

Abstract

The synthesis and self-assembled nanostructures of a series of nucleopeptides (NPs) derived from the dipeptide Phe–Phe and the peptide nucleic acid unit which are covalently attached through an amide or a triazole linker are described. Depending on the variables such as protecting groups, linkers, and nucleobases, spherical nanoparticles were observed through scanning electron microscopy and high-resolution transmission electron microscopy images, and the porous nature of representative NPs was corroborated by carboxyfluorescein entrapment. Hydrophobic substituents on different sites of NPs and solvents employed for peptide self-assembly played a crucial role for corresponding morphologies. The stability of nanoparticles was also probed under external stimuli such as pH, temperature, and enzymatic hydrolysis using proteolytic enzymes. The semiconducting nature of the NP-modified carbon electrodes suggested their potential use as a new capacitor material.
1. Introduction
Self-assembly of the dipeptide diphenylalanine (Phe–Phe) has been widely studied by its crystal and amorphous solid-state structures from mesoscopic to nanometric levels.1−5 Formation of discrete and well-ordered peptide nanotubes by Phe–Phe-derived motifs that represent the key elements of the Alzheimer’s β-amyloid peptide6 combined with their stability, biocompatibility, and water solubility attributes make them ideal for applications in biosensing and catalysis.7−11 Watson–Crick-specific base pairing in a nucleoside domain is well known to unambiguous self-assembling systems.12 Recently, employing peptide nucleic acid (PNA) dimers, Gazit et al. demonstrated that three guanine (G)-containing analogues, viz., CG, GC, and GG, among the 16 possible dimer combinations exhibit wide-range excitation-dependent fluorescence in the visible region and unusual properties such as voltage-dependent electroluminescence. These arise from a combination of both stacking interactions and Watson–Crick base pairing as evident from the single-crystal structure of the GC pair.13 The Fmoc-protected GC PNA dimer also forms highly fluorescent aggregates14 exhibiting aggregation-induced emissions that promise new materials for organic light-emitting diodes. Self-assembly of PNA N-capped G-monomer N-(N-Fmoc-2-aminoethyl)-N-[(N-6-Bhoc-9-guanyl)acetyl]-glycine [Fmoc-GNHBhoc-aeg-OH] leads to nanospheres with unique optical15 and “structure color”16 properties, with formation of photonic crystals useful for optoelectronics. Porphyrin and BODIPY conjugates of PNA monomers have recently been reported as promising candidates for photocatalytic applications.17
These parallel developments in the self-assemblies of dipeptide Phe–Phe and PNA G-monomer prompted us to explore the nucleopeptide (NP) conjugates of Phe–Phe with monomeric PNA motifs to examine their combined influence on mutual self-assembling properties. Previous reports depicted the attachment of purine and pyrimidine nucleobases at the N-terminus of the Phe–Phe motif through an amide (am) linker to design NPs such as (I)18 and (II)19 which behave as hydrogelators that facilitate the delivery of oligonucleotides into human cells.19 Self-assembly of an amphiphilic Phe–Phe (III)20 consisting of a hydrophobic alkyl tail at the N-terminus and a nucleobase at the C-terminus elicited supramolecular nanohelices (Table 1) which were used to generate plasmonic nanomaterials with/without incorporation of AuNPs. Nucleobase peptide amphiphiles on dityrosine backbone (IV) formed helical or curved rodlike micelles leading to gelation and interaction with DNA.21 G-quartet and iron–terpyridine (tpy) complex formation was exhibited simultaneously by tpy-tethered NP V.22 Recently, self-assembly of a series of NPs consisting of purine/pyrimidine nucleoside-conjugated diphenylalanine (VI)23 was shown to form stable spherical nanoparticles in a “molecular necklace” fashion.
Table 1. Self-Assembled Morophology of Phe–Phe Motif Containing NPsa.

B = adenine (A)/cytosine (C)/guanine (G)/thymine (T); R = H, H2PO3; R1 = hexyl, PEG; R2 = C(Me)2.
Nucleobases on a flexible PNA backbone form facile base pairing just as those on rather conformationally constrained ribose/deoxyribose backbones. Hence, PNA monomers were conjugated to the C-terminus of the Phe–Phe motif through different linkers [am and triazole (tz)], and the effects of hydrophobic substituents (Boc, Cbz, iBu, etc.) and nucleobases (A/G/C/T) were investigated on the self-assembly of Phe–Phe NPs. The morphologies of these conjugated NPs were monitored through microscopic techniques [scanning electron microscopy (SEM), high-resolution transmission electron microscopy (HRTEM), and atomic force microscopy (AFM)] and found to form hollow and solid nanospheres. The entrapment of the carboxyfluorescein (CF) molecule in the hollow spherical particles and the stability of these NPs against proteolytic enzymes were investigated. Finally, the tz-modified NPs which showed porosity and excellent stability prompted us to examine their possible semiconducting behavior.
2. Results and Discussion
2.1. Synthesis of Diphenylalanine–PNA-Conjugated Peptides
Four NPs were synthesized through a solid-phase peptide synthesis using a Boc strategy to afford unprotected NPs (Figure 1). For peptide synthesis, Boc-Gly-OH and Boc-PNA monomers with suitable nucleobase protecting groups were commercially available and Boc-Phe-Phe-OH (1) was synthesized according to the literature procedure. Here, Phe–Phe was not directly coupled with a PNA analogue but tethered via a glycyl linker. Unlike previous reports,20,22 the N-termini of these peptides (NP1–4) are uncapped and discriminated structurally by purine and pyrimidine nucleobases (A/C/G/T) being attached. Therefore, these NPs are expected to self-assemble in different patterns. According to the previous reports, substituents present particularly at the N-termini of NPs play an important role for their self-assembly.18−23 Therefore, to check the effect of hydrophobic substituents, PNA monomers 2a–d with free −NH2 groups were coupled directly with Boc-Phe-Phe-OH (1) in the presence of 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide hydrochloride (EDC·HCl) and N-hydroxybenzotriazole (HOBt) (Scheme 1) to afford conjugated peptides 3a–d in moderate to good yields. Boc-Phe-Phe-propyne (4)23 was clicked with PNA-azides (5a–d) using CuAAC-based click chemistry24,25 to yield the tz analogues of the conjugates (Scheme 2). 1,4-Disubstituted tz rings are known bioisosteres of the trans-am bond of peptides26 but devoid of H-bond donor am (CONH) group which may affect the self-assembly process.
Figure 1.

NPs without C-/N-capping and hydrophobic substituents.
Scheme 1. Synthesis of Fully Protected am-Linked NP 3a–d.

Scheme 2. Synthesis of Fully Protected tz-Linked NP 6a–d.

Again, to understand the role of capping groups at the C- and N-termini, partially protected NPs were synthesized. The N-Boc group of fully substituted NPs (6c–d) was deprotected with trifluoroacetic acid (TFA) to afford peptides (7c–d) with free amine groups at the N-termini, whereas ester hydrolysis of 6c–d under basic conditions produced NPs 8c–d with free −CO2H groups. To obtain a control peptide without purine/pyrimidine nucleobase, the intermediate 4 Boc-Phe-Phe-propyne was clicked with the azide ethyl(2-azidoethyl)glycinate to afford Boc-Phe-Phe-tz-aeg-OEt (9) (Scheme 3).
Scheme 3. Synthesis of Partially Protected NPs 7c, 7d, 8c, 8d, and Peptide 9.

Peptides 3a–d, 6a–d, 7c, 7d, 8c, 8d, and 9 were purified by column chromatography over silica gel and structurally characterized by NMR and high-resolution mass spectrometry (HRMS). Peptides NP1–NP4 were purified by reverse-phase high-performance liquid chromatography (HPLC) and characterized by matrix-assisted laser desorption/ionization (MALDI). Intermediate PNA motifs with azide functionality, viz., 5a–e, were characterized by the typical infrared (IR) stretching frequency of azide (νN3) at 2100 cm–1 (Supporting Information). The broad powder X-ray diffraction (PXRD) data observed for the powder forms of NPs indicated their noncrystalline nature (Figure S51 in the Supporting Information).
The characteristic am I bands observed between 1688 and 1643 cm–1 for all NPs (6a–d) indicated the probability of both parallel and antiparallel orientations of peptide chains in a mini β-strand-type structure.27a For NPs 3b–d, a single IR stretching frequency is observed around 1650 cm–1 in the am I region, indicating that β-strand secondary structures may self-assemble in several ways.27 NPs 3a (Boc-Phe-Phe-am-ANHCbz-aeg-OEt), 3b (Boc-Phe-Phe-am-6-Cl-GNHiBu-aeg-OEt), 6c (Boc-Phe-Phe-tz-GNHiBu-aeg-OEt), and 6d (Boc-Phe-Phe-tz-T-aeg-OEt) exhibited strong am II bands around 1500 cm–1.
2.2. Morphologies and Microscopic Architectures
The morphology arising from the self-assembled structures of various NP conjugates was monitored by SEM, HRTEM, and AFM techniques. Peptide samples were prepared as mentioned in the reported experimental procedures.6 NPs were dissolved in 1,1,1,3,3,3-hexafluoro-2-propanol (HFIP) (1 mg/mL) and lyophilized to obtain solid residues which were dissolved further in 1:1 EtOH/H2O (1 mL). These freshly prepared samples were used for microscopic analysis. Self-assembly of completely deprotected NPs was observed first followed by partially and fully protected analogues.
NPs NP1–NP4 without hydrophobic substituents exhibited a wide range of morphologies depending on the attached nucleobases. The G-containing NP NP3 (H-Phe-Phe-gly-G-aeg-NH2) self-assembled into small spherical nanoparticles (Figure 2C) with an average diameter of 15 nm. Although thymine (T)-attached peptide NP4 (H-Phe-Phe-gly-T-aeg-NH2) assembled into nanoparticles (average diameter: 200 nm) (Figure 2D), the adenine (A) analogue NP1 (H-Phe-Phe-gly-A-aeg-NH2) and the cytosine (C) NP2 (H-Phe-Phe-gly-C-aeg-NH2) aggregated to form ill-defined clusters (Figure 2A,B). However, mixing of NP1 and NP2 with their respective complementary NPs (NP4 and NP3) was co-assembled to form better morphologies than individual self-assemblies. A combination of A- and T-analogues (NP1 and NP4, respectively) resulted in aggregated nanoparticles (Figure 2E) while a similar mixture of C- and G-peptides (NP2 and NP3, respectively) produced nanowire-like arrays upon aging (Figure 2F). These phenomena were mostly attributed to the base pair effect. Complex formations by these two pairs of NPs were also confirmed by the electrospray ionization (ESI+)–mass spectroscopy (Figures S47–S50 in the Supporting Information).
Figure 2.

