

Abstract
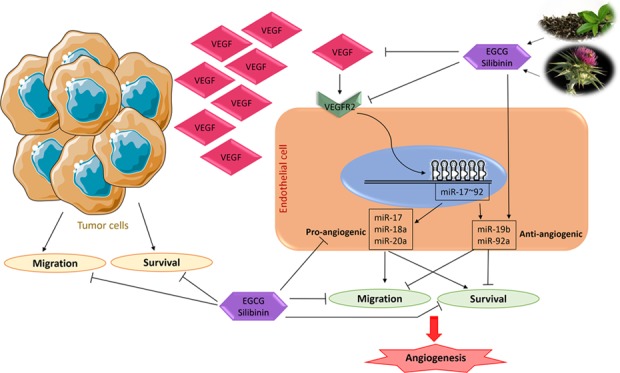
Despite promising benefits, anti-angiogenic strategies have revealed several drawbacks, which necessitate development of novel approaches in cancer therapy strategies including non-small-cell lung cancer, as one of the leading causes of cancer death, all over the world. Combination of flavonoids could be a safe and effective option to synergize their impact on mechanisms controlling tumor angiogenesis. In this study, we have investigated the plausible synergism of epigallocatechin-3-gallate (EGCG) and silibinin on endothelial cells, for the first time. Cell viability and migration were evaluated by survival and wound healing assays, respectively. Then, we assessed the expression of VEGF, VEGFR2, and miR-17–92 cluster using real-time polymerase chain reaction in endothelial–tumor cell and endothelial–fibroblast coculture models. EGCG ± silibinin suppressed endothelial and lung tumor cell migration in lower than 50% toxic doses. VEGF, VEGFR2, and pro-angiogenic members of the miR-17–92 cluster were downregulated upon treatments. Specifically, the combination treatment upregulated an anti-angiogenic member of the cluster, miR-19b. Our data provides evidence to utilize the EGCG and silibinin combination as a novel approach to target tumor angiogenesis in the future.
Introduction
Since its first suggestion in 1971, anti-angiogenic therapy of cancer has been known as an essential approach in treating many types of the disease.1 In non-small-cell lung cancer (NSCLC), like other solid tumors, angiogenesis is accompanied with highly invasive and metastatic properties of disease. Treating approaches including anti-angiogenic agents are among the promising therapies in clinical trials. However, there is a critical need for more studies in this area to overcome observed toxicities and drawbacks.2 Angiogenesis, the process of creating new blood vessels from preexisting ones, is a key physiological and developmental event to maintain homeostasis. Disrupted regulation of angiogenesis is directly linked to different pathologies and life-threatening diseases, particularly cancer.3 Regardless of the angiogenesis-inducing source, endothelial cell functions such as survival and migration are crucial in angiogenesis. Endothelial cell survival and migration directly depend on vascular endothelial growth factor (VEGF)-mediated pathways, the key target in most available anti-angiogenic therapeutic options.4 On targeting this growth factor, its receptors or downstream mediators, by current approaches, not only inhibit tumor angiogenesis but also cause a systemic endothelial cell dysfunction and subsequent toxicity and cardiovascular diseases.5 In addition, resistance to VEGF-targeted therapies has been reported in clinical settings.6 Therefore, development of novel and safe strategies is an unmet health priority to minimize the side effects of anti-angiogenic therapeutics.
Tumor microenvironment is a mixture of different factors, secreted from cancer and stromal cells in response to detrimentally imposed conditions.7 The angiogenic response is a consequence of the interplay among several angiogenesis inducers and inhibitors, secreted by the cells in a tumor microenvironment. However, targeting a single molecular pathway in a specific cell type would not suffice the treatment goals.4b,8 Thus, finding new treatment options targeting multiple target(s) that effectively inhibit the pathological angiogenesis with minimal disruption of physiological angiogenesis is a crucial need.7b,9
MicroRNA (miRNA)s are a class small non-coding RNAs that negatively regulate their downstream target genes either by degrading the mRNA or inhibiting its translation to the protein.10 MiRNAs play diverse roles in endothelial cell integrity and functions. Importantly, they control angiogenesis by regulation of target genes involved in endothelial cell migration and survival.11 Several lines of evidence indicate contribution of the highly expressed polycistronic miR-17–92 cluster to angiogenic properties and tumor development.12 Furthermore, it has been shown that the miR-17–92 cluster regulates the endothelial cell function and angiogenic switch upon VEGF induction.13 This suggests the miR-17–92 cluster to be a potential target in treating malignancies through affecting both tumor and endothelial cells.
A compelling body of evidence support the notion that flavonoids could be considered as promising treatment options to combat cancer. Particularly, in the context of angiogenesis, pleiotropic activity of these phytochemicals via targeting multiple molecular pathways in either tumor or endothelial cells interferes with tumor development and angiogenesis. Combinatorial application of different flavonoids is plausible to strengthen their anti-angiogenic capacity.14 Epigallocatechin-3-gallate (EGCG)15 and silibinin16 are the major flavonoid-type active constituents of two of the most consumed plant products, green tea and milk thistle (Silybum marianum), modulating cell proliferation and apoptosis induction, the anti-angiogenic and anticancer effects of which have been reported in a variety of tumors (Figure 1A).14,17
Figure 1.

Structural properties and the effect of epigallocatechin-gallate (EGCG, C22H18O11) and silibinin (C25H22O10) on cell viability. (A) Structural properties of EGCG and silibinin. Relative cell viability of the human umbilical vein endothelial cell (HUVEC) (B), A549 (C), and human dermal fibroblasts (HDFs) (D) was evaluated in response to EGCG (25, 50, and 75 μgr/mL), silibinin (25, 50, and 75 μM), and combinations (25 μg/mL EGCG + 50 μM silibinin, 50 μg/mL EGCG + 50 μM silibinin, 25 μg/mL EGCG + 75 μM silibinin, 50 μg/mL EGCG + 75 μM silibinin) compared with control, in 24 h. Values represent mean ± standard error of the mean (SEM) of at least three replicates. (Dissimilar letters indicate significant difference with max P < 0.05.)
In this study, we have investigated the impact of combining these flavonoids on cell viability and migration of human umbilical vein endothelial cell (HUVEC) and A549, an epithelial carcinoma cell line, which is a common model for non-small-cell lung cancer (NSCLC) studies. Moreover, we have evaluated the differential effect of single versus combined treatments of EGCG and silibinin on gene expression changes of VEGF and VEGFR2 and their downstream miR-17–92 cluster to find out the potential mechanism underlying the pleiotropic activity of these secondary metabolites. We have focused on the potential additive anti-angiogenic effect of EGCG and silibinin considering the ease of access and extensive consumption worldwide.18 More importantly, EGCG15b and silibinin19 have been previously reported to play anti-angiogenic roles individually. However, the synergistic effect of EGCG and silibinin co-treatment on endothelial cell mechanisms underlying angiogenesis has remained to be addressed.
