

Abstract
Purpose of review
To review the recently published data and provide a practical overview for management of systemic sclerosis-interstitial lung disease (SSc-ILD).
Recent Findings
Published evidence shows considerable practitioner variability in screening patients for ILD. Recent published data supports use of cyclophosphamide or mycophenolate mofetil as first line treatment of SSc-ILD. For patients not responding to first line therapies, consideration is given to rituximab as rescue therapy. Recent trials of hematopoietic autologous stem cell transplantation have demonstrated benefit in patients with progressive SSc-ILD. Anti-fibrotic agents are approved in idiopathic pulmonary fibrosis; studies with anti-fibrotics are underway for SSc-ILD.
Summary
The specter of rapidly progressive lung disease requires clinicians to risk stratify patients according to known predictors for progression and rigorously monitor for symptoms and advancing disease. The above-mentioned therapies promise improved efficacy and favorable side effect profiles compared to cyclophosphamide.
Keywords: Systemic Sclerosis, Interstitial Lung Disease, Management, Treatment
INTRODUCTION
Systemic Sclerosis-Associated Interstitial Lung DiseaseSystemic sclerosis (SSc) is an autoimmune disease that is characterized by immune dysregulation, vasculopathy, and overproduction of collagen leading to skin and internal organ fibrosis1. Systemic sclerosis associated interstitial lung disease (SSc-ILD) is a complex process involving inflammation, alveolar epithelial damage, and the activation of resident fibroblasts resulting in thickening of the pulmonary interstitium2. The reported prevalence varies widely (16-91%) depending on the definition used3,4. The prevalence as determined by high-resolution CT (HRCT) ranges from 47-84%4,5,6. Patients with diffuse cutaneous SSc are more likely to develop SSc-ILD (53%) compared to those with limited cutaneous SSc (35%)7. SSc-ILD has become the leading cause of death (alongside pulmonary arterial hypertension) and accounts for up to 30-35% of SSc mortality8,9. SSc-ILD has heterogeneous disease progression: many patients will have a chronic, indolent course while others may develop progressive, life-threatening disease. Symptoms of cough and exertional dyspnea may be absent or mild early in the disease course. Universal screening is paramount in identifying these patients early. Not all patients require treatment if the disease is detected; monitoring for progression is an essential component of disease management.
Several risk factors for progressive SSc-ILD have been identified (Table 1) and prognostic scoring systems have identified predictors of mortality5,10,11. Considerable variation exists among rheumatologists and SSc experts in terms of practice patterns for SSc-ILD screening and treatment12,13. This article reviews the current literature in terms of screening and treatment recommendations and provides our treatment algorithms for SSc-ILD patients.
Table 1:
Features predictive of disease progression in SSc-ILD.
| FEATURE | Reference |
|---|---|
| DEMOGRAPHIC | |
| Male gender | 60 |
| African-American race | 61 |
| DISEASE STATUS | |
| Diffuse cutaneous systemic sclerosis | 33 |
| BIOMARKERS | |
| Anti-SCL 70 Ab | 33 |
| Nucleolar pattern on ANA (representing anti-Th/To, U3 RNP) | 62 |
| CCL-2 | 64 |
| CCL-18 | 64 |
| Interleukin-6 | 65 |
| C-reactive protein | 38 |
| CXCL-4 | 66 |
| KL-6 | 67 |
| Surfactant protein D | 68 |
| PULMONARY FUNCTION TESTING | |
| Baseline FVC <70% | 69 |
| Baseline DLco <55% | 33 |
| IMAGING | |
| HRCT fibrosis >20%-25% | 21,22 |
| HRCT total lung involvement >20% | 5, 21 |
DISEASE SCREENING AND MONITORING
All patients with SSc should be screened for ILD.
The variable nature of SSc-ILD and lack of robust predictive markers or prediction models makes it challenging to assess who will develop clinically meaningful disease. Although low-risk populations have been identified (e.g., those with anti-centromere antibodies), considerable variability in risk for progression exist2. With the advent of increasingly efficacious treatment with improved toxicity profiles, we recommend that all patients with SSc should be screened with pulmonary function tests (PFTs) and high-resolution chest CT (HRCT) for ILD.
Despite this, a recent global survey13 showed that a wide variability exists in rheumatologists’ use of HRCT in screening practices: only half of general rheumatologists and two-thirds of SSc experts routinely obtain a chest HRCT in newly diagnosed SSc patients. Between the two groups, there were no 100% agreed-upon indications for which an HRCT should be performed (e.g., respiratory symptoms, physical exam finding, oxygen desaturation, or PFT deficit).
Screening and Monitoring.
Screening should involve a careful review of systems, physical examination, complete PFTs with lung volumes, and lung imaging with HRCT (with prone images, see Figure 3)14, and may include functional measures like the 6-minute walk test15 (See Figure 1). Echocardiography and use of the DETECT algorithm should be performed at baseline to exclude concurrent pulmonary hypertension16,17.
Figure 3: HRCT in SSc-ILD.
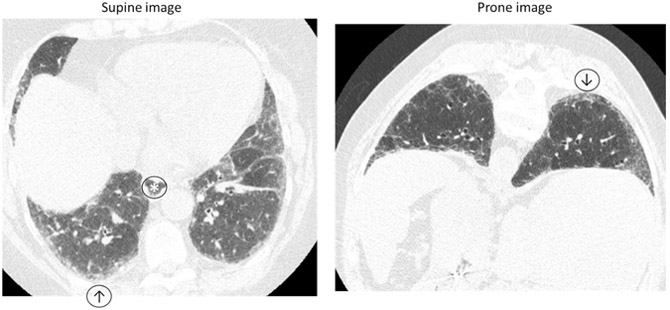
Inspiratory and prone HRCT demonstrating minimal reticulation, subpleural groundglass opacity (↑) that persists on prone imaging (↓) suggestive of interstitial lung abnormalities and an early fibrotic lung disease. No honeycombing or traction bronchiectasis. Note slightly dilated esophagus (*).