SEM images of NP (A) NP1 (H-Phe-Phe-gly-A-aeg-NH2), (B) NP2 (H-Phe-Phe-gly-C-aeg-NH2), (C) NP3 (H-Phe-Phe-gly-G-aeg-NH2), (D) NP4 (H-Phe-Phe-gly-T-aeg-NH2), (E) 1:1 mixture of NP1 and NP4, and (F) 1:1 mixture of NP2 and NP3.
Free amine and carboxylic groups containing NPs (7c, d, and 8c, d) gave poorly self-assembled morphologies (Figure 3). For example, 7c (H-Phe-Phe-tz-GNHiBu-aeg-OEt) and 7d (H-Phe-Phe-tz-T-aeg-OEt) with free −NH2 groups at the N-termini produced small bead-like structures (Figure 3A,B), whereas the free −CO2H groups containing 8c (Boc-Phe-Phe-tz-G-aeg-OH) and 8d (Boc-Phe-Phe-tz-T-aeg-OH) resulted in shapeless agglomeration (Figure 3C,D). To check the effect of chloride (Cl–) as a counter anion for free N-termini containing NPs, the Boc groups of 6c and 6d were removed by 1 M HCl solution. Peptides 7c and 7d thus obtained with Cl– counter anion also did not produce ordered self-assembled morphologies (Figure S61 in the Supporting Information).
Figure 3.

SEM images of NP (A) 7c (H-Phe-Phe-tz-GNHiBu-aeg-OEt), (B) 7d (H-Phe-Phe-tz-T-aeg-OEt), (C) 8c (Boc-Phe-Phe-tz-G-aeg-OH), and (D) 8d (Boc-Phe-Phe-tz-T-aeg-OH).
The SEM images for the hydrophobic group-substituted NPs (3a–d and 6a–d) depicted spherical nanoparticles with subtle variations. The am-linked Phe–Phe PNA conjugates 3a (Boc-Phe-Phe-am-ANHCbz-aeg-OEt), 3b (Boc-Phe-Phe-am-6-Cl-GNHiBu-aeg-OEt), 3c (Boc-Phe-Phe-am-GNHiBu-aeg-OEt), and 3d (Boc-Phe-Phe-am-T-aeg-OEt) all exhibited solid spheres (Figure 4) with average diameters ranging from 0.5 to 1.0 μm, which was quite larger than unprotected G/T-NP analogues described earlier (Figure 2). Evidently, nucleobases have less effect on the self-assembled morphologies for the am-linked NPs.
Figure 4.

SEM images of NP (A) 3a (Boc-Phe-Phe-am-ANHCbz-aeg-OEt), (B) 3b (Boc-Phe-Phe-am-6-Cl-GNHiBu-aeg-OEt), (C) 3c (Boc-Phe-Phe-am-GNHiBu-aeg-OEt), and (D) 3d (Boc-Phe-Phe-am-T-aeg-OEt).
In comparison, most of the tz-linked analogues (6a, b, d) exhibited porous spheres (Figure 5) with an average diameter for nanoparticles ranging from 0.45 to 0.9 μm. The NP 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt) exhibited more perforated nanoparticle surface than 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) perhaps because of the hydrophobic difference between Cbz and Boc. The SEM images of 6c (Boc-Phe-Phe-tz-GNHiBu-aeg-OEt) indicated a larger spherical particle with uneven surface (Figure 5C), which was different from that seen for the rest of the NPs. NP 6d (Boc-Phe-Phe-tz-T-aeg-OEt) formed interlinked, porous spherical nanoparticles (Figure 5D), which were not observed for its am analogue 3d. HRTEM and AFM analyses done on tz peptides affirmed the hollowness of spherical nanoparticles observed in the SEM images. NP 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) showed a pronounced aperture on the nanoparticle surface in HRTEM (Figure 6A), whereas the two other triazolyl NPs, 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt) and 6d (Boc-Phe-Phe-tz-T-aeg-OEt), showed hollowness similar to 6a on spherical self-assembly (Figure 6B,D). The guanine-containing NP 6c (Boc-Phe-Phe-tz-GNHiBu-aeg-OEt) showed nanospheres with uneven surface (Figure 6C). When the PNA moieties were attached to Phe–Phe through am bonds, viz., 3a (Boc-Phe-Phe-am-ANHCbz-aeg-OEt), 3b (Boc-Phe-Phe-am-6-Cl-GNHiBu-aeg-OEt), 3c (Boc-Phe-Phe-am-GNHiBu-aeg-OEt), and 3d (Boc-Phe-Phe-am-T-aeg-OEt), solid spheres without perforation were observed in HRTEM (Figure 6E–H). The hollow spherical self-assembly observed for tz peptides was clearly visible from their height profiles recorded through AFM. AFM images of the nanoparticles from 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) and 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt) clearly showed the hollow nature with the depth found to be 20–60 nm (Figure 6I,J). The rough-surfaced and aggregated nanoparticles were observed for NPs 6c and 6d, respectively (Figure 6K,L). The origin of pore formation on such spherical particles is attributed to the processes involving both solvents and nonsolvents (here, water is the nonsolvent in which the protected NPs are poorly soluble) proposed by Feringa et al.28
Figure 5.

SEM images of NP (A) 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt), (B) 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt), (C) 6c (Boc-Phe-Phe-tz-GNHiBu-aeg-OEt), and (D) 6d (Boc-Phe-Phe-tz-T-aeg-OEt).
Figure 6.

HRTEM images showing morphology for 6a–d (A–D) and 3a–d (E–H); AFM images showing surface topology for 6a–d (I–L) nanoparticles.
The peptide 9 (Boc-Phe-Phe-tz-aeg-OEt) devoid of purine and pyrimidine nucleobase self-organized to form large disk-like particles (Figure 7), which was apparently different form the morphology generated from the rest of the protected and unprotected NP analogues.
Figure 7.

SEM image of peptide 9 (Boc-Phe-Phe-tz-aeg-OEt).
2.3. Contact Angle
The hydrophobic substituents play a crucial role in forming an array of self-assembled structures. The extent of hydrophobicity of these NPs was qualitatively ascertained through contact angle (CA) measurements of water droplets on thin layers of peptides on silicon wafers (Figures 8 and 9). The measured CA values ranged from 70° to 95°, indicating their moderate to high hydrophobicity.23,29 It was observed that the G-containing NPs (3c and 6c) had CA values above 90°, indicating high hydrophobicity while T (3d, 6d) and A-Cbz NPs (3a, 6b) had CA values less than 80°, being less hydrophobic than G-analogues. The bis Boc-A NP 6a (CA = 88°) and 6-chloro-G NP 3b (CA = 84°) were moderately hydrophobic. As expected, the peptide with free amine 7d (H-Phe-Phe-tz-T-aeg-OEt) was less hydrophobic with a lower CA value (70°). However, in the presence of free carboxylic group, 8d (Boc-Phe-Phe-tz-T-aeg-OH) exhibited moderate hydrophobic nature (CA = 81°). The control peptide 9 (Boc-Phe-Phe-tz-aeg-OEt) for this study without nucleobase showed lower hydrophobicity with a CA value of 71°. Corroboration between SEM images and CA measurements clearly indicates the role of hydrophobic substituents to produce ordered spherical nanoparticles.
Figure 8.

Images captured at 0.5 s for CA measurement of (A) bare silicon wafer and silicon wafer coated with NP (B) 3a (Boc-Phe-Phe-am-ANHCbz-aeg-OEt), (C) 3b (Boc-Phe-Phe-am-6-Cl-GNHiBu-aeg-OEt), (D) 3c (Boc-Phe-Phe-am-GNHiBu-aeg-OEt), (E) 3d (Boc-Phe-Phe-am-T-aeg-OEt), (F) 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt), (G) 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt), (H) 6c (Boc-Phe-Phe-tz-GNHiBu-aeg-OEt), (I) 6d (Boc-Phe-Phe-tz-T-aeg-OEt), (J) 7d (H-Phe-Phe-tz-T-aeg-OEt), (K) 8d (Boc-Phe-Phe-tz-T-aeg-OH), and (L) 9 (Boc-Phe-Phe-tz-aeg-OEt).
Figure 9.

CA measured for peptides on silicon wafer surface.
2.4. Effect of External Stimuli on NPs
Representative NPs with prominent and large self-assembled nanoparticles in this series were subjected for their thermal stability and influence of external triggers such as different pHs and enzymes (proteinase K and chymotrypsin). The nanoparticles from tz peptides 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) and 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt) sustained their morphology beyond 100 °C. The thermal stability of representative peptides 3b, 6b, and 6d was confirmed by thermogravimetric analysis (TGA) which confirmed their stabilities up to 220 °C (Figure S62 in the Supporting Information). However, under pH 2 and pH 10, the spherical assembly of 6a was completely disrupted, which was attributed to the cleavage of acid- and base-labile hydrophobic substituent, respectively (Figure 10). Both am- and tz-bridged NPs were proteolytically stable after incubation with proteinase K and chymotrypsin as monitored by MALDI, HPLC trace analysis, and SEM images (Figures 10 and S63–S65 in the Supporting Information). Thus, the NPs are more stable than unmodified diphenylalanine (Phe–Phe) under similar proteolytic conditions.6
Figure 10.

SEM images of 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) (A) at pH 2, (B) at pH 7, (C) at pH 10, (D) after heating at 100 °C for 4 h, and (E) after incubation with proteinase K and (F) SEM image of proteinase K.
2.5. Solvent-Dependent Morphology of Nucleoeptide 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt)
The dependence of NP morphology on different combinations was also investigated. As a representative example, 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) was chosen owing to its discrete porous spherical nature observed thorough different microscopic techniques and therefore easy to follow any deviation from original morphology. Self-assembly of 6a was examined in pure tetrahydrofuran (THF), MeOH, CHCl3, dichloromethane (DCM), and binary solvents, viz., HFIP/water, MeOH/water, CHCl3/MeOH, and THF/water in 1:1 ratio. It was noted that except for MeOH (Figure 11B), other pure solvents failed to produce the spherical morphology (Figure 11A,C,D) reported earlier. However, in binary solvent combinations, NP 6a assembled either into spherical particles (Figure 11E,F,H) or aggregated large bubble (Figure 11G). Notably, such a solvent effect was observed for both the highest and lowest hydrophobic peptide [3c (Boc-Phe-Phe-am-GNHiBu-aeg-OEt) and 7d (H-Phe-Phe-tz-T-aeg-OEt), respectively] (Figure S67 in the Supporting Information) in this series. These observations suggested the crucial role of water as a nonsolvent for the self-assembly process of NP.
Figure 11.

