

Abstract
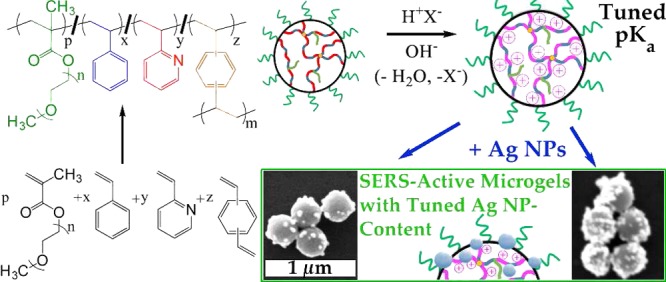
The synthesis of microgels with pH-tunable swelling leads to adjustable and pH-responsive substrates for surface-enhanced Raman scattering (SERS)-active nanoparticles (NPs). Sterically stabilized and cross-linked latexes were synthesized from random copolymers of styrene (S) and 2-vinylpyridine (2VP). The pH-dependent latex-to-microgel transition and swellability were tuned based on their hydrophobic-to-hydrophilic content established by the S/2VP ratio. The electrostatic loading of polystyrene/poly(2-vinylpyridine) microgels [PSxP2VPy (M)] with anions such as tetrachloroaurate (AuCl4–) and borate-capped Ag NPs was quantified. The PSxP2VPy (M) can load ∼0.3 equiv of AuCl4– and the subsequent photoreduction results in Au NP-loaded PSxP2VPy (M) with NPs located throughout the structure. Loading PSxP2VPy (M) with borate-capped Ag NPs produces PSxP2VPy (M) with NPs located on the surface of the microgels, where the Ag content is set by S/2VP. The pH-responsive SERS activity is also reported for these Ag NP-loaded microgels. Analytical enhancement factors for dissolved crystal violet are high (i.e., 109 to 1010) and are set by S/2VP. The Ag NP-loaded microgels with ∼80 wt % 2VP exhibited the most stable pH dependent response.
Introduction
Surface-enhanced Raman scattering (SERS) is a highly sensitive spectroscopic technique for analyzing and detecting simple to complex collections of molecules or biological structures.1,2 The sensitivity arises from a SERS effect that results from a large enhancement of the Raman scattering cross section of molecules that are in close proximity to metal surfaces.3,4 Many theoretical and experimental studies indicate that the excitation of surface plasmons in such metals leads to a localized electromagnetic field that greatly enhances the sensitivity in SERS measurements.5−9 The excitation of surface plasmons depends on factors such as metal type, surface roughness, and for nanostructures, their size, shape, proximity, and packing architecture.10,11 In certain static arrangements with nano- to molecular-scale interparticle gaps, “hot spots” have been reported which have extremely high SERS sensitivity12−15 and the ability to detect single molecules.16−18 The concept of a SERS substrate capable of dynamically tuning SERS-active particle distances is, therefore, attractive. Example systems include the following: the swelling of gels with embedded nanoparticles (NPs),19−21 mechanical stretching of elastomeric substrates bearing silver (Ag) NPs,22 wrinkling of metal and composite films on thermally contracting polymers,23−25 inducing morphology changes of interparticle ligands through pH adjustments,26,27 and aggregation of NPs within magnetic fields.28,29
The simplest SERS experiments are performed by delivering analytes to a suspension of metal NPs of Ag or gold (Au). The main drawback for such experiments is that the NPs are prone to aggregation, which reduces the reproducibility of the SERS effect. Accordingly, anchoring SERS-active NPs to water-dispersible polymer substrates has been explored as a mode for stabilizing the SERS sensitivity. Platforms based on polystyrene (PS),30 polyurethane,31 polyacrylic acid, and polyallylamine32 have shown promise in this regard. Loading stimuli-responsive microgels with NPs has resulted in interesting composites with new properties such as catalytic activity, optical, magnetic, and pH response.33−43 The use of microgels to stabilize the SERS activity of Ag NPs is less well-explored.44 Previously, Armes and Nakamura et al. have studied the pH-responsiveness and Au NP loading of lightly cross-linked and sterically stabilized poly(2-vinylpyridine) (P2VP) microgels.45,46 The Au NP-loaded P2VP microgels studied in their work demonstrated a reversible pH-sensitive plasmon absorbance. Homopolymers, block copolymers, and random copolymers of 4-vinylpyridine and 2VP are widely exploited for their ability to carry a positive charge at a pH lower than 5.0.47−49 Microgels of P2VP are promising candidates for such a role owing to the multifunctionality of the pyridine residues located throughout the macromolecular network. Aqueous protonation of pyridinylenes in lightly cross-linked P2VP results in significant polymer solvation and swelling, while electrostatic and ligand coordination to metal ions can also occur in P2VP materials.50
We present herein the synthesis of sterically stabilized latexes consisting of random copolymers of PS and P2VP [PSxP2VPy (L)]. The dimensions of the latex and its transition to a microgel are studied by titration with monitoring by dynamic light scattering (DLS). The loading of the microgel state with Au ions for subsequent Au NP synthesis is explored. The loading of the copolymer microgels with borate-capped Ag NPs is also described. The Ag NP-loaded microgels exhibit SERS activity demonstrated by detection of pyridinyl groups in the polymer support as well as the detection of a dissolved analyte, crystal violet (CV). A reversible pH-dependent response for the intensity of the SERS spectra for CV is found. The most pH-responsive Ag NP-loaded microgel contains nominal P2VP and PS content values of 80 and 20%, respectively, and operates with a high analytical enhancement factor (AEF) of 1.08 × 1010 and a greater stability than the analogous microgel that lacks the PS content.
Results and Discussion
Synthesis of PSxP2VPy Latexes
An emulsion polymerization approach was selected for the synthesis of sterically stabilized pH-responsive microgels45,46,51 with a tuned hydrophilic: hydrophobic ratio. Scheme 1 outlines the emulsion polymerization that leads to a copolymer latex of S, 2VP, divinylbenzene (DVB), and poly(ethylene glycol)methyl methacrylate (PEGMA). Previous reports have shown that analogous latexes without the PS component are water-dispersible over a large pH range because of the PEGMA-derived PEG-corona on individual particles.45,46 This PEG corona stabilizes the latex and any resulting microgels from aggregation or precipitation by providing a steric repulsion between particles.46 At 60 °C, the monomer reactivity ratios (r) for styrene and 2VP are rs = 0.55 and r2VP = 1.14, respectively.52 The product of rsr2VP is 0.63. Commonly, the range for r1r2 is 0–3, with an ideal copolymerization having r1r2 = 1. The copolymerization of S and 2VP is therefore classified as moderately ideal.53 Also, because the two r values are similar, the two different monomers can be expected to show equal reactivity toward the propagating radical chain ends in the latex.53 Because the reactivity of the monomers is relatively equivalent, the S/2VP ratio in the latex should closely match the nominal S/2VP monomer synthesis ratio, and these two units will likely be randomly included during polymerization.54 Entries 1–5 in Table 1 outline the effect of varying certain synthetic parameters on the mean DH and polydispersity as determined by DLS. Solvent-free diameters, as determined from scanning electron microscopy (SEM) analyses are also reported in this table. Generally, as the nominal weight percent of 2VP is tuned from 40 to 100%, the DH values of the latex (DH,L) at a pH condition of approximately 5 are found to increase from ∼400 to ∼540 nm, while the polydispersity values remained within the range of 0.017–0.063. The increase in diameter with increasing 2VP content suggests that the polarity of neutral P2VP units, and any conjugate acids (P2VP+) units, require additional volume in the colloid to hydrate and accommodate additional counter ions. The DH,L and polydispersity values for these entries in Table 1 are also in agreement with previous and analogous sterically stabilized P2VP latexes.45,46 An interesting deviation in the trend of increasing DH,L with increasing nominal P2VP content was found for the PS40P2VP60 latexes [PS40P2VP60 (L), Table 1, entry 3], which may be because of a latex with polymer chains in an unrelaxed conformation owing to the kinetic conditions of the polymerization. After a transition to a microgel, the diameter of the PS40P2VP60 particle is in agreement with the observed trend for others in the series. The SEM images of solvent-free packed colloids [some exhibiting face-centered cubic (FCC) packing] for the PS0P2VP100 (L), PS20P2VP80 (L), PS40P2VP60 (L), PS50P2VP50 (L), and PS60P2VP40 (L) are shown in Figure 1a–e. The characteristic for low polydispersity in the diameter of the particles is also evident in these images where efficient hexagonal packing of highly uniform particles occurs for all of the materials in this series. Because latex particles are known to pack in an FCC arrangement, the layers in Figure 1a–e represent a (111) plane of the FCC lattice. The number average diameters for the dried latexes (DN,L) were obtained from the SEM images by averaging 5 separate distance measurements, which came from five different and central particles in a close packed seven-particle hexagon. This value is, therefore, determined by measuring the distance between contact points between particles that were in the same plane and row in the packed arrays. In an FCC lattice, these measurements are along the ⟨110⟩ direction (or other geometric equivalents), and hence we provide diameter measurements along this direction with a DN,L,⟨110⟩ label. As expected, the DN,L,⟨110⟩ values were lower than the DH,L values found by DLS. The trend for increasing DN,L,⟨110⟩ with increasing P2VP content was also found in these data (Table 1).
Scheme 1. Generalized Chemical Equation for the Synthesis of Sterically Stabilized pH-Responsive Latex from a Random Copolymer of Styrene and 2-Vinylpyridine [PSxP2VPy (L)] (at Right) with a Tuned a Hydrophilic/Hydrophobic Ratio.

In this work, the molar amounts of the PEGMA steric stabilizer, Aliquat 336 surfactant, and the DVB cross-linker were held constant and in proportion to the total molar amount of the remaining monomers, styrene (S) and 2VP. The hydrophilic/hydrophobic ratio is tuned by adjusting the molar amounts of S and 2VP in the emulsion polymerization.
Table 1. Summary of the Nominal Composition of the PSxP2VPy Particles and the Resulting Hydrodynamic Diameter (DH), Polydispersity, Solvent-Free Number Average Diameter (DN,L), and the Inorganic Loading Values for the Latex/Microgel.
| compositiona |
latex
(L) condition |
microgel (M) condition |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| entry | sample name | S (wt %) | 2VP (wt %) | number average diameter, DN,L in nmb | intensity average diameter DH,L in nm (pH)c | polydispersityc (pH) | intensity average diameter DH,M in nm (pH)c | polydispersityc (pH) | inorganic loading (wt %)d |
| 1 | PS0P2VP100 (L/M) | 0 | 100 | 338 | 542 (5.1) | 0.063 (5.1) | 2409 (3.0) | 0.876 (3.0) | 0 |
| 2 | PS20P2VP80 (L/M) | 20 | 80 | 265 | 476 (5.0) | 0.017 (5.0) | 1897 (2.5) | 0.095 (2.5) | 0 |
| 3 | PS40P2VP60 (L/M) | 40 | 60 | 192 | 318 (5.3) | 0.040 (5.3) | 1498 (2.6) | 0.283 (2.6) | 0 |
| 4 | PS50P2VP50 (L/M) | 50 | 50 | 235 | 424 (5.0) | 0.049 (5.0) | 815 (1.4) | 0.028 (1.4) | 0 |
| 5 | PS60P2VP40 (L/M) | 60 | 40 | 247 | 396 (5.0) | 0.058 (5.0) | 531 (1.0) | 0.139 (1.0) | 0 |
| 6 | PS20P2VP80 (L/M)/AuCl4– | 20 | 80 | 713 (7.0) | 0.168 (7.0) | 959 (2.5) | 0.096 (2.5) | 42.8 | |
| 7 | PS40P2VP60 (L/M)/AuCl4– | 40 | 60 | 567 (7.0) | 0.135 (7.0) | 588 (2.5) | 0.149 (2.5) | 31.5 | |
| 8 | PS60P2VP40 (L/M)/AuCl4– | 60 | 40 | 459 (7.0) | 0.012 (7.0) | 485 (1.1) | 0.010 (1.1) | 19.6 | |
| 9 | PS20P2VP80 (L/M)/Au NPs | 20 | 80 | 653 (4.2) | 0.291 (4.2) | 638 (2.5) | 0.301 (2.5) | 38.3 | |
| 10 | PS40P2VP60 (L/M)/Au NPs | 40 | 60 | 228 | 270 (7.0) | 0.130 (7.0) | 375 (2.5) | 0.218 (2.5) | 24.4 |
| 11 | PS60P2VP40 (L/M)/Au NPs | 60 | 40 | 264 | 428 (7.0) | 0.068 (7.0) | 510 (1.1) | 0.106 (1.1) | 18.9 |
| 12 | PS0P2VP100 (L/M)/Ag NPs | 0 | 100 | 402 | 423 (10.5) | 0.274 (10.5) | 843 (2.0) | 0.309 (2.0) | 39.7 |
| 13 | PS20P2VP80 (L/M)/Ag NPs | 20 | 80 | 385 | 599 (9.1) | 0.247 (9.1) | 896 (1.8) | 0.250 (1.8) | 24.6 |
| 14 | PS40P2VP60 (L/M)/Ag NPs | 40 | 60 | 268 | 582 (9.1) | 0.238 (9.1) | 709 (1.9) | 0.276 (1.9) | 15.1 |
All latexes were nominally synthesized with additional PEGMA (9.0 wt %), Aliquat 336 (3.1 wt %), and DVB (1.0 wt %).
