

Abstract
Biomolecular assembly is a key driving force in nearly all life processes, providing structure, information storage, and communication within cells and at the whole organism level. These assembly processes rely on precise interactions between functional groups on nucleic acids, proteins, carbohydrates, and small molecules, and can be fine tuned to span a range of time, length, and complexity scales. Recognizing the power of these motifs, researchers have sought to emulate and engineer biomolecular assemblies in the laboratory, with goals ranging from modulating cellular function to the creation of new polymeric materials. In most cases, engineering efforts are inspired or informed by understanding the structure and properties of naturally occurring assemblies, which has in turn fueled the development of predictive models that enable computational design of novel assemblies. This Review will focus on selected examples of protein assemblies, highlighting the story arc from initial discovery of an assembly, through initial engineering attempts, toward the ultimate goal of predictive design. The aim of this Review is to highlight areas where significant progress has been made, as well as to outline remaining challenges, as solving these challenges will be the key that unlocks the full power of biomolecules for advances in technology and medicine.
1. INTRODUCTION
Biomolecular assembly plays a central role in nearly all life processes, from short-lived microscopic events such as directing replication of the genetic code, translation of genes into proteins, and signaling within and between cells, to the construction of long-lived macroscopic architectures such as collagen networks and amyloid plaques.1 The ability of these interactions to operate on extraordinarily diverse scales of time, size, and complexity while maintaining exceptional levels of specificity is made possible by the rich chemical toolkit of interactions that are available to biomolecules, along with the ability to scaffold multiple interactions with precise location and directionality. Figure 1a highlights some of the more common interaction motifs utilized in biomolecular assembly, including hydrophobic interactions, π-stacking, cation-π interactions, hydrogen bonding, ionic bonding, and metal-ligand coordination.2 In Figure 1b, the example of the lac repressor is used to demonstrate how precise spatial orientation of these interactions gives rise to molecular recognition, which in turn drives assembly.
Figure 1.
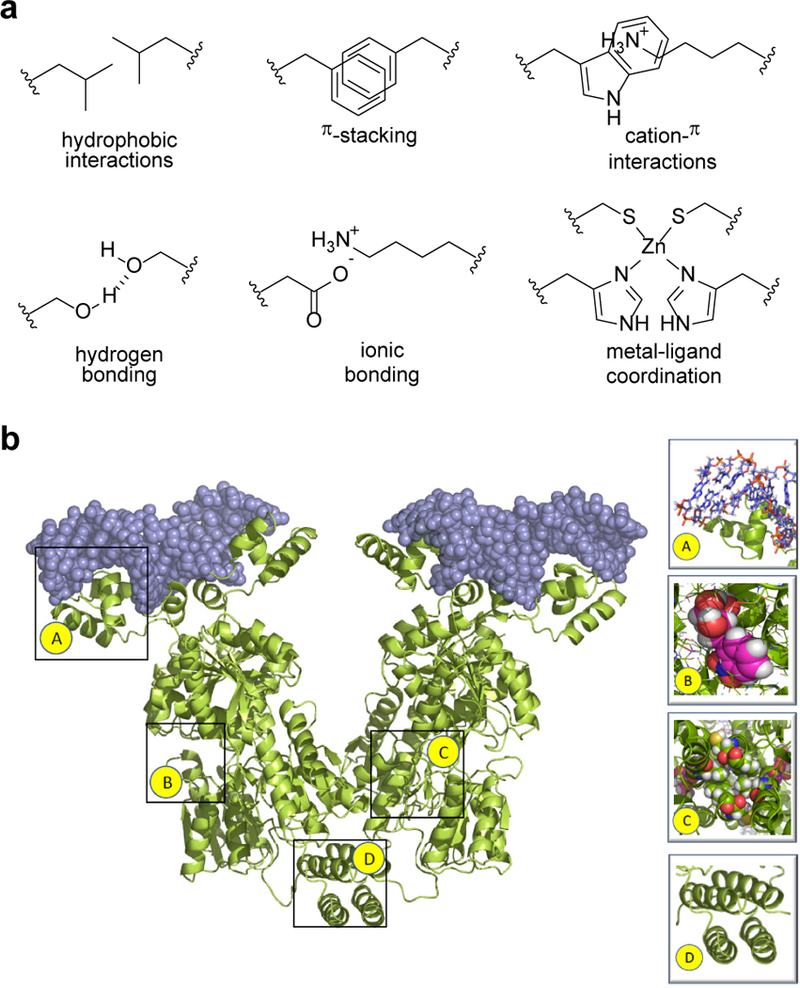
(a) Intermolecular interactions utilized in biomolecular assembly. (b) Lac repressor binding to target DNA, highlighting key interaction motifs: (A) electrostatic, (B) hydrogen bonding, (C) hydrophobic packing, (D) hydrophobic and electrostatic interactions.
Considering the power and versatility of biomolecular assembly, it is not surprising that scientists have long sought to harness these capabilities and redirect them for the creation of modified or synthetic architectures.3 These endeavors often follow the story arc shown in Figure 2 that begins with the discovery or elucidation of an assembly motif in nature, followed by engineering efforts intended to impart desired structures or properties needed for technological applications. Biomolecular engineering can be divided into two complementary strategies, rational design and laboratory evolution. Rational design is driven by hypotheses regarding the impact of changing the identity, location, or orientation of specific interactions along the assembly interface4 and three primary strategies can be employed: (i) intuitive design, (ii) modular design, and (iii) computer-aided design (or computational design). Intuitive design is a form of knowledge-based design, in which the user leverages existing structural and functional knowledge for a given assembly to guide site-specific changes in the scaffold aimed at conferring new properties. Modular design is also a knowledge-based approach, but involves combining existing motifs to create new assemblies that take advantage of the known properties of the modules. Computer-aided design is analogous to civil engineering, but at the atomic level. Using computer-aided design, a “blueprint” of a biomolecule is generated via a molecular-mechanics type model. The designed molecule is then synthesized either biologically or chemically, and characterized structurally and functionally. Computer-aided design typically requires structural knowledge; however, de novo design is possible on a small scale. In cases where the structure or properties of a biomolecule are not sufficiently well-understood to enable rational design, laboratory evolution offers a powerful approach to engineering. Laboratory evolution employs stochastic mutation strategies to generate a large number of variants, and those having the desired properties are identified by screening or possess a survival advantage in the presence of selection pressure.5 Generating variants for laboratory evolution can be achieved via: (i) random mutagenesis or (ii) recombination. Random mutagenesis is typically accomplished using error-prone PCR (EP-PCR) to generate a large library of DNA sequences having single-point mutations, which are then translated into a similarly diverse set of protein variants. Recombination, or “DNA shuffling,” also generates a large sequence space, but instead involves fragmentation followed by reconstruction via overlap extension PCR. This allows fragments of genes (and thus proteins) to be recombined to produce large combinatorial sets. While these engineering efforts can yield success, often the complexity of folding or recognition leads to unintended outcomes, requiring iteration of the design. In these cases, engineering approaches may appear inefficient or at times even futile. However, the information gleaned from these experiments can be used to formulate models for assembly, which in turn can be applied toward the ultimate goal of de novo predictive design of biomolecular assemblies.
Figure 2.
