

Atherosclerosis underlies the leading cause of death in industrialized countries.1 Atherosclerosis is a maladaptive, nonresolving chronic inflammatory disease that occurs predominantly at sites of disturbed laminar flow, notably, arterial branch points and bifurcations, of large and medium‐sized arteries.1 Atherosclerosis is initiated by subendothelial buildup of apolipoprotein B–containing lipoproteins (in particular LDL [low‐density lipoprotein]).1 LDL, particularly following oxidation, has properties of damage‐associated molecular patterns2 and thereby provokes the activation of endothelial cells (ECs)3 and ensuing recruitment of blood‐borne monocytes to the lesion.4 The Ly6Chi subpopulation of monocytes differentiates into macrophages.4 Serving to protect the organism against tissue damage, macrophages have developed plasticity to tailor the inflammatory responses. In short, macrophages have the capacity to trigger inflammation when necessary and to quell the inflammatory response when it is no longer required. In the atherosclerotic milieu, macrophages are predominantly polarized to an inflammatory phenotype.4 Lipid‐loaded cells known as foam cells constitute a hallmark of atherosclerotic lesion, which can be detected at very early stages of atherosclerosis.5 The causal role for metabolic stress factors such as LDL, particularly oxidized LDL (oxLDL) and cholesterol, in promoting vascular inflammation and atherosclerosis has long been appreciated.6 However, the precise mechanism whereby metabolic stress elicits the proinflammatory status and induces atherogenesis remained unclear. When resolution of inflammation goes awry, nonresolving inflammation characterizes the atherosclerotic lesions.7 The mechanisms behind the nonresolving nature of vascular inflammation in the atherosclerotic lesion remain to be entirely defined. The inflammatory environment incurs apoptosis of intimal cells. Defective efferocytosis, a process by which phagocytic cells (predominantly macrophages) engulf the dead cells, promotes the formation of the necrotic core.2 Immune responses are further amplified by relentless production of damage‐associated molecular patterns from necrotic cells in the advanced lesions.5, 7
Innate immunity plays crucial roles in plaque initiation, progression, and rupture.5 It has been well established that the inflammasomes constitute an important component of innate immunity. The term inflammasome was first coined by Jurg Tschopp and his colleagues in 2002 to explain the mechanism for caspase‐1 activation and interleukin‐1β processing.8 To date, diverse inflammasomes have been discovered. Among the various inflammasomes identified, the nucleotide‐binding oligomerization domain, leucine‐rich repeat–containing receptor (NLR) family pyrin domain‐containing 3 (NLRP3) inflammasome is best characterized.8 The groundbreaking findings that the NLRP3 inflammasome is a key player in orchestrating lipid‐driven vascular inflammation and plays a fundamental role in the initiation and progression of atherogenesis9 are transforming the field of atherosclerosis. The focus of this review is on our current understanding of the role, regulation, and mechanisms of NLRP3 inflammasome activation in atherosclerosis. We will also discuss the therapeutic potential of NLRP3 inflammasome intervention.
Two‐Step Activation Model of the NLRP3 Inflammasome
Inflammation is a protective response to infection and injury, which is mounted by the innate immune system. Innate immunity response depends on the recognition of pathogen‐associated molecular patterns and damage‐associated molecular patterns through the pattern‐recognition receptors, such as the Toll‐like receptors and NLRs.10 The NLRs are now recognized as the key sensors of pathogens and danger signals. Engagement of the NLRs elicits downstream signaling cascades and leads to the production of proinflammatory cytokines and type I interferons.10 The core components of the inflammasomes include the adaptor apoptosis‐associated speck‐like protein containing a CARD (ASC), a zymogen pro‐caspase‐1 and an NLR family member, such as the best‐demonstrated NLRP3.8 The NLR is engaged to dictate the assembly of the inflammasomes in response to pathogen‐associated molecular patterns or damage‐associated molecular patterns.8 The NLRP3 inflammasome is a multimeric protein complex that is composed of NLRP3, ASC, and pro‐caspase‐1.8 The NLRP3 contains the N‐terminal pyrin domain responsible for recruitment of ASC, the central nucleotide‐binding oligomerization domain, and the C‐terminal leucine‐rich repeat.10 The nucleotide‐binding oligomerization domain domain enables the activation of the signaling complex via oligomerization, whereas leucine‐rich repeat is thought to function in ligand sensing and autoregulation.8 NEK7, a serine and threonine kinase that is involved in mitosis, can directly bind NLRP3 and control NLRP3 oligomerization.11 NEK7 appears to be a new component of the NLRP3 complex.11
Basal levels of NLRP3 are inadequate for efficient inflammasome formation.12 Meanwhile, NLRP3 is kept in an inactive ubiquitinated state until a priming signal provokes its deubiquitination.13 It has been generally accepted that the activation of the NLRP3 inflammasome requires 2 signals: a priming and an activation signal (Figure 1). A priming signal induces NFκB‐dependent transcriptional up‐regulation of NLRP312 and the deubiquitination of NLRP3 by the Lys63‐specific deubiquitinase BRCC3.13 As the second step in the activation of the NLRP3 inflammasome, an activation signal triggers the assembly and activation of the inflammasome, culminating in the activation of caspase‐1.8 In brief, primed NLRP3 undergoes oligomerization in response to the activation signal. Oligomerized NLRP3 serves as a scaffold to recruit ASC through the pyrin domain–pyrin domain interactions,14 leading to the generation of long ASC filaments, the latter of which recruit pro‐caspase‐1. The close proximity of pro‐caspase‐1 protein then induces autoproteolytic maturation of pro‐caspase‐1 into active caspase‐1.14
Figure 1.
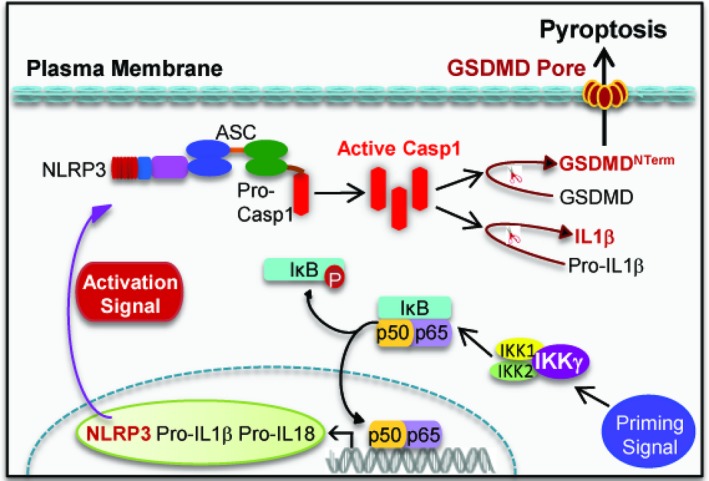
Two‐step activation model of the NLRP3 inflammasome. Activation of the NLRP3 inflammasome requires 2 signals: a priming signal and an activation signal. In unstimulated cells, the transcription factor NFκB is sequestered in the cytoplasm by the IκB family members. IκB degradation is a prerequisite for NFκB activation, which is initiated upon phosphorylation by the IκB kinase (IKK) complex in response to priming signal. The IKK complex consists of 2 catalytic subunits (IKK1 and IKK2) and a regulatory subunit IKKγ. As the second step in inflammasome activation, an activation signal triggers the assembly and activation of the inflammasome, culminating in the activation of caspase‐1 and the production of interleukin‐1β, interleukin‐18 and the GSDMDNT erm. ASC indicates apoptosis‐associated speck‐like protein containing a caspase activation and recruitment domain; GSDMD, gasdermin D; GSDMDNT erm, N‐terminal GSDMD; IL18, interleukin‐18; IL1β, interleukin‐1β; IκB, inhibitor of kappa B; NFκB, nuclear factor kappa‐light‐chain‐enhancer of activated B cells; NLRP3, nucleotide‐binding domain, leucine‐rich–containing family, pyrin domain–containing 3; Pro‐Casp1, pro‐caspase‐1.