The values were estimated by averaging 5 particles in an SEM image. In the case of entries 1–5, the DN,L values correspond to the diameter along the ⟨110⟩ direction of an FCC lattice (DN,L⟨110⟩) and were determined by averaging five central and hexagonally packed particle diameters along the ⟨110⟩ direction of an FCC lattice.
The values were estimated from DLS measurements at 20 °C. The polydispersity = |μ2/Γ2|, where μ2 is the second cumulant and Γ is the average decay rate determined by the CONTIN analysis.
The values were estimated from the char mass at 775 °C in the TGA plot. In the case of Au- and Ag NP-loaded microgels, the mass is assumed to represent the metal-loading values for the composites.
Figure 1.

SEM images of the dried (a) PS0P2VP100 (L), (b) PS20P2VP80 (L), (c) PS40P2VP60 (L), (d) PS50P2VP50 (L), and (e) PS60P2VP40 (L) samples. (f) Plot for the ratio of the number average diameter (DN,L) distance along the ⟨121⟩ to ⟨110⟩ directions versus the nominal P2VP weight content. Representative [110] and [121] directions are shown in (a). The scale bars in (a–e) are 2 μm.
The balance of hydrophobic and hydrophilic (PS and P2VP units, respectively) properties control the water content in the latex particles and consequently may lead to plastic deformation in the dried particles. Cross-linked latexes rich in PS retain their spherical shape when dried because they do not incorporate significant amounts of water at a neutral pH. Latex particles rich in P2VP incorporate small amounts of water that act as a plasticizer to permit polymer flow. This polymer flow can cause a distortion from a spherical shape during the drying process. Inspection of the SEM images in Figure 1 reveals an interesting trend in the evolution of the shape of the solvent-free PSxP2VPy (L). These images depict near-monodisperse particles from dried latexes that were synthesized with a similar amount of PEGMA and Aliquat 336 (9.0 and 3.1 wt %, respectively). As the nominal weight percent of 2VP is tuned from 40 to 100%, the dried and hexagonally packed particles evolve away from a spherical morphology and take on a distorted shape that arises from gradual occupation of the octahedral and tetrahedral void space in the packed monolayer lattices. This distorted shape is a common characteristic of soft or low glass transition (Tg) polymer latexes that dry in packed arrays.55 It is clear that as the nominal weight content of the P2VP increases, the extent of distortion of the spherical latex also increases. This distortion is greatest along the direction that extends from the center of a particle and points toward the voids (for FCC-type packing, the ⟨121⟩ direction bisects octahedral and tetrahedral voids). Conversely, the distortion is minimal along the ⟨110⟩ direction of the lattice where packed spheres contact each other. The distortion is assumed to arise from strong capillary stresses acting on the colloids as water dries from unoccupied space between 3-D packed colloids. The ratio of DN,L,⟨121⟩ to DN,L,⟨110⟩ represents a metric for quantifying the extent of distortion in the packed spheres. The DN,L,⟨121⟩ to DN,L,⟨110⟩ ratio is plotted versus the nominal P2VP weight content and shown in Figure 1f. The trend suggests that when the nominal P2VP content exceeds 60 wt %, distortion along the ⟨121⟩ direction becomes significant and can be tuned according to the P2VP/PS ratio. The uniform trend in the DN,L,⟨121⟩ to DN,L,⟨110⟩ ratio suggests that the synthesis of the copolymer latex is one that permits a high degree of stoichiometric control, which can be used to adjust the ability of the particles to undergo plasticization by water.
pH-Response of the PSxP2VPy Latex Particles
The tuned content of pyridine groups in the latexes contributes to the amount of pH-triggered swelling in these lightly cross-linked materials. Protonation of these groups can result in significant electrostatic forces and an increased volume due to solvation of water and ions within the polymer particles. A representation of the molecular transformation that results from the treatment of the highly neutral PSxP2VPy (L) with a general Brønsted–Lowry acid (HX) is shown in Scheme 2. Measurements by DLS were used to monitor the DH of selected PSxP2VPy (L) in water with 0.1 M NaCl over the course of a titration with 0.1 M HCl. These DH versus pH titration curves are found in Figure 2a. The initial DH values of the latexes are largely in agreement with data reported in Table 1. Most latexes exhibited stable DH values until pH ∼4. Each PSxP2VPy (L) eventually underwent a significant size change upon treatment with acid and consequently transitioned to a microgel state with a larger diameter. The diameter of the microgel also appears to be independent of pH. Previous work has shown that analogous P2VP latexes undergo this swelling transition very rapidly, roughly requiring only 100–150 ms.46 The polydispersity values for the resulting microgels all remained within a range of 0.028–0.876 (Table 1). The DH associated with the microgel state is listed as DH,M in Table 1, where P2VP-rich microgels had the largest DH,M and those that were more PS-rich had the smallest DH,M. A linear correlation was found between the DH,M and the nominal P2VP weight content of the microgels (Figure 2b), which emphasizes the degree of synthetic control for generating a uniform distribution of P2VP+ units throughout the microgels. It is also evident that as the P2VP content in the latex is increased, the degree of change in DH of the resulting microgel also increases. The evolution in the DH,M to DH,L ratio versus the nominal weight percent of P2VP in the latex is reported in Figure 2c. Again, a linear correlation was found for this plot, further emphasizing the degree of synthetic control on the swellability in the microgels made possible by including tailored amounts of PS in the macromolecular network. A volumetric swelling ratio can be calculated from the DH,M and DH,L values to quantify this characteristic. The volumetric swelling factor for the latex with no PS content (PS0P2VP100) was 140, while that for the most PS-rich in the series (PS60P2VP40) was 2.5. The titration curves in Figure 2a can also be used to determine an average pKa value for the pyridinyl groups in each microgel. All pKa values were found to be less than those for P2VP homopolymer (pKa ≈ 4.92).56,57 Armes et al. have previously observed a linear dependence for the pKa of P2VP with a DVB cross-linker.46 The decreasing pKa with an increasing cross-linker content was suggested to arise from a combination of the “polyelectrolyte effect” and macromolecular swelling strain.58 Unique to this work, we have observed that the pKa of the P2VP can be tuned by adjusting the content of hydrophobic PS. Shown in Figure 2d is the plot for the dependency of the pKa versus the nominal weight percent of P2VP in the latex. In this case, a linear trend was found for the correlation of pKa with P2VP content. Enthalpic penalties associated with exposure of PS segments to water are reflected in the overall pKa of the microgel. Utilizing hydrophobic PS content for tuning the pH-responsive properties of pyridinyl groups in microgels represents a novel method and one that may allow for better matching of the conditions and specificity needed for various applications such as pH-reporting,59,60 catalysis,61−63 and molecular detection via SERS.44,64
Scheme 2. Generalized Chemical Equation for the Reversible Transition of Sterically Stabilized PSxP2VPy (L) (at Left) into Sterically Stabilized PSxP2VPy (M) (at Right) upon Addition of a Brønsted–Lowry Acid (HX).

Figure 2.

(a) Titration curves relating DH (by DLS) to pH for PSxP2VPy samples. The large size change in DH in each curve represents the transition from a latex (higher pH) to a microgel (lower pH). (b) Plot for DH,M vs the nominal P2VP weight content in the PSxP2VPy series. The DH,M value was determined by averaging the data for lower pH plateaus in (a). Plots for DH,M to DH,L ratio (c) and pKa (d) vs the nominal P2VP weight content in the PSxP2VPy series.
Loading of PSxP2VPy Microgels with AuCl4– Ions
The electrostatic loading of the selected PSxP2VPy (M) with anionic species such as AuCl4– and borate-caped Ag NPs was investigated. Experiments were designed to probe the effect of loading anionic materials in an aqueous condition with a pH below the pKa values of the microgel. Two routes to metal NP microgels (e.g.,, PSxP2VPy (M)/Au NPs and PSxP2VPy (M)/Ag NPs) are possible, and these are distinguished in Scheme 3. To achieve PSxP2VPy (M)/Au NPs, aurate-loaded microgels (e.g., PSxP2VPy (M)/AuCl4–) were prepared using acidic conditions. Solutions of PS20P2VP80 (M) and PS40P2VP60 (M) had their pH set to 2.5 using 0.1 M HCl, while that for PS60P2VP40 (M) required a pH of 1.2. Measurements by DLS were used to monitor the size of the PS20P2VP80 (M) as the solution was titrated with KAuCl4. In this experiment, the pH of the KAuCl4 was adjusted to match the pH of the PSxP2VPy (M) solution. The plot in Figure 3a reports the evolution in the DH of PS20P2VP80 (M) as increasing amounts of KAuCl4 are introduced (shown as equivalents of KAuCl4 to 2VP units). The initial DH of the microgel was ∼1980 nm and was found to decrease exponentially. The introduction of only 0.05 equiv of KAuCl4 results in a decrease in DH to ∼1420 nm, which corresponds to a volume that is only 37% of that of the initial state. This dramatic contraction confirms a strong electrostatic cross-linking throughout the macromolecular network. This volume contraction continues as more KAuCl4 is delivered, with an ultimate plateau value for a DH of 960 nm once ∼0.30 equivalents are bound (DH,M in entry 6 in Table 1). The final pH of the PS20P2VP80 (M)/AuCl4– solution was similar to that of the initial condition. McPhee et al. have also found that an ultimate DH is achieved in the titration of poly(N-isopropylacrylamide) microgels with a sub-equimolar amount of sodium dodecylsulfate.65 Stable PS40P2VP60 (M)/AuCl4– and PS60P2VP40 (M)/AuCl4– were also produced from the introduction of 0.3 molar equivalents of KAuCl4. The DH values of these samples were also observed to decrease in a similar manner as the KAuCl4 is incorporated into the microgels (entries 7 and 8 in Table 1). The polydispersity values of the PSxP2VPy (M)/AuCl4– samples remained relatively low. Conversion of the aurate-loaded microgels to their latex state [termed PSxP2VPy (L)/AuCl4–] was investigated by raising the pH with 0.1 M NaOH. Only modest adjustments in the DH values were found (Table 1, entries 6–8), further confirming the extensive electrostatic cross-linking present in the samples.
Scheme 3. Loading of the PSxP2VPy (M) with (i) KAuCl4 and the Subsequent Photoreduction (Top Pathway), or (ii) Loading with Borate-Capped Ag NPs (Bottom Pathway).

Figure 3.

(a) Titration curve monitored by DLS for PS20P2VP80 loaded with 24 mM KAuCl4(aq). The pH of the PS20P2VP80 solution and the KAuCl4 solution were both adjusted to 2.5 and the pH of the PS20P2VP80 (M) solution remained 2.5 throughout the titration. (b) TGA curves for unloaded and aurate- and Au NP-loaded PS20P2VP80 (M). (c) TGA curves for Au NP-loaded PS20P2VP80 (M), PS40P2VP60 (M), and PS60P2VP40 (M). Inset: M775°C vs the nominal P2VP weight content for the aurate-loaded (squares) and Au NP-loaded (circles) samples. (d) High-resolution X-ray photoelectron spectroscopy (HRXPS) for PS20P2VP80 (M)/AuCl4–, PS20P2VP80 (M)/Au NPs, PS40P2VP60 (M)/Au NPs, and PS60P2VP40 (M)/Au NPs. The dash vertical line indicates average binding energy of the maxima in the Au 4f7/2 signals for PSxP2VPy (M)/Au NPs. The HRXPS plot has been normalized and offset for clarity. Tracer plots for the absorbance at 1585 cm–1 (e) vs temperature for PS40P2VP60 (M), PS40P2VP60 (M)/AuCl4–, and PS40P2VP60 (M)/Au NPs. In (e), the bottom trace is for a UV-treated PS40P2VP60 (M). (f) Tracer plots for the absorbance at 1585 cm–1 versus temperature for PS20P2VP80 (M)/Au NPs, PS40P2VP60 (M)/Au NPs, and PS60P2VP40 (M)/Au NPs. The absorbance at 1585 cm–1 is associated with pyridine-containing volatiles from P2VP. In (e,f), the absorbance scale has been normalized and plots are offset for clarity.