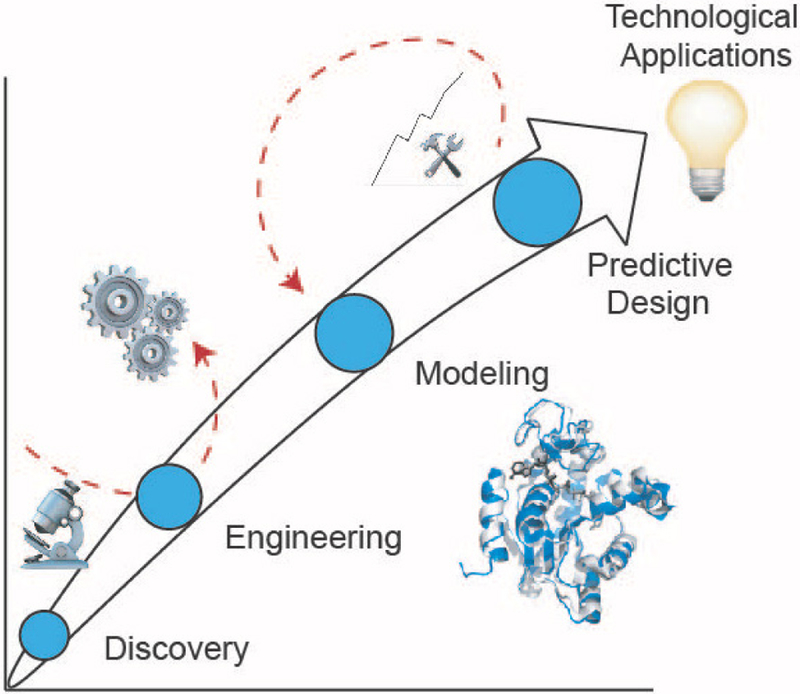
Story arc from initial discovery of an assembly to predictive design. Red arrows depict iteration and the availability of different on-ramps and off-ramps in this process.
For the purposes of simplicity, Figure 2 depicts a linear story arc from discovery to predictive design to technological applications. However, this path can often be more complex. Knowledge from one assembly motif can serve as an on-ramp to the later stages of engineering and design for a different assembly motif. Additionally, iteration between steps is often required to achieve the desired outcome. For example, analyzing the properties of a designed assembly may reveal new information that can be added to the computational model, in turn producing an improved design. We also highlight that while many computational efforts focus on structure, the mechanism and kinetics of assembly also represent important challenges for modeling and predictive design.
A diversity of biopolymer structures exist in nature, primarily comprised of nucleic acids, peptides/proteins, and oligosaccharides. In the case of nucleic acids, assembly is largely governed by Watson-Crick complementarity, and computational tools are already available to design sophisticated assemblies.6 The field of nucleic acid nanotechnology has been extensively reviewed elsewhere,7–9 and thus will not be a focus of the current Review. Oligosaccharides, on the other hand, present a complexity of structures and interactions that researchers are just beginning to grasp. The challenges of studying and engineering this class of molecules are compounded by the lack of universal and robust approaches for their chemical synthesis, though significant progress has been made in this area,10 and the future of this field promises to reveal a wealth of information regarding biomolecular structure and function. Between these two extremes are peptide and protein assemblies, which have been the topic of extensive study, but have yet to yield fully to predictive design. For this reason, we have chosen to focus this Review primarily on peptide- and protein-based assembly motifs, while occasionally exploring their interactions with other biomolecular scaffolds.
While nucleic acids and oligosaccharides are almost universally soluble under biological conditions, the chemical diversity of the amino acid sidechains enables nature to exploit solubility as a means to control the structure and function of peptides and proteins. Actin serves as an excellent example of this, as it exists as a soluble monomer but assembles to form insoluble filaments. These filaments comprise the cytoskeleton, which is critical to a number of cellular processes including motility and division. Moreover, a number of soluble actin-binding proteins can interact with either the soluble or insoluble forms of actin to form, remodel, or disassemble the cytoskeleton.11 The actin example highlights another parameter for characterizing peptide and protein assembly: the similarity of the assembly partners. While most insoluble assemblies (e.g. actin, amyloid) are formed through self-assembly of identical peptides or proteins, biomolecules can also form hetero-assemblies comprised of different peptide or protein subunits. Examples of these include transcription factors12 and signaling complexes,13 which are typically formed from soluble proteins and may be characterized by a wide range of assembly lifetimes. Throughout this Review, we will highlight diverse examples of assemblies that vary along these parameters of solubility and self- versus hetero-assembly.
In the chapters of this Review, we will first explore the mechanics of peptide and protein assembly, which provides a foundation for understanding the process of rational modification, modeling of results, and predictive design. We will then trace the story arc from first discovery to predictive design across multiple types of biomolecular assemblies, including: designer peptides, virus-like particles and other nanoscale protein assemblies, transcription factor complexes, macroscale protein scaffolds, and amyloids and other pathogenic protein aggregates. These examples are not only inspired from biomolecular assemblies found in nature, but share the characteristic of spanning diverse time, size, and complexity scales. We will conclude with a discussion of recent progress in predictive design and a vision for the broader impact that might be realized by advances in this area.
2. MECHANISMS OF NUCLEATION AND PROPAGATION IN BIOPOLYMER CONDENSATION AND ASSEMBLY
2.1. The “Folding Funnel” Model
Proteins are remarkable materials having exceptional structural diversity and catalytic activity,14 and these properties can be harnessed for applications in materials science and bioengineering. For many years, the prevailing paradigm stated that proteins must fold into a specific conformational state to become functionally active.15,16 However, the relatively recent discovery of intrinsically disordered proteins (IDPs) has challenged that paradigm by providing numerous examples of proteins that perform specific biological functions despite existing in a disordered state such as molten globular or random coil.17–20 These proteins pose an intense challenge to the predictive design of biomolecular assembly, which should be viewed as an exciting target for future methods development. However, given the current state of protein assembly design, we will focus this Review on peptides and proteins that adopt defined, folded states.
During the folding process, a protein samples many local intermediate states, some of which are kinetically trapped, in a process that can be generically illustrated by the “folding funnel” (Figure 3).21 The high free energy of the unfolded protein allows it to access many possible intermediate or misfolded states. However, as the protein folds, the number of possible configurations decreases and the barriers to less stable structures increase, such that the funnel width shrinks and converges on the evolved state. At higher concentrations, proteins can condense into assemblies that include amyloids, silks, collagens, and elastins, or co-assemble with other polymers to give actins, tubulins, ribosomes, and nuclear pores. The need to understand the structures of these folded proteins and intermolecular condensates has driven the development of diverse macromolecular spectroscopic methods,22 X-ray crystallography,23,24 and most recently high resolution cryo-EM.25,26 Computational methodologies to systematically predict folded protein structures have also improved;27,28 however, predictive models for biopolymer condensation remain less well developed.
Figure 3.
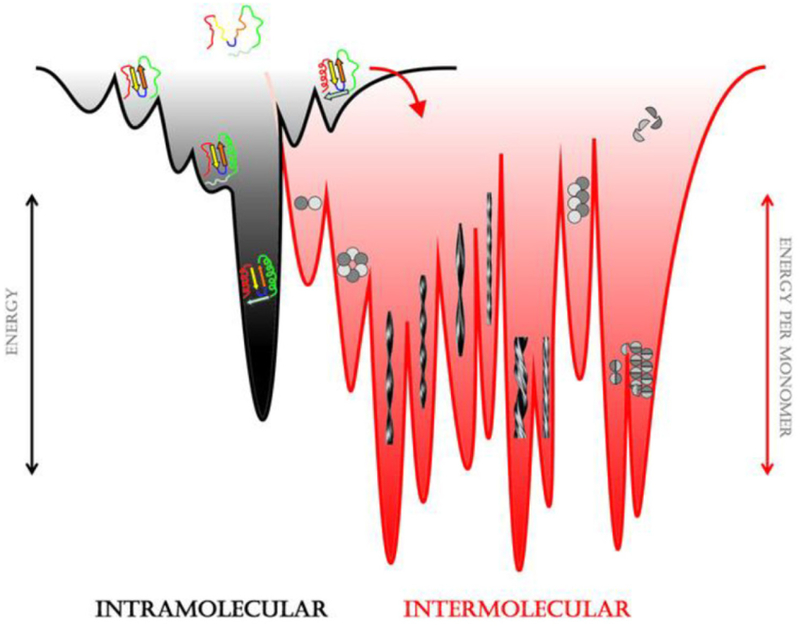
The protein folding funnel. The curved red arrow highlights the transition between protein folding and protein assembly.21 Reprinted with permission from Eichner, T.; Radford, S. E. A diversity of assembly mechanisms of a generic amyloid fold. Mol. Cell 2011, 43, 8–18. Copyright 2011 Elsevier.