Figure 1: Screening and Monitoring Algorithm for ILD in Patients with SSc.

* Lower limit of normal
** Clinically meaningful decline defined as FVC levels of >10% from baseline or decline in FVC ≥ 5% to < 10% and ≥ 15% relative decline in DLCO
@ Other causes of desaturation such as pulmonary hypertension should be ruled out
+ Based on Sircar et al., 201837
Once the diagnosis of SSc-ILD is established, it is important for the treating clinician to identify the constellation of factors that designate one patient to receive routine monitoring (as their risk for progressive ILD may be low), and for another patient to initiate or escalate immunomodulatory treatment (as risk for progressive ILD is high; Table 1)18,19.
HRCT.
HRCT is the only non-invasive way to diagnose ILD. It can characterize the nature and extent of lung involvement. The majority of cases are fibrotic NSIP pattern (figure 4), with a minority (10-15%) with UIP pattern (figure 5); the mortality rates between these two disease patterns do not differ significantly in SSc-ILD20.
Figure 4: Non-Specific Interstitial Pneumonitis Pattern.
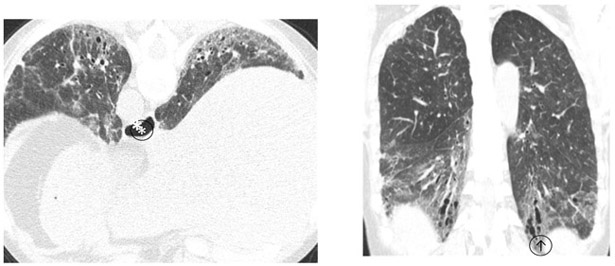
Lower lung predominant homogeneously distributed ground glass opacity, reticulation, traction bronchiectasis(↑) and dilated esophagus (*)without honeycombing. Appearances are compatible with scleroderma related interstitial lung disease (NSIP pattern).
Figure 5: Usual Interstitial Pneumonitis Pattern.
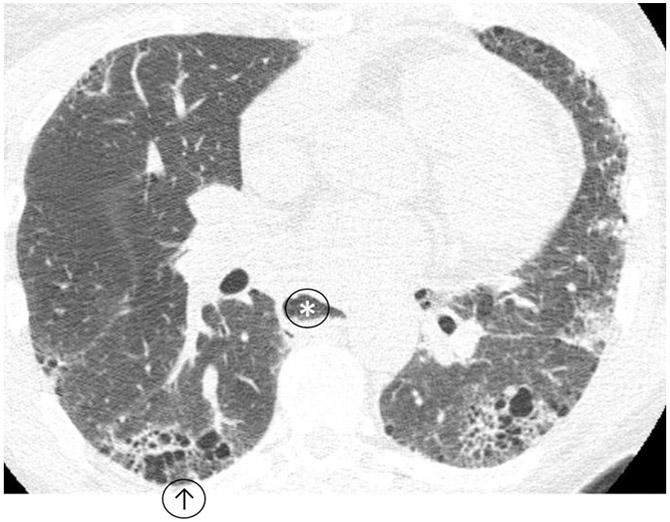
Heterogeneously distributed interstitial lung disease with honeycombing (↑) compatible with usual interstitial pneumonitis due to scleroderma, note dilated esophagus (*).
Quantifying the extent of fibrosis has yielded important information including prediction of pulmonary function decline21 and risk for mortality5,22,23,24. Advances in quantitative measurements of fibrosis25 has allowed HRCT to become a valuable tool for detecting response to treatment. Kim et al., 201626 showed that patients treated with cyclophosphamide in the Scleroderma Lung Study-I had decreasing extents of fibrosis, suggesting quantitative HRCT is a sensitive measure for assessing treatment response. Goldin et al., 201827 also found significant reductions in the extent of fibrosis in response to treatment in the Scleroderma Lung Study-II, which correlated with improving FVC, DLco, and breathlessness.
In clinical practice, we recommend volumetric HRCT with supine and prone images in all patients with SSc. Repeat HRCT should not be routinely performed but considered in patients with progressive symptoms attributable to ILD if it will direct treatment decisions, or in clinically stable patients with declining PFTs to assess for radiographic progression.
Radiation dose from medical imaging has been a growing concern with patients getting scanned repetitively for diagnosis and follow up. For perspective and comparison, the typical effective dose from natural background radiation in North America is 3 millisievert (mSV)28. The effective dose of a PA and lateral radiograph is 0.05 mSV. The effective dose to a 70 kg adult for a chest CT is approximately 5.4 mSV29, and the dose can vary depending on BMI and protocol (HRCT would be more if expiratory and prone imaging are also performed as dose from each would be additive). CT protocols have been tweaked over the last decade in an attempt to reduce radiation exposure with use of tube current modulation, reduced tube voltage, higher pitch and noise reduction filters30. Typically attempts at dose reduction are accompanied with increased image noise which results in decreased spatial resolution and impairment of the ability to see subtle interstitial abnormalities. Lim et al., 201630 demonstrated that types of iterative reconstruction (a newer image reconstruction technique) have been successfully used to decrease image noise in low dose HRCT studies achieving radiation doses around 0.7 mSV versus 3.1 mSV for annual background radiation. However, they noted that these reconstruction techniques tended to overestimate ground glass opacity and underestimate interlobular septal thickening. Dose reduction in HRCT with iterative reconstruction has a lot of potential with larger trials being necessary before it is adopted more widely.
