

Abstract
A fractionation method for technical lignin was developed, combining organic solvent extraction and membrane ultrafiltration of the solvent soluble component. This method was validated on a commercial wheat straw/Sarkanda grass lignin (Protobind 1000) using 2-butanone (MEK) as the solvent for both the extraction and the ultrafiltration operations. The parent lignin and the different obtained fractions were fully characterized in terms of chemical composition and physicochemical properties by gel permeation chromatography, gas chromatography/mass spectrometry (GC/MS), pyrolysis-GC/MS, total phenol contents, 31P nuclear magnetic resonance (31P NMR), thermogravimetric analysis, differential scanning calorimetry analysis, and Fourier-transform infrared spectroscopy. The results show that the proposed process allows a straightforward recovery of the different lignin fractions as well as a selective control over their molecular mass distribution and related dependent properties. Moreover, the operating flexibility of the Soxhlet/ultrafiltration process allows the treatment of lignins from different feedstocks using the same installation just by modulating the choice of the solvent and the membrane porosity with the best characteristics. This is one of the most important features of the proposed strategy, which represents a new fractionation approach with the potential to improve lignin valorization for materials science and preparative organic chemistry applications.
1. Introduction
Lignin is a complex methoxylated phenylpropane polymeric mixture which represents 15–33% of the cell wall of terrestrial plants.1,2 Technical lignin is usually obtained as waste byproduct of the cellulose industry, mainly in paper and biofuel production.3−5 The structure and physicochemical properties of lignin are strongly dependent on its natural origin, on the extraction and the fractionation methods used especially in terms of heterogeneity and molecular size distribution.6,7 Although lignin represents a highly abundant aromatic feedstock with potentially attractive applications from an economic and ecological point of view, thermovalorization seems currently to be the most common exploitation strategy for this cheap and highly available biomass, as up to now around 95% of lignin produced worldwide every year is burnt for energy recovery, typically at the pulping plant itself.8 The industrial use of lignin is limited because of its very complex and bulky structure, its high heterogeneity, and its intrinsic recalcitrance, and only a small amount of the produced biomass is currently utilized as raw industrial material.9,10 The main pathways for lignin valorization typically aim for the production of materials including its use as a filler, in blend with polymers, as a copolymer, and as a water-reducing agent for concrete, allowing its direct exploitation without requiring a preliminary depolymerization step after the original biomass fractionation.11−16
In the production of both chemicals and materials, the full exploitation potential of lignin is strictly related to the availability of efficient and viable depolymerization processes in order to obtain low molecular weight aromatics and building blocks for their further incorporation in higher added value biobased compounds.17,18 This critical process is mainly hindered by the intrinsic lignin resistance to degradation, therefore requiring strong chemical or thermochemical deconstruction methods such as pyrolysis, chemical oxidation, hydrogenolysis, gasification, and hydrolysis under supercritical conditions.19,20 As a suitable alternative, biocatalytic methods are currently investigated because they allow the use of mild conditions and greener reagents.21−24
Despite the potential interest in these valorization approaches, some obstacles to an efficient use of technical lignins for high-value-added applications remain, mainly resulting from their inherent high molecular mass dispersion because of the different biomass fractionation processes used for biomass delignification. The molecular weight of technical lignins plays a fundamental role in their valorization, and the availability of lignins with well-defined mass ranges appears as the key requirement for the whole exploitation of this valuable resource. Within this framework, we recently set up a method of fractionation of a soda pulp lignin with membrane-assisted ultrafiltration in ethanol/water, which represents a particularly interesting technology because of its high separation efficiency and adaptability to different feeds and liquors.25 Nevertheless, one major limitation of this methodology is associated with the poor solubility of most commercial lignins in water-based solutions, which typically leads to membrane fouling, potential reduction of processing throughput because of maintenance for cleaning cycles, and reduction of in-service membrane life time. Furthermore, the recovery of the final products requires additional acidification and extraction operations on the process solutions, resulting in unavoidable mass losses, waste production, and overall yield reduction that can decrease the industrial interest despite its attractiveness due to the use of the environmental friendly water/ethanol mixture.
To bridge this gap, this work presents a new lignin fractionation method based on an initial Soxhlet-mediated extraction of a commercial technical lignin in the organic solvent, directly followed by membrane-assisted cascade ultrafiltrations on the solvent soluble component. It is worth noting that a combined organosolv-ultrafiltration approach for the delignification of non-woody lignocellulosic biomass has been described previously in a biorefinery process.26 In the present work, the focus is on obtaining lignin fractions with controlled properties and on assessing the process performance on the basis of the characteristics of such lignin fractions, which were fully analyzed in terms of their chemical, physical, thermal and structural properties. The approach presented in this study demonstrates a straightforward process to obtain lignin fractions with tailored characteristics using an industrially relevant and toxicologically benign organic solvent with the possibility to be recyclable in a closed circuit plant, in view of a potential reduction of environmental concerns and process costs. In addition, it provides important insights into the structure–property relationships of the resulting materials in view of their potential further exploitation for the development of biobased polymers and chemicals.
2. Results and Discussion
The main goal of this work was to describe a general extraction/mass fractionation process applicable to technical lignins in order to exploit their whole valorization potential by separating the starting raw materials in well-defined and narrowly dispersed molecular size fractions. The fractionation process based on an initial Soxhlet extraction with the organic solvent followed by a membrane-mediated ultrafiltration was carried out in this study on a representative technical lignin (Protobind 1000), using 2-butanone (methyl ethyl ketone, MEK) as the extraction solvent (due to its industrial relevance and its favorable toxicological profile). The selection of a commercially available herbaceous lignin material for this study reflects the great impact of non-woody, agricultural, and crop-derived biomass on the global biobased economy both in terms of economic turnaround and mass availability.27
2.1. Solvent Selection
The selection of a suitable solvent for Soxhlet lignin extraction is a key step for the whole process. The extraction efficiency of Protobind 1000 was tested for a series of six different organic solvents, namely tetrahydrofuran (THF), methanol (MeOH), MEK, ethylacetate (EtOAc), n-butylacetate (BuOAc), and tert-butyl methyl ether (MTBE). The extraction yields are reported in Table 1 together with results obtained from gel permeation chromatography (GPC) analysis on the extracted solutions expressed as number average molecular weight (Mn), weight average molecular weight (Mw), and polydispersity index (Đ).
Table 1. Solvent Extraction of Protobind 1000: Yields, Number Average Molecular Weight (Mn), Weight Number Molecular Weight (Mw), and Polydispersity Index (Đ) as Obtained from GPC Analysis (Molecular Weights Are Relative to Polystyrene Standards)a.
| samples | extraction yield (w/w) (%) | Mn (Da) | Mw (Da) | D̵ |
|---|---|---|---|---|
| Protobind | 1390 | 4660 | 3.3 | |
| THF soluble fraction | 84 | 1500 | 5015 | 3.3 |
| MeOH soluble fraction | 76 | 1355 | 3965 | 2.9 |
| MEK soluble fraction | 75 | 1060 | 2500 | 2.4 |
| EtOAc soluble fraction | 36 | 920 | 1630 | 1.8 |
| BuOAc soluble fraction | 33 | 650 | 1050 | 1.6 |
| MTBE soluble fraction | 12 | 445 | 540 | 1.2 |
Estimated extraction yields standard error ± 1%.