Results and Discussion
EGCG and Silibinin Treatment Regulates Viability in Endothelial and Lung Tumor Cells and Fibroblasts
Previous studies have reported the inhibitory effect of EGCG and silibinin on cell viability of multiple cells.16b,20 However, the potential synergistic effect of EGCG and silibinin treatment on cell viability has remained to be addressed. Our findings indicate that EGCG (25–75 μg/mL) is able to significantly inhibit the viability of HUVEC in 24 h not exceeding 50% of the control group; >60% viability was observed in response to 50 μg/mL (Figure 1B). These results are consistent with previous studies indicating a dose-dependent decrease in cell viability in response to EGCG. In the context of EGCG, regulation of Wnt and Id signaling pathways has been suggested as the anti-proliferative mechanism of action in HUVEC.20b Wnt signaling is involved in angiogenesis through regulation of cell proliferation, survival, migration, differentiation, and apoptosis.21 Regulation of key angiogenesis genes such as VEGF has been shown as a target of Wnt signaling,22 which could be considered in our future investigations. Other studies have reported no significant change in HUVEC viability 24 h after treatment with EGCG (50 μM, ∼23 μg/mL), which are analogous to our data at 25 μg/mL.23
We observed a decreasing but not significant trend in cell viability of HUVEC in response to silibinin treatments (25–75 μM). As previously shown, this reduction could be relevant to a pleiotropic activity of silibinin on endothelial cells. Increase in Cip1/p21, Kip1/p27, and p53 and subsequent cell cycle arrest and apoptosis induction through upregulating BAX and downregulating Mcl1, on one hand, and suppressing Akt and necrosis factor-κB (NF-κB) signaling, on the other hand, are the plausible pathways that are implicated in silibinin effect on endothelial cells.16a The converging result of P53 induction24 and reduction of Akt25 and NF-κB26 is downregulation of VEGF, which can be proposed as the downstream mechanism of silibinin action on endothelial cells. Vakili Zahir et al. have reported a higher tolerance of HUVEC to silibinin treatment compared with the HepG2 (human hepatocellular liver carcinoma) cell line, though treatment with a high level of silibinin leads to a necrotic cell death in HUVEC.27 This indicates that different tumor cell lines, liver versus lung, may differently respond to silibinin.
Interestingly, our results revealed that the combination of EGCG and silibinin at the same concentrations led to no significant reduction of cell viability of HUVEC in comparison with single treatments at equal time point (Figure 1B), and cell viability of HUVEC following the EGCG (50 μg/mL) and silibinin (50 μM) combination treatment was nearby 70%. The importance of this finding is that co-treatment of these two flavonoids enhanced cytotoxicity in lung tumor cells compared with single treatments (Figure 1C).
As shown in Figure 1C, viability of the malignant lung tumor cell line, A549, was not significantly influenced upon 24 h treatment with EGCG (25 and 50 μg/mL) or silibinin (25, 50, and 75 μM). In contrast, the combination of EGCG (50 μg/mL) and silibinin (50 and 75 μM) significantly reduced A549 cell viability, not exceeding 60% of the control group.
A growing number of studies have shown the apoptosis induction and inhibitory activities of EGCG on the growth and development of cancer cells including head and neck,28 breast,29 colorectal,30 prostate,31 hepatocellular carcinoma,32 Kaposi’s sarcoma,33 and lung cancer cells.20a Importantly, it has been shown that A549 cells are extremely resistant to EGCG treatment in vitro.34 However, high concentrations of EGCG are capable of inducing apoptosis in these cells.35 Similarly, there are plenty of studies indicating the suppressive effect of silibinin on hepatocellular carcinoma,36 prostate,37 breast,38 neuroblastoma,39 colorectal,40 and lung16b cancer cells. Silibinin treatment interferes with cell growth prominently through G1 arrest16b and apoptotic induction41 in NSCLC. Our results showed no significant change in cell viability of A549, as a NSCLC model, upon EGCG or silibinin single treatment at lower doses, which was reverted by increasing the concentration (Figure S1). The significant increase in toxicity against A549 cells through co-treatment with both components is suggestive of a direct tumor-killing activity, whose precise underlying mechanism needs to be unfolded.
In parallel, we have evaluated the fibroblast response to relevant concentrations of EGCG or silibinin. Proliferation of fibroblasts is important in wound healing as an example of physiological angiogenesis. They contribute to new vessel formation and integrity by secreting extracellular matrix components.42 We showed that 24 h treatment with EGCG (25 and 50 μg/mL) or silibinin (25, 50, and 75 μM) did not significantly affect the viability of normal fibroblast in comparison with the control group (Figure 1D). However, treatment with mixed concentrations of EGCG (50 μg/mL) and silibinin (50 and 75 μM) revealed a significant decrease in cell viability not exceeding 50%; HDFs revealed higher than 70% viability following treatment with EGCG (50 μg/mL) and silibinin (50 μM). Our results suggest that normal fibroblasts are resistant to EGCG or silibinin; however, combination treatment moderates their viability not more than 50% of untreated cells. It is consistent with the previously reported noncytotoxic effect of EGCG or silibinin on normal fibroblasts.43
As determined by our half-maximal inhibitory concentration (IC50) measurements, HUVEC was the most sensitive cell to EGCG or silibinin treatment (Table 1). In fact, IC50 values of both components were significantly lower in HUVEC than those in A549 and normal fibroblasts in 24 h. Collectively, EGCG and silibinin induced a dose-dependent decrease in cell viability, which was further confirmed at higher concentrations (Figure S1, Supporting Information).
Table 1. IC50 Values of EGCG or Silibinin in HUVEC, A549, and HDF Cells.
| HUVEC | A549 | HDF | |
|---|---|---|---|
| EGCG (μg/mL) | 68.07 ± 1.87 | 444.4 ± 1.71 | 3190 ± 2.50 |
| silibinin (μM) | 91.22 ± 1.64 | 381.8 ± 1.96 | 260.3 ± 1.98 |
Endothelial and Lung Tumor Cell Migration Is Markedly Reduced upon EGCG and Silibinin Treatment
Cell migration is a key process not only in angiogenesis but also in tumor metastasis. It is a process by which endothelial cells undergo massive angiogenesis under not only physiological but also pathological conditions, e.g., tumor growth.44 We conducted the wound healing assay to investigate the potential inhibitory effect of the EGCG and silibinin combination on endothelial and tumor cell migration in concentrations at which beyond 50% cell viability was observed because high toxicity would contradict with the foundation of cell migration estimation. Our results approved the previous reports on the inhibitory effect of EGCG23a or silibinin16a on HUVEC migration. Enumerating migrated cells to the wound area using imageJ revealed that migration of HUVEC is significantly suppressed in response to noncytotoxic doses of EGCG or silibinin in a dose-dependent manner. Importantly, we found that the antimigratory effect of EGCG or silibinin significantly elevated upon combination treatment of these flavonoids in HUVEC, demonstrating a remarkable synergistic effect of EGCG and silibinin on HUVEC migration (Figure 2).
Figure 2.

EGCG and silibinin inhibit HUVEC migration. (A) Cell migration effects of EGCG (25 and 50 μg/mL), silibinin (50 and 75 μM), and their combination (25 μg/mL EGCG + 50 μM silibinin, 50 μg/mL EGCG + 50 μM silibinin, 25 μg/mL EGCG + 75 μM silibinin, 50 μg/mL EGCG + 75 μM silibinin) in 24 h on HUVEC cells. (B) Representative indication of cell migration response of HUVEC to EGCG, silibinin, and their combination. Number of cells within four randomly chosen wound regions were measured using ImageJ and were normalized to the control group (scale bar: 100 μm). Dissimilar Letters indicate significant difference, with max P < 0.05, using statistical analysis by one-way analysis of variance (ANOVA), and values represent mean ± SEM.
Wang et al. have shown that the EGCG-induced antimigratory effect on HUVEC is mediated by suppression of tumor necrosis factor (TNF)-NF-κB axis.23b A downstream mechanism of suppressing NF-κB in cell migration is reduction in the VEGF expression as a regulatory target for EGCG and silibinin treatment in our study.