Because of the vast number of structurally dissimilar agonists for the NLRP3 inflammasome,8 it seems unlikely that these agonists induce the activation of the inflammasome by directly binding to NLRP3. A favorite theory is that NLRP3 responds to a common cellular event that can be initiated by the diverse NLRP3 activators. However, despite years of research, no unified mechanism underlying NLRP3 inflammasome activation has been recognized. To date, different mechanisms behind inflammasome activation have been put forward, involving potassium efflux, calcium influx, mitochondrial dysfunction, the generation of mitochondrial reactive oxygen species (ROS), the release of mitochondrial DNA or cardiolipin, and lysosomal destabilization and rupture.15, 16, 17, 18, 19 Notably, a recent study suggested that diverse NLRP3 stimuli can induce the disassembly of the trans‐Golgi network to the dispersed trans‐Golgi network (dTGN).20 NLRP3 is then recruited to the dTGN through an interaction between a polybasic region within the NLRP3 and phosphatidylinositol‐4‐phosphate on the dTGN. The dTGN functions as a scaffold for NLRP3 aggregation, which is essential for ASC polymerization and ensuing activation of the downstream signaling cascade. The recruitment of NLRP3 to dTGN is thought to be a common event that is required for NLRP3 aggregation and activation in response to different activators.20
Role for the NLRP3 Inflammasome in Atherosclerosis
The components of the NLRP3 inflammasome are dominantly expressed in macrophages and foam cells within human carotid atherosclerotic plaques.21 As such, the majority of studies on the NLRP3 inflammasome were carried out in macrophages. Intriguingly, NLRP3 inflammasome components are also expressed in ECs.22 In resting cells, endogenous NLRP3 in macrophages and ECs is expressed at low levels. In stark contrast, human atherosclerotic plaques have dramatically elevated NLRP3 inflammasome components (including activated caspase‐1) when compared with healthy counterparts.23
Accumulated evidence reveals a causative role for the NLRP3 inflammasome in the initiation and progression of atherosclerosis, although the studies on the role of the NLRP3 inflammasome in this disease have yielded mixed results.9, 24 Remarkably, the impact of inflammasome activation in atherosclerosis is consistent with the earlier findings that genetic depletion of interleukin‐1β or interleukin‐1 receptor (interleukin‐1R) attenuates atherosclerosis in hypercholesterolemic mice.25, 26 Genetic ablation of interleukin‐1R antagonist, an endogenous competitive inhibitor of interleukin‐1R that can block interleukin‐1α and interleukin‐1β responses, aggravates atherogenesis in atherosclerosis‐prone mice.27
Consistent with this proposition, LDL receptor–deficient (Ldlr−/−) mice transplanted with bone marrow from mice deficient in NLRP3, ASC, or interleukin‐1β, respectively, had significantly reduced aortic lesion size and serum interleukin‐18 levels.9 While genetic ablation of caspase‐1 under an ApoE (apolipoprotein E)‐deficient background ameliorated atherosclerosis,28 reconstitution of Ldlr−/− mice with caspase‐1−/− bone marrow significantly thwarted the development of atherosclerotic lesions in comparison with the control bone marrow.29 A recent seminal study indicated that a Western diet induces functional reprogramming of myeloid cells, incites trained immunity, and instigates systemic inflammation in a mouse model of atherosclerosis.30 This study provided further evidence showing that the NLRP3 inflammasome pathway mediates the trained immunity in the context of Western diet feeding and its deleterious effects on inflammatory diseases such as atherosclerosis.30
The major factors that activate the NLRP3 inflammasome in the atherosclerotic scenario has been identified recently (Figure 2). Hypoxia prevails in the atherosclerotic lesion.31 Hypoxia favors plaque angiogenesis,31, 32 promotes foam cell formation,33 and contributes to the formation of the plaque necrotic core.34 It has long been hypothesized that hypoxia may conspire with inflammation to exacerbate atherosclerosis. In support of this notion, a recent study established a direct link between hypoxia and the NLRP3 inflammasome.35 In addition to stabilization of interleukin‐1β protein by restricting its autophagic degradation, hypoxia increased NLRP3 expression and activation in human macrophages and in the plaques.35 This study provided strong evidence that low oxygen tension exacerbates atherosclerosis by aggravating inflammation.
Figure 2.
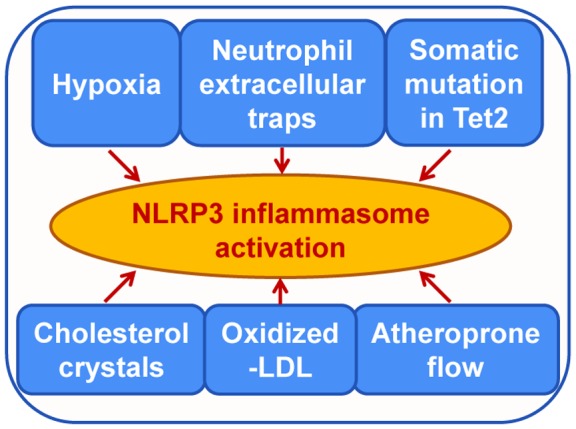
Signals involved in NLRP3 inflammasome activation in atherosclerosis. The activation of the NLRP3 inflammasome drives the initiation and progression of atherosclerosis. Shown are the key stimuli identified to date that induce priming and activating of the NLRP3 inflammasome in the atherosclerotic milieu. LDL indicates low‐density lipoprotein; Tet2, tet methylcytosine dioxygenase 2.
NLRP3 can sense various types of the metabolic stress molecules, as exemplified by cholesterol crystals (CCs)9 and oxLDL.9, 36 We will discuss how CCs and oxLDL drive atherosclerosis later. Different metabolites contribute to the activation of the NLRP3 inflammasome. For instance, the best‐known NLRP3 activator ATP can be released from dying/dead cells into the atherosclerotic necrotic core,37 where it efficiently activates the NLRP3 inflammasome through the engagement of its cognate receptor P2X7R.38 As expected, P2X7R was found to be abundantly expressed in the atherosclerotic plaques.38 Moreover, deletion of P2X7R in Ldlr−/− mice led to declined lesional inflammasome activation and reduced atherosclerotic plaque size.38 These findings implicate lesional ATP in NLRP3 inflammasome activation. Additionally, calcium phosphate crystals were found to participate in the activation of caspase‐1 and the production of active interleukin‐1β in atherosclerotic lesions.39
The oscillatory shear stress likely represents one of the initial triggers for NLRP3 inflammasome activation in atherogenesis.22 Strikingly, this stimulus acts as both priming and activating signals in NLRP3 inflammasome activation in ECs.22 In addition, neutrophil extracellular traps (NETs) can induce the activation of the NLRP3 inflammasome in macrophages.40
Regulation and Mechanisms of CCs‐Triggered NLRP3 Inflammasome Activation
The NLRP3 inflammasome links arterial deposition of lipids and lipoproteins to the inflammatory responses driving the initiation and progression of atherosclerosis. The consumption of high‐fat, high‐cholesterol diets underlies the culprit of hypercholesterolemia, particularly in individuals with genetic predisposition. Cholesterol is an extremely important lipid that has crucial roles in membrane structure; it is essential for maintaining membrane permeability and cell signaling.41 Mammalian cells cannot degrade cholesterol; elimination of excessive cellular cholesterol is primarily mediated by HDL (high‐density lipoprotein).41 Disruption in cholesterol homeostasis leads to CC buildup within the necrotic core causing plaque rupture. The advent of CCs in the atherosclerotic plaques was once considered a late characteristic of atherosclerosis. However, emerging evidence pointed out that the formation of small CCs occurs even at early stage of atherosclerotic lesions.9 A plaque containing abundant CCs is a hallmark of vulnerable plaques. CCs may induce cell death and perforate the fibrous cap.42 Thus, developing agents that dissolve CCs is expected to offer an alternative approach to stabilize vulnerable plaques. Besides their mechanical and toxic effect, CCs induce arterial wall injury through triggering inflammation.
Atherosclerosis begins with deposition of cholesterol into the arterial wall via LDL particles.43 Once LDL is oxidized in the subendothelial region of arterial wall, it can be assimilated by lesion macrophages.42 The lesional macrophages can promote reverse cholesterol transport by producing nascent HDL.44 Impairment of this pathway facilitates the formation of foam cell and CCs.42 Cholesterol within LDL exists in the esterified form.42 Reverse cholesterol transport requires the conversion of esterified cholesterol to free cholesterol (FRC) for it to be mobilized by transporters ATP‐binding cassette A‐1 and G1 (ABCA‐1 and ABCG‐1).45 ABCA1 and ABCG1 transport FRC out of cells to HDL, promoting cholesterol efflux from macrophages onto HDL particles or onto apolipoprotein A1.45 Through this process, cholesterol is transported from peripheral tissues back to the liver, followed by excretion into bile and feces.46 It is known that HDL can partially dissolve CCs.47 Deficiency of ABCA1/G1 in myeloid cells induces significant accumulation of cholesterol in macrophages and ensuing NLRP3 inflammasome activation.48 Mechanistically, myeloid ABCA1/G1 deficiency increases the expression of Nlrp3 and pro‐ interleukin‐1β mRNA likely through a Toll‐like receptor 4–mediated priming effect, and elicits a membrane cholesterol sensing mechanism triggering noncanonical NLRP3 inflammasome activation.48 It is known that noncanonical inflammasome activation induces caspase‐11 cleavage, leading to the activation of the NLRP3 inflammasome.49 Indeed, myeloid Abca1/g1 deficiency enhanced caspase‐11 cleavage in Ly6G+ neutrophils and Ly6G‐CD11b+ macrophages.48 Consistent with the observations made in mouse atherosclerosis model, the patients of Tangier disease with mutations in ABCA1 have higher plasma interleukin‐1β, suggestive of conservation of cholesterol‐mediated inflammasome activation in humans.48
With the progression of atherosclerosis, there is an increase in local and systemic inflammation that can induce HDL dysfunction, as primarily characterized by reduced cholesterol efflux capacity of HDL.50 HDL dysfunction causes FRC accumulation in the cell membrane and extracellular space, which leads to CC formation and cell death.51 In addition to HDL dysfunction, imbalance between esterified cholesterol and FRC contributes to FRC buildup within foam cells.50 It is known that cholesterol ester hydrolase convert esterified cholesterol to FRC, while acyl‐coenzyme A cholesterol acyltransferase 1 (ACAT1) converts FRC to esterified cholesterol.51 Acyltransferase 1 inhibition gives rise to liquid crystalline and cholesterol monohydrate crystals formation in macrophages’ plasma membrane bilayer.51 These membrane cholesterol domains serve as the platforms for CC formation.51 Notably, cells with rich cholesterol membranes including dying foam cells are thought to be the additional sources of FRC.52 The release of cellular contents from dying cells favors CC formation by means of inducing local physical change.53
Cholesterol efflux restrains the production of inflammatory mediators in macrophages.54 Defective cholesterol efflux promotes monocyte and neutrophil production in the bone marrow and the spleen.55 Mouse and human studies revealed that excessive production of inflammatory cells under hypercholesterolemic conditions have a causal role in promoting atherosclerotic cardiovascular disease (CVD).55 Macrophages in atherosclerotic plaques may be derived from blood‐borne monocytes, which are produced in the bone marrow and the spleen.56 As a systemic consequence of inflammasome activation in myeloid cells, neutrophil activation and recruitment into the plaques ensue, leading to the formation of NETs in early atherosclerotic lesions.48
CCs, which are the major driver of atherosclerosis, are now regarded as the most important trigger for NLRP3 inflammasome activation.9 CCs provide both priming and activating signals for the NLRP3 inflammasome. Importantly, HDL can blunt NLRP3 inflammasome activation and interleukin‐1β production incited by CCs in cultured macrophages and in mouse peritonitis model.47 Although more work is needed to unveil the precise mechanism of action, HDL appears to function by antagonizing the transcription of NLRP3 and interleukin‐1β, the activation of caspase‐1, and lysosomal damage imposed by CCs.47 These findings highlight the significance of HDL in the regulation of the NLRP3 inflammasome. Further study is required to explore the impact of HDL on NLRP3 priming.
