

Abstract
Background
Acute respiratory distress syndrome (ARDS) is a life‐threatening condition caused by direct or indirect injury to the lungs. Despite improvements in clinical management (for example, lung protection strategies), mortality in this patient group is at approximately 40%. This is an update of a previous version of this review, last published in 2004.
Objectives
To evaluate the effectiveness of pharmacological agents in adults with ARDS on mortality, mechanical ventilation, and fitness to return to work at 12 months.
Search methods
We searched CENTRAL, MEDLINE, Embase, and CINAHL on 10 December 2018. We searched clinical trials registers and grey literature, and handsearched reference lists of included studies and related reviews.
Selection criteria
We included randomized controlled trials (RCTs) comparing pharmacological agents with control (placebo or standard therapy) to treat adults with established ARDS. We excluded trials of nitric oxide, inhaled prostacyclins, partial liquid ventilation, neuromuscular blocking agents, fluid and nutritional interventions and medical oxygen. We excluded studies published earlier than 2000, because of changes to lung protection strategies for people with ARDS since this date.
Data collection and analysis
Two review authors independently assessed studies for inclusion, extracted data, and assessed risks of bias. We assessed the certainty of evidence with GRADE.
Main results
We included 48 RCTs with 6299 participants who had ARDS; two included only participants with mild ARDS (also called acute lung injury). Most studies included causes of ARDS that were both direct and indirect injuries. We noted differences between studies, for example the time of administration or the size of dose, and because of unclear reporting we were uncertain whether all studies had used equivalent lung protection strategies.
We included five types of agents as the primary comparisons in the review: corticosteroids, surfactants, N‐acetylcysteine, statins, and beta‐agonists. We included 15 additional agents (sivelestat, mesenchymal stem cells, ulinastatin, anisodimine, angiotensin‐converting enzyme (ACE) inhibitor, recombinant human ACE2 (palifermin), AP301, granulocyte‐macrophage colony stimulating factor (GM‐CSF), levosimendan, prostacyclins, lisofylline, ketaconazole, nitroglycerins, L‐2‐oxothiazolidine‐4‐carboxylic acid (OTZ), and penehyclidine hydrochloride).
We used GRADE to downgrade outcomes for imprecision (because of few studies and few participants), for study limitations (e.g. high risks of bias) and for inconsistency (e.g. differences between study data).
Corticosteroids versus placebo or standard therapy
Corticosteroids may reduce all‐cause mortality within three months by 86 per 1000 patients (with as many as 161 fewer to 19 more deaths); however, the 95% confidence interval (CI) includes the possibility of both increased and reduced deaths (risk ratio (RR) 0.77, 95% CI 0.57 to 1.05; 6 studies, 574 participants; low‐certainty evidence). Due to the very low‐certainty evidence, we are uncertain whether corticosteroids make little or no difference to late all‐cause mortality (later than three months) (RR 0.99, 95% CI 0.64 to 1.52; 1 study, 180 participants), or to the duration of mechanical ventilation (mean difference (MD) −4.30, 95% CI −9.72 to 1.12; 3 studies, 277 participants). We found that ventilator‐free days up to day 28 (VFD) may be improved with corticosteroids (MD 4.09, 95% CI 1.74 to 6.44; 4 studies, 494 participants; low‐certainty evidence). No studies reported adverse events leading to discontinuation of study medication, or fitness to return to work at 12 months (FTR).
Surfactants versus placebo or standard therapy
We are uncertain whether surfactants make little or no difference to early mortality (RR 1.08, 95% CI 0.91 to 1.29; 9 studies, 1338 participants), or whether they reduce late all‐cause mortality (RR 1.28, 95% CI 1.01 to 1.61; 1 study, 418 participants). Similarly, we are uncertain whether surfactants reduce the duration of mechanical ventilation (MD −2.50, 95% CI −4.95 to ‐0.05; 1 study, 16 participants), make little or no difference to VFD (MD −0.39, 95% CI −2.49 to 1.72; 2 studies, 344 participants), or to adverse events leading to discontinuation of study medication (RR 0.50, 95% CI 0.17 to 1.44; 2 studies, 88 participants). We are uncertain of these effects because we assessed them as very low‐certainty. No studies reported FTR.
N‐aceytylcysteine versus placebo
We are uncertain whether N‐acetylcysteine makes little or no difference to early mortality, because we assessed this as very low‐certainty evidence (RR 0.64, 95% CI 0.32 to 1.30; 1 study, 36 participants). No studies reported late all‐cause mortality, duration of mechanical ventilation, VFD, adverse events leading to study drug discontinuation, or FTR.
Statins versus placebo
Statins probably make little or no difference to early mortality (RR 0.99, 95% CI 0.78 to 1.26; 3 studies, 1344 participants; moderate‐certainty evidence) or to VFD (MD 0.40, 95% CI −0.71 to 1.52; 3 studies, 1342 participants; moderate‐certainty evidence). Statins may make little or no difference to duration of mechanical ventilation (MD 2.70, 95% CI ‐3.55 to 8.95; 1 study, 60 participants; low‐certainty evidence). We could not include data for adverse events leading to study drug discontinuation in one study because it was unclearly reported. No studies reported late all‐cause mortality or FTR.
Beta‐agonists versus placebo control
Beta‐agonists probably slightly increase early mortality by 40 per 1000 patients (with as many as 119 more or 25 fewer deaths); however, the 95% CI includes the possibility of an increase as well as a reduction in mortality (RR 1.14, 95% CI 0.91 to 1.42; 3 studies, 646 participants; moderate‐certainty evidence). Due to the very low‐certainty evidence, we are uncertain whether beta‐agonists increase VFD (MD −2.20, 95% CI −3.68 to −0.71; 3 studies, 646 participants), or make little or no difference to adverse events leading to study drug discontinuation (one study reported little or no difference between groups, and one study reported more events in the beta‐agonist group). No studies reported late all‐cause mortality, duration of mechanical ventilation, or FTR.
Authors' conclusions
We found insufficient evidence to determine with certainty whether corticosteroids, surfactants, N‐acetylcysteine, statins, or beta‐agonists were effective at reducing mortality in people with ARDS, or duration of mechanical ventilation, or increasing ventilator‐free days. Three studies awaiting classification may alter the conclusions of this review. As the potential long‐term consequences of ARDS are important to survivors, future research should incorporate a longer follow‐up to measure the impacts on quality of life.
Plain language summary
Drugs to treat acute respiratory distress syndrome in adults
We set out to determine, from randomized controlled trials, which drugs improve health outcomes in adults with acute respiratory distress syndrome (ARDS).
Background
ARDS is a life‐threatening condition caused by injury to the lungs, for example from infections such as pneumonia or sepsis, or from trauma. People with ARDS are cared for in an intensive care unit, and need support with breathing from mechanical ventilation. Many people who survive ARDS suffer from muscle weakness, fatigue, reduced quality of life after hospital discharge, and may not be fit for work 12 months later. Despite improvements in techniques to manage ARDS, death rates are still very high. Drugs may help to repair damage to the lung injury, or limit the body's response to the injury (for example, by reducing any excess fluid that may collect around the injured lungs).
Study characteristics
The evidence is current to 10 December 2018. We included 48 studies, of 20 different drug types, involving 6299 people who had ARDS. Three studies are awaiting classification (because we did not have enough details to assess them), and 18 studies are still ongoing. We found differences between included studies, such as the severity of ARDS, or potential differences in clinical management and doses. We excluded studies published before 2000, in order to only include up‐to‐date clinical management of people with ARDS (for example, in the pressure applied during mechanical ventilation). However, we found that many studies did not report these management strategies.
For the main comparisons in this review, we included five types of drugs: corticosteroids, surfactants, N‐acetylcysteine, statins, and beta‐agonists. These were compared to placebo or to standard care.
Key results
Although corticosteroids may reduce the number of people who die within the first three months, and beta‐agonists probably slightly increase these early deaths, we found both an increase and a reduction in deaths in our analyses for these drugs. We found no evidence that surfactants, N‐acetylcysteine, or statins made a difference to the number of people who died within three months. Only two studies (one that assessed steroids, and one surfactants) reported deaths later than three months, but evidence for this was uncertain.
We found that statins or steroids may make little or no difference to the duration of mechanical ventilation, but we were uncertain about the evidence for steroids. Similarly, we were uncertain whether surfactants reduced the use of mechanical ventilation. We found that steroids may improve the number of days that people do not need mechanical ventilation (ventilator‐free days up to day 28), but that beta‐agonists may not improve ventilator‐free days (although we were uncertain about the evidence for beta‐agonists). We found that statins probably make little or no difference to the number of ventilator‐free days; this was also the case for surfactants (although, again, we were uncertain about the evidence for surfactants).
Few studies (and only for surfactants and beta‐agonists) reported whether the study drug was stopped because of serious side effects, and we were uncertain whether either of these drugs led to such serious side effects. No studies reported whether people were fit to return to work 12 months after their illness.
Certainty of the evidence
Most of the findings were supported by low‐ or very low‐certainty evidence, although we were moderately confident in the evidence for some outcomes when statins and beta‐agonists were used. For some outcomes we found too few studies with few participants, and sometimes there were unexplained differences between the studies in their findings. These factors reduced our certainty (or confidence) in our findings. Also, it was not possible to mask some researchers because the study drug was compared to standard therapy (no drug), which may have biased our findings.
Conclusion
We found insufficient evidence to determine confidently whether any type of drug was effective at reducing deaths in people with ARDS, or reducing the length of time that they needed mechanical ventilation. No studies reported fitness to return to work at 12 months. We assessed most outcomes to be low or very low certainty, which reduces our confidence in the findings of the review.
Summary of findings
Background
Description of the condition
Acute respiratory distress syndrome (ARDS), first described in 1967 (Ashbaugh 1967), is characterized by diffuse inflammation of the lung's alveolar‐capillary membrane in response to various pulmonary and extrapulmonary insults (Ware 2000). These insults cause pulmonary injury by direct mechanisms (for example, gastric aspiration, pneumonia, inhalational injury, pulmonary contusion), or indirect mechanisms (for example, sepsis, trauma, pancreatitis, multiple transfusions of blood products). An American‐European Consensus Conference (AECC) formulated a widely‐cited definition of ARDS as follows (Bernard 1994):
an acute onset of hypoxaemia, with a ratio of the partial pressure of arterial oxygen (PaO2) to the inspired fraction of oxygen (FiO2) of 200 mmHg or less;
bilateral infiltrates on a frontal chest radiograph;
no clinical evidence of left atrial hypertension or a pulmonary artery occlusion pressure of 18 mmHg or less in the presence of a pulmonary artery catheter.
This AECC definition defined acute lung injury (ALI) as a milder form of lung injury, with a PaO2/FiO2 ratio of 300 mmHg or less, with ARDS therefore being a subset of ALI. A more recent definition, known as the Berlin definition, provides a time in which ARDS is developed, and categorizes severity into mild, moderate or severe ARDS, thus removing the distinction between ALI and ARDS (ARDS Definition Task Force 2012). This definition includes the following criteria:
within one week of a known clinical insult or new or worsening respiratory symptoms;
bilateral opacities, not fully explained by effusions, lobar or lung collapse, or nodules;
respiratory failure not fully explained by cardiac failure or fluid overload. Objective assessment (e.g. echocardiography) needed to exclude hydrostatic oedema if no risk factor present;
mild ARDS: PaO2/FiO2 > 200 mmHg and ≤ 300 mmHg with positive end‐expiratory pressure (PEEP) or continuous positive airway pressure (CPAP) ≥ 5 cmH2O;
moderate ARDS: PaO2/FiO2 > 100 mmHg and ≤ 200 mmHg with PEEP ≥ 5 cmH2O;
severe ARDS: PaO2/FiO2 ≤ 100 mmHg with PEEP ≥ 5 cmH2O.
An epidemiological study of 50 countries reported the prevalence of ARDS as 10.4% of all intensive care unit (ICU) admissions, with mortality estimated to be approximately 40% (Bellani 2016). Almost all people with ARDS require mechanical ventilation to survive; all require supplemental oxygen. In addition, survivors have a prolonged stay in the ICU and demonstrate significant functional limitations, primarily fatigue and muscle weakness, that reduce their quality of life after hospital discharge; only 50% of survivors are fit to return to work after 12 months (Herridge 2003).
Description of the intervention
Research on therapy for ARDS has focused on both physiological and pharmacological therapies. Lung protection strategies, using lower tidal volumes (Guay 2018), have been widely adopted since the ARDS Network publication (ARDS Network 2000; Petrucci 2013). Other strategies that have been trialed include high‐frequency oscillatory ventilation (Sud 2016), high versus low PEEP (Santa Cruz 2013), pressure‐controlled versus volume‐controlled ventilation (Chacko 2015), recruitment manoeuvres (Hodgson 2016), and use of prone positioning (Bloomfield 2015).
The pathogenesis of ARDS, extensively reviewed elsewhere (Luce 1998; Ware 2000), provides multiple potential targets for pharmacological interventions. Regardless of the inciting insult, the pathology of ARDS features damage to the alveolar‐capillary membrane, with leakage of protein‐rich oedema fluid into alveoli. Epithelial damage involves the basement membrane and types I and II cells. Injury reduces the amount and function of surfactant produced by type II cells. This increases alveolar surface tension, decreases lung compliance, and causes atelectasis. Endothelial damage is associated with numerous inflammatory events. These include neutrophil recruitment, sequestration and activation; formation of oxygen radicals; activation of the coagulation system, leading to microvascular thrombosis with platelet‐fibrin thrombi; and recruitment of mesenchymal cells with production of procollagen (a precursor to fibrosing alveolitis). Within the alveolar space, the balance between pro‐inflammatory (for example, tumour necrosis factor (TNF) alpha and interleukins (IL) 1, 6, and 8) and anti‐inflammatory mediators (for example, IL‐1 receptor antagonist and soluble TNF receptor) favours ongoing inflammation. In summary, initial lung injury is followed by repair, remodelling, and fibrosing alveolitis.
Interventions in this review are pharmacological agents that aim to repair specific damage or response to the lung injury. These agents may be man‐made, natural, or endogenous (from within the body) and include, amongst others: corticosteroids; surfactants; N‐acetylcysteine; statins; and beta‐agonists.
How the intervention might work
The diversity of approaches to pharmacological therapy for ARDS reflects the complex pathophysiology, and each therapy may differ in its proposed mechanism of action. Of just some of the possible pharmacological agents that may be used to treat the symptoms of ARDS: corticosteroids may provide multiple anti‐inflammatory pathways (Pehora 2017; Polderman 2018); surfactants may restore the normal mechanical properties of alveoli (surface tension, alveolar opening) (Spragg 2003); N‐acetylcysteine may be used for its antioxidant properties (Ortolani 2000); statins may reduce pulmonary and systemic inflammatory responses (HARP‐2 2014); and beta‐agonists may reduce pulmonary oedema and improve alveolar fluid clearance (ALTA 2011).
Why it is important to do this review
Recent guidelines have recommended the use of therapies in relation to lung protection strategies, fluid management strategies, neuromuscular blocking agents (Lundstrøm 2017), positive end‐expiratory pressure (PEEP), extra‐corporeal membrane oxygenation (ECMO), extra‐corporeal carbon dioxide removal (ECCOR), and prone positioning (FICM/ICS Guideline Development Group 2018). This guideline provides a research recommendation for corticosteroids because of insufficient evidence; the range of pharmacological agents in this review are not included in the guideline. The effectiveness of pharmacological agents to reduce mortality or mechanical ventilation is still not established (Nanchal 2018).
This review is an update of a previous version (Adhikari 2004). Research interest in this condition continues, and we aim to incorporate new findings for agents included in the previous version, as well as new agents that were not previously included. The outcomes in the previous version remain relevant, but this review includes consideration of the long‐term consequences and includes fitness to return to work after 12 months (Herridge 2003). The methods used to assess the certainty of evidence in Cochrane Reviews has since been updated, and we reflect these methodological changes and now incorporate a GRADE assessment and 'Summary of findings' tables for the primary comparisons.
Objectives
To evaluate the effectiveness of pharmacological agents in adults with ARDS on mortality, mechanical ventilation, and fitness to return to work at 12 months.
Methods
Criteria for considering studies for this review
Types of studies
We included randomized controlled trials (RCTs). We excluded quasi‐randomized trials.
We excluded studies published prior to 2000. We based the decision to restrict studies by publication date on an important study in 2000 advocating the use of lower tidal volumes in people with ARDS, which resulted in changes to standard practice in mechanical ventilation management for these people (ARDS Network 2000).
Types of participants
We included adult participants with ARDS admitted to an ICU. We used authors' definitions of adult, and included studies if the mean age of participants was more than 18 years. In addition, we used authors' definitions of ARDS, and included participants that were diagnosed according to the AECC criteria, with a distinction between ALI and ARDS (Bernard 1994), and the later Berlin definition which includes a distinction between mild, moderate, and severe ARDS (ARDS Definition Task Force 2012), or other criteria. We excluded studies in which ARDS (and ALI, where appropriate) was not reported as a required study inclusion criterion; we therefore excluded studies in which participants with ARDS were reported as a subgroup analysis.
Types of interventions
We included pharmacological agents compared to a placebo or to no therapy for the treatment of established ARDS, including any pharmacological agent given for the treatment of established mild ARDS (or ALI) that may prevent the development of ARDS. We included pharmacological agents that were man‐made, natural, or endogenous (from within the body).
We excluded enteral and intravenous therapies that are either not considered to be pharmacological by regulatory authorities (nutritional or herbal interventions) or are combined with other management strategies (fluid management). We excluded therapies that have been reviewed in other Cochrane Reviews: inhaled nitric oxide (Gebistorf 2016); inhaled prostacyclins (Afshari 2017); and partial liquid ventilation (Galvin 2013). We excluded pharmacological therapies used as part of a strategy of mechanical ventilation (neuromuscular blocking agents), and medical oxygen. We excluded studies of activated protein C (APC), which is now a withdrawn drug.
We excluded any pharmacological therapy started for prophylaxis of mild ARDS, even when continued in people who subsequently developed moderate or severe ARDS. We excluded studies directly comparing two pharmacological therapies without a no‐treatment or placebo control group.
Each type of agent represented a different comparison, and we separately analysed data for each comparison group. We selected five primary comparisons in the review.
Corticosteroids versus control
Surfactants versus control
N‐acetylcysteine versus control
Statins versus control
Beta‐agonists versus control
We analysed other types of agents as secondary comparison groups.
Types of outcome measures
Primary outcomes
Early all‐cause mortality (at or before three months after randomization). We included ICU and hospital mortality in this outcome.
Secondary outcomes
Late all‐cause mortality (more than three months after randomization).
Duration of mechanical ventilation (defined as the time from randomization to extubation, study withdrawal, or death).
Number of ventilator‐free days to day 28 (Schoenfeld 2002).
Adverse events (defined as those leading to discontinuation of the study medication). In studies with a no‐placebo control arm, adverse events were defined as those leading to discontinuation of the study medication, or 'serious adverse events' using study authors' definitions.
Fitness to return to work at 12 months.
Search methods for identification of studies
Electronic searches
We identified RCTs through literature searching with systematic and sensitive search strategies, as outlined in Chapter 6.4 of theCochrane Handbook of Systematic Reviews of Interventions (Cochrane Handbook;Higgins 2011). We applied no restrictions to language or publication status. We searched the following databases for relevant trials:
Cochrane Central Register of Controlled Trials (CENTRAL; 2018; Issue 12);
MEDLINE (Ovid SP; 1946 to 10 December 2018);
Embase (Ovid SP; 1974 to 10 December 2018);
CINAHL (EBSCOhost: 1937 to 10 December 2018).
We developed a subject‐specific search strategy in MEDLINE and other listed databases. We developed the search strategy in consultation with the Information Specialist for the Cochrane Emergency and Critical Group. Search strategies can be found in: Appendix 1; Appendix 2; Appendix 3; Appendix 4.
We scanned the following clinical trials registers for ongoing and unpublished trials:
World Health Organization International Clinical Trials Registry Platform (www.who.int/ictrp/en/) on 18 December 2018);
ClinicalTrials.gov (www.clinicaltrials.gov) on 10 December 2018.
Searching other resources
We carried out citation searching of identified included studies published since 2010 in Web of Science on 7 January 2019 (apps.webofknowledge.com). We conducted a search of grey literature using Opengrey on 7 January 2019 (www.opengrey.eu/). In addition, we scanned reference lists of relevant systematic reviews which were recently published (since 2015). We did not contact content experts to enquire about additional unpublished trails during the 2019 update.
Data collection and analysis
Two review authors (SL and either MP, CT, or AS) independently completed data collection on studies written in English or Spanish before comparing results and reaching consensus. One review author (SL) completed data collection using the English abstract for studies written in Chinese; Dr Henry HL Wu completed full data extraction of the Chinese study reports (Acknowledgements). One review author (AS) was available to resolve conflicts if required.
Selection of studies
We used reference management software to collate the results of searches and to remove duplicates (Endnote). We used Covidence 2018 software to screen results of the search of titles and abstracts and to identify potentially relevant studies. We sourced the full texts of all potentially relevant studies and considered whether they met the inclusion criteria (see Criteria for considering studies for this review). We reviewed abstracts at this stage, and included them in the review only if they provided sufficient information and relevant results that included denominator figures for the intervention and control groups. We recorded the number of papers retrieved at each stage and report this information using a PRISMA flow chart. We reported in the review brief details of closely related but excluded papers.
Data extraction and management
We used Covidence 2018 software to extract data from individual studies. A basic template for data extraction forms is available at www.covidence.org. We adapted this template to include the following information.
Methods: type of study design; setting; dates of study; funding sources and study author declarations of interest.
Participants: number of participants randomized to each group; baseline characteristics (to include: criteria for ARDS diagnosis; time since onset of ARDS; PaO2:FiO2 at baseline; risk factors; Lung Injury Score (LIS) (Murray 1988); and illness severity scores such as 'Acute Physiology and Chronic Health Evaluation II' (APACHE II).
Intervention: details of pharmacological intervention and control (to include dose, timing and duration of administration), details of any other treatment. We attempted to collect data on clinical management of participants during the study period, which may influence results; we based this on the current guidelines for the management of ARDS, which includes information about: lower tidal volumes, fluid management strategies, neuromuscular blocking agents, positive end‐expiratory pressure (PEEP), extra‐corporeal membrane oxygenation (ECMO), extra‐corporeal carbon dioxide removal (ECCOR), and prone positioning (FICM/ICS Guideline Development Group 2018).
Outcomes: all relevant review outcomes as measured and reported by study authors, including time points of measurement.
Outcome data: results of outcome data
We considered the applicability of information from individual studies and the generalizability of data to our intended study population (i.e. the potential for indirectness in our review). If we found associated publications from the same study, we created a composite data set based on all eligible publications.
Assessment of risk of bias in included studies
Two review authors (SL and either MP or CT) independently assessed study quality, study limitations, and the extent of potential bias by using the Cochrane 'Risk of bias' tool (Higgins 2017). We assessed the following domains.
Sequence generation (selection bias);
Allocation concealment (selection bias);
Blinding of participants, personnel, and outcome assessors (performance and detection bias);
Incomplete outcome data (attrition bias);
Selective outcome reporting (reporting bias);
Other potential risks of bias.
In addition, we assessed comparability of baseline characteristics between study groups, as characteristics such as illness severity may influence response to treatment. For each domain, we judged whether study authors had made sufficient attempts to minimize bias in their study design. We made judgements using three measures: high; low; and unclear risk of bias. We recorded this judgement in 'Risk of bias' tables and present a summary 'Risk of bias' figure (see Figure 2).
Measures of treatment effect
We collected dichotomous data for mortality outcomes, adverse events, and fitness to return to work. We collected continuous data for duration of mechanical ventilation, and number of ventilator‐free days up to day 28. We reported dichotomous data as risk ratios (RRs) to compare groups, and continuous data as mean differences (MDs). We reported 95% confidence intervals (CI).
Unit of analysis issues
We noted studies that had more than one intervention group. We combined data in intervention groups and compared these combined data with the control group. We used sensitivity analysis to investigate the effect of combining data from more than one intervention group in a study.
Dealing with missing data
We considered data to be complete if losses were reported and explained by study authors, and we combined no incomplete data in the meta‐analysis. We did not contact study authors to clarify missing information about study characteristics.
Assessment of heterogeneity
We assessed whether evidence of inconsistency was apparent in our results by considering heterogeneity. We assessed clinical and methodological heterogeneity by comparing similarities in our included studies between study designs, participants, interventions, and outcomes, and used the data collected from the full‐text reports (as stated in Data collection and analysis). We assessed statistical heterogeneity by calculating the Chi2 text or I2 statistic and judged any heterogeneity above an I2 value or 60% and a Chi2 P value of 0.05 or less to indicate moderate to substantial statistical heterogeneity (Higgins 2011).
As well as looking at statistical results, we considered point estimates and overlap of CIs. If CIs overlap, then results are more consistent. However, combined studies may show a large consistent effect but with significant heterogeneity. We therefore planned to interpret heterogeneity with caution (Guyatt 2011a).
Assessment of reporting biases
We attempted to source published protocols for each of our included studies by using clinical trials registers. We compared published protocols with published study results, to assess the risk of selective reporting bias. We planned to generate a funnel plot to assess the risk of publication bias if we identified sufficient studies reporting on an outcome (i.e. more than 10 studies (Sterne 2017)). An asymmetrical funnel plot may suggest publication of only positive results (Egger 1997).
Data synthesis
We completed meta‐analyses of outcomes for which we had comparable effect measures from more than one study for each comparison group, and when measures of heterogeneity indicated that pooling of results was appropriate. We did not pool studies that had a high level of methodological or clinical heterogeneity. We used the statistical calculator in Review Manager 5 to perform meta‐analysis (Review Manager 2014).
We used the Mantel‐Haenszel random‐effects model to account for potential variability in participant conditions between studies.
We calculated CIs at 95% and used a P value of 0.05 or less to judge whether a result was statistically significant. We considered imprecision in the results of analyses by assessing the CI around an effect measure; a wide CI would suggest a higher level of imprecision in our results. A small number of studies may also reduce precision (Guyatt 2011b).
Subgroup analysis and investigation of heterogeneity
We did not perform subgroup analysis because we found insufficient studies in each comparison group (i.e. fewer than 10) to enable meaningful analysis (Deeks 2017). If we had found sufficient studies, we planned to perform subgroup analysis as follows:
severity of ARDS: studies with participants meeting the criteria for mild ARDS versus studies with participants meeting the criteria for moderate ARDS; or studies with participants meeting the criteria for moderate ARDS versus studies with participants meeting the criteria for severe ARDS. We used the more recent Berlin definition of ARDS for these cut‐off points (ARDS Definition Task Force 2012);
time since onset of ARDS: early (72 hours or less) versus late (more than 72 hours).
We reported the information collected for these subgroups for each primary comparison group.
Sensitivity analysis
Although we had tried to include studies that only used current guidelines on the clinical management of ARDS by excluding studies published prior to 2000, we found that studies did not consistently or sufficiently report clinical management protocols. The ARDS Network study published in 2000 demonstrated a reduction in mortality when lower tidal volumes were used (ARDS Network 2000). Studies in which this strategy was not used may therefore not be generalizable to current management of people with ARDS, and we excluded these studies during sensitivity analysis in order to explore this potential effect on the results.
We also explored the potential effect of decisions made as part of the review process. We performed the following sensitivity analysis on our primary outcome (early all‐cause mortality) in our primary comparisons in this review.
We excluded studies in which use of lower tidal volumes was not reported.
We excluded studies that we judged at high or unclear risk of selection bias.
We excluded studies that we judged to have high risk of attrition bias because of missing data which were unbalanced between groups, and unexplained.
We conducted meta‐analysis using the alternative meta‐analytic effects model (i.e. fixed‐effect).
In multi‐arm studies in which data from more than one intervention group were combined, we separately included data for each arm in analysis.
In each sensitivity analysis we compared the effect estimate with the main analysis. We reported these effect estimates only if they indicated a difference in interpretation of the effect.
Summary of findings' tables and GRADE
One review author (SL) used the GRADE system to assess the certainty of the body of evidence associated with the following outcomes (Guyatt 2008):
early all‐cause mortality (at or before three months after randomization);
late all‐cause mortality (more than three months after randomization);
duration of mechanical ventilation;
number of ventilator‐free days to day 28;
adverse events;
fitness to return to work at 12 months.
The GRADE approach appraises the certainty of a body of evidence based on the extent to which we can be confident that an estimate of effect or association reflects the item being assessed. Evaluation of the certainty of a body of evidence considers within‐study risk of bias, directness of the evidence, heterogeneity of the data, precision of the effect estimates, and risk of publication bias.
We constructed 'Summary of findings' tables using the GRADE profiler software for the following comparisons in this review (GRADEpro GDT):
corticosteroids versus control (Table 1);
surfactants versus control (Table 2);
N‐acetylcysteine versus control (Table 3);
statins versus control (Table 4);
beta‐agonists versus control (Table 5).
Summary of findings for the main comparison. Corticosteroids compared to control for adults with acute lung injury and acute respiratory distress syndrome.
| Corticosteroids compared to control for adults with acute respiratory distress syndrome | ||||||
|
Population: adults with acute respiratory distress syndrome
Setting: intensive care units in: China; Kuwait; Thailand; and USA Intervention: corticosteroids (methylprednisolone; hydrocortisone; and budesonide Comparison: control (placebo or standard therapy) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with control | Risk with corticosteroids | |||||
|
Early all‐cause mortality (≤ 3 months) Reported at: day 14; day 28; day 60; and in hospital |
Study population | RR 0.77 (0.57 to 1.05) | 574 (6 studies) | ⊕⊕⊝⊝ Lowa |
‐ | |
| 374 per 1000 | 288 per 1000 (213 to 393) | |||||
|
Late all‐cause mortality ( > 3 months) Reported at: 180 days |
Study population | RR 0.99 (0.64 to 1.52) | 180 (1 study) | ⊕⊝⊝⊝ Very lowb |
‐ | |
| 319 per 1000 | 315 per 1000 (204 to 484) | |||||
|
Duration of mechanical ventilation Measured in days |
In the control group, mean values range from 11.6 days to 20.3 days | MD 4.30 days lower (9.72 days lower to 1.12 days higher) | ‐ | 277 (3 studies) | ⊕⊝⊝⊝ Very lowc |
We did not include 1 additional study (91 participants) in the analysis because data were reported as median values; study authors reported a shorter duration of mechanical ventilation with corticosteroids use |
| Ventilator‐free days up to day 28 | In the control group, mean values range from 6.8 days to 12.8 days | MD 4.09 days higher (1.74 higher to 6.44 higher) | ‐ | 494 (4 studies) | ⊕⊕⊝⊝ Lowd |
‐ |
|
Adverse events Defined as leading to discontinuation of study medication; or for studies with standard care control, defined by study authors as "serious adverse events" |
‐ | ‐ | Not estimable | ‐ | ‐ | No studies reported or measured this outcome |
| Fitness to return to work at 12 months | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies reported or measured this outcome |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; MD: mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aWe downgraded one level for study limitations (risks of bias were uncertain or high amongst studies), and by one level for imprecision (evidence was from few studies with few participants). bWe downgraded by one level for study limitations (we were unable to assess risk of reporting bias because of retrospective clinical trial registration and analysis was completed post hoc), and by two levels for imprecision (evidence was from one study with few participants). cWe downgraded by one level for study limitations (risks of bias were uncertain or high amongst studies), by one level for inconsistency (evidence of substantial statistical heterogeneity) and by one level for imprecision (evidence was from few studies with few participants). dWe downgraded by one level for imprecision (evidence was from few studies with few participants), and by one level for inconsistency (we noted a wide confidence interval in the effect estimate).
Summary of findings 2. Surfactants compared to control for adults with acute lung injury and acute respiratory distress syndrome.
| Surfactants compared to control for adults with acute respiratory distress syndrome | ||||||
| Population: adults with acute respiratory distress syndrome Setting: intensive care units in: Austria, Belgium; Canada; Cuba; Europe (one multicentre study did not report countries within Europe); Finland; France; Germany; Greece; Netherlands; Norway; South Africa; Spain; Sweden; UK; USA Intervention: surfactants (surfacen; HL10; venticute; alveofact; calfactant) Comparison: control (placebo or standard therapy) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with control | Risk with surfactants | |||||
|
Early all‐cause mortality (≤ 3 months) Reported at: 28 days; and 90 days. Time point not reported in 4 studies |
Study population | RR 1.08 (0.91 to 1.29) | 1338 (9 studies) | ⊕⊝⊝⊝ Very lowa |
‐ | |
| 284 per 1000 | 307 per 1000 (259 to 367) | |||||
|
Late all‐cause mortality ( > 3 months) Reported at: 180 days |
Study population | RR 1.28 (1.01 to 1.61) | 418 (1 study) | ⊕⊝⊝⊝ Very lowb |
‐ | |
| 362 per 1000 | 463 per 1000 (366 to 583) | |||||
|
Duration of mechanical ventilation Measured in days |
In the control mean duration was 8.1 days | MD 2.5 days lower (4.95 days lower to 0.05 days lower) | ‐ | 16 (1 study) | ⊕⊝⊝⊝ Very lowc |
We did not include 1 additional study (48 participants) in analysis because values of data (mean or median) were unclear; study authors reported little or no difference between groups in duration of mechanical ventilation |
| Ventilator‐free days up to day 28 | In the control group, mean values range from 11.9 days to 13 days | MD 0.39 days lower (2.49 days lower to 1.72 days higher) | ‐ | 344 (2 studies) | ⊕⊝⊝⊝ Very lowd |
We did not include 4 additional studies (512 participants) in analysis because data were not reported sufficiently; 3 studies reported little or no difference between groups, and 1 study reported more ventilator‐free days in the intervention group |
|
Adverse events Defined as leading to discontinuation of study medication; data collected during study period |
Study population | RR 0.50 (0.17 to 1.44) |
88 (2 studies) |
⊕⊝⊝⊝ Very lowe |
‐ | |
| 216 per 1000 |
108 per 1000 (37 to 311) |
|||||
| Fitness to return to work at 12 months | ‐ | Not estimable | No studies reported or measured this outcome | |||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; MD: mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aWe downgraded by three levels; by two levels for study limitations (studies comparing surfactants with standard therapy were all at high risk of performance bias, and we noted other risks of bias that were high or unclear amongst studies), and by one level for inconsistency (we noted some differences in data between studies and we found too few studies to explore these differences through subgroup analyses). bWe downgraded by three levels; by two levels for study limitations (study was at high and unclear risks of bias), and by one level for imprecision (evidence was from a single study). cWe downgraded by three levels; by one level for study limitations (studies were at high risk of performance bias), by one level for inconsistency (we noted differences in data between studies), and by one level for imprecision (evidence was from two studies with few participants). dWe downgraded by three levels; by two levels for study limitations (studies comparing surfactants with standard therapy were all at high risk of performance bias, and we noted other risks of bias that were high or unclear amongst studies), and by one level for inconsistency (we noted differences in data between studies). eWe downgraded by three levels; by two levels for study limitations (studies comparing surfactants with standard therapy were at high risk of performance bias, and we noted other risks of bias that were high or unclear amongst studies), and by one level for imprecision (evidence was from two studies with few participants).
Summary of findings 3. N‐acetylcysteine compared to control for adults with acute lung injury and acute respiratory distress syndrome.
| N‐acetylcysteine compared to control for adults with acute respiratory distress syndrome | ||||||
| Population: adults with acute respiratory distress syndrome Setting: intensive care unit in: Italy Intervention: N‐acetylcysteine Comparison: control (placebo) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with control | Risk with N‐acetylcysteine | |||||
|
Early all‐cause mortality (≤ 3 months) Reported at: 30 days |
Study population | RR 0.64 (0.32 to 1.30) | 36 (1 study) | ⊕⊝⊝⊝ Very lowa |
‐ | |
| 583 per 1000 | 373 per 1000 (187 to 758) | |||||
| Late all‐cause mortality ( > 3 months | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
| Duration of mechanical ventilation | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
| Ventilator‐free days up to day 28 | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
|
Adverse events Defined as leading to discontinuation of study medication |
‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
| Fitness to return to work at 12 months | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aWe downgraded by three levels; by one level for study limitations (unblinded study at high risk of performance bias). and by two levels for imprecision (evidence from one study with few participants).
Summary of findings 4. Statins compared to control for adults with acute lung injury and acute respiratory distress syndrome.
| Statins compared to control for adults with acute respiratory distress syndrome | ||||||
| Population: adults with acute respiratory distress syndrome Setting: intensive care units in: Northern Ireland; UK; USA Intervention: statins (simvastatin; and rosuvastatin) Comparison: control (placebo) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with control | Risk with statins | |||||
|
Early all‐cause mortality (≤ 3 months) Reported at: 60 days; and during hospital stay |
Study population | RR 0.99 (0.78 to 1.26) | 1344 (3 studies) | ⊕⊕⊕⊝ Moderatea |
‐ | |
| 262 per 1000 | 259 per 1000 (204 to 330) | |||||
| Late all‐cause mortality ( > 3 months | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
|
Duration of mechanical ventilation Measured in days |
In the control group, mean duration was 15.9 days | MD 2.70 days higher (0.35 days lower to 8.95 days higher) | ‐ | 60 (1 study) |
⊕⊕⊝⊝ Lowb |
‐ |
| Ventilator‐free days up to day 28 | In the control group, mean values range from 9.1 days to 15.1 days | MD 0.40 days higher (0.71 days lower to 1.52 days higher) | ‐ | 1342 (3 studies) | ⊕⊕⊕⊝ Moderatec |
‐ |
|
Adverse events Defined as leading to discontinuation of study medication |
‐ | ‐ | Not estimable | ‐ | ‐ | One study measured discontinuation of treatment because of an adverse event, but data were not reported by study authors |
| Fitness to return to work at 12 months | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; MD: mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect | ||||||
aWe downgraded by one level for inconsistency; we noted some differences in data between studies and we found too few studies to explore these differences through subgroup analyses. bWe downgraded by two levels for imprecision; evidence was from one study with few participants. cWe downgraded by one level for inconsistency; we noted some differences in data between studies and we found too few studies to conduct subgroup analyses to explore these differences.
Summary of findings 5. Beta‐agonist compared to control for adults with acute lung injury and acute respiratory distress syndrome.
| Beta‐agonist compared to control for adults with acute respiratory distress syndrome | ||||||
| Population: adults with acute respiratory distress syndrome Setting: intensive care units in: UK; and USA Intervention: beta‐agonist (salbutamol; and albuterol) Comparison: control (placebo) | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | Number of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with control | Risk with beta‐agonist | |||||
|
Early all‐cause mortality (≤ 3 months) Reported at: 28 days; 60 days; and during hospital stay |
Study population | RR 1.14 (0.91 to 1.42) | 646 (3 studies) | ⊕⊕⊕⊝ Moderatea |
‐ | |
| 283 per 1000 | 323 per 1000 (258 to 402) | |||||
| Late all‐cause mortality (> 3 months | ‐ | ‐ | ‐ | ‐ | ‐ | No studies measured or reported this outcome |
| Duration of mechanical ventilation | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
| Ventilator‐free days up to day 28 | In the control group, mean values range from 5.3 days to 16.6 days | MD 2.20 days lower (3.68 days lower to 0.71 days lower) | ‐ | 646 (3 studies) | ⊕⊝⊝⊝ Very lowb |
‐ |
|
Adverse events Defined as leading to discontinuation of study medication; data collected during study period |
‐ | ‐ | ‐ | 606 (2 studies) |
⊕⊝⊝⊝ Very lowc |
We did not pool data in 2 studies because of potential differences in types of adverse events. 1 study reported little or no difference between groups; and 1 study reported more adverse events when participants were given beta‐agonists |
| Fitness to return to work at 12 months | ‐ | ‐ | Not estimable | ‐ | ‐ | No studies measured or reported this outcome |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: confidence interval; RR: risk ratio; MD: mean difference | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate. The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited. The true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate. The true effect is likely to be substantially different from the estimate of effect | ||||||
aWe downgraded by one level for imprecision (evidence was from few studies with few participants). bWe downgraded by three levels; by one level for imprecision (evidence was from few studies with few participants), and by two levels for inconsistency (inspection of data showed differences in direction of effect between studies which we could not explain). cWe downgraded by three levels; by one level for imprecision (evidence was from few studies with few participants), and by two levels for inconsistency (inspection of data showed differences in direction of effect between studies, and a high level of statistical heterogeneity, which may be due caused by differences in types of adverse events measured by study authors).