Migration is a critical step in cancer cell invasion and metastasis.45 In the context of lung tumor cells, EGCG46 or silibinin47 is capable of inhibiting cell migration. Similar to HUVEC, treatment with EGCG or silibinin alone inhibited migration of A549 tumor cells compared to the control untreated group. As a novel finding, we report for the first time that the combination of EGCG and silibinin is more potent to attenuate migration of A549 cells, as a typical NSCLC model, compared to either EGCG or silibinin alone. We observed that the combination of EGCG (25 and 50 μg/mL) and silibinin (50 and 75 μM) significantly declined migration of A549 tumor cells compared with the treatment with corresponding concentrations of each flavonoid (Figure 3). It should be noted that co-treatment with EGCG (50 μg/mL) and silibinin (50 μM) led to the highest inhibitory effect on A549 cell migration compared to that of other concentrations examined. Therefore, these doses were utilized in our mechanistic gene expression studies.
Figure 3.

EGCG and silibinin inhibit A549 cell migration. (A) Cell migration effects of silibinin (25, 50, and 75 mM), EGCG (25 and 50 mg/mL), and their combination (25 μg/mL EGCG + 50 μM silibinin, 50 μg/mL EGCG + 50 μM silibinin, 25 μg/mL EGCG + 75 μM silibinin, 50 μg/mL EGCG + 75 μM silibinin) in 24 h on HUVEC cells. (B) Representative indication of cell migration response of A549 to EGCG, silibinin, and their combination. The number of cells within four randomly chosen wound regions were measured using ImageJ and were normalized to the control (scale bar: 100 μm). Dissimilar Letters indicate significant difference, with max P < 0.05, using statistical analysis by one-way ANOVA, and values represent mean ± SEM.
Altogether, our wound healing data suggest that the combinatorial treatment of EGCG and silibinin exerts a synergistic effect on inhibition of migration both in tumor and endothelial cells. While 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) results indicated cell viability around 70% in HUVEC and higher than 80% in A549, following EGCG (50 μg/mL) and silibinin (50 μM) combination administration, the antimigratory capacity of equal treatment was greater than 80% of the control group in both cell lines.
Inhibitory Effect of EGCG and Silibinin Treatment on Endothelial Cell Migration and Viability Is Mediated via Downregulation of VEGF–VEGFR2 Axis
Next, we aimed to determine the mechanisms underlying regulation of cell viability and migration by EGCG and silibinin. It has been previously shown that the VEGF pathway is the key component of angiogenesis in various contexts.3 VEGF is the master mediator in both physiological and pathological angiogenesis.48 VEGF–VEGFR2 signaling critically regulates endothelial cell survival, proliferation, migration, and tube formation.49 The VEGF pathway has emerged as a specific target to minimize elevated angiogenesis in various types of cancer.50 It has been reported that green tea extract is capable of diminishing the expression of VEGF and its receptors VEGFR1 and VEGFR2.51 Deep et al. have shown the downregulation of VEGF and VEGFR2 in HUVEC following treatment with milk thistle-derived flavonolignans including silybin A, silybin B, isosilybin A, and isosilybin B.20c To our knowledge, there is no study on the specific effect of EGCG or silibinin on the VEGF axis in endothelial cells, so far. In this study, we have investigated the impact of EGCG, silibinin, and their combination on the expression of VEGF and VEGFR2 in HUVEC. The novelty of our study is evaluation of gene expression changes provoked by EGCG and silibinin in HUVEC in the presence of A549 tumor cells as robust inducers of angiogenesis. The angiogenic function of endothelial cells is influenced by tumor cells or fibroblasts. Therefore, we examined whether the effect of EGCG and silibinin on endothelial cells is mediated by altering the function of these cells or not. First, HUVECs were cocultured with A549 tumor cells and then treated with EGCG, silibinin, or their combination for 24 h (Figure 4A). Gene expression analysis in isolated HUVEC demonstrated that EGCG, silibinin, and their combination dramatically downregulated VEGF (Figure 4B) and VEGFR2 (Figure 4C) as master mediators of angiogenesis. We further showed that VEGF expression is significantly lowered in HUVEC cocultured with primary human fibroblast obtained from healthy individuals, in response to EGCG, silibinin, or their combination (Figure 4D,E). EGCG treatment was not able to significantly reduce VEGFR2 expression in HUVEC upon coculture with normal fibroblast. In contrary, silibinin reduced VEGFR2 expression, which was reversed after co-treatment with EGCG (Figure 4E).
Figure 4.

Gene expression changes of VEGF and VEGFR2 in HUVEC cells cocultured with A549 or HDF. Representative of HUVEC cocultured differentially with A549 (A) or HDF (D). RNA extraction was performed from HUVEC after 24 h of treatment with EGCG (50 μg/mL), silibinin (50 μM), or the combination (50 μg/mL EGCG + 50 μM silibinin). VEGF (B) or VEGFR2 (C) expression in HUVEC cocultured with A549. VEGF (E) or VEGFR2 (F) expression in HUVEC cocultured with HDF. Gene expression changes were normalized with glyceraldehyde-3-phosphate dehydrogenase (GAPDH). Values represent mean ± SEM of at least two replicates. (Dissimilar letters indicate significant difference with max P < 0.05.)
These data are suggestive of an EGCG ± silibinin-induced VEGF–VEGFR2 signaling in endothelial cells cocultured with tumor cells. However, the nonsignificant effect of the EGCG and silibinin combination on endothelial cells cocultured with normal fibroblasts indicates a selective beneficial impact of this treatment under healthy conditions, which should be mechanistically unfolded in the future.
Expression of miR-17–92 Cluster Is Tightly Regulated by EGCG and Silibinin Treatment
To decipher the consequence of diminished expression of VEGF, we evaluated endothelial changes of miR-17–92 expression in the presence of tumor cells. It has been reported that VEGF induces the expression of the polycistronic miR-17–92 cluster, which is critically involved in endothelial cell function, angiogenesis, and tumor metastasis via regulation of VEGF–VEGFR2 expression.13 The polycystronic miR-17–92 cluster encodes six miRNAs, namely, miR-17, miR-18a, miR-19a, miR-19b, miR-20a, and miR-92a, which are crucially implicated in tumor angiogenesis. Upregulation of the miR-17–92 cluster leads to increased angiogenic and invasive properties of the tumor cells.12 MiR-17–92 is highly expressed in endothelial cells, and it has been revealed that each member acts differentially on angiogenesis.52 For instance, miR-18a and miR-20a53 promote angiogenesis in contrary to miR-19b54 and miR-92a55 that induce anti-angiogenic features. To our knowledge, the regulatory role of EGCG and silibinin in regulation of miR-17–92 expression has not been investigated before. Thus, we performed quantitative real-time polymerase chain reaction (qRT-PCR) to evaluate gene expression changes of the miR-17–92 cluster in HUVEC following treatment with EGCG, silibinin, or their combination cocultured with A549 cells or normal fibroblast.
Gene expression analysis indicated that EGCG, silibinin, and their combination significantly down-regulate miR-18a (Figure 5B), miR-20a (Figure 5D), and miR-92a (Figure 5E) in HUVEC cocultured with A549 cells. Concomitantly, silibinin downregulated miR-17 expression in HUVEC either in single treatment or in combination with EGCG. However, EGCG alone did not alter miR-17 in the same conditions (Figure 5A). On the other hand, miR-19b, as an anti-angiogenic factor, was significantly upregulated specifically after EGCG + silibinin co-treatment in HUVEC. Nevertheless, miR-19b did not significantly change upon EGCG or silibinin treatment compared to the control group (Figure 5C). Interestingly, the combination of EGCG and silibinin did not significantly change the expression of miR-17–92 cluster when HUVEC was cocultured with normal fibroblasts obtained from healthy subjects (data not shown).
Figure 5.

Effects of silibinin, EGCG, and their combination on the miRNA expression level of the miR-17–92 family in HUVECs cocultured with A549. Gene expression changes of miR-17 (A), miR-18a (B), miR-19b (C), miR-20a (D), and miR-92a (E) of HUVECs cocultured with A549 in response to EGCG (50 μg/mL), silibinin (50 μM), or the combination (50 μg/mL EGCG + 50 μM silibinin) were normalized to U6. Values represent mean ± SEM of at least two replicates. (Dissimilar letters indicate significant difference with max P < 0.05.)