2.2. Amyloid as a Model for Intermolecular Condensation and Assembly
The forces that drive intramolecular folding and intermolecular condensation include hydrophobic effects, electrostatic attraction and repulsion, van der Waals interactions, π–π stacking, and hydrogen bonding.2 The primary difference between intramolecular folding and intermolecular condensation, which causes the latter to pose a significantly greater computational challenge, is the difficulty of precisely defining the composition of a stable nucleus and the induction period for its formation.29 Driven by the need to understand protein misfolding diseases, our current knowledge of intermolecular association mechanisms continues to emerge primarily from amyloid structure. Thus, we will utilize amyloid in this Chapter as a model for describing the mechanisms of intermolecular assembly. Importantly, these mechanisms are applicable to the other peptides and proteins that are covered throughout this Review.
Interestingly, amyloid can possess multiple energy minima, giving rise to a diverse series of energetically accessible conformations or paracrystalline polymorphs. As a result, precisely defining the composition of a stable nucleus and the induction period for its formation have remained challenging for both experimental and computational analyses.30 Classic nucleation theory (CNT) has been foundational for defining nucleation processes generally, but recent improvements in spectroscopic and computational analyses have shown that biomaterials generally condense through non-classical nucleation processes,31–33 and these methods often fail to account for the complexities of biomolecular assembly and condensation.
In the assembly process, intermediate metastable phases are formed through liquid-liquid or liquid-solid phase transitions, and provide a favorable environment for subsequent nucleation of crystalline structures or phases. However, the assembly pathways for the intermediate nucleation, sequential phase transitions, and the subsequent propagation mechanisms for lower free energy crystalline phases remain ill-defined. In the sections below, we highlight critical spatial and temporal components that regulate nucleation and propagation of protein condensation. We also use these systems to define general models for the transition of different structures along the condensation pathway. Achieving a deeper understanding of these processes is critical to the development of predictive design tools for non-classical assemblies, which will in turn will advance the many applications of biomolecular assemblies.
2.3. Nucleation Mechanisms
2.3.1. Classical Nucleation.
Classical, or single-step, nucleation assumes the condensation occurs directly from free monomers in solution (Figure 4a). To achieve a stable nucleus, a critical number of peptides are required to overcome the liquid-liquid or liquid-solid interfacial tension, where nucleation is possible only when the free peptide concentration is above a critical value. Both the critical nucleus size and the critical peptide concentration may be approximated with CNT,34,35 which was initially derived to describe the formation of crystal nuclei from supersaturated liquid vapor,34–37 and later applied to amyloid peptide condensation.38–42 The free energy changes for nucleation in CNT are positive, and thus nucleation occurs only because the stochastic fluctuation of the local peptide concentration overcomes the activation energy for a condensation event. The minimum concentration of monomers required for this process to occur is referred to as the critical concentration. While formation of a nucleus is energetically disfavorable due to the high entropic cost, further growth of that nucleus is favorable due to enthalpic contributions. Describing this balance is the critical nucleus size, as nuclei with sizes smaller than this critical value will dissolve, while any larger nuclei remain stable and undergo further growth or propagation (Figure 4a).
Figure 4.
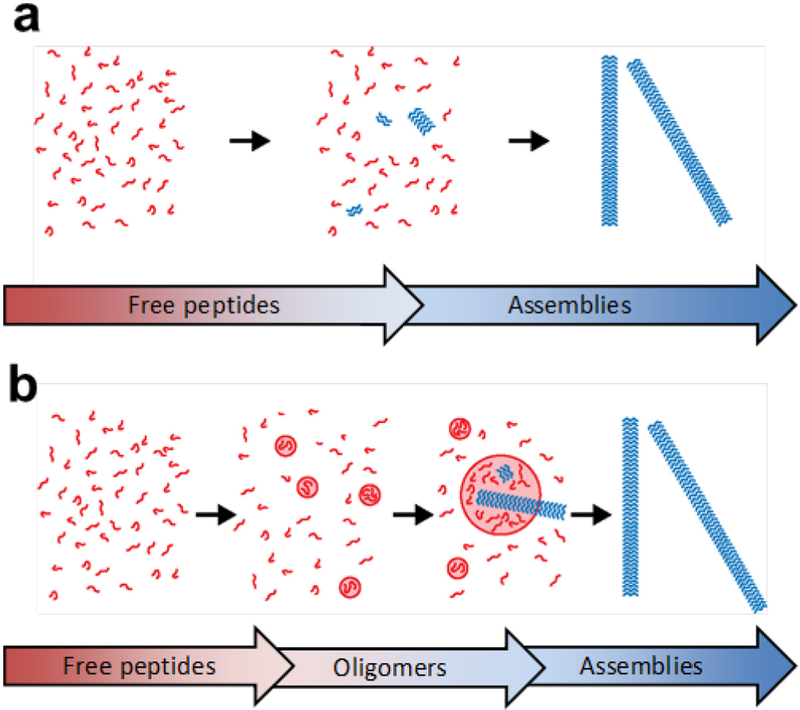
(a) Peptide assembly by classical nucleation. Monomers aggregate directly into structured assemblies and only assemblies above a critical size are propagated. (b) Peptide assembly by non-classical, two-step nucleation. Monomers first undergo liquid-liquid phase separation to form oligomeric particles, which then transition into structured assemblies. Reprinted with permission from Hsieh, M. C.; Lynn, D. G.; Grover, M. A. Kinetic Model for Two-Step Nucleation of Peptide Assembly. J. Phys. Chem. B 2017, 121, 7401–7411. Copyright 2017 American Chemical Society.
Molecular dynamics (MD) simulations have been used to visualize the stabilization of nuclei at an atomic level during the initial stages of condensation. The critical nucleus sizes of some peptides have been defined by these simulations,39,43–48 and some of these assignments have been supported by experimental results.48–52 Hydrophobic forces, electrostatic interactions, and aromatic stacking interactions between hydrophobic residues all appear to play a critical role in the assembly.53,54 For example, removing electrostatic interactions from Aβ(16–22) peptide, the nucleation core of the Aβ peptide K16LVFFAE22 of Alzheimer’s disease (AD), appear to destabilize the assembly nucleus, and these mutants are less likely to propagate into assemblies.53 Additionally, valuable structural information for the nuclei is now available from MD simulations, including structural rearrangements55 and parallel/antiparallel β-sheet transitions.56,57
Although many peptide assemblies do not follow classical nucleation theory, complex nucleation processes can (in some cases) be condensed into a simple rate constant for the sake of developing practical kinetic models. These one-step nucleation models have been successfully applied to fit and explain the growth curves of a variety peptide assembly and protein aggregation processes.58–61 Despite the assumptions made in simplification to a classical nucleation model, this approach is still capable of fitting the kinetics of assemblies having lag phases, which arise from the slowest rate-determining nucleation steps.61,62 Morris and coworkers have proposed a minimal model (Scheme 1) that defines the complex nucleation step with one rate constant (kn). Together with the autocatalytic growth step (ke), this model is capable of describing the assembly growth profiles of multiple peptides (Figure 5), including those with apparent lag phases.62
Scheme 1.
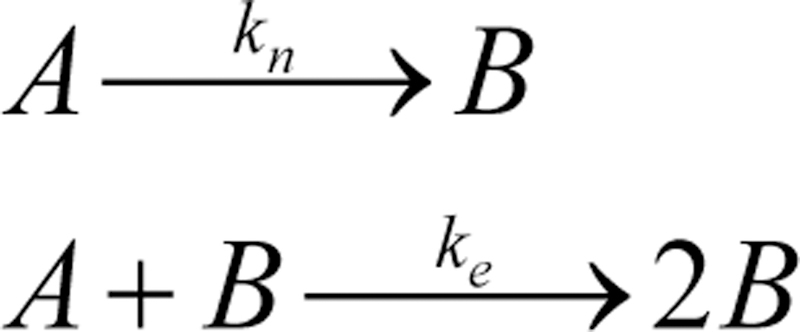
The Finke-Watzky mechanism of nucleation followed by autocatalytic growth [35]. A is the unassembled free peptide, which nucleates into the assembled peptide B with rate constant kn. The unassembled A may also undergo autocatalytic reaction (ke) to produce the assembled
Figure 5.