Because of the broad range of types of pharmacological agents eligible for inclusion in this review, it was not feasible to produce a 'Summary of findings' table for each type of agent. Our choice of comparisons for the 'Summary of findings' tables was based on agents which are most commonly used and recognized in a global clinical setting, and followed advice given by the Cochrane Emergency and Critical Care Group.
Results
Description of studies
Results of the search
We screened 12,574 titles and abstracts, which included forward‐ and backward‐citation searches, clinical trials registers and grey literature. We sourced 352 full‐text reports to assess eligibility (Figure 1).
1.
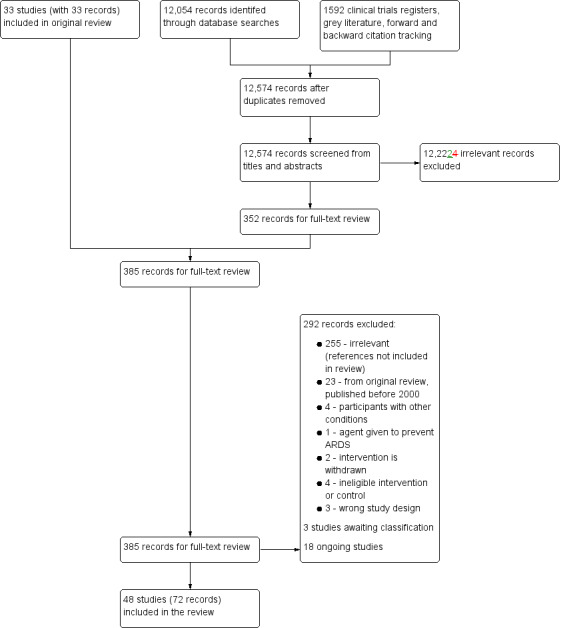
Flow diagram for searches conducted since publication of the last version of the Review up to 10 December 2018
Included studies
See Characteristics of included studies. A summary of study characteristics for the primary comparisons is included in Appendix 5.
We included 48 RCTs (72 publications) with 6299 participants (ALTA 2011; ARDS Network 2002; BALTI 2006; BALTI‐2 2013; Barrese‐Perez 2015; Chen 2017; Endo 2006; Guoshou 2013; HARP 2011; HARP‐2 2014; Huang 2017; Ji 2018; Kadoi 2004; KARE 2017; KARMA 2000; Kesecioglu 2001; Kesecioglu 2009; Khan 2017; Krenn 2017; Li 2010; Liu 2012; Liu 2015; Liu 2017; Meduri 2007; Mohamed 2017; Morelli 2006; Morris 2008; Najafi 2009; Ortolani 2000; Paine 2012; Rezk 2013; Ryugo 2006; SAILS 2014; Soltan‐Sharifi 2007; Spragg 2002a; Spragg 2002b; Spragg 2003; START 2018; Steinberg 2006; STRIVE 2004; Tongyoo 2016; Tsangaris 2007; Vincent 2001; Walmrath 2000; Willson 2015; Wirtz 2017; Zhao 2014; Zheng 2014).
Five studies were conducted by the ARDS Clinical Trials Network (ALTA 2011; ARDS Network 2002; KARMA 2000; SAILS 2014; Steinberg 2006). if the studies were known by acronyms, we used them for study names, rather than study author names. We included five studies for which we could only source the abstract and this limited the details of study characteristics that we were able to extract (Kesecioglu 2001; Spragg 2002a; Spragg 2002b; Walmrath 2000; Wirtz 2017). We sourced the full text of all remaining studies.
Study population
Twenty studies included participants that had ARDS defined by the AECC (ARDS Network 2002; BALTI 2006; BALTI‐2 2013; Barrese‐Perez 2015; HARP 2011; Kadoi 2004; KARE 2017; KARMA 2000; Khan 2017; Krenn 2017; Liu 2012; Liu 2015; Meduri 2007; Paine 2012; Spragg 2003; STRIVE 2004; Tongyoo 2016; Vincent 2001; Willson 2015; Zhao 2014). Only two studies used the more recent Berlin definition (Mohamed 2017; Zheng 2014). Guoshou 2013 used criteria from the Society of Critical Care Medicine of Chinese Medicine Association (SCCMCMA 2006). The remaining studies did not reference the criteria that they used to define ARDS.
We attempted to differentiate the severity of ARDS in the included studies using the Berlin definition of ARDS (ARDS Definition Task Force 2012). However, we found it was not possible to do this precisely for each study because of a lack of detail in study reports. We used only the measure of PaO2/FiO2 to distinguish between severities of ARDS, because these data were most commonly (although not consistently) reported in studies. Two studies included only participants with mild ARDS (defined by the study authors as ALI, with a PaO2/FiO2 ≤ 300 mmHg) (Li 2010; Ryugo 2006). The remaining studies included participants with a mean PaO2/FiO2 value which was ≤ 200 mmHg but was likely to include participants that had mild, moderate, and severe ARDS; we expected from the mean values that most participants had moderate ARDS.
Most studies included ARDS with a risk factor from both direct and indirect causes, with pneumonia and sepsis often reported as the most frequent cause. Five studies reported inclusion of participants with only specific risk factors, which were: trauma (Endo 2006); heatstroke (Chen 2017); sepsis (Liu 2015; Morelli 2006); systematic inflammatory response syndrome after cardiopulmonary bypass surgery (Ryugo 2006); blunt chest trauma (Tsangaris 2007).
Although we hoped to extract sufficient data on clinical management of participants relating to current ICU guidelines on ARDS (FICM/ICS Guideline Development Group 2018), we found that this was generally poorly reported in studies. No studies reported whether practitioners followed all aspects of clinical management that we had hoped to collect during data extraction (lower tidal volumes, fluid management strategies, neuromuscular blocking agents, positive end‐expiratory pressure (PEEP), extra‐corporeal membrane oxygenation (ECMO), extra‐corporeal carbon dioxide removal (ECCOR), and prone positioning). We found that 25 studies reported the use of lower tidal volumes to manage ventilation (ALTA 2011; ARDS Network 2002; BALTI‐2 2013; Chen 2017; HARP‐2 2014; KARE 2017; Kesecioglu 2009; Khan 2017; Krenn 2017; Liu 2012; Liu 2015; Liu 2017; Meduri 2007; Mohamed 2017; Morelli 2006; Paine 2012; SAILS 2014; START 2018; Steinberg 2006; STRIVE 2004; Tongyoo 2016; Tsangaris 2007; Willson 2015; Zhao 2014; Zheng 2014), with many studies using the ARDS Network low tidal volume protocol (ARDS Network 2000). However, compliance with this lung protection strategy was not reported.
Study setting
All studies were conducted in hospital settings; even if not reported, we expected participants to be recruited in ICUs. Twenty‐three were multicentre studies (ALTA 2011; ARDS Network 2002; BALTI‐2 2013; Chen 2017; HARP‐2 2014; KARE 2017; KARMA 2000; Kesecioglu 2001; Kesecioglu 2009; Khan 2017; Liu 2017; Meduri 2007; Morris 2008; Ortolani 2000; Paine 2012; SAILS 2014; Spragg 2003; START 2018; Steinberg 2006; STRIVE 2004; Vincent 2001; Walmrath 2000; Willson 2015). The centre was unclearly reported in four studies (Endo 2006; Spragg 2002a; Spragg 2002b; Wirtz 2017). The remaining studies were conducted in a single centre.
Interventions and comparisons
For the primary comparisons, we found:
seven studies (643 participants) that assessed corticosteroids which were: hydrocortisone (Liu 2012; Tongyoo 2016); methylprednisolone (Meduri 2007; Rezk 2013; Steinberg 2006); or budesonide (Mohamed 2017; Zhao 2014). One study used standard therapy in the control group (Zhao 2014); the remaining studies were compared with a placebo or control agent;
nine studies (1340 participants) compared surfactants with either a placebo (Willson 2015), or standard therapy (Barrese‐Perez 2015; Kesecioglu 2001; Kesecioglu 2009; Spragg 2002a; Spragg 2002b; Spragg 2003; Tsangaris 2007; Walmrath 2000). Spragg 2003 was a three‐arm study which compared a high dose and a low dose of surfactant with a placebo;
three studies (86 participants) compared N‐acetylsysteine with either standard therapy (Najafi 2009; Soltan‐Sharifi 2007), or a placebo (Ortolani 2000). Ortolani 2000 was a three‐arm study that included a N‐acetylcysteine group and a N‐acetylcysteine with rutin group;
three studies (1345 participants) compared statins with a placebo (HARP 2011; HARP‐2 2014; SAILS 2014);
three studies (648 participants) compared a beta‐agonist with a placebo (ALTA 2011; BALTI 2006; BALTI‐2 2013).
For the secondary comparison of other pharmacological agents, we found:
four studies (556 participants) compared sivelestat (Endo 2006; Kadoi 2004; Ryugo 2006; STRIVE 2004). Endo 2006 did not report any information on the control group such as whether a placebo or standard treatment was used as a comparison; the remaining studies compared the agent with a placebo control;
two studies (75 participants) compared mesenchymal stem cells, which were: allogeneic adipose‐derived (Zheng 2014); and allogeneic bone marrow‐derived (START 2018);
two studies (110 participants) compared ulinastatin with standard therapy (Chen 2017; Ji 2018);
one study (50 participants) compared anisodimine with standard therapy (Guoshou 2013);
one study (61 participants) compared ACE inhibitor (Enalaprilat) with a placebo control (Wirtz 2017);
one study (39 participants) compared recombinant human angiotensin‐converting enzyme 2 (rhACE2) with a placebo control (Khan 2017);
one study (60 participants) compared recombinant human keratinocyte growth factor (KGF) (Palifermin) with a placebo control (KARE 2017)
one study (132 participants) compared recombinant human granulocyte‐macrophage colony stimulating factor (GM‐CSF) with a placebo control (Paine 2012);
one study (40 participants) compared AP301 with a placebo control (Krenn 2017);
one study (35 participants) compared levosimendan with a placebo control (Morelli 2006);
two studies (167 participants) compared prostacyclins given intravenously: liposomal prostaglandin‐E1 (PGE1) with a placebo (Vincent 2001), and alprostadil with standard care (Liu 2017);
one study (235 participants) compared lisofylline with a placebo control (ARDS Network 2002);
one study (234 participants) compared ketaconazole with a placebo control (KARMA 2000);
two studies (205 participants) compared nitroglycerin with standard care (Huang 2017; Liu 2015). In Huang 2017, nitroglycerin was given alongside propofol administration;
one study (215 participants) compared L‐2‐oxothiazolidine‐4‐carboxylic acid (OTZ) with a placebo (Morris 2008);
one study (45 participants) compared penehyclidine hydrochloride with standard therapy (Li 2010).
Outcomes
Five studies reported no outcomes relevant to the review (Li 2010; Mohamed 2017; Morelli 2006; Najafi 2009; Soltan‐Sharifi 2007).
Of the remaining studies, only three studies did not report data for early mortality (Huang 2017; Ji 2018; Ryugo 2006). Five studies reported data for late mortality (KARE 2017; Kesecioglu 2009; Paine 2012; Steinberg 2006; STRIVE 2004). Nineteen studies reported duration of mechanical ventilation (Barrese‐Perez 2015; Chen 2017; Endo 2006; Guoshou 2013; HARP 2011; Huang 2017; Ji 2018; Kadoi 2004; KARE 2017; Liu 2015; Liu 2017; Meduri 2007; Paine 2012; Rezk 2013; Ryugo 2006; Tongyoo 2016; Tsangaris 2007; Vincent 2001; Zhao 2014). Twenty‐six studies reported ventilator‐free days up to day 28 (ALTA 2011; ARDS Network 2002; BALTI 2006; BALTI‐2 2013; HARP 2011; HARP‐2 2014; KARE 2017; KARMA 2000; Kesecioglu 2001; Krenn 2017; Liu 2012; Meduri 2007; Morris 2008; Paine 2012; SAILS 2014; Spragg 2002a; Spragg 2002b; Spragg 2003; START 2018; Steinberg 2006; STRIVE 2004; Tongyoo 2016; Walmrath 2000; Willson 2015; Wirtz 2017; Zheng 2014). Nine studies reported adverse events that led to the discontinuation of study medication (ALTA 2011; ARDS Network 2002; BALTI‐2 2013; Barrese‐Perez 2015; Guoshou 2013; HARP‐2 2014; KARMA 2000; Morris 2008; Spragg 2003). We found no studies reporting the long‐term outcome of fitness to return to work at 12 months.
Funding sources
Eight studies were funded by pharmaceutical companies (Kesecioglu 2001; Kesecioglu 2009; Khan 2017; Krenn 2017; Morris 2008; STRIVE 2004; Walmrath 2000; Willson 2015), or by combined funding that included pharmaceutical funding (KARE 2017; Paine 2012; SAILS 2014; Spragg 2003). Of these studies, we noted the involvement of the funders in the study design, implementation, and interpretation of the results in four studies (Kesecioglu 2009; Khan 2017; STRIVE 2004; Willson 2015).
Early stopping
Thirteen studies were stopped early (ALTA 2011; ARDS Network 2002; BALTI‐2 2013; HARP‐2 2014; Kesecioglu 2009; Khan 2017; Morris 2008; Paine 2012; SAILS 2014; STRIVE 2004; Vincent 2001; Willson 2015; Wirtz 2017). Of these, only three studies did not report whether the decision to terminate the trial was made by an independent monitoring board (Khan 2017; Morris 2008; Wirtz 2017). The decision in one study was made by the investigators, who were employees of the funders (Willson 2015). We reported reasons for early stopping in Characteristics of included studies.
Excluded studies
We excluded 292 articles following review of full‐text. We report on 37 studies in the review which we identify as key excluded studies. A brief description of these studies, and the reason for exclusion, is reported in Characteristics of excluded studies.
Twenty‐three of these studies were in the previous version of this review (Adhikari 2004); we excluded them because they were published before 2000 (Abraham 1996; Abraham 1999; Anzueto 1996; Ardizzoia 1993; Bernard 1987; Bernard 1997; Bernard 1999; Bone 1989; Domenighetti 1997; Gottlieb 1994; Gregory 1997; Holcroft 1986; Jepsen 1992; Meduri 1998; Reines 1985; Reines 1992; Rossignon 1990; Shoemaker 1986; Steinberg 1990; Suter 1994; Tuxen 1987; Weg 1994; Weigelt 1985). In addition, we excluded four studies which included participants that did not exclusively have ARDS (Bastin 2010; Bastin 2016; Confalonieri 2005; Presneill 2002), one study in which the agent was given to prevent rather than to treat ARDS (Shyamsundar 2010), two studies that assessed an agent which is now withdrawn from the market (Cornet 2014; Liu 2008), three studies that assessed a neuromuscular blocking agent (Forel 2006; Gainnier 2004; Papazian 2010), one study that did not have a control group (Hua 2013), and three studies that were an ineligible study design (Annane 2006; Markart 2007; Vincent 2009).
Studies awaiting classification
We found three studies awaiting classification (Hegazy 2016; NCT00879606; RPCEC00000126). Hegazy 2016 assessed the use of nebivol; this study is published only as an abstract, with insufficient information to assess eligibility. During our search of clinical trials registers, we found two studies that were completed but for which results have not yet been published; these studies assessed recombinant chimeric anti‐tissue factor antibody ALT‐83 (NCT00879606), and surfactants (RPCEC00000126). See Characteristics of studies awaiting classification
Ongoing studies
We found 18 ongoing studies (ACTRN12612000418875; ACTRN12615000373572; Bellingan 2017; ChiCTR1800014733; ChiCTR1800014998; EUCTR2012‐000775‐17; JPRN‐JapicCTI‐163320; Villar 2016; NCT02326350; NCT02595060; NCT02611609; NCT02895191; NCT03017547; NCT03042143; NCT03202394; NCT03346681; NCT03371498; NCT03608592). Agents assessed in these trials are: heparin; corticosteroids; mesenchymal stem cells; Multistem; rhGM‐CSF; dexamethasone; aspirin; N‐acetylcysteine; ulinastatin; FP‐1201‐lyo; MR11A8; IC‐ 14; and BIO‐11006. Estimated recruitment in these studies is 2068 participants. See Characteristics of ongoing studies.
Risk of bias in included studies
See 'Risk of bias' summary (Figure 2).
2.
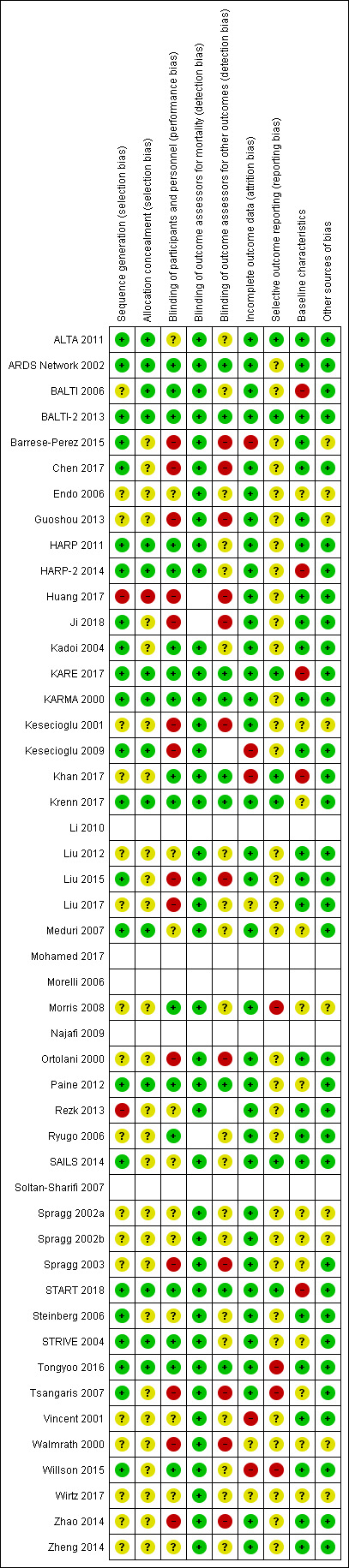
Risk of bias summary: review authors' judgements about each risk of bias item for included studies in which outcome data is reported. Blank spaces in tables indicate that study authors did not report review outcomes.
We did not complete 'Risk of bias' assessments for studies in which no review outcomes were reported (Li 2010; Mohamed 2017; Morelli 2006; Najafi 2009; Soltan‐Sharifi 2007).
We did not assess risk of detection bias for either mortality or other outcomes if these were not reported by study authors.
Blank spaces in the 'Risk of bias' figure indicate that we did not conduct 'Risk of bias' assessments.
Allocation
We judged 23 studies to have low risk of selection bias for sequence generation, because study authors reported sufficient methods for randomization (ALTA 2011; ARDS Network 2002; BALTI‐2 2013; Barrese‐Perez 2015; Chen 2017; HARP 2011; HARP‐2 2014; Ji 2018; Kadoi 2004; KARE 2017; KARMA 2000; Kesecioglu 2009; Krenn 2017; Liu 2015; Meduri 2007; Paine 2012; SAILS 2014; START 2018; Steinberg 2006; STRIVE 2004; Tongyoo 2016; Tsangaris 2007; Willson 2015). One study did not report sufficient methods for randomization; because we noted an unexplained uneven number of participants in each group, we judged this study to have high risk of selection bias for sequence generation (Rezk 2013). We judged one study to have high risk of selection bias because of methods used to randomize participants to groups using folded‐up paper (Huang 2017). Remaining studies reported insufficient methods of sequence generation, and we judged these to be at an unclear risk of bias.
We judged 15 studies to have low risk of bias for allocation concealment, because study authors reported sufficient methods for this judgement (ALTA 2011; ARDS Network 2002; BALTI 2006; BALTI‐2 2013; HARP 2011; HARP‐2 2014; KARE 2017; KARMA 2000; Kesecioglu 2009; Krenn 2017; Meduri 2007; Paine 2012; START 2018; STRIVE 2004; Tongyoo 2016). In Huang 2017, we judged that allocation could not be effectively concealed and we judged this study to have a high risk of bias. Remaining studies reported insufficient details, and we judged them to be at an unclear risk of bias.
Blinding
We judged all studies that used standard therapy control, rather than placebo control, to be at high risk of performance bias (Barrese‐Perez 2015; Chen 2017; Guoshou 2013; Huang 2017; Ji 2018; Kesecioglu 2001; Kesecioglu 2009; Liu 2015; Liu 2017; Spragg 2003; Tsangaris 2007; Walmrath 2000; Zhao 2014). In addition, one placebo‐controlled trial was described as unblinded by the study authors, and we judged this study to be at risk of performance bias (Ortolani 2000). Twelve studies reported insufficient information, and we judged these to have an unclear risk of performance bias (ALTA 2011; Endo 2006; Liu 2012; Meduri 2007; Rezk 2013; SAILS 2014; Spragg 2002a; Spragg 2002b; Steinberg 2006; Vincent 2001; Wirtz 2017; Zheng 2014). We judged the remaining studies to be at a low risk of performance bias.
For detection bias, we did not expect lack of blinding to influence outcome assessment of mortality, and we judged all studies that reported mortality data (including those in which blinding was not possible) to have low risk of detection bias for this outcome. However, we judged studies that used standard therapy control to have a high risk of detection bias for other reported outcomes (Barrese‐Perez 2015; Chen 2017; Guoshou 2013; Ji 2018; Kesecioglu 2001; Li 2010; Liu 2015; Liu 2017; Najafi 2009; Spragg 2003; Tsangaris 2007; Walmrath 2000; Zhao 2014). We judged one placebo‐controlled study to have a high risk of detection bias. Only nine studies adequately reported blinding of outcome assessors for all outcomes, and we judged these studies to have a low risk of detection bias for other reported outcomes (ARDS Network 2002; BALTI‐2 2013; KARE 2017; KARMA 2000; Khan 2017; Krenn 2017; Paine 2012; START 2018; Tongyoo 2016). Remaining studies did not report sufficient information about blinding outcome assessors, and we judged these studies to have an unclear risk of detection bias for other reported outcomes.
Incomplete outcome data
We judged 36 studies to have a low risk of attrition bias because study authors reported no participant losses or few losses (less than 10%) (ALTA 2011; ARDS Network 2002; BALTI 2006; BALTI‐2 2013; Chen 2017; Endo 2006; Guoshou 2013; HARP 2011; HARP‐2 2014; Huang 2017; Ji 2018; Kadoi 2004; KARE 2017; KARMA 2000; Kesecioglu 2001; Krenn 2017; Liu 2012; Liu 2015; Liu 2017; Meduri 2007; Morris 2008; Ortolani 2000; Paine 2012; Rezk 2013; Ryugo 2006; SAILS 2014; Spragg 2002a; Spragg 2002b; Spragg 2003; START 2018; Steinberg 2006; STRIVE 2004; Tongyoo 2016; Tsangaris 2007; Zhao 2014; Zheng 2014).
We judged five studies to have a high risk of attrition bias; in one study we were concerned by uneven numbers of participants who did not complete treatment (Barrese‐Perez 2015), and in four studies we were concerned about the influence of early stopping on the results (Kesecioglu 2009; Khan 2017; Vincent 2001; Willson 2015).
We could not ascertain loss of participants in two studies reported as abstracts because of insufficient information (Walmrath 2000; Wirtz 2017)
Selective reporting
We found only seven studies with prospective clinical trials registration and outcome information reported equivalently in the clinical trials registration documents and the published completed study report (ALTA 2011; BALTI‐2 2013; KARE 2017; Khan 2017; Krenn 2017; SAILS 2014; START 2018).
Three studies were prospectively registered with clinical trials registers, but we noted discrepancies and could not effectively assess risks of reporting bias for these studies (HARP 2011; HARP‐2 2014; Paine 2012); we judged these as having unclear risk of bias.
We judged four studies to have a high risk of reporting bias (Morris 2008; Tongyoo 2016; Tsangaris 2007; Willson 2015). Morris 2008 declared that some data were held by the sponsors who had chosen not to publish the data because of negative results. We were concerned about discrepancies between the clinical trials registration documents and the published completed study reports in Tongyoo 2016 and Willson 2015, In Tsangaris 2007, outcome data were only reported briefly, and some adverse events listed in the Methods section of the study report were omitted from the Results section.
Remaining studies did not report pre‐published protocols or report registration with clinical trials registers, and it was therefore not feasible to assess risk of reporting bias.
Baseline characteristics
We noted some differences baseline characteristics in five studies, which we rated at high risk of bias because differences in these characteristics could influence outcome data (BALTI 2006; HARP‐2 2014; KARE 2017; Khan 2017; START 2018). We also noted differences in the baseline characteristics of six studies, and rated them at unclear risk of bias because we were uncertain whether these characteristics could influence outcome data (Krenn 2017; Meduri 2007; Paine 2012; Spragg 2003; STRIVE 2004; Tsangaris 2007).
Seven studies did not provide sufficient information on baseline characteristics to allow a judgement (Endo 2006; Kesecioglu 2001; Morris 2008; Spragg 2002a; Spragg 2002b; Walmrath 2000; Wirtz 2017).
We judged the remaining studies to have a low risk of bias because baseline characteristics were comparable between groups.
Other potential sources of bias
Five studies were abstracts and, because of insufficient information, it was not possible to assess whether other risks of bias were present (Kesecioglu 2001; Spragg 2002a; Spragg 2002b; Walmrath 2000; Wirtz 2017).
In addition, we judged four studies to have unclear risks of other bias: two did not report details of standard therapy control (Endo 2006; Guoshou 2013), and two studies excluded a large number of participants before randomization (Barrese‐Perez 2015; Morris 2008).
We noted no other potential sources of bias in the remaining studies, and judged these to have low risk of bias.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5
Corticosteroids versus control
Primary outcome
Early all‐cause mortality
Six studies reported early mortality for 574 participants (Liu 2012; Meduri 2007; Rezk 2013; Steinberg 2006; Tongyoo 2016; Zhao 2014). We included data collected at the latest time point which was: day 14 (Rezk 2013); day 28 (Liu 2012; Zhao 2014); day 60 (Steinberg 2006; Tongyoo 2016); and in hospital (Meduri 2007).
Corticosteroids may reduce the number of deaths from any cause within three months by 86 per 1000 patients (with as many as 161 fewer or 19 more deaths). However, we note that the 95% confidence interval (CI) includes the possibility of both increased and reduced deaths (risk ratio (RR) 0.77, 95% CI 0.57 to 1.05; I2 = 27%; low‐certainty evidence; Analysis 1.1). We used GRADE to downgrade the certainty of the evidence by two levels; one level for study limitations (risks of bias were uncertain or high amongst studies) and one level for imprecision (evidence was from few studies with few participants). See Table 1,
1.1. Analysis.
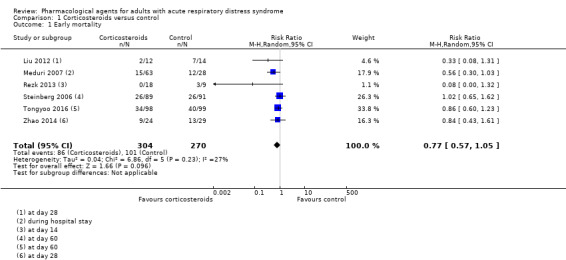
Comparison 1 Corticosteroids versus control, Outcome 1 Early mortality.
Secondary outcomes
Late all‐cause mortality
One study with 180 participants reported mortality at 180 days (Steinberg 2006); this was assessed as a post hoc analysis. We found no evidence of a difference in the number of deaths at 180 days between corticosteroids and the control group in this study (RR 0.99, 95% CI 0.64 to 1.52; very low‐certainty evidence; Table 11). We used GRADE to downgrade the certainty of the evidence by one level for study limitations and by two levels for imprecision; we were unable to assess risk of reporting bias because of retrospective clinical trial registration and analysis was completed post hoc, with evidence from one study with few participants. See Table 1.
1. Main comparisons: single‐study outcome data.
| Corticosteroids versus placebo or standard care: late all‐cause mortality | |||
| Study ID | Intervention group, n/N | Control group, n/N | Effect estimate* |
| Steinberg 2006 | 28/89 | 29/91 | RR 0.99 (95% CI 0.64 to 1.52) |
| Surfactants versus control: late all‐cause mortality | |||
| Study ID | Intervention group, n/N | Control group, n/N | Effect estimate* |
| Kesecioglu 2009 | 96/208 | 76/210 | RR 1.28 (95% CI 1.01 to 1.61) |
| Surfactants versus control: duration of mechanical ventilation | |||
| Study ID | Intervention group, mean (SD)/N | Control group, mean (SD)/N | Effect estimate* |
| Tsangaris 2007 | 5.6 days (± 2.6 days)/8 | 8.1 days (± 2.4 days)/8 | MD −2.50 (95% CI −4.95 to −0.05) |
| N‐acetylcysteine versus placebo: early all‐cause mortality | |||
| Study ID | Intervention group, n/N | Control group, n/N | Effect estimate* |
| Ortolani 2000 | 9/24 | 7/12 | RR 0.64 (95% CI 0.32 to 1.30) |
| Statins versus placebo: duration of mechanical ventilation | |||
| Study ID | Intervention group, mean (SD)/N | Control group, mean (SD)/N | Effect estimate* |
| HARP 2011 | 18.6 (± 14.6) days/30 | 15.9 (± 9.6) days/30 | MD 2.70 (95% CI ‐3.55 to 8.95) |
*effect estimate calculated by review authors using the calculator in RevMan 5 (Review Manager 2014).
CI: confidence interval; MD: mean difference: n: number of participants with an event; N: number of participants in the group; RR: risk ratio; SD: standard deviation
Duration of mechanical ventilation
Four studies reported duration of mechanical ventilation for 368 participants (Tongyoo 2016; Zhao 2014; Meduri 2007; Rezk 2013). Meduri 2007 reported median values which we could not combine in analysis; study authors reported a shorter duration of mechanical ventilation in the corticosteroids group (median 5 days (interquartile range (IQR) 3 to 8 days) compared to the control group (median 9.5 days (IQR 6 to 9.5 days): P = 0.002).
In the remaining three studies, we found no evidence of a difference between groups in duration of mechanical ventilation (MD −4.30 days, 95% CI −9.72 to 1.12; I2 = 93%; 277 participants; very low‐certainty evidence; Analysis 1.2). We note substantial statistical heterogeneity in this effect estimate which we could not explain by methodological or clinical differences between the studies, although we expected that differences may not be evident because of poor reporting. We note that the study with the fewest participants demonstrated shorter duration of ventilation in the corticosteroids group and this was not consistent with the data from the other studies, but this study reported few details and had a high risk of selection bias. We used GRADE to downgrade the certainty of the evidence by three levels; one level for study limitations (risks of bias were uncertain or high amongst studies), one level for inconsistency (evidence of substantial statistical heterogeneity) and one level for imprecision (evidence was from few studies with few participants). See Table 1.
1.2. Analysis.
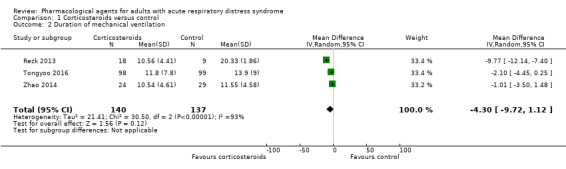
Comparison 1 Corticosteroids versus control, Outcome 2 Duration of mechanical ventilation.
Ventilator‐free days up to day 28
Four studies reported the number of ventilator‐free days up to day 28 for 494 participants (Liu 2012; Meduri 2007; Steinberg 2006; Tongyoo 2016). We found that corticosteroids may increase ventilator‐free days (MD 4.09 days, 95% CI 1.74 to 6.44; I2 = 36%; low‐certainty evidence; Analysis 1.3). We used GRADE to downgrade the evidence by one level for imprecision (evidence was from few studies with few participants) and by one level for inconsistency (we note a wide confidence interval in the effect estimate). See Table 1.
1.3. Analysis.
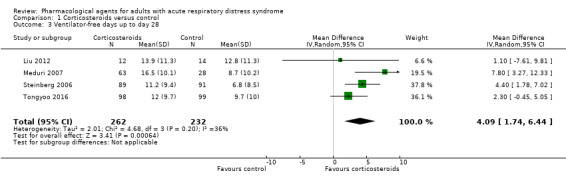
Comparison 1 Corticosteroids versus control, Outcome 3 Ventilator‐free days up to day 28.
Adverse events
No studies reported adverse events defined as leading to discontinuation of study medication (or in the standard care group (Zhao 2014), defined as "serious adverse events").
Fitness to return to work
No studies measured or reported fitness to return to work at 12 months.
Subgroup analysis
We did not perform subgroup analysis on the outcomes because of insufficient studies.
Severity of ARDS: mean values for PaO2/FiO2 suggested that participants in two studies had moderate to severe ARDS (Meduri 2007; Steinberg 2006), and in one study had moderate ARDS (Tongyoo 2016); the remaining studies reported insufficient information to assess severity.
Time since onset of ARDS: one study included participants who had ARDS that had been established for at least seven days (Steinberg 2006); the remaining studies had an onset of within 12 hours (Tongyoo 2016), within 48 hours (Rezk 2013), and within 72 hours (Liu 2012; Meduri 2007). This information was not reported in Zhao 2014.
Sensitivity analysis
Lower tidal volumes: two studies did not report tidal volumes (Rezk 2013; Zhao 2014) and we excluded these studies in analysis of early all‐cause mortality, but this did not alter our interpretation of the effect. We noted that two studies reported a change in clinical management during the study period and that subsequently some participants were managed with higher tidal volumes (Meduri 2007; Steinberg 2006). We performed an additional sensitivity analysis and excluded these studies and those in which tidal volumes were not reported; analysis with the remaining two studies did not alter our interpretation of the effect.
Selection bias: we excluded one study at high risk of selection bias for sequence generation (Rezk 2013) and two studies with an unclear risk of selection bias (Liu 2012; Zhao 2014). This did not alter our interpretation of the effect.
Attrition bias: we judged no studies to have high risk of attrition bias.
Alternative meta‐analytic effects model: we found that the effect showed a slight reduction in all‐cause mortality when we applied the fixed‐effect model (RR 0.78, 95% CI 0.62 to 0.99).
Surfactants versus control
Primary outcome
Early all‐cause mortality
Nine studies reported early mortality for 1338 participants (Barrese‐Perez 2015; Kesecioglu 2001; Kesecioglu 2009; Spragg 2002a; Spragg 2002b; Spragg 2003; Tsangaris 2007; Walmrath 2000; Willson 2015). One study measured mortality data at 90 days (Willson 2015), and four studies measured mortality at 28 days (Kesecioglu 2001; Kesecioglu 2009; Spragg 2003; Tsangaris 2007). The remaining studies did not report a time point for data collection and we have included these data as early mortality.
We could not be certain whether using surfactants affected early all‐cause mortality (RR 1.08, 95% CI 0.91 to 1.29; I2 = 5%; very low‐certainty evidence; Analysis 2.1). We used GRADE to downgrade the certainty of the evidence by three levels; two levels for study limitations (studies comparing surfactants with standard therapy were all at high risk of performance bias, and we noted other risks of bias that were high or unclear amongst studies), and one level for inconsistency (we noted some differences in data between studies and we found too few studies to explore these differences through subgroup analyses). See Table 2,
2.1. Analysis.
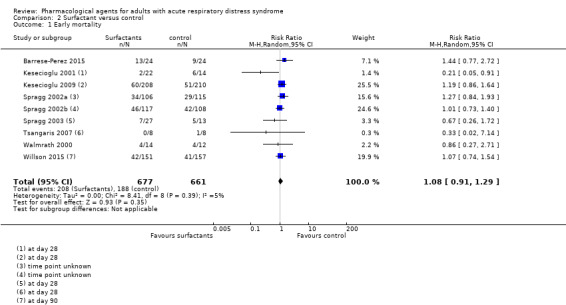
Comparison 2 Surfactant versus control, Outcome 1 Early mortality.
Secondary outcomes
Late all‐cause mortality
One study reported late all‐cause mortality for 418 participants at 180 days (Kesecioglu 2009). We calculated effect estimates using the Review Manager 5 calculator (Review Manager 2014); we could not be certain whether surfactants reduced late all‐cause mortality (RR 1.28, 95% CI 1.01 to 1.61; very low‐certainty evidence; Table 11). We used GRADE to downgrade the certainty of the evidence by three levels: two levels for study limitations (study was at high and unclear risks of bias), and one level for imprecision (evidence was from a single study). See Table 2,
Duration of mechanical ventilation
Two studies reported duration of mechanical ventilation (Barrese‐Perez 2015; Tsangaris 2007). For Tsangaris 2007, we calculated effect estimates using the Review Manager 5 calculator (Review Manager 2014), and found that duration of mechanical ventilation was shorter for participants who were given surfactants (MD −2.50, 95% CI −4.95 to −0.05; very low‐certainty evidence; Table 11). We could not combine data for Barrese‐Perez 2015 in analysis because it was unclear if data were reported as mean of median values; in this study, authors reported little or no difference between groups in duration of mechanical ventilation (14 days in the surfactant group and 16.63 days in the control group: P = 0.36). We used GRADE to downgrade the certainty of the evidence by three levels; one level for study limitations (studies were at high risk of performance bias), one level for inconsistency (we noted differences in data between studies), and one level for imprecision (evidence was from two studies with few participants). See Table 2,
Ventilator‐free days up to day 28
Six studies reported the number of ventilator‐free days up to day 28 for 856 participants (Kesecioglu 2001; Spragg 2002a; Spragg 2002b; Spragg 2003; Walmrath 2000; Willson 2015). We found little or no difference between participants given surfactants or a control for this outcome in two studies (Kesecioglu 2001; Willson 2015) (MD −0.39, 95% CI −2.49 to 1.72; I2 = 0%; 344 participants; very low‐certainty evidence; Analysis 2.2).
2.2. Analysis.
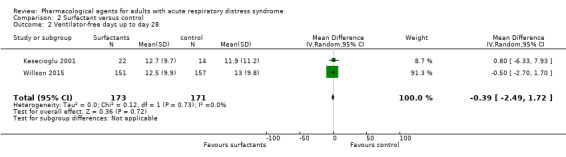
Comparison 2 Surfactant versus control, Outcome 2 Ventilator‐free days up to day 28.
We could not combine data for the remaining four studies; Spragg 2003 reported that differences between groups were "not significant" (high dose surfactant group: median 5 days (IQR 0 to 18) days; low dose surfactant group: median 4 days (IQR 0 to 12 days); control group: median 6 days (IQR 0 to 15 days)), two studies reported that ventilator‐free days were "not different between the groups" (Spragg 2002a; Spragg 2002b), and Walmrath 2000 reported that ventilator‐free days "were improved" in the intervention group with a mean of 10.9 days compared to the control group with a mean of 1.8 days. We used GRADE to downgrade the certainty of the evidence by three levels; two levels for study limitations (studies comparing surfactants with standard therapy were all at high risk of performance bias, and we noted other risks of bias that were high or unclear amongst studies), and one level for inconsistency (we noted differences in data between studies). See Table 2,
Adverse events
Two studies reported adverse events, defined as discontinuation of study medication (Barrese‐Perez 2015; Spragg 2003). We could not be certain whether surfactants had increased adverse events leading to discontinuation of study medication (RR 0.50, 95% CI 0.17 to 1.44; 88 participants; very low‐certainty evidence; Analysis 2.3). We used GRADE to downgrade the certainty of the evidence by three levels; two levels for study limitations (studies comparing surfactants with standard therapy were at high risk of performance bias, and we noted other risks of bias that were high or unclear amongst studies), and one level for imprecision (evidence was from two studies with few participants). See Table 2,
2.3. Analysis.
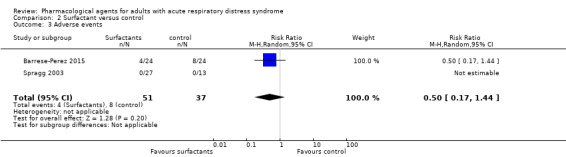
Comparison 2 Surfactant versus control, Outcome 3 Adverse events.