Solvent-dependent morphology in the SEM images of NP 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) in (A) THF, (B) MeOH, (C) CHCl3, (D) DCM, (E) 1:1 HFIP and water, (F) 1:1 MeOH and water, (G) 1:1 CHCl3 and MeOH, and (H) 1:1 THF and water.
2.6. Encapsulation and Release of Dye and Stimuli-Triggered Disruption of Nanospheres
The stimuli-triggered release30 of dye and drug is one of the potent applications for biomaterials. Here, the hollowness of peptide nanoparticles of 6a (Boc-Phe-Phe-tz-AN(Boc)2) allows encapsulation of CF.23,31,32 The confocal images for 6a show that fluorescent molecules entered the cavity of spherical peptide nanoparticles (Figure 12A) upon incubation with CF. A similar experiment carried out with other tz peptides indicated subtle variations in their morphologies (Table 2). The slow release of CF after addition of dicationic dipeptide (Boc-Lys-Lys-OMe)23 into CF-loaded molecules (Figure 12B) confirmed that the fluorescent small molecules were encapsulated inside the hollow nanoparticles and not simply adhered on the outer layer. The amount of CF release reached saturation after 30 h, which was relatively sluggish compared to that seen with core–shell-like nanoparticles.23 A similar experiment was performed with an anticancer drug doxorubicin which showed the same release kinetics under identical condition (Figure S73 in the Supporting Information).
Figure 12.
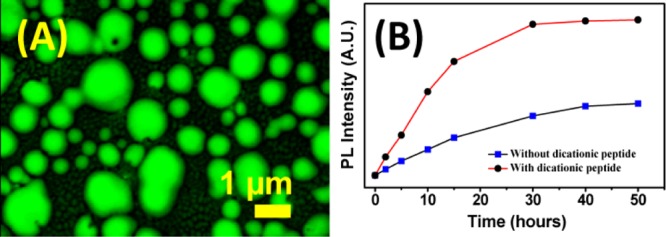
(A) Confocal microscopy images of fluorescent dye-encapsulated 6a (Boc-Phe-Phe-tz-ABoc2-aeg-OEt) and (B) release of encapsulated CF with and without addition of dicationic peptide from peptide nanoparticles.
Table 2. Comparative Microscopic Morphology of Trizolylated NP Nanoparticles.