Suspensions of all PSxP2VPy (M)/AuCl4– were purified by dialysis and subsequently studied using thermogravimetric analysis (TGA)–Fourier transform infrared (FTIR). The TGA data for the unloaded microgels (solid traces in Figure 3b and the Supporting Information, Figure S1a–c) largely exhibit similar mass loss profiles with a single dominant mass loss event at 420 °C and other characteristics such as a temperature for 5% mass loss (TD5%) in the range of 360–380 °C and a char mass at T = 775 °C (M775°C) of ∼0% for the PS20P2VP80 (M). The mass loss at 420 °C arises from a depolymerization degradation mechanism for the PS and P2VP components in the random copolymer network.66,67 The TGA of a AuCl4– compound has been previously published68 and exhibits a single mass loss event at 245 °C with a TD5% of ∼240 °C and a M775°C of ∼35%. The corresponding TGA data for the AuCl4–-loaded microgels are different and are a function of the nominal P2VP weight content (Figures 3b and S1a–c). These PSxP2VPy (M)/AuCl4– materials exhibit a two-step mass-loss profile with main degradation events at ∼260–280 and 400–405 °C. It is expected that as the microgel is enriched in P2VP content that the capacity for loading KAuCl4 should increase. As the nominal P2VP content in the microgel increases in the series of samples, and consequently as the amount of AuCl4– increases, the mass loss associated with the first degradation event increases. On the basis of the mass loss data, and the close correspondence in the temperature of this event to that of the native AuCl4– material, we assign the first degradation event as a mass loss associated with incorporated aurate ions. The second mass loss event would, therefore, correspond to the depolymerization mechanism mentioned above. The M775°C of the AuCl4–-loaded microgels was also found to increase linearly with the nominal P2VP weight content (see inset plot in Figure 3c). The inorganic content of the AuCl4–-loaded microgels is largely responsible for mass at 775 °C.68 The inorganic content of the AuCl4–-loaded microgels is listed in Table 1 (entries 6–8). As the P2VP content is tuned from 40 to 80%, the inorganic content increases from ∼20 to 43%. The high inorganic content and the linear relationship in M775°C with P2VP weight content suggests that the electrostatic binding of aurate into the network proceeds efficiently and utilizes most P2VP+ sites in the microgel. Furthermore, the uniformity in the trend also speaks to a highly uniform distribution of PS and P2VP residues throughout the polymer network.
Microgel Au NP Composites Prepared from AuCl4–-Loaded PSxP2VPy Microgels
The AuCl4–-loaded microgels were investigated as precursors for the preparation of Au NP-loaded PSxP2VPy microgels [termed PSxP2VPy (M)/Au NPs]. Previous investigations on the chemical reduction of similar aurate-loaded P2VP-based microgels using dimethylamineborane as a chemical reducing agent led to microgels with only surface-loaded Au NPs.45 The method of photoreduction of metal ion/polymer solutions takes advantage of ligands coordinated to the metal cation for rapid reduction and NP growth and is, therefore, a reduction method that is not limited by the mass transport of a chemical reducing agent.69 Solutions of yellow AuCl4–-loaded microgels were exposed to 254 nm UV light and produced pink to purple colored solutions, which is indicative of the formation of Au NPs. A control sample of PS20P2VP80 (M) was treated to the same UV exposure and showed similar characteristics to that of the native microgel, confirming that the polymeric scaffold is stable to the UV treatment (see Figure S1d; and later FTIR discussion). The recovered PSxP2VPy (M)/Au NPs were purified by dialysis prior to characterization. The DLS studies revealed that the DH,M values of PSxP2VPy (M)/Au NPs are similar or smaller than the starting PSxP2VPy (M)/AuCl4– materials. Both the PS20P2VP80 (M)/Au NPs and the PS40P2VP60 (M)/Au NPs contracted to 29 and 26% of the original volume, respectively. The PS60P2VP40 (M)/Au NPs had a similar volume to the precursor microgel. We hypothesize that the lower aurate loading and the lower pKa of the PS60P2VP40 (M) is responsible for the different degree of volumetric change when compared to the other two Au NP-loaded microgels. For PS20P2VP80 (M)/Au NPs and PS40P2VP60 (M)/Au NPs, the decrease in DH,M is likely the result of the reduction of Au ions to neutral Au(0). The presence of Au(0) reduces ionic solvation and electrostatic repulsion forces previously at play in the larger DH,M values found for the aurate loaded precursors. Access to the equivalent latex state [termed PSxP2VPy (L)/Au NPs] was attempted with a pH-adjustment using 0.1 M NaOH. Similar to the parent aurate-loaded microgels, relatively small changes in DH were found. A more detailed DH versus pH titration curve for PS20P2VP80 (M)/Au NPs was performed and monitored by DLS, which confirmed this observation (Figure S2a). Riedinger et al. have studied the volume change associated with the latex-to-microgel transition for poly(2-vinylpyridine-co-divinylbenzene) polymer spheres loaded with Au NPs and reported similar stability in DH.70 It was proposed that the NPs induced steric hindrance in the spheres and led to stable diameter values in the more rigid structure. The SEM investigations on PS40P2VP60 (M)/Au NPs and PS60P2VP40 (M)/Au NPs demonstrated that the shape of the Au NP composites were spherical and indicated that the Au NPs were ∼7 nm in diameter (Figure S2b,c). The DN,L values for the solvent-free PS40P2VP60 (M)/Au NPs and PS60P2VP40 (M)/Au NPs samples are also reported in Table 1 (entries 10–11). Similarly, these DN,L values were smaller than any DH value for the Au NP-loaded particles, confirming solvent-based swelling in the Au NP-loaded latex and microgel states.
The TGA mass loss profile for PS20P2VP80 (M)/Au NPs is shown in Figure 3b (dotted trace), and several differences are apparent when the data for the PS20P2VP80 (M)/AuCl4– are compared to that for the parent PS20P2VP80 (M) (dashed and solid traces, respectively). Generally, all Au NP-loaded microgels show a single mass loss event at ∼390–395 °C, which can be assigned to the depolymerization of the microgel polymer network. In NP-loaded samples, the amount of mass loss due to degradation of AuCl4– was negligible (previously discussed mass loss event at ∼T = 250 °C), suggesting that the aurate ions have successfully reduced into the more thermally stable Au(0) metallic state. The exposure of 254 nm UV light to AuCl4– causes multiple electron transfer steps from chloride ligands to the Au(III) center, resulting in a reduction to Au(0) and the liberation of chloride ligands as a byproduct.71,72 In all cases, the char mass of the Au NP-loaded microgels, indicated herein by the M775°C, was found be less than that of the metal anion-loaded precursor. The decrease in char mass can be attributed to the successful reduction of AuCl4– ions to Au NPs and the associated loss of Cl– ions. The TGA mass loss profiles for PS40P2VP60 (M)/Au NPs and PS60P2VP40 (M)/Au NPs are shown alongside that of the PS20P2VP80 (M)/Au NPs in Figure 3c for comparison. As the nominal P2VP weight content increases, no significant differences in the TD5% were found, yet the M775°C increases linearly from 19 to 38% for the Au NP-loaded microgels (see the inset in Figure 3c). The evidence for the loss of Cl– supports the assignment of the M775°C as mass derived primarily from Au NPs and hence inorganic-loading entries 9–11 in Table 1 equate to the Au NP mass content of PSxP2VPy (M)/Au NPs samples. The increasing linear trend in M775°C with increasing P2VP content in the microgels (see the inset plot in Figure 3c) confirms that the P2VP content specifies the ultimate Au NP loading in the PSxP2VPy (M)/Au NPs composites.
The size, location, and the Au-oxidation state of the Au NPs were determined using transmission electron microscopy (TEM) and XPS. The charge-corrected HRXPS plots for the 4f region for Au acquired for PS20P2VP80 (M)/AuCl4– and selected PSxP2VPy (M)/Au NPs are shown in Figure 3d. For the aurate-loaded microgel, the plot reveals two peaks for the 4f core electrons of Au with peak binding energy values for the 4f5/2 and 4f7/2 peaks at 89.8 and 86.1 eV, respectively, and with a peak separation of 3.7 eV. These values confirm that the Au in the ion-loaded microgels has an oxidation state of (III).73 Corresponding plots for PS20P2VP80 (M)/Au NPs, PS40P2VP60 (M)/Au NPs, and PS60P2VP40 (M)/Au NPs reveal the peak binding energy values for the 4f5/2 and 4f7/2 peaks at 87.6 and 84.0 eV, respectively, with a peak separation of 3.6 eV, all of which confirm that the Au in these systems has an oxidation state of (0).74,75 The tomography video (Movie S1) and TEM images (Figure S3a) identify Au NPs in the PS0P2VP100 (M)/Au NP sample. The TEM-estimated diameter for the Au NPs is 7 nm, where ∼150 Au NPs are present in a PS0P2VP100 (M)/Au NP nanosphere. Frames from the 3D tomography dataset have been extracted (Figure S3b) and used to determine that the Au NPs are located throughout the P2VP microgel, not just on their surfaces as previously seen by others.45
To probe at the nature of the bonding associated with pyridinyl groups in PSxP2VPy (M)/AuCl4– and PSxP2VPy (M)/Au NPs, TGA–FTIR studies were conducted. The intensity maps from FTIR for temperature versus wavenumber of the TGA–FTIR datasets are shown in Figure S4a–n. In comparing the data from PS20P2VP80 (M), PS20P2VP80 (M)/AuCl4–, and PS20P2VP80 (M)/Au NPs, all samples show evidence for a methyl-containing volatile (νCH3 = 2973 cm–1) that is liberated with the onset and peak temperatures of 465 and 480 °C, respectively. Similarly, each sample evolves an ether-containing volatile (detected with a νCO at 1136 cm–1; likely from the PEGMA stabilizer) with the onset and peak temperatures of 450 and 475 °C, respectively. Volatiles associated with a νaromatic CH at 3048–3111 cm–1 are liberated at similar onset and peak temperatures (∼445 and ∼480 °C, respectively) as well. The notable differences in the TGA–FTIR studies are found for the vibrational ring-stretching modes for phenyl and pyridinyl groups that are associated with evolved gases from the samples. The intensity tracer plots from the FTIR data relate the intensity of absorbance for the phenyl ring stretch of PS (νbenz = 1635 cm–1) and the pyridinyl ring stretch of P2VP (νpyr = 1585 cm–1); they are reported in Figures S4o and 3e, respectively. For the inert conditions applied, the thermal degradation of PS and P2VP proceeds by a depolymerization mechanism that evolves S and 2VP monomers at temperatures of 436 °C.76 The signals shown in Figures S4o and 3e can therefore be assigned to specific degradation products for PS and P2VP, respectively. From the data in Figure 3e, the flux of pyridine-containing volatiles from PS40P2VP60 (M) occurs with an onset and peak temperature of 410 and 478 °C, respectively. This same flux of pyridine-containing volatiles from PS40P2VP60 (M)/AuCl4– and PS40P2VP60 (M)/Au NPs occurs at lower temperatures (top two traces in Figure 3e). For PS20P2VP80 (M)/Au NPs, the onset and peak temperature (Tonset = 390 °C and Tpeak = 455 °C, respectively) were also ∼20 °C lower than the metal-free analogue. The profiles of the flux of pyridine-containing volatiles from PS20P2VP80 (M)/Au NPs and PS60P2VP40 (M)/Au NPs were also the same as that for PS40P2VP60 (M)/Au NPs (Figure 3f). A control experiment where unloaded PS40P2VP60 (M) was treated to the photoreduction step (bottom trace in Figure 3e) indicates that the chemical changes in the macromolecular network are likely trivial and are not responsible for the decrease in temperature required to volatilize pyridinyl compounds. The decreased thermal stability of P2VP in the aurate- and the Au NP-loaded microgels suggests that there is relatively weak bonding to these pyridinyl groups, which permit a volatilization of the depolymerization products at a lower temperature. However, it is possible that other kinetic factors are at play as well. For example, it is possible that more efficient heat transfer into the microgel network arises from an increase in the thermal conductivity in the samples imparted by the presence of metallic species.77 The temperature required to volatilize degradation products from PS, indicated by the signal associated with the phenyl ring stretch, was also reduced by ∼20 °C (Figure S4o). The lack of significant bonding interactions between pyridine rings, which would have otherwise increased the temperature required to volatilize pyridine-containing molecules, suggests that the surface of the Au NPs is only weakly bonded to the microgel network and is, therefore, available for the binding of analytes for SERS detection.
SERS from PSxP2VPy (M)/Au NPs
The high loading of Au NPs in PSxP2VPy (M)/Au NP composites and the relatively uncoordinated Au surface suggest that the samples may be a candidate material for SERS detection. Figure S5a,b shows the SERS spectra acquired on solutions of PS20P2VP80 (M)/Au NPs, PS40P2VP60 (M)/Au NPs, and PS60P2VP40 (M)/Au NPs using 633 nm excitation. The data suggest that the Raman active vibration modes in the microgel network cannot be excited and/or detected under the conditions tested. Similarly, when the soluble and representative CV analyte was introduced, the SERS spectra did not show any detectable signals for this molecule. The high-resolution TEM data (Figure S3) indicated that the diameter of the Au NPs in this system was ∼7 nm, and hence, their plasmon-excitation absorbance, a requirement for the SERS technique, may be unlikely with the excitation conditions used herein.78 We have further confirmed this by showing that free Au NPs are SERS active for the detection of CV using our experimental conditions provided that the Au NP diameter is greater than or equal to ∼30 nm (Figure S5). While no SERS spectra were acquired using PSxP2VPy (M)/Au NPs, it is possible that the composites would be excellent SERS substrates, provided that a correctly matched plasmon excitation source was employed or that a secondary growth step was used to enlarge the Au NPs. The SERS-activity study that focuses on the application of PSxP2VPy (M)/Au NPs is a topic of future work and is beyond the scope of this current report.