Fitness to return to work at 12 months
No studies measured or reported fitness to return to work at 12 months.
Subgroup analysis
We did not perform subgroup analysis on the outcomes because of insufficient studies.
Severity of ARDS: mean values for PaO2/FiO2 suggested that participants in three studies had moderate ARDS (Kesecioglu 2001; Kesecioglu 2009; Spragg 2003), whilst in one study mean values for PaO2/FiO2 suggested that participants had moderate to severe ARDS (Tsangaris 2007). Five studies reported insufficient information for us to judge severity (Barrese‐Perez 2015; Spragg 2002a; Spragg 2002b; Walmrath 2000; Willson 2015).
Time since onset of ARDS: participants were recruited within 24 hours of onset (Barrese‐Perez 2015); within 48 hours of onset (Spragg 2003; Tsangaris 2007; Willson 2015); and within 60 hours of onset (Kesecioglu 2009). This information was not reported in four studies (Kesecioglu 2001; Spragg 2002a; Spragg 2002b; Walmrath 2000).
Sensitivity analysis
Lower tidal volumes: only three studies reported use of lower tidal volumes (Kesecioglu 2009; Tsangaris 2007; Willson 2015); we excluded the remaining studies from analysis of early all‐cause mortality and this did not alter our interpretation of the effect.
Selection bias: we excluded five studies with unclear risk of selection bias for sequence generation from analysis of early all‐cause mortality (Kesecioglu 2001; Spragg 2002a; Spragg 2002b; Spragg 2003; Walmrath 2000); this did not alter our interpretation of the effect.
Attrition bias: we excluded three studies that were at high risk of attrition bias (Barrese‐Perez 2015; Kesecioglu 2009; Willson 2015), and one study for which attrition bias was unclear (Walmrath 2000), from analysis of early all‐cause mortality; this did not alter our interpretation of the effect.
Alternative meta‐analytic effects model: using a fixed‐effect model did not alter our interpretation of the effect for early all‐cause mortality.
Multi‐arm studies: Spragg 2003 reported data for two intervention groups (high dose and low dose) and we combined data from both groups in the primary analysis. In sensitivity analysis for early all‐cause mortality, we used data for the high‐dose group versus control, and data for the low‐dose group versus control. This did not alter our interpretation of the effect.
N‐acetylcysteine versus control
Primary outcome
Early all‐cause mortality
Only one study reported early all‐cause mortality at 30 days for 36 participants (Ortolani 2000). We calculated effect estimates using the Review Manager 5 calculator (Review Manager 2014) and found no evidence of a difference between groups in early all‐cause mortality (RR 0.64, 95% CI 0.32 to 1.30; very low‐certainty evidence; Table 11). We used GRADE to downgrade the certainty of the evidence by three levels; one level for study limitations because the study was unblinded and at high risk of performance bias, and two levels for imprecision because the evidence was from one study with few participants. See Table 3.
Secondary outcomes
Late all‐cause mortality
No studies measured or reported late all‐cause mortality.
Duration of mechanical ventilation
No studies measured or reported duration of mechanical ventilation.
Ventilator‐free days up to day 28
No studies measured or reported the number of ventilator‐free days up to day 28.
Adverse events
No studies reported adverse events defined as leading to discontinuation of study medication.
Fitness to return to work at 12 months
No studies measured or reported fitness to return to work at 12 months.
Subgroup analysis
We did not perform subgroup analysis on the outcomes because of insufficient studies.
Severity of ARDS: inclusion criteria in this study required that all participants had PaO2/FiO2 ≤ 200 mmHg (or ≤ 250 mmHg if positive‐end expiratory pressure was at least 10 cmH2O).
Time of ARDS onset: participants were recruited within 24 hours of onset.
Sensitivity analysis
This study was a multi‐arm study. In a sensitivity analysis, we analysed data separately for the N‐acetylcysteine group and the N‐acetylcysteine‐with‐rutin group. This did not alter our interpretation of the effect. We did not perform other sensitivity analyses because we included only one study in the primary analysis. Ortolani 2000 did not report whether they used lower tidal volumes. This study had an unclear risk of selection bias, and a low risk of attrition bias.
Statins versus control
Primary outcome
Early all‐cause mortality
Three studies reported early all‐cause mortality for 1344 participants (HARP 2011; HARP‐2 2014; SAILS 2014). Outcomes were reported at 28 days (HARP‐2 2014), at 60 days (SAILS 2014), and during hospital stay (HARP 2011).
Using statins probably makes little or no difference to early all‐cause mortality (RR 0.99, 95% CI 0.78 to 1.26; I2 = 31%; moderate‐certainty evidence; Analysis 3.1). We used GRADE to downgrade the certainty of the evidence by one level for inconsistency; we noted some differences in data between studies, but found too few studies to explore these differences through subgroup analyses. See Table 4,
3.1. Analysis.
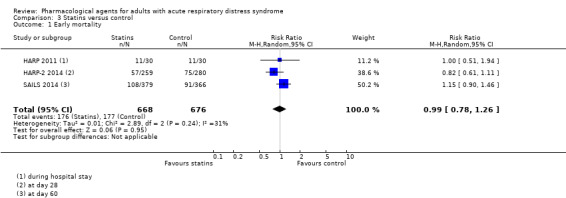
Comparison 3 Statins versus control, Outcome 1 Early mortality.
Secondary outcomes
Late all‐cause mortality
No studies measured or reported late all‐cause mortality.
Duration of mechanical ventilation
One study reported duration of mechanical ventilation for 60 participants (HARP 2011). We calculated effect estimates using the Review Manager 5 calculator (Review Manager 2014), and found that statins may make little or no difference to duration of ventilation (MD 2.70 days, 95% CI −3.55 to 8.95; low‐certainty evidence; Table 11). We used GRADE to downgrade the certainty of the evidence by two levels for imprecision; evidence for this outcome was from one study with few participants. See Table 4,
Ventilator‐free days up to day 28
Three studies reported the number of ventilator‐free days up to day 28 for 1342 participants (HARP 2011; HARP‐2 2014; SAILS 2014). We found that statins probably make little or no difference to the number of ventilator‐free days up to day 28 (MD 0.40 days, 95% CI −0.71 to 1.52; I2 = 0%; moderate‐certainty evidence; Analysis 3.2). We used GRADE to downgrade the certainty of the evidence by one level for inconsistency; we noted some differences in data between studies, but found too few studies to conduct subgroup analyses to explore these differences. See Table 4,
3.2. Analysis.
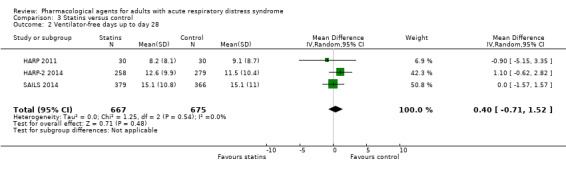
Comparison 3 Statins versus control, Outcome 2 Ventilator‐free days up to day 28.
Adverse events
One study reported that "the most common reasons for discontinuation of the study drug were discharge from critical care, death, and an adverse event that was considered to be related to the study drug" (HARP‐2 2014). No additional details are provided in the study report or the online supplementary appendix to indicate how many participants discontinued study treatment because of an adverse event. We were therefore unable to report data for this study.
Fitness to return to work at 12 months
No studies measured or reported fitness to return to work at 12 months.
Subgroup analysis
We did not perform subgroup analysis on the outcomes because of insufficient studies.
Severity of ARDS: mean values for PaO2/FiO2 suggested that participants in all studies had moderate ARDS.
Time of ARDS onset: all studies included participants with an onset of ARDS within 48 hours.
Sensitivity analysis
Lower tidal volumes: higher mean tidal volumes were reported in one study (HARP 2011); we excluded this from analysis, which did not alter our interpretation of the effect.
Selection bias: we did not conduct sensitivity analysis on selection bias because all studies included in the analysis of early all‐cause mortality had a low risk of bias.
Attrition bias: we did not conduct sensitivity analysis on attrition bias because all studies included in the analysis of early all‐case mortality had a low risk of bias.
Alternative meta‐analytic effects model: use of a fixed‐effect model did not alter our interpretation of the effect for early all‐cause mortality.
Beta‐agonists versus control
Primary outcome
Early all‐cause mortality
Three studies reported all‐cause mortality for 646 participants (ALTA 2011; BALTI 2006; BALTI‐2 2013). Outcomes were reported at: 60 days (ALTA 2011), 28 days (BALTI 2006), and during hospital stay (BALTI‐2 2013).
Beta‐agonists probably slightly increase early all‐cause mortality by 40 per 1000 patients (with as many as 119 more or 25 fewer deaths); however, the 95% CI includes the possibility of an increase as well as a reduction in mortality (RR 1.14, 95% CI 0.91 to 1.42; I2 = 0%; moderate‐certainty evidence; Analysis 4.1). We used GRADE to downgrade the certainty of the evidence by one level for imprecision because we found few studies with few participants. See Table 5,
4.1. Analysis.
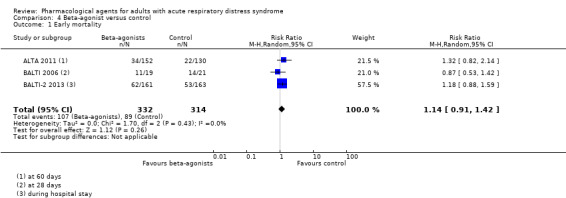
Comparison 4 Beta‐agonist versus control, Outcome 1 Early mortality.
Secondary outcomes
Late all‐cause mortality
No studies measured or reported late all‐cause mortality.
Duration of mechanical ventilation
No studies measured or reported duration of mechanical ventilation.
Ventilator‐free days up to day 28
Three studies reported the number of ventilator‐free days up to day 28 for 646 participants (ALTA 2011; BALTI 2006; BALTI‐2 2013). Although the results of the analysis showed more ventilator‐free days in the control group, this result was uncertain (MD −2.20, 95% CI −3.68 to −0.71; I2 = 0%; very low‐certainty evidence; Analysis 4.2). We used GRADE to downgrade the certainty of the evidence by three levels; one level for imprecision (evidence was from few studies with few participants), and two levels for inconsistency (inspection of data showed differences in direction of effect between studies which we could not explain). See Table 5,
4.2. Analysis.
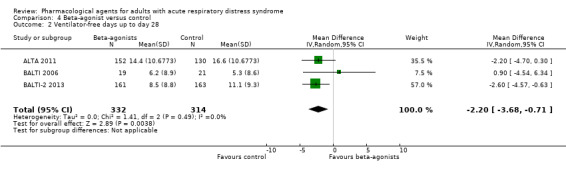
Comparison 4 Beta‐agonist versus control, Outcome 2 Ventilator‐free days up to day 28.
Adverse events
Two studies reported adverse events for 606 participants leading to discontinuation of study medication (ALTA 2011; BALTI‐2 2013). We did not pool data in this analysis because we noted differences in effects between studies which we expected to be caused by differences in the types of adverse events measured and reported by study authors. In ALTA 2011, study authors reported little or no difference between groups in adverse events (which were not described by study authors); we calculated effect estimates using the Review Manager 5 calculator (MD 0.73 days, 95% CI 0.25 to 2.13) (Review Manager 2014). In BALTI‐2 2013, adverse events sufficient to stop treatment were tachycardia, new arrhythmias, and new lactic acidosis; we calculated effect estimates using the Review Manager 5 calculator, which showed an increase in events for participants who were given beta‐agonists (MD 9.52 days, 95% CI 3.89 to 23.31) (Review Manager 2014). These unpooled data are reported in Analysis 4.3.
4.3. Analysis.
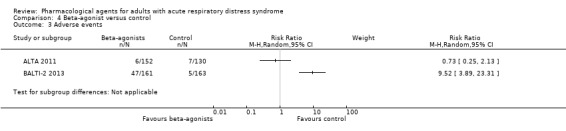
Comparison 4 Beta‐agonist versus control, Outcome 3 Adverse events.
We used GRADE to downgrade the evidence by three levels to very low certainty; one level for imprecision (evidence was from few studies with few participants), and two levels for inconsistency (inspection of data showed differences in direction of effect between studies, and a high level of statistical heterogeneity, which may have been caused by differences in types of adverse events measured by study authors). See Table 5.
Fitness to return to work at 12 months
No studies measured or reported fitness to return to work at 12 months.
Subgroup analysis
We did not perform subgroup analysis on the outcomes because of insufficient studies.
Severity of ARDS: mean values for PaO2/FiO2 suggested that participants in all studies had moderate or moderate‐to‐severe ARDS.
Time of ARDS onset: studies reported onset within 48 hours (BALTI 2006), and within 72 hours (BALTI‐2 2013). Time of onset was not reported in ALTA 2011.
Sensitivity analysis
Lower tidal volumes: only one study did not report use of lower tidal volumes (BALTI 2006); analysis without this study did not alter our interpretation of the effect.
Selection bias: we excluded one study from analysis of early all‐cause mortality because risk of selection bias was unclear (BALTI 2006); this did not alter our interpretation of the effect.
Attrition bias: we did not conduct sensitivity analysis on attrition bias because all studies included in the analysis of early all‐case mortality had a low risk of bias.
Alternative meta‐analytic effects model: use of a fixed‐effect model did not alter our interpretation of the effect for early all‐cause mortality.
Other pharmacological agents versus control
Primary outcome
Early all‐cause mortality
We calculated effect estimates using the Review Manager 5 calculator (Review Manager 2014) for early all‐cause mortality reported in the remaining studies of pharmacological agents in which early all‐cause mortality was measured and reported (Analysis 5.1).
5.1. Analysis.
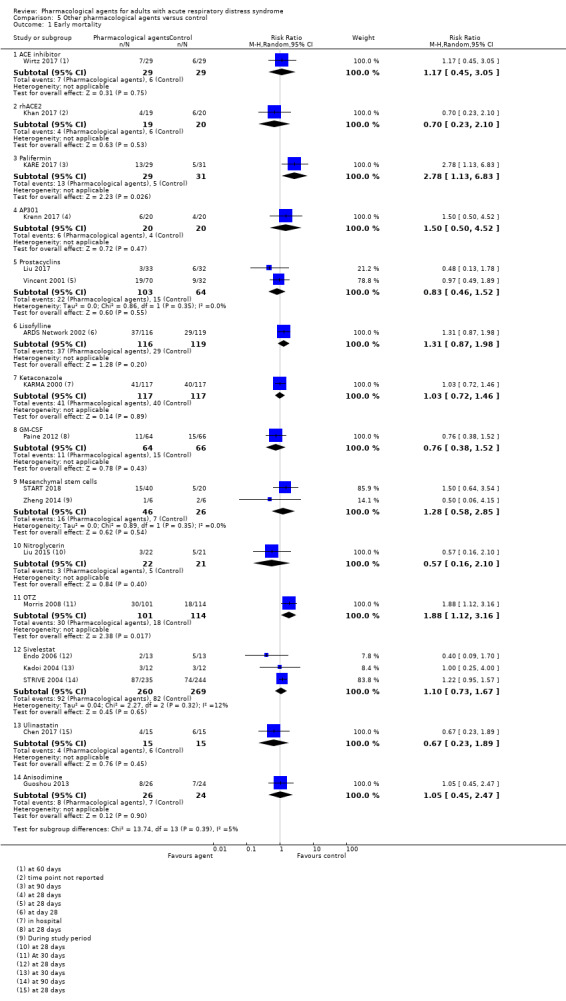
Comparison 5 Other pharmacological agents versus control, Outcome 1 Early mortality.
ACE inhibitor (Wirtz 2017): we found little or no difference between groups in early all‐cause mortality (RR 1.17, 95% CI 0.45 to 3.05; 58 participants)
rhACE2 (Khan 2017): we found little or no difference between groups in early all‐cause mortality (RR 0.70, 95% CI 0.23 to 2.10; 39 participants)
Palifermin (KARE 2017): we found more deaths in the Palifermin group (RR 2.78, 95% CI 1.13 to 6.83; 60 participants); study authors noted that mortality in the placebo group (< 10%) was lower than expected
AP301 (Krenn 2017): we found little or no difference between groups in early all‐cause mortality (RR 1.50, 95% CI 0.50 to 4.52; 40 participants)
Prostacyclins (Liu 2017; Vincent 2001): we found little or no difference between groups in early all‐cause mortality (RR 0.83, 95% CI 0.46 to 1.52; I2 = 0%; 167 participants)
Lisofylline (ARDS Network 2002): we found little or no difference between groups in early all‐cause mortality (RR 1.31, 95% CI 0.87 to 1.98; 235 participants)
Ketaconazole (KARMA 2000): we found little or no difference between groups in early all‐cause mortality (RR 1.02, 95% CI 0.72 to 1.46; 234 participants)
GM‐CSF (Paine 2012): we found little or no difference between groups in early all‐cause mortality (RR 0.76, 95% CI 0.38 to 1.52; 130 participants)
Mensenchymal stem cells (START 2018; Zheng 2014): we found little or no difference between groups in early all‐cause mortality (RR 1.28, 95% CI 0.58 to 2.85; I2 = 0%; 72 participants; )
Nitroglycerin (Liu 2015): we found little or no difference between groups in early all‐cause mortality (RR 0.57, 95% CI 0.16 to 2.10; 43 participants)
OTZ (Morris 2008): we found that more participants died in the OTZ group (RR 1.88, 95% CI 1.12 to 3.16; 215 participants). Study authors noted that mortality in the intervention group (29%) was similar or lower than in other trials.
Sivelestat (Endo 2006; Kadoi 2004; STRIVE 2004): we found little or no difference between groups in early all‐cause mortality (RR 1.10, 95% CI 0.73 to 1.67; 529 participants; I2 = 12%)
Ulinastatin (Chen 2017): we found little or no difference between groups in early all‐cause mortality (RR 0.67, 95% CI 0.23 to 1.89; 30 participants)
Anisodimine (Guoshou 2013): we found little or no difference between groups in early all‐cause mortality (RR 1.05, 95% CI 0.45 to 2.47; 50 participants)
Secondary outcomes
Late all‐cause mortality
We calculated effect estimates using the Review Manager 5 calculator (Review Manager 2014) for late all‐cause mortality reported in the remaining studies of pharmacological agents (Analysis 5.2).
5.2. Analysis.
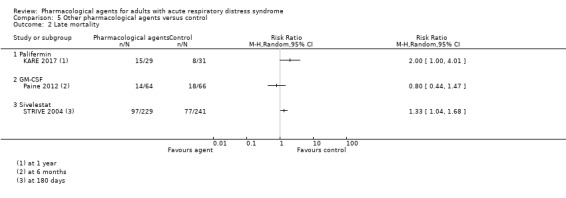
Comparison 5 Other pharmacological agents versus control, Outcome 2 Late mortality.
Palifermin (KARE 2017): we found fewer deaths from any cause at one year when participants where given Palifermin (RR 2.00, 95% CI 1.00 to 4.01; 60 participants)
GM‐CSF (Paine 2012): we found little or no difference between groups in all‐cause mortality at six months (RR 0.80, 95% CI 0.44 to 1.47; 130 participants)
Sivelestat (STRIVE 2004): we found fewer deaths from any cause at 180 days when participants were given sivelestat (RR 1.33, 95% CI 1.04 to 1.68; 470 participants)
Duration of mechanical ventilation
We calculated effect estimates where possible, using the Review Manager 5 calculator (Review Manager 2014) for duration of mechanical ventilation reported in the remaining studies of pharmacological agents (Analysis 5.3).
5.3. Analysis.
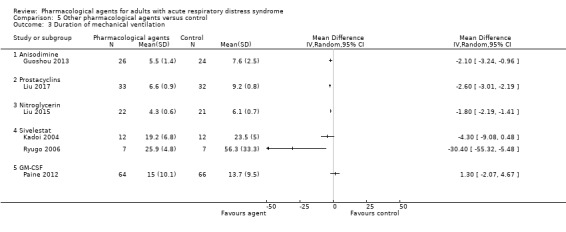
Comparison 5 Other pharmacological agents versus control, Outcome 3 Duration of mechanical ventilation.
Ulinastatin (Chen 2017; Ji 2018): we could not combine data from these studies in analysis because the unit of measure differed. In Chen 2017, we found little of no difference between studies in the duration of mechanical ventilation (MD −33.82 hours, 95% CI −70.76 to 3.12; 30 participants). However, in Ji 2018, we found that duration of mechanical ventilation was shorter for participants given ulinastatin (MD −1.70 days, 05% CI −1.80 to −1.60; 80 participants).
Anisodimine (Guoshou 2013): we found that duration of mechanical ventilation was shorter for participants given anisodimine (MD −2.10 days, 95% CI −3.24 to −0.96; 50 participants).
Prostacyclins (Liu 2017; Vincent 2001): we could not combine data from these studies in analysis because values were reported differently. In Liu 2017, we found that duration of mechanical ventilation was shorter when alprostadil was given (MD −2.60 days, 95% CI −3.01 to −2.19; 65 participants). In Vincent 2001, we could not calculate effect estimates for this study because it was unclear if reported data were mean or median values; study authors reported little or no difference between groups in duration of mechanical ventilation (intervention group 16 days, control group 16.6 days: P = 0.94).
Palifermin (KARE 2017): we could not calculate effect estimates for this study because data were reported as median values; study authors reported a shorter duration of mechanical ventilation in the intervention group (median (IQR) 6 days (13 to 30 days)) compared to the control group (median (IQR) 11 days (8 to 16 days): P = 0.002).
Nitroglycerin (Huang 2017; Liu 2015): we could not combine data from these studies in analysis because values were reported differently. In Huang 2017, data were reported in a graph which we could not clearly interpret; study authors reported shorter duration of ventilation in the intervention group (P < 0.05). In Liu 2015, we found a shorter duration of mechanical ventilation in the intervention group (MD −1.80 days, 95% CI −2.19 to −1.41; 43 participants).
Sivelestat (Endo 2006; Kadoi 2004; Ryugo 2006): data were not available in a format that we could include in Endo 2006, and in the two remaining studies, the units of measurement differed, so that we could not combine data in analysis. In Kadoi 2004, we found little of no difference between groups in the duration of mechanical ventilation (MD −4.30 days, 95% CI −9.08 to 0.48; 24 participants). In Ryugo 2006, we found duration of mechanical ventilation was shorter in participants given sivelestat (MD −30.40 days, 95% CI −55.32 to −5.48; 14 participants).
GM‐CSF (Paine 2012): we found little or no difference between groups in the duration of mechanical ventilation (MD 1.30 days, 95% CI −2.07 to 4.67; 130 participants).
MSCs (START 2018): we could not calculate effect estimates for this study because data were reported as median values; study authors reported little or no difference in duration of mechanical ventilation in participants who survived up to day 28 (intervention group: median 12 days (IQR 4 to 24 days); control group: median 8 days (IQR 4 to 15): P = 0.51).
Number of ventilator‐free days up to day 28
We calculated effect estimates using the Review Manager 5 calculator (Review Manager 2014) for the number of ventilator‐free days up to day 28 reported in the remaining studies of pharmacological agents not included in our primary comparisons (Analysis 5.4).
5.4. Analysis.
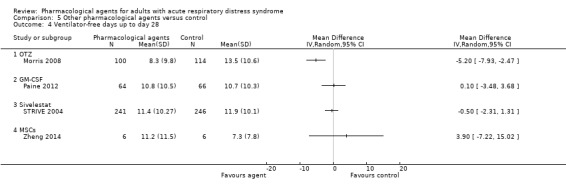
Comparison 5 Other pharmacological agents versus control, Outcome 4 Ventilator‐free days up to day 28.
OTZ (Morris 2008): we found fewer ventilator‐free days up to day 28 when OTZ was used (MD −5.20 days, 95% CI −7.93 to −2.47; 214 participants).
GM‐CSF (Paine 2012): we found little or no difference between groups in the number of ventilator‐free days up to day 28 (MD 0.10 days, 95% CI −3.48 to 3.68; 130 participants).
Palifermin (KARE 2017): we could not calculate effect estimates because data were reported only as median values; study authors reported more ventilator‐free days in the control group (median 20 days (IQR 13 to 22 days) compared to the intervention group (median 1 day (IQR 0 to 17 days)), (P < 0.001).
AP301 (Krenn 2017): we could not calculate effect estimates because data were reported only as median values; study authors reported little or no difference between groups (intervention group: median 15 days (IQR 9 to 21 days); control group: median 12 days (IQR 0 to 20 days): P = 0.22).
ACE inhibitor (Wirtz 2017): study authors reported an effect estimate which showed little or no difference between groups (MD 3.7 days, 95% CI −1.8 to 9.1; 58 participants; P = 0.18).
Sivelestat (STRIVE 2004): we found little or no difference between groups (MD −0.50 days, 95% CI −2.31 to 1.31; 487 participants).
MSCs (Zheng 2014; START 2018): we could not calculate effect estimates for one study because these were reported as median values; study authors reported little or no difference between groups (intervention group: median 2 days (IQR 0 to 56 days); control group: median 17 days (IQR) 0 to 24 days): P = 0.28) (START 2018). In Zheng 2014, we found little or no difference between groups (MD 3.90 days, 95% CI −7.22 to 15.02; 12 participants).
Ketoconazole (KARMA 2000): we could not calculate effect estimates because data were reported only as median values; study authors reported little or no difference between groups (intervention group: 10 days; control group: 9 days:P = 0.89).
Lisofylline (ARDS Network 2002): we could not calculate effect estimates because data were reported only as median values; study authors reported little or no difference between groups (intervention group: 9 days; control group: 11 days: P = 0.62).
Adverse events
Lisofylline (ARDS Network 2002); study authors reported little or no difference in discontinuation of study treatment (P = 0.59).
Anisodimine (Guoshou 2013); study authors reported that no participants discontinued treatment due to adverse events.
Ketoconazole (KARMA 2000); study authors reported that four participants in the ketoconazole group, and one participant in the control group had an adverse event leading to an incomplete course of treatment.
OTZ (Morris 2008): study authors reported that five participants in each group had an adverse event leading to discontinuation of treatment.
Fitness to return to work at 12 months
No studies measured or reported fitness to return to work at 12 months.
Discussion
Summary of main results
We included 48 studies of pharmacological agents in adults with ARDS. We also identified three studies awaiting classification (one is reported only as an abstract and two are completed trials; we await publication of full reports for these three studies), and 18 ongoing studies.
Overall, we found 20 different types of agents. Agents in our primary comparisons were: corticosteroids (seven studies; 643 participants); surfactants (nine studies; 1340 participants); N‐acetylcysteine (three studies; 86 participants); statins (three studies; 1345 participants); and beta‐agonists (three studies; 648 participants).
We found low‐certainty evidence that corticosteroids may reduce early all‐cause mortality (up to three months), although we note that the 95% confidence interval (CI) includes the possibility of both an increase and a decrease in the number of deaths. We are uncertain whether corticosteroids make little or no difference to late all‐cause mortality (after three months) or to the duration of mechanical ventilation; the certainty of the evidence was very low for these outcomes. We found low‐certainty evidence that ventilator‐free days up to day 28 may be improved with corticosteroids.
We are uncertain whether surfactants make little or no difference to early all‐cause mortality, or whether they reduce late all‐cause mortality. Similarly, we are uncertain whether surfactants reduce the duration of mechanical ventilation, make little or no difference to the number of ventilator‐free days up to day 28, or to adverse events leading to discontinuation of study medication. The certainty of the evidence for these outcomes with surfactants was very low.
Only one study reported outcome data for N‐acetylcysteine. We are uncertain whether N‐acetylcysteine makes little or no difference to early all‐cause mortality; the certainty of the evidence was very low. This study did not report other outcomes relevant to the review.
We found moderate‐certainty evidence that statins probably make little or no difference to early all‐cause mortality or to ventilator‐free days up to day 28. Statins may make little or no difference to the duration of mechanical ventilation; the certainty of this evidence was low. We could not include data for adverse events leading to discontinuation of study medication in one study, because it was unclearly reported.
We found moderate‐certainty evidence that beta‐agonists probably slightly increase early all‐cause mortality, although we noted that the 95% CI includes the possibility of both an increase and a decrease in the number of deaths. In addition, we are uncertain whether beta‐agonists increase ventilator‐free days up to day 28, or whether they make little or no difference to adverse events leading to discontinuation of study medication, because the evidence for these outcomes was of very low certainty.
In the primary comparisons, few studies reported adverse events leading to discontinuation of study medication, and no studies reported fitness to return to work at 12 months.
We did not assess the certainty of the evidence in comparisons of other agents in which outcome data were available (ACE inhibitor, rhACE2, AP301, palifermin, prostacyclins, lisofylline, ketaconazole, GM‐CSF, mesenchymal stem cells, nitroglycerin, OTZ, sivelestat, ulinastatin, and anisodimine). Evidence from most of these agents was from single studies with few participants. In summary, one study of palifermin and one study of OTZ found an increase in early all‐cause mortality when participants were given the intervention agent, but this may be explained by differences between participants in study groups. We found no evidence of a difference between each remaining agent and control groups in early all‐cause mortality. Study authors reported lower late all‐cause mortality with palifermin and with sivelestat. Mechanical ventilation was reduced for participants given palifermin and anisodimine, and in one study for participants given nitroglycerin; the remaining studies reported little or no difference in mechanical ventilation, or, for agents with more than one study, reported a difference in findings between studies. For ventilator‐free days, we noted an improvement with OTZ, and fewer ventilator‐free days with palifermin, whilst studies of other agents reported little or no difference between groups. We noted an increase in adverse events leading to discontinuation of treatment for use of ketoconazole. None of these studies reported fitness to return to work at 12 months.
Overall completeness and applicability of evidence
We identified 48 studies with 6299 participants. Despite a large number of included studies, the variety of identified pharmacological agents was broad, and we therefore found few studies for each type of agent. We found 10 agents that were reported only in single studies.
Participants all had ARDS, and from information within the study reports we expected that most participants in most studies had moderate or moderate‐to‐severe ARDS. We excluded studies published prior to 2000, with the intention of increasing the applicability of the evidence to current ICU practices. However, because of insufficient information in study reports we were unable to assess effectively if clinical practices were comparable between groups. Differences in clinical strategies may relate to the use of lower tidal volumes (ARDS Network 2000), or therapies such as fluid management strategies, values of positive end‐expiratory pressure (PEEP), or use of prone positioning. In addition, we could not account for the other pharmacological agents that may have been given to participants based on their primary pathologies.
Because of insufficient studies for each outcome, we were unable to use subgroup analysis to explore the potential effect of differences between studies, in particular the severity of ARDs in participants, and whether interventions had been initiated early (within three days of ARDS onset) or later.
Quality of the evidence
We used GRADE to assess the certainty of the evidence for our main comparisons. For corticosteroids, the certainty of the evidence was low or very low. Evidence for most outcomes included studies with high or unclear risks of bias, and we downgraded for study limitations. In addition, we found few studies with few participants, leading to imprecision. For the duration of mechanical ventilation, we downgraded for inconsistency because we noted substantial statistical heterogeneity.
For surfactants, we judged the certainty of the evidence for all outcomes to be very low. Most evidence was from studies in which the comparison was standard care, and subsequently these studies were at high risk of performance bias, as well as other high or unclear sources of bias. Again, we downgraded for imprecision where evidence was from few studies with few participants, and for inconsistency when we noted unexplained differences between study data.
Evidence for N‐acetylcysteine was from one small study with a high risk of performance bias, and we judged the evidence to be of very low certainty.
For statins, although the evidence for early all‐cause mortality and ventilator‐free days was from only three studies, the sample size was large. However, we were unable to explore any differences in data between studies for these outcomes and we downgraded because of apparent inconsistencies that we noted. Evidence for late mortality was from a smaller sample size and our certainty in the evidence was reduced because of imprecision.
Evidence for beta‐agonists was from few studies and few participants, and the evidence was therefore imprecise. In addition, we noted inconsistencies between some study results and this reduced our certainty in the evidence for these outcomes to very low.
Potential biases in the review process
We conducted a thorough search and used two review authors to assess study eligibility, extract data, and assess risks of bias in the included studies, thereby reducing potential bias in the review process.
This review is an update of a previous version (Adhikari 2004). During the updating process, we made changes to the review to meet current Cochrane standards, which included a more comprehensive assessment of risks of bias and use of GRADE to assess the certainty of the evidence. We also made changes to the review inclusion criteria by excluding studies published prior to 2000. As a consequence of this decision, we excluded 23 studies that had previously been included. Whilst we expected that clinical management in these studies was probably not consistent with current practices, we could not be certain of this. Indeed, despite this decision we still found that several studies did not report sufficient methods for clinical management, and even if they did report them we did not know whether practitioners were compliant with the strategies. We used sensitivity analysis in an attempt to explore the effect of not using lower tidal volumes. In the event of information missing from study reports, we did not attempt to contact authors for clarifications, because the review was completed using funding within a timescale that prohibited this, and this may subsequently have introduced bias.
We added one more outcome during this update ('Fitness to return to work at 12 months'); this attempted to evaluate the long‐term effects for ARDS survivors, although we found no studies reporting it. We did not make other changes to the existing outcomes. In particular, we used the existing definition of adverse events (i.e. leading to the discontinuation of study medication) and did not explore other adverse events relevant to particular agents which were reported by study authors. This review therefore does not evaluate a comprehensive set of potential harms of study agents. We reported when study authors collected adverse events (by study authors' definitions) and when studies were stopped early in Characteristics of included studies. Similarly, this review did not look at outcomes that were specific to included agents (for example, improvements measured using ventilatory parameters, blood gas analysis, or cardiovascular parameters). It is feasible that an agent may not make a difference to mortality, but may improve other symptoms of the condition.
Agreements and disagreements with other studies or reviews
The evidence for corticosteroids in ARDS management was evaluated in the recent Intensive Care Society Guidelines (FICM/ICS Guideline Development Group 2018). Evidence for this agent included studies published prior to changes in lung protection strategies, and similarly reported low‐certainty evidence of little or no difference in mortality. We identified two recently published systematic reviews of corticosteroids (Horita 2015; Yang 2017); these reported no evidence of a difference in mortality, and an improvement in mortality with corticosteroids, respectively. However, evidence for these reviews included studies that were not RCTs, and studies in which the participant population included people without ARDS.
Our evidence for mortality with surfactants, N‐acetylcysteine, statins, and beta‐agonists was also consistent with results in other recently published systematic reviews (Feng 2018; Gao 2016; Meng 2012; Nagendran 2017; Singh 2014; Zhang 2013; Zhang 2017).
Authors' conclusions
Implications for practice.
In our main comparison groups, we found low‐certainty evidence that corticosteroids may reduce mortality within three months of the onset of ARDS, and moderate‐certainty evidence that beta‐agonists probably slightly increase mortality within three months of the onset of ARDS; however, we noted that the 95% confidence interval for these effects suggested both an increase and a reduction in the number of deaths. For surfactants, N‐acetylcysteine and statins, we found no evidence of a difference in early mortality. Only two studies of these agents (one that assessed steroids, and one surfactants) reported deaths later than three months, but evidence for this was uncertain.
In addition, we found that statins may make little or no difference to the duration of mechanical ventilation, and, whilst the evidence was uncertain, we also found little or no difference when steroids were given. Similarly, we were uncertain whether surfactants reduced mechanical ventilation. We found that steroids may increase ventilator‐free days, and we also found a similar effect for beta‐agonists (although the evidence for beta‐agonists was uncertain). We found that statins probably make little or no difference to the number of ventilator‐free days, which we also found for surfactants (although, again, we were uncertain about the evidence for surfactants). Only surfactants and beta‐agonists reported adverse event leading to discontinuation of the study medication, and we were uncertain of any effect. No studies reported fitness to return to work at 12 months.
We describe evidence for some outcomes as uncertain because, during the GRADE assessment, we found the evidence for these outcomes to be of very low certainty.
Evidence from these comparisons is from 25 studies with 4062 participants with a diagnosis of ARDS.
We did not assess the certainty of evidence for the remaining pharmacological agents. The evidence does not include data from three studies awaiting classification, and inclusion of these studies (and ongoing studies) in future updates may increase our certainty in the evidence.
Implications for research.
Research continues in the field of ARDS management, which is reflected in the large number of ongoing studies identified during our search. The outcomes in this review reflect the most important measures of treatment effectiveness in this life‐threatening condition, and we expect that this will be measured and reported in future studies. However, given the importance of the potential long‐term consequences of ARDS to its survivors, we propose that research incorporates a longer follow‐up to measure the impacts on quality of life. We propose that studies ensure that current guidance for lung protection strategies are followed consistently by practitioners, and that compliance with these strategies is clearly reported by study authors.
What's new
| Date | Event | Description |
|---|---|---|
| 30 September 2019 | Amended | Minor typos corrected |
History
Protocol first published: Issue 4, 2003 Review first published: Issue 4, 2004
| Date | Event | Description |
|---|---|---|
| 6 February 2019 | New search has been performed |
|
| 6 February 2019 | New citation required but conclusions have not changed | New studies, with different pharmacological agents, included in the review update. Overall conclusions continue to show little or no difference in outcomes, with uncertainty in the effects |
| 13 December 2018 | Amended | Editorial team changed to Cochrane Emergency and Critical Care |
| 9 June 2010 | Amended | Contact details updated. |
| 2 August 2008 | Amended | Converted to new review format. |
Acknowledgements
We thank the previous review author team (Neill Adhikari, Karen Burns, Maureen Meade, and Mohana Ratnapalan) for their contributions to the previous version of this review (Adhikari 2004). We thank Dr Henry HL Wu for completing data extraction on studies written in Chinese. We would like to thank Nicola Petrucci (Content Editor), Nathan Pace (Statistical Editor), Lowell Ling and Andrew MacDuff (Peer Reviewers), Janet Wale (Consumer Editor), Janne Vendt (Cochrane Information Specialist), Jane Cracknell and Teo Quay (Managing Editors), and Harald Herkner (Co‐ordinating Editor) for their help and editorial advice during the preparation of this systematic review.
Appendices
Appendix 1. CENTRAL search strategy
"acute respiratory distress syndrome":ti,ab,kw
(acute or adult) and (respiratory near distress):ti,ab,kw
acute lung injury:ti,ab,kw
(acute near lung near injur*) or (shock near lung):ti,ab,kw
#1 or #2 or #3 or #4
#5 in Trials
Appendix 2. MEDLINE search strategy
exp Respiratory Distress Syndrome, Adult/
(((acute or adult) and (respiratory adj1 distress)) or ards).mp.
exp Acute Lung Injury/
((acute adj1 lung* adj1 injur*) or (shock adj1 lung*)).mp.
1 or 2 or 3 or 4
((randomized controlled trial or controlled clinical trial).pt. or random*.ab. or placebo.ab. or clinical trials as topic.sh. or trial.ti.) not (exp animals/ not humans.sh.)
5 and 6
Appendix 3. Embase search strategy
exp adult respiratory distress syndrome/
(((acute or adult) and (respiratory adj1 distress)) or ards).mp.
exp acute lung injury/
((acute adj1 lung* adj1 injur*) or (shock adj1 lung*)).mp.
1 or 2 or 3 or 4
(Acyclovir or Albumin* or Anisodomine* or Beta‐agonist* or Corticosteroid* or Dazoxiben or Granulocyte‐macrophage colony‐stimulating factor* or Indomethacin* or Interleukin‐10 or Ketoconazole or Levosimendan or Lisofylline or L02‐oxothiazolidine‐4‐carboxylic acid or Mesenchymal stem cell* or N‐acetylcysteine or procysteine or Neutrophil elastase inhibitor* or Penehyclidine hydrochloride or Pentoxifylline or Prostaglandin E1 or Sivelestat or Statin* or Surfactant* or Xuebijing or drug* or pharmacological agent*).mp.
aciclovir/ or albumin/ or exp beta adrenergic receptor stimulating agent/ or exp corticosteroid/ or dazoxiben/ or granulocyte macrophage colony stimulating factor/ or indometacin/ or interleukin 10/ or ketoconazole/ or levosimendan/ or lisofylline/ or exp mesenchymal stem cell/ or acetylcysteine/ or 2 oxo 4 thiazolidinecarboxylic acid/ or exp leukocyte elastase inhibitor/ or exp cholinergic receptor blocking agent/ or pentoxifylline/ or prostaglandin E1/ or exp hydroxymethylglutaryl coenzyme A reductase inhibitor/ or sivelestat/ or exp surfactant/ or exp drug/ or (drug administration or drug therapy).fs.