PFTs.
Pulmonary function tests represent a cost-effective, clinically feasible, safe and reliable means of early detection of SSc-ILD. The broad range of PFTs considered normal (80-120%) can make interpretation of a cross-sectional evaluation difficult during the first visit. PFTs lack sensitivity and specificity (relative to HRCT) in early or mild disease3, making PFTs more apt for disease monitoring than screening. Baseline FVC abnormalities have been shown to predict mortality at 10 years from disease onset31,32. Patients who reach an FVC of 50-70% of the predicted value within 5 years of onset of disease has been associated with end-stage ILD and increased mortality31,33. As a result, PFTs are routinely performed every 3-6 months for the first 3-5 years of disease34. Goh et al., 201735 found that change in FVC and DLco was predictive of mortality at 1 year (in those with extensive disease); at 2 years, DLco and Kco trends had the greatest prognostic significance. Volkmann et al., 201836 examined patients in SLS-I and SLS-II for significant predictors of long-term mortality and found that the course of FVC and DLCO over 24 months appeared to predict long-term survival better than the baseline measurements of these parameters. This further emphasizes the need for early identification and regular PFTs in early disease.
Our Screening and Monitoring Algorithm
All patients should be evaluated for pulmonary hypertension contemporaneously with ILD screening and prior to pursing ILD treatment. Figure 1 is a proposed screening algorithm for clinical SSc-ILD and for monitoring those with subclinical SSc-ILD or no evidence of ILD at the time of screening. For patients with clinical SSc-ILD (mild-to-severe ILD on HRCT, persisting PFT deficits, and in whom symptoms are attributable to ILD), we initiate treatment. We consider treatment on a case-by-case basis for those who have subclinical ILD with high risk features, including asymptomatic patients with mild ILD on HRCT, mild PFT deficits, and early dcSSc status with high risk biomarkers like a positive anti-SCL 70 antibody33,37 or elevated CRP38,39. In patients without ILD on HRCT we proceed with routine monitoring with PFTs; the development of ILD on HRCT or mild-to-severe deficits on PFTs prompts consideration for treatment.
Figure 2 is a proposed screening algorithm for patients with SSc-ILD who develop worsening respiratory symptoms. Routine and transient causes of cough or mild dyspnea should be considered and excluded (e.g., viral upper respiratory infection, asthma, allergic rhinitis with post nasal drip syndrome, GERD). Sustained or disproportionate decline in DLco prompts simultaneous consideration for development of PAH. We obtain a repeat echocardiogram, serum NT-pro BNP, serum uric acid and calculate DETECT scores to assess if there is developing/advancing pulmonary hypertension, based on 2018 recommendations from the World Pulmonary Hypertension Symposium40,17. Patients should also be assessed for pulmonary and non-pulmonary etiologies for restrictive lung disease, including reduction in chest wall compliance, possible diaphragmatic weakness, deconditioning, and concurrent myopathy. If these measures cannot account for symptoms or changes in PFTs, we repeat HRCT to assess for progressive ILD.
Figure 2: Work-up for SSc-ILD Patients with Worsening Respiratory Symptoms.
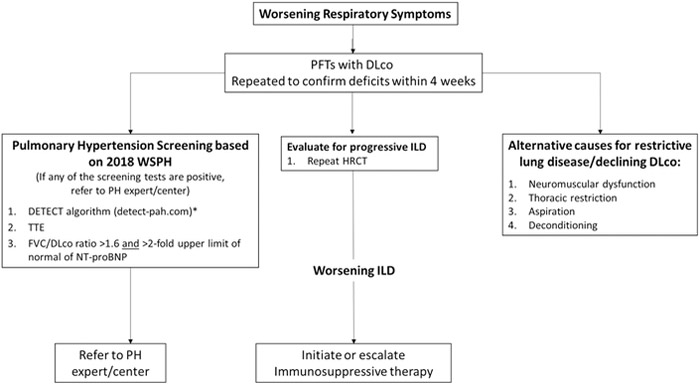
* Based on Coghlan et al., 201516
WSPH World Symposium on Pulmonary Hypertension17
PH Pulmonary Hypertension
DISEASE TREATMENT
Who Requires Treatment
All patients with clinically meaningful SSc-ILD should be offered treatment. Although no consensus definition exists, clinical ILD may be defined as those patients with symptoms attributable to ILD and non-trivial lung involvement seen on HRCT and/or associated decrements in lung physiology and gas exchange41. Disease monitoring should be rigorous in order to detect the subset of clinical ILD patients with progressive disease, operationalized as a decline in FVC levels of >10% from baseline or ≥ 5% to < 10% relative decline in FVC and ≥ 15% relative decline in DLCO42,43 although smaller changes may be clinically meaningful, especially worsening symptoms attributable to ILD43. These patients span a spectrum from mild to rapidly progressive disease, with the latter often having cardinal clues to their risk of progression (e.g., larger extent of fibrosis on HRCT at baseline, shorter duration of SSc, high CRP; see Table 1)44. Decisions to initiate or advance treatment often take into consideration the likelihood of progression, patient comorbidities, risk of toxicities, and current data on efficacy.
Treatment Options
The goal of treating clinical SSc-ILD stabilization or preventing progressive disease45.