Ag NP Microgel Composites from Loading Borate-Capped Ag NPs onto PSxP2VPy Microgels
The low-sensitivity SERS detection nanocomposites from aurate-loaded microgels led us to consider larger, Ag-based NPs that exhibit a red-shifted absorbance instead. The immobilization of Ag NPs onto PSxP2VPy (M) was designed to utilize the cationic sites that are generated at the protonated pyridinyl residues. Ligand-based pyridine–Ag interactions that stabilize bound Ag NPs at the microgel are also possible.79 Borate-capped Ag NPs were selected because of their ease of synthesis, stability, and an inherent negative surface charge.80Figure 4a shows the SEM image of the borate-capped Ag NPs that were used in this work. The Ag NPs are roughly spherical in shape and have a diameter of ∼55 nm, a value that agrees with the DLS data (see the Experimental Section). As described above, the pH of the solution of PSxP2VPy (M) was adjusted below that of the pKa. Introduction of a dilute solution of borate-capped Ag NPs to a solution of PSxP2VPy (M) at pH = 1–2 resulted in the self-assembly of the PSxP2VPy (M)/Ag NP composite. The PSxP2VPy (M)/Ag NP composites were purified by dialysis prior to characterization. The SEM images in Figure 4b–d depict the PS0P2VP100 (M)/Ag NPs, PS20P2VP80 (M)/Ag NPs, and PS40P2VP60 (M)/Ag NPs, respectively. Inspection of these images confirmed that the immobilization procedure led to a single nanostructure composite in each case with neither free Ag NPs on the underlying SEM substrate nor any unloaded PSxP2VPy (M). The SEM-estimated number of Ag NPs per PSxP2VPy (M) was determined by averaging five different PSxP2VPy (M) in each corresponding SEM image (any Ag NPs beneath the colloids are, therefore, omitted). The average number of Ag NPs per PS0P2VP100 (M), PS20P2VP80 (M), and PS40P2VP60 (M) sphere was 29 ± 12, 15 ± 5 and 4 ± 3, respectively (see inset in Figure 5b). On the basis of the diameter values for the Ag NPs and those for the dried latexes, and ignoring any NP–NP overlap, the Ag NP footprints are ∼17, 13, and 9% on the surface of their polymer colloid substrates. This correlation confirms that adjusting the S/2VP ratio at the synthetic stage is a means for controlling the number of Ag NPs per microgel and, therefore, the surface density of Ag NPs for SERS.
Figure 4.

SEM images of (a) Ag NPs, (b) PS0P2VP100 (M)/Ag NPs, (c) PS20P2VP80 (M)/Ag NPs, and (d) PS40P2VP60 (M)/Ag NPs. The scale bars in (b–d) are 2 μm.
Figure 5.

(a) TGA curves of PSxP2VPy (L)/Ag NPs. (b) Mass percent remaining at 775 °C, indicating Ag NP loading. (c) Raman spectra of the PSxP2VPy (M)/Ag NPs. (d) Plot for the intensity of the signal at 1011 cm–1 versus the nominal weight content of the P2VP in PSxP2VPy (M)/Ag NPs.
Further quantification of the Ag content for the immobilization of borate-capped Ag NPs on PSxP2VPy (L) was conducted using TGA. Figure 5a shows the overlaid mass loss profiles for PS0P2VP100 (L)/Ag NPs, PS20P2VP80 (L)/Ag NPs, and PS40P2VP60 (L)/Ag NPs. Each dataset shows that the char plateau is less well-defined relative to the parent and unloaded microgel analogues. Previous TGA analyses on the related borate-loaded polymers attributed the more gradual mass loss in the char region to a slow breakage of B–O bonds for the mass loss.81 Nonetheless, the M775°C metrics can still be extracted from the plots for the Ag NP composites and used for further discussion. The M775°C values for PS0P2VP100 (L)/Ag NPs, PS20P2VP80 (L)/Ag NPs, and PS40P2VP60 (L)/Ag NPs were 40, 25, and 15%, respectively, and based on the near-zero M775°C values for the parent microgels can be assigned as inorganic mass due to Ag. These data are also found in entries 12–14 in Table 1. Figure 5b shows the plot for the M775°C versus the nominal mass percent of P2VP. The increasing trend in Ag mass with P2VP content correlates with conclusions based on the SEM where increased Ag loading occurred as the P2VP content increased. On the basis of the TGA-mass estimate for Ag, the spherical shape and diameter information gleaned from SEM, and the bulk density of Ag,82 the number of Ag NPs per microgel sphere can be calculated using eq 1
| 1 |
where the mass of a single PSxP2VPy colloid (massPSxP2VPy) was determined from the DN,L of the PSxP2VPy (L) and the weighted-average of the bulk density of PS and P2VP;83 the mass of a single Ag NP (massAg NPs) was determined using the SEM estimated diameter for the Ag NPs and the bulk density for Ag. As such, the number of Ag NPs per microgel sphere for PS0P2VP100 (M)/Ag NPs, PS20P2VP80 (M)/Ag NPs, and PS40P2VP60 (M)/Ag NPs was found to be 26, 12, and 5, respectively (see the inset in Figure 5b). These values are in agreement with those determined by SEM particle-counting. The controlled loading of Ag NPs onto sterically stabilized PSxP2VPy (M) offers a potential advantage for tuned SERS activity. Contrary to the Au NP-loaded samples, the PSxP2VPy (M)/Ag NPs analogues exhibit increased pH sensitivity as characterized by DLS (Table 1, entries 12–14). The DH,M of the PSxP2VPy (M)/Ag NP samples are less than the values of their metal-free parent microgels (Table 1, entries 1–3), possibly indicating a contraction due to bonding to the Ag NPs. Interestingly, the DH,M and DH,L for the PS20P2VP80 (M)/Ag NPs subtly deviate from the expected trend of decreasing values with decreasing P2VP content. It is possible that the PS content in this system allows for a reduction in the number of contacts between bound Ag NPs and allows for more flexibility in the polymer network, thus favoring a subtly larger size than the PS0P2VP100 (L/M)/Ag NPs. This effect may be offset by energetic penalties for PS-contacts with water that may dominate the PS40P2VP60 (M)/Ag NP sample. The volumetric swelling factors for the series of samples, however, followed a similar and expected trend to the parent microgels, where the values for PS0P2VP100 (M)/Ag NPs, PS20P2VP80 (M)/Ag NPs, and PS40P2VP60 (M)/Ag NPs, were 7.9, 3.3, and 1.8, respectively. The combination of the tuned Ag NP content and the high pH-sensitivity in the PSxP2VPy (M)/Ag NPs series offer a unique experimental landscape to maximize SERS properties such as sensitivity and stability.
The immobilization of SERS-active NPs onto a polymer structure suggests that a Raman spectrum of that polymer substrate could be acquired. Previous studies on the Raman spectrum of copolymers of PS and P2VP have been reported.79,84 Solutions of dispersed PS0P2VP100 (M)/Ag NPs, PS20P2VP80 (M)/Ag NPs, and PS40P2VP60 (M)/Ag NPs were studied using a Raman spectrometer, and the baseline-corrected 633 nm excitation SERS spectra are presented in Figure 5c. The most prevalent peaks in PS0P2VP100 (M)/Ag NP spectra are found at (i) 1224, (ii) 1049, (iii) 1011, (iv) 751, and (v) 629 cm–1. These correspond to modes relating to (i) the stretching of the C–C bond between the pyridine ring and the P2VP backbone, (ii) a mode for the in-plane C–H bending P2VP, (iii) a symmetric ring breathing mode in P2VP, (iv) the B–O–B bending mode in the borate anions, and (v) in-plane bending of the pyridine ring, respectively.84−86 The signal for the ring-breathing mode at 1011 cm–1 indicates a high content of coordinated pyridine at the silver surface due to the adsorption through its nitrogen atoms.84 The confirmation of the Raman spectra from the PS0P2VP100 (M)/Ag NPs further supports the structure proposed that is shown in Scheme 3. The SERS spectra for PS20P2VP80 (M)/Ag NPs and PS40P2VP60 (M)/Ag NPs are very similar in composition to that of PS0P2VP100 (M)/Ag NPs. The signal at 1049 cm–1 represents a composite C–H bending signal for PS and P2VP. Notably, the signal for the ring-breathing mode in P2VP shifts subtly to 1002 cm–1 once the nominal P2VP is decreased to 60%. This suggests that there is less N-coordinated-pyridine and more free pyridine sites in the P2VP in this system. Figure 5d shows the plot for the intensity for the signal at 1011 cm–1 versus the nominal mass percent of P2VP in the microgel. The exponential decrease in the signal with decreasing P2VP content also reflects this diminished N-coordination. These data suggest that by tailoring the P2VP content in this system, a balanced amount of free P2VP results, which may favor improved acid-based swelling performance in the Ag NP-containing composites.
To assess the potential for PSxP2VPy (M)/Ag NPs to act as substrates for SERS-based detection, CV was used as a model analyte.87Figure 6a (entry i) shows the 633 nm excitation Raman spectra for CV (1.1 × 10–5 M in water) acquired without a SERS method. Because CV has electronic transitions at 633 nm, the observed CV spectra contain an additional resonance Raman enhancement contribution. The spectrum is consistent with previous literature studies88 and consists of five main peaks at (a) 795, (b) 905, (c) 1175, (d) 1385, and (e) 1620 cm–1, which correspond to (i) out-of-plane phenyl C–H bending, (ii) ring-breathing, (iii) asymmetric in-plane phenyl C–H bending, (iv) asymmetric phenyl C–N stretching, and (v) asymmetric phenyl C–C stretching modes for CV, respectively.89 Many of these signature peaks for the analyte do not overlap with those for the PSxP2VPy (M) and, therefore, the background interference is minimal (top most spectra in Figures 5c and S6 in for comparisons). Previous SERS studies with CV under similar conditions concluded that the molecule is normally bonded to the Ag surface by Coulombic and van der Waals interactions.90,91 The Ag NPs used in the synthesis of the PSxP2VPy (M)/Ag NPs are good substrates for SERS-based detection and as such, the 633 nm excitation SERS spectra of 1 × 10–9 M Ag NPs with 1.1 × 10–8 M CV in water are also included in Figure 6a (entry ii). Entries (iii–v) in Figure 6a represent the 633 nm excitation SERS spectra for CV acquired for 1.1 × 10–8 M CV exposed to 1 × 10–10 M PS0P2VP100 (M)/Ag NPs, PS20P2VP80 (M)/Ag NPs, and PS40P2VP60 (M)/Ag NPs, respectively. After comparing these spectra to that acquired using only Ag NPs (entry ii), it is apparent that the Ag NP-loaded microgels function with a similar bonding mode for CV but are more sensitive. Comparison of the intensity from the spectra for the Ag NP-loaded microgels shows that the SERS intensity increases with P2VP content. This increased intensity is presumably due to the increased Ag NP loading that was correlated from TGA and SEM investigations. The signal-to-noise (S/N) ratio for the peak found at 1620 cm–1 was used to further characterize the correlation in intensity with P2VP content, where the S/N value for this peak is plotted versus the nominal P2VP mass percent in the microgel (Figure 6b). A linear dependence of the S/N at 1620 cm–1 with P2VP content was found. Under the conditions investigated herein, the linear trend suggests that the PSxP2VPy (M)/Ag NPs are uniform in their sensitivity and enhancement mechanism. AEFs were calculated using eq 2(87) where
| 2 |
ISERS and IRS are the intensity of the 1620 cm–1 peak for the background-corrected SERS spectra and the non-SERS spectra of CV only, respectively. Parameters cSERS and cRS are the associated concentrations of CV for each test. For free Ag NPs, the AEF value was calculated to be 5.21 × 109. The AEF results are outlined in Figure 6c for the three PSxP2VPy (L)/Ag NP nanocomposite systems (Table 2 also lists the calculated AEFs for the PSxP2VPy (L)/Ag NPs). The highest AEF value was found for the PS0P2VP100 (L)/Ag NPs whose value of 1.11 × 1010 is similar to other systems,87,92,93 but 2.1 fold higher than that found for the control sample containing only Ag NPs (bottom entry in Table 2). As the P2VP content of the system decreased, the AEF values decreased marginally, but remained higher than that of the control sample containing only Ag NPs (Figure 6c). The AEF for the PS20P2VP80 (L)/Ag NPs was 1.08 × 1010, while that for PS40P2VP60 (L)/Ag NPs was 7.08 × 109. Previous work for Au NP-decorated P2VP microgels reported an enhancement factor of 1.3 × 106 for CV with an excitation source of 532 nm.50 The high AEF values reported herein are a consequence of the application of the more SERS-sensitive Ag NPs, additional resonance Raman enhancement at 633 nm excitation for CV, and the controlled loading of these NPs onto well-defined copolymer microgels.
Figure 6.

(a) Initial Raman spectra of the PSxP2VPy (M)/Ag NPs and control comparisons with CV. (b) S/N ratio at 1620 cm–1 for CV for spectra acquired using PSxP2VPy (M)/Ag NPs versus the nominal P2VP weight content. (c) AEFs (for pH cycle 0) of PSxP2VPy (L)/Ag NPs versus the nominal P2VP weight content. (d) Tracking of the S/N of the 1620 cm–1 signal for CV as the PSxP2VPy/Ag NPs are cycled between the latex (integer pH cycles) and microgel (half integer pH cycles) states.