6 or 7
5 and 8
(randomized controlled trial/ or randomization/ or placebo/ or crossover procedure/ or double blind procedure/ or single blind procedure/ or (crossover* or cross over*).ti,ab. or (doubl* adj blind*).ti,ab. or (controlled adj3 (study or design or trial)).ti,ab. or (placebo* or allocat* or trial* or random* or groups).ti,ab.) not ((exp animal/ or animal.hw. or nonhuman/) not (exp human/ or human cell/ or (human or humans).ti,ab.))
9 and 10
Appendix 4. CINAHL search strategy
S1 (MH "Respiratory Distress Syndrome, Acute")
S2 TX acute respiratory distress syndrome
S3 TX ards
S4 (MH "Acute Lung Injury")
S5 TX (acute n3 lung n3 injur*) OR TX (shock n3 lung)
S6 S1 OR S2 OR S3 OR S4 OR S5
S7 (MH "Clinical Trials+")
S8 PT Clinical trial
S9 TX clinic* n1 trial*
S10 TX ((singl* n1 blind*) or (singl* n1 mask*)) or TX ((doubl* n1 blind*) or (doubl* n1 mask*)) or TX ((tripl* n1 blind*) or (tripl* n1 mask*)) or TX ((trebl* n1 blind*) or (trebl* n1 mask*))
S11 TX randomi* control* trial*
S12 (MH "Random Assignment")
S13 TX random* allocat*
S14 TX placebo*
S15 (MH "Placebos")
S16 (MH "Quantitative Studies")
S17 TX allocat* random*
S18 S7 OR S8 OR S9 OR S10 OR S11 OR S12 OR S13 OR S14 OR S15 OR S16 OR S17
S19 S6 AND S18
Appendix 5. Summary of study characteristics: primary comparisons
| Corticosteroids versus control | |||||
| Study ID | Comparison | Dose | Time of ARDS onset |
Baseline PaO2/PaO2 Mean (SD) |
Lower tidal volumes |
| Liu 2012 | Hydrocortisone vs placebo |
100 mg IV; 3 times daily for 7 days |
< 72 h | NR | Yes |
| Meduri 2007 | Methylprednisolone vs placebo |
1mg/kg loading dose; 240 mL normal saline at 10 mL/h of 1 mg/kg/day daily infusion for 14 days; halving of doses on day 15, 22 and 26, up to day 28 |
< 72 h | I: 118.4 (± 51.2) mmHg C: 125.9 (± 38.6) mmHg |
Yes* |
| Rezk 2013 | Methylprednisolone vs placebo |
1 mg/kg loading dose; 240 mL normal saline at 10 mL/h of 1 mg/kg/day daily infusion for 14 days; halving of doses on day 15, 22 and 26, up to day 28 |
< 48 h | NR | NR |
| Steinberg 2006 | Methylprednisolone vs placebo |
2 mg/kg loading dose; 50 mL of 5% dextrose in water of 0.5 mg/kg; daily infusion every 6 h for 14 days; followed by 0.5 mg/kg every 12 h for 7 days, then tapering of dose over 4 days |
7 to 28 days | I: 126 (± 42) mmHg C:126 (± 40) mmHg |
Yes* |
| Tongyoo 2016 | Hydrocortisone vs placebo |
50 mg in 10 mL normal saline, IV bolus; every 6 h for 7 days |
< 12 h | I: 175.4 (± 6.9) mmHg C: 172.4 (± 6.7) mmHg |
Yes |
| Zhao 2014 | Budesonide vs standard care |
2 mg, inhaled; twice a day for 12 days |
NR | NR | Yes |
| Surfactants versus control | |||||
| Study ID | Comparison | Dose | Time of ARDS onset |
Baseline PaO2/FiO2 Mean (SD) |
Lower tidal volumes |
| Barrese‐Perez 2015 | Surfacen vs standard therapy |
100 mg; every 8 h for 3 days |
< 24 h | NR | NR |
| Kesecioglu 2001 | HL‐10 vs standard therapy |
100 ‐ 200 mg/kg of phospholipids; up to 4 doses |
NR | I: 181 (± 91) mmHg C: 175 (± 72) mmHg |
NR |
| Kesecioglu 2009 | HL10 vs standard therapy |
100 mL vials of 3 mg HL10 in 60 mL warm saline; 3 boluses at 0 h, 12 h, and 36 h |
< 60 h | I: 156.7 (± 54.8) mmHg C: 161.4 (± 55.2) mmHg |
Yes |
| Spragg 2002a | Venticute vs placebo |
1 mL/kg; up to 4 doses in 12 to 24 h |
NR | NR | NR |
| Spragg 2002b | Venticute vs placebo |
1 mL/kg; up to 4 doses in 12 to 24 h |
NR | NR | NR |
| Spragg 2003 | venticute (high dose) vs venticute (low dose) vs standard therapy |
High dose: 1 mL/kg; up to 4 doses in 24 h Low dose: 0.5 mL/kg; up to 4 doses in 24 h |
< 48 h | I (high): 133.6 (± 8.9) mmHg C (low): 113.9 (± 8.3) mmHg C: 120.9 (± 6.5) mmHg |
NR |
| Tsangaris 2007 | Alveofact vs standard therapy |
(200/19) mg/kg body weight | < 48 h | I: 100 (± 20) mmHg C: 103 (± 14) mmHg |
Yes |
| Walmrath 2000 | Venticute vs standard therapy |
1 mL/kg; up to 4 doses in 24 h |
NR | NR | NR |
| Willson 2015 | Pneumasurf vs placebo |
30 mg/cm of height; up to 3 doses, 12 h apart |
< 48 h | NR | Yes |
| N‐acetylcysteine versus control | |||||
| Study ID | Comparison | Dose | Time of ARDS onset |
Baseline PaO2/FiO2 mean (SD) |
Lower tidal volumes |
| Ortolani 2000 | NAC vs NAC + rutin vs placebo |
50 mg/kg NAC (+ 5 mg/kg rutin); IV every 8 h | < 24 h | NR | NR |
| Statins versus control | |||||
| Study ID | Comparison | Dose | Time of ARDS onset |
Baseline PaO2/FiO2 mean (SD) |
Lower tidal volumes |
| HARP 2011 | Simvastatin vs placebo |
80 mg; daily for 14 days |
< 48 h | I: 173 (± 47) mmHg C: 166 (± 60) mmHg |
No (mean tidal volumes, 8.5 mmHg) |
| HARP‐2 2014 | Simvastatin vs placebo |
80 mg; daily for up to 28 days |
< 48 h | I: 123 (± 54.8) mmHg C: 132.4 (± 55.4) mmHg |
Yes |
| SAILS 2014 | Rosuvastatin vs placebo |
40 mg loading dose; then 20 mg daily; for up to 28 days |
< 48 h | I: 170 (± 71) mmHg C: 170 (± 67) mmHg |
Yes |
| Beta‐agonists versus control | |||||
| Study ID | Comparison | Dose | Time of ARDS onset | Baseline PaO2/FiO2 mean (SD) | Lower tidal volumes |
| ALTA 2011 | Albuterol vs placebo |
5.0 mg in saline; aerosolized; very 4 h for up to 10 days |
NR | I: 170 (± 84) mmHg C: 170 (± 84) mmHg |
Yes |
| BALTI 2006 | Salbutamol vs placebo |
0.2 mg/mL; IV 0.075 mL/kg/h; for 7 days |
< 48 h | I: 15.6 (± 6.6) kPa C: 13.7 (± 4.9) kPa |
NR |
| BALTI‐2 2013 | Salbutamol vs placebo |
IV; 0.075 mL/kg/h; for 7 days |
< 72 h | I: 103.5 (± 36.75) mmHg I: 103.5 (± 36.75) mmHg |
Yes |
| Footnotes: *Lung protection strategies were altered part‐way through the study following a publication (ARDS Network 2000); participants recruited after this publication were managed with lower tidal volumes ARDS: acute respiratory distress syndrome; C: control group; I: intervention group; IV: intravenous; NAC: N‐acetylcysteine; NR: not reported; PaO2/FiO2: ratio of partial pressure of arterial oxygen to fraction of inspired oxygen; SD: standard deviation | |||||
Data and analyses
Comparison 1. Corticosteroids versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Early mortality | 6 | 574 | Risk Ratio (M‐H, Random, 95% CI) | 0.77 [0.57, 1.05] |
| 2 Duration of mechanical ventilation | 3 | 277 | Mean Difference (IV, Random, 95% CI) | ‐4.30 [‐9.72, 1.12] |
| 3 Ventilator‐free days up to day 28 | 4 | 494 | Mean Difference (IV, Random, 95% CI) | 4.09 [1.74, 6.44] |
Comparison 2. Surfactant versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Early mortality | 9 | 1338 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.91, 1.29] |
| 2 Ventilator‐free days up to day 28 | 2 | 344 | Mean Difference (IV, Random, 95% CI) | ‐0.39 [‐2.49, 1.72] |
| 3 Adverse events | 2 | 88 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.17, 1.44] |
Comparison 3. Statins versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Early mortality | 3 | 1344 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.78, 1.26] |
| 2 Ventilator‐free days up to day 28 | 3 | 1342 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐0.71, 1.52] |
Comparison 4. Beta‐agonist versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Early mortality | 3 | 646 | Risk Ratio (M‐H, Random, 95% CI) | 1.14 [0.91, 1.42] |
| 2 Ventilator‐free days up to day 28 | 3 | 646 | Mean Difference (IV, Random, 95% CI) | ‐2.20 [‐3.68, ‐0.71] |
| 3 Adverse events | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only |
Comparison 5. Other pharmacological agents versus control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Early mortality | 18 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 ACE inhibitor | 1 | 58 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.45, 3.05] |
| 1.2 rhACE2 | 1 | 39 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.23, 2.10] |
| 1.3 Palifermin | 1 | 60 | Risk Ratio (M‐H, Random, 95% CI) | 2.78 [1.13, 6.83] |
| 1.4 AP301 | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 1.5 [0.50, 4.52] |
| 1.5 Prostacyclins | 2 | 167 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.46, 1.52] |
| 1.6 Lisofylline | 1 | 235 | Risk Ratio (M‐H, Random, 95% CI) | 1.31 [0.87, 1.98] |
| 1.7 Ketaconazole | 1 | 234 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.72, 1.46] |
| 1.8 GM‐CSF | 1 | 130 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.38, 1.52] |
| 1.9 Mesenchymal stem cells | 2 | 72 | Risk Ratio (M‐H, Random, 95% CI) | 1.28 [0.58, 2.85] |
| 1.10 Nitroglycerin | 1 | 43 | Risk Ratio (M‐H, Random, 95% CI) | 0.57 [0.16, 2.10] |
| 1.11 OTZ | 1 | 215 | Risk Ratio (M‐H, Random, 95% CI) | 1.88 [1.12, 3.16] |
| 1.12 Sivelestat | 3 | 529 | Risk Ratio (M‐H, Random, 95% CI) | 1.10 [0.73, 1.67] |
| 1.13 Ulinastatin | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 0.67 [0.23, 1.89] |
| 1.14 Anisodimine | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.45, 2.47] |
| 2 Late mortality | 3 | Risk Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2.1 Palifermin | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.2 GM‐CSF | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 2.3 Sivelestat | 1 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3 Duration of mechanical ventilation | 6 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Anisodimine | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.2 Prostacyclins | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.3 Nitroglycerin | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.4 Sivelestat | 2 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 3.5 GM‐CSF | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Ventilator‐free days up to day 28 | 4 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4.1 OTZ | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.2 GM‐CSF | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.3 Sivelestat | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4.4 MSCs | 1 | Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
ALTA 2011.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 282 Inclusion criteria: patients had to be intubated and receiving mechanical ventilation; have bilateral pulmonary infiltrates consistent with oedema on frontal chest radiograph; have a ratio of PaO2/FiO2 ≤ 300 mmHg; no clinical evidence of left atrial hypertension Exclusion criteria: chronic lung disease; unable to obtain consent; time window exceeded; acute myocardial infarction; high risk of 6 month mortality; chronic liver disease; physician refusal; not committed to full support; neuromuscular disease. Details in on‐line supplement; mean exclusion of patients < 13 years of age Baseline characteristics Intervention group (beta‐agonist)
Control group (placebo)
Country: USA Setting: multicentre; 33 hospitals |
|
| Interventions |
Intervention group (beta‐agonist)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: number of ventilator‐free days (up to 28 days); mortality (at day 60 and day 90); number of ICU‐free days; number of organ failure‐free days; adverse events (to include events leading to discontinuation of study medication) Outcomes relevant to the review: mortality (day 90), ventilator‐free days up to day 28; adverse events (tachycardia sufficient to stop study treatment; new arrhythmia sufficient to stop study treatment; adverse events sufficient to stop study treatment) |
|
| Notes |
Funding/declarations of interest: supported by National Heart, Lung, and Blood Institute
Study dates: August 2007 to July 2008 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "A centralized web‐based system was used to randomize patients to receive aerosolized albuterol or placebo" |
| Allocation concealment (selection bias) | Low risk | External randomization, and we assumed allocation was concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant in each group withdrew after randomization but before drug administration. Small number unlikely to influence data. Study authors used an intention‐to‐treat analysis on all outcomes. We noted early stopping before intended recruitment targets; this decision was made by an independent DSMB |
| Selective outcome reporting (reporting bias) | Low risk | Prospective clinical trials registration (NCT00434993). Outcomes all reported |
| Baseline characteristics | Low risk | Largely comparable |
| Other sources of bias | Low risk | No other sources of bias identified |
ARDS Network 2002.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 235 Inclusion criteria: mechanical ventilation; AECC definition for ALI and ARDS (Bernard 1994); duration of ALI or ARDS < 36 hours Exclusion criteria: neurologic disease impairing weaning; chronic lung disease; morbid obesity; liver disease; immunocompromised; burns; increased intracranial pressure Baseline characteristics Intervention group (lisofylline)
Control group (saline)
Country: USA Setting: multicentre; ICUs at 21 hospitals at 10 centres constituting the ARDS Clinical Trials Network |
|
| Interventions |
Intervention group (lisofylline)
Control group (saline)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28); unassisted breathing for > 48 hours; organ failure‐free days; serious infections; infection‐related mortality Outcomes relevant to the review: mortality (at day 28); ventilator free‐days (to day 28); adverse events leading to discontinuation of treatment |
|
| Notes |
Funding/declarations of interest: in part, by National Institutes of Health/National Heart, Lung and Blood Institute Study dates: not reported Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | External computer‐generated randomization |
| Allocation concealment (selection bias) | Low risk | Study drugs were prepared in blinded kits with serial numbers. Randomization code for a 'kit' was held externally |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The local research pharmacist, patients, investigators, study co‐ordinators, and all clinical personnel were blinded to the randomization" Quote: "Lisofylline was packaged in individual blinded patient kits that had enough medication for the entire treatment course" |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Quote: "The local research pharmacist, patients, investigators, study co‐ordinators, and all clinical personnel were blinded to the randomization" |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | Quote: "The local research pharmacist, patients, investigators, study co‐ordinators, and all clinical personnel were blinded to the randomization" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Largely comparable |
| Other sources of bias | Low risk | Study included some participants who were managed with higher tidal volumes, but this was balanced between groups |
BALTI 2006.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 40 Inclusion criteria: mechanically ventilated adult patients; within 48 hours of onset of ALI or ARDS; defined according to AECC definition (Bernard 1994) Exclusion criteria: < 18 years of age; participation in another intervention trial; severe obstructive airway disease requiring nebulized or intravenous beta‐agonist; treatment with beta‐blockers within 48 hours; neutrophil count < 0.3 x 109 L; brainstem death; treatment withdrawal within 24 hours; immunosuppression; lobectomy/pneumonectomy; burns > 40% of body surface area; consent declined from next of kin Baseline characteristics Intervention group (beta‐agonist)
Control group (placebo)
Country: UK Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (beta‐agonist)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: extravascular lung water; LIS; PaO2/FiO2; plateau pressure (at day 7); ventilator‐free days up to day 28; mortality (at day 28); adverse events (safety and tolerability) Outcomes relevant to the review: mortality (at day 28); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declarations of interest: grant from West Midlands Intensive Care Society. "None of the authors have a financial relationship with a commercial entity that has an interest in the subject of this manuscript." Study dates: January 2001 to December 2003 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Randomization by blocks of four, conducted by one of the authors. Size of blocks unknown to other researchers, but method of sequence generation not described. |
| Allocation concealment (selection bias) | Low risk | Concealed in opaque, sequentially‐numbered, sealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Drug preparations made by nurses who were not involved in the study |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 participants did not receive treatment but included in ITT analysis |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | High risk | Participants in intervention group were older. More participants in placebo group had direct lung injury |
| Other sources of bias | Low risk | None identified |
BALTI‐2 2013.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 326 Inclusion criteria: intubated and mechanically ventilated; ≥16 years of age; within 72 hours of ARDS onset; ARDS according to AECC (Bernard 1994) Exclusion criteria: pregnancy; current treatment with intravenous beta‐2‐agonist or need for continuous, regular aerosolized beta‐2‐agonists; current treatment with beta‐adrenergic antagonists; imminent withdrawal of medical treatment; chronic liver disease; enrolment in another trial of an investigational medicinal product within previous 28 days Baseline characteristics Intervention group (beta‐agonists)
Control group (placebo)
Country: UK Setting: multicentre; 46 ICUs |
|
| Interventions |
Intervention group (beta‐agonists)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28); mortality in ICU or hospital; ventilator‐free and organ failure‐free days (up to day 28); length of stay in ICU and hospital; tachycardia; new arrhythmia; other side effects sufficient to stop treatment of trial drug Outcomes relevant to the review: mortality (at day 28, in ICU, in hospital); ventilator‐free days up to day 28; adverse events (tachycardia sufficient to stop study treatment; new arrhythmia sufficient to stop study treatment; new lactic acidosis sufficient to stop study treatment; serious adverse events related to study drug) |
|
| Notes |
Funding/declarations of interest: UK Medical Research Council, UK Department of Health, UK Intensive Care Foundation; some authors have received investigator‐led research grants, and fees for lectures etc. from pharmaceutical companies ‐ all unrelated to beta‐agonists. The sponsor of the study had no role in study design, data collection, data analysis, data interpretation, or writing of the report Study dates: December 2006 to March 2010 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "We used a computer‐generated randomisation sequence with a block size of eight" |
| Allocation concealment (selection bias) | Low risk | Use of external randomization service |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Participants, care providers, and investigators were masked to group assignment" |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | We assumed that outcome assessors were blinded |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | We assumed that outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 2 withdrawals, and 2 not given study drug but small number of losses and ITT analysis used. Trial was stopped early due to increased mortality in salbutamol group; decision made by Data Monitoring and Ethics Committee |
| Selective outcome reporting (reporting bias) | Low risk | Prospective clinical trials registration (ISRCTN38366450). All outcomes presented in full report |
| Baseline characteristics | Low risk | All comparable |
| Other sources of bias | Low risk | None identified |
Barrese‐Perez 2015.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 48 Inclusion criteria: 18 to 75 years of age; AECC diagnosis of ARDS (Bernard 1994) within 24 hours; PEEP > 5 cm H2O; consent from family Exclusion criteria: pregnancy; COPD; hypersensitivity to surfacen Baseline characteristics Intervention group (surfactants)
Control group (standard therapy)
Country: Cuba Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (surfactants)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: PaO2/FiO2; chest x‐ray changes; duration of mechanical ventilation; mortality; length of stay in ICU Outcomes relevant to the review: mortality (time point not reported); duration of mechanical ventilation; interruption of treatment (adverse events) |
|
| Notes |
Funding/declarations of interest: funding source not reported but 2 authors reported to be involved in a national clinical trial centre; these authors were not involved in participant recruitment etc., only in interpretation of the results Study dates: not reported Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Use of computer‐generated random‐number tables |
| Allocation concealment (selection bias) | Unclear risk | Sealed envelopes used but no additional details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding not possible as no placebo was used; comparison against standard treatment only |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect that blinding of outcome assessors would influence mortality outcome data |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | There were 25% of participants who did not complete treatment. Although these were included in ITT analysis, we noted that the number of participants who completed treatment was unbalanced between groups. |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Unclear risk | Relative small number of participants recruited from screening; unclear if this was due to exclusion criteria, other factors, or potential bias at recruitment stage |
Chen 2017.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 30 Inclusion criteria: with acute lung injury induced by severe heatstroke; fulfilling criteria listed in Society of Critical Care Medicine of PLAs 2015 Guideline for standardized diagnosis of heatstroke Exclusion criteria: < 16 years of age; hospital stay > 24 hours; brain stem death; COPD patients Baseline characteristics Intervention group (Ulinastatin)
Control group (standard therapy)
Country: China Setting: multicentre; 2 hospitals |
|
| Interventions |
Intervention group (ulinastatin)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: duration of mechanical ventilation; length of ICU stay; mortality (at 28 days); concentration of TNF‐alpha and IL‐6 in BALF and alveolar macrophage supernatant before and after treatment; expression levels of TREM‐1 protein and mRNA in alveolar macrophage after treatment Outcomes relevant to the review: duration of mechanical ventilation; mortality (at 28 days) |
|
| Notes |
Funding/declarations of interest: National Natural Science Youth Fund; Guangdong Province Medical Research Fund; Dongguan Social Science and Technology Project Fund Study dates: January 2013 to October 2016 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Investigators used computer randomization |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Control is standard therapy and therefore blinding not possible |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding of outcome assessors. However, we did not expect this to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Study authors did not report a pre‐published protocol or clinical trials registration number and therefore we were unable to assess risk of selective outcome reporting bias |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Endo 2006.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 26 Inclusion criteria: patients with sepsis who were diagnosed with SIRS as well as ALI Exclusion criteria: ≥ 76 years of age; < 16 years of age; presence of multiple organ dysfunction involving 4 organs or more; presence of underlying cancer; steroid administration Baseline characteristics Table of characteristics not reported; study authors report "no significant differences" Risk factor: indirect ‐ all participants had sepsis Country: Japan Setting: not stated |
|
| Interventions |
Intervention group (sivelestat)
Control group
|
|
| Outcomes |
Outcomes measured/reported: duration of mechanical ventilation and pulmonary oxygenation ability; mortality (at day 28); concentrations of PMN‐E, SP‐D, TRN‐a and IL‐8 in blood Outcomes relevant to the review: mortality (at day 28); duration of mechanical ventilation |
|
| Notes |
Funding/declarations of interest: Mutual Aid Corporation for Private School of Japan and the Ministry of Education, Culture, Sports, Science, and Technology of Japan Study dates: not reported Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Participants were described as "randomly assigned" but no additional details |
| Allocation concealment (selection bias) | Unclear risk | Assigned using the "envelope method". No additional details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details of what treatment the control group received and whether blinding was feasible or attempted |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect that blinding of outcome assessors would influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Unclear risk | Baseline characteristics table is referred to but not included in the full report. Study authors report that there were no significant differences observed between the groups in relation to any of the background characteristics |
| Other sources of bias | Unclear risk | Details of control group are not described which means that other risks of bias are unknown |
Guoshou 2013.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 50 Inclusion criteria: traumatic ALI/ARDS patients whose diagnoses were in accordance with the diagnostic criteria on ALI/ARDS of the Society of Critical Care Medicine of Chinese Medical Association (SCCMCMA 2006) Exclusion criteria: no details Baseline characteristics Intervention group (anisodimine)
Control (standard therapy)
Country: China Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (anisodimine)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: oxygenation and respiratory variables; mechanical ventilation time; mortality; adverse reactions Outcomes relevant to the review: duration of mechanical ventilation; mortality (time point not reported); adverse reactions (leading to termination of study treatment) |
|
| Notes |
Funding/declarations of interest: not reported Study dates: January 2009 to October 2011 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Participants were "randomly classified". No additional details. |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Compared with standard therapy, therefore not possible to blind personnel to treatment groups |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement. |
| Baseline characteristics | Low risk | Limited detail but appear balanced between groups |
| Other sources of bias | Unclear risk | Insufficient detail in paper regarding methodology. No details of 'conventional' therapy used as comparison. |
HARP 2011.
| Methods | RCT Parallel design |
|
| Participants |
Total numbers of randomized participants: 60 Inclusion criteria: mechanically‐ventilated patients within 48 hours of a diagnosis of ALI and ARDS according to AECC (Bernard 1994) Exclusion criteria: creatine kinase > 10 times upper limit normal range; liver transaminases > 3 times upper limit normal range; severe renal impairment not receiving RRT; severe liver disease; known lactose intolerance; current treatment with any lipid‐lowering agent including statins; contraindication to enteral drug administration; < 18 years of age; pregnancy; participation in a clinical trial with an investigational medicinal product within 30 days; unlikely to survive beyond 48 hours; declined consent Baseline characteristics Intervention group (statins)
Control group (placebo)
Country: Northern Ireland, UK Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (statins)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: reduction in extravascular lung water indexed to actual body weight (EVLWI); oxygenation index; plateau pressure; SOFA score; adverse reactions (not defined as leading to discontinuation of study medication); additional ventilation parameters; haemodynamic variables; need for renal replacement; VAP; duration of mechanical ventilation in ICU survivors; ventilator‐free days up to day 28; ICU and hospital length of stay; ICU and hospital survival Outcomes relevant to the review: mortality (in hospital), ventilator‐free days up to day 28 |
|
| Notes |
Funding/declarations of interest: supported by the HSC R&D Division, Public Health Agency Northern Ireland and REVIVE. 2 authors received fees from various pharmaceutical companies, full details reported in full text Study dates: September 2006 to March 2009 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "performed by an independent clinical trials statistician. The block size was unknown to the investigators. An independent clinical trials pharmacist performed treatment randomization." |
| Allocation concealment (selection bias) | Low risk | Randomization completed externally and drugs "encapsulated" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Blinding of the simvastatin and placebo tablets was achieved by encapsulation" |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Prospective clinical trials registration in March 2006 (ISRCTN70127774). However outcome section was edited in clinical trials register in April 2012 (after publication of full text). It is therefore not possible to judge whether a priori outcomes have been reported fully |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | No other sources of bias identified |
HARP‐2 2014.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 540 Inclusion criteria: intubated and mechanically ventilated and within 48 hours after onset of ARDS, defined as PaO2/FiO2 ≤ 300 mmHg; if bilateral pulmonary oedema was present on chest radiography; if there was no evidence of left atrial hypertension Exclusion criteria: < 16 years of age; not intubated and ventilated; assessed ≥ 48 hours after onset of ARDS; pregnant; elevated creatine kinase level; elevated aminotransferase level; interaction with concomitant drug; severe renal impairment and not receiving RRT; severe liver disease; received statins within previous 2 weeks; required statins for a proven indication; contraindication to enteral drug administration; enrolled in another drug trial; having treatment imminently withdrawn; declined consent Baseline characteristics Intervention group (statins)
Control group (placebo)
Country: UK and Ireland Setting: multicentre; 40 ICUs |
|
| Interventions |
Intervention group (statins)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: number of ventilator‐free days (up to day 28); change in oxygenation index and SOFA score; number of days free of non‐pulmonary organ failure; death from any cause (at 28 days); death before discharge from critical care or hospital; adverse events and serious adverse events related to the study drug Outcomes relevant to the review: mortality (at 28 days), ventilator‐free days up to day 28; adverse events (see note) |
|
| Notes |
Funding/declarations of interest: UK Efficacy and Mechanisim Evaluation Programme, a Medical Research Council and NIHR partnership. 3 authors in receipt of funding from pharmaceutical manufacturer. "Funders had no role in the study design, data acquisition, data analysis, or manuscript preparation." Study dates: December 2010 to March 2014 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Randomization performed using automated, centralized 24‐hour service |
| Allocation concealment (selection bias) | Low risk | External randomization, in which allocation can be concealed |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients received once‐daily simvastatin (at a dose of 80 mg) or identical placebo tablets enterally for up to 28 days." |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant lost to follow‐up in intervention group, and 2 participants withdrew consent in control group. Data reported with different denominator figures not always consistent with these losses, but very small number and unlikely to influence results |
| Selective outcome reporting (reporting bias) | Unclear risk | Prospective trial registration ISRCTN88244364. Some outcomes not reported in full publication as stated in trial registration document, to include mortality at 12 months |
| Baseline characteristics | High risk | Differences apparent in ratio of PaO2/FiO2 (less in simvastatin group), reported by authors to be statistically significant |
| Other sources of bias | Low risk | No other sources of bias identified |
Huang 2017.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 140 Inclusion criteria: acute onset respiratory frequency or respiration distress; hypoxaemia defined as PaO2 < 60 mmHg or FiO2 ≤ 200 mmHg; invasive shallows observed in both lungs; pulmonary artery wedge pressure ≤ 18 mmHg Exclusion criteria: cardiogenic pulmonary oedema induced by other causes; disorders in cardiac, hepatic, and renal function; or incomplete medical records Baseline characteristics Intervention group (propofol + nitroglycerin)
Control group (standard care)
Country: China Setting: single‐centre |
|
| Interventions |
Intervention group (propofol + nitroglycerin)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: levels of inflammatory markers; blood and gas analysis; duration of mechanical ventilation; disease remission Outcomes relevant to the review: duration of mechanical ventilation |
|
| Notes |
Funding/declarations of interest: not reported Study dates: January 2015 to January 2016 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | High risk | Pieces of paper (labelled as group A or group B) were folded and placed in a box. Participants selected a piece of paper to assign group allocation |
| Allocation concealment (selection bias) | High risk | Possible that allocation could be manipulated because of methods of randomization |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding not possible because control group is standard care |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | Unblinded trial |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported; it is therefore not feasible to assess reporting bias |
| Baseline characteristics | Low risk | Baseline characteristics appear comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Ji 2018.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 80 Inclusion criteria: with ARDS (diagnosed by clinical manifestations and pulmonary blood gas analysis) Exclusion criteria: acute heart failure; accompanied with hypovolaemic shock; other organ system failure; systemic immune system diseases; malignant tumour; estimated survival time was within 24 hours; refused to be enrolled in the study Baseline characteristics Intervention group (ulinastatin)
Control group (standard care)
Country: China Setting: single‐centre |
|
| Interventions |
Intervention group (ulinastatin)
Control group (standard care)
|
|
| Outcomes |
Outcomes measured/reported: changes in relevant indices of oxygen metabolism; lung function; time of ventilator treatment; total hospital stay; SGRQ score; changes in inflammatory cytokine levels; dopamine receptor‐related hormone levels; SOD; MDA; total antioxidant capacity (before intervention and at 1 and 4 weeks after intervention) Outcomes relevant to the review: duration of mechanical ventilation |
|
| Notes |
Funding/declarations of interest: not reported. Study authors report that they have no competing interests Study dates: January 2015 to December 2016 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "Patients were divided into the observation (n=40) and control (n=40) groups according to a random number table." |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Because study design does not include a placebo control agent, no blinding is possible |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | Outcome assessors are unblinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Study does not report clinical trials registration or published protocol and it is therefore not possible to assess risk of selective outcome reporting |
| Baseline characteristics | Low risk | Baseline characteristic appear to be comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Kadoi 2004.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 24 Inclusion criteria: > 20 years of age; ARDS according to AECC (Bernard 1994) within the preceding 72 hours; PaO2/FiO2 < 200 mmHg; bilateral CXR infiltrates; pulmonary artery occlusion pressure ratio ≤ 18 mmHg or no clinical evidence of left arterial hypertension; received immediately before randomization; FiO2 of ≥ 0.8 for ≥ 12 hours or FiO2 of ≥ 0.6 for ≥ 24 hours Exclusion criteria: history of immunosuppression; use of prednisone within last 30 days; chemotherapy or radiotherapy within the last 30 months; persistent hypotension; low survival expectancy because of underlying disease, such as malignancy, significant hepatic or renal failure; head trauma; pregnant Baseline characteristics Intervention group (sivelestat)
Control group (saline)
Country: Japan Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (sivelestat)
Control group (saline)
|
|
| Outcomes |
Outcomes measured/reported: ARDS during 30 days after intervention; duration of mechanical ventilation; oxygenation variables; cytokine levels; number of participants alive at 30 days who did not receive mechanical ventilation; mortality (at 30 days) Outcomes relevant to the review: mortality at 30 days; duration of mechanical ventilation |
|
| Notes |
Funding/declarations of interest: supported by a grant from the Japanese Ministry of Science, Education and Culture Study dates: October 2002 to May 2003 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "blocked randomization algorithm" |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Neither the patients nor the medical personnel responsible for their care were aware of which treatment was being administered" |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | None identified |
KARE 2017.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 60 Inclusion criteria: patients with ARDS according to AECC (Bernard 1994) Exclusion criteria: presence of ARDS for > 48 hours; current treatment with palifermin; known hypersensitivity to palifermin or Escherichia coli‐derived proteins (palifermin is produced in an E coli‐based protein production system, so hypersensitivity to E coli‐derived protein is an exclusion criterion); previous adverse reaction to palifermin; imminent withdrawal of medical treatment; chronic liver disease (Child‐Pugh score > 12); history of active malignancy; < 18 years of age; pregnancy; enrolment in another clinical trial of an investigational medicinal product within the previous 30 days; inability to obtain informed consent Baseline characteristics Intervention group (palifermin)
Control group (placebo)
Country: UK Setting: multicentre; 2 ICUs |
|
| Interventions |
Intervention group (palifermin)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: OI; physiological indices of pulmonary function; change in SOFA score; adverse events (but not reported as leading to discontinuation of study medication); ventilator‐free days (to day 28); duration of ventilation and ICU stay; hospital mortality; mortality (28 days, 90 days, 1 year) Outcomes relevant to the review: mortality (at 90 days); mortality (at ≥ 1 year); ventilator‐free days; duration of mechanical ventilation |
|
| Notes |
Funding/declarations of interest: The Northern Ireland Public Health Agency Research and Development Division. "The funder or Swedish Orphan Biovitrum AB [supplied palifermin] had no role in the study design, data collection, data analysis, data interpretation, or writing of the report." Authors declare no competing interests. Study dates: February 2011 to February 2014 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "The randomisation schedule was computer generated by the trial biostatistician" |
| Allocation concealment (selection bias) | Low risk | Quote: "An independent clinical trials pharmacist allocated the patient to the designated treatment group, and was the only person with access to the randomisation schedule" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote:"Patients and investigators were both masked to treatment" |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Quote: "Investigators were masked to treatment allocation" |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | Quote: "Investigators were masked to treatment allocation" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Low risk | Prospective clinical trials registration (ISRCTN95690673). Outcomes reported according to registration documents |
| Baseline characteristics | High risk | Some differences in baseline characteristics (higher PEEP and oxygenation index, and a lower PaO2/FiO2 in the placebo group). It is unclear whether this influenced results. Study authors also note difference in mortality data, in particular that the placebo group has fewer then average deaths which could justify the apparent larger number of deaths in the KGF group |
| Other sources of bias | Low risk | No source of bias identified |
KARMA 2000.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 234 Inclusion criteria: mechanical ventilation; AECC definition for ALI and ARDS (Bernard 1994); duration of ALI or ARDS < 36 hours Exclusion criteria: < 18 years of age; neurologic disease impairing weaning; chronic lung disease; morbid obesity; liver disease; immunocompromised; burns; increased intracranial pressure Additonal participant information: ALI and ARDS; study authors report the number of participants per group according to PaO2/FiO2 ratio; these were balanced between groups Baseline characteristics Intervention group (ketoconazole)
Control group (placebo)
Country: USA Setting: multicentre; ICUs in 24 hospitals associated with 10 network centres |
|
| Interventions |
Intervention group (ketoconazole)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: ventilator‐free days up to day 28; hospital mortality; unassisted breathing for ≥ 48 hours; organ failure‐free days; days meeting "commence weaning" criteria; liver toxicity; occurrence of barotrauma Outcomes relevant to the review: ventilator‐free days up to day 28; hospital mortality; adverse events (leading to discontinuation of treatment) |
|
| Notes |
Funding/declarations of interest: supported by National Institutes of Health/National Heart, Lung, and Blood Institute Study dates: March 1996 to January 1997 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "The data coordinating center provided assignment using a computer‐generated randomization" |
| Allocation concealment (selection bias) | Low risk | Group allocation was prepared externally. Also, study drugs were prepared externally by pharmacist to ensure concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The patients, investigators, study coordinators, and all clinical personnel remained blinded to the randomization" |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | We assumed outcome assessors were blinded |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | We assumed outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Large number of participants did not receive complete course (main reason due to hepatic injury), although balanced between groups. No participants were lost to follow‐up. We noted that the study was stopped early, but the decision was made by an independent DSMB |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | All comparable |
| Other sources of bias | Low risk | None identified |
Kesecioglu 2001.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 36 Inclusion criteria: patients with ALI/ARDS (not described) Exclusion criteria: no details Baseline Characteristics Intervention group (surfactant)
Control group (standard therapy)
Country: Europe (countries not reported in abstract) Setting: multicentre |
|
| Interventions |
Intervention group (surfactant)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: ventilator‐free days; mortality (at day 28) Outcomes relevant to the review: ventilator‐free days up to day 28; mortality (at day 28) |
|
| Notes |
Funding/declarations of interest: funded by Leo Pharmaceutical Products Study dates: not reported Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as "randomized" but no further details. Abstract only |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding not possible because control is standard therapy |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Unclear risk | No details |
| Other sources of bias | Unclear risk | Not possible to judge other risks of bias because of insufficient information in abstract report |
Kesecioglu 2009.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 418 Inclusion criteria: intubated and mechanically‐ventilated patients with a diagnosis of ALI/ARDS; < 60 hours from start of mechanical ventilation to first large bolus of surfactant; an expected requirement of mechanical ventilation of > 24 hours; ≥ 18 years of age Exclusion criteria: acute bronchial asthma attack or suspected pulmonary thrombo‐embolism; lung fibrosis; COPD; pneumonectomy or lobectomy; pneumothorax; tracheostomy; GCS ≤ 10 before sedation; life expectancy < 3 months due to primary disease; known or suspected hypersensitivity to investigational product Additional participant information: study authors report the number of participants per group according to P/F ratio; these were balanced between groups Baseline characteristics Intervention group (surfactant)
Control group (standard therapy)
Country: Austria, Belgium, Canada, Denmark, Finland, France, Germany, Netherlands, Norway, Spain, Sweden, UK Setting: multicentre; 67 medical units |
|
| Interventions |
Intervention group (surfactant)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28 and at day 180); days alive and out of ICU (at day 28); changes in PaO2/FiO2 ratio and other lung parameters; change in SOFA score; adverse events (these were not defined as "serious adverse events" or leading to discontinuation of study medication) Outcomes relevant to the review: mortality (at day 28 and day 180) |
|
| Notes |
Funding/declarations of interest: funded by LEO Pharma A/S. Sponsor had responsibility for trial design and interpretation of results Study dates: January 2003 to May 2004. Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Central telephone randomization system |
| Allocation concealment (selection bias) | Low risk | Quote: "To avoid allocation bias a central telephone randomization procedure was used" |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "The study could not be performed as a double‐blind trial because control patients could not safely have a placebo instilled" |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 3 participants did not receive treatment in HL‐10 group but analysed as ITT. There are some discrepancies in the number of deaths in the HL‐10 group. Figure 1 in the study report states 66 deaths, the text in study report states 60 participants. Study terminated early due to increased trend towards mortality in the intervention group at days 60 and days 90 (decision made by independent monitoring board). However, authors do not report data for these time points |
| Selective outcome reporting (reporting bias) | Unclear risk | Retrospective clinical trials registration (NCT00742482). Not possible to assess selective reporting bias against these reports |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | No other sources of bias identified |
Khan 2017.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 39 Inclusion criteria: between 18 and 80 years of age; either gender; diagnosed with ARDS within 48 hours of randomization that was associated with infection, sepsis, pneumonia, aspiration, or similar disease, based on AECC (Bernard 1994); haemodynamically stable in the 4 to 6 hours preceding initiation of treatment: stable pressor requirements; on mechanical ventilation for < 72 hours before dosing began; and were managed with low tidal volume mechanical ventilation Exclusion criteria: haemodynamically unstable; positive hepatitis B surface antigen, hepatitis C antibody or HIV antibody; liver disease, or known hepatic or biliary abnormalities; known history of substance abuse or alcohol abuse Baseline characteristics Intervention group (rhACE2)
Control group (placebo)
Country: USA and Canada Setting: multicentre; 10 ICUs |
|
| Interventions |
Intervention group (rhACE2)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality; clinical laboratory tests and immunogenicity; vital signs; electrocardiograms; adverse events (events possibly related to study drugs; not defined as leading to discontinuation of study medication) Outcomes relevant to the review: mortality (follow‐up in clinical trials register states follow‐up at 7 days) |
|
| Notes |
Funding/declarations of interest: GlaxoSmithKline. Study authors are employees of GlaxoSmithKline Study dates: September 2012 to October 2014 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Quote: "Subjects were randomized using a 1:1 allocation." Comment: No additional details. |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "double‐blind (sponsor unblinded) investigation". Comment: All personnel were blinded, except the sponsor. |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Only the sponsor was unblinded |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | Only the sponsor was unblinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Loss of participants was similar in each group and explained by study authors. However, losses were high (overall 25% loss). We noted that the study was stopped early for futility; it was not clear whether this decision was made by an independent monitoring board, and we noted that the study authors were employees of a pharmaceutical company |
| Selective outcome reporting (reporting bias) | Low risk | Study prospectively registered on clinical trials register (NCT01597635). Outcomes have been reported according to pre‐published registration documents |
| Baseline characteristics | High risk | Some differences in baseline characteristics between groups. Higher SOFA scores and lower P/F ratios in the rhACE2 group may influence outcome data |
| Other sources of bias | Low risk | No other sources of bias identified |
Krenn 2017.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 40 Inclusion criteria: patients ≥ 18 years of age with ARDS within 48 hours of diagnosis who required intubation and mechanical ventilation; ARDS diagnosed by AECC (Bernard 1994); had an EVLWI ≥ 8 mL/kg PBW; had a negative pregnancy test (for women of child‐bearing potential); and presented with stable haemodynamics for ≥ 8 hours Exclusion criteria: brainstem death; cardiogenic pulmonary oedema; current evidence of septic shock; neutrophil count < 0.3 × 109 L; immunosuppression (i.e. high‐dose steroids: prednisolone > 80 mg/day or hydrocortisone > 300 mg/day, cancer treatment including chemotherapy or biological or immunosuppressive therapy for organ transplantation within 2 weeks); BMI < 18.5 or > 35 kg/m2; active pregnancy; and participation in other interventional trials Baseline characteristics Intervention group (AP301)
Control group (placebo)
Country: Austria Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (AP301)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: EVLWI; ventilation parameters; blood gas analysis; changes in LIS; ventilator‐free days up to day 28; duration of mechanical ventilation; adverse events (tracheostomy, anaemia, worsening of existing anaemia, cardiac arrest, fever, thrombopenia, leucocytosis, atrial flutter/fibrillation, pleural effusion; none defined as leading to discontinuation of study medication); mortality (up to day 28) Outcomes relevant to the review: mortality (at day 28); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declarations of interest: funded by Apeptico GmbH (Vienna, Austria), Apeptico GmbH provided the study drug AP301 and all available information on the peptide AP301. Study authors reported receipt of a grant by Apeptico GmbH (Vienna, Austria) to their institution during the conduct of the study and outside the submitted work, and one author reports receipt of personal fees from this pharmaceutical company Study dates: August 2012 to February 2014 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | "The randomization method for both strata was block randomization using random computer‐generated permuted blocks with block sizes of one to three patients." |
| Allocation concealment (selection bias) | Low risk | "Randomization was performed using separate randomization lists for strata A and B that were prepared by Bioconsult GmbH (Breitenfurt, Austria) and known only to the local pharmacy at the Medical University of Vienna where enrolled patients were assigned to treatment groups, and blinded study drugs were prepared." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinded study. Drugs prepared by external pharmacist in equivalent containers |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Only pharmacist aware of treatment groups |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | Only pharmacist aware of treatment groups |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low number of losses which were explained |
| Selective outcome reporting (reporting bias) | Low risk | Prospective clinical trials registration (NCT01627613). Outcomes reported according to registration documents |
| Baseline characteristics | Unclear risk | Baseline characteristics reported in detail. Characteristics are mostly comparable, although some differences in number of participants who had received ECMO at screening; unclear whether this influenced results |
| Other sources of bias | Low risk | No other sources of bias identified |
Li 2010.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 45 Inclusion criteria: participants with ALI; acute onset of symptoms; PaO2/FiO2 ≤ 300 mmHg regardless of PEEP level; bilateral CXR infiltrations; pulmonary arterial wedge pressure ≤ 18 mmHg or no clinical evidence of left atrial hypertension Exclusion criteria: participants with ARDS (PaO2/FiO2 ≤ 200 mmHg), > 65 years or < 16 years of age; chronic lung diseases; heart dysfunction; liver and kidney functions damage; contraindication to penehyclidine hydrochloride Baseline characteristics Intervention group (penehyclidine hydrochloride)
Control group (standard therapy)
Country: China Setting: single‐centre; hospital |
|
| Interventions |
Intervention group (penehyclidine hydrochloride)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: improvement in APACHE II scores; development of ARDS; length of hospital and ICU stay; arterial blood gases; TLR4 expression on surface of peripheral blood monocytes; change of serum cytokines Outcomes relevant to the review: none |
|
| Notes |
Funding/declarations of interest: no details Study dates: September 2007 to August 2008 |
|
Liu 2012.