Cytotoxic DMARDs
Scleroderma Lung Study I (1-year course of oral cyclophosphamide (CYC) up to 2 mg/kg/day) showed a statistically significant but small improvement in FVC (2.5% improvement) vs. placebo and a paucity of sustained benefit after the medication was discontinued. Scleroderma Lung Study II (head-to-head comparison of oral CYC up to 2 mg/kg/day for 1 year to mycophenolate mofetil (MMF) at 1.5 grams twice a day for 2 years) showed MMF to have a positive treatment effect on FVC similar to those treated with oral CYC at 2 years (MMF 2.2%, CYC 2.9%), improved measures of dyspnea, and MMF had a superior safety profile.
Biologic DMARDs
Phase III clinical data suggest the role of the IL-6 pathway in SSc-ILD and treatment of early SSc with elevated CRP led to stabilization of FVC% in tocilizumab group vs. a clinically meaningful decline in the placebo group over 48 weeks: treatment difference of 4.2%; p = 0.0002. The mean [SD] FVC% was 82.1% [14.8%] at baseline and this trial highlights the benefit of treating patients with subclinical ILD with high risk features (early dcSSc, and elevated CRP) 39, further emphasizing the need for early detection of the disease.
Rituximab (RTX) therapy in SSc has gained favor in response to promising effects on both ILD and skin thickening. A recent open-label, randomized, controlled trial of head-to-head RTX vs. monthly pulse CYC analyzed a population of 60 early, treatment naïve, anti-SCL-70+, dcSSc with ILD patients receiving either arm. Patients in the CYC group received 500mg/m2 CYC IV pulses every 4 weeks for 24 weeks; patients in the RTX group received two RTX pulses of 1000mg at 0 and 15 days. They found the RTX group to have improved FVC% at the end of 6 months (RTX group improved, 61.3% to 67.5% while the CYC group did not, 59.3% to 58.1%). The efficacy and safety demonstrated in this trial argues that RTX may be considered as a first line therapy.
Autologous Hematopoietic Stem Cell Transplant
Hematopoietic stem cell transplant (HSCT) represents an emerging treatment option for those patients with SSc-ILD that is severe and refractory to standard therapy, and who are likely to benefit from the procedure while unlikely to develop post-transplant complications. Three key trials (ASSIST, ASTIS, and SCOT) have shown improved survival compared to CYC, in addition to improved quality of life, skin thickening, and FVC46. Due to limited space, we will only discuss the recently published SCOT trial47.
SCOT was a multi-center randomized phase III trial including 75 patients with early dcSSc; 100% of patients in the HSCT group had ILD. HSCT patients (n= 36) were conditioned with CYC (120mg/kg), anti-thymocyte globulin, received total body irradiation (800cGy) and received a stem cell transplant (CD34+ selected); the comparator arm received CYC (750mg/m2) x12 months (n= 39). At baseline, the two groups had similar averages [SD] on FVC: 74.5% [14.8] in the HSCT group compared to 73.8 [17.0] in the CYC group. The two groups also had similar averages on DLco: 53.9% [7.6] compared to 52.7 [8.2], respectively.
Overall, the trial demonstrated that HSCT significantly improved event-free survival compared with CYC, where ‘event-free’ was operationalized as survival without respiratory, renal, or cardiac failure. With specific focus on SSc-ILD and respiratory outcomes, more patients receiving HSCT improved in FVC than those in the CYC group at 54 months: 36% of the HSCT patients improved (relative increase of FVC by ≥10%) compared to 23% of the CYC patients. Conversely, fewer patients in the HSCT group worsened (relative decrease by ≥10%) compared to the CYC group (17% vs. 41%, respectively). HSCT was also associated with improvement on the HRCT compared to CYC on computer-based quantitative image analysis48.
Lung Transplant
Lung Transplant remains a therapy for appropriately selected candidates with treatment-refractory lung disease49. Advancing disease should prompt an early referral, as these patients require a multi-disciplinary evaluation prior to transplant consideration and optimization prior to procedure. One nationwide cohort study found an increased 1-year mortality rate in SSc-ILD patients compared to those with non-SSc-ILD50. Outcomes of mortality up to 5 years suggest similar outcomes to those with non-SSc fibrotic lung disease51.
Our Treatment Practice
In clinical SSc-ILD, we initiate induction therapy using MMF with a goal of 3 grams/day in divided dose. If not tolerated, we ensure the patient is taking MMF with food as this may decrease nausea/vomiting. If intolerance remains, we recommend comparable dosing for mycophenolic acid (720mg three times daily). In clinical SSc-ILD patients with rapidly progressive disease or those with significant GI dysmotility, we advance to pulse monthly CYC at 500-750 mg/m2. For those not responding, we consider the addition of RTX 1000 mg for 2 doses. In patients with early dcSSc and progressive ILD (on PFT, HRCT, and/or symptoms), who are not responding to MMF or other immunosuppressive therapy, we consider HSCT52. Based on recent data53, we now consider tocilizumab as first line therapy in early dcSSc and subclinical ILD.
Non-pharmacologic Therapy
All patients with SSc-ILD should have a multidisciplinary team. Comorbid gastrointestinal disease, especially GERD and aspiration should be considered. Every SSc-ILD patient should be educated to sleep at an angle and aggressively control ongoing GERD symptoms. All patients with SSc and especially those with SSc-ILD should be encouraged to quit smoking and tobacco products. Supplemental oxygen should be provided to those patients requiring it to maintain SpO2 ≥88% during ambulation. All patients should be given inactive influenza and pneumococcal vaccines, administered before starting B cell depleting biological therapy54. Finally, pulmonary rehabilitation 55 may be effective. For those patients who may be considered a transplant candidate, pulmonary rehabilitation will be a necessary step in their evaluation.
