2009). [DOI] [PubMed] [Google Scholar]
- 234.Safo MK et al. Vzhe-039, a Novel Structurally-Enhanced Allosteric Hemoglobin Effector Inhibits Sickling of SS Erythrocytes In Vitro, and Exhibits Improved Pharmacologic Properties In Vivo. Blood 128, 3645–3645 (2016). [Google Scholar]
- 235.Nakagawa A et al. Identification of a small molecule that increases hemoglobin oxygen affinity and reduces SS erythrocyte sickling. ACS chemical biology 9, 2318–2325, doi: 10.1021/cb500230b (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Zhang Y et al. Elevated sphingosine-1-phosphate promotes sickling and sickle cell disease progression. The Journal of clinical investigation 124, 2750–2761, doi: 10.1172/JCI74604 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.De Franceschi L, Brugnara C, Rouyer-Fessard P, Jouault H & Beuzard Y Formation of dense erythrocytes in SAD mice exposed to chronic hypoxia: evaluation of different therapeutic regimens and of a combination of oral clotrimazole and magnesium therapies. Blood 94, 4307–4313 (1999). [PubMed] [Google Scholar]
- 238.Brugnara C et al. Therapy with oral clotrimazole induces inhibition of the Gardos channel and reduction of erythrocyte dehydration in patients with sickle cell disease. The Journal of clinical investigation 97, 1227–1234, doi: 10.1172/jci118537 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Ataga KI et al. Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). British journal of haematology 153, 92–104, doi: 10.1111/j.1365-2141.2010.08520.x (2011). [DOI] [PubMed] [Google Scholar]
- 240.Hebbel RP et al. The HDAC inhibitors trichostatin A and suberoylanilide hydroxamic acid exhibit multiple modalities of benefit for the vascular pathobiology of sickle transgenic mice. Blood 115, 2483–2490, doi: 10.1182/blood-2009-02-204990 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Kaul DK et al. Robust vascular protective effect of hydroxamic acid derivatives in a sickle mouse model of inflammation. Microcirculation 13, 489–497, doi: 10.1080/10739680600778456 (2006). [DOI] [PubMed] [Google Scholar]
- 242.Zhao Y, Schwartz EA, Palmer GM & Zennadi R MEK1/2 inhibitors reverse acute vascular occlusion in mouse models of sickle cell disease. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 30, 1171–1186, doi: 10.1096/fj.15-278481 (2016). [DOI] [PubMed] [Google Scholar]
- 243.Mahaseth H et al. Polynitroxyl albumin inhibits inflammation and vasoocclusion in transgenic sickle mice. Journal of Laboratory and Clinical Medicine 145, 204–211, doi: 10.1016/j.lab.2005.02.008 (2005). [DOI] [PubMed] [Google Scholar]
- 244.Martins VD, Manfredini V, Peralba MC & Benfato MS Alpha-lipoic acid modifies oxidative stress parameters in sickle cell trait subjects and sickle cell patients. Clin Nutr 28, 192–197, doi: 10.1016/j.clnu.2009.01.017 (2009). [DOI] [PubMed] [Google Scholar]
- 245.El-Beshlawy A et al. Diastolic dysfunction and pulmonary hypertension in sickle cell anemia: is there a role for L-carnitine treatment? Acta Haematol 115, 91–96, doi:AHA20061151_2091 [pii] 10.1159/000089472 (2006). [DOI] [PubMed] [Google Scholar]
- 246.Serjeant BE, Harris J, Thomas P & Serjeant GR Propionyl-L-carnitine in chronic leg ulcers of homozygous sickle cell disease: a pilot study. J Am Acad Dermatol 37, 491–493 (1997). [DOI] [PubMed] [Google Scholar]
- 247.Musicki B, Liu T, Sezen SF & Burnett AL Targeting NADPH oxidase decreases oxidative stress in the transgenic sickle cell mouse penis. The journal of sexual medicine 9, 1980–1987, doi: 10.1111/j.1743-6109.2012.02798.x (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Field JJ et al. A Phase I Single Ascending Dose Study Of NKTT120 In Stable Adult Sickle Cell Patients. Blood 122, 977 (2013). [Google Scholar]