| Methods | RCT Parallel group |
|
| Participants |
Total number of randomized participants: 26 Inclusion criteria: 18 to 80 years of age; fulfils criteria of ARDS according to the AECC (Bernard 1994); ARDS diagnosis within 3 days of admission; fulfils CIRCI diagnosis according to Society of Critical Care Medicine of PLAs Guidelines 2006 Exclusion criteria: pregnant or breastfeeding; malignant disease; using immunosuppressing medications; history of bone marrow or lung transplant; history of primary or secondary adrenal disease; use of steroids in previous 3 months; rejects conventional treatment as stated in this study; took part in clinical trial in prior 30 days of this study Baseline characteristics Intervention group (corticosteroids)
Control group (placebo)
Country: China Setting: single‐centre |
|
| Interventions |
Intervention group (corticosteroids)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: APACHE II; SOFA; GCS (pre‐ and post‐treatment); number of ventilator‐free days (at day 28); number of coma‐free days (at day 28); number of coma events (at day 28); mortality (at day 28); length of ICU stay; arterial blood gas (pretreatment and 7 days after treatment) Outcomes relevant to the review: number of ventilator‐free days (at day 28); mortality (at day 28) |
|
| Notes |
Funding/declarations of interest: Nanjing Technology Development Fund; Jiangsu Medical Development Learning Fund Study dates: June 2009 to December 2011 Note: study reported in Chinese. Data extraction and 'Risk of bias' assessment completed by Dr Henry HL Wu |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as randomized but no additional details |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Liu 2015.
| Methods | RCT Parallel group |
|
| Participants |
Total number of randomized participants: 43 Inclusion criteria: > 18 years of age; meets criteria for septic shock according to 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference (Levy 2003); systemic immune response present; systolic/diastolic blood pressure 90/40 mmHg or lower; positive blood culture results; meets criteria of ARDS according to AECC (Bernard 1994) Exclusion criteria: neurogenic shock; neurovascular injury or head trauma patients; use of other nitric oxide‐related products within 24 hours of inpatient admission; predicted to have high mortality within 24 hours clinically Baseline characteristics Intervention group (nitroglycerin)
Control group (standard therapy)
Country: China Setting: single centre |
|
| Interventions |
Intervention group (nitroglycerin)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: haemodynamic measurements; serum inflammatory marker measurements; mortality (at day 28); duration of mechanical ventilation; length of ICU stay; length of hospital stay Outcomes relevant to the review: mortality (at day 28); duration of mechanical ventilation |
|
| Notes |
Funding/declarations of interest: First Hospital of Lanzhou University Hospital Fund Study dates: January 2013 to January 2014 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Investigators used computer randomization |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Control is standard therapy and therefore blinding not possible |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Liu 2017.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 65 Inclusion criteria: aged > 18 years of age, septic shock associated with ARDS defined according to AECC (Bernard 1994) Exclusion criteria: coagulation‐related pathology; allergy to alprostadil; mortality prognosis of < 24 hours from first ARDS presentation Baseline characteristics Intervention group (alprostadil)
Control group (standard therapy)
Country: China Setting: multicentre |
|
| Interventions |
Intervention group (alprostadil)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: APACHE II scores; ventilator conditions; duration of mechanical ventilation; duration of ICU stay; mortality (at day 28; in hospital); immune index; inflammatory markers Outcomes relevant to the review: mortality (at day 28); duration of mechanical ventilation |
|
| Notes |
Funding/declarations of interest: supported by the Natural Science Foundation of Gansu and First Hospital of Lanzhou University Study dates: January 2015 to June 2016 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as "randomized" but no additional details |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Control is standard therapy and therefore blinding not possible |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Meduri 2007.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 91 Inclusion criteria: adult intubated patients receiving mechanical ventilation; meeting criteria for ARDS according to AECC (Bernard 1994) within 72 hours Exclusion criteria: from online supplementary paper: enrolment in another study; extensive burns; organ transplant recipients; active life‐threatening fungal infection; moribund state (not expected to live > 6 hours); terminal illness with life expectancy < 3 months; positive HIV status; cytotoxic therapy within 3 weeks; malignancy with estimated 6‐month mortality > 50%; severe chronic liver disease; pre‐hospitalization Karnofsky Performance Status Scale ≤ 50; > 200% of ideal body weight; major gastrointestinal bleeding within last 3 months; underlying disease requiring > 0.5 mg/kg/day of methylprednisolone equivalent (e.g. asthma); primary care physician not fully committed to aggressive support of patient at time of randomization Baseline characteristics Intervention group (corticosteroids)
Control group (saline)
Country: USA Setting: multicentre; 5 ICUs |
|
| Interventions |
Intervention group (corticosteroids)
Control group (saline)
|
|
| Outcomes |
Outcomes measured/reported: LIS; PaO2/FiO2; PEEP; ventilator‐free days up to day 28; MODS score; C‐reactive protein level; cortisol level; participants with new infection, VAP, pneumothorax, neuromuscular weakness, hyperglycaemia requiring insulin, pancreatitis, or gastrointestinal bleeding requiring transfusion; survivors (at day 7); duration of mechanical ventilation; length of ICU stay; survivors of ICU admission; length of hospital stay; survivors of hospital admission Outcomes relevant to the review: mortality (during hospital stay); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declarations of interest: supported by Baptist Memorial Health Care Foundation and Assisi Foundation of Memphis; study authors reported that there were no conflicts of interest Study dates: April 1997 to April 2002 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Use of a random‐number generator (2:1 randomization). Information taken from additional online supplement |
| Allocation concealment (selection bias) | Low risk | Quote: "randomization assignments were provided in sealed envelopes for each institution, and the pharmacist maintained records on a study log." Comment: Information taken from additional online supplement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Some protocol violations and discontinuation of intervention, however all data analysed as ITT |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Unclear risk | Baseline characteristics were largely comparable with the exception of a higher proportion of participants with catecholamine‐dependent shock in the control group |
| Other sources of bias | Low risk | None identified |
Mohamed 2017.
| Methods | RCT Parallel group |
|
| Participants |
Total number of randomized participants: 60 Inclusion criteria: with ALI/ARDS according to 2012 Berlin definition Exclusion criteria: refusal of consent (by relatives); < 18 years of age; > 65 years of age; COPD; restrictive respiratory insufficiency; pneumonia; increased intracranial pressure; bronchopleural fistula; the persistence of unstable postsurgical haemodynamics despite appropriate supportive therapy; liver cell failure (Child‑Pugh Class B or C); end‐stage chronic renal failure on haemodialysis; acute myocardial infarction; neuromuscular disease Baseline characteristics Intervention group (corticosteroid)
Control group (saline)
Country: Egypt Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (corticosteroid)
Control group (saline)
|
|
| Outcomes |
Outcomes measured/reported: cardiorespiratory and ventilator parameters; level of serum inflammatory cytokines Outcomes relevant to the review: relevant outcomes not reported |
|
| Notes |
Funding/declarations of interest: financial support and sponsorship from South Valley University and Qena University Hospital, Egypt Study dates: January 2014 to January 2016 |
|
Morelli 2006.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 35 Inclusion criteria: septic shock; requiring mechanical respiratory support due to ARDS within first 3 days from onset of acute respiratory failure Exclusion criteria: presence of COPD; cardiogenic pulmonary oedema; pregnancy; < 18 years of age; or presence of a leaking chest tube Baseline characteristics Intervention group (levosimendan)
Control group (placebo)
Country: Italy Setting: single‐centre; ICU, university hospital |
|
| Interventions |
Intervention group (levosimendan)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: variables to assess pulmonary vascular resistance and right ventricular function Outcomes relevant to the review: relevant outcomes not reported |
|
| Notes |
Funding/declarations of interest: in part, by an independent research grant from the Department of Anaesthesiology and Intensive Care at the University of Rome; study authors do not disclose any potential conflicts of interest Study dates: not reported |
|
Morris 2008.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 215 Inclusion criteria: meeting criteria for ARDS; informed consent Exclusion criteria: pregnant or lactating women; < 17 years of age in USA and Canada; < 18 years of age in other countries; severe chronic lung disease; pulmonary oedema due to congestive heart failure; ventilator failure due to neurologic disease; presence of an acute myocardial infarction in the past 6 weeks; severe hepatic dysfunction; severe head trauma or stroke; moribund (not expected to live > 24 hours); physician, family, or patient not committed to full medical support; renal failure requiring dialysis; neutrophil count < 1000 attributed to cancer chemotherapy, leukaemia, lymphoma, or other haematologic malignancy not in remission; use of prednisone or other glucocorticoid in doses exceeding prednisone 0.5 mg/kg a day for > 2 weeks; primary immune deficiency diseases; known immunodeficiency virus positive; metastatic or inoperable solid malignancy; bone marrow transplantation with past 6 months; use of another investigational drug within 30 days of enrolment; receipt of NAC within 12 hours of study entry; known hypersensitivity to OTZ; enrolment time window has been exceeded Baseline characteristics Overall
Intervention group (OTZ)
Control group (placebo)
Country: USA, Canada, other countries not stated Setting: multi‐centre |
|
| Interventions |
Intervention group (OTZ)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: number of ventilator‐free days (for first 30 days); mortality (at day 30); new onset of organ dysfunction; length of ICU stay; length of hospital stay; adverse events Outcomes relevant to the review: mortality (at day 30); ventilator‐free days up to day 30; adverse events (leading to discontinuation of study medication) |
|
| Notes |
Funding/declarations of interest: supported by Transcend Therapeutics. 2 authors received grants from Transcend Therapeutics and from Clintec Technologies; study authors do not disclose any potential conflicts of interest Study dates: May 1997 to March 1998 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as randomized study but no details reported |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Intervention and placebo are identical in appearance. Study described as double‐blind, and we have assumed that personnel are blinded |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Study was terminated early. However, all the endpoints seem to have been analysed |
| Selective outcome reporting (reporting bias) | High risk | Clinical trials registration or pre‐published protocol not reported. Authors declare that not all study data are reported. Some data held by sponsors, who chose not to publish the data due to the negative results |
| Baseline characteristics | Unclear risk | No baseline characteristics table presented and characteristics were not reported by group |
| Other sources of bias | Unclear risk | Length of time between study completion and publication is unusually long, although explanation presented by study authors. Uneven number of participants in each group which is not explained |
Najafi 2009.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 23 Inclusion criteria: with ARDS; required mechanical ventilation; had a PaO2/FiO2 of < 200 mmHg; pulmonary capillary wedge pressure < 18 mmHg Exclusion criteria: PaO2/FiO2 > 200 mmHg; left ventricular failure; presence of regional and global left ventricular hypokinesia; left ventricular fractional area contraction of < 4 under inotropic support; chronic respiratory failure; chronic renal failure; known allergy to NAC; diabetes mellitus; < 18 years of age; pregnancy; any medical condition considered to be irreversible or lethal within 48 hours after ICU admission Baseline characteristics Intervention group (NAC)
Control group (standard therapy)
Country: Iran Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (NAC)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: microalbumin creatine ratios; survival (but not reported by group); PaO2/FiO2; MAP; APACHE II scores Outcomes relevant to the review: none |
|
| Notes |
Funding/declarations of interest: no details Study dates: September 2006 to September 2007. |
|
Ortolani 2000.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 36 Inclusion criteria: mechanical ventilation; bilateral CXR infiltrates; PaO2/FiO2 ≤ 200 mmHg or ≤ 250 mmHg if PEEP at least 10 cm H2O Exclusion criteria: duration of ARDS > 24 hours; haemodynamic instability; severe heart or liver disease; "septic complications during trial" Baseline characteristics Intervention group (NAC)
Intervention group (NAC and rutin)
Control group (placebo)
Country: Italy Setting: 2 centres, ICU |
|
| Interventions |
Intervention group (NAC)
Intervention group (NAC and rutin)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 30) Outcomes relevant to the review: mortality (at day 30) |
|
| Notes |
Funding/declarations of interest: no details Study dates: May 1995 to October 1997
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Participants described as randomly assigned but no further details |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Described as not blinded |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Baseline characteristics were comparable |
| Other sources of bias | Low risk | None identified |
Paine 2012.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 132 Inclusion criteria: patients meeting AECC criteria for ALI or ARDS (Bernard 1994) Exclusion criteria: < 18 years of age; > 7 days had elapsed since onset of ALI/ARDS; evidence of pre‐existing chronic respiratory failure; neutropenic; history of haematologic malignancy or bone marrow transplantation; entered other therapeutic trial; decision by patient or physician to forego aggressive care Baseline characteristics Intervention group (rhGM‐CSF)
Control group (placebo)
Country: USA Setting: multicentre; 3 ICUs |
|
| Interventions |
Intervention group (rhGM‐CSF)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: ventilator‐free days (up to day 28); all‐cause mortality (at day 28 day and 6 months); organ failure‐free days (within first 28 days); oxygenation index; serious adverse events (not defined as leading to discontinuation of study medication) Outcomes relevant to the review: ventilator‐free days (up to day 28); all‐cause mortality (at day 28 day and 6 months) |
|
| Notes |
Funding/declarations of interest: supported by a specialized Center for Clinical Research award from the National Heart Lung and Blood Institute. Study drug provided from pharmaceutical company free of charge but with no involvement in study Study dates: July 2004 to March 2009 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "randomized block design at each site, generated by the biostatistics core" |
| Allocation concealment (selection bias) | Low risk | Sealed opaque envelopes were used to conceal allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The investigators, study coordinators, and clinicians involved in patient care all were blinded to treatment and outcomes for the duration of the study." Comment: Study drug and placebo were identical in appearance. |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | We assumed that outcome assessors were blinded |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | We assumed that outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss of only 1 participant in each group, with reasons presented. Low number unlikely to influence outcome assessment. We noted the trial was stopped early, but this decision was made by an independent monitoring board |
| Selective outcome reporting (reporting bias) | Unclear risk | Trial prospectively registered NCT00201409. Additional mortality outcomes are reported in the final publication which are not listed in the protocol |
| Baseline characteristics | Unclear risk | Baseline characteristics were comparable except for MAP which was slightly yet statistically significantly lower in the placebo group |
| Other sources of bias | Low risk | No other sources of bias identified |
Rezk 2013.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 27 Inclusion criteria: patients with ARDS; mechanically ventilated; start of treatment in first 48 hours Exclusion criteria: PaO2/FiO2 > 200 mmHg; patients not mechanically ventilated Baseline characteristics Intervention group (corticosteroids)
Control group (placebo)
Country: Kuwait Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (corticosteroids)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: respiratory variables; oxygenation variables; haemodynamic variables; extubation time; mortality (at day 14) Outcomes relevant to the review: mortality (at day 14) |
|
| Notes |
Funding/declarations of interest: no details Study dates: October 2011 to October 2012 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | High risk | Quote: "we divided the 27 patients with ARDS randomly into two groups" Comment: Insufficient detail on methods of randomization. We noted an unexplained uneven number of participants in each group which could be caused by possible inadequate randomization methods |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Low risk | Appear largely comparable |
| Other sources of bias | Low risk | None identified |
Ryugo 2006.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 14 Inclusion criteria: undergone cardiovascular surgery within > 2 hours; cardiopulmonary bypass; development of SIRS and ALI Exclusion criteria: no details Baseline characteristics Intervention group (sivelestat)
Control group (placebo)
Country: Japan Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (sivelestat)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: oxygenation and respiratory variables; inflammatory markers; intubation times (in hours); adverse events (not defined as leading to discontinuation of study medication) Outcomes relevant to the review: duration of mechanical ventilation |
|
| Notes |
Funding/declaration of interests: no details Study dates: not reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as randomized but no details |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | ICU doctors not informed of group allocation. Description of intervention and control suggests that both agents were masked |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trial registration or pre‐published protocol not reported and therefore not feasible to assess risk of reporting bias |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | None identified |
SAILS 2014.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 745 Inclusion criteria: receiving positive‐pressure mechanical ventilation through an endotracheal tube; ratio of PaO2/FiO2 of ≤ 300 mmHg; had bilateral infiltrates on chest radiography consistent with pulmonary oedema; without evidence of left atrial hypertension; known or suspected infection; either of the following criteria for a systemic inflammatory response: a white‐cell count of > 12,000/mm3 or < 4000/mm3 or a differential count with > 10% band forms, or a core body temperature of > 38 °C or < 36 °C Exclusion criteria: ARDS for > 48 hours; chronic conditions that could adversely affect survival; impaired weaning from the ventilator; or compromise adherence to the protocol; serum levels of creatine kinase; aspartate aminotransferase; alanine aminotransferase of > 5 times the upper limit of the normal range; ingestion of a statin (on an inpatient or outpatient basis) in the 48 hours before randomization; inability to obtain consent Additional participant information: study authors report the number of participants by group according to PaO2/FiO2 ratio; these were balanced between groups Baseline characteristics Intervention group (statins)
Control group (placebo)
Country: USA Setting: multicentre; ICUs |
|
| Interventions |
Intervention group (statins)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality (before hospital discharge home or until study day 60); ventilator‐free days (up to day 28); ICU‐free days (to day 28); organ failure‐free days (to day 14); adverse events (not defined as leading to discontinuation of study medication) Outcomes relevant to the review: mortality (at day 60); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declaration of interests: funded by National, Heart, Lung, and Blood Institute; and by AstraZeneca. AstraZeneca supplied study drugs and resources to measure blood levels but had no role in study design, conduct, data analysis, or interpretation Study dates: March 2010 to September 2013 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Use of permuted blocks |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 participant lost to follow‐up, participants not treated with study drug after randomization, but all participants included in ITT analysis. We note that this trial was stopped early and that the decision was reached by an independent monitoring board |
| Selective outcome reporting (reporting bias) | Low risk | Prospectively clinical trials registration NCT00979121. All outcomes reported (additional adverse outcomes in protocol are reported in online supplementary appendix) |
| Baseline characteristics | Low risk | All comparable |
| Other sources of bias | Low risk | No other sources of bias identified |
Soltan‐Sharifi 2007.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 27 Inclusion criteria: required mechanical ventilation; PaO2/FiO2 ≤ 200 mmHg Exclusion criteria: PaO2/FiO2 > 200 mmHg; cardiovascular disease; < 18 years of age; pregnancy Baseline characteristics Intervention group (NAC)
Control group (standard therapy)
Country: Iran Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (NAC)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: clinical improvement of participants (change to APACHE II scores); intracellular GSH assay; total anti‐oxidant power Outcomes relevant to the review: none |
|
| Notes |
Funding/declarations of interest: grant from Tehran University of Medical Sciences Study dates: July 2005 to April 2006 |
|
Spragg 2002a.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 221 Inclusion criteria: patients with ARDS (not described) Exclusion criteria: unknown Baseline characteristics No baseline characteristics reported Country: unknown Setting: unknown |
|
| Interventions |
Intervention group (surfactant)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28); ventilator‐free days up to day 28 Outcomes relevant to the review: mortality (at day 28); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declarations of interest: unknown Study dates: unknown Notes
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Unknown. Information taken from original review in which full 'Risk of bias' assessment was not completed (Adhikari 2004). Original study reported only as an abstract |
| Allocation concealment (selection bias) | Unclear risk | No details. Information taken from original review (Adhikari 2004). |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unknown |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Unknown. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | Unknown |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No losses. Information taken from original review (Adhikari 2004) |
| Selective outcome reporting (reporting bias) | Unclear risk | Unknown |
| Baseline characteristics | Unclear risk | Unknown |
| Other sources of bias | Unclear risk | Unknown |
Spragg 2002b.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 227 Inclusion criteria: patients with ARDS Exclusion criteria: unknown Baseline characteristics Unknown Country: unknown Setting: unknown |
|
| Interventions |
Intervention group (surfactant)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28); ventilator‐free days (to day 28) Outcomes relevant to the review: mortality (at day 28); ventilator‐free days (to day 28) |
|
| Notes |
Funding/declarations of interest: unknown Study dates: unknown Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Unknown. Information taken from original review in which full 'Risk of bias' assessment was not completed (Adhikari 2004). Original study reported only as an abstract |
| Allocation concealment (selection bias) | Unclear risk | Unknown |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Unknown |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Unknown. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | Unknown |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | 1 loss reported in each group |
| Selective outcome reporting (reporting bias) | Unclear risk | Unknown |
| Baseline characteristics | Unclear risk | Unknown |
| Other sources of bias | Unclear risk | Unknown |
Spragg 2003.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 40 Inclusion criteria: risk factor for ARDS; AECC definition of ARDS (Bernard 1994); duration of ARDS not > 48 hours; PEEP of ≥ 5 cmH20; ≥ 48 hours of antimicrobial therapy if pneumonia present Exclusion criteria: haemodynamic instability; severe hypoxaemia; lung cancer; AIDS Baseline characteristics Intervention group (surfactant ‐ high dose)
Intervention group (surfactant ‐ low dose)
Control group (standard therapy)
Country: USA Setting: multicentre; 11 centres |
|
| Interventions |
Intervention group (surfactant ‐ high dose)
Intervention group (surfactant ‐ low dose)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28); ventilator‐free days; adverse events; safety data Outcomes relevant to the review: mortality (at day 28); ventilator‐free days up to day 28; adverse events |
|
| Notes |
Funding/declarations of interest: supported by ATLANTA Pharma AG. Analytic efforts supported in by part by grant from National Heart, Lung, and Blood Institute Study dates: not reported Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as randomized. No additional details. |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Control is standard treatment and therefore blinding not possible |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Unclear risk | Largely comparable, although participants in control group have a shorter time from diagnosis to treatment |
| Other sources of bias | Low risk | None identified |
START 2018.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 63 Inclusion criteria: moderate to severe ARDS; endotracheally intubated; had a PaO2/FiO2 < 27 kPa; mechanically ventilated with ≥ 8 cm H2O PEEP; had bilateral pulmonary infiltrates consistent with pulmonary oedema on chest radiographs; had no clinical evidence of left‐heart failure or volume overload as the primary cause of the pulmonary oedema. A protocol amendment was made to allow enrolment of patients with PEEP of 5 cm H2O if they had evidence of barotrauma Exclusion criteria: < 18 years of age; ARDS present for more than 96 hours; pregnancy or breastfeeding; being an inmate in the prison system; having received treatment for cancer in the past 2 years (except non‐melanoma skin cancer); having an underlying medical status with life expectancy < 6 months; moderate to severe liver disease (Child‐Pugh score > 12); severe chronic lung disease with the use of home oxygen; or partial arterial pressure of carbon dioxide > 7 kPa; not being committed to full support (i.e. had 'do not resuscitate' or limit on life support orders) Baseline characteristics Intervention group (MSC)
Control group (placebo)
Country: USA Setting: multicentre; 5 ICUs |
|
| Interventions |
Intervention group (MSC)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: safety of the MSC infusion (assessed with prespecified infusion‐associated adverse events focused on acute haemodynamic or respiratory compromise; not defined as leading to discontinuation of study medication); all‐cause mortality (at day 28 and day 60); number of ventilator‐free days (to day 28); duration of ventilation in participants (at day 28), number of ICU‐free days (to day 28), number of days free from organ failure (to day 28); SOFA score Outcomes relevant to the review: mortality (at day 60); duration of mechanical ventilation; ventilator‐free days up to day 28 |
|
| Notes |
Funding/declarations of interest: funded by National Heart, Lung, and Blood Institute. The funder had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. Authors report receipt of grants and personnel fees from some pharmaceutical companies Study dates: March 2014 to Feb 2017 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "The randomisation had a variable block design, was stratified by site, and the sequence was generated by computer." |
| Allocation concealment (selection bias) | Low risk | Quote: "The allocation sequence could be accessed by each cell laboratory through a dedicated website" Personnel in the cell laboratories were not masked, but patients, clinical staff, and investigators were unaware of treatment assignment." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Study products and intravenous tubing had opaque coverings applied in the cell laboratories." |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Outcome assessors blinded to group allocation |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | Oucome assessors blinded to group allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss of only 3 participants (in the MSC group) |
| Selective outcome reporting (reporting bias) | Low risk | Prospective clinical trials registration (NCT02097641). Outcomes reported according to registration documents |
| Baseline characteristics | High risk | Some baseline differences (APACHE III and SOFA scores) were higher in the MSC group, as were respiratory parameters (min ventilation and PEEP) |
| Other sources of bias | Low risk | No other sources of bias identified |
Steinberg 2006.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 180 Inclusion criteria: intubated and receiving mechanical ventilation; 7 to 28 days after onset of ARDS; on day of study entry PaO2/FiO2 had to be < 200 mmHg Exclusion criteria: undrained abscess; intravascular infection; disseminated fungal infection; new nosocomial pneumonia with < 72 hours of antibiotics; ongoing septic shock; < 13 years of age; participation in other trials within 30 days; pregnancy; burns requiring skin grafting; AIDS; treatment with corticosteroids (> 300 mg prednisone (or its equivalent) cumulative dose within 21 days or > 15 mg/day within 7 days prior to enrolment); cytotoxic therapy within 3 weeks; pre‐existing condition with estimated 6‐month mortality > 50%; severe chronic respiratory disease; bone marrow or lung transplantation; severe chronic liver disease; known or suspected adrenal insufficiency; vasculitis or diffuse alveolar haemorrhage; or refusal of the attending physician Baseline characteristics Intervention group (corticosteroid)
Control group (D5W)
Country: USA Setting: multicentre; 25 ICUs |
|
| Interventions |
Intervention group (corticosteroid)
Control group (D5W)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 60); number of ventilator‐free days (at day 28), number of days without organ failure; infectious complications and changes in marker of inflammation and fibroproliferation (on study day 7); serious adverse events associated with myopathy or neuropathy (not defined as leading to discontinuation of study medication); suspected or probable pneumonia; serious infections; post hoc analysis at 180 days to include mortality Outcomes relevant to the review: mortality (at day 60); mortality (at day 180); number of ventilator‐free days (at day 28) |
|
| Notes |
Funding/declaration of interest: National Heart, Lung, and Blood Institute Study dates: August 1997 to November 2003 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned with the use of permuted blocks" |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Insufficient information for both mortality time points. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Retrospective clinical trials registration NCT00295269. Not feasible to assess outcomes against clinical trials document. Post hoc analysis of participant outcome data at day 180 |
| Baseline characteristics | Low risk | Largely comparable. More men in the placebo group but unlikely to influence outcome data |
| Other sources of bias | Low risk | None identified |
STRIVE 2004.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 492 Inclusion criteria: ≥ 18 years of age with ALI on invasive mechanical ventilation for < 48 hours; ALI defined using AECC definition; must have had a PaO2/FiO2 of ≤ 300 mmHg on invasive mechanical ventilation regardless of the level of PEEP; no clinical evidence of congestive heart failure; and bilateral pulmonary infiltrates not explained by effusions, masses, or atelectasis Exclusion criteria: moribund and not expected to survive ≥ 24 hours or whose family or physician were not committed to aggressive support for ≥ 72 hours; severe pre‐existing heart, liver, or lung disease and those with uncontrolled malignancies Baseline characteristics Intervention group (sivelestat)
Control group (placebo)
Country: USA, Canada, Belgium, Spain, Australia, New Zealand Setting: multicentre; 105 ICUs |
|
| Interventions |
Intervention group (sivelestat)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: ventilator‐free days (at day 28); mortality (at day 28; also reported at day 90 and 180); circulatory, renal, hepatic and coagulation failure‐free days; ICU‐ and hospital‐free days; adverse events (not defined as leading to discontinuation of study medication) Outcomes relevant to the review: mortality (at day 90, and at day 180); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declaration of interests: Eli Lilly and Company. Study authors include 3 employees from pharmaceutical company Study dates: August 2001 to January 2003 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Use of central randomization source |
| Allocation concealment (selection bias) | Low risk | Unblinded pharmacist obtained individual participant treatment group and prepared drugs in masked bags for clinicians |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Study drugs concealed in sealed opaque bags, prepared by pharmacist |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Some loss‐to‐follow up in analysis at 90 and 180 days. ITT analysis completed. We noted that the trial was stopped early, at 28 days. However, this decision was made by the DSMB and we did not expect that this introduced bias |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement |
| Baseline characteristics | Unclear risk | All comparable, except interleukin‐6 which had a higher level in the placebo group; unclear whether this may influence outcome results |
| Other sources of bias | Low risk | No other sources of bias identified |
Tongyoo 2016.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 206 Inclusion criteria: ≥ 18 years of age; with severe sepsis or septic shock; receiving mechanical ventilation for hypoxaemic respiratory failure; within 12 hours of study entry; meeting the diagnostic criteria for ALI/ARDS according to the AECC definition (Bernard 1994) Exclusion criteria: moribund state (i.e. not expected to live > 24 hours); advanced malignancy with life expectancy < 6 months; pregnancy; immunosuppressive therapy; underlying disease requiring long‐term glucocorticoid treatment within the last 6 months or short‐term glucocorticoid treatment within the past 4 weeks; difficult‐to‐control diabetes Baseline characteristics Intervention group (corticosteroid)
Control group (placebo)
Country: Thailand Setting: single centre; ICU |
|
| Interventions |
Intervention group (corticosteroid)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28 and 60); duration of mechanical ventilation (up to day 28); ventilator‐free days (up to day 28); duration of vasopressor treatment; renal replacement therapy; duration of renal replacement therapy dependence; alive on day 28 without organ support; organ support‐free days (up to day 28); adverse events (nosocomial infection, lung infection, catheter‐related BSI, UTI, other nosocomial infection, hyperglycaemia, new‐onset AF, reintubation within 28 days, GI bleeding) ‐ adverse events not defined as leading to discontinuation of study medication Outcomes relevant to the review: duration of mechanical ventilation (up to day 28); number of ventilator‐free days; mortality (at 60 days); adverse events |
|
| Notes |
Funding/declarations of interest: supported by Siriraj critical care research funding Study dates: December 2010 to December 2014 Note:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned without restriction in a 1:1 ratio (hydrocortisone to placebo) according to a computer‐generated randomization table." |
| Allocation concealment (selection bias) | Low risk | Principal investigator was responsible for randomization, and had no involvement in participant care |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Research nurse not otherwise involved in the study prepared both the study drug and placebo." |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | We assumed that outcome assessors were blinded |
| Blinding of outcome assessors for other outcomes (detection bias) | Low risk | We assumed that outcome assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss of < 10% participants because of withdrawal of consent during treatment. Balanced between groups |
| Selective outcome reporting (reporting bias) | High risk | Clinical trials registration completed in the month following start of recruitment (NCT01284452). Additional outcomes are reported which are not listed in clinical trials register (mortality at longer time point, duration of mechanical ventilation up to 28 days, adverse outcomes). It is unclear whether this has introduced selective outcome reporting |
| Baseline characteristics | Low risk | Baseline characteristics appear to be comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Tsangaris 2007.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 16 Inclusion criteria: ventilated patients with blunt chest trauma at ARDS; severe hypoxaemia (PaO2/FiO2 < 150 mmHg 24 to 48 hours after initiation of mechanical ventilation); intact heart function; lung contusions manifested as areas of non‐aerated lung parenchyma on computed tomography. Exclusion criteria: high intracranial pressure or need for neurosurgical intervention, or both; haemodynamic instability; massive transfusion ( > 4 blood units); massive haemoptysis and tracheobronchial tree rupture Baseline characteristics Intervention group (surfactant)
Control group (standard therapy)
Country: Greece Setting: single centre; ICU |
|
| Interventions |
Intervention group (surfactant)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: oxygenation and compliance; response to recruitment; changes in ventilator parameters; duration of mechanical ventilation; adverse events (to include desaturation, hypotension and arrhythmias), and mortality (at day 28). Events were not described as "serious adverse events" and were not reported as leading to discontinuation of study medication), pulmonary complications and undesired effects Outcomes relevant to the review: mortality (at day 28); duration of mechanical ventilation |
|
| Notes |
Funding/declaration of interests: no details Study dates: no details (preliminary results presented at annual congress of ESICM in 2005) |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Computer‐generated list |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Not possible due to procedure required to administer surfactant and lack of placebo or control agent |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | High risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make judgement. Outcomes reported briefly in study report, e.g. insufficient detail reported on adverse effects |
| Baseline characteristics | Unclear risk | Mainly comparable. Statistically significant difference noted in end‐inspiratory pressure, but we could not be certain whether this would influence outcome data |
| Other sources of bias | Low risk | No other sources identified |
Vincent 2001.