Table 2. Summary of Calculated Analytical Enhancement Factors for the 1620 cm–1 Raman-Active Mode of CV.
| PSxP2VPy (M)/Ag NPs + CV ([CV] = 1.1 × 10–8 M) | average AEF |
|---|---|
| PS0P2VP100 (M)/Ag NPs + CV | 1.11 × 1010 |
| PS20P2VP80 (M)/Ag NPs + CV | 1.08 × 1010 |
| PS40P2VP60 (M)/Ag NPs + CV | 7.08 × 109 |
| Ag NPs + CV | 5.15 × 109 |
The demonstration of a successful transition of the PSxP2VPy (M) hybrids to their latex-like state suggests that the sensitivity of the SERS-substrate may be pH-tuned. Previous work on Au NPs immobilized on grafted P2VP brushes correlated the pH-dependent SERS activity to the interparticle spacing.94 The SERS-based spectra for CV were therefore iteratively acquired using PS0P2VP100 (M)/Ag NPs, PS20P2VP80 (M)/Ag NPs, and PS40P2VP60 (M)/Ag NPs upon shifting the pH of the media from values of 7 to 2. We, therefore, define a pH cycle value to track the performance of the SERS substrate. Integer values for the pH cycle value represent the number of times that a PSxP2VPy (M)/Ag NP sample was cycled back to pH = 7 for the acquisition of the SERS spectra of CV. Half-integer values of the pH cycle value represent intermediate experiments where the SERS spectra of CV were acquired at the intermediate condition of pH = 2. In all cases, the average intensity of the spectra increased with an increase in pH. The S/N value for the asymmetric phenyl C–C stretching modes at 1620 cm–1 was used as a representative metric for this intensity change and is plotted against the pH cycle number, n, (Figure 6d). During the period of n = 0–4, the PS0P2VP100 (M)/Ag NP system showed an overall evolution to a condition where the sets of S/N values become more self-consistent for the high and low pH conditions. After this 4 cycle break-in period, the S/N values alternate between ∼35 and ∼20 for high and low pH, respectively. Interestingly, the PS20P2VP80 (M)/Ag NP and PS40P2VP60 (M)/Ag NP systems function with a reduced break-in period (Figure 6d). The number of pH cycles required to stabilize the S/N at 1620 cm–1 for the SERS spectra of CV for PS20P2VP80 (M)/Ag NP and PS40P2VP60 (M)/Ag NP systems was 1.5 and 2.5, respectively. After break-in, the S/N values for the SERS spectra of CV for PS20P2VP80 (M)/Ag NPs alternate between ∼77 and ∼30 for high and low pH, respectively. The corresponding values for the PS40P2VP60 (M)/Ag NP system were 44 and 18, respectively. The PS20P2VP80 (M)/Ag NP system, therefore, appears to have the greatest sensitivity for pH-tuned SERS activity and operates with the least amount of break-in.
The stability of the intensity of the SERS spectra of CV was evaluated using PS0P2VP100 (L)/Ag NPs and PS20P2VP80 (L)/Ag NPs. Spectra were acquired over a multiday period with pH cycling on each day, and the value of the S/N for the 1620 cm–1 vibration mode is plotted versus time and reported in Figure S7. When PS0P2VP100 (L)/Ag NPs were employed, the S/N for the 1620 cm–1 for CV dropped from ∼50 to an ultimate value of 4 over three days. The corresponding CV SERS signal derived from the PS20P2VP80 (L)/Ag NPs statistically increased during the first 3 days (from 82 to 237) followed by a decline to ultimate values within the range of 13 to 30 after 1 week. The observation that the CV SERS signal derived from PS20P2VP80 (L)/Ag NPs is greater than that derived from PS0P2VP100 (L)/Ag NPs further emphasizes the importance of the PS component in the microgel/latex. We hypothesize that the decline in the CV SERS signal in the PS20P2VP80 (L)/Ag NP system is due to oxidation of the Ag NPs with ongoing exposure to the ambient atmosphere. Confirming this and further stabilizing the SERS active NPs by way of the addition of secondary metals and/or refining the size and shape control of the NPs represents the subject of current work in this area.
We hypothesize that the inclusion of some PS into the structure of the PSxP2VPy (M)/Ag NPs improves the pH-sensitive SERS performance by balancing swellability with interfacial bonding strain. By reducing the number of pyridine sites in the microgel, the swellability is reduced (Figure 2b) and the bonding strain is reduced at the Ag NP–microgel interface during the pH-triggered iterative expansion and contraction is reduced. If this interfacial bonding strain is too high, the bonds to the Ag NPs break and the SERS activity and pH-sensitivity will decrease because of the loss of Ag NPs and an overall loss in the number of SERS-active interparticle gaps. Figure S8 shows the STEM images of the PS0P2VP100 (M)/Ag NPs and PS20P2VP80 (M)/Ag NPs prior to any pH cycling and after 3 pH cycles, respectively. It is clear that the number of Ag NPs on the PS0P2VP100 (M)/Ag NP sample decreases during the first 3 pH cycles, a finding that explains the decrease in the intensity of the Raman spectra (i.e., the decrease in the S/N at 1620 cm–1 in Figure 6d). The population of Ag NPs on the PS20P2VP80 (M)/Ag NPs after 3 pH cycles is similar to that of uncycled analogue. This supports the theory that a composite with balanced swellability and a high number of pyridine sites for anchoring Ag NPs will exhibit improved pH-sensitive SERS performance. To further confirm the relationship between the change in the SERS signal and a related variation of the distance of the gap between Ag NPs, a UV–vis study was also conducted. The UV–vis spectra for PS0P2VP100 (M/L)/Ag NPs and PS20P2VP80 (M/L)/Ag NPs in Figures S9 and S10 were acquired over the course of the first three pH cycles. In the initial microgel condition (spectra I), both materials only show a broad plasmon absorbance band at ∼405 nm that is derived from roughly spherical and nonagglomerated Ag NPs. Upon treatment with acid, the latex condition (spectra II) evolved and included an additional plasmon-coupled absorbance feature at ∼610 nm because of a decreased distance between the Ag NPs for both the PS0P2VP100 (L)/Ag NPs and PS20P2VP80 (L)/Ag NPs. In both systems, the plasmon-coupled absorbance value reversibly alternates in size as the two PSxP2VPy (M/L)/Ag NP samples are taken through pH cycling until the final test with n = 3 (spectra VI). This strongly suggests that the alternating SERS signal intensity reported in Figure 6d is related to the number and the size of the nanoscale gaps between the Ag NP. Additionally, it is also apparent that the ultimate plasmon-coupled absorbance for PS20P2VP80 (L)/Ag NPs is stronger than that found in PS0P2VP100 (L)/Ag NPs (spectra VI in Figures S9 and S10). This finding also confirms that the PS20P2VP80 (L)/Ag NPs are the more SERS-sensitive material after break-in.
Conclusions
A controlled methodology for the synthesis of microgels with programmed properties such as pH sensitivity, swellability, and capacity for metal-loading is of great importance to many fields. In this work, sterically stabilized and covalently cross-linked latexes based on random copolymers of S and 2VP are synthesized. The latexes were found to be monodisperse by DLS and by SEM and exhibited a pH-triggered latex-to-microgel transition in the range of pH = 1.2–3.8, a characteristic that is set by the S/2VP ratio that is defined at the synthesis stage. The volumetric swelling factor was tuned from 2.5 to 140 with this ratio. The charged PSxP2VPy (M) was loaded with metal-containing anions such as AuCl4–- and borate-capped Ag NPs. Loading of PSxP2VPy microgels by DLS with KAuCl4 showed a diameter reduction with increased binding where a loading limit of ∼0.3 molar equivalents of AuCl4– to pyridinyl groups was determined. The aurate-loaded PSxP2VPy (M) was converted to Au NP-loaded PSxP2VPy (M) by photoreduction. Tomography by TEM showed that Au NPs were located throughout the microgel structure. The loading of PSxP2VPy (M) with borate-capped Ag NPs produced PSxP2VPy (M) with surface-located Ag NPs. The Ag NP content of this nanocomposite was determined by the molecular ratio of S/2VP corresponding to a range of ∼5–30 NPs per microgel particle when the P2VP content ranged from 60 to 100%. The Ag NP-loaded PSxP2VPy (M) composites exhibited SERS activity for the macromolecular structure in the microgel, as well as for the representative solution-phase analyte, CV. A pH-responsive SERS activity for the Ag NP-loaded PSxP2VPy (M) was found where the AEF for dissolved CV was tunable from ∼5.21 × 109 to 1.11 × 1010. The Ag NP-loaded PSxP2VPy (M) with ∼80 wt % P2VP exhibited the most stable pH response and a high AEF of 1.08 × 1010.
Experimental Section
Materials
Styrene (S; 99% purity), 2VP (97% purity), and 2,2′-azobis(2-methylpropionamidine) dihydrochloride (AIBA; 98% purity) were acquired from Acros Organics. PEGMA (average Mn = 2000, 50 wt % in H2O) and DVB (55% purity) were purchased from Sigma-Aldrich. N-Methyl-N,N,N-trioctyloctan-1-ammonium chloride (Aliquat 336) was used as received from Alfa Aesar. Potassium tetrachloroaurate (KAuCl4), hydrogen tetrachloroaurate (HAuCl4), sodium borohydride (NaBH4), silver nitrate (AgNO3), and dry alumina oxide were obtained through Fischer Scientific, Inc. Hydrochloric acid (HCl) was used as received from Macron Fine Chemicals. Ultrapure water was produced using a Barnstead Nanopure water purifier and had an overall resistivity of 18 MΩ·cm. Regenerated cellulose dialysis tubing with a MWCO of 8000 or 12 000–14 000 were used as received from Fisher Scientific, Inc. The purification of S, 2VP, and DVB was done under Schlenk conditions by passing each monomer through dry alumina oxide powder to remove the polymerization inhibitors. The purified monomers were used within 24 h, and if short-term storage was required, they were sealed in round bottom flasks with an inert atmosphere and stored in the dark at −18 °C.
Synthesis of PSxP2VPy Latexes
The preparation of latex particles based on random copolymers of PS and P2VP (PSxP2VPy) follows a modified procedure outlined by Dupin et al.46 The emulsion polymerization was carried out in a 100 mL single-necked round-bottom reaction flask. The PEGMA stabilizer (0.420 mL) and Aliquat 336 surfactant (0.15 g) were both dissolved in ultrapure water (40.00 g) in the reaction flask and sealed with a rubber septum. The reaction flask contents were deaerated with 30 min of nitrogen bubbling or five freeze, pump, thaw cycles. The flask, containing an inert atmosphere, was placed in a hot oil bath, and the contents stirred at 500 rpm until the temperature stabilized at 60 °C. The monomer mixture of 2VP (2.00–5.00 g), S (0.00–3.00 g) and DVB (0.03 g) was added to a scintillation vial under inert atmosphere for a total targeted amount of 5.03 g. The contents of the monomer vial were transferred to the reaction flask using a syringe equipped with a filter (PTFE, 0.45 μm, Millex). An emulsified condition was established by continual stirring (500 rpm, 60 °C) for approximately 30 min. Deaerated, ultrapure water (5.00 g) was used to pre-dissolve the AIBA initiator (0.050 g) in a separate scintillation vial with an inert atmosphere. Once dissolved, the AIBA solution was rapidly delivered to the reaction flask via a syringe to initiate the polymerization reaction. Stirring continued for 24 h. Purification of the resulting latex was achieved by centrifuging the crude latex for 15 min at 19 000 rpm in high-density polycarbonate tubes (Thermo Scientific). After decanting the supernatant liquid, an addition of ultrapure water was made to bring the volume of the latex solution back to its original level. Redispersion was carried out by sonication and/or stirring until the solution appeared uniform. This centrifugation/decanting process was repeated a minimum of three times. The final solution contained 8–9 wt % solids (with yields of 72–81%).
Preparation of Au NP-Loaded PSxP2VPy Microgels
The procedure for the preparation of Au ion-loaded colloids was adapted from Nakamura et al.45 The pH of the PSxP2VPy (L) was adjusted to transform the latex into its microgel state, (i.e., to pH = 2.5 for PS0P2VP100, PS20P2VP80, and PS40P2VP60, and pH = 1.2 for PS60P2VP40) using 0.1 M HCl(aq). The PSxP2VPy (M) was further diluted with HCl(aq) (1:20) using the appropriate HCl solution. The KAuCl4 was dissolved in 0.0032 M HCl(aq) (i.e., pH = 2.5 for PS20P2VP80 and PS40P2VP60) and 0.063 M HCl(aq) (i.e., pH = 1.2 for PS60P2VP40) to make a 24 mM KAuCl4 solution. This pH-matched KAuCl4 solution was added slowly to the diluted PSxP2VPy (M) until the desired level of ion-loading was achieved. In some cases, the loading was monitored by DLS. The final solution was stirred for 30 min and purified by dialysis with a MWCO of 8000 for 48 h. This afforded the ion-doped colloidal microgels. The ion-doped microgels were subjected to photolysis with stirring (500 rpm) for ∼18 h using a Rayonet model RPR-600 UV photochemical reactor (Southern New England Ultraviolet, Inc.) equipped with eight light sources, each with a 253.7 nm maximum emission and a power of 8 W. The resulting Au NP-loaded PSxP2VPy (M) were purified by three sequential 8 h dialysis steps using a MWCO of 12 000–14 000 membrane and fresh ultrapure water.