The highest extraction yields were obtained with THF, MeOH, and MEK as the extracting solvents. In order to achieve an efficient recovery of the higher molecular mass components (and enhance the overall process yields), it is thus mandatory to select a solvent with high extraction capability. This is also confirmed from GPC data that demonstrate an evident shift toward low molecular mass values of solutions with low extraction yields. In the present study, MEK was selected considering other aspects such as easy evaporation, low viscosity of the extracts (to avoid clogging in filtration membranes), good recyclability, uncritical handling, and low toxicity. It is important, however, to emphasize that this is not to be considered a general indication because technical lignins can have very different solubility behaviors as a result of the specific delignification process used for their recovery and of the biomass origin. To provide a comparative example, the solubility of another commercial lignin (softwood Kraft lignin) was determined for the same solvent series and the results are reported in the Supporting Information. In this case, the best solvent seems to be methanol. The operating flexibility of the Soxhlet/ultrafiltration process, which allows to treat lignins from very different sources using the same installation just by modulating the choice of the most suitable solvent from a broad range of possibilities, is one of the most important features of the proposed strategy.
2.2. Fractionation Process
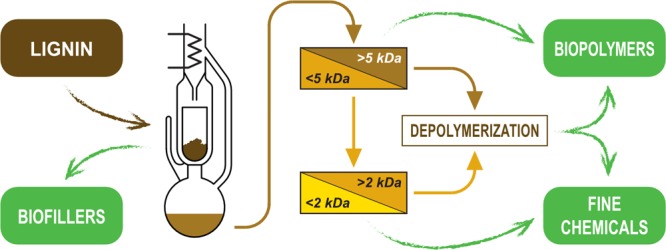
The sequential process of solvent extraction and mass fractionation of technical lignins is reported in Figure 1. It represents the basis of an idealized total valorization process (including the downstream depolymerization and purification/upgrading operations that are not part of the present study). An essential part of the whole process concept is the recovery of the outcome lignin fractions by solvent evaporation and recycle (not illustrated here for clarity).
Figure 1.

Overview of the fractionation process depicted as basis for an idealized total lignin valorization workflow (plain line: fractionation process, dashed line: downstream potential use of the isolated fractions; solvent recycling pathway not illustrated).
The first step of the process is a Soxhlet solvent extraction of the target technical lignin (LF-0). MEK was selected as the suitable organic solvent and the initial concentration in the extractor loading was 105 g/L. The Soxhlet extraction allowed the transfer of the soluble components of the starting lignin to the liquid phase, leaving the impurities as the solid residual matter. The desired compounds dissolved in the warm solvent were concentrated in the collecting flask. The solvent reflux was kept for 8 h. From the Soxhlet extraction, an insoluble solid residual fraction (LF-I) and a soluble extracted fraction (LF-S) were recovered. LF-S was then directly submitted to the downstream cascade ultrafiltration process: the first ultrafiltration step (with membrane cutoff of 5 kDa) generated two fractions, LF-R5 (retentate) and LF-P5 (permeate). This latter was processed with a second ultrafiltration step (with a 2 kDa cutoff membrane) to give LF-R2 (retentate) and finally LF-P2 (permeate). The mass yields after the different steps (obtained from solvent evaporation) were reported in Table 2. As it appears clearly from these data, the overall mass balance of the process was close to 75%.
Table 2. Process Fractions and w/w Yields Relative to the Previous Step and to the Starting Material (Protobind 1000)a.
| fraction | description | cumulative relative yield (% w/w initial LF-0) | relative step yield (% w/w previous step) |
|---|---|---|---|
| LF-I | unextracted solid residual | 18.1 | 19.4 |
| LF-S | extracted material | 75.0 | 80.6 |
| LF-R5 | 5 kDa cutoff retentate | 66.3 | 88.4 |
| LF-P5 | 5 kDa cutoff permeate | 8.70 | 11.6 |
| LF-R2 | 2 kDa cutoff retentate | 7.02 | 81.4 |
| LF-P2 | 2 kDa cutoff permeate | 1.60 | 18.6 |
LF-S and LF-P5 are intermediate fractions not recovered in the nominal process.
Process parameters were optimized for the selected solvent (MEK) as previously described (see Materials and Methods and Supporting Information for details). The Soxhlet repeatability was assessed performing 20 extraction operations in the optimized process conditions, obtaining ±1.7% maximum deviation of the extraction yields. Furthermore, unlike what recently reported with aqueous solutions,25 the use of MEK allowed to work with very high lignin concentrations starting solutions (105 g/L vs 15 g/L) and also prevented the fouling of the stabilized cellulose-based membranes during ultrafiltration. Indeed, the filtration procedure was repeated up to 10 complete consecutive cycles, maintaining a very stable area-specific air diffusion rate through the membranes.
The obtained mass fractionation products, with particular reference to fractions LF-R5, LF-R2 and LF-P2, were fully characterized by GPC, gas chromatography (GC)/mass spectrometry (MS), Py-GC/MS, phenol titration, 31P NMR, Fourier-transform infrared (FTIR), thermogravimetric analysis (TGA), differential scanning calorimetry (DSC). The LF-I fraction (solid residual of Soxhlet extraction) was not further characterized because, although being a fully legitimate process outcome, it was not considered relevant in this context.
2.3. Mass Distribution Based on GPC
In order to evaluate the effect of the combined solvent fractionation and ultrafiltration steps on the molecular weights and molecular weight distribution of the technical lignin considered in this work, GPC was performed on all fractions in order to obtain the characteristic values of number-average molecular weight (Mn), weight-average molecular weight (Mw), and polydispersity index (Đ). Analyses were performed using monodispersed polystyrene standards as the reference. The chromatograms of all analyzed materials are reported in Figure S3, and the numerical results are listed in Table 3.
Table 3. GPC Analysis (Molecular Weights Mn and Mw, Polydispersity Index Đ of All Examined Soluble Lignin Fractions)a.
| sample | Mn (Da) | Mw (Da) | D̵ |
|---|---|---|---|
| LF-0 | 1390 | 4660 | 3.3 |
| LF-S | 1060 | 2500 | 2.4 |
| LF-R5 | 1150 | 1950 | 1.7 |
| LF-P5 | 770 | 1180 | 1.5 |
| LF-R2 | 790 | 1190 | 1.5 |
| LF-P2 | 570 | 700 | 1.2 |
Samples have been eluted after acetylation. The reported values for molecular weights are relative to polystyrene standards.
The preliminary solvent-assisted extraction process allows the recovery of the first soluble fraction LF-S with a slightly narrower molecular weight distribution compared to that found in the parent lignin LF-0 (Đ = 2.4 and 3.3 for LF-S and LF-0, respectively). Similarly, upon ultrafiltration through a 5 kDa membrane, the retentate fraction LF-R5 shows a further slight reduction in Đ, resulting from a decrease in Mw. These results are in line with the extraction/recovery yields presented in Table 2, where LF-R5 was found to account for approximately 90% of the soluble fraction recovered after MEK extraction. This trend indicates that such soluble fraction is relatively homogenous in terms of molecular size, and can therefore be potentially exploited as the starting material for lignin-based polymers. Conversely, the permeate fraction LF-P5 (that accounts for approximately 9% of the total mass of the parent material) is found to possess a significantly reduced Mn and Mw compared with LF-0 (770 and 1180 g/mol vs 1390 and 4660 g/mol, respectively). This evidence indicates that the first membrane-assisted ultrafiltration step (5 kDa cutoff) enables the recovery of a permeate fraction enriched in lower molecular weight molecules that can be further considered as a potential source of valuable biobased small molecules (as will be further discussed in the next sections). It is worth noticing that by further processing, the resulting permeate fraction LF-P5 through a 2 kDa membrane, an additional reduction in Mn and Mw can be achieved along with a significant narrowing of Đ, ultimately leading to values of 570, 700 g/mol, and 1.2, respectively. Based on these results, the proposed multistep fractionation process appears to be very effective in narrowing the molecular weight distribution of the obtained fractions and more generally in allowing easy and precise control of the molecular weight of the starting material.