Extensive studies have been conducted to uncover the mechanisms whereby CCs activate the NLRP3 inflammasome. As discussed below, CCs initiate inflammation via triggering the activation of the NLRP3 inflammasome through multiple mechanisms (Figure 3).
Figure 3.
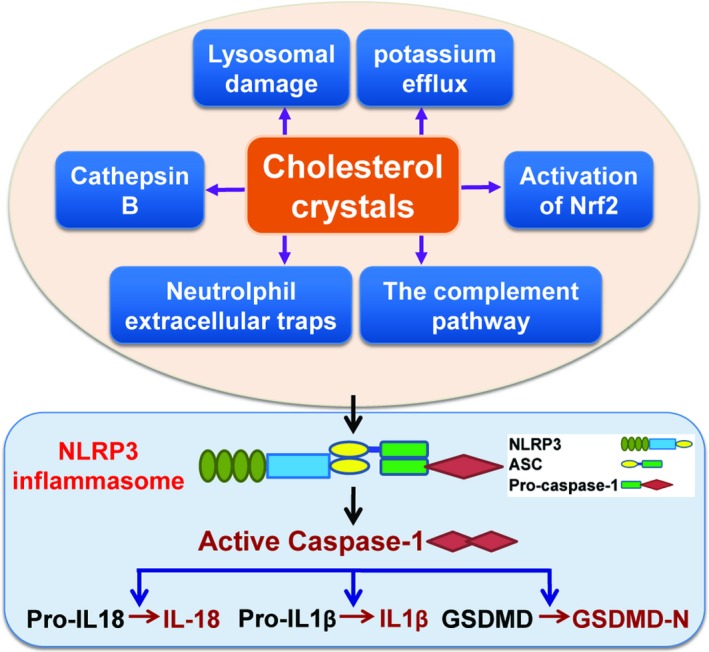
Mechanisms underlying the activation of the NLRP3 inflammasome by cholesterol crystals. The causative role for cholesterol in vascular inflammation and atherosclerosis is undeniable, yet the precise mechanism by which cholesterol elicits the inflammatory response and dictates atherogenesis remained mysterious. The newly identified mechanisms whereby cholesterol crystals license the activation of the NLRP3 inflammasome are summarized. GSDMD‐N indicates the amino terminus of GSDMD; IL‐18, interleukin‐18; IL1β, interleukin‐1β; Nrf2, nuclear factor E2‐related factor 2.
CCs prime macrophage NLRP3 inflammasome in atherosclerosis through inciting the formation of NETs, a process known as NETosis.40 NETosis has been implicated in atherothrombosis.57 NETs are large extracellular weblike structures containing DNA, histones, proteases, and myeloperoxidase,58 which have been discovered in atherosclerotic plaques.59 There is a potential link between the NLRP3 inflammasome and plaque NETs. A recent study indicated that CCs promote the release of NETs by neutrophils, which further triggers the activation of the NLRP3 inflammasome in lesional macrophages.40 The mechanism whereby NETs activate the NLRP3 inflammasome needs to be delineated in the future. CCs induce NETosis likely through inflammasome activation.60 However, the finding that the NETs cause the activation of the NLRP3 inflammasome is under debate.61 Further uncovering the mechanisms linking CCs to NETs’ release may lead to the development of novel therapeutics specifically targeting atherosclerotic inflammation.60
It is well demonstrated that CCs fuel NLRP3 inflammasome activation by inducing lysosomal damage. CC uptake or formation in macrophages induces lysosomal damage and ensuing NLRP3 inflammasome activation, leading to the production of atherogenic cytokines interleukin‐1β and interleukin‐18.9, 36 CCs can be degraded in macrophage lysosomes.62 Inefficient solubilization of CCs causes lysosomal dysfunction,62 which may interfere with autophagy.63 Autophagy has been reported to restrain the activity of the NLRP3 inflammasome by promoting their degradation in lysosomes.64 Macrophage autophagy plays an atheroprotective role by abrogating NLRP3 inflammasome activation incited by CCs. Defective autophagy potentiates CCs‐mediated inflammasome activation in macrophages.63 Consistent with this proposition, macrophages loaded with atherogenic lipids showed an increase in the autophagy chaperone p62, a response that is caused predominantly by disruption of the autophagy‐lysosome system,65 boosting the formation of the NLRP3 inflammasome in atherosclerotic macrophages.65 Apoe−/− mice with autophagy deficiency in macrophages exhibited enhanced inflammasome activation in macrophages and enlarged atherosclerotic plaques with increased the content of CCs.63 Further support for this proposition comes from a gain‐of‐function study carried out by forced expression of the transcription factor EB, which is known as the master regulator of lysosomal biogenesis that elicits the expression of lysosomal and autophagy genes in response to lysosomal stress.66 As expected, overexpression of transcription factor EB was shown to rescue CC‐mediated lysosomal dysfunction and to block interleukin‐1β secretion.66 Additionally, cathepsin B and potassium efflux are required for CC‐triggered activation of the NLRP3 inflammasome.67
CCs can trigger the activation of the NLRP3 inflammasome through a mechanism associated with the oxidative stress‐responsive transcription factor Nrf2.68 Mechanistically, CCs trigger Nrf2‐dependent interleukin‐1β expression in macrophages in a manner that is dependent on NLRP3 inflammasome‐mediated caspase‐1 activation.68 Importantly, the NLRP3 inflammasome integrates inflammation and oxidative stress, both of which are known to contribute significantly to atherogenesis.
CCs trigger NLRP3 inflammasome activation by increasing CD36 expression.69 Elevated CD36 levels potentiate oxLDL uptake into macrophages,69 promoting intracellular cholesterol crystallization and NLRP3 inflammasome activation.36 In addition, macrophages can be efficiently primed by oxLDL for CC‐mediated NLRP3 inflammasome activation.9 These results demonstrate the dual role of oxLDL as NLRP3 priming and activating signals.