Synthesis of Borate-Capped Ag NPs
The borate-capped Ag NPs were synthesized according to modified procedures.80,95 The reducible Ag ion solution (5 mM) was prepared by dissolving 21.2 mg of AgNO3 in 25 mL of ultrapure water. This solution was added to an ice bath-chilled 75 mL aqueous solution of NaBH4 (2 mM) and stirred for a minimum of 1 h. The reaction mixture gradually turned yellow-green over the first hour. This solution was aged in the dark for a minimum of 2 weeks prior to use. An average diameter of 59.8 nm (polydispersity = 0.361) for a pH = 8 solution of borate-capped Ag NPs was determined by DLS.
Complexation of PSxP2VPy Microgels with Ag NPs
A 10 μL volume of the purified PSxP2VPy (L) was diluted to a final volume of 10 mL with ultrapure water. The latexes were converted to the microgel state as indicated above. The borate-capped Ag NP solution was evaporated to half the volume (3 mL) and then was added slowly to the stirred microgel solution. The pH of the final solution was adjusted back to the starting value with the addition of HCl(aq). The Ag NP-loaded PSxP2VPy microgels were purified by three consecutive 8 h dialysis steps using a MWCO of 12 000–14 000 membrane and fresh ultrapure water.
Particle Size Characterization by DLS
DLS measurements were conducted using a DelsaNano HC instrument (Beckman Coulter, Inc., Fullerton, CA) equipped with a 658 nm laser diode. The photomultiplier and aperture attenuator were set to collect scattered light at an angle of 15° with a photon counting rate of ∼10 kcps. Size measurements were conducted on ∼2–3 mL of the sample in disposable PS cuvettes (4 mL capacity). The properties of pH-adjusted solutions were assumed to be the same as water (n = 1.3329, η = 0.890 cP at 25 ± 1 °C). Samples (0.2 wt % solids) were filtered using 5 μm cellulose syringe filters (Millex) and allowed to equilibrate at 25 °C for ∼5 min before each measurement, followed by a 35–60 s acquisition time (1 s per acquisition). The calculation of the particle size and size distribution was performed using CONTIN particle size analysis routines. The intensity distribution plots were averaged, and the data are reported in Table 1 as the average hydrodynamic diameter (DH) of the particles. A DLS-monitored titration with 0.1 M HCl was conducted on diluted PSxP2VPy colloids (original solution/ultrapure water = 1:20 v/v), with DLS size measurements occurring every 0.2 pH units after 1 min of equilibration.
Thermal Analysis
A TA Instruments Q500 TGA instrument equipped with an inline Thermo Scientific iS10 FTIR gas cell spectrometer was used for both the TGA and TGA–FTIR experiments.96 To monitor the mass changes during thermal degradation and to determine the NP-loading, TGA experiments were conducted with a 15 min isotherm at 100 °C followed by a 20 °C/min ramp from 100 to 800 °C. An isotherm before and after this temperature program was held for 5 min to monitor the background signal of FTIR spectra. The effluent from the sample was carried to the FTIR spectrometer via an isothermal transfer line (225 °C) with dry nitrogen gas flowing at 90 mL/min. The FTIR spectra (4 scans at 8 cm–1 resolution per spectra) were acquired at a 2.7 s interval for the duration of the TGA and were corrected using a background dataset created from a time averaged signal (5 min) from the blank nitrogen carrier gas.
General Instrumentation
XPS data were acquired using a Kratos M-Probe instrument equipped with an Al Kα monochromatic X-ray source and a hemispherical analyzer (with an outer diameter of 145 mm and an inner diameter of 88 mm). The analysis spot size was 15.71 μm2 in area and the take-off angle was 55° with respect to the vertical direction of samples. The spectra were fit using 30% Gaussian–Lorentzian components and a Shirley background subtraction. All binding energies were corrected to the C 1s peak for the background hydrocarbon component (C–C/C–Hx) at 284.9 eV.97 Electron tomography was conducted using a Talos F200X transmission electron microscope. The tilted view inspections of the samples were conducted with Inspect 3D software. SEM images were acquired using either Vega TS 5136MM operated with an accelerating voltage of 15 kV or JEOL JSM-7200F/LV operated with an accelerating voltage of 30 kV. With an exception of the Ag NPs, samples for SEM analysis were coated with a thin layer of Au and palladium (Pd) to dissipate charge (nominal thickness ≈ 1 nm). Scanning TEM (STEM) images were acquired with the JEOL JSM-7200F/LV instrument. Samples investigated by STEM were applied to a lacey carbon copper grid (Ted Pella) and not coated with charge dissipation materials prior to microscopy. The UV–vis spectroscopy was performed with a JASCO V670 spectrophotometer. The wavelength resolution was set to 1.0 nm and the scan speed was 400 nm/min. Absorbance was monitored for Ag-loaded PSxP2VPy solutions following dialysis and a 1:50 v/v dilution with pH = 2.5 aqueous HCl. The UV–vis spectra were acquired at each pH condition for a total of three pH cycles. The pH was cycled with the addition of either 82 μL of 3.0 M NaOH or 41 μL of 1.71 M HCl to 2.5 mL of the diluted Ag-loaded PSxP2VPy. The UV–vis spectra were normalized to their peak maxima.
Surface-Enhanced Raman Scattering
The SERS data were acquired using a DeltaNu Advantage 200A spectrometer equipped with a 633 nm helium–neon laser (5 mW). The spectral acquisition time was 60 s. The system was calibrated using bulk PS and neat cyclohexane as standards for the Raman frequency shifts (peaks for PS at 1001 and 621 cm–1; peaks for cyclohexane at 1444 and 1266 cm–1). All of the spectra were interpreted after background correction to exclude interference due to fluorescence.98 Error bars correspond to the 95% confidence interval for 4–5 replicates.
Acknowledgments
This work was supported in part by external grants from the American Chemical Society Petroleum Research Fund (ACS-PRF UNI #51559-UNI10 and ACS-PRF UR #54780-UR10) and Research Corporation (#21109). Equipment in the STS Electron Microscopy Laboratory at WWU was partially funded by a grant from the M. J. Murdock Charitable Trust (#2015117). The authors also acknowledge the use of facilities in Washington State that are part of the Joint Center for Deployment and Research in Earth Abundant Materials (JCDREAM). Michael Kraft and Dan Carnevale are acknowledged for their assistance with SEM characterization. Lee Pullan at Thermo Fisher Scientific is acknowledged for TEM and TEM-tomography studies. T.C., A.K.T., and S.E.A. are grateful for research grants from the Vice Provost for Research at WWU.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b01561.
TGA–FTIR data for selected PSxP2VPy (M) samples and their AuCl4– and Au NP-loaded equivalents; TGA–FTIR data for a control PSxP2VPy (M) sample treated to a UV photoreduction condition; TGA–FTIR data for selected PSxP2VPy (M)/Ag NPs; DLS-monitored pH titration for PS20P2VP80 (M)/Au NPs; SEM images for PS40P2VP60 (M)/Au NPs and PS60P2VP40 (M)/Au NPs; TEM-tomography video (AVI), micrographs, and tilt frames for PS0P2VP100 (M)/Au NPs; Raman spectra for selected PSxP2VPy (M)/Au NPs and equivalents exposed to solution state CV; S/N of the 1620 cm–1 signal for CV versus the diameter of free Au NPs; background comparison from an overlay plot for the Raman spectra of the PS20P2VP80 (M)/Ag NPs without and with CV in the solution; study for the S/N at 1620 cm–1 for CV versus time using selected PSxP2VPy (M)/Ag NPs; STEM images of PS0P2VP100/Ag NPs (L) and PS20P2VP80/Ag NPs (L) after 0 pH cycles and after 3 pH cycles; and UV–vis spectra for selected PSxP2VPy (L)/Ag NPs during sequential pH cycling (PDF)
Author Present Address
∥ Department of Chemistry, Simon Fraser University, 8888 University Drive, Burnaby, Canada, V5A 1S6.
Author Present Address
⊥ Department of Chemistry, Indiana University, 800 E. Kirkwood Avenue, Bloomington, IN 47405-7102.
The authors declare no competing financial interest.
Supplementary Material
References
- Van Duyne R. P.Chemical and Biochemical Applications of Lasers; Academic Press: New York, 1979; Vol. 4, pp 101–185. [Google Scholar]
- Moskovits M. Surface-enhanced spectroscopy. Rev. Mod. Phys. 1985, 57, 783–826. 10.1103/revmodphys.57.783. [DOI] [Google Scholar]
- Michaels A. M.; Nirmal M.; Brus L. E. Surface Enhanced Raman Spectroscopy of Individual Rhodamine 6G Molecules on Large Ag Nanocrystals. J. Am. Chem. Soc. 1999, 121, 9932–9939. 10.1021/ja992128q. [DOI] [Google Scholar]
- Tian Z. Q. Surface-enhanced Raman spectroscopy: advancements and applications. J. Raman Spectrosc. 2005, 36, 466–470. 10.1002/jrs.1378. [DOI] [Google Scholar]
- Wang D.-S.; Kerker M. Enhanced Raman scattering by molecules adsorbed at the surface of colloidal spheroids. Phys. Rev. B: Condens. Matter Mater. Phys. 1981, 24, 1777–1790. 10.1103/physrevb.24.1777. [DOI] [Google Scholar]
- Metal Nanoparticles Synthesis, Characterization and Applications; Feldheim D. L., Foss C. A. Jr., Eds.; Marcel Dekker Inc.: New York, USA, 2002; pp 1–338. [Google Scholar]
- Eustis S.; El-Sayed M. A. Why gold nanoparticles are more precious than pretty gold: Noble metal surface plasmon resonance and its enhancement of the radiative and nonradiative properties of nanocrystals of different shapes. Chem. Soc. Rev. 2006, 35, 209–217. 10.1039/b514191e. [DOI] [PubMed] [Google Scholar]
- Stiles P. L.; Dieringer J. A.; Shah N. C.; Van Duyne R. P. Surface-Enhanced Raman Spectroscopy. Annu. Rev. Anal. Chem. 2008, 1, 601–626. 10.1146/annurev.anchem.1.031207.112814. [DOI] [PubMed] [Google Scholar]
- Halas N. J.; Lal S.; Chang W.-S.; Link S.; Nordlander P. Plasmons in Strongly Coupled Metallic Nanostructures. Chem. Rev. 2011, 111, 3913–3961. 10.1021/cr200061k. [DOI] [PubMed] [Google Scholar]
- Cao Y.; Li D.; Jiang F.; Yang Y.; Huang Z. Engineering Metal Nanostructure for SERS Application. J. Nanomater. 2013, 2013, 123812. 10.1155/2013/123812. [DOI] [Google Scholar]
- Kumar G. V. P. Plasmonic nano-architectures for surface enhanced Raman scattering: a review. J. Nanophotonics 2012, 6, 064503. 10.1117/1.jnp.6.064503. [DOI] [Google Scholar]
- Su X.; Zhang J.; Sun L.; Koo T.-W.; Chan S.; Sundararajan N.; Yamakawa M.; Berlin A. A. Composite Organic–Inorganic Nanoparticles (COINs) with Chemically Encoded Optical Signatures. Nano Lett. 2005, 5, 49–54. 10.1021/nl0484088. [DOI] [PubMed] [Google Scholar]