The mass fractionation results were compared with a further test (process B) carried out by fractionating the same technical lignin (Protobind 1000) in an aqueous solvent (ethanol/basic water 60:40 v/v) with a different first stage process (0.7 μm membrane microfiltration instead of the Soxhlet extraction), as we described in a previous work with different membranes.25Figure 2 reports Mw, Mn, and Đ of the output isolated fractions (LF-R5, LF-R2, and LF-P2) obtained in both process conditions. In this case, we expected a different size distribution for the first, heavier fractions (here LF-R5) because of the modified solvent and first-stage process that result in a different extracted quantity of high molecular mass material (i.e., the most critical to be solubilized and thus the most sensible to the extraction process parameters).
Figure 2.
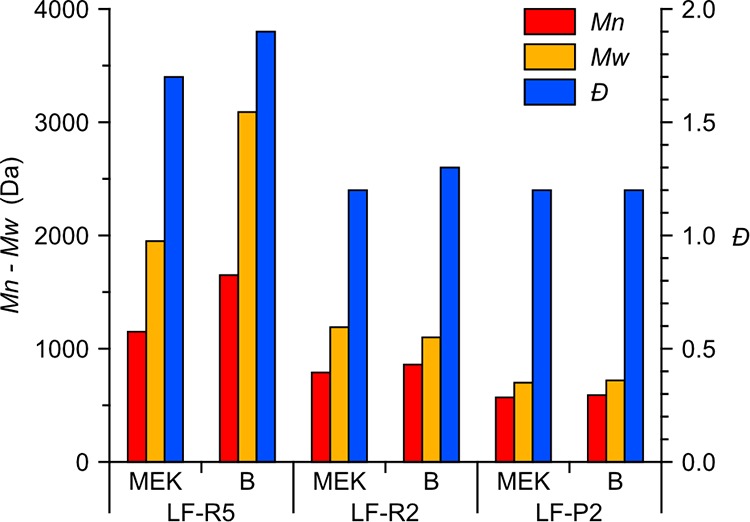
Mn, Mw, and Đ values obtained via GPC analysis on the output isolated fractions (LF-R5, LF-R2, and LF-P2) recovered upon treating lignin with the process described in this work (MEK) and with a different fractionation process (B) based on the ethanol/basic water solvent and the microfiltration first stage (see text for details).
On the other hand, the results of the lower molecular weight fractions appeared to be quite similar, proving that the impact of the solvent and of the first step of the process (microfiltration vs. Soxhlet extraction) is most critical for the higher MW fractions. Moreover, the data also confirm that the filtration membranes performance is not affected by the solvent used, indicating a good independence between the extraction operation (controlled by the solvent) and the downstream fractionation (controlled by the ultrafiltration membrane). This is a very important feature from an industrial scale-up perspective because it allows an easy process optimization with different lignins (e.g., soda vs Kraft pulping).
It is worth it to point out that the accurate determination of the molecular weight of lignin samples has been widely discussed in the literature.28−30 In particular, it is widely acknowledged that the molecular weight determination by GPC analysis only provides a relative quantification of the mass distribution with respect to the standard used for GPC calibration. In this work, monodisperse polystyrene standards were employed for GPC calibration, in line with common practice in this field. In this respect, unavoidable structural differences between the most commonly employed polymer standards (typically linear polymers) and lignin macromolecules (known to be highly branched) are a very well-known source of error in the determination of molecular weight.31,32 Based on these considerations, it is not uncommon to find limited correspondence between the cut-off dimensions of the membranes used for the ultrafiltration process in lignin fractionation and the average molecular weights found by means of GPC analysis on the resulting lignin fractions, with membrane cut-off dimensions frequently overestimating the obtained molecular weight values with respect to GPC.33−36 Therefore, the molecular weight characterization of the obtained lignin fractions is to be intended as a proof of the beneficial effect of membrane-assisted ultrafiltration in isolating lignins with decreasing the molecular weight and polydispersity as the membrane cut-off decreases.
2.4. Determination of Hydroxylation Levels
2.4.1. Phenolic Hydroxyl Group Determination
Folin−Ciocalteu (FC) assay has been carried out to determine the total phenolic content in the original lignin and all process fractions. It is based on the reaction of phenolic hydroxyl groups with a specific redox reagent (FC reagent) which leads to the formation of a blue chromophore which is however sensitive and unstable in strong bases.37 Therefore, in this work, dimethylsulfoxide (DMSO) was used as the solvent for the samples in order to obtain complete sample solubilization in neutral conditions.
The results are reported in Table 4 as gallic acid equivalents (GAEs, μg gallic acid per mL of working solution) and as total phenolic content in mmol phenolic groups per g of lignin. It is worth noting that with the FC method, only two phenolic groups of gallic acid are reactive, so one GAE must be considered as two phenolic group equivalents.38 LF-P2 appeared to be the fraction with a higher content in phenolic hydroxyl groups as it will be confirmed below with 31P NMR experiments.
Table 4. Results of Phenolic Hydroxyl Group Determination in the Lignin Fractions as GAE in the Analytical Working Solution (Expressed in μg/mL) and as Total Phenolic Content in the Dry Lignin (Expressed in mmol/g)a.
| fraction | GAE (μg/mL) | phenolic −OH content (mmol/g) |
|---|---|---|
| LF-0 | 357 | 4.2 |
| LF-S | 333 | 3.9 |
| LF-R5 | 394 | 4.6 |
| LF-P5 | 367 | 4.3 |
| LF-R2 | 374 | 4.4 |
| LF-P2 | 422 | 5.0 |
Estimated standard errors (1 σ, from calibration data): GAE ±20 μg/mL and phenolic OH content ±0.2 mmol/g.
2.4.2. Total Hydroxyl Groups Quantification with 31P NMR Analysis
In the last years, 31P NMR constitutes the most recognized method for the quantification of hydroxyl groups in lignins because the use of the phosphorylating agent 2-chloro-4,4,5,5-tetramethyl-1,3-2-dioxaphospholane leads to the complete phosphorylation of all the OH-groups present in the analyzed samples.39 Furthermore, the clear differentiation in chemical shifts of aliphatic, aromatic, and carboxylic groups allows their quantification using an internal standard as the reference (here, N-hydroxy-5-norbornene-2,3-dicarboxylic acid diimide). Figure 3 shows a comparison of the 31P NMR spectra of the starting material LF-0 with the different fractions obtained in the process of fractionation, allowing to distinguish the different hydroxyl groups present in the lignin backbone.
Figure 3.

31P NMR spectra of lignin fractions.
In detail, the region between 151 and 146 ppm represents the aliphatic hydroxyl groups, whereas the region between 145 and 138 ppm accounts for the aromatic hydroxyl groups. The signals of carboxylic acids groups are centered on 136 ppm. The peak integration in the three main portions of the spectrum leads to the quantification of the total hydroxyl groups as reported in Table 5.
Table 5. Detailed Hydroxyl/Carboxyl Quantification by 31P NMR.
| lignin | –OH aliphatic [mmol/g] | –OH phenolic [mmol/g] | –COOH [mmol/g] |
|---|---|---|---|
| LF-0 | 1.83 | 3.54 | 0.92 |
| LF-S | 0.74 | 3.29 | 0.66 |
| LF-R5 | 1.24 | 4.19 | 0.09 |
| LF-R2 | 1.27 | 4.52 | 1.29 |
| LF-P2 | 1.48 | 5.2 | 1.52 |
Both FC assay and 31P NMR data indicate a slight phenolic hydroxyls increment in the lower MW fractions LF-R2 and LF-P2. 31P NMR data indicate that this trend is extended also to the aliphatic hydroxyl and carboxyl groups.