FUTURE DIRECTIONS
Emerging Therapies
Idiopathic pulmonary fibrosis is a fibrotic lung condition which shares pathological features with SSc-ILD of fibroblast proliferation, migration, and differentiation56. Nintedanib is an intracellular tyrosine kinase inhibitor approved for the treatment of IPF and is currently being investigated in a phase III, multicenter, double-blind, placebo-controlled trial (SENSCIS; )57. The final results are anticipated in 2019.
The LOTUSS trial58 was a phase II international, multicenter, open-label assessment of Pirfenidone, an anti-fibrotic/anti-inflammatory agent also approved for IPF. This study showed tolerability and safety with concurrent use of MMF. The Scleroderma Lung Study-III is an on-going trial since 11/2017 comparing MMF and pirfenidone vs. MMF and placebo (). The goal enrollment is 150 patients with SSc-ILD and the primary endpoint is FVC at 18 months. There are several other ongoing clinical trials for SSc-ILD59.
CONCLUSION
ILD is common in SSc. At present we lack the granularity in predicting which subsets of patients will develop organ and potentially life-threatening disease. The potential risk for morbidity and mortality is the impetus for rigorous monitoring for symptoms and signs of ILD development and progression. At this time, the standards of care include cyclophosphamide and mycophenolate mofetil, both of which have shown modest improvements in FVC; treatment supports the attenuation of disease progression. Newer therapies include biologics, stem cell transplant, and anti-fibrotics; preliminary data suggest improved efficacy and safety profiles compared to cyclophosphamide therapy.
Key Points.
-
Point 1
Diagnosis of SSc-ILD is determined by HRCT; prognosis depends on a constellation of risk for progressive SSc-ILD (disease type, auto-antibody status, extent of involvement on HRCT, pulmonary function impairment).
-
Point 2
There is a large disparity in screening practices by rheumatologists in terms of who gets screened for ILD.
-
Point 3
Not all patients with SSc-ILD require treatment. Patients should be routinely monitored earlier in the disease. Early and aggressive treatment with non-pharmacologic and pharmacologic therapies are warranted.
-
Point 4
Anti-fibrotic agents and repositioned biologic therapies are currently being studied in clinical trials for SSc-ILD.
Acknowledgments
Financial Support and Sponsorship: Dr. D. Khanna was supported by the NIH/NIAMS K24AR063120
Footnotes
Conflicts of Interest: Consultancies: Actelion, Astra Zeneca, Bayer, BMS, Boehringer-Ingelheim, Corbus, Cytori, Galapagos, Genentech/Roche, GSK, Sanofi-Aventis/Genzyme, UCB Pharma. Stock ownership or options: Eicos Sciences, Inc/ CiviBioPharma, Inc. Employment: University of Michigan and CiviBioPharma, Inc.
References
- 1.Denton CP, Khanna D. Systemic sclerosis. Lancet. 2017;390(10103):1685–1699. doi: 10.1016/S0140-6736(17)30933-9 [DOI] [PubMed] [Google Scholar]
- 2.Wells AU. Interstitial lung disease in systemic sclerosis. Press Medicale. 2014;43(10):329–343. doi: 10.1016/j.lpm.2014.08.002 [DOI] [PubMed] [Google Scholar]
- 3.Suliman YA, Dobrota R, Huscher D, et al. Pulmonary function tests: High rate of false-negative results in the early detection and screening of scleroderma-related interstitial lung disease. Arthritis Rheumatol. 2015;67(12):3256–3261. doi: 10.1002/art.39405 [DOI] [PubMed] [Google Scholar]
- 4.Steele R, Hudson M, Lo E, Baron M. Clinical decision rule to predict the presence of interstitial lung disease in systemic sclerosis. Arthritis Care Res. 2012;64(4):519–524. doi: 10.1002/acr.21583 [DOI] [PubMed] [Google Scholar]
- 5.Goh NSL, Desai SR, Veeraraghavan S, et al. Interstitial Lung Disease in Systemic Sclerosis. Am J Respir Crit Care Med. 2008;177(11):1248–1254. doi: 10.1164/rccm.200706-877OC [DOI] [PubMed] [Google Scholar]
- 6.Vonk MC, Broers B, Heijdra YF, et al. Systemic sclerosis and its pulmonary complications in The Netherlands: An epidemiological study. Ann Rheum Dis. 2009;68(6):961–965. doi: 10.1136/ard.2008.091710 [DOI] [PubMed] [Google Scholar]
- 7.Walker UA, Tyndall A, Czirják L, et al. Clinical risk assessment of organ manifestations in systemic sclerosis: A report from the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis. 2007;66(6):754–763. doi: 10.1136/ard.2006.062901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Steen VD. Severe organ involvement in systemic sclerosis. ARTHRITIS Rheum. 2000;43(11):2437–2444. doi: [DOI] [PubMed] [Google Scholar]
- 9.Tyndall AJ, Bannert B, Vonk M, et al. Causes and risk factors for death in systemic sclerosis: A study from the EULAR Scleroderma Trials and Research (EUSTAR) database. Ann Rheum Dis. 2010;69(10):1809–1815. doi: 10.1136/ard.2009.114264 [DOI] [PubMed] [Google Scholar]
- 10.Elhai M, Meune C, Boubaya M, et al. Mapping and predicting mortality from systemic sclerosis. Ann Rheum Dis. 2017;76(11):1897–1905. doi: 10.1136/annrheumdis-2017-211448 [DOI] [PubMed] [Google Scholar]