- Xu S.; Ji X.; Xu W.; Zhao B.; Dou X.; Bai Y.; Ozaki Y. Surface-enhanced Raman scattering studies on immunoassay. J. Biomed. Opt. 2005, 10, 031112. 10.1117/1.1915487. [DOI] [PubMed] [Google Scholar]
- Jeong D. H.; Zhang Y. X.; Moskovits M. Polarized Surface Enhanced Raman Scattering from Aligned Silver Nanowire Rafts. J. Phys. Chem. B 2004, 108, 12724–12728. 10.1021/jp037973g. [DOI] [Google Scholar]
- Lee S. J.; Morrill A. R.; Moskovits M. Hot Spots in Silver Nanowire Bundles for Surface-Enhanced Raman Spectroscopy. J. Am. Chem. Soc. 2006, 128, 2200–2201. 10.1021/ja0578350. [DOI] [PubMed] [Google Scholar]
- Pieczonka N. P. W.; Aroca R. F. Single molecule analysis by surfaced-enhanced Raman scattering. Chem. Soc. Rev. 2008, 37, 946–954. 10.1039/b709739p. [DOI] [PubMed] [Google Scholar]
- Nie S.; Emory S. R. Probing Single Molecules and Single Nanoparticles by Surface-Enhanced Raman Scattering. Science 1997, 275, 1102–1106. 10.1126/science.275.5303.1102. [DOI] [PubMed] [Google Scholar]
- Kneipp K.; Wang Y.; Kneipp H.; Perelman L. T.; Itzkan I.; Dasari R. R.; Feld M. S. Single molecule detection using surface-enhanced Raman scattering (SERS). Phys. Rev. Lett. 1997, 78, 1667–1670. 10.1103/physrevlett.78.1667. [DOI] [Google Scholar]
- Aldeanueva-Potel P.; Faoucher E.; Alvarez-Puebla R. A.; Liz-Marzán L. M.; Brust M. Recyclable Molecular Trapping and SERS Detection in Silver-Loaded Agarose Gels with Dynamic Hot Spots. Anal. Chem. 2009, 81, 9233–9238. 10.1021/ac901333p. [DOI] [PubMed] [Google Scholar]
- Wu Y.; Zhou F.; Yang L.; Liu J. A shrinking strategy for creating dynamic SERS hot spots on the surface of thermosensitive polymer nanospheres. Chem. Commun. 2013, 49, 5025–5027. 10.1039/c3cc40875b. [DOI] [PubMed] [Google Scholar]
- Manikas A. C.; Romeo G.; Papa A.; Netti P. A. Highly Efficient Surface-Enhanced Raman Scattering Substrate Formulation by Self-Assembled Gold Nanoparticles Physisorbed on Poly(N-isopropylacrylamide) Thermoresponsive Hydrogels. Langmuir 2014, 30, 3869–3875. 10.1021/la4048616. [DOI] [PubMed] [Google Scholar]
- Alexander K. D.; Skinner K.; Zhang S.; Wei H.; Lopez R. Tunable SERS in Gold Nanorod Dimers through Strain Control on an Elastomeric Substrate. Nano Lett. 2010, 10, 4488–4493. 10.1021/nl1023172. [DOI] [PubMed] [Google Scholar]
- Liu H.; Zhang L.; Lang X.; Yamaguchi Y.; Iwasaki H.; Inouye Y.; Xue Q.; Chen M. Single molecule detection from a large-scale SERS-active Au79Ag21 substrate. Sci. Rep. 2011, 1, 112. 10.1038/srep00112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lang X.; Hirata A.; Chen M. Wrinkled Nanoporous Gold Films with Ultrahigh Surface-Enhanced Raman Scattering Enhancement. ACS Nano 2011, 5, 4407–4413. 10.1021/nn201443p. [DOI] [PubMed] [Google Scholar]
- Wei H.; Xu H. Hot spots in different metal nanostructures for plasmon-enhanced Raman spectroscopy. Nanoscale 2013, 5, 10794–10805. 10.1039/c3nr02924g. [DOI] [PubMed] [Google Scholar]
- Taladriz-Blanco P.; Buurma N. J.; Rodríguez-Lorenzo L.; Pérez-Juste J.; Liz-Marzán L. M.; Hervés P. Reversible assembly of metal nanoparticles induced by penicillamine. Dynamic formation of SERS hot spots. J. Mater. Chem. 2011, 21, 16880–16887. 10.1039/c1jm12175h. [DOI] [Google Scholar]
- Guo Q.-H.; Zhang C.-J.; Wei C.; Xu M.-M.; Yuan Y.-X.; Gu R.-A.; Yao J.-L. Controlling dynamic SERS hot spots on a monolayer film of Fe3O4@Au nanoparticles by a magnetic field. Spectrochim. Acta, Part A 2016, 152, 336–342. 10.1016/j.saa.2015.07.092. [DOI] [PubMed] [Google Scholar]
- Jun B.-H.; Kim G.; Baek J.; Kang H.; Kim T.; Hyeon T.; Jeong D. H.; Lee Y.-S. Magnetic field induced aggregation of nanoparticles for sensitive molecular detection. Phys. Chem. Chem. Phys. 2011, 13, 7298–7303. 10.1039/c0cp02055a. [DOI] [PubMed] [Google Scholar]
- Kim K.; Choi J.-Y.; Lee H. B.; Shin K. S. Silanization of Ag-Deposited Magnetite Particles: An Efficient Route to Fabricate Magnetic Nanoparticle-Based Raman Barcode Materials. ACS Appl. Mater. Interfaces 2010, 2, 1872–1878. 10.1021/am1002074. [DOI] [PubMed] [Google Scholar]
- Ahijado-Guzmán R.; Gómez-Puertas P.; Alvarez-Puebla R. A.; Rivas G.; Liz-Marzán L. M. Surface-enhanced Raman scattering-based detection of the interactions between the essential cell division FtsZ protein and bacterial membrane elements. ACS Nano 2012, 6, 7514–7520. 10.1021/nn302825u. [DOI] [PubMed] [Google Scholar]
- Yang M.; Alvarez-Puebla R.; Kim H.-S.; Aldeanueva-Potel P.; Liz-Marzán L. M.; Kotov N. A. SERS-active gold lace nanoshells with built-in hotspots. Nano Lett. 2010, 10, 4013–4019. 10.1021/nl101946c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abalde-Cela S.; Ho S.; Rodríguez-González B.; Correa-Duarte M. A.; Álvarez-Puebla R. A.; Liz-Marzán L. M.; Kotov N. A. Loading of exponentially grown LBL films with silver nanoparticles and their application to generalized SERS detection. Angew. Chem., Int. Ed. 2009, 48, 5326–5329. 10.1002/anie.200901807. [DOI] [PubMed] [Google Scholar]
- Lu Y.; Liu G. L.; Lee L. P. High-Density Silver Nanoparticle Film with Temperature-Controllable Interparticle Spacing for a Tunable Surface Enhanced Raman Scattering Substrate. Nano Lett. 2005, 5, 5–9. 10.1021/nl048965u. [DOI] [PubMed] [Google Scholar]
- Mitsuishi M.; Koishikawa Y.; Tanaka H.; Sato E.; Mikayama T.; Matsui J.; Miyashita T. Nanoscale Actuation of Thermoreversible Polymer Brushes Coupled with Localized Surface Plasmon Resonance of Gold Nanoparticles. Langmuir 2007, 23, 7472–7474. 10.1021/la701215t. [DOI] [PubMed] [Google Scholar]
- Xu H.; Xu J.; Jiang X.; Zhu Z.; Rao J.; Yin J.; Wu T.; Liu H.; Liu S. Thermosensitive Unimolecular Micelles Surface-Decorated with Gold Nanoparticles of Tunable Spatial Distribution. Chem. Mater. 2007, 19, 2489–2494. 10.1021/cm070088g. [DOI] [Google Scholar]
- Li D.; Cui Y.; Wang K.; He Q.; Yan X.; Li J. Thermosensitive Nanostructures Comprising Gold Nanoparticles Grafted with Block Copolymers. Adv. Funct. Mater. 2007, 17, 3134–3140. 10.1002/adfm.200700427. [DOI] [Google Scholar]
- Yusa S.-i.; Fukuda K.; Yamamoto T.; Iwasaki Y.; Watanabe A.; Akiyoshi K.; Morishima Y. Salt Effect on the Heat-Induced Association Behavior of Gold Nanoparticles Coated with Poly(N-isopropylacrylamide) Prepared via Reversible Addition–Fragmentation Chain Transfer (RAFT) Radical Polymerization. Langmuir 2007, 23, 12842–12848. 10.1021/la702741q. [DOI] [PubMed] [Google Scholar]
- Mei Y.; Lu Y.; Polzer F.; Ballauff M.; Drechsler M. Catalytic Activity of Palladium Nanoparticles Encapsulated in Spherical Polyelectrolyte Brushes and Core–Shell Microgels. Chem. Mater. 2007, 19, 1062–1069. 10.1021/cm062554s. [DOI] [Google Scholar]
- Kroll E.; Winnik F. M.; Ziolo R. F. In Situ Preparation of Nanocrystalline γ-Fe2O3in Iron(II) Cross-Linked Alginate Gels. Chem. Mater. 1996, 8, 1594–1596. 10.1021/cm960095x. [DOI] [Google Scholar]
- Pich A.; Bhattacharya S.; Lu Y.; Boyko V.; Adler H.-J. P. Temperature-Sensitive Hybrid Microgels with Magnetic Properties. Langmuir 2004, 20, 10706–10711. 10.1021/la040084f. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S.; Eckert F.; Boyko V.; Pich A. Temperature-, pH-, and Magnetic-Field-Sensitive Hybrid Microgels. Small 2007, 3, 650–657. 10.1002/smll.200600590. [DOI] [PubMed] [Google Scholar]
- Rubio-Retama J.; Zafeiropoulos N. E.; Serafinelli C.; Rojas-Reyna R.; Voit B.; Lopez Cabarcos E.; Stamm M. Synthesis and Characterization of Thermosensitive PNIPAM Microgels Covered with Superparamagnetic γ-Fe2O3Nanoparticles. Langmuir 2007, 23, 10280–10285. 10.1021/la7009594. [DOI] [PubMed] [Google Scholar]
- Mohammadi Z.; Cole A.; Berkland C. J. In Situ Synthesis of Iron Oxide within Polyvinylamine Nanoparticle Reactors. J. Phys. Chem. C 2009, 113, 7652–7658. 10.1021/jp901036b. [DOI] [Google Scholar]
- Chen H.; You T.; Jiang L.; Gao Y.; Yin P. Creating dynamic SERS hotspots on the surface of pH-responsive microgels for direct detection of crystal violet in solution. RSC Adv. 2017, 7, 32743–32748. 10.1039/c7ra05567f. [DOI] [Google Scholar]
- Akamatsu K.; Shimada M.; Tsuruoka T.; Nawafune H.; Fujii S.; Nakamura Y. Synthesis of pH-Responsive Nanocomposite Microgels with Size-Controlled Gold Nanoparticles from Ion-Doped, Lightly Cross-Linked Poly(vinylpyridine). Langmuir 2010, 26, 1254–1259. 10.1021/la902450c. [DOI] [PubMed] [Google Scholar]
- Dupin D.; Fujii S.; Armes S. P.; Reeve P.; Baxter S. M. Efficient synthesis of sterically stabilized pH-responsive microgels of controllable particle diameter by emulsion polymerization. Langmuir 2006, 22, 3381–3387. 10.1021/la053258h. [DOI] [PubMed] [Google Scholar]
- Martin T. J.; Procházka K.; Munk P.; Webber S. E. pH-Dependent Micellization of Poly(2-vinylpyridine)-block-poly(ethylene oxide). Macromolecules 1996, 29, 6071–6073. 10.1021/ma960629f. [DOI] [Google Scholar]
- Shen H.; Zhang L.; Eisenberg A. Multiple pH-Induced Morphological Changes in Aggregates of Polystyrene-block-poly(4-vinylpyridine) in DMF/H2O Mixtures. J. Am. Chem. Soc. 1999, 121, 2728–2740. 10.1021/ja983712m. [DOI] [Google Scholar]
- Satoh M.; Yoda E.; Hayashi T.; Komiyama J. Potentiometric titration of poly(vinylpyridines) and hydrophobic interaction in the counterion binding. Macromolecules 1989, 22, 1808–1812. 10.1021/ma00194a051. [DOI] [Google Scholar]
- Bronstein L. M.; Sidorov S. N.; Valetsky P. M.; Hartmann J.; Cölfen H.; Antonietti M. Induced Micellization by Interaction of Poly(2-vinylpyridine)-block-poly(ethylene oxide) with Metal Compounds. Micelle Characteristics and Metal Nanoparticle Formation. Langmuir 1999, 15, 6256–6262. 10.1021/la990146f. [DOI] [Google Scholar]
- Cook J. P.; Riley D. J. pH induced swelling of PVP microgel particles - A first order phase transition?. J. Colloid Interface Sci. 2012, 370, 67–72. 10.1016/j.jcis.2011.12.064. [DOI] [PubMed] [Google Scholar]
- Handbook of Vinyl Polymers: Radical Polymerization, Process, and Technology, 2nd ed.; Mishra M. K., Yagci Y., Ed.; CRC Press: Boca Raton, FL, 2016; pp 1–784. [Google Scholar]
- Principles of Polymerization, 4th ed.; Odian G. G., Ed.; Wiley-Interscience: Hoboken, New Jersey, 2004; pp 1–832. [Google Scholar]
- Batich C. D.; Yan J.; Bucaria C.; Elsabee M. Swelling behavior of pH-sensitive copolymers based on styrene and 4-(or 2-)vinylpyridine. Macromolecules 1993, 26, 4675–4680. 10.1021/ma00069a040. [DOI] [Google Scholar]