2.7. Turbidity Assay
The turbidity assay33 was performed for hydrophobic group-substituted NPs following their absorbance at 405 nm (Figure 13 and Supporting Information), which showed that A-peptides 3a (Boc-Phe-Phe-am-ANHCbz-aeg-OEt), 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt), and 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt) have greater tendency to self-assemble than other NPs with high absorbance values. Guanine peptides (3c, 6c) also showed the aggregation property while T-peptides 3d, 6d, and 6-Cl-GNHiBu peptide (3b) were least prone toward aggregation. This is attributed to better H-bonding and π–π stacking abilities of purine nucleobases than pyrimidine analogue thymine. The turbidity assay measured at 570 nm33b wavelength showed an identical pattern of absorbance values which is used to check aggregation properties of Phe–Phe containing NPs. In addition, the high aggregation tendency of 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt) and 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt) was also confirmed from their increased polydispersity index values (from 0.03 to 0.34 and 0.07 to 0.57 for NP 6a and 6b, respectively) in DLS experiments and visualized in AFM and SEM images recorded after 10 days of incubation of peptide solutions (Figure S68 in the Supporting Information). The increment of average particle size for 6a (0.45–1.8 μm) and 6b (0.32–0.77 μm) was noticed after incubation.
Figure 13.
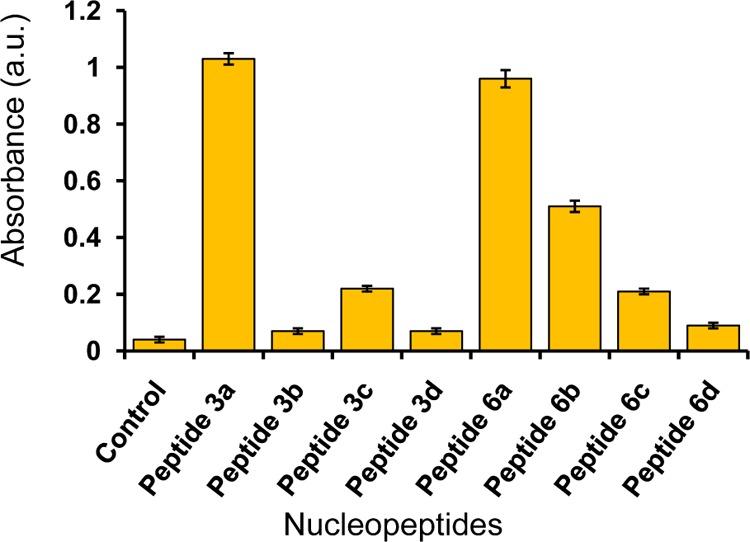
Absorbance values measured from turbidity assay of NPs at 405 nm.
2.8. Cyclic Voltammetry of Modified Electrodes
We opined that the porous self-assembled morphology of these NPs might furnish semiconducting properties as bioinspired doping material.34 The porous nature of the peptide films may modify electrode properties as it increases the effective surface area to allow better interaction of electrolyte with the electrodes and reduce the poor wettability of semiconducting materials like graphite and carbon nanotubes. By employing vapor deposition technique to vertically align diphenylalanine-peptide nanotubes, on modified electrodes, Gazit and co-workers35 observed that such modifications lead to better efficacy of hydrophobic graphite electrodes. These electrodes had an advantage of longevity even after several cycles.36 Expecting such stability from porous Boc-Phe-Phe-tz-PNA, conductivity measurements were performed by cyclic voltammetry (CV) on Torey carbon electrodes coated with peptides.
The current density response versus voltage for the trizole peptide-modified electrode was found to be higher than that of an unmodified carbon electrode. The CV profiles for all tested electrodes presented quite rectangular (Figure 14) and symmetric shape, consistent even after 500 cycles, which is a clear evidence of a double-layer capacitance charge–discharge process with no faradaic effects.37 The value of capacitance (Table 3) suggests that these peptides can be used as efficient supercapacitors. The capacitance of the unmodified Torey carbon electrode was 90 μF/cm2. After modification, the G-peptide 6c (Boc-Phe-Phe-tz-GNHiBu-aeg-OEt) exhibited the highest capacitance value of 617 μF/cm2, almost 7 times higher than that for the uncoated carbon electrode. Electrodes coated with adenine- and thymine-modified peptides (6a, b, and 6d, respectively) displayed lower capacitance values than that of G-analogue, probably attributed to the nature of self-assembly. As a control for NPs, peptide 9 (Boc-Phe-Phe-tz-aeg-OEt) produced a capacitance value of 500 μF/cm2. However, for peptide 9, a perturbed rectangular shape of current density response curve was observed unlike the symmetric nature observed for nuclopeptides (Supporting Information) indicating a faradaic process.
Figure 14.
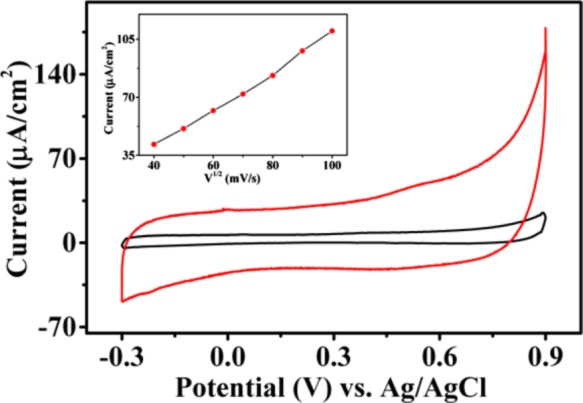
Cyclic voltammograms of Boc-Phe-Phe-tz-GNHiBu-supported Toray carbon electrode in 0.1 M potassium phosphate buffer electrolytes at pH = 7.4 and 0.1 M KCl at a scan rate of 50 mV/s. Inset: scan rate effect at different scan rates from 10 to 90 mV/s.
Table 3. Capacitance Values of Peptide-Modified Torey Carbon Electrodes.
| peptide used for electrode modification | capacitance (μF/cm2) |
|---|---|
| Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt (6a) | 260.0 |
| Boc-Phe-Phe-tz-ANHCbz-aeg-OEt (6b) | 205.8 |
| Boc-Phe-Phe-tz-GNHiBu-aeg-OEt (6c) | 617.0 |
| Boc-Phe-Phe-tz-T-aeg-OEt (6d) | 342.0 |
| Boc-Phe-Phe-tz-aeg-OEt (9) | 500.0 |
| none (unmodified electrode) | 90.0 |
We proposed that the porous nature of tz-linked NPs enhances the electrode–electrolyte contact, which leads to the increment of the effective activation area of the modified Torey carbon electrodes for electrical conductivity. Therefore, it is expected that these self-assembled NPs with perforated morphology to follow a similar mechanism as reported by Gazit et al.34−36 enhancing the capacitance of modified electrodes. Thus, without a sophisticated vapor deposition method, simple thin layer coating of these peptides resulted in attaining good capacitor efficacy (Experimental Section). Self-assembled peptide nanoparticles can potentially create well-defined porous membranes, which can interact with the electrolyte efficiently compared to bare and hydrophobic Torey carbon electrodes used in this study.
3. Conclusions
A new series of NPs was synthesized and their self-assembled morphologies were monitored systematically. Self-assembly of these conjugates are influenced by both Phe–Phe and PNA moieties. NPs without C-/N-termini capping and free nucleobases did not show very good self-assembled morphologies, whereas solid and porous nanospheres were obtained from protected forms of Phe-Phe-PNA conjugates. Partial removal of capping groups also leads to poorly organized agglomerated structures. Thus, importance of hydrophobic substituents on nucleobases and C-/N-termini of NP for self-assembled structures were observed. The extent of hydrophobicity was assessed qualitatively by CA measurement. An equal mixture of NPs with complementary base pairs elicited better nanoarchitectures among completely deprotected analouges. Effects of linkers (tz/am) for protected NPs were evident from self-assembled morphologies, albeit the reason is not clearly understood. Effect of mixed solvent was crucial for consistent and regular morphologies from these peptides. Adenine-containing NPs were more prone toward self-aggregation as observed form the turbidity assay and DLS studies. Representative hydrophobic NPs were found stable under a wide range of pH, high temperature, and proteolytic enzymes, which showed their ability to behave as stable biocompatible templates. Noncovalent encapsulation of CF inside the porous nanospheres was evident from the confocal images. Slow release of dye from peptide nanoparticle upon treatment of dicationic dipeptide as an external stimulus. To utilize such porous nanoparticles in the field of bioelectronics, Torey carbon electrodes were fabricated with tz-NPs and enhanced capacitance was measured. The guanine nucleobase containing NP furnished the best result with a 7-fold increment of the capacitance value compared to the bare, unmodified electrode. Thus, NPs consisting of Phe–Phe and PNA motifs can be tuned to engineer hybrid peptides with potential application as biomaterials.
4. Experimental Section
4.1. Materials and Methods
All reagents and proteolytic enzymes were commercially purchased from Sigma-Aldrich. Column chromatographic separations were done using silica gel (230–400 mesh). Solvents were dried and distilled following standard procedures. Thin layer chromatography (TLC) was carried out on precoated plates (Merck silica gel 60, F254) and the spots were visualized with UV light or by charring the plates by dipping in 5% ninhydrin in n-butanol solutions. 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Bruker NMR spectrometer. All 1H and 13C NMR spectra were recorded in CDCl3, CD3OD, and DMSO-d6. Chemical shifts are reported in parts per million (ppm, δ scale). Mass spectroscopy data were obtained from a Synapt G2QTof mass spectrometer in ESI+ mode. Melting points were determined in open-end capillary tubes and uncorrected. UV–vis measurements were made using a UV–vis spectrophotometer (model Lambda 25).
The self-assembly process of the peptides was monitored after 1 mg/mL was dissolved in HFIP, lyophilized, and then diluted with 50% ethanol in water.
4.2. Synthesis of NPs on Solid Phase
The Boc-PNA monomers Boc-Gly-OH and Boc-Phe-Phe-OH were incorporated into a peptide sequence by solid-phase synthesis on an MBHA resin having 0.67 mmol/g loading value without further reduction of loading capacity. After swelling of resin beads in DCM overnight, these were washed with 15% N,N-diisopropylethylamine (DIPEA) in DCM (3 × 10 min) to generate free amines from the commercially available salt form. Free amine groups were then coupled with carboxylic acid of PNA monomers first in dry dimethylformamide (DMF) (0.5 mL) using HOBt (3 equiv), 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (3 equiv), and DIPEA (3 equiv). The reagents were removed by filtration and washed with DMF (3 × 10 mL) and DCM (3 × 10 mL). Deprotection of Boc group from the N-terminus of the resin-bound PNA monomers with 50% TFA in DCM (3 × 15 min) was followed by washing with DCM and DMF (3 × 10 mL) to give a TFA salt of amine which was neutralized using 10% DIPEA in DCM (3 × 10 min) to generate free amine. After washing with DCM and DMF (3 × 10 mL), the free amine was coupled with carboxylic acid of incoming monomers (Boc-Gly-OH and Boc-Phe-Phe-OH) in dry DMF (0.5 mL) under an aforementioned am coupling condition. The protocol for washing and regeneration of free amine was repeated to afford the desired peptides. The resin-bound peptides were cleaved from solid support by TFA and trifluoromethanesulfonic acid.