- 11.Morisset J, Vittinghoff E, Elicker BM, et al. Mortality Risk Prediction in Scleroderma-Related Interstitial Lung Disease: The SADL Model. Chest. 2017;152(5):999–1007. doi: 10.1016/j.chest.2017.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Walker KM, Pope J. Treatment of Systemic Sclerosis Complications: What to Use When First-Line Treatment Fails-A Consensus of Systemic Sclerosis Experts. Semin Arthritis Rheum. 2012;42(1):42–55. doi: 10.1016/j.semarthrit.2012.01.003 [DOI] [PubMed] [Google Scholar]
- 13.Bernstein EJ, Khanna D, Lederer DJ. Screening High-Resolution Computed Tomography of the Chest to Detect Interstitial Lung Disease in Systemic Sclerosis: A Global Survey of Rheumatologists. Arthritis Rheumatol. 2018;70(6):971–972. doi: 10.1002/art.40441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White B Interstitial lung disease in scleroderma. Rheum Dis Clin North Am. 2003;29(2):371–390. doi: 10.1016/S0889-857X(03)00025-5 [DOI] [PubMed] [Google Scholar]
- 15.Rizzi M, Radovanovic D, Santus P, et al. Usefulness of six-minute walk test in systemic sclerosis. Clin Exp Rheumatol. 2018;36(4):161–167. [PubMed] [Google Scholar]
- 16.Coghlan JG, Denton CP, Grünig E, et al. Evidence-based detection of pulmonary arterial hypertension in systemic sclerosis: The DETECT study. Ann Rheum Dis. 2014;73(7):1340–1349. doi: 10.1136/annrheumdis-2013-203301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Frost A, Badesch D, Gibbs JSR, et al. Diagnosis of pulmonary hypertension. Eur Respir J. 2018:1801904. doi: 10.1183/13993003.01904-2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doyle TJ, Dellaripa PF. Lung Manifestations in the Rheumatic Diseases. Chest. 2017;152(6):1283–1295. doi: 10.1016/j.chest.2017.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kouranos V, Miranda G, Corte TJ, Renzoni EA. New treatment paradigms for connective tissue disease-associated interstitial lung disease. Curr Opin Pulm Med. 2018. doi: 10.1097/MCP.0000000000000508 [DOI] [PubMed] [Google Scholar]
- 20.Bouros D, Wells AU, Nicholson AG, et al. Histopathologic subsets of fibrosing alveolitis in patients with systemic sclerosis and their relationship to outcome. Am J Respir Crit Care Med. 2002;165(12):1581–1586. doi: 10.1097/MOL.0000000000000453 [DOI] [PubMed] [Google Scholar]
- 21.Khanna D, Nagaraja V, Tseng C hong, et al. Predictors of lung function decline in scleroderma-related interstitial lung disease based on high-resolution computed tomography: Implications for cohort enrichment in systemic sclerosis-associated interstitial lung disease trials. Arthritis Res Ther. 2015. doi: 10.1186/s13075-015-0872-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moore OA, Goh N, Corte T, et al. Extent of disease on high-resolution computed tomography lung is a predictor of decline and mortality in systemic sclerosis-related interstitial lung disease. Rheumatol (United Kingdom). 2013;52(1):155–160. doi: 10.1093/rheumatology/kes289 [DOI] [PubMed] [Google Scholar]
- 23.Takei R, Arita M, Kumagai S, et al. Radiographic fibrosis score predicts survival in systemic sclerosis-associated interstitial lung disease. Respirology. 2018. doi: 10.1111/resp.13175 [DOI] [PubMed] [Google Scholar]
- 24.Hax V, Bredemeier M, Didonet Moro AL, et al. Clinical algorithms for the diagnosis and prognosis of interstitial lung disease in systemic sclerosis. Semin Arthritis Rheum. 2017. doi: 10.1016/j.semarthrit.2017.03.019 [DOI] [PubMed] [Google Scholar]
- 25.Kim HJ, Tashkin DP, Clements P, et al. A computer-aided diagnosis system for quantitative scoring of extent of lung fibrosis in scleroderma patients. Clin Exp Rheumatol. 2010;28(5 SUPPL. 62):S-26–S-35. doi: 10.1016/j.surg.2006.10.010.Use [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim HJ, Tashkin DP, Gjertson DW, et al. Transitions to different patterns of interstitial lung disease in scleroderma with and without treatment. Ann Rheum Dis. 2016;75(7):1367–1371. doi: 10.1136/annrheumdis-2015-208929 [DOI] [PubMed] [Google Scholar]
- 27.Goldin JG, Kim GHJ, Tseng C-H, et al. Longitudinal Changes in Quantitative Interstitial Lung Disease on CT after Immunosuppression in the Scleroderma Lung Study II. Ann Am Thorac Soc. 2018;15(11):1286–1295. doi: 10.1513/AnnalsATS.201802-079OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.(NCRP) National Council on Radiation Protection and Measurements. Ionizing radiation exposure of the population of the United States. NCRP report no. 93. Natl Counc Radiat Prot Meas. 1987;93:1–10. [Google Scholar]
- 29.Huda W, Scalzetti EM, Roskopf M. Effective doses to patients undergoing thoracic computed tomography examinations. Med Phys. 2000;27:838–844. [DOI] [PubMed] [Google Scholar]