2.4.3. GC/MS Results
The distribution of small molecules such as aromatic monomers and aliphatic carboxylic acids isolated from the different fractions was determined by GC/MS. The analyzed samples were prepared with a small scale chromatography on silica gel in order to eliminate all the polymeric/oligomeric fractions and to recover only the suitable fraction for GC/MS analysis (% of mass recovered after chromatography are reported in Table 6). It is clearly noticeable that, going through the fractionation process, the percentages are congruent with the composition of each fraction. In particular, LF-R5 which is the higher MW retentate contains only 7% of small molecules which can be analyzed by GC/MS, whereas LF-P2 which is the last permeate of the process is considerably enriched in small molecules (63%).
Table 6. Summary of GC/MS Results of Fractions LF-S, R5, R2, P2 (#% of the Fraction Analyzed) Illustrating the Estimated Amount of Volatile Compounds, Divided in Three Main Groups, Reported as Total Mass of Compounds/Mass of Initial Fraction × 100 (the Values Were Rounded up to the Nearest Two Significant Figures with an Estimated Relative Error of ±0.01).
| fractions |
||||
|---|---|---|---|---|
| LF-S (36%)# (%) | LF-R5 (7.0%)# (%) | LF-R2 (36%)# (%) | LF-P2 (63%)# (%) | |
| Compounds | ||||
| ArCHO, ArCORa | 19 | 14 | 13 | 31 |
| Ar-COOH+ ArCHCHCOOHa | 11 | 5.6 | 16 | |
| R-COOHb | 2.6 | 2.2 | ||
| total monomers | 33 | 14 | 21 | 47 |
Ar = aromatic residue.
R = aliphatic chain.
The results are reported in Figure 4 and Table 6 and describe the distribution of the small main compounds present in each fraction: they are divided into three main classes such as benzaldehydes and acetophenone derivatives (ArCHO, ArCOR), benzoic and coumaric acids (ArCOOH, ArCHCHCOOH), and aliphatic long chain carboxylic acids (RCOOH). The distribution trends highlighted in Table 6 confirm the efficacy of the fractionation procedure, as the percentages of total monomers in each sample increases when proceeding through the process, passing from 14% (w/w) of monomers in the analyzed higher molecular weight fraction LF-R5 to a maximum of 47% (w/w) in the lower molecular weight fraction LF-P2.
Figure 4.

GC/MS chromatograms for the fractions LF-S, LF-R5, LF-R2, and LF-P2 with the assignment of the main compounds signals.
The chromatogram profiles of the different fractions and the structures of the most abundant identified compounds are reported in Figure 4, where the enrichment in smaller MW products clearly appears when moving throughout the fractionation process, from fraction LF-S to fraction LF-P2 as the number of peaks in the chromatogram becomes much more relevant. The complete characterization of all peaks is reported in the Supporting Information.
2.4.4. Pyrolysis-GC/MS
Py-GC/MS represents a very useful tool for lignin structure elucidation.40,41 The so-called pulsed pyrolysis offers a wide temperature range for the applied pyrolysis temperature by platinum resistive heating in comparison to a Curie point pyrolysator.42 The aim of the pyrolysis evaluation in lignin is to see if changes in the so-called S-/G-/H-units distribution of the obtained fractions occurred going through the fractionation sequence. Py-GC/MS of lignin results usually in different phenolic compounds with p-hydroxyphenyl (H), guaiacyl (G), and syringyl (S) aromatic moieties with or without alkyl substituents.
The aromatic (H), (G), and (S) moieties differ in the amount of methoxy groups (zero, one, or two groups, respectively). Depending on the pyrolysis conditions, the methoxy groups may be largely unaffected and the distribution of pyrolysates from each of the monomers in the liquid fraction may reflect the relative abundance of the monomers in the starting feedstock. Therefore, it is possible to measure the S/G ratio in lignin polymers based on pyrolysate distributions. For the pyrolysis, two potential temperatures were selected after a TGA of the lignins LF-0 and LF-S (obtained curves are reported in Figure S16). The first one was 600 °C because at this temperature, the pyrolytic degradation of the lignin stops in the obtained TGA thermograms. The second one was 450 °C because this value corresponds to the middle of the degradation process of the samples.
The impact of the temperature choice is obvious in the two selected samples. In the case of starting lignin LF-0 and the solvent-extracted LF-S (Figure 5 panels A/B, and panels C/D, respectively), the amount of small signals is reduced and the intensity of other signals became increased at the lower temperature. This is probably caused by the reduction of secondary reactions in the gas phase of the pyrolyzed lignin.43,44 Furthermore, the quality of the signals increased at 450 °C.
Figure 5.

Py-GC/MS TIC chromatograms of LF-0 at 600 °C (panel A) and 450 °C (panel B), and LF-S at 600 °C (panel C) and 450 °C (panel D).
The identification of the peaks was performed in combination with the NIST Database (NIST MS Search 2.0g, data reported in the Supporting Information). The identification was necessary to assign the (H), (G), and (S) subunits to the obtained fragments for the determination of the S/G ratio (see Table 7).
Table 7. S/G Ratio of the Obtained Fractions after Ultrafiltration (C = Carbohydrate).
| sample | C | S | G | H | S/G-ratio |
|---|---|---|---|---|---|
| LF-0 Protobind 1000 | 5.055 | 56.36 | 29.29 | 9.29 | 1.92 |
| LF-S | 4.566 | 53.01 | 34.32 | 8.106 | 1.54 |
| LF-R5 | 5.683 | 46.97 | 29.12 | 18.23 | 1.61 |
| LF-P5 | 6.783 | 45.02 | 38.80 | 9.393 | 1.16 |
| LF-R2 | 7.891 | 40.38 | 30.94 | 20.79 | 1.31 |
| LF-P2 | 3.282 | 41.25 | 41.14 | 14.33 | 1.00 |
Remarkable changes in the S/G ratio are reported during the different steps of the filtration process, which is found to decrease with the size of the ultrafiltration membrane. The pristine material LF-0 exhibits a S/G ratio of 1.92, whereas in the solvent-extracted LF-S fraction, S/G is found to reach 1.54. With each filtration step, the ratio goes down to 1.00. This means that the smaller lignin molecules possess a different average composition if compared to the raw material or the bulky fraction and that this fractionation process has a strong impact on the molecular distribution of the lignin extract, reflected by the decrease of polydispersity Đ.
2.5. Fourier Transform Infrared Spectroscopy
FTIR spectroscopy was employed to analyze the chemical characteristics of the lignin fractions recovered after solvent extraction and subsequent membrane-assisted ultrafiltration. The spectra of each material are reported in Figure 6 together with the spectrum of the parent materials LF-0.
Figure 6.

(a) FTIR spectra of all lignin fractions analyzed in this work. (b) Zoom in the 1850–700 cm–1 spectral region.
All samples present the characteristic peaks of soda-pulp lignin with clearly identifiable signals in the 3800–2700 and 1850–700 cm–1 spectral regions.45,46 Some differences among the obtained fractions appear upon solvent/membrane-assisted filtration. In particular, a reduction of the intensity of the −OH stretching signal at 3390 cm–1 is registered after Soxhlet extraction compared with the pristine material, as evidenced in the FTIR spectrum of the recovered soluble fraction LF-S. This evidence suggests that a decrease in hydroxyl group concentration is experienced upon solvent extraction, likely due to the poor hydrogen bonding power of the solubilizing solvent (MEK in this case) toward lignin macromolecules. Upon filtration through membranes of decreasing cut off, the signal at 3390 cm–1 is generally found to be higher for permeate fractions (LF-P5 and LF-P2) than for the corresponding retentate fractions (LF-R5 and LF-R2). Considering that permeate fractions are typically characterized by lower molecular weights (viz., chain lengths) than the corresponding retentate fractions (as also observed in GPC analysis, Figure S3), these trends observed on FTIR analysis may indicate that shorter chains are characterized by a lower concentration of −OH groups. Therefore, in addition to a fine control over the average molecular weight of the resulting fractions during membrane-assisted ultrafiltration, a modulation of concentration of reactive groups can also be obtained.