CCs can elicit the activation of the NLRP3 inflammasome by their potential to activate the complement pathways, specifically the C5a and the C5aR pathways,70 which can be engaged to activate the NFκB pathway.70 C5a is important for reactive oxygen species production and caspase‐1 activation. Thus, the activated complement factors provide the priming signal for the NLRP3 inflammasome and also promote CC phagocytosis.70
Role and Mechanism for Shear Stress in Regulating Endothelial NLRP3 Inflammasome
ECs are considered as the atypical immune cells.71 ECs express pattern‐recognition receptors such as Toll‐like receptors and CD36 scavenger receptor, engagement of which leads to the activation of NFκB signaling.71 ECs also express NLRs such as NLRP3.72 Recent studies demonstrated that the activation of the NLRP3 inflammasome in ECs occurs in response to diverse insults such as disturbed flow.22 Mimicking disturbed flow and oscillatory shear stress substantially augments the production of active caspase‐1 and interleukin‐1β in ECs.22 In line with the in vitro finding, the activation of the NLRP3 inflammasome is evident in mouse aortic arch, as evidenced by elevated levels of active caspase‐1 and interleukin‐1β. A study showed that macrophage‐derived microparticles increase the expression of cell adhesion molecules in ECs via the activation of endothelial NLRP3 inflammasome.73 In addition to provoking the processing of caspase‐1 and proinflammatory cytokines, activated caspase‐1 triggers pyroptosis, the inflammatory cell death that appears to precipitate the development of atherosclerosis.22, 72
In contrast to the well‐defined model for inflammasome activation in macrophages, much less is known regarding the 2‐step mechanism underlying NLRP3 inflammasome activation in ECs. SREBP2 (Sterol regulatory element‐binding protein 2) was identified as a potent activator of the NLRP3 inflammasome in ECs. SREBP2 is known as a master regulator in cholesterol biosynthesis.72 In support of the above finding, SREBP2 exacerbates the initiation and progression of atherosclerosis. Intriguingly, disturbed flow‐activated SREBP2 can induce the NLRP3 inflammasome via transactivation of NLRP3 in ECs.22 Taken together, SREBP2 is involved in both priming and activation of the NLRP3 inflammasome. A recent study pointed out that SREBP2 is a transcription factor that regulates tumor necrosis factor‐α receptor–associated factor (TRAF)‐interacting protein with a forkhead‐associated domain (TIFA).74 TIFA was shown to activate NFκB signaling, at least partly, through interacting with tumor necrosis factor‐α receptor–associated factor 2 or tumor necrosis factor‐α receptor–associated factor 6.75 Remarkably, TIFA is involved in the induction of inflammasome components through the SREBP2–TIFA–NFκB axis.74 Notably, oxidative and/or inflammatory stimuli, including oscillatory shear stress, tumor necrosis factor‐α and oxLDL, elicit a robust activation of Akt, which can target TIFA for phosphorylation. Phosphorylated TIFA undergoes oligomerization and subsequently interacts with caspase‐1, thereby potentiating the assembly and activation of the NLRP3 inflammasome. Collectively, TIFA is involved in both priming and activation of the NLRP3 inflamamsome in ECs.74
Role and Mechanism for the NLRP3 Inflammasome in Tet2 Somatic Mutation‐Driven Atherosclerosis
Although the contribution of hypercholesterolemia to atherosclerosis is undeniable, the impact of hypercholesterolemia on cardiovascular risk, however, was found to reduce gradually with aging.76 Recent studies showed that most individuals at low cardiovascular risk were affected with substantial subclinical atherosclerosis.77 Furthermore, emerging evidence suggests that unidentified age‐related factors contribute to the development of atherosclerosis.78 Accumulated evidence emphasizes that somatic mutations in hematopoietic cells may drive atherosclerotic CVD.79 The somatic mutation may confer a competitive advantage to the cell, leading to clonal expansion,80 which is particularly true for the hematopoietic stem/progenitor cell. Somatic mutation‐driven clonal hematopoiesis, whose role in the development of atherosclerosis is poorly defined, is common in the elderly population.81 The impact of somatic mutation‐driven clonal hematopoiesis on atherosclerosis is attracting increasing attention. Most somatic mutations associated with clonal hematopoiesis occur in 4 genes: tet methylcytosine dioxygenase 2 (TET2), DNA methyltransferase 3α (DNMT3A), additional sex combs like 1 and Janus kinase 2.82 However, their relevance in CVD remained unexplored until a recent discovery demonstrating the causative role of somatic mutation‐driven clonal hematopoiesis in exacerbating atherosclerosis.83, 84
Ldlr−/− mice acquired a small number of Tet2+/−, Tet2−/−, or myeloid cell‐specific Tet2−/− cells through competitive bone marrow transplantation, which largely recapitulates the scenario of clonal hematopoiesis linked to somatic mutations in human hematopoietic Tet2. These cells expanded progressively in bone marrow with a preferential differentiation into the Ly6Chi monocyte population.84 The clonal hematopoiesis robustly accelerated atherosclerosis.84 These findings were independently replicated in mice with full hematopoietic ablation of Tet2.83 Mechanistically, somatic mutation or defect of Tet2 leads to the overproduction of proinflammatory cytokines (particularly interleukin‐1β) in macrophages.83, 84, 85 Tet2 is known as a DNA demethylating enzyme that enhances transcriptional activation by catalyzing the oxidation of 5‐methylcytosine in DNA to 5‐hydroxymethylcytosine.84 Intriguingly, Tet2 suppresses pro‐interleukin‐1β transcription through nucleating histone deacetylases to its promoter region, a function that is independent of Tet2 catalytic activity.84 More importantly, Tet2 deficiency promotes NLRP3 inflammasome activation, facilitating the processing of pro‐interleukin‐1β.84 In support of this finding, pharmacologic blockade of the NLRP3 inflammasome significantly ameliorated atherosclerosis development incited by somatic mutation in Tet2. These observations highlight the fundamental importance of the NLRP3 inflammasome in mediating Tet2 mutation‐driven atherosclerosis.84 Excessive interleukin‐1β production by Tet2‐deficient cells promotes the expression of P‐selectin and the activation of ECs in the plaque, leading to increased monocyte recruitment to the lesion.84 Since interleukin‐1β is known to stimulate its own expression,43 it is likely that overproduction of interleukin‐1β by Tet2‐deficient cells promotes further expression of interleukin‐1β in both Tet2‐deficient and wild‐type cells. This finding demonstrated that the recruitment of small number of Tet2‐mutant cells in the plaque may be adequate to fuel atherosclerosis by promoting NLRP3/pro‐interleukin‐1β‐driven vascular inflammation.84 It is reasonable to speculate that individuals carrying somatic mutations in Tet2 may respond much more favorably than general population to NLRP3/interleukin‐1β‐targeted therapeutic, which will furnish the basis for a personalized therapy for atherosclerotic CVD patients with somatic mutations in Tet2.
Mechanism for the NLRP3 Inflammasome to Drive Atherosclerosis
Since the NLRP3 inflammasome plays critical roles in the initiation and progression of atherosclerosis, it is important to unveil how NLRP3 inflammasome activation drives atherogenesis. The link between NLRP3 inflammasome activation and atherosclerosis remains to be completely elucidated. It has been well established that NLRP3 inflammasome activation results in the maturation of interleukin‐1β and interleukin‐18, both of which are considered to be the major contributors of atherogenesis.86, 87 Both cytokines have been documented to be highly expressed in human atherosclerotic plaques compared with normal arteries and positively correlated to disease severity.88, 89 Interleukin‐1β deficiency ameliorates atherosclerosis in ApoE−/− mice,25 while atherosclerotic lesion size in mice with partial deletion of interleukin‐1R antagonist (interleukin‐1Ra) (interleukin‐1Ra+/−/ApoE−/−) is significantly elevated compared with that in interleukin‐1Ra+/+/ApoE−/− mice.90 Notably, interleukin‐1Ra is the structural homologue of interleukin‐1 that competes with interleukin‐1 for binding to the interleukin‐1R but fails to initiate interleukin‐1R activation.91 Likewise, genetic ablation of interleukin‐18 mitigated the development of atherosclerosis, whereas administration of recombinant interleukin‐18 significantly increased the size of the atherosclerotic lesions in hypercholesterolemic mice.86, 92 These findings indicate that interleukin‐18 plays a proatherogenic role in the development of atherosclerosis.
In addition to inducing the production of interleukin‐1β and interleukin‐18, the activation of the NLRP3 inflammasome boosts the migratory capacity of macrophages and augments lipids deposition in lysosomes in macrophages.93 These events facilitate entry of macrophages into the arterial wall, stimulate foam cell formation, and ultimately promote atherosclerosis.93 The major role and mechanism for interleukin‐1β to drive vascular inflammation and atherosclerosis are outlined in Figure 4.
Figure 4.
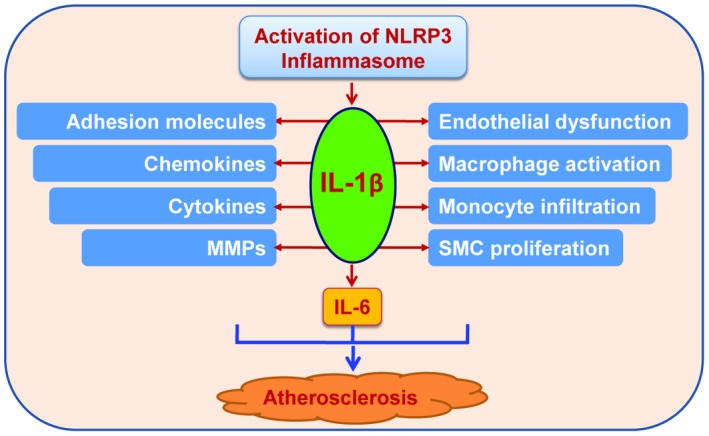
Selected functions of interleukin‐1β linked to atherosclerosis. Interleukin‐1β acts on different cell types and organs, including those involved in atherosclerosis such as endothelial cells, vascular smooth muscle cells, macrophages and the liver. Interleukin‐1β exerts diverse biochemical and biological functions shown in autocrine, paracrine, and endocrine manners. IL‐1β indicates interleukin‐1β; IL‐6, interleukin‐6; MMPs, matrix metalloproteinases; SMC, smooth muscle cell.
Interleukin‐1β is a primordial proinflammatory cytokine94 and has been shown to be involved in a broad spectrum of inflammatory disorders.95 The experimental and clinical evidence demonstrates interleukin‐1β as both a local vascular and systemic contributor to atherogenesis.43 Numerous studies support causality of interleukin‐1β in coronary heart disease. Interleukin‐1β exerts its functions through the autocrine, paracrine, or endocrine mechanisms.94
Interleukin‐1β is the apical proinflammatory mediator and among the most powerful inducers of innate immunity.95 Interleukin‐1β induces its own gene expression in various cell types that are major driver of atherogenesis, an amplification loop termed autoinduction.43 Also, interleukin‐1β triggers and/or amplifies endothelial dysfunction.43 Interleukin‐1β stimulates the expression of adhesion molecules such as intercellular adhesion molecule‐1 and vascular cell adhesion molecule‐1. Interleukin‐1β incites the expression of chemokines such as monocyte chemoattractant protein‐1.43 Accordingly, interleukin‐1β promotes leukocyte adhesion to vascular ECs and leads to procoagulant activity and recruitment of leukocytes to the lesions.96 Interleukin‐1β also boosts the recruitment of neutrophils to atherosclerotic lesions.97 Neutrophils are present in all stages of atherosclerotic plaque promoting atherogenesis and plaque rupture.98 Interleukin‐1β stimulates the production of NETs in a vicious circle.99
Interleukin‐1β lies in the upstream in the pathway and is central in shaping the proinflammatory response by strongly inducing diverse cells, including smooth muscle cells to elaborate secondary inflammatory mediators involving interleukin‐6.43, 100 Interleukin‐6 acts as the primary cytokine to incite the acute phase response with hepatic production of C‐reactive protein, a risk marker for atherothrombosis.100 Accordingly, interleukin‐1β is critical for the activation of the humoral arm of innate immunity. Interleukin‐1β also has multiple effects on smooth muscle cells.43 For example, interleukin‐1β can induce the production of platelet‐derived growth factor, a crucial growth factor that can stimulate the proliferation of smooth muscle cells.43
NLRP3 inflammasome activation causes pyroptosis, a proinflammatory programmed cell death that stimulates the pathological ion efflux and release of inflammatory substances, further amplifying local inflammation.101 Recent studies demonstrated that mature caspase‐1 mediates proteolytic cleavage of gasdermin D, which in turn triggers pyroptosis through formation of membrane pores.102 Therefore, caspase‐1‐dependent pyroptosis may very well participate in atherosclerosis.