- Yu J.; Geng C.; Zheng L.; Ma Z.; Tan T.; Wang X.; Yan Q.; Shen D. Preparation of High-Quality Colloidal Mask for Nanosphere Lithography by a Combination of Air/Water Interface Self-Assembly and Solvent Vapor Annealing. Langmuir 2012, 28, 12681–12689. 10.1021/la3026182. [DOI] [PubMed] [Google Scholar]
- Tantavichet N.; Pritzker M. D.; Burns C. M. Proton uptake by poly(2-vinylpyridine) coatings. J. Appl. Polym. Sci. 2001, 81, 1493–1497. 10.1002/app.1577. [DOI] [Google Scholar]
- Ripoll C.; Muller G.; Selegny E. Polyelectrolytes basiques faibles-II. Determination du pKa de la poly(vinyl-2 pyridine) et des coefficients d’activite des petits ions de la solution. Discussion et conclusion. Eur. Polym. J. 1971, 7, 1393–1409. 10.1016/0014-3057(71)90034-6. [DOI] [Google Scholar]
- Graham S.; Cormack P. A. G.; Sherrington D. C. One-Pot Synthesis of Branched Poly(methacrylic acid)s and Suppression of the Rheological “Polyelectrolyte Effect”. Macromolecules 2005, 38, 86–90. 10.1021/ma048651t. [DOI] [Google Scholar]
- Kocak G.; Tuncer C.; Bütün V. pH-Responsive polymers. Polym. Chem. 2017, 8, 144–176. 10.1039/c6py01872f. [DOI] [Google Scholar]
- Thaiboonrod S.; Berkland C.; Milani A. H.; Ulijn R.; Saunders B. R. Poly(vinylamine) microgels: pH-responsive particles with high primary amine contents. Soft Matter 2013, 9, 3920–3930. 10.1039/c3sm27728c. [DOI] [Google Scholar]
- Zhang J.; Zhang M.; Tang K.; Verpoort F.; Sun T. Polymer-Based Stimuli-Responsive Recyclable Catalytic Systems for Organic Synthesis. Small 2014, 10, 32–46. 10.1002/smll.201300287. [DOI] [PubMed] [Google Scholar]
- Cera G.; Biffis A.; Canton P.; Villa A.; Prati L. Metal nanoclusters stabilized by pH-responsive microgels: Preparation and evaluation of their catalytic potential. React. Funct. Polym. 2017, 115, 81–86. 10.1016/j.reactfunctpolym.2017.04.002. [DOI] [Google Scholar]
- Liu G.; Wang D.; Zhou F.; Liu W. Electrostatic Self-Assembly of Au Nanoparticles onto Thermosensitive Magnetic Core-Shell Microgels for Thermally Tunable and Magnetically Recyclable Catalysis. Small 2015, 11, 2807–2816. 10.1002/smll.201403305. [DOI] [PubMed] [Google Scholar]
- Fernández-López C.; Polavarapu L.; Solís D. M.; Taboada J. M.; Obelleiro F.; Contreras-Cáceres R.; Pastoriza-Santos I.; Pérez-Juste J. Gold Nanorod-pNIPAM Hybrids with Reversible Plasmon Coupling: Synthesis, Modeling, and SERS Properties. ACS Appl. Mater. Interfaces 2015, 7, 12530–12538. 10.1021/am5087209. [DOI] [PubMed] [Google Scholar]
- Thorne J. B.; Vine G. J.; Snowden M. J. Microgel applications and commercial considerations. Colloid Polym. Sci. 2011, 289, 625–646. 10.1007/s00396-010-2369-5. [DOI] [Google Scholar]
- Lyons A. M.; Pearce E. M.; Mujsce A. M. Thermal decomposition of poly(2-vinylpyridine): Effect of complexation with copper chloride. J. Polym. Sci., Part A: Polym. Chem. 1990, 28, 245–259. 10.1002/pola.1990.080280202. [DOI] [Google Scholar]
- Madorsky S. L. Rates of thermal degradation of polystyrene and polyethylene in a vacuum. J. Polym. Sci. 1952, 9, 133–156. 10.1002/pol.1952.120090203. [DOI] [Google Scholar]
- Tonelli M.; Turrell S.; Cristini-Robbe O.; El Hamzaoui H.; Capoen B.; Bouazaoui M.; Gazzano M.; Cassani M. C. Synthesis of gold nanoparticles within silica monoliths through irradiation techniques using Au(I) and Au(III) precursors. RSC Adv. 2014, 4, 26038–26045. 10.1039/c4ra01315h. [DOI] [Google Scholar]
- Borodko Y.; Ercius P.; Pushkarev V.; Thompson C.; Somorjai G. From Single Pt Atoms to Pt Nanocrystals: Photoreduction of Pt2+ Inside of a PAMAM Dendrimer. J. Phys. Chem. Lett. 2012, 3, 236–241. 10.1021/jz201599u. [DOI] [Google Scholar]
- Riedinger A.; Pernia Leal M.; Deka S. R.; George C.; Franchini I. R.; Falqui A.; Cingolani R.; Pellegrino T. Nanohybrids Based on pH-Responsive Hydrogels and Inorganic Nanoparticles for Drug Delivery and Sensor Applications. Nano Lett. 2011, 11, 3136–3141. 10.1021/nl2011627. [DOI] [PubMed] [Google Scholar]
- Pérez-Juste J.; Liz-Marzán L. M.; Carnie S.; Chan D. Y. C.; Mulvaney P. Electric-Field-Directed Growth of Gold Nanorods in Aqueous Surfactant Solutions. Adv. Funct. Mater. 2004, 14, 571–579. 10.1002/adfm.200305068. [DOI] [Google Scholar]
- Miranda O. R.; Ahmadi T. S. Effects of Intensity and Energy of CW UV Light on the Growth of Gold Nanorods. J. Phys. Chem. B 2005, 109, 15724–15734. 10.1021/jp0514832. [DOI] [PubMed] [Google Scholar]
- Kitagawa H.; Kojima N.; Nakajima T. Studies of mixed-valence states in three-dimensional halogen-bridged gold compounds, Cs2Au I Au III X6, (X = Cl, Br or I). Part 2. X-Ray photoelectron spectroscopic study. J. Chem. Soc., Dalton Trans. 1991, 3121–3125. 10.1039/dt9910003121. [DOI] [Google Scholar]
- Turner N. H.; Single A. M. Determination of peak positions and areas from wide-scan XPS spectra. Surf. Interface Anal. 1990, 15, 215–222. 10.1002/sia.740150305. [DOI] [Google Scholar]
- van Attekum P. M. T. M.; Trooster J. M. Bulk- and surface-plasmon-loss intensities in photoelectron, Auger, and electron-energy-loss spectra of Mg metal. Phys. Rev. B: Condens. Matter Mater. Phys. 1979, 20, 2335–2340. 10.1103/physrevb.20.2335. [DOI] [Google Scholar]
- Beyler C. L.; Hirschler M. M.. SFPE Handbook of Fire Protection Engineering, 3rd ed.; National Fire Protection Association and Society of Fire Protection Engineers: Quincy, MA, 2002; pp 1–131. [Google Scholar]
- Yoon S.-M.; Kim U. J.; Benayad A.; Lee I. H.; Son H.; Shin H.-J.; Choi W. M.; Lee Y. H.; Jin Y. W.; Lee E.-H.; Lee S. Y.; Choi J.-Y.; Kim J. M. Thermal Conversion of Electronic and Electrical Properties of AuCl3-Doped Single-Walled Carbon Nanotubes. ACS Nano 2011, 5, 1353–1359. 10.1021/nn103055u. [DOI] [PubMed] [Google Scholar]
- Yoon J. K.; Kim K.; Shin K. S. Raman Scattering of 4-Aminobenzenethiol Sandwiched between Au Nanoparticles and a Macroscopically Smooth Au Substrate: Effect of Size of Au Nanoparticles. J. Phys. Chem. C 2009, 113, 1769–1774. 10.1021/jp8089243. [DOI] [Google Scholar]
- Roth P. G.; Boerio F. J. Surface-enhanced raman scattering from poly(4-vinyl pyridine). J. Polym. Sci., Part B: Polym. Phys. 1987, 25, 1923–1933. 10.1002/polb.1987.090250912. [DOI] [Google Scholar]
- Mulfinger L.; Solomon S. D.; Bahadory M.; Jeyarajasingam A. V.; Rutkowsky S. A.; Boritz C. Synthesis and Study of Silver Nanoparticles. J. Chem. Educ. 2007, 84, 322. 10.1021/ed084p322. [DOI] [Google Scholar]
- Gao J.; Jiang C.; Su X. Synthesis and Thermal Properties of Boron-Nitrogen Containing Phenol Formaldehyde Resin/MMT Nanocomposites. Int. J. Polym. Mater. 2010, 59, 544–552. 10.1080/00914031003760659. [DOI] [Google Scholar]
- CRC Materials Science and Engineering Handbook, 4th ed.; CRC Press, 2015; pp 3–338. [Google Scholar]
- Han S. H.; Kim J. K.; Pryamitsyn V.; Ganesan V. Phase Behavior of Binary Blends of Block Copolymers Having Hydrogen Bonding. Macromolecules 2011, 44, 4970–4976. 10.1021/ma200747d. [DOI] [Google Scholar]
- Hong P. P.; Boerio F. J.; Tirrell M.; Dhoot S.; Guenoun P. An investigation of the adsorption of polystyrene/poly(2-vinylpyridine) diblock copolymers onto silver substrates using surface-enhanced Raman scattering. Macromolecules 1993, 26, 3953–3959. 10.1021/ma00067a034. [DOI] [Google Scholar]
- Tsai W. H.; Boerio F. J.; Clarson S. J.; Parsonage E. E.; Tirrell M. Characterization of adsorbed 2-vinylpyridine/styrene diblock copolymers on silver surfaces using surface-enhanced Raman scattering. Macromolecules 1991, 24, 2538–2545. 10.1021/ma00009a062. [DOI] [Google Scholar]
- Venkatachalam R. S.; Boerio F. J.; Roth P. G.; Tsai W. H. Surface-enhanced Raman scattering from bilayers of polystyrene, diglycidyl ether of bisphenol-A, poly(4-vinyl pyridine), and poly(4-styrene sulfonate). J. Polym. Sci., Part B: Polym. Phys. 1988, 26, 2447–2461. 10.1002/polb.1988.090261205. [DOI] [Google Scholar]
- Le Ru E. C.; Blackie E.; Meyer M.; Etchegoin P. G. Surface Enhanced Raman Scattering Enhancement Factors: A Comprehensive Study. J. Phys. Chem. C 2007, 111, 13794–13803. 10.1021/jp0687908. [DOI] [Google Scholar]
- Kneipp K.; Kneipp H. Single molecule Raman scattering. Appl. Spectrosc. 2006, 60, 322A–334A. 10.1366/000370206779321418. [DOI] [PubMed] [Google Scholar]
- Sunder S.; Bernstein H. J. Resonance Raman spectrum of a deuterated crystal violet: [p(CH3)2NC6D4]3C+Cl–. Can. J. Chem. 1981, 59, 964–967. 10.1139/v81-140. [DOI] [Google Scholar]
- Persaud I.; Grossman W. E. L. Surface-enhanced Raman scattering of triphenylmethane dyes on colloidal silver. J. Raman Spectrosc. 1993, 24, 107–112. 10.1002/jrs.1250240209. [DOI] [Google Scholar]
- Cialla D.; Hübner U.; Schneidewind H.; Möller R.; Popp J. Probing innovative microfabricated substrates for their reproducible SERS activity. ChemPhysChem 2008, 9, 758–762. 10.1002/cphc.200700705. [DOI] [PubMed] [Google Scholar]
- Lee P. C.; Meisel D. Adsorption and surface-enhanced Raman of dyes on silver and gold sols. J. Phys. Chem. 1982, 86, 3391–3395. 10.1021/j100214a025. [DOI] [Google Scholar]
- Le Ru E. C.; Meyer M.; Etchegoin P. G. Proof of Single-Molecule Sensitivity in Surface Enhanced Raman Scattering (SERS) by Means of a Two-Analyte Technique. J. Phys. Chem. B 2006, 110, 1944–1948. 10.1021/jp054732v. [DOI] [PubMed] [Google Scholar]
- Tokareva I.; Minko S.; Fendler J. H.; Hutter E. Nanosensors Based on Responsive Polymer Brushes and Gold Nanoparticle Enhanced Transmission Surface Plasmon Resonance Spectroscopy. J. Am. Chem. Soc. 2004, 126, 15950–15951. 10.1021/ja044575y. [DOI] [PubMed] [Google Scholar]
- Lee P. C.; Meisel D. Adsorption and surface-enhanced Raman of dyes on silver and gold sols. J. Phys. Chem. 1982, 86, 3391–3395. 10.1021/j100214a025. [DOI] [Google Scholar]
- Hackler R. A.; Hollcraft A. T.; Kirkness T. A.; Larson N. S.; Hoekstra N. L.; Rider D. A. Relief of Cure Stress in Prepreg Composites with Engineered Voids: A Solution to Warping in Composite Phenolic Resin/Fiberglass Laminates. Ind. Eng. Chem. Res. 2016, 55, 3568–3578. 10.1021/acs.iecr.5b02852. [DOI] [Google Scholar]
- High Resolution XPS of Organic Polymers—The Scienta ESCA300 Database; John Wiley & Sons: Chichester, 1992. [Google Scholar]
- Kim H.; Kosuda K. M.; Van Duyne R. P.; Stair P. C. Resonance Raman and Surface- And Tip-Enhanced Raman Spectroscopy Methods to Study Solid Catalysts and Heterogeneous Catalytic Reactions. Chem. Soc. Rev. 2010, 39, 4820–4844. 10.1039/c0cs00044b. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.