| Methods | RCT Parallel design |
|
| Participants |
Total number of participants randomized: 102 Included criteria: AECC definition for ARDS (Bernard 1994); duration of ARDS < 24 hours Excluded criteria: recent MI; chronic congestive heart failure; liver or renal failure; pneumonectomy; neurogenic pulmonary oedema; neutropenia Baseline characteristics Intervention group (PGE1)
Control group (D5W)
Country: Belgium, France, Germany, Switzerland, Netherlands, UK Setting: multicentre; 31 hospitals |
|
| Interventions |
Intervention group (PGE1)
Control group (D5W)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at day 28); duration of ventilation; adverse events (not defined as leading to discontinuation of study medication) Outcomes relevant to the review: mortality (at day 28); duration of ventilation |
|
| Notes |
Funding/declarations of interest: no details Study dates: not reported Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as randomized (2:1 ratio) but no additional details |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Large number of participants did not complete the trial and reasons are provided by group. Baseline characteristics reported for number of participants initially randomized, it is unclear if outcome data include all participants. In addition, we noted that the study was terminated early and study authors do not report whether this decision was made by an independent monitoring board |
| Selective outcome reporting (reporting bias) | Unclear risk | Clinical trials registration or pre‐published protocol not reported. Insufficient information to make a judgement |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | None identified |
Walmrath 2000.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 26 Inclusion criteria: ARDS, not further described Exclusion criteria: no details Baseline characteristics Intervention group (surfactant) No baseline characteristics reported Control group (standard therapy) No baseline characteristics reported Country: Europe (countries not specified) and South Africa Setting: multicentre |
|
| Interventions |
Intervention group (surfactant)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: mortality; ventilator‐free days up to day 28 Outcomes relevant to the review: mortality (time point not reported); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declarations of interest: supported by Byk Gulden, Konstanz, Germany Study dates: not reported Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Participants randomized; no additional details. Abstract only |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Intervention compared to standard therapy and therefore not possible to blind personnel |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | Open‐label trial, and we have judged outcome assessors to be unblinded. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | Unblinded outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | We noted that outcome data were not reported for a group of participants given a higher dose of surfactant. We did not include these participants in the review and we made the 'Risk of bias' judgement on the remaining intervention group and control for which no details were reported |
| Selective outcome reporting (reporting bias) | Unclear risk | No details |
| Baseline characteristics | Unclear risk | No details |
| Other sources of bias | Unclear risk | Not feasible to assess other risks of bias from the abstract |
Willson 2015.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 308 Inclusion criteria: between 18 and 65 years of age; met AECC definition of ALI/ARDS (Bernard 1994) due to direct lung injury; were within 48 hours of initiation of mechanical ventilation; did not have significant other organ failure or chronic lung disease; care not limited Exclusion criteria: intubated after 48 hours; pre‐existing lung disease; other organ failure; care limited/do not resuscitate; patient/surrogate refusal; GCS < 8; congestive heart failure Additional participant information: study authors report distribution of participants at higher risk (PaO2/FiO2 <100) by group Baseline characteristics Intervention group (surfactant)
Control group (placebo)
Country: 6 countries (not listed in study report; list of countries included in an online appendix which were unable to source) Setting: multicentre; ICUs |
|
| Interventions |
Intervention group (surfactant)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: all‐cause mortality (at day 90); ventilator‐free days (at day 90); durations of ICU and hospital stay; duration of oxygen use; changes in oxygenation after the study intervention; adverse events (to include possibly related, probably related, and related; however adverse events not defined as "serious adverse events" or leading to discontinuation of study medication) Outcomes relevant to the review: all‐cause mortality (at day 90) |
|
| Notes |
Funding/declaration of interests: supported by Pneuma Pharmaceuticals. 3 authors are employees of pharmaceutical company Study dates: July 2008 to July 2010 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Low risk | Use of study web site for randomization. Participants stratified by PaO2:FiO2 |
| Allocation concealment (selection bias) | Unclear risk | Investigators naïve to randomization scheme but no details provided |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Blinding was accomplished by having the intervention performed by a nurse and/or respiratory therapist not otherwise involved in the subject’s care and who agreed to not divulge treatment assignment." Comment: Use of a sham treatment with air |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 7 participants were lost to analysis as they were found to be ineligible after randomization. The 7 participants were not treated and were excluded from analysis. In addition, trial was stopped early and decision was made by investigators (employed by a pharmaceutical company) rather than an independent monitoring board |
| Selective outcome reporting (reporting bias) | High risk | Prospective clinical trials registration NCT00682500. We noted discrepancies between the clinical trials registration documents and the published study report, with additional outcomes included in the published study report. |
| Baseline characteristics | Low risk | Appear comparable between groups |
| Other sources of bias | Low risk | No other sources of bias identified |
Wirtz 2017.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 61 Inclusion criteria: patients with ALI/ARDS on mechanical ventilation Exclusion criteria: no details Baseline characteristics "Baseline characteristics appear balanced". No further details in abstract Country: not reported Setting: not reported |
|
| Interventions |
Intervention group (enalaprilat)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: mortality (at 60 days); number of days without RRT; fluid balance; vasoactive therapy; SOFA scores; number of ventilator‐free days (up to day 28); days alive outside the ICU Outcomes relevant to the review: mortality (at day 60); number of ventilator‐free days (up to day 28) |
|
| Notes |
Funding/declarations of interest: not reported Study dates: May 2012 to October 2015 Notes:
|
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Study described as randomized. Only available as an abstract, with limited information to assess how random sequence was generated |
| Allocation concealment (selection bias) | Unclear risk | No details. Abstract only |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Described as a placebo‐controlled trial, but no additional details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Loss of 3 participants in analysis. Reason for losses not explained in abstract, and unclear to which groups these losses belonged. We noted that the trial was stopped early due to slow accrual (decision made by independent monitoring board) |
| Selective outcome reporting (reporting bias) | Unclear risk | No details of clinical trials registration or pre‐published protocol, and therefore not feasible to assess risk of selective outcome reporting bias |
| Baseline characteristics | Unclear risk | Baseline characteristics not reported in abstract |
| Other sources of bias | Unclear risk | Not feasible to assess other sources of bias because of insufficient information in abstract |
Zhao 2014.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 53 Inclusion criteria: fulfils criteria of ARDS according to AECC (Bernard 1994) Exclusion criteria: history of idiopathic pulmonary fibrosis; history of peptic ulcer disease; use of glucocorticoids in the past 1 month; contraindication to steroid medication and steroid‐related products; receiving haemodialysis; with other respiratory or cardiac diseases Baseline characteristics Intervention group (corticosteroid) Baseline characteristics no reported Control group (standard therapy) Baseline characteristics no reported Country: China Setting: single centre |
|
| Interventions |
Intervention group (corticosteroid)
Control group (standard therapy)
|
|
| Outcomes |
Outcomes measured/reported: pulmonary fibrosis index (post‐treatment comparisons); mottling area; adverse events; duration of mechanical ventilation; length of ICU stay; mortality (at day 28); incidence of MODS Outcomes relevant to the review: adverse events; duration of mechanical ventilation; mortality (at day 28); |
|
| Notes |
Funding/declarations of interest: internally funded by the Shanghai Songjiang District Center Hospital Study dates: December 2011 to June 2013 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Described as randomized but no further details |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Control is standard therapy and therefore blinding not possible |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No blinding. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | No details of clinical trials registration or pre‐published protocol, and therefore not feasible to assess risk of selective outcome reporting bias |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | No other source of bias identified |
Zheng 2014.
| Methods | RCT Parallel design |
|
| Participants |
Total number of randomized participants: 12 Inclusion criteria: ≥ 18 years of age; diagnosed within 48 hours with a PaO2/FiO2 ratio of < 200; ARDS defined and classified according to the Berlin definition (ARDS Definition Task Force 2012). Exclusion criteria: pre‐existing severe disease of any major organs; pregnancy; pulmonary hypertension; malignant disease; HIV infection; informed consent could not be obtained Baseline characteristics Intervention group (MSCs)
Control group (placebo)
Country: China Setting: single‐centre; ICU |
|
| Interventions |
Intervention group (MSCs)
Control group (placebo)
|
|
| Outcomes |
Outcomes measured/reported: adverse events (not defined as leading to discontinuation of study medication); PaO2/FiO2 ratio; hospital indices (length of hospital stay, ventilator‐free days and ICU‐free days at day 28); and serum biomarkers of ARDS including IL‐6, IL‐8 and SP‐D; mortality during the study period Outcomes relevant to the review: mortality (during study); ventilator‐free days up to day 28 |
|
| Notes |
Funding/declaration of interests: National Natural Science Foundation of China and Shaoxing 330 Plan to JX, and the National Natural Science Foundation of China and the Zhejiang Province Science and Technology Program to QS; authors declare that they have no competing interests Study dates: January 2013 to April 2013 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Sequence generation (selection bias) | Unclear risk | Participants were described as randomized but no details |
| Allocation concealment (selection bias) | Unclear risk | No details |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No details |
| Blinding of outcome assessors for mortality (detection bias) | Low risk | No details. However, we did not expect blinding of outcome assessors to influence outcome data for mortality |
| Blinding of outcome assessors for other outcomes (detection bias) | Unclear risk | No details |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No apparent losses |
| Selective outcome reporting (reporting bias) | Unclear risk | Retrospective clinical trials registration NCT01902082. Therefore not feasible to assess selective outcome reporting The protocol states that TNF‐alpha will be reported as an outcome but the study does not appear to have included this. All other outcomes were reported. |
| Baseline characteristics | Low risk | Appear comparable |
| Other sources of bias | Low risk | None apparent |
AECC: American‐European Consensus Conference (Bernard 1991); AF: atrial fibrillation; AIDS: acquired immunodeficiency syndrome; ALI: acute lung injury; APACHE: Acute Physiology and Chronic Health Evaluation; ARDS: acute respiratory distress syndrome; BALF: bronchoalveolar lavage; BSI: bloodstream infection; CI: confidence interval; CIRCI: critical illness‐related corticosteroid insufficiency; COPD: chronic obstructive pulmonary disease; CXR: chest radiograph; D5W: 5% dextrose in water; DPPC: dipalmitoylphosphatidylcholine; ECMO: extracorporeal membrane oxygenation; ESICM: European Society of Intensive Care Medicine; EVLWI: extravascular lung water index; FiO2: fraction of inspired oxygen; GCS: Glasgow Coma Scale; GI: gastrointestinal; GSH: glutathione; HIV: human immunodeficiency virus; HL‐10: freeze‐dried natural surfactant isolated from pig lungs; ICU: intensive care unit; IL‐6: interleukin 6; IL‐8: interleukin 8; IQR: interquartile range; ITT: intention‐to‐treat; IV: intravenous; KGF: keratinocyte growth factor; kPa: kilopascal; LAH: left atrial hypertension; LIS: lung injury score; LODS: Logistic Organ Dysfunction System; MAP: mean arterial blood pressure; MD: difference (mean in treatment group ‐ mean in control group); MDA: malondialdehyde; M/F: male/female; MI: myocardial infarction; MODS: multiple organ dysfunction score (see Critical Care Medicine 1995;23(10):1638‐52); mRNA: messenger ribonucleic acid; MSC: mesenchymal stromal cells; n: number of randomized participants; NAC: N‐acetylcysteine; NIHR: National Institute for Health Research; OI: oxygenation index; OTZ: L‐2‐oxothiazolidine‐4‐carboxylic acid; PaO2: partial pressure of arterial oxygen; PAOP: pulmonary artery occlusion pressure; PBW: predicted body weight; PEEP: positive end‐expiratory pressure; P/F ratio: arterial oxygen partial pressure to fractional inspired oxygen ratio; PGE1: prostaglandin E1;PLA: People's Liberation Army; PMN‐E: neutrophil elastase; RCT: randomized control trial; rhACE2: recombinant human angiotensin‐converting enzyme 2; rhGM‐CSF: recombinant human granulocyte‐macrophage colony stimulating factor; rSP‐C: recombinant surfactant protein C; RR: relative risk; RRT: renal replacement therapy; SAPS: simplified acute physiology score (see Critical Care Medicine 1984;12(11):975‐7); SD: standard deviation; SGRQ: St. George's Respiratory Questionnaire; SIRS: systemic inflammatory response syndrome; SOFA: Sequential Organ Failure Assessment; SOD: superoxide dismutase; SP‐D: surfactant protein D; TLC C‐53: liposomes and prostaglandin E1;TLR4: toll‐like receptor 4; TNF‐alpha: tumour necrosis factor alpha; TREM‐1: triggering receptor expressed on myeloid cells; TRN‐a: transfer ribonucleic acid; UTI: urinary tract infection; VAP: ventilator‐associated pneumonia
NOTE: Blank spaces in the 'Risk of bias' figure indicate that we did not conduct 'Risk of bias' assessment.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abraham 1996 | RCT, liposomal PGE1 versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Abraham 1999 | RCT, liposomal PGE1 versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Annane 2006 | Retrospective analysis of previous clinical trial of corticosteroids versus placebo. Retrospective analysis includes only participants with ARDS. Excluded because of ineligible study design |
| Anzueto 1996 | RCT, Exosurf versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Ardizzoia 1993 | RCT, PTX versus standard therapy. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Bastin 2010 | RCT, NAC versus control, adults undergoing lung resection with open‐lung ventilation for lung cancer, but not specifically patients with ALI or ARDS |
| Bastin 2016 | RCT, NAC versus control, adults undergoing lung resection with open‐lung ventilation, but not specifically patients with ALI or ARDS |
| Bernard 1987 | RCT, corticosteroids (methylprednisolone) versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Bernard 1997 | RCT, multi‐arm study. NAC; OTZ; control. Included in previous version of the review (Adhikari 2004).Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Bernard 1999 | RCT, IL‐10 (use of control not described). Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Bone 1989 | PGE1 versus placebo. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Confalonieri 2005 | RCT, hydrocortisone versus control. Inclusion criterion is for community‐acquired pneumonia rather than specifically ARDS. |
| Cornet 2014 | RCT, participants with ARDS, recombinant human activated protein C versus saline. This drug is now withdrawn from the market and it is not feasible to include this study in the review |
| Domenighetti 1997 | RCT, NAC versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Forel 2006 | RCT, participants with ARDS. Trial of neuromuscular blocking agent for mechanical ventilation of patients, rather than treatment of ARDS |
| Gainnier 2004 | RCT, participants with ARDS. Trial of neuromuscular blocking agent for mechanical ventilation of patients, rather than treatment of ARDS |
| Gottlieb 1994 | RCT, neutrophil elastase inhibitor versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Gregory 1997 | RCT, surfactant (at 2 doses) versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Holcroft 1986 | RCT, PGE1 versus control. Included in previous version of the Review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Hua 2013 | RCT, participants with ARDS, terlipressin versus dopamine, but does not include a placebo or control as a comparison agent and therefore excluded |
| Jepsen 1992 | RCT, NAC versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Liu 2008 | RCT, participants with ALI, recombinant human activated protein C versus control. This drug is now withdrawn from the market and it is not feasible to include this study in the review |
| Markart 2007 | Phase I/II pilot study of recombinant surfactant protein C‐based surfactant (venticute), study design not relevant for review, no relevant outcomes reported |
| Meduri 1998 | RCT, corticosteroids (methylprednisolone) versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Papazian 2010 | RCT, participants with ARDS. Trial of neuromuscular blocking agent for mechanical ventilation of patients, rather than treatment of ARDS |
| Presneill 2002 | RCT, granulocyte‐macrophage colony‐stimulating factor versus placebo. Included in previous version of the review (Adhikari 2004). Excluded because ALI or ARDS is not a specific inclusion criteria. |
| Reines 1985 | RCT, thromboxane synthase inhibitor (Dazoxiben) versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Reines 1992 | RCT, surfactant versus placebo. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Rossignon 1990 | RCT, PGE1 versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Shoemaker 1986 | RCT, PGE1 versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Shyamsundar 2010 | RCT, recombinant human keratinocyte growth factor versus control, but given to prevent not treat ALI and therefore excluded |
| Steinberg 1990 | RCT, indomethacin versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Suter 1994 | RCT, NAC versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Tuxen 1987 | RCT, acyclovir versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Vincent 2009 | RCT, inactivated recombinant factor VIIa versus control. Dose escalation study, not relevant for review without agreed appropriate dosing for this agent |
| Weg 1994 | RCT, surfactant versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
| Weigelt 1985 | RCT, methylprednisolone versus control. Included in previous version of the review (Adhikari 2004). Excluded because publication date was prior to 2000 (see Differences between protocol and review) |
ALI: acute lung injury; ARDS: acute respiratory distress syndrome; IL‐10: interleukin 10; NAC: N‐acetylcysteine; OTZ: L‐2‐oxothiazolidine‐4‐carboxylic acid; PGE1: prostaglandin E1;PTX: pentoxifylline; RCT: randomized controlled trial
Characteristics of studies awaiting assessment [ordered by study ID]
Hegazy 2016.
| Methods | RCT |
| Participants |
Total number of randomized participants: 100 Inclusion criteria: mechanically ventilated patients of both genders with severe ARDS Exclusion criteria: not given Country: Egypt Setting: single‐centre; ICU |
| Interventions |
Intervention group
Control group
|
| Outcomes | Primary outcomes: mortality; length of hospital stay; length of ICU stay; ventilator‐free days |
| Notes | The study is reported as an abstract only, with insufficient information to assess eligibility. We await publication of the full report to assess eligibility |
NCT00879606.
| Methods | RCT |
| Participants |
Target participant recruitment: 150 Inclusion criteria: suspected or proven infection; hypoxaemia: PaO2/FiO2 ≤ 300 mmHg; bilateral infiltrates consistent with pulmonary oedema; positive‐pressure mechanical ventilation through an endotracheal tube; no clinical evidence of left atrial hypertension to explain bilateral infiltrates; presence of ≥ 3 of the 4 SIRS criteria. If only 2 criteria are evidenced, 1 must be temperature or WBC Exclusion criteria: < 18 years of age; inability to obtain consent; patient, surrogate, or physician not committed to full support; moribund state in which death was perceived to be imminent; morbid obesity; malignancy or other irreversible disease or condition for which 6‐month mortality is estimated to be > 50%; known HIV‐positive with known end‐stage processes; prior cardiac arrest requiring CPR without fully demonstrated neurological recovery; or NYHA Class IV; pregnant or nursing; ALI/ARDS induced by mechanical or chemical injury directly to the lung (including burns, trauma, and near drowning); > 48 hours since all inclusion criteria are met; neuromuscular disease that impairs ability to ventilate without assistance; severe chronic respiratory disease, severe pulmonary hypertension, or ventilator dependency; chest wall deformity resulting in severe exercise restriction, secondary polycythaemia, or respirator‐dependent; history of organ transplant (including bone marrow); severe chronic liver disease, as determined by a Child‐Pugh Score > 10; haemoglobin persistently < 7.0 g/dL; platelet count < 50,000/mm3; prolonged INR > 3; bleeding disorders unless corrective surgery has been performed; active internal bleeding; major surgery within 24 hours before study drug infusion, or evidence of active bleeding postoperatively, or plan for any major surgery within 3 days after study drug infusion; diffuse alveolar haemorrhage from vasculitis; known bleeding diathesis; presence of an epidural catheter or lumbar puncture within 48 hours before study drug infusion or anticipation of receiving an epidural catheter or a lumbar puncture within 48 hours after study drug infusion; stroke within 3 months of study entry; trauma with an increased risk of life‐threatening bleeding; a history of severe head trauma that required hospitalization, or intracranial surgery within 2 months of study entry; any history of intracerebral arteriovenous malformation, cerebral aneurysm, or central nervous system mass lesion; uses of certain medications or treatment regimens such as chemotherapy, unfractionated heparin, low‐molecular‐weight heparin, warfarin, antithrombin III, acetylsalicylic acid, glycoprotein IIb/IIIa antagonists, thrombolytic therapy, and activated Protein C are restricted; participation in another experimental medication study within 30 days of study entry Country: USA Setting: multicentre; ICU |
| Interventions |
Intervention group
Control group
|
| Outcomes |
Primary outcomes: safety profile of the study drug (throughout 28 days following treatment); number of ventilator‐free days (at day 28) Secondary outcomes: mortality (at day 7, 14, 21, 28 and 60); length of hospitalization (at day 28); length of ICU stay (at day 28); number of non‐pulmonary organ failure‐free days (at day 28); changes in physiological variables of lung injury (throughout the 28 days following treatment); changes in disease severity and lung injury scores (throughout the 28 days following treatment); effects of the study drug and the aetiology of the disease (at day 28); pharmacokinetics and pharmacodynamics (throughout the 28 days following treatment); immunogenicity (throughout the 28 days following treatment) |
| Notes |
Funding/declarations of interest: Altor BioScience; National Heart, Lungs, and Blood Institute Study dates: April 2009 to January 2013 Note:
|
RPCEC00000126.
| Methods | RCT |
| Participants |
Target participant recruitment: 72 Inclusion criteria: presence of ARDS in the first 24 hours of diagnosis; PEEP > 5 cmH2O; patients whose relatives give written consent to participate; between 18 and 75 years of age Exclusion criteria: pregnant women, breastfeeding or post partum; COPD; hypersensitivity to surfacen or other component of the formulation Country: Cuba Setting: ICU |
| Interventions |
Intervention group
Control group
|
| Outcomes |
Primary outcome: PaO2/FiO2 ratio (favourable when ≥ 200) (group A: 1 hour, 4 hours and 8 hours after each dose. Group B: 1 hour, 4 hours and 8 hours after haemodynamic stabilization) Secondary outcomes: gasometric evaluation (PaO2, PaCO2, pH); ventilatory evaluation; clinic evaluation; radiographic evaluation; mechanical ventilation days; total hospital days; condition of the participant; all‐cause mortality (at 28 days); adverse events (at 28 days) |
| Notes |
Funding/declarations of interest: CENSA; MINSAP Study dates: March 2006 |
ALI: acute lung injury; ALT‐836: anti‐TF monoclonal antibody; ARDS: acute respiratory distress syndrome; CENSA: National Center for Animal and Plant Health; COPD: chronic obstructive pulmonary disease; CPR: cardiopulmonary resuscitation; FiO2: fraction of inspired oxygen; HIV: human immunodeficiency virus; ICU: intensive care unit; INR: international normalized ratio; MINSAP: Ministry of Public Health; NYHA: New York Heart Association; PaO2: partial pressure of arterial oxygen; PEEP: positive‐end expiratory pressure; RCT: randomized control trial; SIRS: systemic inflammatory response syndrome; WBC: white blood cell count
Characteristics of ongoing studies [ordered by study ID]
ACTRN12612000418875.
| Trial name or title | A multi‐centre randomized, placebo controlled trial of nebulized heparin in participants with or at risk of developing Acute Respiratory Distress Syndrome, to determine if nebulized heparin improves long term physical function |
| Methods | RCT |
| Participants |
Target participant recruitment: 256 Inclusion criteria: ≥ 18 years of age; receiving ventilation through an endotracheal tube; started ventilation yesterday or today; expected to require invasive ventilation for at least all of today and all of tomorrow; PaO2 to FiO2 ratio < 300; active ventilator circuit humidification Exclusion criteria: allergy to heparin; history of heparin‐induced thrombocytopenia; platelet count < 50 x 109/L; APTT prolonged to > 80 seconds, not due to anticoagulant therapy; uncontrolled bleeding; pulmonary bleeding during hospital admission; history of intracranial, spinal or epidural haemorrhage; neurosurgical procedures during hospital admission or such procedures planned; epidural catheter is in place; hepatic encephalopathy or history of gastrointestinal bleeding due to portal hypertension or biopsy proven cirrhosis with documented portal hypertension; tracheostomy in place; usually receives home oxygen; usually receives any type of assisted ventilation at home; cervical spinal cord injury associated with reduced long‐term ability to breathe independently; spinal or peripheral nerve disease with a likely prolonged reduction in the ability to breathe independently; receiving high‐frequency oscillation ventilation or extracorporeal membrane oxygenation; pregnant; treatment limits restrict the provision of renal replacement therapy, inotropes, vasopressors or prolonged invasive ventilation; usually treated with haemodialysis or peritoneal dialysis for end‐stage renal failure; dementia; death is deemed imminent or inevitable or there is underlying disease with a life expectancy of less than 90 days Country: Australia Setting: ICU |
| Interventions |
Intervention group (nebulized liquid heparin)
Control group (placebo)
|
| Outcomes |
Primary outcome: physical function assessed using the physical function component of the SF‐36 health survey Secondary outcomes: change in Murray Lung Injury Score assessed by review of medical records; change in plasma thrombin time, D‐Dimer, antithrombin thrombin levels and serum cytokines assessed by blood analysis; development of ALI or ARDS assessed by review of medical records; healthcare utilisation assessed by review of medical records; hospital stay duration; ICU stay duration; lung rescue therapies; major bleeding or other complications; mechanical VFDs; mortality; quality of life assessed by contact with participant or next of kin and undertaking EQ5D survey |
| Starting date | 1 August 2012 |
| Contact information | Email: barry.dixon@svhm.org.au |
| Notes | Funding/declarations of interest: St.Vincents Hospital/Institute |
ACTRN12615000373572.
| Trial name or title | Effect of nebulized budesonide on respiratory mechanics and oxygenation in patients with acute lung injury/acute respiratory distress syndrome (ALI/ARDS) |
| Methods | RCT |
| Participants |
Target participant recruitment: 60 Inclusion criteria: patients fulfilling the criteria of ALI/ARDS according to 2012 Berlin definition of ALI/ARDS Exclusion criteria: refusal of consent by relatives; < 18 or > 65 years of age; chronic obstructive pulmonary disease; restrictive respiratory insufficiency; increased intracranial pressure; bronchopleural fistula; acute myocardial infarction; neuromuscular disease Country: Egypt Setting: ICU |
| Interventions |
Intervention group (budesonide)
Control group (placebo)
|
| Outcomes |
Primary outcome: PaO2/FiO2, calculated from the measured PaO2 (from arterial blood gas analysis) and the inspired oxygen concentration. FiO2 is the inspired fraction of oxygen given by anaesthesiologist whether pure oxygen (100% oxygen) or in combination with air (60% oxygen in 40% air) Secondary outcome: PIP; plateau pressure; both parameters are assessed from the screen of Binnette respirometer |
| Starting date | 2014 |
| Contact information | Email: dr.hatem_saber@hotmail.com |
| Notes | Funding/declarations of interest: South Valley University and Qena University Hospital |
Bellingan 2017.
| Trial name or title | Comparison of the efficacy and safety of FP‐1201‐lyo and placebo in the treatment of patients with moderate or severe acute respiratory distress syndrome: study protocol for a randomized controlled trial |
| Methods | RCT |
| Participants |
Target participant recruitment: 300 Inclusion criteria: ≥ 18 years of age; trachea is intubated; receiving mechanical ventilation; diagnosis of moderate or severe ARDS according to the Berlin definition of ARDS; acute onset of respiratory failure within 1 week of a known clinical insult or new or worsening respiratory symptoms; respiratory failure associated with known ARDS risk factors and not fully explained by either cardiac failure or fluid overload; radiological abnormalities on chest X‐ray or on CT scan; radiological and hypoxaemia criteria must occur within the same 24‐hour period; time of onset of ARDS is defined as the time when the last of these 2 ARDS criteria is met; administration of the first dose of study drug planned to take place within 48 hours of moderate or severe ARDS diagnosis; signed written informed consent form from the participant or the participant’s personal legal representative or a professional legal representative Exclusion criteria: woman known to be pregnant or lactating; simultaneously taking part in another pharmacotherapy protocol; not expected to survive for 24 hours; has underlying clinical condition where, in the opinion of the investigator, it would be extremely unlikely that the patient would be able to come off ventilation; severe COPD requiring long‐term home oxygen therapy or mechanical ventilation except for CPAP or BIPAP used solely for sleep‐disordered breathing; congestive heart failure, defined as NYHA class IV; acute left ventricular failure; liver failure (Child‐Pugh grade C); received any prior IFN; known hypersensitivity to natural or recombinant IFN beta or to any of the excipients; receiving renal dialysis therapy for chronic renal failure; receiving extracorporeal membrane oxygenation, HFOV or any form of extracorporeal lung support; has had any form of mechanical ventilation (invasive or non‐invasive, excluding CPAP alone) for > 48 hours prior to the diagnosis of ARDS; noninvasive ventilation has to be continuously applied for at least 12 hours a day in these 48 hours; burns to ≥ 15% of their TBSA Country: 9 European countries Setting: multicentre; ICU |
| Interventions |
Intervention group (FP‐1201‐lyo)
Control group (placebo)
|
| Outcomes |
Primary outcomes: composite of death (at 28 days); days free of mechanical ventilation (at 28 days) Secondary outcomes: all‐cause mortality (at 28, 90, 180 and 360 days); days free of organ failure (at 28 days); length of hospital stay; efficacy, safety and exploratory variables |
| Starting date | 28 December 2015 |
| Contact information | Email: geoff.bellingan@uclh.nhs.uk |
| Notes | Funding/declarations of interest: Faron Pharmaceuticals Ltd.; European Union Seventh Framework Program; Mikael Maksimow, Markku Jalkanen and Ilse Piippo are employed by Faron Pharmaceuticals and hold Faron shares and/or options for shares. The other authors are members of the INTEREST trial Steering Committee and have received expenses only for participation in required study meetings |
ChiCTR1800014733.
| Trial name or title | Clinical study of rhGM‐CSF in the treatment of pulmonary extraneous acute respiratory distress syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: 90 Inclusion criteria: between 18 and 70 years of age; the primary disease of ARDS is a non‐pulmonary sepsis, which is in line with the definition of sepsis in 2016; patient or family members agree to join the clinical study Exclusion criteria: pregnant women; patients with malignant tumour, immunodeficiency or autoimmune disease; white blood cell count is > 40 x 109/L or the oxygenation index is less than 80; patient or family members need to withdraw from the study Country: China Setting: single‐centre; ICU |
| Interventions |
Intervention group (thymosin)
Intervention group (GM‐CSF)
Control group (standard therapy)
|
| Outcomes |
Primary outcomes: oxygenation index; HLA‐D expression rate of CD14 positive mononuclear cells Secondary outcomes: concentration of HMGB‐1, TNF‐a, PCT and GM‐CSF in plasma; concentration of HMGB‐1, TNF‐a and GM‐CSF in bronchoalveolar lavage fluid; APACHE II score, SOFA score, and lung injury score; duration of ventilator use, incidence of ventilator‐associated pneumonia; mortality (at 28 days) |
| Starting date | 2 January 2018 |
| Contact information | Email: phoenix413@163.com |
| Notes |
ChiCTR1800014998.
| Trial name or title | Human umbilical cord derived mesenchymal stem cells therapy in acute respiratory distress syndrome: a pilot study |
| Methods | RCT |
| Participants |
Target participant recruitment: 12 Inclusion criteria: invasive ventilation, OI PaO2/FiO2 < 200; PEEP = 8 cmH2O; bilateral infiltration of lung in X‐ray or CT; in first week after onset; still OI < 200 after protective ventilation or conservative fluid management Exclusion criteria: any malignant disease; cardiogenic pulmonary oedema; > 50% atelectasis either lung lobe in X‐ray; pregnancy or perinatal or lactation; previous end‐stage respiratory disease; > 3 organs failure; liver failure with MELD score > 40; stage III or IV pulmonary hypertension; noninvasive arterial and central venous catheter; concurrent deep venous thrombus or pulmonary embolism in 3 months; cerebral hernia; > 96 hours after ARDS onset; < 18 years of age Country: China Setting: single‐centre; University Hospital |
| Interventions |
Intervention group (MSC)
Control group (placebo)
|
| Outcomes |
Primary outcome: safety Secondary outcomes: ventilator‐free days; oxygenation index change |
| Starting date | 27 February 2018 |
| Contact information | Email: haijinlv@163.com |
| Notes |
EUCTR2012‐000775‐17.
| Trial name or title | A comparative, randomized controlled trial for evaluating the efficacy of dexamethasone administration in the treatment of patients with the Acute Respiratory Distress Syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: not given Inclusion criteria: ≥ 18 years of age; have acute onset of ARDS, as defined by the AECC criteria for ARDS (Bernard 1994); intubated and mechanically ventilated; provided signed written informed consent from the patient or the patient's personal legal representative Exclusion criteria: woman known to be pregnant or lactating; participating in another experimental treatment protocol; brain death; terminal‐stage cancer or other terminal disease; do‐not‐resuscitate orders; immune‐compromised; receiving corticosteroids or immunosuppressive drugs; > 24 hours elapsed after initially meeting the AECC ARDS criteria (Bernard 1994) before consent and results of initial standard ventilator settings could be obtained; severe COPD; congestive heart failure Country: Spain Setting: ICU |
| Interventions |
Intervention group (dexamethasone)
Control group (standard treatment)
|
| Outcomes |
Primary outcomes: VFDs (at day 28) Secondary outcomes: all‐cause mortality (at Day 60); days on mechanical ventilation; number of extra‐pulmonary organ failures (at day 60) |
| Starting date | 21 November 2012 |
| Contact information | Email: osalidia.fernandez@gmail.com |
| Notes | Funding/declarations of interest: Fundación Mutua Madrileña |
JPRN‐JapicCTI‐163320.
| Trial name or title | A phase 3 clinical study to evaluate the efficacy and safety of intravenous MR11A8 in the treatment of patients with moderate or severe acute respiratory distress syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: 120 Inclusion criteria: ≥ 20 years of age; Japanese patient with a diagnosis of moderate or severe ARDS according to the Berlin definition of ARDS; acute onset of respiratory failure within 1 week of a known clinical insult or new or worsening respiratory symptoms; respiratory failure associated with known ARDS risk factors and not fully explained by either cardiac failure or fluid overload; radiological abnormalities on chest X‐ray or on computerized tomography scan; hypoxaemia; moderate ARDS 100 mmHg = 5 cmH2O; severe ARDS PaO2/FiO2 ≤ 100 mmHg with PEEP ≥ 5 cmH2O; the radiological and hypoxaemia criteria in ARDS criteria must be met within the same 24‐hour period. The time of onset of ARDS is when the last of the 2 specified ARDS criteria is met; administration of the first dose of study drug must be planned to take place within 48 hours of first fulfilling the above inclusion criteria; patient is intubated and mechanically ventilated; signed informed consent form from the patient or the patient's personal legal representative or a professional legal representative Exclusion criteria: woman known to be pregnant, lactating or with a positive or indeterminate pregnancy test; patient is simultaneously taking part in another pharmacotherapy protocol; not expected to survive for 24 hours; has an underlying clinical condition where, in the opinion of the Investigator, it would be extremely unlikely that the patient would come off ventilation; severe chronic obstructive pulmonary disease requiring long‐term home oxygen therapy or mechanical ventilation (non‐invasive ventilation or tracheotomy) except for CPAP or BIPAP used solely for sleep‐disordered breathing; has congestive heart failure, defined as NYHA class 4; acute left ventricular failure; liver failure (Child‐Pugh grade C); received any prior IFN; known hypersensitivity to natural or recombinant IFN beta or to any of the excipients; receiving renal dialysis therapy for chronic renal failure; receiving extra‐corporeal membrane oxygenation, high‐frequency oscillatory ventilation or any form of extracorporeal lung support; had any form of mechanical ventilation (invasive or non‐invasive, excluding CPAP alone) for > 48 hours prior to the diagnosis of ARDS. Non‐invasive ventilation has to be continuously applied for ≥ 12 hours a day in these 48 hours; burns to ≥ 15% of their TBSA; receiving Sho‐saiko‐to or is scheduled to receive Sho‐saiko‐to during the study drug administration period Country: Japan Setting: multicentre; ICUs |
| Interventions |
Intervention group (MR11A8)
Control group (placebo)
|
| Outcomes |
Primary outcome: all‐cause mortality (at day 28) Secondary outcomes: composite endpoint including any‐cause death (at day 28); days free of mechanical ventilation (within 28 days); all‐cause mortality (at day 90); mortality in ICU (up to day 28); mortality in hospital (up to day 28) |
| Starting date | 16 July 2016 |
| Contact information | Name: Maruishi Pharmaceutical Co., Ltd, Clinical Development Department, Pharmaceutical Research and Development Division |
| Notes | Funding/declarations of interest: Maruishi Pharmaceutical Co., Ltd. |
NCT02326350.
| Trial name or title | ASpirin as a Treatment for ARDS (STAR): a phase 2 randomized control trial |
| Methods | RCT |
| Participants |
Target participant recruitment: 60 Inclusion criteria: ≥ 16 years of age; receiving invasive mechanical ventilation; ARDS as defined by the Berlin definition Exclusion criteria: > 72 hours from the onset of ARDS; < 16 years of age; pregnant; participation in a clinical trial of an investigational medicinal product within 30 days; current treatment with aspirin or within the past 4 weeks; platelet count < 50 x 109/L; haemophilia or other haemorrhagic disorder or concurrent therapeutic anticoagulant therapy; history of aspirin‐sensitive asthma or nasal polyps associated with asthma; active or history of recurrent peptic ulcer and/or gastric/intestinal haemorrhage or other kinds of bleeding such as cerebrovascular haemorrhage; traumatic brain injury; active gout; currently receiving methotrexate; severe chronic liver disease with Child‐Pugh score > 12; known hypersensitivity or previous adverse reaction to salicylic acid compounds or prostaglandin synthetase inhibitors; physician decision that aspirin is required for proven indication; contraindication to enteral drug administration; treatment withdrawal imminent within 24 hours; consent declined Country: UK Setting: ICU |
| Interventions |
Intervention Group (aspirin)
Control group (placebo)
|
| Outcomes |
Primary outcome: OI (at day 7) Secondary outcomes: OI (at days 4 and 14); SOFA score (at days 4, 7 and 14); Crs (at days 4, 7 and 14); P/F ratio (at days 4, 7 and 14); safety and tolerability as assessed by the occurrence of serious adverse events and suspected unexpected serious adverse reactions (up to 28 days after completion of study drug) |
| Starting date | January 2015 |
| Contact information | Professor Danny F McAuley |
| Notes | Funding/declarations of interest: Belfast Health and Social Care Trust; Queen's University, Belfast; Northern Ireland Clinical Trials Unit |
NCT02595060.
| Trial name or title | GM‐CSF Inhalation to Improve Host Defense and Pulmonary Barrier Restoration (GI‐HOPE). A Randomized, Double‐blind, Parallel Group, Multicenter, Phase II Study |
| Methods | RCT |
| Participants |
Target participant recruitment: 45 Inclusion criteria: signed informed consent form by the patient or a legal representative; between 18 and 75 years of age; women who have been post‐menopausal for > 1 year or women of childbearing potential period using a highly efficient method of contraception (i.e. a method with < 1% failure rate such as combined hormonal contraception, progesterone‐only hormonal contraception; diagnosis of pneumonia‐associated ARDS, where the underlying condition is CAP or HAP in patients not on invasive ventilation upon diagnosis of HAP; diagnosis of ARDS according to the Berlin ARDS definition; requirement for positive pressure ventilation (non‐invasive or endotracheal tube) for > 72 hours in total with inspiratory oxygen concentration (FiO2) ≥ 50% (or less when on additional ECMO therapy) not > 14 days Exclusion criteria: receiving vasopressors of > 100 µg/min; history of liver cirrhosis Child Pugh C, chronic haemodialysis (before severe pneumonia/ARDS), lung cancer; malignancy with expected survival time of < 6 months; history of or listing for lung transplantation; highly immunosuppressive therapy or anti‐malignant combination chemotherapy within 3 weeks prior to first dose of study drug; any anti‐malignant chemotherapy within 24 hours prior to first dose of study drug; AIDS or known history of HIV infection; pregnancy; autoimmune thrombocytopenia, myelodysplastic syndromes with > 20% marrow blast cells; history or presence of hypersensitivity or idiosyncratic reaction to molgramostim or to related compounds; participation in another clinical trial within 90 days prior to the first dose of study drug Country: Germany Setting: multicentre; ICU |
| Interventions |
Intervention group (rhGM‐CSF; 150 mcg)
Intervention group (rhGM‐CSF; 450 mcg)
Control group (placebo)
|
| Outcomes |
Primary outcome: GI‐HOPE score representing changes at day 4/5 with respect to baseline (at baseline and day 4 and 5) Secondary outcomes: number of participants with adverse events, serious adverse events and adverse drug reactions (at baseline to 28 days); oxygenation (at baseline to day 11); APACHE (at baseline to day 11); SOFA (at baseline to day 11); extravascular lung water index (at baseline to day 11); C‐reactive Protein (at baseline to day 11); days on vasoactive drugs (at baseline to day 28); all‐cause mortality (at baseline to day 28); serum GM‐CSF (at baseline and days 1 to 4) |
| Starting date | January 2016 |
| Contact information | Email: susanne.herold@innere.med.uni‐giessen.de |
| Notes | Funding/declarations of interest: University of Giessen |
NCT02611609.
| Trial name or title | A phase 1/2 study to assess the safety and efficacy of MultiStem® therapy in subjects with acute respiratory distress syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: 36 Inclusion criteria: between 18 and 90 years of age; moderate to severe ARDS, as defined by the Berlin definition, requiring an endotracheal or tracheal tube; able to receive investigational medicinal product within 96 hours of meeting the last of the ARDS diagnosis criteria Exclusion criteria: concurrent illness that shortens life expectancy to < 6 months; other serious medical or psychiatric illness Country: USA and UK Setting: ICU |
| Interventions |
Intervention group (low dose MultiStem)
Intervention group (high dose MultiStem)
Control group
|
| Outcomes |
Primary outcomes: frequency of sustained hypoxaemia or hypotension (within 4 hours); SUSARs (within 24 hours) Secondary outcomes: frequency of adverse events (up to 365 days); changes in vital signs (up to 7 days); changes in blood safety laboratories (up to 7 days); VFDs (28 days); ICU‐free days (28 days); total length of hospital stay (28 days); all‐cause mortality (28 days); changes in levels of oxygenation (up to 28 days); changes in PEEP (up to 28 days); changes in respiratory physiologic measures including lung compliance and airway resistance (peak and plateau pressures) (up to day 365); all‐cause mortality (up to day 365) |
| Starting date | January 2016 |
| Contact information | Email: ating@athersysltd.co.uk |
| Notes | Funding/declarations of interest: Athersys Inc; Athersys Limited; Cell Therapy Catapult |
NCT02895191.