Targeting the NLRP3 Inflammasome for Treatment of Atherosclerotic Disease
Elevated levels of blood cholesterol, more precisely LDL cholesterol, account for the major risk factor and have been causally linked to the occurrence of atherogenesis.103 To date, LDL cholesterol–lowering statins remain the mainstay for treatment of atherosclerosis. However, atherosclerotic plaques still undergo progression to a great extent in a large proportion of individuals whose blood cholesterol levels dramatically decline upon treatment with cholesterol‐lowering drugs.104 Thus, a large burden of residual disorder in individuals treated with statins demonstrates an unmet need for new therapies.46 Given that atherosclerosis is a chronic inflammatory disorder, various clinical trials are currently being conducted to assess anti‐inflammatory agents for atherosclerosis treatment. Both LDL cholesterol and C‐reactive protein should be measured to evaluate an individual's residual cholesterol or inflammatory risk for recurrent cardiovascular events. The patients will definitely benefit from personalized biomarker‐based therapeutic approach.
Numerous studies have established the critical role for the NLRP3 inflammasome in driving atherogenesis.9, 72 From a translational point of view, the NLRP3 inflammasome is an attractive drug target for atherosclerosis. Indeed, targeting the NLRP3 inflammasome or its products in susceptible patients is emerging as an important topic in atherosclerosis field. Because of its well‐demonstrated causality in atherosclerosis,105 interleukin‐1β has been selected as a valuable therapeutic target for atherosclerosis. Canakinumab is a human monoclonal antibody that can antagonize the interaction of interleukin‐1β with interleukin‐1R by binding interleukin‐1β, which blunts subsequent proinflammatory signaling events.105 The CANTOS (Canakinumab Anti‐inflammatory Thrombosis Outcomes Study) trial evaluated the impact of canakinumab on the occurrence/recurrence of cardiovascular events among 17 200 patients with stable coronary artery disease.105 Compared with placebo, canakinumab treatment reduced the risk of major adverse cardiovascular events (including myocardial infarction, stroke, and cardiovascular death).105 Notably, administration of canakinumab significantly reduces plasma inflammatory markers (such as C‐reactive protein and interleukin‐6) without affecting the levels of LDL cholesterol or HDL cholesterol.105 Statin‐treated patients with residual inflammatory risk will benefit from canakinumab administration. Importantly, canakinumab selectively inhibits interleukin‐1β, while leaving other interleukin‐1 family members unaffected. In this regard, long‐term treatment with canakinumab might be safer compared with treatment with anakinra, a recombinant interleukin‐1R antagonist.
The data from the CANTOS trial provide strong support for the critical role of the NLRP3 inflammasome in the initiation and progression of atherosclerosis. However, caution should still be used since targeting inflammatory pathway systemically may bear the risk of interfering with immune homeostasis. Direct targeting the NLRP3 inflammasome is expected to mitigate local inflammatory responses through the retardation of maturation and secretion of interleukin‐1β and interleukin‐18 as well as by the suppression of pyroptotic cell death. It is tempting to hypothesize that direct intervention of NLRP3 inflammasome function may be beneficial for reducing cardiovascular risk. For instance, arglabin, a natural product that has an anti‐inflammatory capacity, restrains atherosclerotic development in atherosclerosis‐prone mice.106 Mechanistically, arglabin impedes the activation of the NLRP3 inflammasome in macrophages.106 Consistent with this observation, arglabin administration led to the reduction of plasma interleukin‐1β levels.106 This study indicated that inhibition of the NLRP3 inflammasome potently restricts the initiation and progression of atherosclerosis, making the NLRP3 inflammasome a promising therapeutic target for the treatment of atherosclerosis. In addition, remarkable attempts were made with atorvastatin107 and colchicine108 to restrain atherosclerosis by targeting the NLRP3 inflammasome. There is a considerable interest in developing small molecule inhibitors for the NLRP3 inflammasome.109 MCC950, a potent and selective inhibitor of the NLRP3 inflammasome,110 dramatically prevents the production of mature interleukin‐1β upon exposure of macrophages and dendritic cells to lipopolysaccharide and CCs.110 Strikingly, this work showed that MCC950 ameliorates the development of atherosclerotic lesions,110 highlighting the therapeutic potential of these small molecule inhibitors for CVD.
NLRP3 inflammasome becomes activated in response to several key triggers (in particular CCs) that have been well characterized to fuel the development of atherosclerosis. An alternative strategy for the treatment of atherosclerosis is to remove the atherogenic NLRP3 inflammasome triggers. Importantly, animal studies showed that 2‐hydroxypropyl‐β‐cyclodextrin, a cholesterol‐solubilizing substance, blunts vascular CCs deposition, mitigates atherosclerotic development and promotes the regression of atherosclerosis.111 In addition to removing CCs, 2‐hydroxypropyl‐β‐cyclodextrin induces cholesterol efflux and reverse cholesterol transport.111 These findings highlight that removing the trigger of the NLRP3 inflammasome is a valuable therapeutic approach to intervention of atherosclerosis development. Targeting the NLRP3 inflammasome will usher in a new era of anti‐inflammatory therapies for atherosclerosis.
Concluding Remarks and Future Perspectives
Years of studies have brought rapid progress in our understanding of NLRP3 inflammasome biology. While some old mysteries remain unsolved, these developments have raised new questions. Unraveling the regulation and molecular mechanisms responsible for NLRP3 inflammasome activation is critical for improving our understanding of the pathogenesis of atherosclerosis. To provide reliable therapeutic strategies for atherosclerosis that target the NLRP3 inflammasome, extensive studies need to delineate the molecular mechanisms for this inflammasome. Obviously, our understanding of NLRP3 inflammasome activation needs to be integrated with information for the molecular structure of the NLRP3 inflammasome. These advances and questions set the stage for future studies to achieve unprecedented understanding of the NLRP3 inflammasome at a structural and biochemical level. We have just begun to understand the negative regulators and their mechanisms that finely control and prevent excessive inflammasome activation (for a detailed review, see Broz and Dixit112). Extensive analysis of the negative regulators and signals should help us manipulate NLRP3 inflammasome activation therapeutically. Identifying and characterizing specific binding partners modulating NLRP3 inflammasome activation in vitro and in vivo may be interesting and challenging.
These advances in the NLRP3 inflammasome are transforming the field of atherosclerosis. How will this newly obtained knowledge be translated into treatment for atherosclerosis? CCs license the activation of the NLRP3 inflammasome and thereby trigger vascular inflammation, governing the initiation and progression of atherosclerosis. Understanding how CCs and other danger signals can induce the activation of the NLRP3 inflammasome remains a topic of considerable interest. Given that the formation of CCs occurs even at early stage of atherosclerotic lesions, CCs are now regarded as one of the initiating inflammatory insults for atherosclerosis. In the future, targeting cholesterol crystallization is a promising therapeutic approach for atherosclerosis and is worthwhile to investigate. We are entering an exciting era in which we may reap therapeutic benefits from mechanistic findings in the NLRP3 inflammasome and inflammatory pathways in atherosclerosis. However, the extrapolation of discoveries from a mouse model to human disease always requires caution. In addition, it will be important to define the interplay between NLRP3 inflammasome signaling with other inflammatory pathways involved in atherosclerotic development.
Somatic mutation in TET2 is the first to be causally linked to atherosclerotic CVD.83, 84 It has been appreciated that somatic mutations of additional genes, as exemplified by DNA methyltransferase 3α, additional sex combs like 1, and Janus kinase 2, may be implicated in atherosclerosis.83 Based upon their unique functions and underlying mechanisms, it is plausible to speculate that these mutated genes may differentially impact atherosclerotic CVD. Future studies will need to uncover the contribution of somatic mutations in these genes to atherosclerotic CVD.
Sources of Funding
This work was supported by grants from the National Natural Science Foundation of China (81872381) and from Health Commission of Hubei Province Scientific Research Projects (WJ2019Z003 and WJ2019M047). This work was also supported by start‐up funds from the Hubei University of Medicine and the Hubei University of Medicine Renmin Hospital (to Drs Fu and Jin).
Disclosures
None.
(J Am Heart Assoc. 2019;8:e012219 DOI: 10.1161/JAHA.119.012219.)
Contributor Information
Ying Jin, Email: jianfu0001@yahoo.com.
Jian Fu, Email: jdyj0001@163.com.