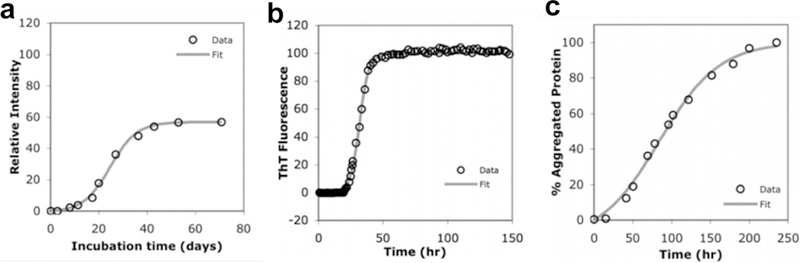
Assembly of (a) amyloid β, (b) α-synuclein and (c) polyglutamine peptides kinetically fit using the Finke-Watzky mechanism (Scheme 1).62 This simplified mechanism is capable of fitting assemblies having lag phases. Reprinted with permission from Morris, A. M.; Watzky, M. A.; Agar, J. N.; Finke, R. G. Fitting Neurological Protein Aggregation Kinetic Data via a 2-Step, Minimal/”Ockham’s Razor” Model: The Finke-Watzky Mechanism of Nucleation Followed by Autocatalytic Surface Growth. Biochemistry 2008, 47, 2413–2427. Copyright 2008 American Chemical Society.
2.3.2. Non-Classical Nucleation.
The CNT models described above assume that condensates are directly formed in solution without precursors or intermediates. However, this single-step nucleation may be prevented by the strong desolvation energy barriers required to overcome the interfacial tension for the nucleus.63,64 In these cases, non-classical, or two-step nucleation models propose that liquid-liquid phase separation occurs first to give oligomeric particles, which can then undergo transition to more ordered assemblies (Figure 4b). Based on Ostwald’s rule of stages,65 the less stable phase nucleates first through a lower energy barrier and sets the environment for nucleation of the more ordered phases. For peptide assembly, the peptides may form metastable intermediates that help the final assembly to nucleate,38 instead of generating the final structure directly from the free peptides. In this way, assemblies that follow the two-step model circumvent the strong desolvation energy barrier associated with direct nucleation.
The crystallization process for many materials, including some proteins,31,32,66 minerals,67 and colloids68 preferentially follows non-classical nucleation mechanisms. Even for a simple peptide condensation, the TEM images shown in Figure 6 indicate that oligomeric particles appear prior to the final ordered structures, consistent with two-step non-classical assembly pathway for Boc-FF,65 KLVFFAE,69,70 and KLVFFAQ.71,72 The peptide VEALYL also aggregates via a dense liquid phase before transforming into stable fibers.73 Large proteins can also assemble via non-classical nucleation mechanisms. For example, deoxy sickle cell hemoglobin initially forms dense protein droplets,74 and as detailed in Chapter 7, the yeast prion Sup35 forms an initial intermediate phase during assembly.75 Simulations of the condensation energies of these dehydrated, disordered, and metastable oligomeric intermediate phases support non-classical nucleation mechanisms,76 and most recently, low dimensionality coarse grain protein models predict low-energy initial particle condensation of α-synuclein, a critical initiating event in Parkinson’s disease.77
Figure 6.

Two-step nucleation mechanism for (a) Boc-FF,65 (b) Aβ(16–22),70 and (c) Aβ(16–22)E22L peptides.71 Peptide particles are observed before the emergence of ordered fibers. Scale bars = 100 nm for (b) and 200 nm for (c). Reprinted with permission from Levin, A.; Mason, T. O.; Adler-Abramovich, L.; Buell, A. K.; Meisl, G.; Galvagnion, C.; Bram, Y.; Stratford, S. A.; Dobson, C. M.; Knowles, T. P.; Gazit, E. Ostwald’s rule of stages governs structural transitions and morphology of dipeptide supramolecular polymers. Nat. Commun. 2014, 5, 5219. Copyright 2014 Springer Nature. Reprinted with permission from Hsieh, M. C.; Lynn, D. G.; Grover, M. A. Kinetic Model for Two-Step Nucleation of Peptide Assembly. J. Phys. Chem. B 2017, 121, 7401–7411. Copyright 2017 American Chemical Society. Reprinted with permission from Liang, C.; Ni, R.; Smith, J. E.; Childers, W. S.; Mehta, A. K.; Lynn, D. G. Kinetic intermediates in amyloid assembly. J. Am. Chem. Soc. 2014, 136, 15146–15149. Copyright 2014 American Chemical Society.
Kinetic models for assembly have generally focused on the nucleation rate of the final assemblies from the intermediate metastable condensate;31,32,69,78,79 however, the reversible growth of the metastable oligomer phase is crucial to assembly nucleation kinetics. Temperature, mass, and viscosity of the metastable intermediates have also proven to be critical factors contributing to the rate-limiting step for nucleation of the ordered assembly. Additionally, models have been developed in which the sizes of the individual oligomeric phases dictate the assembly nucleation rate (Scheme 2).38,78,80 To demonstrate the importance of the nucleation rates, Auer et al. compared the one-step and two-step nucleation models for assembly of amyloid fibrils, and concluded that while two-step nucleation best explains the observed assembly kinetics, one-step nucleation may contribute to the nucleation kinetics in some assemblies.38
Scheme 2.
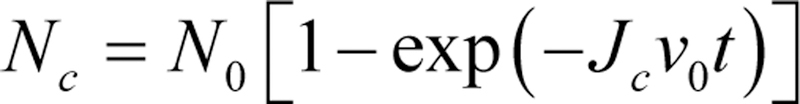
Number of nuclei (Nc) as a function of the volume (v0) of individual intermediate particles. N0 is the number of particles, Jc is the nucleation rate, and t is the reaction time.78
2.4. Propagation and Secondary Nucleation
After stable nuclei emerge, growth occurs through the process of templated propagation. The termini of these assemblies have been modeled as organizing free peptides through a “dock and lock” mechanism46 in which free peptides are initially “docked” loosely onto the assembly ends in a condensate much like the initial oligomer, and become “locked” when they adopt the conformation of the template. Several factors contribute to the propagation rate, including temperature,81 pH,82 and free peptide concentration.61 However, in some cases the rate may plateau or become concentration-independent when the peptide concentration is greater than a threshold value. Under these conditions, propagation is controlled by the rate-limiting structural rearrangement required in the “lock” step.83 Although most propagating species have low solubility and undergo irreversible propagation,42,84 reversible elongation can be observed when peptide “docking” to the template is weak.59,85,86
The density of nuclei termini also impacts elongation kinetics, and additional sources of nuclei can greatly enhance propagation rates.60,87 For example, fiber fragmentation can occur when the assemblies are weak and vulnerable to shear forces, generating additional termini which can undergo propagation.48,88 Similarly, the concentration of active termini may be increased artificially by seeding the peptide solution with pre-formed assemblies.89 The number of nuclei may also increase via surface nucleation, in which the assembly surface templates the formation of new nuclei. While fragmentation is a monomer-independent process, surface nucleation is a monomer-dependent process, leading to different kinetic signatures for these two processes.59
2.5. Kinetic Models for Nucleation and Assembly
Kinetic models have now been constructed for assembly pathways including particle formation, paracrystallization, propagation, and the conformational transitions leading to the final state. In some of these models, micelles can be used as a surrogate for the intermediate particle where crystallization occurs.84,90 Sauter et al. proposed a two-step nucleation model to address the kinetic conformational transition through both metastable and final phases.32 Most recently, Hsieh et al. proposed a model to describe the development of linear fiber assemblies that nucleate inside of spherical oligomer colloids, but grow differently in solution.70 This model not only provides insight into how the metastable species transition into ordered assemblies, but also simulates possible final states of this multi-step process. In the model, the intermediate phases may either (i) dissolve as the assembly phases accumulate;64,82 (ii) coexist with the final assemblies;91,92 or (iii) remain stable and predominate in the system if final assembly nucleation is extremely slow. The nature of the combined kinetic constants determines which of these three options is most likely to occur.82,93,94 Although this model is designed for transitions from spherical intermediates to linear assembly phases, it can be modified to enable investigation of other systems following similar two-step nucleation processes, including drug delivery systems having a coacervate-vesicle transition95 or Boc-FF assembly with particle-fiber-tube transitions.65
2.6. Structural Evolution of Assemblies
The final structure of an assembly may be significantly different from the structure of the initial nucleus, or even that of the propagated assembly. This complexity is significantly impacted by the nucleation mechanism. Under the single-step nucleation mechanism, the assembly structure is inherited from direct nucleation events in solution. Although the kinetically selected nuclei could give rise to structurally different assemblies, pathways for this to occur are limited by the fact that all nuclei exist in the solution phase. In contrast, in the two-step nucleation mechanism, the oligomeric phases may serve as distinct desolvated microenvironments having high peptide concentration, which may favor the nucleation of kinetic products that are inaccessible in the solution phase. Since these products may not be thermodynamically stable upon entering the solution phase, they may undergo further structural transitions before forming the final assembly.69,71
This process of structural evolution has been observed with many different peptides. The AD associated peptide Aβ(16–22) forms antiparallel out-of-register ribbons from the initial particles, but later transitions into in-register fibers (Figure 7), which are stabilized by electrostatic cross-strand pairing between the positively charged lysines and the negatively charged glutamic acids.69 The congener of Aβ(16–22), the Dutch mutant Aβ(16–22)E22Q or Ac-KLVFFAQ-NH2, follows similar assembly pathways, initially assembling in the particles as anti-parallel out-of-register ribbons, but later transitioning into parallel in-register fibers (Figure 7c). Somewhat conversely, the BAM β-sheet mimetic peptide initially forms an anti-parallel out-of-register β-barrel having alternating strong and weak interfaces, but over time, the weak interface opens and re-closes to yield flat β-sheets within anti-parallel out-of-register fibers.96 In some cases of structural evolution, the final assembly is present in the early stages as a minor component, and becomes predominant over time through conversion of the less stable assemblies. For example, the Aβ(1–42) peptide initially condenses as both parallel and anti-parallel hairpins, but the anti-parallel hairpins diminish with time and the final mature fibers consist only of parallel β-sheets.97
Figure 7.