| Trial name or title | A randomized, blind, placebo‐controlled, parallel group, multicenter study to evaluate the safety and dose response relationship of ulinastatin for acute respiratory distress syndrome (ARDS) |
| Methods | RCT |
| Participants |
Target participant recruitment: 60 Inclusion criteria: provided signed written informed consent form from the patient or the patient's legal representative; men or women ≥ 18 years of age; ARDS defined using 2012 Berlin Criteria; ARDS diagnosed ≤ 7 days; mechanically ventilated (invasive or noninvasive or both); 100 mmHg < PaO2/FiO2 < 250 mmhg with CPAP/PEEP ≥ 5 cmH2O Exclusion criteria: patients with known hypersensitivity to ulinastatin/adjuvant or patient with allergic constitution; with artificial organ replacement therapy for liver or kidney; GCS ≤ 8; cardiogenic pulmonary oedema as the only or primary reason for respiratory failure; ARDS caused by burning, drowning, poisoning; presence of severe chronic liver diseases (Child‐Pugh score 12 ‐ 15) or severe chronic respiratory disease with a PaCO2 > 50 mmHg or the use of home oxygen; neutrophils < 1.5 × 109/L; moribund patients, or with recent cardiopulmonary arrest; needing long‐term glucocorticoid treatment or need to be treated with immunosuppressive drugs; no intent/unwillingness to follow lung protective ventilation strategy or fluid management protocol; lung transplant; with malignancy, expected to live no longer than 6 months; pregnant or breastfeeding; have participated in any clinical study within 3 months prior to the screening; with any condition that in the opinion of the investigator would add to the patient's risk or jeopardize the operation of the study Country: China Setting: ICU |
| Interventions |
Intervention group (Ulinastatin; 4.8 million units per day)
Intervention group (Ulinastatin; 2.4 million units per day)
Intervention group (Ulinastatin; 1.2 million units per day)
Control group
|
| Outcomes |
Primary outcome: incidence of adverse events (day 1 to 90) Secondary outcomes: changes of PaO2/FiO2 ratio (day 0 and 1 to within 24 hours after last treatment); days alive and off ventilator (day 1 to 28); days in the ICU (day 1 to 14); changes of pulmonary compliance (day 1, 3, 7, and within 24 hours after last treatment); rate of new organ failure (day 1 to 90); changes of APACHE Ⅱ score from baseline (day 3 ,7 and within 24 hours after last treatment); changes of Murray Lung Injury Score from baseline (day 3 ,7 and within 24 hours after last treatment); changes of SOFA score from baseline (day 3 ,7 and within 24 hours after last treatment); all‐cause mortality (day 28 ,90 and day 1 to 14) |
| Starting date | August 2016 |
| Contact information | Name: Dr Yimin Li |
| Notes | Funding/declarations of interest: First Affiliated Hospital of Guangzhou Medical University |
NCT03017547.
| Trial name or title | A phase 2, randomized, double‐blind, placebo‐controlled, preliminary efficacy study of IC14 in acute respiratory distress syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: 160 Inclusion criteria: ICU admission; between 18 and 70 years of age; presence of a known ARDS clinical risk within 7 days of onset; anticipated duration of mechanical ventilation > 48 hours Exclusion criteria: treatment with a drug or device within the last 30 days that has not received regulatory approval at the time of study entry; intubation for cardiopulmonary arrest; DNAR status; intubation for status asthmaticus, pulmonary embolus, myocardial infarction; anticipated survival < 48 hours from intubation; anticipated survival < 28 days due to pre‐existing medical condition; significant pre‐existing organ dysfunction; currently receiving home oxygen therapy as documented in medical record; pre‐existing congestive heart failure; chronic renal failure requiring renal replacement therapy; severe chronic liver disease; organ transplant recipient; chronic high‐dose corticosteroids; oncolytic drug therapy within the past 14 days; known HIV positive; current treatment with enbrel (etanercept), remicade (infliximab), humira (adalimumab), cimzia (certolizumab), or simponi (golimumab), kineret (anakinra), or arcalyst (rilonacept); pregnancy; history of hypersensitivity or idiosyncratic reaction to IC14; deprivation of freedom by administrative or court order Country: USA Setting: ICU |
| Interventions |
Intervention group (IC14)
Control group (placebo)
|
| Outcomes |
Primary outcomes: safety (28 days); VFDs (28 days) Secondary outcomes: change in ARDS biologic markers (28 days) |
| Starting date | 11 January 2017 |
| Contact information | Email: garry.redlich@implicitbioscience.com |
| Notes | Funding/declarations of interest: Implicit Bioscience |
NCT03042143.
| Trial name or title | Repair of acute respiratory distress syndrome by stromal cell administration (REALIST): an open label dose escalation phase 1 trial followed by a randomized, double‐blind, placebo‐controlled phase 2 trial |
| Methods | RCT |
| Participants |
Target participant recruitment: 75 Inclusion criteria: ≥ 16 years of age; ARDS as defined by the Berlin definition; onset within 1 week of identified insult; within the same 24 hours time period; hypoxic respiratory failure (PaO2/ FiO2 ratio ≤ 27 kPa on PEEP ≥ 5 cmH2O); bilateral infiltrates on chest X‐ray consistent with pulmonary oedema not explained by another pulmonary pathology; respiratory failure not fully explained by cardiac failure or fluid overload; receiving invasive mechanical ventilation Exclusion criteria: > 48 hours from the onset of ARDS; < 16 years of age; pregnant; participation in clinical trial of an investigational medicinal product within 30 days; major trauma in the prior 5 days; presence of any active malignancy (other than non‐melanoma skin cancer) that required treatment within the last year; WHO Class III or IV pulmonary hypertension; venous thromboembolism currently receiving anticoagulation or within the past 3 months; currently receiving ECLS; severe chronic liver disease with Child‐Pugh score > 12; DNAR order in place; treatment withdrawal imminent within 24 hours; consent declined; prisoners; non‐English‐speaking patients or those who do not adequately understand verbal or written information unless an interpreter is available; previously enrolled in the REALIST trial Country: UK Setting: ICU |
| Interventions |
Intervention group (MSC)
Control group (placebo)
|
| Outcomes |
Primary outcomes: OI (at day 7); incidence of serious adverse events (at 28 days) Secondary outcomes: OI (days 4 and 14); SOFA score (days 4, 7 and 14); Crs (days 4, 7 and 14); P/F ratio (days 4, 7 and 14) |
| Starting date | 22 November 2018 |
| Contact information | Email: d.f.mcauley@qub.ac.uk |
| Notes | Funding/declarations of interest: Belfast Health and Social Care Trust; Queen's University, Belfast; Northern Ireland Clinical Trials Unit; NHS Blood and Transplant |
NCT03202394.
| Trial name or title | A phase IIa, placebo controlled, multicenter pilot study to evaluate the safety and efficacy of BIO‐11006 inhalation solution in patients with acute respiratory distress syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: 60 Inclusion criteria: patients between 18 and 75 years of age; provided (or relative has) written informed consent and authorisation for use and disclosure of protected health information; has a clinical diagnosis of sepsis or septic shock; enrolment must occur within 48 hours of first meeting ARDS criteria according to the Berlin definition of ARDS (ARDS Definition Task Force 2012) and no more than 72 hours from the initiation of mechanical ventilation Exclusion criteria: < 18 or > 75 years of age; > 48 hours since first meeting ARDS criteria according to the Berlin definition of ARDS; pregnant or breastfeeding; prisoner; any other irreversible disease or condition for which 6‐month mortality is estimated to be > 50%; moderate to severe liver failure (Child Pugh Score > 12); severe chronic respiratory disease with a PaCO2 > 50 mmHg or the use of home oxygen; patient, surrogate, or physician not committed to full support; major trauma in the prior 5 days; lung transplant patient; no consent/inability to obtain consent; moribund patient not expected to survive 24 hours; WHO Functional; class III or IV pulmonary hypertension; no intent/unwillingness to follow lung protective ventilation strategy or fluid management protocol; currently receiving extracorporeal life support or high‐frequency oscillatory ventilation; known hypersensitivity to BIO 11006; burn victims > 20% TBSA or with known airway inhalation injury Country: USA Setting: multicentre; ICU |
| Interventions |
Intervention group (BIO‐11006)
Control group (placebo)
|
| Outcomes |
Primary outcome: incidence of treatment‐emergent adverse events (at 28 days) Secondary outcomes: mortality (at 28 days and 180 days); number of ICU‐free days (at 28 days); VFDs (at 28 days); change in S/F ratio (at 28 days); change in pro‐inflammatory biomarkers from baseline to end of treatment (pretreatment and end of treatment period) |
| Starting date | 5 August 2017 |
| Contact information | Email: bdickson@biomarck.com |
| Notes | Funding/declarations of interest: BioMarck Pharmaceuticals, Ltd. |
NCT03346681.
| Trial name or title | NAC in early acute respiratory distress syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: 52 Inclusion criteria: adult patients admitted to the medical intensive care unit or coronary care unit; mechanically ventilated with a positive end expiratory pressure > 5 cmH2O, with noncardiogenic pulmonary oedema on chest x‐ray within 48 hours of being noted to have a P/F ratio < 150 Exclusion criteria: patients < 18 years of age; patients for whom no aggressive measures are desired; already receiving "rescue methods" (prone positioning, advanced ventilator modes, paralytics); trauma patients; vulnerable patient groups (pregnant, prisoners); have undergone a surgical operation during their time on the ventilator; with end‐stage liver disease; on chronic ventilators; asthmatics Country: USA Setting: ICU |
| Interventions |
Intervention group (NAC)
Control group
|
| Outcomes |
Primary outcome: ventilator days (from time of intubation until one of predefined endpoints (up to 60 days)) Secondary outcome: ICU days (from time of admission to the ICU until transfer out of the unit (up to 60 days)); mortality (up to 60 days); P/F ratio (daily until predefined endpoints (up to 60 days)); use of "rescue" manoeuvre (daily until the predefined endpoints (up to 60 days)) |
| Starting date | 1 February 2018 |
| Contact information | Email: drjudlewis@gmail.com |
| Notes |
NCT03371498.
| Trial name or title | Procollagen‐3 driven corticosteroids for persistent acute respiratory distress syndrome |
| Methods | RCT |
| Participants |
Target participant recruitment: 356 Inclusion criteria: ≥ 18 years of age; continuous endotracheal ventilation; moderate to severe ARDS according to Berlin definition with PaO2/FiO2 ≤ 200 with PEEP ≥ 5 cmH2O; date of ARDS onset: ≥ day 5 and ≤ day 14 after the onset of ARDS criteria (regardless of ARDS severity); procollagen III > 9 µg/L in a bronchoalveolar lavage performed by the attending physician between days 3 and 13 after the onset of ARDS and realized within 5 days prior to randomization Exclusion criteria: known pregnancy or breastfeeding; participation to another interventional trial within 30 days with mortality or VFDs as the main endpoint; clinical evidence of active untreated infection; known, undrained abscess; intravascular nidus of infection; disseminated fungal infection Country: France Setting: ICU |
| Interventions |
Intervention group (methylprednisolone)
Control group (placebo)
|
| Outcomes |
Primary outcome: ventilator‐free days (at 60 days) Secondary outcome: ICU and hospital mortality (at 90 days) |
| Starting date | 15 January 2018 |
| Contact information | Email: jean‐marie.forel@ap‐hm.fr |
| Notes | Funding/declarations of interest: Assistance Publique Hopitaux De Marseille |
NCT03608592.
| Trial name or title | Human umbilical cord derived mesenchymal stem cells therapy in acute respiratory distress syndrome: a pilot study |
| Methods | RCT |
| Participants |
Target participant recruitment: 12 Inclusion criteria: ≥ 18 years of age; invasive ventilation, OI (PaO2/FiO2) < 200; PEEP ≥ 8 cmH2O; bilateral infiltration of lung in X‐ray or CT; 1 week after onset; still OI < 200 after protective ventilation or conservative fluid management Exclusion criteria: any malignant disease; cardiogenic pulmonary oedema; > 50% atelectasis either lung lobe in X‐ray; pregnancy or perinatal or lactation; previous end‐stage respiratory disease; > 3 organs failure; liver failure with MELD score > 40; stage III or IV pulmonary hypertension; noninvasive arterial and central venous catheter; concurrent deep venous thrombus or pulmonary embolism in 3 months; cerebral hernia; > 96 hours after ARDS onset Country: China Setting: ICU |
| Interventions |
Intervention group (UCMSCs)
Control group (placebo)
|
| Outcomes |
Primary outcome: infusion‐associated events (from infusion beginning to the second day, 24 hours) Secondary outcomes: VFDs (from the day of UCMSCs use to day 28); OI changes (day 0 to 7); LIS (day 0, 1, 3, 7); PEEP (day 0 to 7); plateau pressure (day 0 to 7); driving pressure (day 0 to 7); static compliance (day 0 to 7) |
| Starting date | August 2018 |
| Contact information | Email: ylhmin@hotmail.com |
| Notes | Funding/declarations of interest: Sun Yat‐sen University |
Villar 2016.
| Trial name or title | Evaluating the efficacy of dexamethasone in the treatment of patients with persistent acute respiratory distress syndrome: Study protocol for a randomized controlled trial |
| Methods | RCT |
| Participants |
Target participant recruitment: 314 Inclusion criteria: ≥18 years of age; acute onset of ARDS, as defined by the AECC criteria for ARDS (Bernard 1994); be intubated and mechanically ventilated; signed written informed consent from the patient or the patient's personal legal representative Exclusion criteria: pregnant or lactating; involved in another experimental treatment protocol; brain death; terminal‐stage cancer or other terminal disease; do‐not‐resuscitate orders; immune‐compromised; receiving corticosteroids or immunosuppressive drugs; > 24 hours had elapsed after initially meeting the AECC ARDS criteria (Bernard 1994) before consent and results of initial standard ventilator settings could be obtained; severe COPD; congestive heart failure Country: Spain Setting: ICU |
| Interventions |
Intervention group (dexamethasone)
Control group (standard therapy)
|
| Outcomes |
Primary outcome: VFDs (at 28 days) Secondary outcomes: mortality (at 60 days); organ failure (at ICU discharge) |
| Starting date | April 2013 |
| Contact information | Email: jesus.villar54@gmail.com |
| Notes | Funding/declarations of interest: Dr Negrin University Hospital; Fundación Mutua Madrileña; Asociación Científica Pulmón y Ventilación Mecánica |
AECC: American‐European consensus conference (Bernard 1991); AIDS: acquired immunodeficiency syndrome; ALI: acute lung injury; APACHE: acute physiology and chronic health evaluation; aPC: activated protein C; APTT: activated partial thromboplastin time; ARDS: acute respiratory distress syndrome; BIO‐11006: drug name (10‐amino acid peptide developed as a potential treatment for COPD); BIPAP: bi‐level positive airway pressure; CAP: community‐acquired pneumonia; COPD: chronic obstructive pulmonary disease; CPAP: continuous positive airway pressure; Crs: respiratory compliance; CT: computed tomography; DNAR: do not attempt resuscitation; ECLS: extracorporeal life support; ECMO: extracorporeal membrane oxygenation; EQ5D: EuroQOL five dimensions questionnaire; FiO2: fraction of inspired oxygen; FP‐1201‐lyo: intravenously administered recombinant human interferon beta‐1a; GCS: Glasgow coma scale; GI‐HOPE: Host defence and pulmonary barrier restoration; GM‐CSF: granulocyte‐macrophage colony stimulating factor; HAP: hospital‐acquired pneumonia; HFOV: high‐frequency oscillatory ventilation; HLA‐D: human leucocyte antigen – antigen D related; HMGB‐1: high mobility group box 1 protein; hucMSC: human derived mesenchymal stromal cells; IC14: drug name (recombinant chimeric human/murine monoclonal antibody); ICU: intensive care unit; IFN: interferon; IU: international unit; IV: intravenous; LIS: lung injury score; MELD: model for end‐stage liver disease; MSC: mesenchymal stem cell; NS: normal saline; NYHA: New York Heart Association; OI: oxygenation index; PaCO2: partial pressure of carbon dioxide in the arterial blood; PaO2: partial pressure of arterial oxygen; PCT: procalcitonin; PEEP: positive end‐expiratory pressure; P/F ratio: partial pressure of arterial oxygen to the fraction of inspired oxygen ratio; PIP: peak inspiratory pressure; RCT: randomized control trial; rhGM‐CSF: molgramostim; SOFA: sequential organ failure assessment; S/F ratio: oxygen saturation to fraction of inspired oxygen ratio; SF‐36: short form health survey; SUSARs: suspected unexpected serious adverse reactions; TBSA: total body surface area; TNF‐a: tumour necrosis factor UCMSCs: umbilical cord derived mesenchymal stem cells suspension; VFDs: ventilation‐free days; WHO: World Health Organization
Differences between protocol and review
Differences between the updated review and the previous version (Adhikari 2004)
We changed the title of the review to reflect the more recent Berlin definition of ARDS (ARDS Definition Task Force 2012).
We changed the author team of the review. Authors of the previous version of this review were no longer involved in the review conduct (Adhikari 2004).
We added an extra outcome (fitness to return to work after 12 months) to reflect the long‐term consequences of ARDS (Herridge 2003).
Types of interventions: we excluded studies of inhaled prostacyclins because these are reviewed elsewhere. We clarified exclusion of neuromuscular blocking agents which are used as part of a mechanical ventilation strategy. We excluded studies that were published before 2000, in order to reflect current guidelines for lung protection strategies (FICM/ICS Guideline Development Group 2018).
Search strategies: we amended our strategies to include additional MeSH terms, and to incorporate current RCT filters. We did not include a search of Healthstar database because we believed that the remaining databases were sufficiently broad in scope for this review.
Sensitivity analysis: we expanded the number of factors in sensitivity analyses in order to explore more comprehensively the decisions we made during the review process. We explored the effects of excluding studies with high or unclear risk of selection bias (rather than allocation concealment), and also included sensitivity analyses regarding use of lower tidal volumes, studies with a high risk of attrition bias, using the alternative meta‐analytic effects model, and the use of data in multi‐arm studies. However, we did not explore the effect of whether co‐interventions were balanced between intervention and control groups.
All sections of the review: we re‐wrote all sections using current MECIR standards, and incorporating standard Cochrane subheadings. The review now also incorporates GRADE assessments and 'Summary of findings' tables.
Excluded studies: we have removed studies previously included. We have reported only key excluded studies identified during the 2018 search.
Contributions of authors
Conceiving the review: previous review author team (Adhikari 2004) Co‐ordinating the review: SL Undertaking manual searches: SL, MP, CT Screening search results: SL, MP, CT Organizing retrieval of papers: SL. CT, MP Screening retrieved papers against inclusion criteria: SL, CT, MP Appraising quality of papers: SL, CT, MP, AS Abstracting data from papers: SL, CT, MP, AS Managing data for the review: SL Entering data into Review Manager 5 (Review Manager 2014): SL, MP, CT Analysing Review Manager 5 statistical data: SL Interpreting data: SL Writing the review: SL, MP, CT Securing funding for the Review: AS Guarantor for the review: AS
Sources of support
Internal sources
No sources of support supplied
External sources
NIHR Cochrane Incentive Awards Scheme 2018, UK.
Declarations of interest
Sharon R Lewis: none known Michael W Pritchard: none known Carmel M Thomas: none known Andrew F Smith: none known
Edited (no change to conclusions)
References
References to studies included in this review
ALTA 2011 {published data only}
- Matthay MA, Brower RG, Carson S, Douglas IS, Eisner M, Hite D. Randomized, placebo‐controlled clinical trial of an aerosolized β2‐agonist for treatment of acute lung injury. American Journal of Respiratory and Critical Care Medicine 2011;184(5):561‐8. [PUBMED: 21562125] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Brower RG, Carson S, Douglas IS, Eisner M, Hite D, et al. Erratum: randomized, placebo‐controlled clinical trial of an aerosolized β2‐agonist for treatment of acute lung injury. American Journal of Respiratory and Critical Care Medicine 2013;188(11):1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
ARDS Network 2002 {published data only}
- The ARDS Clinical Trials Network, National Heart, Lung and Blood Institute, National Institutes of Health. Randomized, placebo‐controlled trial of lisofylline for early treatment of acute lung injury and acute respiratory distress syndrome. Critical Care Medicine 2002;30(1):1‐6. [PUBMED: 11902249] [DOI] [PubMed] [Google Scholar]
BALTI 2006 {published data only}
- Gao F, Thickett DR. In vivo and in vitro effects of salbutamol on alveolar epithelial repair in acute lung injury. Thorax 2008;63(3):215‐20. [PUBMED: 17951278] [DOI] [PubMed] [Google Scholar]
- Perkins GD, McAuley DF, Thickett DR, Gao F. The ß‐agonist lung injury trial (BALTI). American Journal of Respiratory and Critical Care Medicine 2006;173(3):281‐7. [PUBMED: 16254268] [DOI] [PubMed] [Google Scholar]
BALTI‐2 2013 {published data only}
- Gao Smith F, Perkins GD, Gates S, Young D, McAuley DF, Tunnicliffe W, et al. Effect of intravenous beta‐2 agonist treatment on clinical outcomes in acute respiratory distress syndrome (BALTI‐2): a multicentre, randomised controlled trial. Lancet 2012;379(9812):229‐35. [PUBMED: 22166903] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gates S, Perkins GD, Lamb SE, Kelly C, Thickett DR, Young JD, et al. Beta‐agonist lung injury trIal‐2 (BALTI‐2): a multicentre, randomised, double‐blind, placebo‐controlled trial and economic evaluation of intravenous infusion of salbutamol versus placebo in patients with acute respiratory distress syndrome. Health Technology Assessment 2013;17(38):v‐vi: 1‐87. [PUBMED: 24028755] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins GD, Gates S, Lamb SE, McCabe C, Young D, Gao F. Beta agonist lung injury trIal‐2 (BALTI‐2) trial protocol: a randomised, double‐blind, placebo‐controlled of intravenous infusion of salbutamol in the acute respiratory distress syndrome. Trials 2011;12:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barrese‐Perez 2015 {published data only}
- Barrese‐Perez Y, Hidalgo‐Sanchez AO, Avila‐Albuerne Y, Uranga‐Pina R, Diaz‐Casanas E, Fernandez‐Limia O. Pulmonary surfactant exogenous in adults with acute respiratory distress syndrome. Revista del Instituto Nacional de Enfermedades Respiratorias 2015;74(3):176‐8. [Google Scholar]
Chen 2017 {published data only}
- Chen Y, Luo JJ, Jiang DX, Lin YP, Tong HS, Su L. Effect of ulinastatin on acute lung injury induced by severe heat stroke and its mechanism. Medical Journal of Chinese People's Liberation Army 2017;42(4):301‐6. [Embase: 616874253] [Google Scholar]
Endo 2006 {published data only}
- Endo S, Sato N, Yaegashi Y, Suzuki Y, Kojika M, Yamada Y, et al. Sivelestat sodium hydrate improves septic acute lung injury by reducing alveolar dysfunction. Research Communications in Molecular Pathology and Pharmacology 2006;119(1‐6):53‐65. [PUBMED: 17974096] [PubMed] [Google Scholar]
Guoshou 2013 {published data only}
- Guoshou Z, Chengye Z, Zhihui L, Jinlong L. Effects of high dose of anisodamine on the respiratory function of patients with traumatic acute lung injury. Cell Biochemistry and Biophysics 2013;66(2):365‐9. [PUBMED: 23504631] [DOI] [PubMed] [Google Scholar]
HARP 2011 {published data only}
- Brealey D, Hargreaves I, Land J, Singer M, McAuley D. Plasma ubiquinone levels are unaffected by simvastatin in acute lung injury. Intensive Care Medicine 2012;38:S64. [Google Scholar]
- Craig TR, Duffy MJ, Shyamsundar M, McDowell C, O'Kane CM, Elborn JS, et al. A randomized clinical trial of hydroxymethylglutaryl‐coenzyme a reductase inhibition for acute lung injury (the HARP study). American Journal of Respiratory and Critical Care Medicine 2011;183(5):620‐6. [PUBMED: 20870757] [DOI] [PubMed] [Google Scholar]
- Craig TR, Duffy MJ, Shyamsundar M, McDowell C, O'Kane CM, Elborn JS, et al. Erratum: a randomized clinical trial of hydroxymethylglutaryl‐coenzyme a reductase inhibition for acute lung injury (the HARP study). American Journal of Respiratory and Critical Care Medicine 2014;190(10):1199‐200. [DOI] [PubMed] [Google Scholar]
- Craig TR, Duffy MJ, Shyamsundar M, O'Kane C, Elborn JS, McAuley DF. Simvastatin reduces inflammation and improves clinical outcomes in ALI: results of The HARP Study. Thorax 2009;64:A2. [Google Scholar]
- Craig TR, Duffy MJ, Shyamsundar M, O'Kane CM, Elborn JS, McAuley DF. Results of the HARP study: a randomized double blind phase II trial of 80mg simvastatin in acute lung injury. American Journal of Respiratory and Critical Care Medicine. Conference: American Thoracic Society International Conference, ATS. 2010; Vol. 181:no pagination.
- McAuley DF, O'Kane CM, Craig TR, Shyamsundar M, Herwald H, Dib K. Simvastatin decreases the level of heparin‐binding protein in patients with acute lung injury. BMC Pulmonary Medicine 2013;13:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
HARP‐2 2014 {published data only}
- Laffey JG, O'Kane CM, Cross M, Perkins GD, Murphy L, McNally C, et al. Hydroxymethylglutaryl‐CoA reductase inhibition with simvastatin in acute lung injury to reduce pulmonary dysfunction (HARP‐2) trial: study protocol for a randomized controlled trial. Trials 2012;13(170):1‐9. [PUBMED: 22985805] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley DF, Laffey JG, O'Kane CM, Perkins GD, Mullan B, Trinder T, et al. Simvastatin in the acute respiratory distress syndrome. New England Journal of Medicine 2014;371(18):1695‐703. [PUBMED: 25268516] [DOI] [PubMed] [Google Scholar]
- McAuley DF, Laffey JG, O'Kane CM, Perkins GD, Mullan B, Trinder TJ, et al. SImvastatin to reduce pulmonary dysfunction in patients with acute respiratory distress syndrome: the HARP‐2 RCT. Efficacy and Mechanism Evaluation 2018;5(1):no pagination. [PUBMED: 29400921] [PubMed] [Google Scholar]
Huang 2017 {published data only}
- Huang SR, Ma AY, Liu Y, Qu Y. Effects of inflammatory factors including plasma tumor necrosis factor‐alpha in the clinical treatment of acute respiratory distress syndrome. Oncology Letters 2017;13(6):5016‐20. [PUBMED: 28599503] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ji 2018 {published data only}
- Ji M, Chen T, Wang B, Chen M, Ding Q, Chen L, et al. Effects of ulinastatin combined with mechanical ventilation on oxygen metabolism, inflammation and stress response and antioxidant capacity of ARDS. Experimental and Therapeutic Medicine 2018;15(6):4665‐70. [PUBMED: 29805484] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kadoi 2004 {published data only}
- Kadoi Y, Hinohara H, Kunimoto F, Saito S, Goto F, Kosaka T, et al. Pilot study of the effects of ONO‐5046 in patients with acute respiratory distress syndrome. Anesthesia and Analgesia 2004;99(3):872‐7. [PUBMED: 15333424] [DOI] [PubMed] [Google Scholar]
KARE 2017 {published data only}
- Cross LJ, O'Kane CM, McDowell C, Elborn JJ, Matthay MA, McAuley DF. Erratum: keratinocyte growth factor in acute lung injury to reduce pulmonary dysfunction ‐ a randomised placebo‐controlled trial (KARE): study protocol. Trials 2017;18(1):214. [PUBMED: 28494790] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross LJ, O'Kane CM, McDowell C, Elborn JJ, Matthay MA, McAuley DF. Keratinocyte growth factor in acute lung injury to reduce pulmonary dysfunction ‐ a randomised placebo‐controlled trial (KARE): study protocol. Trials 2013;14:51. [PUBMED: 23419093] [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAuley DF, Cross LJ, Hamid U, Gardner E, Elborn JS, Cullen KM, et al. Keratinocyte growth factor for the treatment of the acute respiratory distress syndrome (KARE): a randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Respiratory Medicine 2017;5(6):484‐91. [PUBMED: 28526233] [DOI] [PubMed] [Google Scholar]
KARMA 2000 {published data only}
- National Heart Lung and Blood Institute, The ARDS Clinical Trials Network. Ketoconazole for early treatment of acute lung injury and acute respiratory distress syndrome: a randomized controlled trial. JAMA 2000;283(15):1995‐2002. [PUBMED: 10789668] [DOI] [PubMed] [Google Scholar]
Kesecioglu 2001 {published data only}
- Kesecioglu J, Schultz MJ, Lundberg D, Lauven PM, Lachmann B. Treatment of acute lung injury (ALI/ARDS) with surfactant. American Journal of Respiratory and Critical Care Medicine 2001;163(5 Pt. 2):A819. [Google Scholar]
Kesecioglu 2009 {published data only}
- Kesecioglu J, Beale R, Stewart TE, Findlay GP, Rouby JJ, Holzapfel L, et al. Exogenous natural surfactant for treatment of acute lung injury and the acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine 2009;180(10):989‐94. [PUBMED: 19713451] [DOI] [PubMed] [Google Scholar]
Khan 2017 {published data only}
- Khan A, Benthin C, Zeno B, Albertson T, Boyd J, Christie J, et al. A pilot clinical trial of recombinant human angiotensin‐converting enzyme 2 in acute respiratory distress syndrome. Critical Care 2017;21(1):234. [PUBMED: 28877748] [DOI] [PMC free article] [PubMed] [Google Scholar]
Krenn 2017 {published data only}
- Krenn K, Lucas R, Croizé A, Boehme S, Klein KU, Hermann R, et al. Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: a phase IIa randomized placebo‐controlled trial. Critical Care 2017;21(1):194. [PUBMED: 28750677] [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2010 {published data only}
- Li BQ, Sun HC, Nie SN, Shao DB, Liu HM, Qian XM. Effect of penehyclidine hydrochloride on patients with acute lung injury and its mechanisms. Chinese Journal of Traumatology ‐ English Edition 2010;13(6):329‐35. [PUBMED: 21126389] [PubMed] [Google Scholar]
Liu 2012 {published data only}
- Liu L, Li J, Huang YZ, Liu SQ, Yang CS, Guo FM, et al. The effect of stress dose glucocorticoid on patients with acute respiratory distress syndrome combined with critical illness‐related corticosteroid insufficiency. Chinese Journal of Internal Medicine 2012;51(8):599‐603. [PUBMED: 23158856] [PubMed] [Google Scholar]
Liu 2015 {published data only}
- Liu LP, Li B, Zhu L, Dou ZM, Li YM. Clinical efficacy of nitroglycerin in patients with septic shock with ARDS. Medical Journal of Chinese People's Liberation Army 2015;40(8):647‐51. [EMBASE: 2015326178] [Google Scholar]
Liu 2017 {published data only}
- Liu LP, Hu SW, Shuai TK, Deng YY, Wang LJ, Li YM. Clinical effect of alprostadil in patients with septic shock associated with acute respiratory distress syndrome. Medical Journal of Chinese People's Liberation Army 2017;42(9):805‐9. [Embase: 20170777094] [Google Scholar]
Meduri 2007 {published data only}
- Meduri GU, Golden E, Freire AX, Taylor E, Zaman M, Carson SJ, et al. Methylprednisolone infusion in early severe ARDS results of a randomized controlled trial. Chest 2009;136(5 Suppl):e30. [Embase: 358384513] [DOI] [PubMed] [Google Scholar]
- Meduri GU, Golden E, Freire AX, Taylor E, Zaman M, Carson SJ, et al. Methylprednisolone infusion in early severe ARDS: results of a randomized controlled trial. Chest 2007;131(4):954‐63. [PUBMED: 17426195] [DOI] [PubMed] [Google Scholar]
Mohamed 2017 {published data only}
- Mohamed HS, Meguid MM. Effect of nebulized budesonide on respiratory mechanics and oxygenation in acute lung injury/acute respiratory distress syndrome: randomized controlled study. Saudi Journal of Anaesthesia 2017;11(1):9‐14. [PUBMED: 28217046] [DOI] [PMC free article] [PubMed] [Google Scholar]
Morelli 2006 {published data only}
- Morelli A, Teboul JL, Maggiore SM, Vieillard‐Baron A, Rocco M, Conti G, et al. Effects of levosimendan on right ventricular afterload in patients with acute respiratory distress syndrome: a pilot study. Critical Care Medicine 2006;34(9):2287‐93. [PUBMED: 16791109] [DOI] [PubMed] [Google Scholar]
Morris 2008 {published data only}
- Morris PE, Papadakos P, Russell JA, Wunderink R, Schuster DP, Truwit JD, et al. A double‐blind placebo‐controlled study to evaluate the safety and efficacy of L‐2‐oxothiazolidine‐4‐carboxylic acid in the treatment of patients with acute respiratory distress syndrome. Critical Care Medicine 2008;36(3):782‐8. [PUBMED: 18209670] [DOI] [PubMed] [Google Scholar]
Najafi 2009 {published data only}
- Najafi A, Mojtahedzadeh M, Mahmoodpoor A, Aghamohammadi M, Ahmadi A, Nahreini S, et al. Effect of N‐acetylcysteine on microalbuminuria in patients with acute respiratory distress syndrome. Archives of Medical Science 2009;5(3):408‐4. [EMBASE: 358052253] [Google Scholar]
Ortolani 2000 {published data only}
- Ortolani O, Conti A, Gaudio AR, Masoni M, Novelli G. Protective effects of N‐acetylcysteine and rutin on the lipid peroxidation of the lung epithelium during the adult respiratory distress syndrome. Shock 2000;13(1):14‐8. [PUBMED: 10638663] [DOI] [PubMed] [Google Scholar]
Paine 2012 {published data only}
- Coley C, Dechert R, Harding S, Paine R, Moss M, Martin GS, et al. Granulocyte macrophage colony stimulating factor is associated with decreased quality of life at 180 days post‐event when administered to patients with acute lung injury. American Journal of Respiratory and Critical Care Medicine. Conference: American Thoracic Society International Conference, ATS 2012;185:no pagination. [Embase: 71986815] [Google Scholar]
- Paine R, Standiford TJ, Dechert RE, Moss M, Martin GS, Rosenberg AL, et al. A randomized trial of recombinant human granulocyte‐macrophage colony stimulating factor for patients with acute lung injury. Critical Care Medicine 2012;40(1):90‐7. [PUBMED: 21926600] [DOI] [PMC free article] [PubMed] [Google Scholar]
Rezk 2013 {published data only}
- Rezk NA, Ibrahim AM. Effects of methyl prednisolone in early ARDS. Egyptian Journal of Chest Diseases and Tuberculosis 2013;62(1):167‐72. [EMBASE: 369315254] [Google Scholar]
Ryugo 2006 {published data only}
- Ryugo M, Sawa Y, Takano H, Matsumiya G, Iwai S, Ono M, et al. Effect of a polymorphonuclear elastase inhibitor (sivelestat sodium) on acute lung injury after cardiopulmonary bypass: findings of a double‐blind randomized study. Surgery Today 2006;36(4):321‐6. [PUBMED: 16554988] [DOI] [PubMed] [Google Scholar]
SAILS 2014 {published data only}
- Dinglas VD, Hopkins RO, Wozniak AW, Hough CL, Morris PE, Jackson JC, et al. One‐year outcomes of rosuvastatin versus placebo in sepsis‐associated acute respiratory distress syndrome: prospective follow‐up of SAILS randomised trial. Thorax 2016;71(5):401‐10. [PUBMED: 26936876] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu RK, Truwit JD, Matthay MA, Levitt JE, Thompson BT, Liu KD. Effect of rosuvastatin on acute kidney injury in sepsis‐associated acute respiratory distress syndrome. Canadian Journal of Kidney Health and Disease 2018;5:1‐8. [PUBMED: 30116543] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needham DM, Colantuoni E, Dinglas VD, Hough CL, Wozniak AW, Jackson JC, et al. Rosuvastatin versus placebo for delirium in intensive care and subsequent cognitive impairment in patients with sepsis‐associated acute respiratory distress syndrome: an ancillary study to a randomised controlled trial. Lancet Respiratory Medicine 2016;4(3):203‐12. [PUBMED: 26832963] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers A, Guan J, Hunninghake GM, Trtchounian A, Liu K, Matthay MA, et al. Inflammasome activation is associated with ARDS mortality, particularly in patients randomized to statin therapy. American Journal of Respiratory and Critical Care Medicine 2017;195:no pagination. [Embase: 617705951] [Google Scholar]
- Truwit JD, Bernard GR, Steingrub J, Matthay MA, Liu K, Albertson TE, et al. Sails: Statins for acutely injured lungs (ARDS) from sepsis. American Journal of Respiratory and Critical Care Medicine. Conference: American Thoracic Society International Conference, ATS 2014;189:no pagination. [Embase: 72047035] [Google Scholar]
- Truwit JD, Bernard GR, Steingrub J, Matthay MA, Liu KD, Albertson TE, et al. Rosuvastatin for sepsis‐associated acute respiratory distress syndrome. New England Journal of Medicine 2014;370(23):2191‐200. [PUBMED: 24835849] [DOI] [PMC free article] [PubMed] [Google Scholar]
Soltan‐Sharifi 2007 {published data only}
- Soltan‐Sharifi MS, Mojtahedzadeh M, Najafi A, Reza Khajavi M, Reza Rouini M, Moradi M, et al. Improvement by N‐acetylcysteine of acute respiratory distress syndrome through increasing intracellular glutathione, and extracellular thiol molecules and anti‐oxidant power: evidence for underlying toxicological mechanisms. Human and Experimental Toxicology 2007;26(9):697‐703. [PUBMED: 17984140] [DOI] [PubMed] [Google Scholar]
Spragg 2002a {published data only}
- Spragg RG, Lewis JF, Rathgeb F, Hafner D, Seeger W. Intratracheal instillation of rSP‐C surfactant improves oxygenation in patients with ARDS. American Journal of Respiratory and Critical Care Medicine 2002;165(5 Pt. 2):A22. [Google Scholar]
Spragg 2002b {published data only}
- Spragg RG, Lewis JF, Rathgeb F, Hafner D, Seeger W. Intratracheal instillation of rSP‐C surfactant improves oxygenation in patients with ARDS. American Journal of Respiratory and Critical Care Medicine 2002;165(5 Pt. 2):A22. [Google Scholar]
Spragg 2003 {published data only}
- Spragg RG, Lewis JF, Wurst W, Häfner D, Baughman RP, Wewers MD. Treatment of acute respiratory distress syndrome with recombinant surfactant protein C surfactant. American Journal of Respiratory and Critical Care Medicine 2003;185(1):1562‐6. [PUBMED: 12649125] [DOI] [PubMed] [Google Scholar]
START 2018 {published data only}
- Liu KD, Wilson JG, Zhuo H, Caballero L, McMillan ML, Fang X, et al. Design and implementation of the START (stem cells for ARDS treatment) trial, a phase 1/2 trial of human mesenchymal stem/stromal cells for the treatment of moderate‐severe acute respiratory distress syndrome. Annals of Intensive Care 2014;4(1):1‐9. [PUBMED: 25593740] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthay MA, Calfee CS, Zhuo H, Thompson BT, Wilson JG, Levitt JE, et al. Treatment with allogeneic mesenchymal stromal cells for moderate to severe acute respiratory distress syndrome (START study): a randomised phase 2a safety trial. Lancet Respiratory Medicine 2019; Vol. 7, issue 2:154‐62. [PUBMED: 30455077] [DOI] [PMC free article] [PubMed]
Steinberg 2006 {published data only}
- Meduri GU, Bridges L, Siemieniuk RA, Kocak M. An exploratory reanalysis of the randomized trial on efficacy of corticosteroids as rescue therapy for the late phase of acute respiratory distress syndrome. Critical Care Medicine 2018;46(6):884‐91. [PUBMED: 29432350] [DOI] [PubMed] [Google Scholar]
- Steinberg KP, Hudson LD, Goodman RB, Hough CL, Lanken PN, Hyzy R, et al. Efficacy and safety of corticosteroids for persistent acute respiratory distress syndrome. New England Journal of Medicine 2006;354(16):1671‐84. [PUBMED: 16625008] [DOI] [PubMed] [Google Scholar]
STRIVE 2004 {published data only}
- Zeiher BG, Artigas A, Vincent JL, Dmitrienko A, Jackson K, Thompson BT, et al. Neutrophil elastase inhibition in acute lung injury: results of the STRIVE study. Critical Care Medicine 2004;32(8):1695‐702. [PUBMED: 15286546] [DOI] [PubMed] [Google Scholar]
Tongyoo 2016 {published data only}
- Tongyoo S, Permpikul C, Mongkolpun W, Vattanavanit V, Udompanturak S, Kocak M, et al. Hydrocortisone treatment in early sepsis‐associated acute respiratory distress syndrome: results of a randomized controlled trial. Critical Care 2016;20(1):329. [PUBMED: 27741949] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tongyoo S, Permpikul C, Vattanavanit V, Mongkolpun W. Hydrocortisone in treatment of severe sepsis and septic shock with acute respiratory distress syndrome: a randomised controlled trial. Intensive Care Medicine Experimental 2015;3:no pagination. [Embase: 611853166] [Google Scholar]
Tsangaris 2007 {published data only}
- Tsangaris I, Galiatsou E, Kostanti E, Nakos G. The effect of exogenous surfactant in patients with lung contusions and acute lung injury. Intensive Care Medicine 2007;33(5):851‐5. [PUBMED: 17377767] [DOI] [PubMed] [Google Scholar]
Vincent 2001 {published data only}
- Vincent JL, Brase R, Santman F, Suter PM, McLuckie A, Dhainaut JF, et al. A multi‐centre, double‐blind, placebo‐controlled study of liposomal prostaglandin E1 (TLC C‐53) in patients with acute respiratory distress syndrome. Intensive Care Medicine 2001;27(10):1578‐83. [PUBMED: 11685297] [DOI] [PubMed] [Google Scholar]
Walmrath 2000 {published data only}
- Walmrath D, Vaal JB, Bruining HA, Kilian JG, Papazian L, Hohlfeld J, et al. Treatment of ARDS with a recombinant SP‐C (rSP‐C) based synthetic surfactant. American Journal of Respiratory and Critical Care Medicine 2000;161:A379. [Google Scholar]
Willson 2015 {published data only}
- Willson DF, Truwit JD, Conaway MR, Traul CS, Egan EE. The adult calfactant in acute respiratory distress syndrome trial. Chest 2015;148(2):356‐64. [PUBMED: 25855884] [DOI] [PubMed] [Google Scholar]
Wirtz 2017 {published data only}
- Wirtz H, Hasenclever D, Schwabe K, Jaschinski U, Weyland A, Kuhnt E, et al. ACE inhibitor for lung protection during mechanical ventilation for acute lung injury‐results of the double‐blind, placebo controlled, randomised ACEmeVent pilot study. American Journal of Respiratory and Critical Care Medicine. Conference: American Thoracic Society International Conference, ATS 2017. United states 2017;195:no pagination. [Embase: 617707751] [Google Scholar]
Zhao 2014 {published data only}
- Zhao WB, Wan SX, Gu DF, Shi B. Therapeutic effect of glucocorticoid inhalation for pulmonary fibrosis in ARDS patients. Medical Journal of Chinese People's Liberation Army 2014;39(9):741‐5. [EMBASE: 2014842327] [Google Scholar]
Zheng 2014 {published data only}
- Zheng G, Huang L, Tong H, Shu Q, Hu Y, Ge M, et al. Treatment of acute respiratory distress syndrome with allogeneic adipose‐derived mesenchymal stem cells: a randomized, placebo‐controlled pilot study. American Journal of Respiratory and Critical Care Medicine. Conference: American Thoracic Society International Conference, ATS 2014;189:no pagination. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Huang L, Tong H, Shu Q, Hu Y, Ge M, et al. Treatment of acute respiratory distress syndrome with allogeneic adipose‐derived mesenchymal stem cells: a randomized, placebo‐controlled pilot study. Respiratory Research 2014;15(1):39. [PUBMED: 24708472] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
Abraham 1996 {published data only}
- Abraham E, Park YC, Covington P, Conrad SA, Schwartz M. Liposomal prostaglandin E1 in acute respiratory distress syndrome: a placebo‐controlled, randomized, double‐blind, multicenter clinical trial. Critical Care Medicine 1996;24(1):10‐5. [PUBMED: 8565513] [DOI] [PubMed] [Google Scholar]
Abraham 1999 {published data only}
- Abraham E, Baughman R, Fletcher E, Heard S, Lamberti J, Levy H, et al. Liposomal prostaglandin E1 (TLC C‐53) in acute respiratory distress syndrome: a controlled, randomized, double‐blind, multicenter clinical trial. TLC C‐53 ARDS Study Group. Critical Care Medicine 1999;27(8):1478‐85. [PUBMED: 10470753] [DOI] [PubMed] [Google Scholar]
Annane 2006 {published data only}
- Annane D, Sebille V, Bellissant E. Effect of low doses of corticosteroids in septic shock patients with or without early acute respiratory distress syndrome. Critical Care Medicine 2006;34(1):22‐30. [PUBMED: 16374152] [DOI] [PubMed] [Google Scholar]
Anzueto 1996 {published data only}
- Anzueto A, Baughman RP, Guntupalli KK, Weg JG, Wiedemann HP, Raventos AA, et al. Aerosolized surfactant in adults with sepsis‐induced acute respiratory distress syndrome. Exosurf Acute Respiratory Distress Syndrome Sepsis Study Group. New England Journal of Medicine 1996;334(22):1417‐21. [PUBMED: 8618579] [DOI] [PubMed] [Google Scholar]
Ardizzoia 1993 {published data only}
- Ardizzoia A, Lissoni P, Tancini G, Paolorossi F, Crispino S, Villa S, et al. Respiratory distress syndrome in patients with advanced cancer treated with pentoxifylline: a randomized study. Support Care in Cancer 1993;1(6):331‐3. [PUBMED: 8156252] [DOI] [PubMed] [Google Scholar]
Bastin 2010 {published data only}
- Bastin AJ, Lagan AL, Mumby S, Quinlan GJ, Griffiths MJD. Effect of N‐acetylcysteine in preventing inflammation after lung resection and one‐lung ventilation. A randomised controlled trial. American Journal of Respiratory and Critical Care Medicine 2010;181(1):no pagination. [Google Scholar]
Bastin 2016 {published data only}
- Bastin AJ, Davies N, Lim E, Quinlan GJ, Griffiths MJ. Systemic inflammation and oxidative stress post‐lung resection: Effect of pretreatment with N‐acetylcysteine. Respirology 2016;21(1):180‐7. [PUBMED: 26503312] [DOI] [PubMed] [Google Scholar]
Bernard 1987 {published data only}
- Bernard GR, Luce JM, Sprung CL, Rinaldo JE, Tate RM, Sibbald WJ, et al. High‐dose corticosteroids in patients with the adult respiratory distress syndrome. New England Journal of Medicine 1987;317(25):1565‐70. [PUBMED: 3317054] [DOI] [PubMed] [Google Scholar]
Bernard 1997 {published data only}
- Bernard GR, Wheeler AP, Arons MM, Morris PE, Paz HL, Russell JA, et al. A trial of antioxidants N‐acetylcysteine and procysteine in ARDS. The Antioxidant in ARDS Study Group. Chest 1997;112(1):164‐72. [PUBMED: 9228372] [DOI] [PubMed] [Google Scholar]
Bernard 1999 {published data only}
- Bernard GR, Wheeler AP, Naum CC, Morris PE, Nelson L, Schein R, et al. A placebo‐controlled, randomized trial of IL‐10 in acute lung injury (ALI). Chest. 1999; Vol. 116, issue 4 Suppl 2:206S.