References
- 1. Tabas I, Garcia‐Cardena G, Owens GK. Recent insights into the cellular biology of atherosclerosis. J Cell Biol. 2015;209:13–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gimbrone MA Jr, Garcia‐Cardena G. Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc Pathol. 2013;22:9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Swirski FK, Robbins CS, Nahrendorf M. Development and function of arterial and cardiac macrophages. Trends Immunol. 2016;37:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114:1867–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yin Y, Yan Y, Jiang X, Mai J, Chen NC, Wang H, Yang XF. Inflammasomes are differentially expressed in cardiovascular and other tissues. Int J Immunopathol Pharmacol. 2009;22:311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fredman G, Tabas I. Boosting inflammation resolution in atherosclerosis: the next frontier for therapy. Am J Pathol. 2017;187:1211–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. [DOI] [PubMed] [Google Scholar]
- 9. Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, Espevik T, Lien E, Fitzgerald KA, Rock KL, Moore KJ, Wright SD, Hornung V, Latz E. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature. 2010;464:1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, Hoffman HM, Hugot JP, Inohara N, Mackenzie A, Maltais LJ, Nunez G, Ogura Y, Otten LA, Philpott D, Reed JC, Reith W, Schreiber S, Steimle V, Ward PA. The NLR gene family: a standard nomenclature. Immunity. 2008;28:285–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. He Y, Zeng MY, Yang D, Motro B, Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature. 2016;530:354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, Fernandes‐Alnemri T, Wu J, Monks BG, Fitzgerald KA, Hornung V, Latz E. Cutting edge: NF‐kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183:787–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Py BF, Kim MS, Vakifahmetoglu‐Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–338. [DOI] [PubMed] [Google Scholar]
- 14. Lamkanfi M, Dixit VM. Mechanisms and functions of inflammasomes. Cell. 2014;157:1013–1022. [DOI] [PubMed] [Google Scholar]
- 15. Lee GS, Subramanian N, Kim AI, Aksentijevich I, Goldbach‐Mansky R, Sacks DB, Germain RN, Kastner DL, Chae JJ. The calcium‐sensing receptor regulates the NLRP3 inflammasome through Ca2+ and CAMP. Nature. 2012;492:123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Petrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–1589. [DOI] [PubMed] [Google Scholar]
- 17. Munoz‐Planillo R, Kuffa P, Martinez‐Colon G, Smith BL, Rajendiran TM, Nunez G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Okada M, Matsuzawa A, Yoshimura A, Ichijo H. The lysosome rupture‐activated TAK1‐JNK pathway regulates NLRP3 inflammasome activation. J Biol Chem. 2014;289:32926–32936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gurung P, Lukens JR, Kanneganti TD. Mitochondria: diversity in the regulation of the NLRP3 inflammasome. Trends Mol Med. 2015;21:193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen J, Chen ZJ. Ptdins4P on dispersed trans‐Golgi network mediates NLRP3 inflammasome activation. Nature. 2018;564:71–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shi X, Xie WL, Kong WW, Chen D, Qu P. Expression of the NLRP3 inflammasome in carotid atherosclerosis. J Stroke Cerebrovasc Dis. 2015;24:2455–2466. [DOI] [PubMed] [Google Scholar]
- 22. Xiao H, Lu M, Lin TY, Chen Z, Chen G, Wang WC, Marin T, Shentu TP, Wen L, Gongol B, Sun W, Liang X, Chen J, Huang HD, Pedra JH, Johnson DA, Shyy JY. Sterol regulatory element binding protein 2 activation of NLRP3 inflammasome in endothelium mediates hemodynamic‐induced atherosclerosis susceptibility. Circulation. 2013;128:632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zheng F, Xing S, Gong Z, Xing Q. NLRP3 inflammasomes show high expression in aorta of patients with atherosclerosis. Heart Lung Circ. 2013;22:746–750. [DOI] [PubMed] [Google Scholar]
- 24. Menu P, Pellegrin M, Aubert JF, Bouzourene K, Tardivel A, Mazzolai L, Tschopp J. Atherosclerosis in ApoE‐deficient mice progresses independently of the NLRP3 inflammasome. Cell Death Dis. 2011;2:e137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kirii H, Niwa T, Yamada Y, Wada H, Saito K, Iwakura Y, Asano M, Moriwaki H, Seishima M. Lack of interleukin‐1beta decreases the severity of atherosclerosis in ApoE‐deficient mice. Arterioscler Thromb Vasc Biol. 2003;23:656–660. [DOI] [PubMed] [Google Scholar]
- 26. Chamberlain J, Evans D, King A, Dewberry R, Dower S, Crossman D, Francis S. Interleukin‐1beta and signaling of interleukin‐1 in vascular wall and circulating cells modulates the extent of neointima formation in mice. Am J Pathol. 2006;168:1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Devlin CM, Kuriakose G, Hirsch E, Tabas I. Genetic alterations of IL‐1 receptor antagonist in mice affect plasma cholesterol level and foam cell lesion size. Proc Natl Acad Sci U S A. 2002;99:6280–6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gage J, Hasu M, Thabet M, Whitman SC. Caspase‐1 deficiency decreases atherosclerosis in apolipoprotein E‐null mice. Can J Cardiol. 2012;28:222–229. [DOI] [PubMed] [Google Scholar]
- 29. Hendrikx T, Jeurissen ML, van Gorp PJ, Gijbels MJ, Walenbergh SM, Houben T, van Gorp R, Pottgens CC, Stienstra R, Netea MG, Hofker MH, Donners MM, Shiri‐Sverdlov R. Bone marrow–specific caspase‐1/11 deficiency inhibits atherosclerosis development in Ldlr(‐/‐) mice. FEBS J. 2015;282:2327–2338. [DOI] [PubMed] [Google Scholar]
- 30. Christ A, Gunther P, Lauterbach MAR, Duewell P, Biswas D, Pelka K, Scholz CJ, Oosting M, Haendler K, Bassler K, Klee K, Schulte‐Schrepping J, Ulas T, Moorlag S, Kumar V, Park MH, Joosten LAB, Groh LA, Riksen NP, Espevik T, Schlitzer A, Li Y, Fitzgerald ML, Netea MG, Schultze JL, Latz E. Western diet triggers NLRP3‐dependent innate immune reprogramming. Cell. 2018;172:162–175.e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Parathath S, Mick SL, Feig JE, Joaquin V, Grauer L, Habiel DM, Gassmann M, Gardner LB, Fisher EA. Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res. 2011;109:1141–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Moulton KS, Heller E, Konerding MA, Flynn E, Palinski W, Folkman J. Angiogenesis inhibitors endostatin or TNP‐470 reduce intimal neovascularization and plaque growth in apolipoprotein E–deficient mice. Circulation. 1999;99:1726–1732. [DOI] [PubMed] [Google Scholar]
- 33. Bostrom P, Magnusson B, Svensson PA, Wiklund O, Boren J, Carlsson LM, Stahlman M, Olofsson SO, Hulten LM. Hypoxia converts human macrophages into triglyceride‐loaded foam cells. Arterioscler Thromb Vasc Biol. 2006;26:1871–1876. [DOI] [PubMed] [Google Scholar]
- 34. Folco EJ, Sheikine Y, Rocha VZ, Christen T, Shvartz E, Sukhova GK, Di Carli MF, Libby P. Hypoxia but not inflammation augments glucose uptake in human macrophages: implications for imaging atherosclerosis with 18fluorine‐labeled 2‐deoxy‐D‐glucose positron emission tomography. J Am Coll Cardiol. 2011;58:603–614. [DOI] [PubMed] [Google Scholar]
- 35. Folco EJ, Sukhova GK, Quillard T, Libby P. Moderate hypoxia potentiates interleukin‐1beta production in activated human macrophages. Circ Res. 2014;115:875–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sheedy FJ, Grebe A, Rayner KJ, Kalantari P, Ramkhelawon B, Carpenter SB, Becker CE, Ediriweera HN, Mullick AE, Golenbock DT, Stuart LM, Latz E, Fitzgerald KA, Moore KJ. CD36 coordinates NLRP3 inflammasome activation by facilitating intracellular nucleation of soluble ligands into particulate ligands in sterile inflammation. Nat Immunol. 2013;14:812–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose‐Girma M, Lee WP, Weinrauch Y, Monack DM, Dixit VM. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–232. [DOI] [PubMed] [Google Scholar]
- 38. Stachon P, Heidenreich A, Merz J, Hilgendorf I, Wolf D, Willecke F, von Garlen S, Albrecht P, Hardtner C, Ehrat N, Hoppe N, Reinohl J, von Zur Muhlen C, Bode C, Idzko M, Zirlik A. P2X7 deficiency blocks lesional inflammasome activity and ameliorates atherosclerosis in mice. Circulation. 2017;135:2524–2533. [DOI] [PubMed] [Google Scholar]
- 39. Usui F, Shirasuna K, Kimura H, Tatsumi K, Kawashima A, Karasawa T, Hida S, Sagara J, Taniguchi S, Takahashi M. Critical role of caspase‐1 in vascular inflammation and development of atherosclerosis in Western diet–fed apolipoprotein E–deficient mice. Biochem Biophys Res Commun. 2012;425:162–168. [DOI] [PubMed] [Google Scholar]
- 40. Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V. Inflammation. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science. 2015;349:316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Grebe A, Latz E. Cholesterol crystals and inflammation. Curr Rheumatol Rep. 2013;15:313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Janoudi A, Shamoun FE, Kalavakunta JK, Abela GS. Cholesterol crystal induced arterial inflammation and destabilization of atherosclerotic plaque. Eur Heart J. 2016;37:1959–1967. [DOI] [PubMed] [Google Scholar]