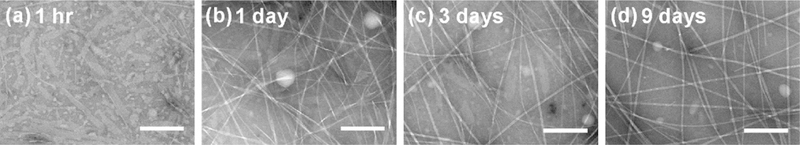
Morphological evolution Aβ(16–22) peptide assemblies from 1 h to 9 days. Ribbon intermediates are initially observed, but are later replaced by fibers.69 Reprinted with permission from Hsieh, M. C.; Liang, C.; Mehta, A. K.; Lynn, D. G.; Grover, M. A. Multistep Conformation Selection in Amyloid Assembly. J. Am. Chem. Soc. 2017, 139, 17007–17010. Copyright 2017 American Chemical Society.
As described above, a variety of mechanisms may contribute to the structural evolution of peptide assemblies. One mechanism for this is shown in Figure 8a, where structural mutations at the assembly termini occur when the incoming peptide adopts a more stable configuration (green) than the original template (blue). This templating mechanism has been observed within a single assembled peptide fiber,98 and it is hypothesized that the Aβ(16–22) and Aβ(16–22)E22Q assemblies also exhibit structural transition due to mutation at the assembly termini.69,71 Another possible mechanism for structural evolution is surface nucleation, in which the surface of the intermediate assemblies (blue) can serve as a nucleation site for the formation of new nuclei, allowing different nuclei (green) to emerge and even dominate the phase network (Figure 8b). Watanabe-Nakayama et al. observed that in the case of Aβ(1–42) peptide, a new structural species emerges from surface nucleation on the existing assemblies and later fuses with its parental fiber, resulting in wider fibers in the final assembly.99
Figure 8.
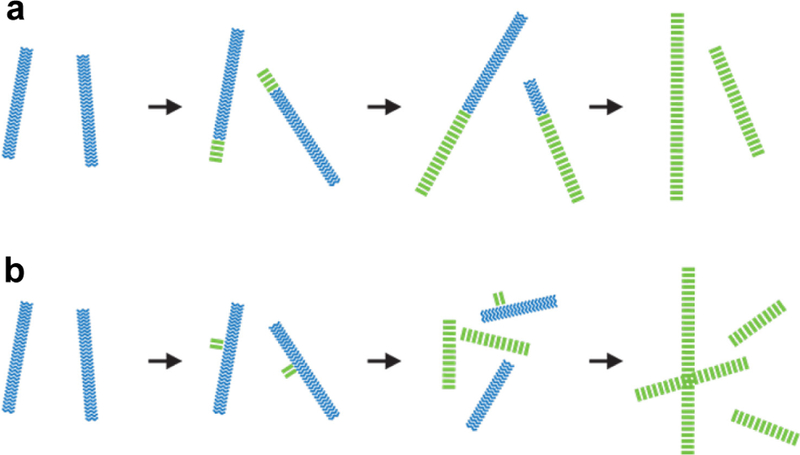
Structural evolution arising from (a) configuration mutation at the assembly termini and (b) surface nucleation. Blue assemblies represent the kinetic intermediates and green assemblies represent the thermodynamically stable final assembly state.
Clearly the environmental conditions which favor conformational mutations and secondary nucleation events contribute significantly to polymorphism and structural evolution of these dynamic, low-dimensional crystalline phase networks. Fortunately, many atomic-resolution biophysical methods to characterize these processes are emerging, including solid-state NMR, cryo-EM, and oriented diffraction. While these approaches require frozen or dried samples, and thus do not fully capture the dynamic evolution process, significant progress is being made to determine the energetic folding codes for peptide and protein condensation. This is in turn anticipated to unlock new potential for the prediction, control, and design of peptide and protein assembly.
2.7. Conclusion and future perspective
This Chapter outlines models for understanding and recapitulating the mechanisms for nucleation and propagation of peptide and protein self-assembly. We specifically highlight non-classical processes including phase changes, two-step nucleation, surface nucleation, and propagation mutation as mechanisms that access diverse areas of the condensation energy landscape and enable assembly of new functional mesoscale structures. Recent discoveries increasingly reveal the dynamic complexity of protein assembly, emphasizing the need for more comprehensive simulation models and predictive approaches as we strive to achieve functional specificity. From the disease-associated amyloids outlined in Chapters 3 and 7, to the framework silks, collagens, elastins, transcription factors, and viral coat proteins outlined in Chapters 4–6, there are common elements underlying the assembly mechanism that can now be exploited to create new functional assemblies and expand that function into alternative environments.
Understanding single-component self-assembly pathways and mechanisms is a critical first step toward engineering and predictive design of protein- and peptide-based assemblies. Compared to DNA structures, which may be precisely predicted by Watson-Crick pairing, protein folding and peptide assembly are affected by many forces as outlined in Chapter 2.2. These forces contribute to the complex reaction pathway for intramolecular folding and intermolecular condensation, and thus, as outlined in Chapter 3.3 and 3.4, make any precise predictions for the final structure challenging. Also, many of the possible polymorphic structures and the kinetic intermediates on the assembly pathway remain to be clearly defined, further complicating these predictions.
The perspective developed for the folding pathway in Chapter 2 is not only important for single component systems, but also for co-assembly of different polymers, as highlighted in the more complex architectures of actins, tubulins, ribosomes, transcriptional complexes, and nuclear pores. We remain encouraged by the biophysical methods now available for determining the 3-dimensional structures of peptide and protein assemblies. These methods not only reveal the molecular details of the final assemblies, but also provide insight into the energetic codes for condensation and assembly, generating constraints for learning algorithms necessary for robust predictive design of new complex materials.