Our data suggest that the expression of pro-angiogenic miRNAs in endothelial cells elevated in the presence of tumor cells could be modulated upon EGCG, silibinin, or combination treatment. More interestingly, the combination of silibinin and EGCG treatment in endothelial cells is required to provoke a miRNA-mediated anti-angiogenic response, which was abolished by tumor cells.
Differential impact of these compounds on pro- versus anti-angiogenic miRNAs along with their suppressive effect on the VEGF–VEGFR2 axis provides mechanistic evidence to support the notion that the combination of EGCG and silibinin could effectively minimize angiogenesis in solid tumors. Nevertheless, extensive in vivo pharmacokinetic and mechanistic studies on animal models of cancer will determine the potential future application of this combinatorial therapy in clinical settings. In addition, in silico modeling investigations will further reveal the mode of interaction between EGCG and silibinin and specific molecular targets on endothelial cells.
Conclusions
Targeting multiple pathways of tumor angiogenesis along with minimal side effects on normal tissues is a demanding factor in the development of anticancer therapeutic strategies. A variety of flavonoids exhibit anti-angiogenic effects by targeting a wide range of molecular targets in both tumor and endothelial cells.9 Herein, we have addressed the inhibitory effect of the EGCG and silibinin combination on endothelial cell migration, survival, VEGF–VEGFR2, and miR-17–92 expression, as essential events in angiogenesis. Altogether, our results suggest that the EGCG and silibinin combination may not only beneficially modulate endothelial cell functions but also directly target tumor cells. It could further the anti-angiogenic antitumor properties by widening the target cells, which deserves detailed investigations in the future.
Anti-angiogenic therapy has extensive benefits, which is based on the critical reliance of solid tumors on neoangiogenesis.7b,56 However, there are a number of challenges in front that encourage researchers to develop more efficient and less toxic approaches.6 High distribution of flavonoids in fruits and vegetables and widespread consumption of plant products containing a variety of flavonoids as food or beverage all over the world, in parallel with growing evidence of their antioxidant and anticancer capacity, have made them promising alternatives for anti-angiogenic therapies.57
In addition, a variety of in vivo studies and human clinical trials on whether oral administration or intravenous injection of some flavonoids introduces an inconsistency with in vitro results which specify a severe concern about the nonsoluble flavonoids in water and the stability of these compounds in physiological conditions.57,58 This would affect not only the bioavailability of the flavonoid but also degradation by enzymatic reactions, starting from mixing with saliva, and is capable of forming pro-oxidant molecules with possible side effects.59 As an example, in the case of EGCG, the peak plasma level of orally administered flavonoid is in sub-micromolar range,60 which is very low compare to approved active concentrations in an in vitro situation. To enhance the exploitation of the compound, there are some solutions, among which increasing the intestinal absorption by a nano-drug delivery system using polymeric micelles has been reported to exhibit a variety of benefits.61 Sustained drug release, increased drug load, enhanced tumor accumulation, and high stability61,62 are among the welfares of using the polymeric micelle approach, and improved efficacy has been reported for a number of flavonoids including EGCG63 and quercetin.62 Low solubility of silibinin in water, however, can be overcome by increasing the administered doses because highly tolerable characteristics of its consumption have been approved in a variety of in vivo and clinical studies. Cumulative uptake amount of this flavonoid in parallel with introducing the novel silibinin formulation can intensify bioavailability and plasma absorption.19 However, it is beneficial to be cautious about using silibinin in combination with other drugs. In a clinical study of using oral administration of a commercial formula of silibinin, silybin-phytosome, in prostate cancer patients, an improvement in the bioavailability and plasma absorbance of silibinin was observed; however, variability in inter- or intrapatient responses emphasizes the impact of complexity of physiological conditions on its functionality and necessitates wide and detailed preclinical studies prior to using flavonoids in clinical conditions. Altogether, these are suggestive of evaluating promising drug delivery approaches for future studies on EGCG + silibinin in in vivo and further clinical trials.
Experimental Section
Cell Culture
Human umbilical vascular endothelial cell (obtained from the Medical Biology Research Center of Kermanshah University of Medical Sciences) and the A549 cell line (ATCC CCL-185) (obtained from the Pasture Institute of Iran) were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Bioidea) supplemented with 10% fetal bovine serum (FBS) (Gibco), penicillin (100 U/mL), and streptomycin (100 μg/mL) (Bioidea) in a humidified incubator at 37 °C and 5% CO2.
Human Dermal Fibroblast Isolation
Human dermal fibroblasts (HDFs) were isolated from the obtained foreskin tissue samples of children (age range between 10 days and 2 months) immediately after circumcision by modifying and setting up the method reported by Nejaddehbashi et al.64 Briefly, tissue samples obtained from a private clinic were transferred to the laboratory on ice-cold phosphate-buffered saline (PBS) containing penicillin (200 U/mL), streptomycin (200 μg/mL), and 0.3% amphotericin B. After sterilizing the samples with 70% ethanol and washing with PBS (200 U/mL penicillin, 200 μg/mL streptomycin, and 0.3% amphotericin B) three to five times, the hypodermis layer and related blood vessels were removed from the tissues. Samples were cut into 1 cm pieces and incubated in 0.25% trypsin–ethylenediaminetetraacetic acid (Bioidea) at 4 °C overnight. After incubation, the epidermis was set apart from the dermis, the dermis was chapped into very small pieces, and collagenase IV (1 mg/mL) was allowed to the tissue pieces for 1 h in an incubator at 37 °C, 5% CO2, and 95% humidity, shaking every 5 min. After neutralizing with an FBS-containing medium, the suspension was centrifuged at 1600 rpm for 5 min and the supernatant was cultivated in cell culture flasks with 20% FBS.
Treatment Preparation
EGCG (Sigma-Aldrich, CAS number: 989-51-5, purity (high-performance liquid chromatography (HPLC) area %): 94%) and silibinin (Sigma-Aldrich, CAS number: 22888-70-6, purity (HPLC area %): 99.1%) were obtained from Sigma. EGCG and silibinin high-concentration stock solutions were prepared by dissolving the compounds in appropriate solvents, water for EGCG and dimethyl sulfoxide (DMSO) for silibinin. Treatment solutions were prepared freshly just before the experiment by diluting the appropriate amount of stock solutions in 1% FBS-containing medium. The final concentration of DMSO did not exceed 0.1% in culture medium.
Cell Viability Assay
Cell viability in response to different treatments was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, cells were seeded in a 96-well plate (0.5 × 104 cells/well) and treated with different concentrations of EGCG, silibinin, or their combination in DMEM containing 1% FBS. After 24 h, cell viability was compared with the control group using the MTT assay (Autocell) at 570 nm with the reference wavelength of 630 nm.
Wound Healing Assay
The migration capacity of different cells was evaluated via the wound healing assay. Briefly, cells were grown in 24-well plates at high density in DMEM containing 10% FBS and abovementioned antibiotics. Next day, a scratch was created across the confluent cell layer using a tip. After gently removing the old medium and detached cells, fresh medium supplemented with 2% FBS and different concentrations of treatments including EGCG (25 and 50 μg/mL), silibinin (50 and 75 μM), and their combination (25 μg/mL EGCG + 50 μM silibinin, 50 μg/mL EGCG + 50 μM silibinin, 25 μg/mL EGCG + 75 μM silibinin, 50 μg/mL EGCG + 75 μM silibinin) was added to each well. After 24 h incubation at 37 °C, 95% humidity, and 5% CO2, the number of migrated cells was calculated in each treatment in four randomly chosen microscopic fields and compared with the nontreated control using ImageJ software.