Bone 1989 {published data only}
- Bone RC, Slotman G, Maunder R, Silverman H, Hyers TM, Kerstein MD, et al. Randomized double‐blind, multicenter study of prostaglandin E1 in patients with the adult respiratory distress syndrome. Prostaglandin E1 Study Group. Chest 1989;96(1):114‐9. [PUBMED: 2661155] [DOI] [PubMed] [Google Scholar]
- Slotman GJ, Kerstein MD, Bone RC, Silverman H, Maunder R, Hyers TM, et al. The effects of prostaglandin E1 on non‐pulmonary organ function during clinical acute respiratory failure. The Prostaglandin E1 Study Group. Journal of Trauma 1992;32(4):480‐8. [PUBMED: 1569622] [PubMed] [Google Scholar]
Confalonieri 2005 {published data only}
- Confalonieri M, Urbino R, Potena A, Piattella M, Parigi P, Puccio G, et al. Hydrocortisone infusion for severe community‐acquired pneumonia: a preliminary randomized study. American Journal of Respiratory and Critical Care Medicine 2005;171(3):242‐8. [PUBMED: 15557131] [DOI] [PubMed] [Google Scholar]
Cornet 2014 {published data only}
- Cornet AD, Groeneveld AB, Hofstra JJ, Vlaar AP, Tuinman PR, Lingen A, et al. Recombinant human activated protein C in the treatment of acute respiratory distress syndrome: a randomized clinical trial. PLOS One 2014;9(3):e90983. [PUBMED: 24632673] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cornet AD, Hofstra JJ, Vlaar AP, Tuinman PR, Levi M, Girbes AR, et al. Activated protein C attenuates pulmonary coagulopathy in patients with acute respiratory distress syndrome. Journal of Thrombosis and Haemostasis 2013;11(5):894‐901. [PUBMED: 23433188] [DOI] [PMC free article] [PubMed] [Google Scholar]
Domenighetti 1997 {published data only}
- Domenighetti G, Suter PM, Schaller MD, Ritz R, Perret C. Treatment with N‐acetylcysteine during acute respiratory distress syndrome: a randomized, double‐blind, placebo‐controlled clinical study. Journal of Critical Care 1997;12(4):177‐82. [PUBMED: 9459113] [DOI] [PubMed] [Google Scholar]
Forel 2006 {published data only}
- Forel JM, Roch A, Marin V, Michelet P, Demory D, Blache JL, et al. Neuromuscular blocking agents decrease inflammatory response in patients presenting with acute respiratory distress syndrome. Critical Care Medicine 2006;34(11):2749‐57. [PUBMED: 16932229] [DOI] [PubMed] [Google Scholar]
Gainnier 2004 {published data only}
- Gainnier M, Roch A, Forel JM, Thirion X, Arnal JM, Donati S, et al. Effect of neuromuscular blocking agents on gas exchange in patients presenting with acute respiratory distress syndrome. Critical Care Medicine 2004;32(1):113‐9. [PUBMED: 14707568] [DOI] [PubMed] [Google Scholar]
Gottlieb 1994 {published data only}
- Gottlieb JE, Elmer M, Steiner R. Efficacy of intravenous ICI 200,880, a neutrophil elastase inhibitor, in the treatment of adult respiratory distress syndrome (ARDS). Chest 1994;106(Suppl 2):67S. [Google Scholar]
Gregory 1997 {published data only}
- Gregory TJ, Steinberg KP, Spragg R, Gadek JE, Hyers TM, Longmore WJ, et al. Bovine surfactant therapy for patients with acute respiratory distress syndrome. American Journal of Respiratory and Critical Care Medicine 1997;155(4):1309‐15. [PUBMED: 9105072] [DOI] [PubMed] [Google Scholar]
Holcroft 1986 {published data only}
- Holcroft JW, Vassar MJ, Weber CJ. Prostaglandin E1 and survival in patients with the adult respiratory distress syndrome. A prospective trial. Annals of Surgery 1986;203(4):371‐8. [PUBMED: 3516085] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holcroft JW, Vassar MJ, Weber CJ. Prostaglandin E1 and survival in patients with the adult respiratory distress syndrome. A prospective trial. Seminars in Respiratory Medicine 1986;7(5):40‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hua 2013 {published data only}
- Hua F, Wang X, Zhu L. Terlipressin decreases vascular endothelial growth factor expression and improves oxygenation in patients with acute respiratory distress syndrome and shock. Journal of Emergency Medicine 2013;44(2):434‐9. [PUBMED: 22921855] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jepsen 1992 {published data only}
- Jepsen S, Herlevsen P, Knudsen P, Bud MI, Klausen NO. Antioxidant treatment with N‐acetylcysteine during adult respiratory distress syndrome: a prospective, randomized, placebo‐controlled study. Critical Care Medicine 1992;20(7):918‐23. [PUBMED: 1617983] [DOI] [PubMed] [Google Scholar]
Liu 2008 {published data only}
- Liu KD, Levitt J, Zhuo H, Kallet RH, Brady S, Steingrub J, et al. Randomized clinical trial of activated protein C for the treatment of acute lung injury. American Journal of Respiratory and Critical Care Medicine 2008;178(6):618‐23. [PUBMED: 18565951] [DOI] [PMC free article] [PubMed] [Google Scholar]
Markart 2007 {published data only}
- Markart P, Ruppert C, Wygrecka M, Colaris T, Dahal B, Walmrath D, et al. Patients with ARDS show improvement but not normalisation of alveolar surface activity with surfactant treatment: putative role of neutral lipids. Thorax 2007;62(7):588‐94. [PUBMED: 17287298] [DOI] [PMC free article] [PubMed] [Google Scholar]
Meduri 1998 {published data only}
- Meduri GU, Headley AS, Golden E, Carson SJ, Umberger RA, Kelso T, et al. Effect of prolonged methylprednisolone therapy in unresolving acute respiratory distress syndrome: a randomized controlled trial. JAMA 1998;280(2):159‐65. [PUBMED: 9669790] [DOI] [PubMed] [Google Scholar]
Papazian 2010 {published data only}
- Papazian L, Forel JM, Gacouin A, Penot‐Ragon C, Perrin G, Loundou A, et al. Neuromuscular blockers in early acute respiratory distress syndrome. New England Journal of Medicine 2010;363(12):1107‐16. [PUBMED: 20843245] [DOI] [PubMed] [Google Scholar]
- Papazian L, Forel JM, Gacouin A, Perrin G, Jaber S, Arnal JM, et al. Systematic two‐day muscle relaxants course in the early phase of severe acute respiratory distress syndrome. A multicenter randomized controlled trial. Intensive Care Medicine 2009;35:S6. [Embase: 70190221] [Google Scholar]
- Papazian L, Forel JM, Morange S, Barrau K, Penot‐Ragon C. Interest of a 48 h systematic paralysis at the early phase, in patients presenting with ARDS. The ACURASYS study. Protocol presentation [Interet d'une curarisation systematique precoce de 48 heures sur le pronostic des patients presentant un syndrome de detresse respiratoire aigue. Etude ACURASYS. Presentation du protocole]. Reanimation 2007;16(1):88‐95. [Google Scholar]
Presneill 2002 {published data only}
- Presneill JJ, Harris T, Stewart AG, Cade JF, Wilson JW. A randomized phase II trial of granulocyte‐macrophage colony‐stimulating factor therapy in severe sepsis with respiratory dysfunction. American Journal of Respiratory and Critical Care Medicine 2002;166(2):138‐43. [PUBMED: 12119223] [DOI] [PubMed] [Google Scholar]
Reines 1985 {published data only}
- Reines HD, Halushka PV, Olanoff LS, Hunt PS. Dazoxiben in human sepsis and adult respiratory distress syndrome. Clinical Pharmacology and Therapeutics 1985;37(4):391‐5. [PUBMED: 3979000] [DOI] [PubMed] [Google Scholar]
Reines 1992 {published data only}
- Reines HD, Sliverman H, Hurst J, Warren J, Williams J, Rotello L, et al. Effects of two concentrations of nebulized surfactant (Exosurf) in sepsis‐induced adult respiratory distress syndrome (ARDS). Critical Care Medicine 1992;20(4 Suppl):S61. [Google Scholar]
- Wiedermann H, Baughman R, DeBoisblanc B, Schuster D, Caldwell E, Weg J, et al. A multicenter trial in human sepsis‐induced ARDS of an aerosolized synthetic surfactant (Exosurf). American Review of Respiratory Disease 1992;145(4 part 2):A184. [Google Scholar]
Rossignon 1990 {published data only}
- Rossignon MD, Khayat D, Royer C, Rouby JJ, Jacquillat C, Viars P. Functional and metabolic activity of polymorphonuclear leukocytes from patients with adult respiratory distress syndrome: results of a randomized double‐blind placebo‐controlled study on the activity of prostaglandin E1. Anesthesiology 1990;72(2):276‐81. [PUBMED: 2405741] [DOI] [PubMed] [Google Scholar]
Shoemaker 1986 {published data only}
- Shoemaker WC. Effectiveness of prostaglandin E1 in adult respiratory distress syndrome. Progress in Clinical and Biological Research 1987;236A:361‐81. [PUBMED: 3615440] [PubMed] [Google Scholar]
- Shoemaker WC, Appel PL. Effects of prostaglandin E1 in adult respiratory distress syndrome. Surgery 1986;99(3):275‐83. [PUBMED: 3513357] [PubMed] [Google Scholar]
Shyamsundar 2010 {published data only}
- Shyamsundar M, O'Kane CM, Calfee C, McKeown ST, Taggart C, Matthay MA, et al. KGF enhances pulmonary production of proepithelial repair factors in a human in vivo model of acute lung injur y. Thorax 2010;65:A47. [Embase: 70325792] [Google Scholar]
Steinberg 1990 {published data only}
- Steinberg SM, Rodriguez JL, Bitzer LG, Rhee JW, Kelley KA, Flint LM. Indomethacin treatment of human adult respiratory distress syndrome. Circulatory Shock 1990;30(4):375‐84. [PUBMED: 2350875] [PubMed] [Google Scholar]
Suter 1994 {published data only}
- Suter PM, Domenighetti G, Schaller MD, Laverriere MC, Ritz R, Perret C. N‐acetylcysteine enhances recovery from acute lung injury in man. A randomized, double‐blind, placebo‐controlled clinical study. Chest 1994;105(1):190‐4. [PUBMED: 8272731] [DOI] [PubMed] [Google Scholar]
Tuxen 1987 {published data only}
- Tuxen DV, Wilson JW, Cade JF. Prevention of lower respiratory herpes simplex virus infection with acyclovir in patients with the adult respiratory distress syndrome. American Review of Respiratory Distress 1987;136(2):402‐5. [PUBMED: 3039882] [DOI] [PubMed] [Google Scholar]
Vincent 2009 {published data only}
- Vincent JL, Artigas A, Petersen LC, Meyer C. A multicenter, randomized, double‐blind, placebo‐controlled, dose‐escalation trial assessing safety and efficacy of active site inactivated recombinant factor VIIa in subjects with acute lung injury or acute respiratory distress syndrome. Critical Care Medicine 2009;37(6):1874‐80. [PUBMED: 19384216] [DOI] [PubMed] [Google Scholar]
Weg 1994 {published data only}
- Doig C, Weg JG. Aerosolized surfactant in sepsis‐induced adult respiratory distress syndrome. JAMA 1995;273(16):1259‐60. [PUBMED: 7715036] [PubMed] [Google Scholar]
- Weg JG, Balk RA, Tharratt RS, Jenkinson SG, Shah JB, Zaccardelli D, et al. Safety and potential efficacy of an aerosolized surfactant in human sepsis‐induced adult respiratory distress syndrome. JAMA 1994;272(18):1433‐8. [PUBMED: 7933425] [PubMed] [Google Scholar]
Weigelt 1985 {published data only}
- Weigelt JA, Norcross JF, Borman KR, Snyder WH 3rd. Early steroid therapy for respiratory failure. Archives of Surgery 1985;120(5):536‐40. [PUBMED: 3885915] [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Hegazy 2016 {published data only}
- Hegazy S, Helmy T, Salah M. Role of nebivolol therapy in patients with severe acute respiratory distress syndrome. Critical Care Medicine 2016;44(12 Supplement 1):314. [Google Scholar]
NCT00879606 {published data only}
- NCT00879606. Efficacy and safety evaluation of ALT‐836 in patients with sepsis and acute lung injury/acute respiratory distress syndrome. www.clinicaltrials.gov/ct2/show/NCT00879606 (first received 10 April 2009).
RPCEC00000126 {published data only}
- RPCEC00000126. Evaluation of the effect and safety of SURFACEN in the treatment of acute respiratory distress syndrome in adults. rpcec.sld.cu/en/trials/RPCEC00000126‐En (first received 11 April 2012).
References to ongoing studies
ACTRN12612000418875 {published data only}
- ACTRN12612000418875. A multi‐centre randomised, placebo controlled trial of nebulised heparin in patients with or at risk of developing acute respiratory distress syndrome, to determine if nebulised heparin improves long term physical function. www.anzctr.org.au/ACTRN12612000418875.aspx (first received 6 April 2012).
ACTRN12615000373572 {published data only}
- ACTRN12615000373572. Effect of nebulized budesonide on respiratory mechanics and oxygenation in patients with acute lung injury/acute respiratory distress syndrome (ALI/ARDS). www.anzctr.org.au/ACTRN12615000373572.aspx (first received 13 January 2015).
Bellingan 2017 {published data only}
- Bellingan G, Brealey D, Mancebo J, Mercat A, Patroniti N, Pettila V, et al. Comparison of the efficacy and safety of FP‐1201‐lyo (intravenously administered recombinant human interferon beta‐1a) and placebo in the treatment of patients with moderate or severe acute respiratory distress syndrome: study protocol for a randomized controlled trial. Trials 2017;18(1):no pagination. [DOI] [PMC free article] [PubMed] [Google Scholar]
ChiCTR1800014733 {published data only}
- ChiCTR1800014733. Clinical study of rhGM‐CSF in the treatment of pulmonary extraneous acute respiratory distress syndrome. www.chictr.org.cn/showproj.aspx?proj=25020 (first received 31 January 2018).
ChiCTR1800014998 {published data only}
- ChiCTR1800014998. Human umbilical cord derived mesenchymal stem cells therapy in acute respiratory distress syndrome: a pilot study. www.chictr.org.cn/showproj.aspx?proj=25576 (first received 27 February 2018).
EUCTR2012‐000775‐17 {published data only}
- EudraCT2012‐000775‐17. A comparative, randomised controlled trial for evaluating the efficacy of dexamethasone administration in the treatment of patients with the acute respiratory distress syndrome. www.clinicaltrialsregister.eu/ctr‐search/search?query=eudract_number:2012‐000775‐17 (first received 21 November 2012).
JPRN‐JapicCTI‐163320 {published data only}
- JPRN‐JapicCTI‐163320. A phase 3 clinical study to evaluate the efficacy and safety of intravenous MR11A8 in the treatment of patients with moderate or severe acute respiratory distress syndrome. www.clinicaltrials.jp/user/showCteDetailE.jsp?japicId=JapicCTI‐163320 (first received 19 July 2016).
NCT02326350 {published data only}
- NCT02326350. Aspirin as a treatment for ARDS (STAR): a phase 2 randomised control trial. clinicaltrials.gov/ct2/show/NCT02326350 (first received 29 December 2014).
NCT02595060 {published data only}
- NCT02595060. GM‐CSF inhalation to improve host defense and pulmonary barrier restoration (GI‐HOPE). A randomized, double‐blind, parallel group, multicenter, phase II study. www.clinicaltrials.gov/ct2/show/NCT02595060 (first received 3 November 2015).
NCT02611609 {published data only}
- NCT02611609. A phase 1/2 study to assess the safety and efficacy of MultiStem® therapy in subjects with acute respiratory distress syndrome. clinicaltrials.gov/ct2/show/NCT02611609 (first received 23 November 2015).
NCT02895191 {published data only}
- NCT02895191. A randomized, blind, placebo‐controlled, parallel group, multi‐center study to evaluate the safety and dose response relationship of ulinastatin for acute respiratory distress syndrome (ARDS). clinicaltrials.gov/ct2/show/NCT02895191 (first received 9 September 2016).
NCT03017547 {published data only}
- NCT03017547. A phase 2, randomized, double‐blind, placebo‐controlled, preliminary efficacy study of IC14 in acute respiratory distress syndrome. clinicaltrials.gov/ct2/show/NCT03017547 (first received 11 January 2017).
NCT03042143 {published data only}
- NCT03042143. Repair of acute respiratory distress syndrome by stromal cell administration (REALIST): an open label dose escalation phase 1 trial followed by a randomized, double‐blind, placebo‐controlled phase 2 trial. clinicaltrials.gov/ct2/show/NCT03042143 (first received 3 February 2017).
NCT03202394 {published data only}
- NCT03202394. A phase IIa, placebo controlled, multicenter pilot study to evaluate the safety and efficacy of BIO‐11006 inhalation solution in patients with acute respiratory distress syndrome. clinicaltrials.gov/ct2/show/NCT03202394 (first received 28 June 2017).
NCT03346681 {published data only}
- NCT03346681. N‐acetylcysteine in early acute respiratory distress syndrome (NARDS). clinicaltrials.gov/show/NCT03346681 (first received 17 November 2017).
NCT03371498 {published data only}
- NCT03371498. Procollagen‐3 driven corticosteroids for persistent acute respiratory distress syndrome (ProCoCo). clinicaltrials.gov/ct2/show/nct03371498 (first received 13 December 2017).
NCT03608592 {published data only}
- NCT03608592. Human umbilical cord derived mesenchymal stem cells therapy in acute respiratory distress syndrome: a pilot study. clinicaltrials.gov/ct2/show/NCT03608592 (first received 1 August 2018).
Villar 2016 {published data only}
- NCT01731795. A comparative, randomised controlled trial for evaluating the efficacy of dexamethasone in the treatment of patients with acute respiratory distress syndrome. clinicaltrials.gov/ct2/show/NCT01731795 (first received 22 November 2012).
- Villar J, Belda J, Añón JM, Blanco J, Pérez‐Méndez L, Ferrando C, et al. Evaluating the efficacy of dexamethasone in the treatment of patients with persistent acute respiratory distress syndrome: Study protocol for a randomized controlled trial. Trials 2016 2016;17(342):no pagination. [DOI] [PMC free article] [PubMed] [Google Scholar]
Additional references
Afshari 2017
- Afshari A, Bastholm Bille A, Allingstrup M. Aerosolized prostacyclins for acute respiratory distress syndrome (ARDS). Cochrane Database of Systematic Reviews 2017, Issue 7. [DOI: 10.1002/14651858.CD007733.pub3; PUBMED: 28806480] [DOI] [PMC free article] [PubMed] [Google Scholar]
ARDS Definition Task Force 2012
- The ARDS Defintion Task Force. Acute respiratory distress syndrome; the Berlin definition. JAMA 2012;307(23):2526‐33. [PUBMED: 22797452] [DOI] [PubMed] [Google Scholar]
ARDS Network 2000
- The Acute Respiratory Distress Syndrome Network. Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. New England Journal of Medicine 2000;342(18):1301‐8. [PUBMED: 10793162] [DOI] [PubMed] [Google Scholar]
Ashbaugh 1967
- Ashbaugh DG, Bigelow DB, Petty TL, Levine BE. Acute respiratory distress in adults. Lancet 1967;2(7511):319‐23. [PUBMED: 4143721] [DOI] [PubMed] [Google Scholar]
Bellani 2016
- Bellani G, Laffey JG, Pham T, Fan E, Brochard L, Esteban A, et al. Epidemiology, patterns of care, and mortality for patients with acute respiratory distress syndrome in intensive care units in 50 countries. JAMA 2016;315(8):788‐800. [PUBMED: 26903337] [DOI] [PubMed] [Google Scholar]
Bernard 1991
- Bernard GR. N‐acetylcysteine in experimental and clinical acute lung injury. American Journal of Medicine 1991;91(3C):54S‐9S. [PUBMED: 1928212] [DOI] [PubMed] [Google Scholar]
Bernard 1994
- Bernard GR, Artigas A, Brigham KL, Carlet J, Falke K, Hudson L, et al. The American‐European Consensus Conference on ARDS. Definitions, mechanisms, relevant outcomes, and clinical trial coordination. American Journal of Respiratory and Critical Care Medicine 1994;149(3 Pt 1):818‐24. [PUBMED: 7509706] [DOI] [PubMed] [Google Scholar]
Bloomfield 2015
- Bloomfield R, Noble DW, Sudlow A. Prone position for acute respiratory failure in adults. Cochrane Database of Systematic Reviews 2015, Issue 11. [DOI: 10.1002/14651858.CD008095.pub2; PUBMED: 26561745] [DOI] [PMC free article] [PubMed] [Google Scholar]
Brower 2004
- Brower RG, Lanken PN, MacIntyre N, Matthay MA, Morris A, Ancukiewicz M, et al. Higher versus lower positive end‐expiratory pressures in patients with the acute respiratory distress syndrome. New England Journal of Medicine 2004;351(4):327‐36. [PUBMED: 15269312] [DOI] [PubMed] [Google Scholar]
Chacko 2015
- Chacko B, Peter JV, Tharyan P, John G, Jeyaseelan L. Pressure‐controlled versus volume‐controlled ventilation for acute respiratory failure due to acute lung injury (ALI) or acute respiratory distress syndrome (ARDS). Cochrane Database of Systematic Reviews 2015, Issue 1. [DOI: 10.1002/14651858.CD008807.pub2; PUBMED: 25586462] [DOI] [PMC free article] [PubMed] [Google Scholar]
Covidence 2018 [Computer program]
- Veritas Health Innovation. Covidence. Version accessed 10 December 2018. Melbourne, Australia: Veritas Health Innovation.
Deeks 2017
- Deeks JJ, Higgins JP, Altman DG, editor(s) on behalf of the Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking metaanalyses. In: Higgins JP, Churchill R, Chandler J, Cumpston MS editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Dellinger 2008
- Dellinger RP, Levy MM, Carlet JM, Bion J, Parker MM, Jaeschke R, et al. Surviving Sepsis Campaign: international guidelines for management of severe sepsis and septic shock: 2008. Critical Care Medicine 2008;36(1):296‐327. [PUBMED: 18158437] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ (Clinical research ed.) 1997;315(7109):629‐34. [PUBMED: 9310563] [DOI] [PMC free article] [PubMed] [Google Scholar]
Endnote [Computer program]
- Thomson Reuters. Endnote X5. Philadelphia (PA): Thomson Reuters, 2011.
Feng 2018
FICM/ICS Guideline Development Group 2018
- The Faculty of Intensive Care Medicine and the Intensive Care Society. Guidelines on the management of acute respiratory distress syndrome. www.ics.ac.uk. Version 1 July 2018 (accessed 10 December 2018).
Galvin 2013
- Galvin IM, Steel A, Pinto R, Ferguson ND, Davies MW. Partial liquid ventilation for preventing death and morbidity in adults with acute lung injury and acute respiratory distress syndrome. Cochrane Database of Systematic Reviews 2013, Issue 7. [DOI: 10.1002/14651858.CD003707.pub3; PUBMED: 23881653] [DOI] [PMC free article] [PubMed] [Google Scholar]
Gao 2016
Gebistorf 2016
- Gebistorf F, Karam O, Wetterslev J, Afshari A. Inhaled nitric oxide for acute respiratory distress syndrome (ARDS) in children and adults. Cochrane Database of Systematic Reviews 2016, Issue 6. [DOI: 10.1002/14651858.CD002787.pub3; PUBMED: 27347773] [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version accessed 25 January 2019. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Guay 2018
- Guay J, Ochroch EA, Kopp S. Intraoperative use of low volume ventilation to decrease postoperative mortality, mechanical ventilation, lengths of stay and lung injury in adults without acute lung injury. Cochrane Database of Systematic Reviews 2018, Issue 7. [DOI: 10.1002/14651858.CD011151.pub3; PUBMED: 26641378] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2008
- Guyatt GH, Oxman AD, Kunz R, Vist GE, Falck‐Ytter Y, Schunemann HJ. What is "quality of evidence" and why is it important to clinicians?. BMJ (Clinical research ed.) 2008;336(7651):995‐8. [PUBMED: 18456631] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2011a
- Guyatt GH, Oxman AD, Kunz R, Woodcock J, Brozek J, Helfand M, et al. GRADE guidelines: 7. Rating the quality of evidence‐‐inconsistency. Journal of Clinical Epidemiology 2011;64(12):1294‐302. [PUBMED: 21803546] [DOI] [PubMed] [Google Scholar]
Guyatt 2011b
- Guyatt GH, Oxman AD, Kunz R, Brozek J, Alonso‐Coello P, Rind D, et al. GRADE guidelines 6. Rating the quality of evidence‐‐imprecision. Journal of Clinical Epidemiology 2011;64(12):1283‐93. [PUBMED: 21839614] [DOI] [PubMed] [Google Scholar]
Herridge 2003
- Herridge MS, Cheung AM, Tansey CM, Matte‐Martyn A, Diaz‐Granados N, Al Saidi F, et al Canadian Critical Care Trials Group. One‐year outcomes in survivors of the acute respiratory distress syndrome. New England Journal of Medicine 2003;348(8):683‐93. [PUBMED: 12594312] [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2017
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Hodgson 2016
- Hodgson C, Goligher EC, Young ME, Keating JL, Holland AE, Romero L, et al. Recruitment manoeuvres for adults with acute respiratory distress syndrome receiving mechanical ventilation. Cochrane Database of Systematic Reviews 2016, Issue 11. [DOI: 10.1002/14651858.CD006667.pub3; PUBMED: 27855477] [DOI] [PMC free article] [PubMed] [Google Scholar]
Horita 2015
Levy 2003
- Levy MM, Fink MP, Marshall JC, Abraham E, Angus D, Cook D, et al. 2001 SCCM/ESICM/ACCP/ATS/SIS International Sepsis Definitions Conference. Critical Care Medicine 2003;31(4):1250‐6. [PUBMED: 12682500] [DOI] [PubMed] [Google Scholar]
Luce 1998
- Luce JM. Acute lung injury and the acute respiratory distress syndrome. Critical Care Medicine 1998;26(2):369‐76. [PUBMED: 9468178] [DOI] [PubMed] [Google Scholar]
Lundstrøm 2017
- Lundstrøm LH, Duez CH, Nørskov AK, Rosenstock CV, Thomsen JL, Møller AM, et al. Avoidance versus use of neuromuscular blocking agents for improving conditions during tracheal intubation or direct laryngoscopy in adults and adolescents. Cochrane Database of Systematic Reviews 2017, Issue 5. [DOI: 10.1002/14651858.CD009237.pub2; PUBMED: 28513831] [DOI] [PMC free article] [PubMed] [Google Scholar]
Meng 2012
- Meng H, Sun Y, Lu J, Fu S, Meng Z, Scott M, et al. Exogenous surfactant may improve oxygenation but not mortality in adult patients with acute lung injury/acute respiratory distress syndrome: a meta‐analysis of 9 clinical trials. Journal of Cardiothoracic & Vascular Anesthesia 2012; Vol. 26, issue 5:849‐56. [PUBMED: 22265270] [DOI] [PMC free article] [PubMed]
Murray 1988
- Murray JF, Matthay MA, Luce JM, Flick MR. An expanded definition of the adult respiratory distress syndrome. American Review of Respiratory Disease 1988;138(3):720‐3. [PUBMED: 3202424] [DOI] [PubMed] [Google Scholar]
Nagendran 2017
Nanchal 2018
- Nanchal RS, Truwit JD. Recent advances in understanding and treating acute respiratory distress syndrome. F1000 Research 2018;7:no pagination. [PUBMED: 30210781] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pehora 2017
- Pehora C, Pearson AM, Kaushal A, Crawford MW, Johnston B. Dexamethasone as an adjuvant to peripheral nerve block. Cochrane Database of Systematic Reviews 2017, Issue 11. [DOI: 10.1002/14651858.CD011770.pub2; PUBMED: 29121400] [DOI] [PMC free article] [PubMed] [Google Scholar]
Petrucci 2013
- Petrucci N, Feo C. Lung protective ventilation strategy for the acute respiratory distress syndrome. Cochrane Database of Systematic Reviews 2013, Issue 2. [DOI: 10.1002/14651858.CD003844.pub4; PUBMED: 23450544] [DOI] [PMC free article] [PubMed] [Google Scholar]
Polderman 2018
- Polderman JA, Farhang‐Razi V, Dieren S, Kranke P, DeVries JH, Hollmann MW, et al. Adverse side effects of dexamethasone in surgical patients. Cochrane Database of Systematic Reviews 2018, Issue 8. [DOI: 10.1002/14651858.CD011940.pub2; PUBMED: 30152137 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Santa Cruz 2013
- Santa Cruz R, Rojas JI, Nervi R, Heredia R, Ciapponi A. High versus low end‐expiratory pressure (PEEP) levels for mechanically ventilated adult patients with acute lung injury and acute respiratory distress syndrome. Cochrane Database of Systematic Reviews 2013, Issue 6. [DOI: 10.1002/14651858.CD009098.pub2; PUBMED: 23740697] [DOI] [PMC free article] [PubMed] [Google Scholar]
SCCMCMA 2006
- Society of Critical Care Medicine of Chinese Medical Association. Guideline of the diagnosis and treatment of ALI/ARDS. Chinese Journal of Critical Care Medicine 2006;18(12):706‐10. [PUBMED: 17166345] 17166345 [Google Scholar]
Schoenfeld 2002
- Schoenfeld DA, Bernard GR. Statistical evaluation of ventilator‐free days as an efficacy measure in clinical trials of treatments for acute respiratory distress syndrome. Critical Care Medicine 2002;30(8):1772‐7. [PUBMED: 12163791] [DOI] [PubMed] [Google Scholar]
Singh 2014
Sterne 2017
- Sterne JA, Egger M, Moher D, Boutron I, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Sud 2016
- Sud S, Sud M, Friedrick JO, Wunsch H, Meade MO, Ferguson ND, et al. High‐frequency oscillatory ventilation versus conventional ventilation for acute respiratory distress syndrome. Cochrane Database of Systematic Reviews 2016, Issue 4. [DOI: 10.1002/14651858.CD004085.pub4; PUBMED: 27043185] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ware 2000
- Ware LB, Matthay MA. The acute respiratory distress syndrome. New England Journal of Medicine 2000;342(18):1334‐49. [PUBMED: 10793167] [DOI] [PubMed] [Google Scholar]
Wiedemann 2006
- Wiedemann HP, Wheeler AP, Bernard GR, Thompson BT, Hayden D, deBoisblanc B, et al. Comparison of two fluid‐management strategies in acute lung injury. New England Journal of Medicine 2006;354(24):2564‐75. [PUBMED: 16714767] [DOI] [PubMed] [Google Scholar]
Yang 2017
- Yang ZG, Lei XL, Li XL. Early application of low‐dose glucocorticoid improves acute respiratory distress syndrome: A meta‐analysis of randomized controlled trials. Experimental and Therapeutic Medicine 2017; Vol. 13, issue 4:1215‐24. [PUBMED: 28413460] [DOI] [PMC free article] [PubMed]
Zhang 2013
- Zhang LN, Sun JP, Xue XY, Wang JX. Exogenous pulmonary surfactant for acute respiratory distress syndrome in adults: A systematic review and meta‐analysis. Experimental and Therapeutic Medicine 2013; Vol. 5, issue 1:237‐42. [PUBMED: 23251275] [DOI] [PMC free article] [PubMed]
Zhang 2017
- Zhang Y, Ding S, Li C, Wang Y, Chen Z, Wang Z. Effects of N‐acetylcysteine treatment in acute respiratory distress syndrome: A meta‐analysis. Experimental and Therapeutic Medicine 2017; Vol. 14, issue 4:2863‐8. [PUBMED: 28928799] [DOI] [PMC free article] [PubMed]
References to other published versions of this review
Adhikari 2002
- Adhikari NK, Burns KEA, Meade MO. Pharmacologic therapies for adults with acute lung injury and the acute respiratory distress syndrome. Cochrane Database of Systematic Reviews 2002, Issue 4. [DOI: 10.1002/14651858.CD004477] [DOI] [PMC free article] [PubMed] [Google Scholar]
Adhikari 2004
- Adhikari NKJ, Burns KEA, Meade MO, Ratnapalan M. Pharmacologic therapies for adults with acute lung injury and acute respiratory distress syndrome. Cochrane Database of Systematic Reviews 2004, Issue 4. [DOI: 10.1002/14651858.CD004477.pub2; PUBMED: 15495113] [DOI] [PMC free article] [PubMed] [Google Scholar]