3. DESIGNER α-HELICAL AND β-STRAND PEPTIDE ASSEMBLIES
3.1. Introduction to Designer Peptide Assemblies
While the assembly of higher order nucleic acid structures is dominated by the energetics of base-pairing, providing a digital code for storing information and structure design, protein backbones contain many interactions whose force constants are context dependent and change along the folding pathway (see Chapter 2 above). This analog-like folding behavior of proteins allows their functions to be far more environmentally responsive, enabling critical adaptations in evolution.100 Nevertheless, in the hierarchical folding process, small peptides having α-helical or β-strand secondary structures are capable of assembly to form nanoscale architectures, and here we review progress toward the rational molecular design of these structures.
The central design challenge in this field is to formulate peptide primary structures that will produce predetermined secondary structures, specific intermolecular arrangements within nanoscale structures, and specific mesoscale interactions in protein and mixed biopolymer assemblies. The peptides investigated by recent studies are usually composed of fewer than 50 amino acids and are likely to be characterized by a single secondary structure. In this molecular size range, intermolecular interactions contribute significantly to molecular conformational stability, such that peptide folding and assembly are closely related. The design challenge bears some resemblance to the protein folding problem, which aims to predict detailed 3-dimensional molecular structures of folded proteins based on their primary structures; the mechanistic connection between peptide assembly and protein folding is discussed in Chapter 2 above. The design challenge of peptide assembly similarly relates to efforts to predict structures of naturally-occurring networks such as amyloid fibrils, which are the topic of Chapter 7 in this Review. The scope of our discussion in this Chapter will include not only self-assembling peptides, but also binary systems of complementary peptides that form nanostructures through co-assembly. While some rational design effort has been applied to peptides that do not form α-helices or β-strands, the majority of work on rational design of assembling peptides has focused on α-helical coiled-coil systems and amyloid-like β-sheet forming peptides. As we explore this topic, we will build on what is known in natural systems to set the ground work for the de novo design of assembling polypeptides.
Figure 9 illustrates the different levels of molecular structure that can be controlled for directed peptide assembly. Peptide primary structure refers to the amino acid sequence along the polypeptide backbone. Ultimately, this sequence is a central target of our design challenge. Peptide secondary structure (most dominantly β-strand or α-helix) describes the conformation of the peptide backbone and the 3-dimensional arrangement of sidechain groups (Figure 9a). Single β-strands and α-helices are domains of uniform secondary structure within a single molecule, but each molecule may contain more than one secondary structural domain potentially in dynamic exchange. Nanostructured assemblies are established when multiple secondary structural units organize to form structures such as cross-β assemblies or α-helical coiled coils (Figure 9b). This level of structure is likely to require 2-step nucleation of peptide assembly, as highlighted in Chapter 2, and once nucleated, these minimal supramolecular structural units often undergo further assembly (e.g., stacking of β-sheets, bundling of coiled coils), as illustrated in Figure 9c. Further hierarchical assembly produces architectures having micron length scales. As with nucleic acids, the structural features outlined in Figure 9 show that molecular organization at every length scale is influenced through the dynamic interactions between the backbone and the amino acid sidechains as set by the primary amino acid sequence. Control of structure at all levels of the molecular structural hierarchy therefore requires an understanding of these dynamics at multiple interfaces. Figure 10 includes examples of the growing number of designer peptide assemblies for which molecular level structural details are known.
Figure 9.
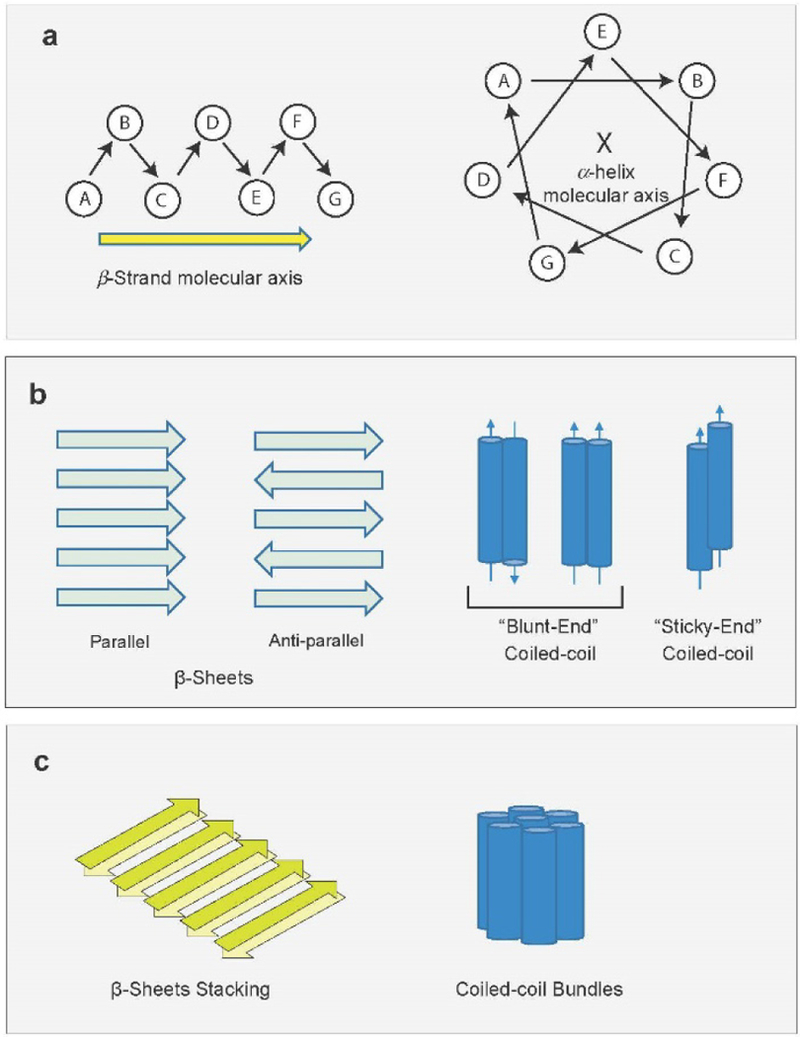
Levels of peptide structure within assemblies for (a) conformation of the peptide backbone and the 3-dimensional arrangement of sidechain groups for β-strand and α-helix, (b) multiple secondary structural units organizing to form cross-β assemblies or α-helical coiled coils, (c) stacking of β-sheets and bundling of coiled coils.
Figure 10.
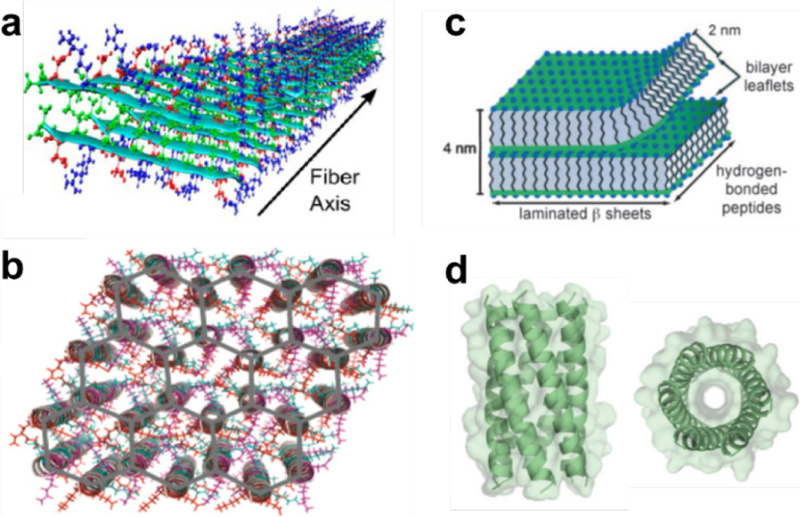
Sample models for molecular organization with (a) a peptide nanofiber, (b) a nanosheet, (c) a nanotube, and (d) a nanoparticle. Reprinted with permission from Cormier, A. R.; Pang, X.; Zimmerman, M. I.; Zhou, H.-X.; Paravastu, A. K. Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano 2013, 7, 7562–7572. Copyright 2013 American Chemical Society. Reprinted with permission from Magnotti, E. L.; Hughes, S. A.; Dillard, R. S.; Wang, S.; Hough, L.; Karumbamkandathil, A.; Lian, T.; Wall, J. S.; Zuo, X.; Wright, E. R. Self-Assembly of an α-Helical Peptide into a Crystalline Two-Dimensional Nanoporous Framework. J. Am. Chem. Soc. 2016, 138, 16274–16282. Copyright 2016 American Chemical Society. Reprinted with permission from Childers, W. S.; Mehta, A. K.; Ni, R.; Taylor, J. V.; Lynn, D. G. Peptides Organized as Bilayer Membranes. Angew. Chem. Int. Ed. 2010, 49, 4104–4107. Copyright 2010 John Wiley and Sons. Reprinted with permission from Thomson, A. R.; Wood, C. W.; Burton, A. J.; Bartlett, G. J.; Sessions, R. B.; Brady, R. L.; Woolfson, D. N. Computational design of water-soluble α-helical barrels. Science 2014, 346, 485–488. Copyright 2014 American Association for the Advancement of Science.