- 249.Kalish BT et al. Dietary omega-3 fatty acids protect against vasculopathy in a transgenic mouse model of sickle cell disease. Haematologica 100, 870–880, doi: 10.3324/haematol.2015.124586 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Daak AA et al. Effect of omega-3 (n-3) fatty acid supplementation in patients with sickle cell anemia: randomized, double-blind, placebo-controlled trial. Am J Clin Nutr 97, 37–44, doi: 10.3945/ajcn.112.036319 (2013). [DOI] [PubMed] [Google Scholar]
- 251.Beckman JD et al. Inhaled carbon monoxide reduces leukocytosis in a murine model of sickle cell disease. Am J Physiol Heart Circ Physiol 297, H1243–1253, doi: 10.1152/ajpheart.00327.2009 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Misra H et al. A Phase Ib open label, randomized, safety study of SANGUINATE in patients with sickle cell anemia. Rev Bras Hematol Hemoter 39, 20–27, doi: 10.1016/j.bjhh.2016.08.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Haynes J Jr., Baliga BS, Obiako B, Ofori-Acquah S & Pace B Zileuton induces hemoglobin F synthesis in erythroid progenitors: role of the L-arginine-nitric oxide signaling pathway. Blood 103, 3945–3950, doi: 10.1182/blood-2003-08-2969 (2004). [DOI] [PubMed] [Google Scholar]
- 254.Eiymo Mwa Mpollo M. S. et al. Placenta growth factor augments airway hyperresponsiveness via leukotrienes and IL-13. J Clin Invest 126, 571–584, doi: 10.1172/JCI77250 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Opene M, Kurantsin-Mills J, Husain S & Ibe BO Sickle erythrocytes and platelets augment lung leukotriene synthesis with downregulation of anti-inflammatory proteins: relevance in the pathology of the acute chest syndrome. Pulmonary circulation 4, 482–495, doi: 10.1086/677363 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Belcher JD et al. The fucosylation inhibitor, 2-fluorofucose, inhibits vaso-occlusion, leukocyte-endothelium interactions and NF-kB activation in transgenic sickle mice. PLoS One 10, e0117772, doi: 10.1371/journal.pone.0117772 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Zhang D et al. Neutrophil ageing is regulated by the microbiome. Nature 525, 528–532, doi: 10.1038/nature15367 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Schaid TR et al. Complement Activation in a Murine Model of Sickle Cell Disease: Inhibition of Vaso-Occlusion By Blocking C5 Activation. Blood 128, 158–158 (2016). [Google Scholar]
- 259.Valverde Y, Benson B, Gupta M & Gupta K Spinal glial activation and oxidative stress are alleviated by treatment with curcumin or coenzyme Q in sickle mice. Haematologica 101, e44–47, doi: 10.3324/haematol.2015.137489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Semple MJ, Al-Hasani SF, Kioy P & Savidge GF A double-blind trial of ticlopidine in sickle cell disease. Thrombosis and haemostasis 51, 303–306 (1984). [PubMed] [Google Scholar]
- 261.Cabannes R et al. Clinical and biological double-blind-study of ticlopidine in preventive treatment of sickle-cell disease crises. Agents and actions. Supplements 15, 199–212 (1984). [PubMed] [Google Scholar]
- 262.Eke FU, Obamyonyi A, Eke NN & Oyewo EA An open comparative study of dispersible piroxicam versus soluble acetylsalicylic acid for the treatment of osteoarticular painful attack during sickle cell crisis. Tropical medicine & international health : TM & IH 5, 81–84 (2000). [DOI] [PubMed] [Google Scholar]
- 263.Desai PC et al. A pilot study of eptifibatide for treatment of acute pain episodes in sickle cell disease. Thrombosis research 132, 341–345, doi: 10.1016/j.thromres.2013.08.002 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Schnog JB et al. Low adjusted-dose acenocoumarol therapy in sickle cell disease: a pilot study. American journal of hematology 68, 179–183 (2001). [DOI] [PubMed] [Google Scholar]
- 265.van Zuuren EJ & Fedorowicz Z Low-molecular-weight heparins for managing vaso-occlusive crises in people with sickle cell disease. The Cochrane database of systematic reviews, Cd010155, doi: 10.1002/14651858.CD010155.pub3 (2015). [DOI] [PubMed] [Google Scholar]