At 1705 cm–1, a clear sharp peak is observed in all spectra, attributable to the stretching vibration of C=O bonds in conjugated aldehydes and carboxylic acids. It is observed that after solvent extraction (LF-S), a notable increase in this signal is observed compared with the parent material, likely indicating a higher concentration of carbonyl and carboxylic groups. After ultrafiltration, a broadening of the signal at 1705 cm–1 toward lower frequencies is registered, more noticeable for the permeate fraction recovered downstream of the 2 kDa membrane (LF-P2). This is a further indication of the enrichment in oxidized species for fractions of lower molecular weight, as also confirmed by the trends observed on 31P NMR (Figure 3 and Table 5). In the 1400–1000 cm–1 spectral region, signals of variable intensity are observed, to be attributed to bending vibrations of phenolic O–H and aliphatic C–H in methyl groups (1370 cm–1), C–O, C–C, and C–O stretching vibrations (1270 and 1210 cm–1), C–H in plane deformations (1125 cm–1), and C–O deformations in primary (1030 cm–1) alcohols. In general, an overall reduction of the intensity of such signals is registered after solvent extraction and ultrafiltration, indicating a decrease of concentration of the corresponding species in the resulting fractions when compared with the parent material.
2.6. Thermal Behavior of Lignin Samples
The thermal transitions in the recovered lignin fractions were evaluated by means of DSC analysis and compared with the parent system LF-0. As evidenced from Figure 7a, where DSC traces of all samples are reported, the solvent extraction step leads to a notable decrease in glass transition temperature (Tg) of the recovered material, found to be 99 °C for LF-S as opposed to 139 °C for LF-0. Considering the significant decrease of hydroxyl (especially aliphatic) group concentration experienced by LF-S compared with the parent material (as observed by means of 31P NMR, Table 5), this reduction in Tg may be associated with an increased mobility of the macromolecular chains in LF-S resulting from the reduction of intra- and interchain hydrogen bonding interactions.
Figure 7.

(a) DSC traces and (b) TGA thermograms of all lignin fractions analyzed in this work (the solid arrow indicates the increase in intensity of the low-temperature mass loss event, the dashed arrow indicates the shift of TDTGmax at lower temperatures).
By successive ultrafiltration of LF-S through the membrane of decreasing cutoff (5 and 2 kDa, respectively), a further reduction in Tg is observed in the permeate fractions LF-P5 (53 °C) and LF-P2 (50 °C). Conversely, a slightly higher Tg compared with the input material LF-S is found for the retentate fraction recovered after the first ultrafiltration (LF-R5, Tg = 114 °C). Such trends may be correlated with the outcomes of GPC (Figure S3 and Table 3) analyses discussed previously, in which a general decrease of molecular weight (and reduction of polydispersity) was found in lignin fractions obtained after filtration through increasingly finer membranes. As previously observed also on other types of lignin,47 these results highlight the strong dependence of Tg on the molecular weight of lignin.
The investigation of the thermal stability of the lignin samples considered in this work was performed by means of TGA measurements conducted under N2 flow. As shown in Figure 7b, where TGA thermograms (mass loss curves and corresponding mass loss derivatives) of all lignin fractions are reported, all systems exhibit a major mass loss event in the 300–450 °C temperature range, which may be attributed to the rupture of carbon–carbon bonds between lignin structural units and functional groups.48 In particular, all fractions exhibit a maximum mass loss derivative temperature (TDTGmax) centered at around 300–400 °C, which is, however, found to decrease slightly for fractions recovered after progressively finer membranes. Similarly, a reduction of the final char residue at 760 °C is also observed. In addition, the appearance of an additional mass loss event centered at 200 °C of increasing intensity is reported for such materials, attributable to the breaking of α- and β- alkyl−aryl−ether links, aliphatic chains, and decarboxylation reactions. These trends well correlate with the results from DSC analysis and further confirm the major impact of the molecular weight on the thermal stability of lignin, the latter being found to be relatively poorer in lower molecular weight fractions.
3. Conclusions
Fractionation of an industrial wheat straw/Sarkanda grass lignin obtained by the soda pulp process was successfully carried out by means of a new process based on the combination of a Soxhlet solvent extraction and two sequential membrane-mediated ultrafiltrations.
This reported two-step process represents a valuable approach toward the optimized valorization of technical lignins, allowing the access to lignin fractions with well-defined physico-chemical properties (including mass distribution, Tg, aliphatic and phenolic hydroxyl groups concentration, syringyl/guaiacyl unit ratio). Moreover, the use of a solvent like MEK permitted to work with very high starting lignin concentrations (up to 105 g/L), allowed the direct coupling of the Soxhlet extraction outcome with the membrane-assisted ultrafiltration system, prevented fouling of the stabilized cellulose-based membranes and ultimately led to an easy product recovery by a simple solvent evaporation step. All these characteristics may enable the implementation of a scalable approach with potential to reduce environmental impact and process fractionation costs. The availability of technical lignin fractions with tailored and reproducible characteristics potentially allows enhanced lignin valorization strategies to be put in place for the development of biobased polymers and for the preparation of key platform chemicals, thereby paving the way for an effective exploitation of this remarkable resource.
4. Materials and Methods
4.1. Materials
All chemicals and analytical grade solvents such as THF, methanol (MeOH), ethylacetate (EtOAc), n-butylacetate (BuOAc), and tert-butyl methyl ether (MTBE) were purchased from Sigma-Aldrich. 2-Butanone (methyl ethyl ketone, MEK) was provided by BCD Chemie GmbH (Hamburg, Germany). Lignin (Protobind 1000, a mixed wheat straw/Sarkanda grass lignin from soda pulping of non-woody biomass) was provided by Green Value (Orbe, Switzerland). Softwood Kraft lignin (Indulin AT) was provided by Meadwestvaco (USA).
All analyses were carried out at least in duplicates unless otherwise stated.
4.2. Lignin Solubility in Organic Solvents
Lignin solubility in the organic solvent was determined treating 10 g of the starting lignin with 100 mL of the solvent stirring at 400 rpm. Each test was carried out overnight at room temperature. The suspension was then filtered and the solvent was evaporated at reduced pressure, and the final residue was dried until a constant weight was achieved prior to quantification.
4.3. Fractionation Process
The fractionation process comprises a Soxhlet extraction followed by a two membrane cascade ultrafiltration (shown in Figure 1 as part of an idealized lignin valorization scheme) and a final fraction recovery step, as described below (see also the Results and Discussion section).
4.4. Soxhlet Extraction
Soxhlet extractions were performed using a standard glass apparatus (Buchi extraction system B-811) which allows the realization of four extractions at the same time. Each extraction unit is composed of a 150 mL working volume bottom solvent flask, a 330 mL capacity thimble holder, and a water-cooled condenser. For each extraction process, 150 mL of MEK were placed in the solvent flask and about 15 g of lignin were inserted inside a 41 mm diameter and 123 mm height cellulose paper thimble (FiltraTECH). The solvent reflux was kept for 8 h, adjusting the heating power in order to have 4 extraction cycles/h.