Cell Coculture in Transwell Plates
Transwell plates (Corning, Cat# 3493) were used to evaluate the effect of different treatments on gene expression changes. After seeding and attachment of HUVECs in lower chambers, A549 cells were cultured in upper chambers of 12-well transwell plates and supplemented with DMEM, 10% FBS, and antibiotics. Coculture cells were treated with freshly prepared EGCG (50 μg/mL), silibinin (50 μM), or their combination (50 μg/mL EGCG + 50 μM silibinin) in 2% FBS for 24 h. The experiments were performed in triplicate and in two independent repeats.
RNA Extraction and RT-PCR
Total RNA of cocultured cells was extracted using RNX-plus (CinnaGen, Iran) according to the manufacturer’s instructions. The yield and purity of the extracted RNAs were assessed by 2% agarose gel and NanoDrop 1000 (Termo Scientific), and complementary DNAs were synthesized using the PrimeScript RT reagent kit (Takara Bio, Japan) according to the manufacturer’s protocol.
Quantitative Real-Time PCR
Gene expression changes of VEGF, VEGFR1, and VEGFR2 were evaluated in cocultured HUVECs after different treatments using an Applied Biosystem StepOne instrument (Applied Biosystem) and SYBR Premix Ex Taq II (Takara Bio, Japan) according to the manufacturer’s protocol. Quantitative real-time PCR conditions were as follows: 95 °C for 30 s, followed by 40 cycles at 95 °C for 5 s and 60 °C for 30 s. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was used as the endogenous control to normalize changes of target genes through the 2–ΔΔCt method. All samples were duplicated and repeated at least in two different biological repeats. Primer sequences are mentioned in Table 2.
Table 2. Primer Sequences Used in RT-PCR.
| gene ID | name | strand | primers 5′–3′ | product size | annealing Tm |
|---|---|---|---|---|---|
| 7422 | VEGF | forward | CTACCTCCACCATGCCAAGT | 174 | 56 |
| reverse | CACACAGGATGGCTTGAAGA | ||||
| 3791 | VEGFR2 | forward | GCGATTGAAAGAAGGAACTAGA | 166 | 54 |
| reverse | TAGTCTTTGCCATCCTGCTG | ||||
| 2597 | GAPDH | forward | ACTCTGGTAAAGTGGATATTGTTGC | 162 | 54 |
| reverse | GGAAGATGGTGATGGGATTTC |
Statistical Analysis
All data were obtained from at least two independent experiments and expressed as the mean ± SEM. One-way analysis of variance (ANOVA) with the Tukey post-hoc test was used to determine the effectiveness of different treatments compared with the control group. Two-way ANOVA with Tukey post-hoc analysis was utilized in assessment of the difference between treatment groups. The P-value less than 0.05 was considered as statistically significant.
Acknowledgments
This work has been supported by the Iran National Science Foundation (INSF) (research project 95844786). We gratefully acknowledge Dr. Hesamaldin Movassagh for kindly editing the manuscript.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00224.
Decreased cell viability of HUVEC, A549, and HDF in a dose-dependent manner in response to higher concentrations of EGCG and silibinin (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Kerbel R. S. Tumor Angiogenesis. N. Engl. J. Med. 2008, 358, 2039–2049. 10.1056/NEJMra0706596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Alshangiti A.; Chandhoke G.; Ellis P. M. Antiangiogenic therapies in non-small-cell lung cancer. Curr. Oncol. 2018, 25, S45–S58. 10.3747/co.25.3747. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Janning M.; Loges S. Anti-Angiogenics: Their Value in Lung Cancer Therapy. Oncol. Res. Treat. 2018, 41, 172–180. 10.1159/000488119. [DOI] [PubMed] [Google Scholar]
- Carmeliet P.; Jain R. K. Molecular mechanisms and clinical applications of angiogenesis. Nature 2011, 473, 298–307. 10.1038/nature10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ellis L. M.; Hicklin D. J. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat. Rev. Cancer 2008, 8, 579–591. 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]; b Shojaei F. Anti-angiogenesis therapy in cancer: Current challenges and future perspectives. Cancer Lett. 2012, 320, 130–137. 10.1016/j.canlet.2012.03.008. [DOI] [PubMed] [Google Scholar]; c Hicklin D. J.; Ellis L. M. Role of the Vascular Endothelial Growth Factor Pathway in Tumor Growth and Angiogenesis. J. Clin. Oncol. 2005, 23, 1011–1027. 10.1200/JCO.2005.06.081. [DOI] [PubMed] [Google Scholar]
- Morbidelli L.; Donnini S.; Ziche M. Targeting endothelial cell metabolism for cardio-protection from the toxicity of antitumor agents. Cardio-Oncology 2016, 2, 3 10.1186/s40959-016-0010-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gacche R. N.; Meshram R. J. Angiogenic factors as potential drug target: efficacy and limitations of anti-angiogenic therapy. Biochim. Biophys. Acta, Rev. Cancer 2014, 1846, 161–179. 10.1016/j.bbcan.2014.05.002. [DOI] [PubMed] [Google Scholar]
- a Hanahan D.; Coussens L. M. Accessories to the Crime: Functions of Cells Recruited to the Tumor Microenvironment. Cancer Cell 2012, 21, 309–322. 10.1016/j.ccr.2012.02.022. [DOI] [PubMed] [Google Scholar]; b Albini A.; Tosetti F.; Li V. W.; Noonan D. M.; Li W. W. Cancer prevention by targeting angiogenesis. Nat. Rev. Clin. Oncol. 2012, 9, 498–509. 10.1038/nrclinonc.2012.120. [DOI] [PubMed] [Google Scholar]
- a Carmeliet P. Angiogenesis in health and disease. Nat. Med. 2003, 9, 653–660. 10.1038/nm0603-653. [DOI] [PubMed] [Google Scholar]; b Bergers G.; Hanahan D. Modes of resistance to anti-angiogenic therapy. Nat. Rev. Cancer 2008, 8, 592–603. 10.1038/nrc2442. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Loges S.; Schmidt T.; Carmeliet P. Mechanisms of Resistance to Anti-Angiogenic Therapy and Development of Third-Generation Anti-Angiogenic Drug Candidates. Genes Cancer 2010, 1, 12–25. 10.1177/1947601909356574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Dabrosin C.; Yin X.; Fuster M. M.; Arreola A.; Rathmell W. K.; Generali D.; Nagaraju G. P.; El-Rayes B.; Ribatti D.; Chen Y. C.; Honoki K.; Fujii H.; Georgakilas A. G.; Nowsheen S.; Amedei A.; Niccolai E.; Amin A.; Ashraf S. S.; Helferich B.; Yang X.; Guha G.; Bhakta D.; Ciriolo M. R.; Aquilano K.; Chen S.; Halicka D.; Mohammed S. I.; Azmi A. S.; Bilsland A.; Keith W. N.; Jensen L. D. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. 10.1016/j.semcancer.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ambros V. The functions of animal microRNAs. Nature 2004, 431, 350–355. 10.1038/nature02871. [DOI] [PubMed] [Google Scholar]