- 266.Gladwin MT et al. Nitric oxide for inhalation in the acute treatment of sickle cell pain crisis: a randomized controlled trial. Jama 305, 893–902, doi: 10.1001/jama.2011.235 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Silva FH et al. Beneficial Effect of the Nitric Oxide Donor Compound 3-(1,3-Dioxoisoindolin-2-yl)Benzyl Nitrate on Dysregulated Phosphodiesterase 5, NADPH Oxidase, and Nitrosative Stress in the Sickle Cell Mouse Penis: Implication for Priapism Treatment. The Journal of pharmacology and experimental therapeutics 359, 230–237, doi: 10.1124/jpet.116.235473 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Katusic ZS, d’Uscio LV & Nath KA Vascular protection by tetrahydrobiopterin: progress and therapeutic prospects. Trends Pharmacol Sci 30, 48–54, doi: 10.1016/j.tips.2008.10.003 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Machado RF et al. Hospitalization for pain in patients with sickle cell disease treated with sildenafil for elevated TRV and low exercise capacity. Blood 118, 855–864, doi: 10.1182/blood-2010-09-306167 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Minniti CP et al. Topical sodium nitrite for chronic leg ulcers in patients with sickle cell anaemia: a phase 1 dose-finding safety and tolerability trial. The Lancet. Haematology 1, e95–e103 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Belcher JDN,J.; Chen C.; Abdulla.; Nguyen P.; Nguyen M.; Zhang P.; Xu P.; Slipek NJ.; Vercellotti GM. Dimethyl Fumarate Induces Cytoprotection and Inhibits Vaso-Occlusion in Transgenic Sickle Mice. American Society of Hematology 124, 219 (2014). [Google Scholar]
- 272.Keleku-Lukwete N et al. Amelioration of inflammation and tissue damage in sickle cell model mice by Nrf2 activation. Proc Natl Acad Sci U S A 112, 12169–12174, doi: 10.1073/pnas.1509158112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Ghosh S et al. Nonhematopoietic Nrf2 dominantly impedes adult progression of sickle cell anemia in mice. JCI Insight 1, doi: 10.1172/jci.insight.81090 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Belcher JD et al. Heme oxygenase-1 gene delivery by Sleeping Beauty inhibits vascular stasis in a murine model of sickle cell disease. J Mol Med (Berl) 88, 665–675, doi: 10.1007/s00109-010-0613-6 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Shi PA et al. Sustained treatment of sickle cell mice with haptoglobin increases HO-1 and H-ferritin expression and decreases iron deposition in the kidney without improvement in kidney function. Br J Haematol 175, 714–723, doi: 10.1111/bjh.14280 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Vinchi F et al. Hemopexin therapy reverts heme-induced proinflammatory phenotypic switching of macrophages in a mouse model of sickle cell disease. Blood 127, 473–486, doi: 10.1182/blood-2015-08-663245 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Burnette AD et al. RNA aptamer therapy for vaso-occlusion in sickle cell disease. Nucleic acid therapeutics 21, 275–283, doi: 10.1089/nat.2010.0270 (2011). [DOI] [PubMed] [Google Scholar]
- 278.Gutsaeva DR et al. Inhibition of cell adhesion by anti-P-selectin aptamer: a new potential therapeutic agent for sickle cell disease. Blood 117, 727–735, doi: 10.1182/blood-2010-05-285718 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.De Franceschi L et al. Protective effects of phosphodiesterase-4 (PDE-4) inhibition in the early phase of pulmonary arterial hypertension in transgenic sickle cell mice. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 22, 1849–1860, doi: 10.1096/fj.07-098921 (2008). [DOI] [PubMed] [Google Scholar]
- 280.Aoki RY & Saad ST Enalapril reduces the albuminuria of patients with sickle cell disease. The American journal of medicine 98, 432–435, doi: 10.1016/S0002-9343(99)80341-6 (1995). [DOI] [PubMed] [Google Scholar]
- 281.Foucan L et al. A randomized trial of captopril for microalbuminuria in normotensive adults with sickle cell anemia. The American journal of medicine 104, 339–342 (1998). [DOI] [PubMed] [Google Scholar]