4.5. Membrane Ultrafiltration
Lignin fractionation was performed in MEK directly on the Soxhlet extraction solution by means of an ultrafiltration apparatus (Sartoflow Advanced filtration module purchased from Sartorius Stedim) equipped with flowmeters and pressure sensors to control permeate flow, trans-membrane pressure, and filtration time (see the Supporting Information for details). The membranes (Hydrosart membranes, Sartorius Stedim) in stabilized cellulose had nominal molecular-weight cutoffs of 5 and 2 kDa and a filtration area of 0.1 m2 each. The membrane regeneration and storage were performed at 40 °C using 1 and 0.1 M NaOH solution, respectively.
4.6. Fraction Recovery Procedure
The recovered fractions from the sequential process of fractionation were evaporated under reduced pressure without further processing. The final solid residues were dried until a constant weight was achieved prior to analysis and quantification.
4.7. Gas Chromatography/Mass Spectrometry
The GC/MS apparatus used was an Agilent GC System 7890A, with an inert MSD with the Triple-Axis Detector 7975C. The separation was performed on a DB-5MS column (30 m × 250 μm × 0.25 μm, Phenomenex) with a helium flow rate of 1.18 mL/min, a temperature program of 50 °C (1 min) to 280 °C at 10 °C/min, 280 °C at 15 min (total run time 39 min, temperature of the injector 250 °C, injection volume 1.00 μL, injection mode split, split ratio 5:1). A solvent delay of 4 min was selected. The samples were prepared by derivatization and dissolved in methanol or acetone in a concentration around 0.5–1 mg/mL as previously described.25 Compound identification was performed by means of NIST 2008 mass spectral library search.
4.8. Pyrolysis GC/MS
Py-GC/MS experiments were conducted using a pyrolysis unit (GERSTEL PYRO with thermal desorption unit GERSTEL TDU) which used a pulsed pyrolysis at temperatures ranging from 350 to 1000 °C coupled with GC/MS Thermo Scientific Trace 1310 with a Thermo Scientific ISQ mass spectrometer using a TG-5SILMS column (30 m × 250 μm × 0.25 μm). The pyrolysis of the sample was performed in a quartz tube inside the heating chamber under the helium atmosphere. The obtained pyrolysis gas was directly transferred to the GC using a heated transfer line. No concentration methods such as thermal desorption or cryo focusing were used. About 0.5 mg of lignin particles were loaded into the quartz pyrolysis tube and the pyrolysis temperature was set to 450 or 600 °C. In the pulse mode, the sample was placed on a cold pyrolysis probe, which was rapidly (typically in the milliseconds range) heated to a predetermined pyrolysis temperature and maintained for 0.33 min at that temperature. The thermal desorption unit temperature was set to 280 °C like the transfer temperature. GC column temperature program started at 45 °C for 5 min, followed by a gradient of 4 °C to the final temperature of 280 °C and held for 15 min.
4.9. Gel Permeation Chromatography
A Waters 510 HPLC system was used equipped with a refractive index detector. THF was used as the eluent. The analyzed lignin sample (volume 200 μL, concentration 1 mg/mL in THF) was injected into a system of three columns connected in series (Ultrastyragel HR, Waters—dimensions 7.8 mm × 300 mm) and the analysis was performed at 30 °C at a flow rate of 0.5 mL/min. The GPC system was calibrated against polystyrene standards in the 102 to 104 g/mol molecular weight range. To allow complete solubility in the THF eluent, before the analysis, the parent lignin and the fractions were acetylated following a standard literature procedure.49 The estimation of the number-average and weight-average molecular weights of the obtained lignin fractions was performed excluding the signals related to the solvent (THF) and the solvent stabilizer (butylated hydroxytoluene), visible at long elution times (>29.5 min).
4.10. FC Assay
Total phenolic contents of the different fractions were determined by the classical FC method with some modifications in the sample preparation step.38,50 The samples were prepared by dissolving lignin in DMSO with a final concentration of 1 mg/mL. DMSO was chosen because it is completely miscible in water, it allows a complete lignin solubilization and does not interfere with the FC assay.
For each determination, 5 μL of the working solution (or the standard solution) were then mixed with 120 μL of deionized water, 125 μL of the FC reagent (Sigma 47641), and kept for 6 min at r.t. after 30 s of vortex stirring. Then, after addition of 1.25 mL of 5% sodium carbonate and mixing, the vial was incubated on a thermoshaker at 40 °C for 30 min. The reaction mixture absorbance was measured using a UV/vis spectrophotometer (Jasco V-560) equipped with a temperature-controlled cuvette holder and a thermostatic water bath (Haake K10). All spectrophotometric measurements were carried out at 760 nm, 25 °C using a 1 cm optical path cuvette. Gallic acid was chosen as the reference standard. The calibration curve has been constructed with nine different gallic acid solutions in DMSO with concentration in the range 0–800 μg/mL. Each FC assay determination was carried out in triplicate.
4.11. 31P NMR Analysis
31P NMR spectroscopic analysis was recorded on a Bruker Instrument AVANCE400 spectrometer. Acquisition and data treatment were performed with Bruker TopSpin 3.2 software. The spectra were collected at 29 °C with 4 s acquisition time, 5 s relaxation delay, and 256 scans. Prior to analysis, samples were dried for 24 h under vacuum and then derivatized according to a procedure described in the literature with a few modifications.39 All chemical shifts reported are related to the reaction product of the phosphorylating agent with water which gives a signal at 132.2 ppm.
The lignin sample (40 mg) was completely dissolved in 300 μL of N,N-dimethylformamide. To this solution, the following components were added: 200 μL of dry pyridine, 100 μL of solution of internal standard (10 mg of endo-N-hydroxy-5-norbornene-2,3-dicarboximide (Sigma 226378) dissolved in 0.5 mL of a mixture of pyridine and CDCl3 1.6:1 v/v), 50 μL of solution of the relaxation agent [5.7 mg of chromium(III)acetylacetonate (Sigma 574082) dissolved in 0.5 mL of a mixture of pyridine and CDCl3 1.6:1 v/v], 100 μL of 2-chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane (Sigma 447536), and at the end, 130 μL of CDCl3.
4.12. Differential Scanning Calorimetry
DSC was performed on solid-state samples (∼10–15 mg) by means of a Mettler-Toledo DSC/823e instrument at a scan rate of 20 °C/min under nitrogen flux.
4.13. Thermogravimetric Analysis
TGA was carried out on solid-state samples (∼10–15 mg) with a Q500 TGA system (TA Instruments) from room temperature to 800 °C at a scan rate of 20 °C/min under nitrogen flow.
4.14. Fourier Transform Infrared Spectroscopy
FTIR spectra of all lignin fractions were recorded in the transmission mode on films spin-cast onto KBr disks. The analysis was performed by means of a Nicolet 760-FTIR spectrophotometer at room temperature in air in the 4000–700 cm–1 wavenumber range with 64 accumulated scans and a resolution of 2 cm–1.
Acknowledgments
This project was funded by the European Commission’s Seventh Framework Programme for research, technological development, and demonstration under grant agreement no. FP7-KBBE-2013-7–613802 (“ValorPlus—Valorisation of biorefinery byproducts, leading to closed-loop systems with improved economic and environmental performance”). The calculations of the S/G ratios were performed by Dr. Andreas Klingberg and Dr. Philip Wenig from Lablicaze GmbH, Hamburg, Germany, with the software OpenChrom and ChromIdent.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.8b02851.