4.3. Purification of NPs by Reverse-Phase HPLC
Purification of NPs was performed through a semipreparative BEH130 C18 (10 × 250 mm) column with the following gradient elution method: 100% A to 100% B in 33 min; A = 0.1% TFA in CH3CN/H2O (5:95); B = 0.1% TFA in CH3CN/H2O (1:1) with a flow rate of 2 mL/min. Oligomers were monitored at 220 and 260 nm wavelengths during purification.
4.4. Synthesis of NPs in Solution Phase
4.4.1. NP 3a (Boc-Phe-Phe-am-ANHCbz-aeg-OEt)
To a mixture of compound 1 (1.00 g, 2.43 mmol) in dry DMF (10 mL), EDC·HCl (0.46 g, 2.43 mmol) and HOBT (0.16 g, 1.22 mmol) were added at 0 °C. To the reaction mixture, DIPEA (0.85 mL, 4.86 mmol) was added. After 0.25 h, the TFA salt of 2a (1.43 g, 2.43 mmol) dissolved in dry DMF (5 mL) was added slowly into the reaction mixture and stirred at room temperature (rt) under inert atmosphere. After 12 h, brine solution (40 mL) was added into it and an aqueous (aq) layer was washed with ethyl acetate (EtOAc) (3 × 25 mL). An organic layer was washed with sat. NaHCO3, 10% citric acid, and brine solutions (2 × 50 mL). The organic layer was separated, dried over anhyd. Na2SO4, filtered, and the filtrate was concentrated under reduced pressure. The crude mass thus obtained was purified by column chromatography [eluent: 50–80% of EtOAc in pet ether] to afford peptide 3a (1.44 g, 70% with respect to 1). White hygroscopic solid; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 8.94 (s, 1H), 8.22 (d, J = 10.7 Hz, 1H), 7.41–7.08 (m, 20H), 7.06–6.84 (m, 3H), 6.66–6.23 (m, 2H), 5.09 (ddd, J = 26.8, 11.3, 5.8 Hz, 4H), 4.99–4.86 (m, 1H), 4.63–4.52 (m, 1H), 4.37–3.87 (m, 7H), 3.56–3.20 (m, 4H), 3.13–2.73 (m, 5H), 1.31–1.19 ppm (m, 12H); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 171.6, 171.3, 171.1, 170.8, 170.1, 169.4, 169.1, 160.0, 159.5, 156.0, 155.6, 154.6, 154.4, 153.6, 151.2, 136.0, 135.5, 129.3, 129.3, 129.2, 128.8, 128.6, 128.6, 128.5, 128.5, 128.4, 128.0, 127.1, 114.1, 80.9, 80.7, 67.9, 67.5, 62.1, 61.7, 60.5, 56.3, 55.8, 54.2, 49.7, 48.7, 47.8, 47.5, 42.7, 37.4, 37.3, 37.2, 37.1, 37.1, 33.9, 32.0, 29.7, 28.2, 28.1, 14.2, 14.1 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C44H52N9O9, 850.3888; found, 850.3899.
4.4.2. NP 3b (Boc-Phe-Phe-am-6-Cl-GNHiBu-aeg-OEt)
Compound 1 (0.50 g, 1.21 mmol) was coupled with the TFA salt of 2b (0.63 g, 1.21 mmol) following the method described for peptide 3a to afford peptide 3b (0.69 g, 70% with respect to 1), which was purified by column chromatography [eluent: 60–80% of EtOAc in pet ether]. Yellowish white solid; mp 110–113 °C; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 12.36 (s, 1H), 10.64 (s, J = 23.8 Hz, 1H), 7.94 (s, 1H), 7.65 (s, J = 8.5 Hz, 1H), 7.55 (s, 1H), 7.28–7.03 (m, 10H), 5.95 (s, 1H), 5.04–4.81 (m, 2H), 4.30–4.17 (m, 2H), 4.16–4.08 (m, 2H), 4.00 (d, J = 17.4 Hz, 1H), 3.82 (d, J = 7.6 Hz, 1H), 3.53–3.42 (m, 1H), 3.21 (dd, J = 13.8, 4.5 Hz, 1H), 3.12–2.84 (m, 3H), 2.69–2.52 (m, 1H), 2.19 (d, J = 29.8 Hz, 2H), 1.26–1.15 ppm (m, 18H). 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 180.2, 172.6, 172.2, 169.4, 166.8, 156.3, 156.2, 149.4, 148.5, 140.4, 137.0, 136.7, 129.5, 129.3, 128.8, 128.7, 128.5, 127.1, 126.9, 120.2, 114.19, 80.3, 61.7, 56.9, 54.4, 47.8, 47.3, 44.2, 37.9, 37.5, 37.1, 35.9, 33.9, 32.0, 29.8, 29.8, 29.7, 22.8, 19.4, 19.0, 14.3, 14.2 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C40H51ClN9O8, 820.3549; found, 820.3539.
4.4.3. NP 3c (Boc-Phe-Phe-am-GNHiBu-aeg-OEt)
Compound 1 (0.41 g, 1.00 mmol) was coupled with the TFA salt of 2c (0.50 g, 1.00 mmol) following the method described for peptide 3a to obtain peptide 3c (0.48 g, 60% with respect to 1), which was purified by column chromatography [eluent: 0–3% of MeOH in DCM]. White solid; mp 105–108 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C): δ 12.07 (s, 1H), 11.57 (s, 1H), 8.17 (t, J = 12.7 Hz, 1H), 7.89 (s, 0.6H), 7.85 (s, 0.4H), 7.32–7.02 (m, 12H), 6.89 (d, J = 8.5 Hz, 1H), 5.18–4.89 (m, 2H), 4.64–4.51 (m, 1H), 4.40 (s, 1H), 4.22 (q, J = 7.0 Hz, 1H), 4.10 (dt, J = 14.1, 6.1 Hz, 2H), 3.45 (dd, J = 24.8, 18.2 Hz, 2H), 3.26–3.13 (m, 1H), 3.06–2.62 (m, 6H), 1.30–1.20 (m, 9H), 1.19–1.13 (m, 3H), 1.11 ppm (dd, J = 6.8, 3.6 Hz, 6H); 13C NMR (100 MHz, DMSO-d6, 25 °C): δ 180.1, 171.7, 169.1, 167.0, 166.6, 154.9, 149.1, 147.9, 140.8, 138.0, 137.5, 129.2, 129.1, 128.1, 127.9, 126.3, 126.1, 119.5, 78.2, 61.3, 60.6, 55.8, 54.1, 47.7, 46.7, 44.0, 37.6, 37.5, 37.2, 34.7, 33.2, 31.5, 29.4, 29.0, 28.1, 27.7, 18.9, 14.0 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C40H52N9O9, 802.3888; found, 802.3889.
4.4.4. NP 3d (Boc-Phe-Phe-am-T-aeg-OEt)
Compound 1 (0.21 g, 0.51 mmol) was coupled with the TFA salt of 2d (0.21 g, 0.51 mmol) following the procedure described for peptide 3a to afford peptide 3b (0.29 g, 80% with respect to 1), which was purified by column chromatography [eluent: 3–6% of MeOH in DCM]. White solid; mp 92–95 °C; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 10.03 (s, 1H), 7.77 (d, J = 9.2 Hz, 1H), 7.34–6.97 (m, 11H), 6.49 (s, 1H), 5.42–5.14 (m, 1H), 4.86–4.61 (m, 2H), 4.36–4.18 (m, 1H), 4.16–3.91 (m, 4H), 3.77 (t, J = 21.3 Hz, 1H), 3.69–3.41 (m, 1H), 3.37–3.13 (m, 2H), 3.09–2.99 (m, 1H), 2.93–2.73 (m, 3H), 1.90 (d, J = 12.0 Hz, 3H), 1.44–1.26 (m, 9H), 1.23–1.13 (m, 3H); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 172.4, 172.0, 171.0, 169.3, 167.1, 164.3, 155.6, 151.2, 140.9, 137.1, 136.6, 130.0, 129.7, 129.5, 129.3, 128.6, 128.3, 128.2, 126.4, 126.2, 110.9, 80.6, 61.7, 55.3, 53.5, 48.6, 48.3, 47.3, 38.2, 36.9, 36.6, 28.3, 14.0, 12.4 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C36H47N6O9, 707.3404; found, 707.3395.
4.4.5. NP 6a (Boc-Phe-Phe-tz-AN(Boc)2-aeg-OEt)
To a mixture of peptide 4 (0.25 g, 0.56 mmol) in THF (5.0 mL) and 10 mL of distilled water, CuSO4·5H2O (7 mg, 0.03 mmol) and sodium-l-(+)-ascorbate (0.06 g, 0.28 mmol) were added and stirred. To the resulting suspension, azide 5a (0.33 g, 0.60 mmol) dissolved in THF (5.0 mL) was added and stirred at rt. After 12 h, EtOAc (20 mL) was added into it and an aq layer was further washed with EtOAc (3 × 20 mL). The organic layer was separated, dried over anhyd. Na2SO4, filtered, and the filtrate was concentrated under reduced pressure. The crude mass thus obtained was purified by column chromatography [eluent: 0–3% of MeOH in DCM] to afford peptide 6a (0.44 g, 80% with respect to 4). White hygroscopic solid; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 8.80 (t, J = 5.5 Hz, 1H), 8.28 (d, J = 2.3 Hz, 1H), 7.90–7.77 (m, 1H), 7.49 (d, J = 14.2 Hz, 1H), 7.36–7.03 (m, 10H), 6.96 (d, J = 21.6 Hz, 2H), 6.40 (d, J = 24.3 Hz, 1H), 5.31–4.88 (m, 2H), 4.68 (d, J = 25.0 Hz, 3H), 4.54–4.41 (m, 2H), 4.27–4.16 (m, 3H), 4.08 (dd, J = 14.9, 11.7 Hz, 2H), 3.96–3.74 (m, 2H), 3.16 (s, 1H), 3.05–2.74 (m, 3H), 1.42 (dd, J = 13.0, 1.9 Hz, 18H), 1.33–1.23 ppm (m, 12H); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 171.9, 171.6, 171.1, 170.9, 169.0, 168.8, 167.2, 156.1, 153.7, 152.2, 150.5, 146.2, 136.0, 129.3, 129.2, 129.0, 128.9, 128.9, 128.4, 127.3, 123.8, 83.9, 83.9, 83.9, 81.3, 80.9, 62.4, 61.9, 56.7, 54.0, 50.6, 49.7, 47.9, 47.8, 44.2, 37.6, 37.1, 35.5, 35.4, 28.2, 28.0, 14.2 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C49H65N12O11, 997.4896; found, 997.4894.
4.4.6. NP 6b (Boc-Phe-Phe-tz-ANHCbz-aeg-OEt)
Boc-Phe-Phe-propyne 4 (0.25 g, 0.56 mmol) was clicked with azide 5b (0.29 g, 0.60 mmol) following the procedure described for peptide 6a to afford peptide 6b (0.39 g, 75% with respect to 4), which was purified by column chromatography [eluent: 0–3% of MeOH in DCM]. White hygroscopic solid; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 8.83–8.61 (m, 2H), 8.09 (d, J = 5.3 Hz, 0.7H), 7.83 (s, 0.3H), 7.52–7.31 (m, 5H), 7.27–7.05 (m, 8H), 7.03–6.88 (m, 2H), 6.60 (dd, J = 23.7, 6.9 Hz, 1H), 5.37–5.20 (m, 2H), 5.05 (dt, J = 32.8, 16.5 Hz, 2H), 4.73–4.60 (m, 2H), 4.58–4.32 (m, 3H), 4.26–4.11 (m, 3H), 4.03 (d, J = 23.8 Hz, 2H), 3.94–3.70 (m, 2H), 3.16–2.76 (m, 4H), 1.34–1.19 ppm (m, 12H); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 172.0, 171.7, 171.1, 170.9, 169.0, 168.8, 167.3, 156.0, 152.9, 151.7, 151.2, 149.5, 145.7, 144.2, 138.0, 136.2, 135.6, 129.2, 129.2, 129.1, 128.9, 128.8, 128.8, 128.7, 128.7, 128.6, 128.3, 127.2, 127.2, 125.4, 124.3, 123.7, 121.4, 81.1, 80.9, 80.7, 67.8, 62.4, 61.9, 56.5, 54.1, 50.5, 49.4, 48.4, 47.7, 44.1, 43.1, 37.7, 37.2, 35.3, 28.2, 14.2 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C47H55N12O9, 931.4215; found, 931.4214.
4.4.7. NP 6c (Boc-Phe-Phe-tz-GNHiBu-aeg-OEt)