- 30.Lim HJ, Chung MJ, Shin KE, Hwang HS, Lee KS. The impact of iterative reconstruction in low-dose computed tomography on the evaluation of diffuse interstitial lung disease. Korean J Radiol. 2016;17(6):950–960. doi: 10.3348/kjr.2016.17.6.950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Steen VD, Conte C, Owens GR, Medsger T a. Severe restrictive lung disease in systemic sclerosis. Arthritis Rheum. 1994;37(9):1283–1289. doi: 10.1002/art.1780370903 [DOI] [PubMed] [Google Scholar]
- 32.Steen V Predictors of end stage lung disease in systemic sclerosis. Ann Rheum Dis. 2003;62(2):97–99. doi: 10.1136/ard.62.2.97 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nihtyanova SI, Schreiber BE, Ong VH, et al. Prediction of pulmonary complications and long-term survival in systemic sclerosis. Arthritis Rheumatol. 2014;66(6):1625–1635. doi: 10.1002/art.38390 [DOI] [PubMed] [Google Scholar]
- 34.Schoenfeld SR, Castelino FV. Evaluation and management approaches for scleroderma lung disease. Ther Adv Respir Dis. 2017;11(8):327–340. doi: 10.1177/1753465817713680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Goh NS, Hoyles RK, Denton CP, et al. Short-Term Pulmonary Function Trends Are Predictive of Mortality in Interstitial Lung Disease Associated With Systemic Sclerosis. Arthritis Rheumatol. 2017. doi: 10.1002/art.40130 [DOI] [PubMed] [Google Scholar]
- 36.Volkmann ER, Tashkin DP, Sim M, et al. Short-term progression of interstitial lung disease in systemic sclerosis predicts long-term survival in two independent clinical trial cohorts. Ann Rheum Dis. 2019;78(1):122–130. doi: 10.1136/annrheumdis-2018-213708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sircar G, Goswami RP, Sircar D, Ghosh A, Ghosh P. Intravenous cyclophosphamide vs rituximab for the treatment of early diffuse scleroderma lung disease: open label, randomized, controlled trial. Rheumatology (Oxford). 2018;57(12):2106–2113. doi: 10.1093/rheumatology/key213 [DOI] [PubMed] [Google Scholar]
- 38.Liu X, Mayes MD, Pedroza C, et al. Does C-reactive protein predict the long-term progression of interstitial lung disease and survival in patients with early systemic sclerosis? Arthritis Care Res. 2013;65(8):1375–1380. doi: 10.1002/acr.21968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Khanna D, Denton CP, Jahreis A, et al. Safety and efficacy of subcutaneous tocilizumab in adults with systemic sclerosis (faSScinate): a phase 2, randomised, controlled trial. Lancet. 2016;387(10038):2630–2640. doi: 10.1016/S0140-6736(16)00232-4 [DOI] [PubMed] [Google Scholar]
- 40.Galie N, Humbert M, Vachiery J, et al. ESC/ERS guidelines for the diagnosis and treat ment of pulmonary hypert ension. Eur Respir J2. 2015;46(1):879–882. doi: 10.1093/eurheartj/ehv317 [DOI] [Google Scholar]
- 41.Wells AU, Denton CP. Interstitial lung disease in connective tissue disease—mechanisms and management. Nat Rev Rheumatol. 2014. doi: 10.1038/nrrheum.2014.149 [DOI] [PubMed] [Google Scholar]
- 42.Khanna D, Mittoo S, Aggarwal R, et al. Connective tissue disease-associated interstitial lung diseases (CTD-ILD) - Report from OMERACT CTD-ILD working group. J Rheumatol. 2015;42(11):2168–2171. doi: 10.3899/jrheum.141182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Flaherty KR, Brown KK, Wells AU, et al. Design of the PF-ILD trial: a double-blind, randomised, placebo-controlled phase III trial of nintedanib in patients with progressive fibrosing interstitial lung disease. BMJ open Respir Res. 2017;4(1):1–7. doi: 10.1136/bmjresp-2017-000212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Winstone TA, Assayag D, Wilcox PG, et al. Predictors of mortality and progression in scleroderma-associated interstitial lung disease: A systematic review. Chest. 2014. doi: 10.1378/chest.13-2626 [DOI] [PubMed] [Google Scholar]
- 45.George PM, Wells AU. Disease staging and sub setting of interstitial lung disease associated with systemic sclerosis: impact on therapy. Expert Rev Clin Immunol. 2018;14(2):127–135. doi: 10.1080/1744666X.2018.1427064 [DOI] [PubMed] [Google Scholar]
- 46.Walker UA, Saketkoo LA, Distler O. Haematopoietic stem cell transplantation in systemic sclerosis. RMD Open. 2018;4(1):1–7. doi: 10.1136/rmdopen-2017-000533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sullivan K, Goldmuntz E, Keyes-Elstein L, et al. Myeloablative Autologous Stem-Cell Transplantation for Severe Scleroderma — NEJM. N Engl J Med. 2018:35–47. doi: 10.21430/M3SM4LTLH [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goldin J, Keyes-Elstein L, Crofford L, et al. Changes in Quantitative Scleroderma Lung CT Measures in Patients Treated with Cyclophosphamide or Transplantation. In: Arthritis Rheumatol. Vol 70 ; 2018:Suppl 10 https://acrabstracts.org/abstract/changes-in-quantitative-scleroderma-lung-ct-measures-in-patients-treated-with-cyclophosphamide-or-transplantation/. [Google Scholar]
- 49.Jablonski R, Dematte J, Bhorade S. Lung transplantation in scleroderma. Curr Opin Rheumatol. 2018;30(6):1. doi: 10.3109/03008207.2014.984804 [DOI] [PubMed] [Google Scholar]