- a Chamorro-Jorganes A.; Araldi E.; Suarez Y. MicroRNAs as pharmacological targets in endothelial cell function and dysfunction. Pharmacol. Res. 2013, 75, 15–27. 10.1016/j.phrs.2013.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Kuehbacher A.; Urbich C.; Dimmeler S. Targeting microRNA expression to regulate angiogenesis. Trends Pharmacol. Sci. 2008, 29, 12–15. 10.1016/j.tips.2007.10.014. [DOI] [PubMed] [Google Scholar]; c Kuninty P. R.; Schnittert J.; Storm G.; Prakash J. MicroRNA Targeting to Modulate Tumor Microenvironment. Front. Oncol. 2016, 6, 3 10.3389/fonc.2016.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Tréguer K.; Heinrich E. M.; Ohtani K.; Bonauer A.; Dimmeler S. Role of the MicroRNA-17–92 Cluster in the Endothelial Differentiation of Stem Cells. J. Vasc. Res. 2012, 49, 447–460. 10.1159/000339429. [DOI] [PubMed] [Google Scholar]; e Wang S.; Olson E. N. AngiomiRs—Key regulators of angiogenesis. Curr. Opin. Genet. Dev. 2009, 19, 205–211. 10.1016/j.gde.2009.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Würdinger T.; Tannous B. A.; Saydam O.; Skog J.; Grau S.; Soutschek J.; Weissleder R.; Breakefield X. O.; Krichevsky A. M. miR-296 Regulates Growth Factor Receptor Overexpression in Angiogenic Endothelial Cells. Cancer Cell 2008, 14, 382–393. 10.1016/j.ccr.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Mogilyansky E.; Rigoutsos I. The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20, 1603–1614. 10.1038/cdd.2013.125. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Zhang K.; Zhang L.; Zhang M.; Zhang Y.; Fan D.; Jiang J.; Ye L.; Fang X.; Chen X.; Fan S.; Chao M.; Liang C. Prognostic value of high-expression of miR-17-92 cluster in various tumors: evidence from a meta-analysis. Sci. Rep. 2017, 7, 8375 10.1038/s41598-017-08349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chamorro-Jorganes A.; Lee M. Y.; Araldi E.; Landskroner-Eiger S.; Fernández-Fuertes M.; Sahraei M.; Quiles del Rey M.; van Solingen C.; Yu J.; Fernández-Hernando C.; Sessa W. C.; Suárez Y. VEGF-Induced Expression of miR-17–92 Cluster in Endothelial Cells Is Mediated by ERK/ELK1 Activation and Regulates Angiogenesis. Circ. Res. 2016, 118, 38–47. 10.1161/CIRCRESAHA.115.307408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.; Dabrosin C.; Yin X.; Fuster M. M.; Arreola A.; Rathmell W. K.; Generali D.; Nagaraju G. P.; El-Rayes B.; Ribatti D.; Chen Y. C.; Honoki K.; Fujii H.; Georgakilas A. G.; Nowsheen S.; Amedei A.; Niccolai E.; Amin A.; Ashraf S. S.; Helferich B.; Yang X.; Guha G.; Bhakta D.; Ciriolo M. R.; Aquilano K.; Chen S.; Halicka D.; Mohammed S. I.; Azmi A. S.; Bilsland A.; Keith W. N.; Jensen L. D. Broad targeting of angiogenesis for cancer prevention and therapy. Semin. Cancer Biol. 2015, 35, S224–S243. 10.1016/j.semcancer.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Tang F. Y.; Nguyen N.; Meydani M. Green tea catechins inhibit VEGF-induced angiogenesis in vitro through suppression of VE-cadherin phosphorylation and inactivation of Akt molecule. Int. J. Cancer 2003, 106, 871–878. 10.1002/ijc.11325. [DOI] [PubMed] [Google Scholar]; b Singh B. N.; Shankar S.; Srivastava R. K. Green tea catechin, epigallocatechin-3-gallate (EGCG): mechanisms, perspectives and clinical applications. Biochem. Pharmacol. 2011, 82, 1807–1821. 10.1016/j.bcp.2011.07.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Singh R. P.; Dhanalakshmi S.; Agarwal C.; Agarwal R. Silibinin strongly inhibits growth and survival of human endothelial cells via cell cycle arrest and downregulation of survivin, Akt and NF-κB: implications for angioprevention and antiangiogenic therapy. Oncogene 2005, 24, 1188. 10.1038/sj.onc.1208276. [DOI] [PubMed] [Google Scholar]; b Mateen S.; Tyagi A.; Agarwal C.; Singh R. P.; Agarwal R. Silibinin inhibits human nonsmall cell lung cancer cell growth through cell-cycle arrest by modulating expression and function of key cell-cycle regulators. Mol. Carcinog. 2010, 49, 247–258. 10.1002/mc.20667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Rady I.; Mohamed H.; Rady M.; Siddiqui I. A.; Mukhtar H. Cancer preventive and therapeutic effects of EGCG, the major polyphenol in green tea. Egypt. J. Basic Appl. Sci. 2018, 5, 1–23. 10.1016/j.ejbas.2017.12.001. [DOI] [Google Scholar]; b Siegel A. B.; Stebbing J. Milk thistle: early seeds of potential. Lancet Oncol. 2013, 14, 929–930. 10.1016/S1470-2045(13)70414-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Yang C. S.; Wang X.; Lu G.; Picinich S. C. Cancer prevention by tea: animal studies, molecular mechanisms and human relevance. Nat. Rev. Cancer 2009, 9, 429. 10.1038/nrc2641. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Tiwari P.; Mishra K. P. Silibinin in cancer therapy: A promising prospect. Cancer Research Frontiers. Cancer Res. Front. 2015, 1, 303–318. 10.17980/2015.303. [DOI] [Google Scholar]
- Deep G.; Agarwal R. Antimetastatic efficacy of silibinin: molecular mechanisms and therapeutic potential against cancer. Cancer Metastasis Rev. 2010, 29, 447–463. 10.1007/s10555-010-9237-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Sakamoto Y.; Terashita N.; Muraguchi T.; Fukusato T.; Kubota S. Effects of Epigallocatechin-3-gallate (EGCG) on A549 Lung Cancer Tumor Growth and Angiogenesis. Biosci., Biotechnol., Biochem. 2013, 77, 1799–1803. 10.1271/bbb.120882. [DOI] [PubMed] [Google Scholar]; b Liu L.; Lai C. Q.; Nie L.; Ordovas J.; Band M.; Moser L.; Meydani M. The modulation of endothelial cell gene expression by green tea polyphenol-EGCG. Mol. Nutr. Food Res. 2008, 52, 1182–1192. 10.1002/mnfr.200700499. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Deep G.; Gangar S. C.; Rajamanickam S.; Raina K.; Gu M.; Agarwal C.; Oberlies N. H.; Agarwal R. Angiopreventive Efficacy of Pure Flavonolignans from Milk Thistle Extract against Prostate Cancer: Targeting VEGF-VEGFR Signaling. PLoS One 2012, 7, e34630 10.1371/journal.pone.0034630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olsen J. J.; Pohl S. Ö.-G.; Deshmukh A.; Visweswaran M.; Ward N. C.; Arfuso F.; Agostino M.; Dharmarajan A. The Role of Wnt Signalling in Angiogenesis. Clin. Biochem. Rev. 2017, 38, 131–142. [PMC free article] [PubMed] [Google Scholar]
- Zhang X.; Gaspard J. P.; Chung D. C. Regulation of Vascular Endothelial Growth Factor by the Wnt and K-ras Pathways in Colonic Neoplasia. Cancer Res. 2001, 61, 6050. [PubMed] [Google Scholar]