Alkyne 4 (0.25 g, 0.56 mmol) was reacted with azide 5c (0.26 g, 0.60 mmol) following the procedure described for peptide 6a to afford peptide 6c (0.34 g, 70% with respect to 4), which was purified by column chromatography [eluent: 0–10% of MeOH in CHCl3]. White solid; mp 100–103 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C): δ 12.09 (d, J = 10.9 Hz, 1H), 11.64 (d, J = 14.1 Hz, 1H), 8.62–8.47 (m, 1H), 8.38–8.33 (m, 0.3H), 8.04–7.98 (m, 0.7H), 7.96 (s, 0.5H), 7.83 (s, 0.5H), 7.74 (d, J = 10.1 Hz, 1H), 7.30–7.05 (m, 11H), 6.93–6.87 (m, 0.7H), 6.71 (t, J = 9.2 Hz, 0.3H), 5.00 (s, 1H), 4.94 (s, 1H), 4.70 (s, 1H), 4.60–4.50 (m, 1H), 4.48–4.26 (m, 4H), 4.18 (dt, J = 16.3, 8.1 Hz, 1H), 4.13–4.02 (m, 3H), 3.97 (s, 1H), 3.73 (t, J = 6.1 Hz, 1H), 2.99 (d, J = 13.8 Hz, 1H), 2.89–2.72 (m, 3H), 2.62 (d, J = 11.8 Hz, 1H), 1.26 (t, J = 4.4 Hz, 9H), 1.16 (t, J = 7.1 Hz, 3H), 1.11 ppm (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6, 25 °C): δ 180.2, 180.1, 171.4, 170.9, 170.8, 169.4, 168.9, 167.4, 166.8, 155.1, 154.9, 149.3, 149.2, 147.9, 145.0, 144.6, 140.5, 140.4, 138.0, 137.7, 137.5, 129.3, 129.2, 129.1, 128.0, 128.0, 127.9, 126.3, 126.1, 123.7, 123.2, 119.6, 79.2, 78.2, 61.4, 60.7, 55.9, 53.7, 49.1, 47.9, 47.6, 47.4, 46.6, 44.1, 43.7, 37.8, 37.5, 34.7, 34.3, 28.1, 18.9, 14.0 ppm. HRMS (ESI+): m/z calcd for (M + Na)+ C43H54N12O9Na, 905.4034; found, 905.4028.
4.4.8. NP 6d (Boc-Phe-Phe-tz-T-aeg-OEt)
Alkyne 4 (0.25 g, 0.56 mmol) was clicked with azide 5d(38) (0.20 g, 0.60 mmol) following the procedure described for peptide 6a to afford peptide 6d (0.39 g, 90% with respect to 4), which was purified by column chromatography [eluent: 0–6% of MeOH in CHCl3]. White solid; mp 132–135 °C; 1H NMR (400 MHz, CDCl3 + DMSO-d6, 2:1, 25 °C, TMS): δ 10.82 (s, 0.5H), 10.72 (s, 0.5H), 7.85 (s, 1H), 7.66 (d, J = 8.4 Hz, 1H), 7.63 (dd, J = 20.9, 8.1 Hz, 0.5H), 7.61 (d, J = 7.7 Hz, 0.5H), 7.28–7.10 (m, 10H), 7.06 (d, J = 1.2 Hz, 0.5H), 6.94 (d, J = 1.2 Hz, 0.5H), 6.00 (d, J = 7.3 Hz, 0.4H), 5.85 (d, J = 7.8 Hz, 0.6H), 4.77–4.68 (m, 1H), 4.62 (d, J = 5.2 Hz, 1H), 4.55–4.42 (m, 1H), 4.40–4.24 (m, 3H), 4.23–4.15 (m, 2H), 4.10 (s, 1H), 3.92 (d, J = 20.2 Hz, 3H), 3.62 (d, J = 19.0 Hz, 1H), 3.17–3.07 (m, 1H), 3.04–2.96 (m, 3H), 1.91 (d, J = 0.7 Hz, 1.5H), 1.86 (d, J = 0.7 Hz, 1.5H), 1.34 (d, J = 11.7 Hz, 9H), 1.26 ppm (dt, J = 14.0, 5.7 Hz, 3H); 13C NMR (100 MHz, CDCl3 + DMSO-d6, 2:1, 25 °C, TMS): δ 171.6, 171.3, 170.8, 170.6, 168.7, 168.4, 167.6, 167.1, 164.3, 164.2, 155.3, 155.2, 151.2, 150.9, 140.6, 140.4, 136.8, 136.6, 128.9, 128.8, 127.9, 126.2, 124.0, 123.0, 110.2, 79.5, 61.6, 61.1, 55.8, 55.4, 53.6, 49.6, 48.9, 48.8, 48.3, 47.9, 47.7, 46.8, 39.6, 37.7, 37.6, 37.0, 36.8, 35.0, 34.7, 27.9, 13.7, 11.9 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C39H50N9O7, 788.3731; found, 788.3722.
4.4.9. NP 7c (H-Phe-Phe-tz-GiBu-aeg-OEt)
To an ice-cold mixture of peptide 6c (0.2 g, 0.23 mmol) in DCM (2 mL), TFA (2 mL) was added slowly and stirred for 4 h at rt. Completion of reaction was monitored by TLC. The volatile portion of the reaction mixture was completely removed by coevaporation with toluene and carbon tetrachloride. The residue was washed with CHCl3 first and then triturated well with chilled Et2O to obtain peptide 7c (0.15 g, 85%) as a yellowish white hygroscopic solid. 1H NMR (400 MHz, DMSO-d6, 25 °C): δ 12.18–11.99 (m, 1H), 11.71–11.54 (m, 1H), 8.94–8.79 (m, 1H), 8.78–8.59 (m, 1H), 8.02 (d, J = 6.7 Hz, 2H), 7.96 (s, 2H), 7.85–7.78 (m, 1H), 7.69 (d, J = 3.5 Hz, 1H), 7.34–7.12 (m, 11H), 6.98 (d, J = 3.4 Hz, 1H), 6.51 (s, 1H), 5.00 (s, 1H), 4.85 (dd, J = 15.4, 7.8 Hz, 1H), 4.63 (dd, J = 36.2, 9.1 Hz, 3H), 4.44 (t, J = 12.4 Hz, 2H), 4.33 (dd, J = 18.4, 8.0 Hz, 2H), 4.20 (dt, J = 7.2, 5.8 Hz, 2H), 4.11–3.93 (m, 5H), 3.73 (s, 1H), 3.00 (ddd, J = 31.9, 23.2, 11.5 Hz, 2H), 2.86–2.58 (m, 3H), 1.26 (td, J = 7.1, 1.3 Hz, 1.5H), 1.19–1.13 (m, 1.5H), 1.11 ppm (d, J = 6.8 Hz, 6H); 13C NMR (100 MHz, DMSO-d6, 25 °C): δ 170.7, 170.5, 170.4, 169.4, 168.9, 167.9, 167.8, 167.4, 166.7, 166.7, 154.9, 154.8, 149.3, 149.2, 148.0, 144.8, 144.4, 144.3, 140.4, 137.3, 134.7, 129.6, 129.5, 129.3, 129.2, 128.5, 128.5, 128.2, 127.1, 126.5, 123.9, 123.2, 119.5, 61.4, 60.7, 54.2, 54.0, 53.3, 53.2, 47.4, 46.6, 44.1, 43.7, 38.2, 37.8, 36.9, 36.8, 34.7, 34.2, 18.9, 14.0 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C38H47N12O7, 783.3691; found, 783.3686.
4.4.10. NP 7d (H-Phe-Phe-tz-T-aeg-OEt)
Peptide 6d (0.5 g, 0.64 mmol) was hydrolyzed with TFA in DCM as described for 7c to afford peptide 7d (0.39 g, 90%) as a white solid. mp 98–100 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C): δ 11.36–11.19 (m, 1H), 8.92 (dd, J = 8.6, 2.6 Hz, 0.5H), 8.82 (d, J = 8.1 Hz, 0.5H), 8.74 (dt, J = 10.9, 5.4 Hz, 0.5H), 8.66–8.59 (m, 0.5H), 8.08 (s, 1H), 8.01 (s, 1H), 7.97 (s, 0.5H), 7.89 (s, 0.5H), 7.82 (s, 0.5H), 7.70 (s, 0.5H), 7.31–7.16 (m, 11H), 6.97 (dd, J = 6.4, 2.8 Hz, 1H), 4.75–4.55 (m, 3H), 4.52–4.23 (m, 6H), 4.19–4.03 (m, 5H), 3.87 (d, J = 3.5 Hz, 1H), 3.81–3.65 (m, 1H), 3.16–3.07 (m, 1H), 3.04–2.53 (m, 4H), 1.74 (ddd, J = 6.6, 4.9, 1.2 Hz, 3H), 1.23 (td, J = 7.1, 1.2 Hz, 1.5H), 1.19–1.15 ppm (m, 1.5H); 13C NMR (100 MHz, DMSO-d6, 25 °C): δ 170.8, 170.5, 170.5, 170.2, 170.2, 170.2, 169.2, 168.9, 168.1, 167.9, 167.7, 167.5, 164.4, 158.3, 158.0, 151.5, 151.0, 150.9, 150.9, 144.7, 144.7, 144.4, 144.3, 142.0, 141.9, 137.7, 137.4, 137.3, 134.7, 134.6, 129.6, 129.5, 129.3, 129.2, 129.2, 129.1, 128.5, 128.4, 128.2, 128.1, 128.0, 127.1, 126.5, 126.4, 123.7, 123.2, 108.3, 107.7, 61.3, 60.6, 60.0, 54.2, 54.0, 53.3, 53.1, 49.0, 48.0, 47.9, 47.8, 47.5, 47.4, 47.3, 46.6, 38.2, 37.8, 37.0, 36.9, 34.2, 34.2, 22.5, 14.0, 14.0, 11.9, 11.8 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C34H42N9O7, 688.3207; found, 688.3210.
4.4.11. NP 8c (Boc-Phe-Phe-tz-G-aeg-OH)
To a solution of peptide 6c (0.10 g, 0.11 mmol) in THF (4 mL), 1 M NaOH solution (3 mL) was added at 0 °C and the resulting mixture was stirred for 8 h. The volatile part of the mixture was evaporated under reduced pressure. The residue was neutralized with cold 1 M HCl solution and the resulting aq layer was extracted with EtOAc (20 × 3 mL). The combined organic layer was separated, dried over anhyd. Na2SO4, filtered, and the filtrate was concentrated under reduced pressure. The crude mass thus obtained was purified by column chromatography [eluent: 5–50% of MeOH in CHCl3] to afford peptide 8c (0.07 g, 75%) as a white hygroscopic solid. 1H NMR (400 MHz, DMSO-d6, 25 °C): δ 11.04 (s, 1H), 8.76 (s, 0.5H), 8.48 (d, J = 21.1 Hz, 1.5H), 7.83 (s, 0.5H), 7.94 (s, 0.5H), 7.51 (d, J = 3.3 Hz, 1H), 7.30–6.92 (m, 12H), 6.76 (d, J = 23.2 Hz, 2H), 4.82 (s, J = 50.2 Hz, 2H), 4.48 (dd, J = 16.5, 10.4 Hz, 3H), 4.32 (dd, J = 33.5, 7.4 Hz, 2H), 4.12 (dd, J = 26.3, 22.1 Hz, 1H), 3.75–3.56 (m, 3H), 3.07–2.96 (m, 1H), 2.90–2.78 (m, 2H), 2.74–2.60 (m, 9H), 2.13 (t, J = 6.9 Hz, 1H), 1.24 ppm (d, J = 4.8 Hz, 9H); 13C NMR (150 MHz, DMSO-d6, 25 °C): δ 171.7, 171.5, 170.9, 170.7, 170.5, 167.6, 166.2, 157.1, 155.3, 155.1, 153.8, 151.8, 144.3, 138.3, 138.1, 137.9, 137.7, 129.3, 128.0, 127.9, 126.3, 126.0, 123.6, 123.5, 115.9, 79.2, 78.0, 56.1, 55.8, 54.2, 53.9, 53.1, 48.6, 48.4, 47.3, 47.1, 43.6, 34.5, 28.1, 27.7 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C37H45N12O8, 785.3483; found, 785.3474.
4.4.12. NP 8d (Boc-Phe-Phe-tz-T-aeg-OH)
Peptide 6d (0.25 g, 0.32 mmol) was ester-hydrolyzed under basic condition as described for peptide 8c to afford peptide 8d (0.19 g, 80%) as a white solid. mp 120–123 °C; 1H NMR (400 MHz, DMSO-d6, 25 °C): δ 11.28 (d, J = 2.5 Hz, 1H), 8.51 (t, J = 9.5 Hz, 1H), 8.32 (s, 1H), 8.04–7.95 (m, 0.7H), 7.89 (s, 0.5H), 7.81 (s, 0.3H), 7.73 (s, 0.5H), 7.32–7.05 (m, 11H), 6.90 (d, J = 6.8 Hz, 0.75H), 6.75 (d, J = 7.1 Hz, 0.25H), 4.63 (s, 1H), 4.50 (dd, J = 24.6, 13.8 Hz, 3H), 4.39–4.23 (m, 4H), 4.20–4.05 (m, 2H), 3.96 (s, 1H), 3.84 (s, 1H), 3.70 (s, 1H), 3.06–2.94 (m, 1H), 2.90–2.60 (m, 3H), 1.74 (d, J = 3.9 Hz, 2H), 1.27 (d, J = 3.7 Hz, 9H) ppm; 13C NMR (100 MHz, DMSO-d6, 25 °C): δ 171.4, 171.0, 170.8, 170.7, 170.4, 168.0, 167.4, 164.5, 164.4, 155.3, 155.2, 151.1, 151.0, 144.9, 144.6, 142.1, 142.0, 138.1, 137.8, 137.6, 129.4, 129.2, 129.2, 128.1, 128.0, 127.9, 126.3, 126.2, 123.8, 123.3, 108.3, 79.3, 78.2, 78.1, 55.9, 55.6, 53.8, 49.0, 47.9, 47.7, 47.5, 46.8, 37.9, 37.5, 34.4, 28.2, 27.8, 12.0 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C37H46N9O9, 760.3418; found, 760.3417.
4.4.13. Peptide 9 (Boc-Phe-Phe-tz-aeg-OEt)
Alkyne 4 (0.20 g, 0.44 mmol) was reacted with ethyl(2-azidoethyl)glycinate38 (0.09 g, 0.50 mmol) following the method described for peptide 6a to afford peptide 9 (0.25 g, 89% with respect to 4), which was purified by column chromatography [eluent: 0–5% of MeOH in DCM]. Hygroscopic solid; 1H NMR (400 MHz, CDCl3, 25 °C, TMS): δ 7.43 (d, J = 8.9 Hz, 1H), 7.26–6.86 (m, 11H), 6.60 (s, 1H), 5.29 (s, 0.5H), 4.99 (s, 0.5H), 4.63 (dt, J = 14.1, 6.9 Hz, 1H), 4.47–4.19 (m, 5H), 4.09 (tt, J = 11.7, 5.7 Hz, 2H), 3.63 (s, 2H), 3.41 (s, 1H), 3.31 (s, 2H), 3.15 (t, J = 6.3 Hz, 1H), 3.08–2.75 (m, 5H), 1.23 (s, J = 5.5 Hz, 9H), 1.21–1.15 ppm (m, 3H); 13C NMR (100 MHz, CDCl3, 25 °C, TMS): δ 172.6, 171.6, 171.3, 171.2, 171.2, 170.8, 170.7, 155.8, 136.6, 136.3, 129.4, 129.4, 129.3, 128.8, 128.7, 128.6, 127.2, 127.0, 123.1, 80.6, 80.4, 61.0, 60.9, 56.6, 56.5, 56.1, 56.1, 55.9, 55.8, 55.0, 54.9, 54.4, 54.0, 53.9, 53.9, 52.0, 50.4, 49.3, 48.7, 38.2, 38.0, 37.7, 37.5, 35.2, 35.0, 28.3, 28.2, 14.3 ppm. HRMS (ESI+): m/z calcd for (M + H)+ C32H44N7O6, 622.3353; found, 622.3354.
4.5. Fourier Transform Infrared Spectroscopy
Fourier transform infrared (FT-IR) spectra were recorded from a FT-IR (Bruker alpha-E) spectrophotometer.