Important aspects of the assembling peptide design challenge are highlighted when systems are viewed in terms of nanoscale dimensions. As illustrated in Figure 11, peptides could assemble into nanofibers (Figure 11a), nanosheets (Figure 11b), nanotubes (Figure 11c) or nanoparticles (Figure 11d). This topological framework for the description of structure is less focused on molecular structural details and emphasizes features that can be directly observed using imaging techniques such as electron and atomic force microscopy. Nanofibers are 1-dimensional nanostructures that are limited to molecular dimensions (or small multiples of molecular dimensions) in two out of three dimensions. Nanofiber lengths can extend beyond the nanoscale (microns). Nanosheets and nanotubes are largely defined by growth in two dimensions. Nanoparticles are approximately spherical or even cubic in shape, and can have dimensions ranging from molecular (small oligomers) to hundreds of nanometeres. Design of these nanoscale dimensions do not always build on detailed knowledge of molecular structure. Rather, the challenge of achieving a specific nanoscale morphology could be described in terms of factors that govern limited growth in at least one dimension. Furthermore, with experimental limitations related to nuances of imaging and sample preparation, it is not always clear how nanoscale dimensions of peptide assemblies are related to their underlying molecular structures.
Figure 11.
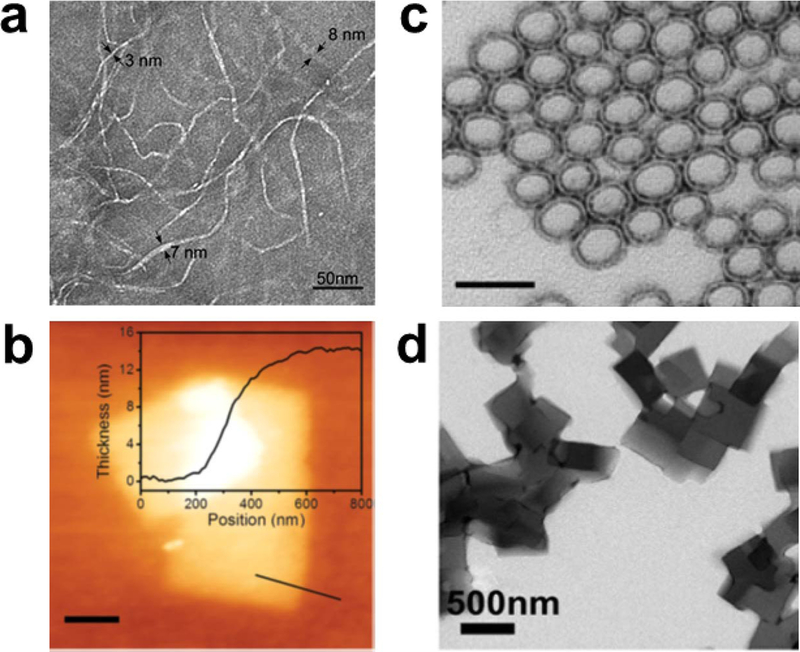
Nanoscale morphologies that are possible for peptide assemblies: (a) nanofibers, (b) nanosheets, and (c) nanoparticles, imaged by TEM (a and c) or AFM (b). The cross-section in Panel b shows the nanosheet thickness. Scale bars in Panels a, b, and c correspond to 50 nm, 200 nm, and 500 nm, respectively. Reprinted with permission from Reprinted with permission from Cormier, A. R.; Pang, X.; Zimmerman, M. I.; Zhou, H.-X.; Paravastu, A. K. Molecular structure of RADA16-I designer self-assembling peptide nanofibers. ACS Nano 2013, 7, 7562–7572. Copyright 2013 American Chemical Society. Reprinted with permission from Jiang, T.; Xu, C.; Liu, Y.; Liu, Z.; Wall, J. S.; Zuo, X.; Lian, T.; Salaita, K.; Ni, C.; Pochan, D.; Conticello, V. P. Structurally defined nanoscale sheets from self-assembly of collagen-mimetic peptides. J. Am. Chem. Soc. 2014, 136, 4300–4308. Copyright 2014 American Chemical Society. Tian, Y.; Zhang, H. V.; Kiick, K. L.; Saven, J. G.; Pochan, D. J. Transition from disordered aggregates to ordered lattices: kinetic control of the assembly of a computationally designed peptide. Org. Biomol. Chem. 2017, 15, 6109–6118. Copyright 2017 Royal Society of Chemistry.
The remaining sections of this chapter attempt to synthesize our present capacity for designing assembling peptides in the context of the structural features highlighted in Figures 9–11. To provide an experimental basis for this discussion, we first summarize the methods available for characterizing the structure of designed peptide assemblies. We subsequently discuss selected examples of de novo peptide designs in which different structural features are successfully controlled. We do not attempt to comprehensively discuss all relevant literature because much of this work is well-summarized in previous review articles.101–130
3.2. Experimental Methods for Characterizing the Structure of Peptide Assemblies
The ability to probe molecular structure of peptide assemblies is essential to the development of design methodologies. When a design strategy is implemented, its efficacy can only be assessed by using structural analysis to determine if the desired structure is achieved. With the current state of the art in protein structure determination, it is highly feasible to evaluate nanoscale structures using microscopy and scattering techniques.131–138 If structural order is high, cryogenic transmission electron microscopy can be employed to probe molecular structure.139–141 Spectroscopic techniques can also be used to determine peptide secondary structure.142–145 However, these methods only provide information on the global architecture of the assembly, and detailed evaluation of molecular structure within peptide assemblies poses a significant challenge. The primary obstacle to high-resolution molecular-level evaluation of nanoscale assemblies is their incompatibility with the most powerful established techniques in protein structure determination, namely X-ray crystallography and solution-state NMR spectroscopy. Although nanoscale peptide assemblies are often characterized by uniform molecular conformations and schemes of intermolecular packing, they can be paracrystalline. Thus, these assemblies are inaccessible to X-ray crystallography, as they generally do not produce X-ray diffraction patterns at a sufficient resolution.146 All but the smallest of assemblies (MW <1000 kDa)147 are too large for detailed solution-state NMR spectroscopy, as rotational correlation times exceed the measurement timescale and cause prohibitive orientation-dependent spectral broadening.148,149 As described below, the field of designer peptide assembly has benefitted greatly from solid-state NMR structural methodologies that have been developed to study disease-related protein aggregation.150
While atomic-resolution structures may be difficult to obtain for mesoscale assemblies, direct observation of nanostructure can be accomplished using microscopy and particle size measurements. Electron and atomic force microscopy can provide detailed information on nanoscale dimensions (e.g., nanofiber widths) and features within nanostructures (e.g., bundled sub-filaments).134,151–153 Particle sizes in solution can also be measured using techniques such as light scattering, size exclusion chromatography, and analytical ultracentrifugation.154–160 Nanoscale assembly in solution can be observed indirectly via turbidity measurements or solution-state NMR, in the latter case via loss of signal from soluble peptide.160,161 Hydrogel formation, which is often associated with nanostructure formation, can be detected through rheological measurements of fluid mechanical properties.162–165 Finally, neutron and X-ray scattering can report on supramolecular arrangements.162,166–168
Beyond nanoscale morphology, the molecular secondary structure of assembled peptides can be studied using spectroscopic techniques, including circular dichroism and infrared spectroscopy.69–72,138,142,163,166,169,170 Since these peptides are normally designed to adopt a single secondary structure, interpretation of spectroscopic data is often straightforward. Furthermore, infrared spectroscopy can report on dynamic intermolecular association through specific spectroscopic shifts.69,71,72,135,143,171–173 Observation of β-strand secondary structure infers the presence of β-sheet nanostructures, as β-strands themselves are unstable in isolation. Although α-helices may be stable without assembly, interactions between designer α-helical peptides can be detected via increase of α-helical spectral signatures when concentration is increased, or upon mixing of complementary co-assembling peptides.143,146,155,156,174,175 For β-sheet forming peptides, assembly can be readily detected via optical measurements on β-sheet-binding dyes, such as the fluorescent thioflavin-T or Congo Red135,176,177 Recently, designer peptide assemblies have been analyzed using solid-state NMR methodologies that were originally designed for analysis of naturally occurring (e.g., amyloid) protein self-assemblies.145,178,179 When solid-state NMR methods are applied to samples with selectively incorporated isotopic labels (13C and 15N), it is possible to construct detailed structural models constrained by experimental data on secondary structures (NMR peak positions), inter-residue proximities (cross-peaks in 2-dimensional spectra), and alignments of neighboring peptide backbones (dipolar coupling methods).180–183