- 43. Libby P. Interleukin‐1 beta as a target for atherosclerosis therapy: biological basis of CANTOS and beyond. J Am Coll Cardiol. 2017;70:2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kruth HS, Skarlatos SI, Gaynor PM, Gamble W. Production of cholesterol‐enriched nascent high density lipoproteins by human monocyte‐derived macrophages is a mechanism that contributes to macrophage cholesterol efflux. J Biol Chem. 1994;269:24511–24518. [PubMed] [Google Scholar]
- 45. Wang N, Lan D, Chen W, Matsuura F, Tall AR. ATP‐binding cassette transporters G1 and G4 mediate cellular cholesterol efflux to high‐density lipoproteins. Proc Natl Acad Sci U S A. 2004;101:9774–9779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tall AR, Yvan‐Charvet L. Cholesterol, inflammation and innate immunity. Nat Rev Immunol. 2015;15:104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Thacker SG, Zarzour A, Chen Y, Alcicek MS, Freeman LA, Sviridov DO, Demosky SJ Jr, Remaley AT. High‐density lipoprotein reduces inflammation from cholesterol crystals by inhibiting inflammasome activation. Immunology. 2016;149:306–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Westerterp M, Fotakis P, Ouimet M, Bochem AE, Zhang H, Molusky MM, Wang W, Abramowicz S, la Bastide‐van Gemert S, Wang N, Welch CL, Reilly MP, Stroes ES, Moore KJ, Tall AR. Cholesterol efflux pathways suppress inflammasome activation, NETosis, and atherogenesis. Circulation. 2018;138:898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, Newton K, Qu Y, Liu J, Heldens S, Zhang J, Lee WP, Roose‐Girma M, Dixit VM. Non‐canonical inflammasome activation targets caspase‐11. Nature. 2011;479:117–121. [DOI] [PubMed] [Google Scholar]
- 50. de la Llera Moya M, McGillicuddy FC, Hinkle CC, Byrne M, Joshi MR, Nguyen V, Tabita‐Martinez J, Wolfe ML, Badellino K, Pruscino L, Mehta NN, Asztalos BF, Reilly MP. Inflammation modulates human HDL composition and function in vivo. Atherosclerosis. 2012;222:390–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Kellner‐Weibel G, Jerome WG, Small DM, Warner GJ, Stoltenborg JK, Kearney MA, Corjay MH, Phillips MC, Rothblat GH. Effects of intracellular free cholesterol accumulation on macrophage viability: a model for foam cell death. Arterioscler Thromb Vasc Biol. 1998;18:423–431. [DOI] [PubMed] [Google Scholar]
- 52. Kolodgie FD, Burke AP, Nakazawa G, Cheng Q, Xu X, Virmani R. Free cholesterol in atherosclerotic plaques: where does it come from? Curr Opin Lipidol. 2007;18:500–507. [DOI] [PubMed] [Google Scholar]
- 53. Vedre A, Pathak DR, Crimp M, Lum C, Koochesfahani M, Abela GS. Physical factors that trigger cholesterol crystallization leading to plaque rupture. Atherosclerosis. 2009;203:89–96. [DOI] [PubMed] [Google Scholar]
- 54. Yvan‐Charvet L, Pagler T, Gautier EL, Avagyan S, Siry RL, Han S, Welch CL, Wang N, Randolph GJ, Snoeck HW, Tall AR. ATP‐binding cassette transporters and HDL suppress hematopoietic stem cell proliferation. Science. 2010;328:1689–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Swirski FK, Nahrendorf M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science. 2013;339:161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Westerterp M, Bochem AE, Yvan‐Charvet L, Murphy AJ, Wang N, Tall AR. ATP‐binding cassette transporters, atherosclerosis, and inflammation. Circ Res. 2014;114:157–170. [DOI] [PubMed] [Google Scholar]
- 57. Franck G, Mawson TL, Folco EJ, Molinaro R, Ruvkun V, Engelbertsen D, Liu X, Tesmenitsky Y, Shvartz E, Sukhova GK, Michel JB, Nicoletti A, Lichtman A, Wagner D, Croce KJ, Libby P. Roles of PAD4 and NETosis in experimental atherosclerosis and arterial injury: implications for superficial erosion. Circ Res. 2018;123:33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Jorch SK, Kubes P. An emerging role for neutrophil extracellular traps in noninfectious disease. Nat Med. 2017;23:279–287. [DOI] [PubMed] [Google Scholar]
- 59. Stakos DA, Kambas K, Konstantinidis T, Mitroulis I, Apostolidou E, Arelaki S, Tsironidou V, Giatromanolaki A, Skendros P, Konstantinides S, Ritis K. Expression of functional tissue factor by neutrophil extracellular traps in culprit artery of acute myocardial infarction. Eur Heart J. 2015;36:1405–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Van Avondt K, Maegdefessel L, Soehnlein O. Therapeutic targeting of neutrophil extracellular traps in atherogenic inflammation. Thromb Haemost. 2019;119:542–552. [DOI] [PubMed] [Google Scholar]
- 61. Doring Y, Soehnlein O, Weber C. Neutrophil extracellular traps in atherosclerosis and atherothrombosis. Circ Res. 2017;120:736–743. [DOI] [PubMed] [Google Scholar]
- 62. Jerome WG. Lysosomes, cholesterol and atherosclerosis. Clin Lipidol. 2010;5:853–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB, Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shi CS, Shenderov K, Huang NN, Kabat J, Abu‐Asab M, Fitzgerald KA, Sher A, Kehrl JH. Activation of autophagy by inflammatory signals limits IL‐1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Sergin I, Bhattacharya S, Emanuel R, Esen E, Stokes CJ, Evans TD, Arif B, Curci JA, Razani B. Inclusion bodies enriched for p62 and polyubiquitinated proteins in macrophages protect against atherosclerosis. Sci Signal. 2016;9:ra2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Emanuel R, Sergin I, Bhattacharya S, Turner J, Epelman S, Settembre C, Diwan A, Ballabio A, Razani B. Induction of lysosomal biogenesis in atherosclerotic macrophages can rescue lipid‐induced lysosomal dysfunction and downstream sequelae. Arterioscler Thromb Vasc Biol. 2014;34:1942–1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Rajamaki K, Lappalainen J, Oorni K, Valimaki E, Matikainen S, Kovanen PT, Eklund KK. Cholesterol crystals activate the NLRP3 inflammasome in human macrophages: a novel link between cholesterol metabolism and inflammation. PLoS One. 2010;5:e11765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Freigang S, Ampenberger F, Spohn G, Heer S, Shamshiev AT, Kisielow J, Hersberger M, Yamamoto M, Bachmann MF, Kopf M. Nrf2 is essential for cholesterol crystal–induced inflammasome activation and exacerbation of atherosclerosis. Eur J Immunol. 2011;41:2040–2051. [DOI] [PubMed] [Google Scholar]
- 69. Kotla S, Singh NK, Rao GN. ROS via BTK‐p300‐STAT1‐PPARgamma signaling activation mediates cholesterol crystals–induced CD36 expression and foam cell formation. Redox Biol. 2017;11:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Samstad EO, Niyonzima N, Nymo S, Aune MH, Ryan L, Bakke SS, Lappegard KT, Brekke OL, Lambris JD, Damas JK, Latz E, Mollnes TE, Espevik T. Cholesterol crystals induce complement‐dependent inflammasome activation and cytokine release. J Immunol. 2014;192:2837–2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. [DOI] [PubMed] [Google Scholar]
- 72. Chen Z, Martin M, Li Z, Shyy JY. Endothelial dysfunction: the role of sterol regulatory element–binding protein–induced NOD‐like receptor family pyrin domain–containing protein 3 inflammasome in atherosclerosis. Curr Opin Lipidol. 2014;25:339–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Wang JG, Williams JC, Davis BK, Jacobson K, Doerschuk CM, Ting JP, Mackman N. Monocytic microparticles activate endothelial cells in an IL‐1beta‐dependent manner. Blood. 2011;118:2366–2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Lin TY, Wei TW, Li S, Wang SC, He M, Martin M, Zhang J, Shentu TP, Xiao H, Kang J, Wang KC, Chen Z, Chien S, Tsai MD, Shyy JY. TIFA as a crucial mediator for NLRP3 inflammasome. Proc Natl Acad Sci U S A. 2016;113:15078–15083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Ea CK, Sun L, Inoue J, Chen ZJ. TIFA activates IkappaB kinase (IKK) by promoting oligomerization and ubiquitination of TRAF6. Proc Natl Acad Sci U S A. 2004;101:15318–15323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Sniderman AD, Islam S, McQueen M, Pencina M, Furberg CD, Thanassoulis G, Yusuf S. Age and cardiovascular risk attributable to apolipoprotein B, low‐density lipoprotein cholesterol or non–high‐density lipoprotein cholesterol. J Am Heart Assoc. 2016;5:e003665 DOI: 10.1161/JAHA.116.003665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Fernandez‐Friera L, Fuster V, Lopez‐Melgar B, Oliva B, Garcia‐Ruiz JM, Mendiguren J, Bueno H, Pocock S, Ibanez B, Fernandez‐Ortiz A, Sanz J. Normal LDL‐cholesterol levels are associated with subclinical atherosclerosis in the absence of risk factors. J Am Coll Cardiol. 2017;70:2979–2991. [DOI] [PubMed] [Google Scholar]
- 78. Sniderman AD, Furberg CD. Age as a modifiable risk factor for cardiovascular disease. Lancet. 2008;371:1547–1549. [DOI] [PubMed] [Google Scholar]