- a Cao L.; Liu H.; Lam D. S.-C.; Yam G. H.-F.; Pang C.-P. In Vitro Screening for Angiostatic Potential of Herbal Chemicals. Invest. Ophthalmol. Visual Sci. 2010, 51, 6658–6664. 10.1167/iovs.10-5524. [DOI] [PubMed] [Google Scholar]; b Wang Z. M.; Gao W.; Wang H.; Zhao D.; Nie Z. L.; Shi J. Q.; Zhao S.; Lu X.; Wang L. S.; Yang Z. J. Green Tea Polyphenol Epigallocatechin-3-Gallate Inhibits TNF-a-Induced Production of Monocyte Chemoattractant Protein-1 in Human Umbilical Vein Endothelial Cells. Cell. Physiol. Biochem. 2014, 33, 1349–1358. 10.1159/000358702. [DOI] [PubMed] [Google Scholar]
- Ghahremani M. F.; Goossens S.; Nittner D.; Bisteau X.; Bartunkova S.; Zwolinska A.; Hulpiau P.; Haigh K.; Haenebalcke L.; Drogat B.; Jochemsen A.; Roger P. P.; Marine J. C.; Haigh J. J. p53 promotes VEGF expression and angiogenesis in the absence of an intact p21-Rb pathway. Cell Death Differ. 2013, 20, 888. 10.1038/cdd.2013.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karar J.; Maity A. PI3K/AKT/mTOR Pathway in Angiogenesis. Front. Mol. Neurosci. 2011, 4, 51 10.3389/fnmol.2011.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T. X.; Xia Z.; Zhang N.; Gong W.; Huang S. Constitutive NF-kappaB activity regulates the expression of VEGF and IL-8 and tumor angiogenesis of human glioblastoma. Oncol. Rep. 2010, 23, 725–732. 10.3892/or_00000813. [DOI] [PubMed] [Google Scholar]
- Zahir N. V.; Nakhjavani M.; Hajian P.; Shirazi F. H.; Mirzaei H. Evaluation of Silibinin Effects on the Viability of HepG2 (Human hepatocellular liver carcinoma) and HUVEC (Human Umbilical Vein Endothelial) Cell Lines. Iran. J. Pharm. Res. 2018, 17, 261–267. 10.22037/ijpr.2018.2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M.; Suzui M.; Lim J. T. E.; Deguchi A.; Soh J.-W.; Weinstein I. B. Epigallocatechin-3-gallate decreases VEGF production in head and neck and breast carcinoma cells by inhibiting EGFR-related pathways of signal transduction. J. Exp. Ther. Oncol. 2002, 2, 350–359. 10.1046/j.1359-4117.2002.01062.x. [DOI] [PubMed] [Google Scholar]
- Gu J.-W.; Makey K. L.; Tucker K. B.; Chinchar E.; Mao X.; Pei I.; Thomas E. Y.; Miele L. EGCG, a major green tea catechin suppresses breast tumor angiogenesis and growth via inhibiting the activation of HIF-1α and NFκB, and VEGF expression. Vasc. Cell 2013, 5, 9. 10.1186/2045-824X-5-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin H.; Gong W.; Zhang C.; Wang S. Epigallocatechin gallate inhibits the proliferation of colorectal cancer cells by regulating Notch signaling. OncoTargets Ther. 2013, 6, 145–153. 10.2147/OTT.S40914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L. Y.; Cheung T. C.; Kong S. K.; Fung K. P.; Choy Y. M.; Chan Z. Y.; Kwok T. T. Induction of apoptosis by green tea catechins in human prostate cancer DU145 cells. Life Sci. 2001, 68, 1207–1214. 10.1016/S0024-3205(00)01020-1. [DOI] [PubMed] [Google Scholar]
- Shirakami Y.; Shimizu M.; Adachi S.; Sakai H.; Nakagawa T.; Yasuda Y.; Tsurumi H.; Hara Y.; Moriwaki H. (-)-Epigallocatechin gallate suppresses the growth of human hepatocellular carcinoma cells by inhibiting activation of the vascular endothelial growth factor-vascular endothelial growth factor receptor axis. Cancer Sci. 2009, 100, 1957–1962. 10.1111/j.1349-7006.2009.01241.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassina G.; Venè R.; Morini M.; Minghelli S.; Benelli R.; Noonan D. M.; Albini A. Mechanisms of Inhibition of Tumor Angiogenesis and Vascular Tumor Growth by Epigallocatechin-3-Gallate. Clin. Cancer Res. 2004, 10, 4865–4873. 10.1158/1078-0432.CCR-03-0672. [DOI] [PubMed] [Google Scholar]
- Kweon M. H.; Adhami V. M.; Lee J. S.; Mukhtar H. Constitutive overexpression of Nrf2-dependent heme oxygenase-1 in A549 cells contributes to resistance to apoptosis induced by epigallocatechin 3-gallate. J. Biol. Chem. 2006, 281, 33761–33772. 10.1074/jbc.M604748200. [DOI] [PubMed] [Google Scholar]
- Li M.; Li J. J.; Gu Q. H.; An J.; Cao L. M.; Yang H. P.; Hu C. P. EGCG induces lung cancer A549 cell apoptosis by regulating Ku70 acetylation. Oncol. Rep. 2016, 35, 2339–2347. 10.3892/or.2016.4587. [DOI] [PubMed] [Google Scholar]
- Momeny M.; Khorramizadeh M. R.; Ghaffari S. H.; Yousefi M.; Yekaninejad M. S.; Esmaeili R.; Jahanshiri Z.; Nooridaloii M. R. Effects of silibinin on cell growth and invasive properties of a human hepatocellular carcinoma cell line, HepG-2, through inhibition of extracellular signal-regulated kinase 1/2 phosphorylation. Eur. J. Pharmacol. 2008, 591, 13–20. 10.1016/j.ejphar.2008.06.011. [DOI] [PubMed] [Google Scholar]
- Deep G.; Kumar R.; Nambiar D. K.; Jain A. K.; Ramteke A. M.; Serkova N. J.; Agarwal C.; Agarwal R. Silibinin inhibits hypoxia-induced HIF-1α-mediated signaling, angiogenesis and lipogenesis in prostate cancer cells: In vitro evidence and in vivo functional imaging and metabolomics. Mol. Carcinog. 2017, 56, 833–848. 10.1002/mc.22537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molavi O.; Narimani F.; Asiaee F.; Sharifi S.; Tarhriz V.; Shayanfar A.; Hejazi M.; Lai R. Silibinin sensitizes chemo-resistant breast cancer cells to chemotherapy. Pharm. Biol. 2017, 55, 729–739. 10.1080/13880209.2016.1270972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi M.; Ghaffari S. H.; Soltani B. M.; Nafissi S.; Momeny M.; Zekri A.; Behmanesh M.; Alimoghaddam K.; Ghavamzadeh A. Therapeutic efficacy of silibinin on human neuroblastoma cells: Akt and NF-kappaB expressions may play an important role in silibinin-induced response. Neurochem. Res. 2012, 37, 2053–2063. 10.1007/s11064-012-0827-9. [DOI] [PubMed] [Google Scholar]
- Raina K.; Kumar S.; Dhar D.; Agarwal R. Silibinin and colorectal cancer chemoprevention: a comprehensive review on mechanisms and efficacy. J. Biomed. Res. 2016, 30, 452–465. 10.7555/jbr.30.20150111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma G.; Singh R. P.; Chan D. C.; Agarwal R. Silibinin induces growth inhibition and apoptotic cell death in human lung carcinoma cells. Anticancer Res 2003, 23, 2649–2655. [PubMed] [Google Scholar]
- Darby I. A.; Laverdet B.; Bonté F.; Desmoulière A. Fibroblasts and myofibroblasts in wound healing. Clin., Cosmet. Invest. Dermatol. 2014, 7, 301–311. 10.2147/ccid.s50046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Park G.; Yoon B. S.; Moon J.-H.; Kim B.; Jun E. K.; Oh S.; Kim H.; Song H. J.; Noh J. Y.; Oh C.; You S. Green Tea Polyphenol Epigallocatechin-3-Gallate Suppresses Collagen Production and Proliferation in Keloid Fibroblasts via Inhibition of the STAT3-Signaling Pathway. J. Invest. Dermatol. 2008, 128, 2429–2441. 10.1038/jid.2008.103. [DOI] [PubMed] [Google Scholar]; b Guillermo-Lagae R.; Deep G.; Ting H.; Agarwal C.; Agarwal R. Silibinin enhances the repair of ultraviolet B-induced DNA damage by activating p53-dependent nucleotide excision repair mechanism in human dermal fibroblasts. Oncotarget 2015, 6, 39594–39606. 10.18632/oncotarget.5519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamalice L.; Le Boeuf F.; Huot J. Endothelial Cell Migration During Angiogenesis. Circ. Res. 2007, 100, 782–794. 10.1161/01.RES.0000259593.07661.1e. [DOI] [PubMed] [Google Scholar]