Apparatus for lignin fractionation; operational parameters for the ultrafiltration process; extraction yields of Kraft and Protobind; GC/MS chromatograms; calibration curve of FC phenol titration; peak identification in Py-GC/MS (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Lewis N. G.; Yamamoto E. Lignin: occurrence, biogenesis and biodegradation. Annu. Rev. Plant Physiol. Plant Mol. Biol. 1990, 41, 455–496. 10.1146/annurev.pp.41.060190.002323. [DOI] [PubMed] [Google Scholar]
- Boerjan W.; Ralph J.; Baucher M. Lignin Biosynthesis. Annu. Rev. Plant Biol. 2003, 54, 519–546. 10.1146/annurev.arplant.54.031902.134938. [DOI] [PubMed] [Google Scholar]
- Berlin A.; Balakshin M.. Industrial Lignins. Bioenergy Research: Advances and Applications; Elsevier, 2014; Vol. 18, pp 315–336. [Google Scholar]
- Tuck C. O.; Perez E.; Horvath I. T.; Sheldon R. A.; Poliakoff M. Valorization of Biomass: Deriving More Value from Waste. Science 2012, 337, 695–699. 10.1126/science.1218930. [DOI] [PubMed] [Google Scholar]
- Schutyser W.; Renders T.; Van den Bosch S.; Koelewijn S.-F.; Beckham G. T.; Sels B. F. Chemicals from lignin: an interplay of lignocellulose fractionation, depolymerisation, and upgrading. Chem. Soc. Rev. 2018, 47, 852–908. 10.1039/c7cs00566k. [DOI] [PubMed] [Google Scholar]
- Aro T.; Fatehi P. Production and Application of Lignosulfonates and Sulfonated Lignin. Chemsuschem 2017, 10, 1861–1877. 10.1002/cssc.201700082. [DOI] [PubMed] [Google Scholar]
- Isikgor F. H.; Becer C. R. Lignocellulosic biomass: a sustainable platform for the production of bio-based chemicals and polymers. Polym. Chem. 2015, 6, 4497–4559. 10.1039/C5PY00263J. [DOI] [Google Scholar]
- Ragauskas A. J.; Beckham G. T.; Biddy M. J.; Chandra R.; Chen F.; Davis M. F.; Davison B. H.; Dixon R. A.; Gilna P.; Keller M.; Langan P.; Naskar A. K.; Saddler J. N.; Tschaplinski T. J.; Tuskan G. A.; Wyman C. E. Lignin Valorization: Improving Lignin Processing in the Biorefinery. Science 2014, 344, 1246843. 10.1126/science.1246843. [DOI] [PubMed] [Google Scholar]
- Stewart D. Lignin as a base material for materials applications: chemistry, application and economics. Ind. Crops Prod. 2008, 27, 202–207. 10.1016/j.indcrop.2007.07.008. [DOI] [Google Scholar]
- Upton B. M.; Kasko A. M. Strategies for the Conversion of Lignin to High-Value Polymeric Materials: Review and Perspective. Chem. Rev. 2016, 116, 2275–2306. 10.1021/acs.chemrev.5b00345. [DOI] [PubMed] [Google Scholar]
- Griffini G.; Passoni V.; Suriano R.; Levi M.; Turri S. Polyurethane Coatings Based on Chemically Unmodified Fractionated Lignin. ACS Sustainable Chem. Eng. 2015, 3, 1145–1154. 10.1021/acssuschemeng.5b00073. [DOI] [Google Scholar]
- Bonini C.; D’Auria M.; Emanuele L.; Ferri R.; Pucciariello R.; Sabia A. R. Polyurethanes and polyesters from lignin. J. Appl. Polym. Sci. 2005, 98, 1451–1456. 10.1002/app.22277. [DOI] [Google Scholar]
- El Mansouri N. E.; Yuan Q.; Huang F. Synthesis and characterization of kraft lignin-based epoxy resins. BioResources 2011, 6, 2492–2503. [Google Scholar]
- Ismail T. N. M. T.; Hassan H. A.; Hirose S.; Taguchi Y.; Hatakeyama T.; Hatakeyama H. Synthesis and thermal properties of ester-type crosslinked epoxy resins derived from lignosulfonate and glycerol. Polym. Int. 2009, 59, 181–186. 10.1002/pi.2705. [DOI] [Google Scholar]
- Scarica C.; Suriano R.; Levi M.; Turri S.; Griffini G. Lignin Functionalized with Succinic Anhydride as Building Block for Biobased Thermosetting Polyester Coatings. ACS Sustainable Chem. Eng. 2018, 6, 3392–3401. 10.1021/acssuschemeng.7b03583. [DOI] [Google Scholar]
- Garcia Gonzalez M. N.; Levi M.; Turri S.; Griffini G. Lignin nanoparticles by ultrasonication and their incorporation in waterborne polymer nanocomposites. J. Appl. Polym. Sci. 2017, 134, 45318. 10.1002/app.45318. [DOI] [Google Scholar]
- Zakzeski J.; Bruijnincx P. C. A.; Jongerius A. L.; Weckhuysen B. M. The catalytic valorization of lignin for the production of renewable chemicals. Chem. Rev. 2010, 110, 3552–3599. 10.1021/cr900354u. [DOI] [PubMed] [Google Scholar]
- Pandey M. P.; Kim C. S. Lignin Depolymerization and Conversion: A Review of Thermochemical Methods. Chem. Eng. Technol. 2011, 34, 29–41. 10.1002/ceat.201000270. [DOI] [Google Scholar]
- Güvenatam B.; Heeres E. H. J.; Pidko E. A.; Hensen E. J. M. Lewis-acid catalyzed depolymerization of Protobind lignin in supercritical water and ethanol. Catal. Today 2016, 259, 460–466. 10.1016/j.cattod.2015.03.041. [DOI] [Google Scholar]
- Lange H.; Decina S.; Crestini C. Oxidative upgrade of lignin - Recent routes reviewed. Eur. Polym. J. 2013, 49, 1151–1173. 10.1016/j.eurpolymj.2013.03.002. [DOI] [Google Scholar]
- Tonin F.; Vignali E.; Pollegioni L.; D’Arrigo P.; Rosini E. A novel, simple screening method for investigating the properties of lignin oxidative activity. Enzyme Microb. Technol. 2017, 96, 143–150. 10.1016/j.enzmictec.2016.10.013. [DOI] [PubMed] [Google Scholar]
- Rosini E.; Allegretti C.; Melis R.; Cerioli L.; Conti G.; Pollegioni L.; D’Arrigo P. Cascade enzymatic cleavage of the β-O-4 linkage in a lignin model compound. Catal. Sci. Technol. 2016, 6, 2195–2205. 10.1039/c5cy01591j. [DOI] [Google Scholar]
- Rosini E.; D’Arrigo P.; Pollegioni L. Demethylation of vanillic acid by recombinant LigM in a one-pot cofactor regeneration system. Catal. Sci. Technol. 2016, 6, 7729–7737. 10.1039/c6cy01402j. [DOI] [Google Scholar]
- Zhao C.; Xie S.; Pu Y.; Zhang R.; Huang F.; Ragauskas A. J.; Yuan J. S. Synergistic enzymatic and microbial lignin conversion. Green Chem. 2016, 18, 1306–1312. 10.1039/C5GC01955A. [DOI] [Google Scholar]
- Allegretti C.; Fontanay S.; Krauke Y.; Luebbert M.; Strini A.; Troquet J.; Turri S.; Griffini G.; D’Arrigo P. Fractionation of Soda Pulp Lignin in Aqueous Solvent through Membrane-Assisted Ultrafiltration. ACS Sustainable Chem. Eng. 2018, 6, 9056–9064. 10.1021/acssuschemeng.8b01410. [DOI] [Google Scholar]
- Alriols M. G.; García A.; Llano-Ponte R.; Labidi J. Combined organosolv and ultrafiltration lignocellulosic biorefinery process. Chem. Eng. J. 2010, 157, 113–120. 10.1016/j.cej.2009.10.058. [DOI] [Google Scholar]