- 50.Bernstein EJ, Peterson ER, Sell JL, et al. Survival of adults with systemic sclerosis following lung transplantation: A nationwide cohort study. Arthritis Rheumatol. 2015;67(5):1314–1322. doi: 10.1002/art.39021 [DOI] [PubMed] [Google Scholar]
- 51.Miele CH, Schwab K, Saggar R, et al. Lung transplant outcomes in systemic sclerosis with significant esophageal dysfunction: A comprehensive single-center experience. Ann Am Thorac Soc. 2016;13(6):793–802. doi: 10.1513/AnnalsATS.201512-806OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fernández-Codina A, Walker KM, Pope JE. Treatment Algorithms for Systemic Sclerosis According to Experts. Arthritis Rheumatol. 2018;70(11):1820–1828. doi: 10.1002/art.40560 [DOI] [PubMed] [Google Scholar]
- 53.Khanna D, Lin CJF, Kuwana M, et al. Efficacy and Safety of Tocilizumab for the Treatment of Systemic Sclerosis: Results from a Phase 3 Randomized Controlled Trial. Arthritis Rheumatol. 2018;70(Suppl 10):Suppl 10. [Google Scholar]
- 54.Van Assen S, Agmon-Levin N, Elkayam O, et al. EULAR recommendations for vaccination in adult patients with autoimmune inflammatory rheumatic diseases. Ann Rheum Dis. 2011;70(3):414–422. doi: 10.1136/ard.2010.137216 [DOI] [PubMed] [Google Scholar]
- 55.Dowman LM, McDonald CF, Hill CJ, et al. The evidence of benefits of exercise training in interstitial lung disease: a randomised controlled trial. Thorax. 2017;72(7):610–619. doi: 10.1136/thoraxjnl-2016-208638 [DOI] [PubMed] [Google Scholar]
- 56.Bagnato G, Harari S. Cellular interactions in the pathogenesis of interstitial lung diseases. Eur Respir Rev. 2015;24(135):102–114. doi: 10.1183/09059180.00003214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Distler O, Brown KK, Distler JHW, et al. SENSCIS study 2017 Nintednaib in SSC ILD. Clin Exp Rheumatol. 2017;35(6):75–81. [Google Scholar]
- 58.Khanna D, Albera C, Fischer A, et al. An open-label, phase II study of the safety and tolerability of pirfenidone in patients with scleroderma-associated interstitial lung disease: The LOTUSS trial. J Rheumatol. 2016;43(9):1672–1679. doi: 10.3899/jrheum.151322 [DOI] [PubMed] [Google Scholar]
- 59.Khanna D, Tashkin DP, Denton CP, Lubell MW, Vazquez-Mateo C, Wax S. Ongoing clinical trials and treatment options for patients with systemic sclerosis–associated interstitial lung disease. Rheumatology (Oxford). 2018;1:1–13. doi: 10.1093/rheumatology/key151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mayes MD, Lacey JV., Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum. 2003;48(8):2246–2255. doi: 10.1002/art.11073. [DOI] [PubMed] [Google Scholar]
- 61.Greidinger EL, Flaherty KT, White B, Rosen A, Wigley FM, Wise RA. African-American race and antibodies to topoisomerase I are associated with increased severity of scleroderma lung disease. Chest. 1998;114(3):801–807. doi: 10.1378/chest.114.3.801. [DOI] [PubMed] [Google Scholar]
- 62.Steen VD. Autoantibodies in systemic sclerosis. Bull Rheum Dis. 1996;45(10):6. [PubMed] [Google Scholar]
- 63.Carulli MT, Handler C, Coghlan JG, Black CM, Denton CP. Can CCL2 serum levels be used in risk stratification or to monitor treatment response in systemic sclerosis? Ann Rheum Dis. 2008;67(1):105–109. doi: 10.1136/ard.2006.067967. [DOI] [PubMed] [Google Scholar]
- 64.Hoffmann-Vold AM, Tennøe AH, Garen T, et al. High Level of Chemokine CCL18 Is Associated With Pulmonary Function Deterioration, Lung Fibrosis Progression, and Reduced Survival in Systemic Sclerosis. Chest. 2016;150(2):299–306. doi: 10.1016/j.chest.2016.03.004. [DOI] [PubMed] [Google Scholar]
- 65.De Lauretis A, Sestini P, Pantelidis P, et al. Serum Interleukin 6 Is Predictive of Early Functional Decline and Mortality in Interstitial Lung Disease Associated with Systemic Sclerosis. J Rheumatol. 2013;40(4):435–446. doi: 10.3899/jrheum.120725. [DOI] [PubMed] [Google Scholar]
- 66.Volkmann ER, Tashkin DP, Roth MD, et al. Changes in plasma CXCL4 levels are associated with improvements in lung function in patients receiving immunosuppressive therapy for systemic sclerosis-related interstitial lung disease. Arthritis Res Ther. 2016;18(1):305. doi: 10.1186/s13075-016-1203-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Salazar GA, Kuwana M, Wu M, et al. KL-6 But Not CCL-18 Is a Predictor of Early Progression in Systemic Sclerosis-related Interstitial Lung Disease. J Rheumatol. 2018:jrheum.170518. doi: 10.3899/jrheum.170518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hasegawa M, Fujimoto M, Hamaguchi Y, et al. Use of serum Clara cell 16-kDa (CC16) Levels as a potential indicator of active pulmonary fibrosis in systemic sclerosis. J Rheumatol. 2011;38(5):877–884. doi: 10.3899/jrheum.100591. [DOI] [PubMed] [Google Scholar]
- 69.Tashkin DP, Elashoff R, Clements PJ, et al. Effects of 1-year treatment with cyclophosphamide on outcomes at 2 years in scleroderma lung disease. Am J Respir Crit Care Med. 2007;176(10):1026–1034. doi: 10.1164/rccm.200702-326OC. [DOI] [PMC free article] [PubMed] [Google Scholar]