- Polacheck W. J.; Zervantonakis I. K.; Kamm R. D. Tumor cell migration in complex microenvironments. Cell. Mol. Life Sci. 2013, 70, 1335–1356. 10.1007/s00018-012-1115-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J.; Chen S.; Shi Y.; Li C. H.; Wang X. J.; Li F. J.; Wang C. H.; Meng Q. H.; Zhong J. N.; Liu M.; Wang Z. M. Epigallocatechin gallate from green tea exhibits potent anticancer effects in A-549 non-small lung cancer cells by inducing apoptosis, cell cycle arrest and inhibition of cell migration. J. B.U.ON: Off. J. Balk. Union Oncol. 2017, 22, 1422–1427. [PubMed] [Google Scholar]
- Chu S. C.; Chiou H. L.; Chen P. N.; Yang S. F.; Hsieh Y. S. Silibinin inhibits the invasion of human lung cancer cells via decreased productions of urokinase-plasminogen activator and matrix metalloproteinase-2. Mol. Carcinog. 2004, 40, 143–149. 10.1002/mc.20018. [DOI] [PubMed] [Google Scholar]
- Carmeliet P. VEGF as a key mediator of angiogenesis in cancer. Oncology 2005, 69, 4–10. 10.1159/000088478. [DOI] [PubMed] [Google Scholar]
- Abhinand C. S.; Raju R.; Soumya S. J.; Arya P. S.; Sudhakaran P. R. VEGF-A/VEGFR2 signaling network in endothelial cells relevant to angiogenesis. J. Cell Commun. Signaling 2016, 10, 347–354. 10.1007/s12079-016-0352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Ferrara N. VEGF as a therapeutic target in cancer. Oncology 2005, 69, 11–16. 10.1159/000088479. [DOI] [PubMed] [Google Scholar]; b Zhao Y.; Adjei A. A. Targeting Angiogenesis in Cancer Therapy: Moving Beyond Vascular Endothelial Growth Factor. Oncologist 2015, 20, 660–673. 10.1634/theoncologist.2014-0465. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Rapisarda A.; Melillo G. Role of the VEGF/VEGFR axis in cancer biology and therapy. Adv. Cancer Res. 2012, 114, 237–267. 10.1016/B978-0-12-386503-8.00006-5. [DOI] [PubMed] [Google Scholar]
- Kojima-Yuasa A.; Hua J. J.; Kennedy D. O.; Matsui-Yuasa I. Green tea extract inhibits angiogenesis of human umbilical vein endothelial cells through reduction of expression of VEGF receptors. Life Sci. 2003, 73, 1299–1313. 10.1016/S0024-3205(03)00424-7. [DOI] [PubMed] [Google Scholar]
- a Bonauer A.; Carmona G.; Iwasaki M.; Mione M.; Koyanagi M.; Fischer A.; Burchfield J.; Fox H.; Doebele C.; Ohtani K.; Chavakis E.; Potente M.; Tjwa M.; Urbich C.; Zeiher A. M.; Dimmeler S. MicroRNA-92a Controls Angiogenesis and Functional Recovery of Ischemic Tissues in Mice. Science 2009, 324, 1710. 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]; b Yin R.; Bao W.; Xing Y.; Xi T.; Gou S. MiR-19b-1 inhibits angiogenesis by blocking cell cycle progression of endothelial cells. Biochem. Biophys. Res. Commun. 2012, 417, 771–776. 10.1016/j.bbrc.2011.12.032. [DOI] [PubMed] [Google Scholar]
- Dews M.; Homayouni A.; Yu D.; Murphy D.; Sevignani C.; Wentzel E.; Furth E. E.; Lee W. M.; Enders G. H.; Mendell J. T.; Thomas-Tikhonenko A. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat. Genet. 2006, 38, 1060–1065. 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R.; Bao W.; Xing Y.; Xi T.; Gou S. MiR-19b-1 inhibits angiogenesis by blocking cell cycle progression of endothelial cells. Biochem. Biophys. Res. Commun. 2012, 417, 771–776. 10.1016/j.bbrc.2011.12.032. [DOI] [PubMed] [Google Scholar]
- Gallant-Behm C. L.; Piper J.; Dickinson B. A.; Dalby C. M.; Pestano L. A.; Jackson A. L. A synthetic microRNA-92a inhibitor (MRG-110) accelerates angiogenesis and wound healing in diabetic and nondiabetic wounds. Wound Repair Regener. 2018, 26, 311–323. 10.1111/wrr.12660. [DOI] [PubMed] [Google Scholar]
- Schmid-Bindert G. Update on antiangiogenic treatment of advanced non-small cell lung cancer (NSCLC). Targeted Oncol. 2013, 8, 15–26. 10.1007/s11523-013-0261-1. [DOI] [PubMed] [Google Scholar]
- Thilakarathna S. H.; Rupasinghe H. P. Flavonoid bioavailability and attempts for bioavailability enhancement. Nutrients 2013, 5, 3367–3387. 10.3390/nu5093367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu M.; Wu B.; Liu Z. Bioavailability of Polyphenols and Flavonoids in the Era of Precision Medicine. Mol. Pharmaceutics 2017, 14, 2861–2863. 10.1021/acs.molpharmaceut.7b00545. [DOI] [PubMed] [Google Scholar]
- Krupkova O.; Ferguson S. J.; Wuertz-Kozak K. Stability of (-)-epigallocatechin gallate and its activity in liquid formulations and delivery systems. J. Nutr. Biochem. 2016, 37, 1–12. 10.1016/j.jnutbio.2016.01.002. [DOI] [PubMed] [Google Scholar]
- Narumi K.; Sonoda J.; Shiotani K.; Shigeru M.; Shibata M.; Kawachi A.; Tomishige E.; Sato K.; Motoya T. Simultaneous detection of green tea catechins and gallic acid in human serum after ingestion of green tea tablets using ion-pair high-performance liquid chromatography with electrochemical detection. J. Chromatogr. B: Anal. Technol. Biomed. Life Sci. 2014, 945–946, 147–153. 10.1016/j.jchromb.2013.11.007. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Huang Y.; Li S. Polymeric micelles: nanocarriers for cancer-targeted drug delivery. AAPS PharmSciTech 2014, 15, 862–871. 10.1208/s12249-014-0113-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra A.; Satpathy S.; Shenoy A. K.; Bush J. A.; Kazi M.; Hussain M. D. Formulation and evaluation of mixed polymeric micelles of quercetin for treatment of breast, ovarian, and multidrug resistant cancers. Int. J. Nanomed. 2018, 13, 2869–2881. 10.2147/IJN.S153094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haratifar S.; Meckling K. A.; Corredig M. Antiproliferative activity of tea catechins associated with casein micelles, using HT29 colon cancer cells. J. Dairy Sci. 2014, 97, 672–678. 10.3168/jds.2013-7263. [DOI] [PubMed] [Google Scholar]
- Nejaddehbashi F.; Hashemitabar M.; Bayati V.; Abbaspour M.; Moghimipour E.; Orazizadeh M. Application of polycaprolactone, chitosan, and collagen composite as a nanofibrous mat loaded with silver sulfadiazine and growth factors for wound dressing. Artif. Organs 2018, 413–423. 10.1111/aor.13369. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.