- Scarlat N.; Dallemand J.-F.; Monforti-Ferrario F.; Nita V. The role of biomass and bioenergy in a future bioeconomy: Policies and facts. Environ. Dev. 2015, 15, 3–34. 10.1016/j.envdev.2015.03.006. [DOI] [Google Scholar]
- Constant S.; Wienk H. L. J.; Frissen A. E.; Peinder P. d.; Boelens R.; van Es D. S.; Grisel R. J. H.; Weckhuysen B. M.; Huijgen W. J. J.; Gosselink R. J. A.; Bruijnincx P. C. A. New insights into the structure and composition of technical lignins: a comparative characterisation study. Green Chem. 2016, 18, 2651–2665. 10.1039/c5gc03043a. [DOI] [Google Scholar]
- Baumberger S.; Abaecherli A.; Fasching M.; Gellerstedt G.; Gosselink R.; Hortling B.; Li J.; Saake B.; de Jong E. Molar mass determination of lignins by size-exclusion chromatography: towards standardisation of the method. Holzforschung 2007, 61, 459–468. 10.1515/hf.2007.074. [DOI] [Google Scholar]
- Lange H.; Rulli F.; Crestini C. Gel Permeation Chromatography in Determining Molecular Weights of Lignins: Critical Aspects Revisited for Improved Utility in the Development of Novel Materials. ACS Sustainable Chem. Eng. 2016, 4, 5167–5180. 10.1021/acssuschemeng.6b00929. [DOI] [Google Scholar]
- Ringena O.; Lebioda S.; Lehnen R.; Saake B. Size-exclusion chromatography of technical lignins in dimethyl sulfoxide/water and dimethylacetamide. J. Chromatogr. A 2006, 1102, 154–163. 10.1016/j.chroma.2005.10.037. [DOI] [PubMed] [Google Scholar]
- Costa C. A. E.; Pinto P. C. R.; Rodrigues A. E. Lignin fractionation from E. Globulus kraft liquor by ultrafiltration in a three stage membrane sequence. Sep. Purif. Technol. 2018, 192, 140–151. 10.1016/j.seppur.2017.09.066. [DOI] [Google Scholar]
- Alriols M. G.; García A.; Llano-ponte R.; Labidi J. Combined organosolv and ultrafiltration lignocellulosic biorefinery process. Chem. Eng. J. 2010, 157, 113–120. 10.1016/j.cej.2009.10.058. [DOI] [Google Scholar]
- Shao L.; Zhang X.; Chen F.; Xu F. Fast pyrolysis of Kraft lignins fractionated by ultrafiltration. J. Anal. Appl. Pyrolysis 2017, 128, 27–34. 10.1016/j.jaap.2017.11.003. [DOI] [Google Scholar]
- Toledano A.; García A.; Mondragon I.; Labidi J. Lignin separation and fractionation by ultrafiltration. Sep. Purif. Technol. 2010, 71, 38–43. 10.1016/j.seppur.2009.10.024. [DOI] [Google Scholar]
- Toledano A.; Serrano L.; Garcia A.; Mondragon I.; Labidi J. Comparative study of lignin fractionation by ultrafiltration and selective precipitation. Chem. Eng. J. 2010, 157, 93–99. 10.1016/j.cej.2009.10.056. [DOI] [Google Scholar]
- Box J. D. Investigation of the Folin-Ciocalteau phenol reagent for the determination of polyphenolic substances in natural waters. Water Res. 1983, 17, 511–525. 10.1016/0043-1354(83)90111-2. [DOI] [Google Scholar]
- Sricharoen P.; Techawongstein S.; Chanthai S. A high correlation indicating for an evaluation of antioxidant activity and total phenolics content of various chilli varieties. J. Food Sci. Technol. 2015, 52, 8077–8085. 10.1007/s13197-015-1931-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granata A.; Argyropoulos D. S. 2-Chloro-4,4,5,5-tetramethyl-1,3,2-dioxaphospholane, a Reagent for the Accurate determination of the Uncondensed and Condensed Phenolic Moieties in Lignins. J. Agric. Food Chem. 1995, 43, 1538–1544. 10.1021/jf00054a023. [DOI] [Google Scholar]
- Constant S.; Wienk H. L. J.; Frissen A. E.; et al. New insights into the structure and composition of technical lignins: a comparative characterisation study. Green Chem. 2016, 18, 2651–2665. 10.1039/C5GC03043A. [DOI] [Google Scholar]
- Nunes C. A.; Lima C. F.; Barbosa L. C. A.; Colodette J. L.; Gouveia A. F. G.; Silvério F. O. Determination of Eucalyptus spp lignin S/G ratio: A comparison between methods. Bioresour. Technol. 2010, 101, 4056–4061. 10.1016/j.biortech.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Ohra-aho T.; Gomes F. J. B.; Colodette J. L.; Tamminen T. S/G ratio and lignin structure among Eucalyptus hybrids determined by Py-GC/MS and nitrobenzene oxidation. J. Anal. Appl. Pyrolysis 2013, 101, 166–171. 10.1016/j.jaap.2013.01.015. [DOI] [Google Scholar]
- Zhan S.; Chenguang W.; Kang B.; Xinghua Z.; Chiling Y.; Renje D.; Longlong M.; Changle P. Py-GC/MS study of lignin pyrolysis and effect of catalysts on product distribution. Int. J. Agric. Biol. Eng. 2017, 10, 214–225. 10.25165/j.ijabe.20171005.2852. [DOI] [Google Scholar]
- Jegers H. E.; Klein M. T. Primary and Secondary Lignin Pyrolysis Reaction Pathways. Ind. Eng. Chem. Process Des. Dev. 1985, 24, 173–183. 10.1021/i200028a030. [DOI] [Google Scholar]
- Domínguez-Robles J.; Tamminen T.; Liitiä T.; Peresin M. S.; Rodríguez A.; Jääskeläinen A.-S. Aqueous acetone fractionation of kraft, organosolv and soda lignins. Int. J. Biol. Macromol. 2018, 106, 979–987. 10.1016/j.ijbiomac.2017.08.102. [DOI] [PubMed] [Google Scholar]
- Agarwal U. P.; Atalla R. H. In Vibrational Spectroscopy; Heitner C., Dimmel D. R., Schmidt J. A., Eds.; Lignin and Lignans: Advances in Chemistry; CrC Press: Boca Raton, 2010. [Google Scholar]
- Passoni V.; Scarica C.; Levi M.; Turri S.; Griffini G. Fractionation of Industrial Softwood Kraft Lignin: Solvent Selection as a Tool for Tailored Material Properties. ACS Sustainable Chem. Eng. 2016, 4, 2232–2242. 10.1021/acssuschemeng.5b01722. [DOI] [Google Scholar]
- Domínguez J. C.; Oliet M.; Alonso M. V.; Gilarranz M. A.; Rodríguez F. Thermal stability and pyrolysis kinetics of organosolv lignins obtained from Eucalyptus globulus. Ind. Crops Prod. 2008, 27, 150–156. 10.1016/j.indcrop.2007.07.006. [DOI] [Google Scholar]
- Lange H.; Rulli F.; Crestini C. Gel Permeation Chromatography in Determining Molecular Weights of Lignins: Critical Aspects Revisited for Improved Utility in the Development of Novel Materials. ACS Sustainable Chem. Eng. 2016, 4, 5167–5180. 10.1021/acssuschemeng.6b00929. [DOI] [Google Scholar]
- Singleton V. L.; Orthofer R.; Lamuela-Raventós R. M.. Analysis of Total Phenols and Other Oxidation Substrates and Antioxidants by Means of Folin–Ciocalteu Reagents; Methods in Enzymology; Academic Press, 1999; Vol. 299, pp 152–178. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.