4.6. PXRD Study
The crystalline nature of the peptides was characterized by PXRD using a Bruker D8 Advance Series-II PXRD. Cu Kα radiation and a Lynxeye 1-D detector were used to record powder diffraction data. A goniometer was configured in theta–theta Bragg Brentano geometry optimized to obtain best intensity and resolution on bulk powder samples. The peptides were dried under vacuum before PXRD data were taken.
4.7. Dynamic Light Scattering
Dynamic light scattering (DLS) experiments was performed through a Nano ZS-90 apparatus (633 nm red laser beam at 90° angle) from Malvern instruments to find the size and dispersity of NP nanoparticles. The average size values are reported after at least three consecutive measurements.
4.8. Microscopy Studies
Microscopy experiments were performed to characterize the size of the self-assembled nanoparticles. Samples were prepared carefully to minimize the effect of aggregation. Then, 3–4 μL of peptide solutions was drop-casted on different substrates followed by drying under high vacuum [Formvar coated Cu grid for HRTEM, silicon wafer substrate for AFM, and field emission SEM (FESEM)]. FESEM images were recorded using Zeiss Ultra Plus scanning electron microscope after gold coating. AFM images were recorded using Agilent instruments. The imaging was carried out in tapping mode using a TAP-190AL-G50 probe from Budget sensors which had a nominal spring constant of 48 N/m and the resonance frequency of 190. HRTEM images were captured through a 1024 × 1024 digital CCD camera using a Tecnai-G220-TWIN microscope instrument.
SEM samples were prepared by drop-casting NP and control peptide solutions (3 μL, concentration: 1 mg/mL in 50:50 EtOH/H2O after lyophilization in 1 mL HFIP) on silicon wafers. Silicon wafers were then dried at rt for 2 days and imaged. Similarly, HRTEM samples were prepared by depositing NP solutions (3 μL, concentration: 1 mg/mL in 50:50 EtOH/H2O) on a copper grid, dried at rt for 1 day, and then imaged. For AFM, samples were drop-casted on freshly cleaved silicon wafers, air-dried at rt for 2–3 days. Tapping mode was applied for AFM imaging according to the reported procedures.
4.9. Thermogravimetric Analysis
Thermogravimetric analyses were carried out in a PerkinElmer STA 6000 simultaneous thermal analyzer. The exact parameters of the physical stability of self-assembled peptide vesicles were determined through TGA. The weight loss is monitored as a function of increasing temperature. Analysis was conducted isothermally for 30 min at rt followed by a scan rate of 5 °C/min up to 750 °C. Experiments were performed under dry condition and ultrahigh purity argon atmosphere.
4.10. CF Encapsulation Study
CF (1.3 mM) and doxorubicin (1.8 μM) solutions were added to the peptide vesicle solution of peptide 6a to make the final concentration of 0.1 mM and 0.2 μM, respectively, kept it overnight, and dialyzed with a 500 D cutoff dialysis tube (Float-A-Lyzer G2 Dialysis Device, Spectrum Labs). CF and doxorubicin-entrapped vesicles solutions were drop-casted on glass slides, dried, and then imaged using Carl Zeiss LSM-710 laser scanning confocal microscopy. A laser scanning confocal microscope reveals the green and red fluorescence emissions from the CF and doxorubicin-entrapped nanostructures, respectively, suggesting the encapsulation of dye molecules in the nanospheres.
4.11. Dicationic Dipeptide-Mediated Release
Porous peptide nanoparticles of 6a (200 μL) loaded with CF/doxorubicin was sealed in a dialysis membrane. Dicationic dipeptide (Boc-Lys-Lys-OMe) (200 μL, 5 mM) was then added to it. The dialysis bag was then suspended in agitating ethanol/water (1:1) solution. Aliquot (300 μL) of solution outside the dialysis bag was collected at different intervals for the quantification of released CF/doxorubicin. Dye/drug release measurement experiments were carried out using Fluorolog-3 HORIBA Jobin Yvon fluorescence fluorimeter, with 417 and 480 nm excitation and 437–750 nm emission range using 1/1 slit and 1 nm data interval.
4.12. CA Measurement
To check the hydrophobicity of the synthetic peptides, CA measurements were carried out. CAs on the peptide-coated silicon wafers were measured by using a contact angle meter (model ID: HO-IAD-CAM-01, Holmarc Opto-Mechatronics Pvt. Ltd.), which was followed by LBADSA drop analysis (ImageJ Software). For these measurements, a 2 × 2 cm2 uniform area of silicon wafers was accurately cut, washed with Milli Q water and methanol, and dried well. Uniform thin layers of NPs were prepared after slow drop-casting of 4 μL per droplet of samples (dissolved in CHCl3/MeOH) and dried well at rt. The CA measurements were recorded four consecutive times from different areas of the sample and captured at 0.5 s, following which the average CA value was calculated.
4.13. Turbidity Assay Experiment
Turbidity assay experiment was done in the 96-well microtest plate flat-bottom wells (Tarsons, CAT no. 941196) by using VARIOSKAN FLASH (Thermo scientific). Peptide samples were placed (400 μL, 1 mg/400 μL in 50:50 EtOH/H2O after lyophilization in 1 mL HFIP) in 96-well microtest plate flat-bottom wells (Tarsons, CAT no. 941196).
4.14. Electrochemical Analysis
Electrochemical analysis was done in the Biologic VMP3 multichannel potentiostat. A standard three-electrode cell consisting of a Toray carbon paper having 1 cm2 as the working electrode, Ag/AgCl (sat. KCl) as the reference electrode, and a Pt foil as the counter electrode were used for electrochemical measurements. The solutions were purged with Ar during the experiments. The potential values are in the standard hydrogen electrode scale.
All solutions were prepared using triple-distilled water and the cell temperature was maintained at 27 °C. Currents are normalized with respect to geometric area (1 cm2). All the electrolytic solutions were prepared by 0.1 M potassium phosphate buffer electrolytes at pH = 7.4 and 0.1 M KCl. Working electrodes were made by drop-casting a suspension of those peptides (2 mg/400 μL in a 10 wt % aq dispersion of polytetrafluoroethylene) on a Toray carbon paper electrode.
Acknowledgments
D.D. thanks the Department of Chemistry, Department of Physics, IISER Pune, and Savitribai Phule Pune University for instrumental facilities. D.D. is also thankful to DST SERB, India, for National Post Doctoral Fellowship (Project Code: PDF/2016/002510). M.K.G. thanks the University Grant Commission, New Delhi, for his fellowship.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsomega.9b00047.
Solution-phase synthesis, NMR spectra, IR spectra, ESI–MS data, HPLC traces, MALDI-Tof mass spectra, PXRD data, CA, transmission electron microscopy, SEM, AFM, EDAX, confocal images, DLS, UV, fluorescence, and CV data (PDF)
Author Contributions
† D.D. and O.T. contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Görbitz C. H. Nanotube formation by hydrophobic dipeptides. Chem.—Eur. J. 2001, 7, 5153–5159. . [DOI] [PubMed] [Google Scholar]
- Hartgerink J. D.; Beniash E.; Stupp S. I. Self-assembly and mineralization of peptide-amphiphile nanofibers. Science 2001, 294, 1684–1688. 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- Aggeli A.; Fytas G.; Vlassopoulos D.; McLeish T. C. B.; Mawer P. J.; Boden N. Structure and Dynamics of Self-Assembling β-Sheet Peptide Tapes by Dynamic Light Scattering. Biomacromolecules 2001, 2, 378–388. 10.1021/bm000080z. [DOI] [PubMed] [Google Scholar]
- Lee S.-W.; Mao C.; Flynn C. E.; Belcher A. M. Ordering of quantum dots using genetically engineered viruses. Science 2002, 296, 892–895. 10.1126/science.1068054. [DOI] [PubMed] [Google Scholar]
- Zhang S. Fabrication of novel biomaterials through molecular self-assembly. Nat. Biotechnol. 2003, 21, 1171–1178. 10.1038/nbt874. [DOI] [PubMed] [Google Scholar]
- Reches M.; Gazit E. Casting Metal Nanowires Within Discrete Self-Assembled Peptide Nanotubes. Science 2003, 300, 625–627. 10.1126/science.1082387. [DOI] [PubMed] [Google Scholar]
- Reches M.; Gazit E. Formation of Closed-Cage Nanostructures by Self-Assembly of Aromatic Dipeptides. Nano Lett. 2004, 4, 581–585. 10.1021/nl035159z. [DOI] [Google Scholar]
- Reches M.; Gazit E. Controlled patterning of aligned self-assembled peptide nanotubes. Nat. Nanotechnol. 2006, 1, 195–200. 10.1038/nnano.2006.139. [DOI] [PubMed] [Google Scholar]
- Adler-Abramovich L.; Reches M.; Sedman V. L.; Allen S.; Tendler S. J. B.; Gazit E. Thermal and Chemical Stability of Diphenylalanine Peptide Nanotubes: Implications for Nanotechnological Applications. Langmuir 2006, 22, 1313–1320. 10.1021/la052409d. [DOI] [PubMed] [Google Scholar]
- Tamamis P.; Adler-Abramovich L.; Reches M.; Marshall K.; Sikorski P.; Serpell L.; Gazit E.; Archontis G. Self-assembly of phenylalanine oligopeptides: insights from experiments and simulations. Biophys. J. 2009, 96, 5020–5029. 10.1016/j.bpj.2009.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azuri I.; Adler-Abramovich L.; Gazit E.; Hod O.; Kronik L. Why Are Diphenylalanine-Based Peptide Nanostructures so Rigid? Insights from First Principles Calculations. J. Am. Chem. Soc. 2014, 136, 963–969. 10.1021/ja408713x. [DOI] [PubMed] [Google Scholar]