- 79. Ju YS, Martincorena I, Gerstung M, Petljak M, Alexandrov LB, Rahbari R, Wedge DC, Davies HR, Ramakrishna M, Fullam A, Martin S, Alder C, Patel N, Gamble S, O'Meara S, Giri DD, Sauer T, Pinder SE, Purdie CA, Borg A, Stunnenberg H, van de Vijver M, Tan BK, Caldas C, Tutt A, Ueno NT, van ‘t Veer LJ, Martens JW, Sotiriou C, Knappskog S, Span PN, Lakhani SR, Eyfjord JE, Borresen‐Dale AL, Richardson A, Thompson AM, Viari A, Hurles ME, Nik‐Zainal S, Campbell PJ, Stratton MR. Somatic mutations reveal asymmetric cellular dynamics in the early human embryo. Nature. 2017;543:714–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Forsberg LA, Gisselsson D, Dumanski JP. Mosaicism in health and disease—clones picking up speed. Nat Rev Genet. 2017;18:128–142. [DOI] [PubMed] [Google Scholar]
- 81. Zink F, Stacey SN, Norddahl GL, Frigge ML, Magnusson OT, Jonsdottir I, Thorgeirsson TE, Sigurdsson A, Gudjonsson SA, Gudmundsson J, Jonasson JG, Tryggvadottir L, Jonsson T, Helgason A, Gylfason A, Sulem P, Rafnar T, Thorsteinsdottir U, Gudbjartsson DF, Masson G, Kong A, Stefansson K. Clonal hematopoiesis, with and without candidate driver mutations, is common in the elderly. Blood. 2017;130:742–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Fuster JJ, Walsh K. Somatic mutations and clonal hematopoiesis: unexpected potential new drivers of age‐related cardiovascular disease. Circ Res. 2018;122:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S, Ebert BL. Clonal hematopoiesis and risk of atherosclerotic cardiovascular disease. N Engl J Med. 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Fuster JJ, MacLauchlan S, Zuriaga MA, Polackal MN, Ostriker AC, Chakraborty R, Wu CL, Sano S, Muralidharan S, Rius C, Vuong J, Jacob S, Muralidhar V, Robertson AA, Cooper MA, Andres V, Hirschi KK, Martin KA, Walsh K. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science. 2017;355:842–847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cull AH, Snetsinger B, Buckstein R, Wells RA, Rauh MJ. Tet2 restrains inflammatory gene expression in macrophages. Exp Hematol. 2017;55:56–70.e13. [DOI] [PubMed] [Google Scholar]
- 86. Whitman SC, Ravisankar P, Daugherty A. Interleukin‐18 enhances atherosclerosis in apolipoprotein E(‐/‐) mice through release of interferon‐gamma. Circ Res. 2002;90:E34–E38. [DOI] [PubMed] [Google Scholar]
- 87. Merhi‐Soussi F, Kwak BR, Magne D, Chadjichristos C, Berti M, Pelli G, James RW, Mach F, Gabay C. Interleukin‐1 plays a major role in vascular inflammation and atherosclerosis in male apolipoprotein E–knockout mice. Cardiovasc Res. 2005;66:583–593. [DOI] [PubMed] [Google Scholar]
- 88. Galea J, Armstrong J, Gadsdon P, Holden H, Francis SE, Holt CM. Interleukin‐1 beta in coronary arteries of patients with ischemic heart disease. Arterioscler Thromb Vasc Biol. 1996;16:1000–1006. [DOI] [PubMed] [Google Scholar]
- 89. Mallat Z, Corbaz A, Scoazec A, Besnard S, Leseche G, Chvatchko Y, Tedgui A. Expression of interleukin‐18 in human atherosclerotic plaques and relation to plaque instability. Circulation. 2001;104:1598–1603. [DOI] [PubMed] [Google Scholar]
- 90. Isoda K, Sawada S, Ishigami N, Matsuki T, Miyazaki K, Kusuhara M, Iwakura Y, Ohsuzu F. Lack of interleukin‐1 receptor antagonist modulates plaque composition in apolipoprotein E–deficient mice. Arterioscler Thromb Vasc Biol. 2004;24:1068–1073. [DOI] [PubMed] [Google Scholar]
- 91. Nicklin MJ, Hughes DE, Barton JL, Ure JM, Duff GW. Arterial inflammation in mice lacking the interleukin 1 receptor antagonist gene. J Exp Med. 2000;191:303–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Elhage R, Jawien J, Rudling M, Ljunggren HG, Takeda K, Akira S, Bayard F, Hansson GK. Reduced atherosclerosis in interleukin‐18 deficient apolipoprotein E–knockout mice. Cardiovasc Res. 2003;59:234–240. [DOI] [PubMed] [Google Scholar]
- 93. Li X, Zhang Y, Xia M, Gulbins E, Boini KM, Li PL. Activation of Nlrp3 inflammasomes enhances macrophage lipid‐deposition and migration: implication of a novel role of inflammasome in atherogenesis. PLoS One. 2014;9:e87552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Dinarello CA, Simon A, van der Meer JW. Treating inflammation by blocking interleukin‐1 in a broad spectrum of diseases. Nat Rev Drug Discov. 2012;11:633–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Dinarello CA. Interleukin‐1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, Gimbrone MA Jr. Interleukin‐1 activation of vascular endothelium. Effects on procoagulant activity and leukocyte adhesion. Am J Pathol. 1985;121:394–403. [PMC free article] [PubMed] [Google Scholar]
- 97. Rotzius P, Thams S, Soehnlein O, Kenne E, Tseng CN, Bjorkstrom NK, Malmberg KJ, Lindbom L, Eriksson EE. Distinct infiltration of neutrophils in lesion shoulders in ApoE‐/‐ mice. Am J Pathol. 2010;177:493–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Soehnlein O. Multiple roles for neutrophils in atherosclerosis. Circ Res. 2012;110:875–888. [DOI] [PubMed] [Google Scholar]
- 99. Kahlenberg JM, Carmona‐Rivera C, Smith CK, Kaplan MJ. Neutrophil extracellular trap–associated protein activation of the NLRP3 inflammasome is enhanced in lupus macrophages. J Immunol. 2013;190:1217–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Ridker PM. From C‐reactive protein to interleukin‐6 to interleukin‐1: moving upstream to identify novel targets for atheroprotection. Circ Res. 2016;118:145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Shi J, Gao W, Shao F. Pyroptosis: gasdermin‐mediated programmed necrotic cell death. Trends Biochem Sci. 2017;42:245–254. [DOI] [PubMed] [Google Scholar]
- 103. Ference BA, Ginsberg HN, Graham I, Ray KK, Packard CJ, Bruckert E, Hegele RA, Krauss RM, Raal FJ, Schunkert H, Watts GF, Boren J, Fazio S, Horton JD, Masana L, Nicholls SJ, Nordestgaard BG, van de Sluis B, Taskinen MR, Tokgozoglu L, Landmesser U, Laufs U, Wiklund O, Stock JK, Chapman MJ, Catapano AL. Low‐density lipoproteins cause atherosclerotic cardiovascular disease. 1. Evidence from genetic, epidemiologic, and clinical studies. A consensus statement from the European Atherosclerosis Society Consensus Panel. Eur Heart J. 2017;38:2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Nicholls SJ, Puri R, Anderson T, Ballantyne CM, Cho L, Kastelein JJ, Koenig W, Somaratne R, Kassahun H, Yang J, Wasserman SM, Scott R, Ungi I, Podolec J, Ophuis AO, Cornel JH, Borgman M, Brennan DM, Nissen SE. Effect of evolocumab on progression of coronary disease in statin‐treated patients: the GLAGOV randomized clinical trial. JAMA. 2016;316:2373–2384. [DOI] [PubMed] [Google Scholar]
- 105. Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, Kastelein JJP, Cornel JH, Pais P, Pella D, Genest J, Cifkova R, Lorenzatti A, Forster T, Kobalava Z, Vida‐Simiti L, Flather M, Shimokawa H, Ogawa H, Dellborg M, Rossi PRF, Troquay RPT, Libby P, Glynn RJ; CANTOS Trial Group . Antiinflammatory therapy with canakinumab for atherosclerotic disease. N Engl J Med. 2017;377:1119–1131. [DOI] [PubMed] [Google Scholar]
- 106. Abderrazak A, Couchie D, Mahmood DF, Elhage R, Vindis C, Laffargue M, Mateo V, Buchele B, Ayala MR, El Gaafary M, Syrovets T, Slimane MN, Friguet B, Fulop T, Simmet T, El Hadri K, Rouis M. Anti‐inflammatory and antiatherogenic effects of the NLRP3 inflammasome inhibitor arglabin in ApoE2.Ki mice fed a high‐fat diet. Circulation. 2015;131:1061–1070. [DOI] [PubMed] [Google Scholar]
- 107. Kong F, Ye B, Lin L, Cai X, Huang W, Huang Z. Atorvastatin suppresses NLRP3 inflammasome activation via TLR4/MyD88/NF‐kappaB signaling in PMA‐stimulated THP‐1 monocytes. Biomed Pharmacother. 2016;82:167–172. [DOI] [PubMed] [Google Scholar]
- 108. Dalbeth N, Lauterio TJ, Wolfe HR. Mechanism of action of colchicine in the treatment of gout. Clin Ther. 2014;36:1465–1479. [DOI] [PubMed] [Google Scholar]
- 109. Baldwin AG, Brough D, Freeman S. Inhibiting the inflammasome: a chemical perspective. J Med Chem. 2016;59:1691–1710. [DOI] [PubMed] [Google Scholar]
- 110. van der Heijden T, Kritikou E, Venema W, van Duijn J, van Santbrink PJ, Slutter B, Foks AC, Bot I, Kuiper J. NLRP3 inflammasome inhibition by MCC950 reduces atherosclerotic lesion development in apolipoprotein E–deficient mice—brief report. Arterioscler Thromb Vasc Biol. 2017;37:1457–1461. [DOI] [PubMed] [Google Scholar]
- 111. Zimmer S, Grebe A, Bakke SS, Bode N, Halvorsen B, Ulas T, Skjelland M, De Nardo D, Labzin LI, Kerksiek A, Hempel C, Heneka MT, Hawxhurst V, Fitzgerald ML, Trebicka J, Bjorkhem I, Gustafsson JA, Westerterp M, Tall AR, Wright SD, Espevik T, Schultze JL, Nickenig G, Lutjohann D, Latz E. Cyclodextrin promotes atherosclerosis regression via macrophage reprogramming. Sci Transl Med. 2016;8:333ra350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–420. [DOI] [PubMed] [Google Scholar]