- Berger O.; Gazit E. Molecular self-assembly using peptide nucleic acids. Peptide Sci. 2017, 108, e22930 10.1002/bip.22930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger O.; Adler-Abramovich L.; Levy-Sakin M.; Grunwald A.; Liebes-Peer Y.; Bachar M.; Buzhansky L.; Mossou E.; Forsyth V. T.; Schwartz T.; Ebenstein Y.; Frolow F.; Shimon L. J. W.; Patolsky F.; Gazit E. Light-emitting self-assembled peptide nucleic acids exhibit both stacking interactions and Watson-Crick base pairing. Nat. Nanotechnol. 2015, 10, 353–360. 10.1038/nnano.2015.27. [DOI] [PubMed] [Google Scholar]
- Avitabile C.; Diaferia C.; Della Ventura B.; Mercurio F. A.; Leone M.; Roviello V.; Saviano M.; Velotta R.; Morelli G.; Accardo A.; Romanelli A. Self-Assembling of Fmoc-GC Peptide Nucleic Acid Dimers into Highly Fluorescent Aggregates. Chem.—Eur. J. 2018, 24, 4729–4735. 10.1002/chem.201800279. [DOI] [PubMed] [Google Scholar]
- Berger O.; Yoskovitz E.; Adler-Abramovich L.; Gazit E. Spectral Transition in Bio-Inspired Self-Assembled Peptide Nucleic Acid Photonic Crystals. Adv. Mater. 2016, 28, 2195–2200. 10.1002/adma.201504160. [DOI] [PubMed] [Google Scholar]
- a Kinoshita S.; Yoshioka S. Structural colors in nature: The role of regularity and irregularity in the structure. ChemPhysChem 2005, 6, 1442–1459. 10.1002/cphc.200500007. [DOI] [PubMed] [Google Scholar]; b Tadepalli S.; Slocik J. M.; Gupta M. K.; Naik R. R.; Singamaneni S. Bio-Optics and Bio-Inspired Optical Materials. Chem. Rev. 2017, 117, 12705–12763. 10.1021/acs.chemrev.7b00153. [DOI] [PubMed] [Google Scholar]
- Nikoloudakis E.; Karikis K.; Han J.; Kokotidou C.; Charisiadis A.; Folias F.; Douvas A. M.; Mitraki A.; Charalambidis G.; Yan X.; Coutsolelos A. G. A self-assembly study of PNA–porphyrin and PNA–BODIPY hybrids in mixed solvent systems. Nanoscale 2019, 11, 3557–3566. 10.1039/c8nr05667f. [DOI] [PubMed] [Google Scholar]
- Li X.; Kuang Y.; Lin H.-C.; Gao Y.; Shi J.; Xu B. Supramolecular Nanofibers and Hydrogels of Nucleopeptides. Angew. Chem., Int. Ed. 2011, 50, 9365–9369. 10.1002/anie.201103641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.; Yi Kuang Y.; Shi J.; Gao Y.; Lin H.-C.; Xu B. Multifunctional, Biocompatible Supramolecular Hydrogelators Consist Only of Nucleobase, Amino Acid, and Glycoside. J. Am. Chem. Soc. 2011, 133, 17513–17518. 10.1021/ja208456k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y.; Pashuck E. T.; Thomas M. R.; Amdursky N.; Wang S.-T.; Chow L. W.; Stevens M. M. Plasmonic Chirality Imprinting on Nucleobase-Displaying Supramolecular Nanohelices by Metal-Nucleobase Recognition. Angew. Chem., Int. Ed. 2017, 56, 2361–2365. 10.1002/anie.201610976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serpell C. J.; Barłóg M.; Basu K.; Fakhoury J. F.; Bazzi H. S.; Sleiman H. F. Nucleobase peptide amphiphiles. Mater. Horiz. 2014, 1, 348–354. 10.1039/c3mh00154g. [DOI] [Google Scholar]
- Zhu X.; Zou R.; Sun P.; Wang Q.; Wu J. A supramolecular peptide polymer from hydrogen-bond and coordination-driven self-assembly. Polym. Chem. 2018, 9, 69–76. 10.1039/c7py01901g. [DOI] [Google Scholar]
- Datta D.; Tiwari O.; Ganesh K. N. New archetypes in self-assembled Phe-Phe motif induced nanostructures from nucleoside conjugated-diphenylalanines. Nanoscale 2018, 10, 3212–3224. 10.1039/c7nr08436f. [DOI] [PubMed] [Google Scholar]
- Tornøe C. W.; Christensen C.; Meldal M. Peptidotriazoles on Solid Phase: [1,2,3]-Triazoles by Regiospecific Copper(I)-Catalyzed 1,3-Dipolar Cycloadditions of Terminal Alkynes to Azides. J. Org. Chem. 2002, 67, 3057–3064. 10.1021/jo011148j. [DOI] [PubMed] [Google Scholar]
- Kolb H. C.; Finn M. G.; Sharpless K. B. Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed. 2001, 40, 2004–2021. . [DOI] [PubMed] [Google Scholar]
- a Angell Y. L.; Burgess K. Peptidomimetics via copper-catalyzed azide-alkyne cycloadditions. Chem. Soc. Rev. 2007, 36, 1674–1689. 10.1039/b701444a. [DOI] [PubMed] [Google Scholar]; b Pedersen D. S.; Abell A. 1,2,3-Triazoles in peptidomimetic chemistry. Eur. J. Org. Chem. 2011, 2011, 2399–2411. 10.1002/ejoc.201100157. [DOI] [Google Scholar]; c Tiwari V. K.; Mishra B. B.; Mishra K. B.; Mishra N.; Singh A. S.; Chen X. Cu-Catalyzed Click Reaction in Carbohydrate Chemistry. Chem. Rev. 2016, 116, 3086–3240. 10.1021/acs.chemrev.5b00408. [DOI] [PubMed] [Google Scholar]
- a Mayans E.; Casanovas J.; Gil A. M.; Jiménez A. I.; Cativiela C.; Puiggalí J.; Alemán C. Diversity and Hierarchy in Supramolecular Assemblies of Triphenylalanine: From Laminated Helical Ribbons to Toroids. Langmuir 2017, 33, 4036–4048. 10.1021/acs.langmuir.7b00622. [DOI] [PubMed] [Google Scholar]; b Brown N.; Lei J.; Zhan C.; Shimon L. J. W.; Adler-Abramovich L.; Wei G.; Gazit E. Structural polymorphism in a self-assembled tri-aromatic peptide system. ACS Nano 2018, 12, 3253–3262. 10.1021/acsnano.7b07723. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Kong J.; Yu S. Fourier transform infrared spectroscopic analysis of protein secondary structures. Acta Biochim. Biophys. Sin. 2007, 39, 549–559. 10.1111/j.1745-7270.2007.00320.x. [DOI] [PubMed] [Google Scholar]; d Khurana R.; Fink A. L. Do Parallel β-Helix Proteins Have a Unique Fourier Transform Infrared Spectrum?. Biophys. J. 2000, 78, 994–1000. 10.1016/s0006-3495(00)76657-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Haris P. I.; Chapman D. The conformational analysis of peptides using Fourier transform IR spectroscopy. Biopolymers 1995, 37, 251–263. 10.1002/bip.360370404. [DOI] [PubMed] [Google Scholar]; f Qian W.; Bandekar J.; Krimm S. Vibrational analysis of crystalline tri-L-alanine. Biopolymers 1991, 31, 193–210. 10.1002/bip.360310208. [DOI] [PubMed] [Google Scholar]; g Toniolo C.; Palumbo M. Solid-state infrared absorption spectra and chain arrangement in some synthetic homooligopeptides in the intermolecularly hydrogen-bonded pleated-sheet β-conformation. Biopolymers 1977, 16, 219–224. 10.1002/bip.1977.360160116. [DOI] [PubMed] [Google Scholar]
- Franken L. E.; Wei Y.; Chen J.; Boekema E. J.; Zhao D.; Stuart M. C. A.; Feringa B. L. Solvent Mixing To Induce Molecular Motor Aggregation into Bowl-Shaped Particles: Underlying Mechanism, Particle Nature, and Application To Control Motor Behavior. J. Am. Chem. Soc. 2018, 140, 7860–7868. 10.1021/jacs.8b03045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu C.; Gao Y.; Li H.; Meng S.; Li L.; Francisco J. S.; Zeng X. C. Characterizing hydrophobicity of amino acid side chains in a protein environment via measuring contact angle of a water nanodroplet on planar peptide network. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 12946–12951. 10.1073/pnas.1616138113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Baral A.; Roy S.; Dehsorkhi A.; Hamley I. W.; Mohapatra S.; Ghosh S.; Banerjee A. Assembly of an Injectable Noncytotoxic Peptide-Based Hydrogelator for Sustained Release of Drugs. Langmuir 2014, 30, 929–936. 10.1021/la4043638. [DOI] [PubMed] [Google Scholar]; b Basu K.; Baral A.; Basak S.; Dehsorkhi A.; Nanda J.; Bhunia D.; Ghosh S.; Castelletto V.; Hamley I. W.; Banerjee A. Peptide based hydrogels for cancer drug release: modulation of stiffness, drug release and proteolytic stability of hydrogels by incorporating D-amino acid residue(s). Chem. Commun. 2016, 52, 5045–5048. 10.1039/c6cc01744d. [DOI] [PubMed] [Google Scholar]
- Misra R.; Reja R. M.; Narendra L. V.; George G.; Raghothama S.; Gopi H. N. Exploring structural features of folded peptide architectures in the construction of nanomaterials. Chem. Commun. 2016, 52, 9597–9600. 10.1039/c6cc04502b. [DOI] [PubMed] [Google Scholar]
- Ingole T. S.; Kale S. S.; Santhosh Babu S.; Sanjayan G. J. Self-assembled vesicles of urea-tethered foldamers as hydrophobic drug carriers. Chem. Commun. 2016, 52, 10771–10774. 10.1039/c6cc05079d. [DOI] [PubMed] [Google Scholar]
- a Wang S. S.-S.; Chen Y.-T.; Chou S.-W. Inhibition of amyloid fibril formation of β-amyloid peptides via the amphiphilic surfactants. Biochim. Biophys. Acta 2005, 1741, 307–313. 10.1016/j.bbadis.2005.05.004. [DOI] [PubMed] [Google Scholar]; b Carny O.; Gazit E. Creating Prebiotic Sanctuary: Self-Assembling Supramolecular Peptide Structures Bind and Stabilize RNA. Life Evol. Biosph. 2011, 41, 121–132. 10.1007/s11084-010-9219-. [DOI] [PubMed] [Google Scholar]; c Zhao R.; So M.; Maat H.; Ray N. J.; Arisaka F.; Goto Y.; Carver J. A.; Hall D. Measurement of amyloid formation by turbidity assay-seeing through the cloud. Biophys. Rev. 2016, 8, 445–471. 10.1007/s12551-016-0233-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao K.; Makam P.; Aizen R.; Gazit E. Self-assembling peptide semiconductors. Science 2017, 358, 885. 10.1126/science.aam9756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler-Abramovich L.; Aronov D.; Beker P.; Yevnin M.; Stempler S.; Buzhansky L.; Rosenman G.; Gazit E. Self-assembled arrays of peptide nanotubes by vapour deposition. Nat. Nanotechnol. 2009, 4, 849. 10.1038/nnano.2009.298. [DOI] [PubMed] [Google Scholar]
- Beker P.; Koren I.; Amdursky N.; Gazit E.; Rosenman G. Bioinspired peptide nanotubes as supercapacitor electrodes. J. Mater. Sci. 2010, 45, 6374–6378. 10.1007/s10853-010-4624-z. [DOI] [Google Scholar]
- Conway B. E.Electrochemical Supercapacitors: Scientific Fundamentals and Technological Applications; Kluwer Academic/Plenum Publishers: New York, 1999. [Google Scholar]
- Efthymiou T. C.; Desaulniers J.-P. Synthesis and properties of oligonucleotides that contain a triazole-linked nucleic acid dimer. J. Heterocyclic Chem. 2011, 48, 533–539. 10.1002/jhet.532. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.