3.3. Rational Design of α-Helical Peptide Assemblies
The α-helix is a common structural motif within proteins, and is characterized by a backbone conformation having a pitch of 3.6 amino-acid residues per turn and stabilized by intramolecular hydrogen bonding between backbone amine (N-H) groups and carbonyl groups (C=O) separated by 4 amino acid units.108 The coiling of a single α-helical backbone, which can bring hydrophobic and hydrophilic amino acid sidechains into proximity, does not result in protection of hydrophobic groups from exposure to water. Similar to the role of hydrophobic collapse in protein folding, the stability of an α-helix can be highly dependent on interactions with hydrophobic elements on other protein domains or biological membranes.130 Bioinformatic analysis of known protein structures has revealed that some amino acids are more likely than others to be involved in α-helical conformations.184 Additionally, analysis of α-helical domains in proteins of known structure has revealed that these helices may form an α-helical coiled-coil, a motif in which multiple α-helices align about their long axes and twist around one another in an extended conformation. This coiled-coil motif is associated with heptad repeat patterning in the peptide sequence, denoted (HPPHPPP)n, in which (H) are amino acids having hydrophobic sidechains and (P) are amino acids having polar or charged sidechains (Figure 9a).108,155,174,185 A variety of α-helical designer peptides have been discovered by evaluating multiple sequences that conform to the heptad repeat pattern. The amino acid sequences of selected α-helical designer peptides are shown in Table 1.
Table 1.
Selected α-helix forming self-assembling or binary co-assembling designer peptides, color-coded to show amino acid sidechain patterning. Black, green, blue, and red letters indicate amino acids having hydrophobic, polar, positively charged, and negatively charged sidechains, respectively. Indicated charges correspond to neutral pH.
| SAF-p1 | K | IAALKQK | IASLKQE | IDALEYE | NDALEQ |
| SAF-p2a | K | IRRLKQK | NARLKQE | IAALEYE | IAALEQ |
| SAF-p2 | K | IRALKAK | NAHLKQE | IAALEQE | IAALEQ |
| hSAFAAQ-p1 | K | IAALKQK | IASLKQE | IAALEQE | NAALEQ |
| hSAFAAQ-p2 | K | IAALKQK | NAALKQE | IAALEQE | IAALEQ |
| heptad repeat | g | abcdefg | abcdefg | abcdefg | abcdef |
3.3.1. Sidechain patterning for α-Helical Designer Assembling Peptides.
The α-helical secondary structure is promoted by amino acid sequences having the (HPPHPPP)n heptad repeat. Upon folding, this patterning forms an amphipathic helical structure, in which the hydrophobic residues align on one flank of the coiled helix to create a hydrophobic patch. Amphipathic α-helices are of special interest to peptide designers, due to their propensity to aggregate predictably via hydrophobic interactions between these hydrophobic patches. In Figure 9a, a helical wheel is shown to represent the 3-dimensional organization of sidechains in an α-helix. In this diagram, the nanofiber axis is perpendicular to (goes into) the page and the 7 letters a-g correspond to positions within the heptad repeat subunit. Interactions between α-helices are promoted by hydrophobic interactions between sidechains at positions a and d and Coulombic interactions between charged sidechains strategically placed at positions e and g.130,186–188 Although charged sidechains are typically placed to promote specific intermolecular arrangements, it has also been shown that individual α-helices can be stabilized by formation of salt bridges between oppositely charged sidechains that align vertically on the same molecule.144
3.3.2. Controlling the Arrangement of Adjacent α-Helices.
Unlike β-sheets, which rely on intermolecular hydrogen bonding for assembly, the assembly of α-helices is governed by intermolecular sidechain interactions, which can be optimized to pack hydrophobic residues together and pair polar or oppositely-charged residues.133,134,136,163,186,189–191 Interestingly, specific combinations of sidechains on adjacent molecules tend to promote specific intermolecular alignments.192–194 When small numbers of α-helices align with one another in a “blunt-ended” manner (Figure 9b), they can assemble to form nanoparticle structures (Figures 10a and 11a).133,143,161,185,195 Nanosheets can be generated by arranging α-helices into 2-dimensional arrays (Figures 10c and 11c).133,196–199 When neighboring molecules do not align in a way that maximizes overlap, they leave “sticky-ends” that can promote assembly along the axis of the helix to form nanofibers (Figure 10b).187,190
3.3.3. Controlling Higher-Order Assembly of α-Helices.
Design of α-helical nanostructures has proven to be one of the most successful applications of computational design to biomolecular nanostructure engineering. While the ability to predict peptide and protein structures purely from first principles remains beyond reach, remarkable progress has been made in the area of semi-empirical computer-aided design.105,157,200,201 Computational tools have proven to be effective for the prediction and optimization of helix-helix interfaces, enabling rational design of α-helical peptide assemblies to form nanoparticles (or oligomeric structures), nanofibers, and nanosheets.133,136,137,200,201
An α-helical assembly can form nanofiber structure through sticky-end association of molecular structural units. In early work, Pandya et al. engineered the first self-assembled fiber (SAF) binary peptide system by placing charged amino acids (positions e and g in the α-helix drawn in Figure 9a) such that each α-helix had a net positive surface charge on the first half of its length and a net negative charge on the second half of its length; this configuration promoted sticky-end coiled-coil heterodimers.187 Interactions between oppositely charged sidechains promoted head-to-tail assembly of these sticky-ended heterodimers into protofilaments, which themselves bundled to further assemble into nanofibers. For the next generation of this system (SAF-p1/p2a), a hypothesized intermolecular arrangement was posited based on light-to-dark striation patters observed in negative-stain transmission electron microscopy images.134,139,146 The dimensions and periodicity of these striations were consistent with laterally aligned coiled coils made from 28-residue α-helices arranged with the helical axis parallel to the nanofiber axis. Additional X-ray fiber diffraction experiments supported the hypothesized arrangement and indicated a highly ordered scheme for lateral association of coiled-coil protofilaments to form nanofibers, although it is worth noting that lateral association was not predicted in the original design of first-generation SAF-p1/p2.146,187 In other variants of SAF peptides, hydrophilic residues at coiled-coil interfaces were used to promote specific alignments of α-helices.187,202 In subsequent studies of SAF variants, the hydrophobicity of residues at positions b, c, and f (Figure 9a) were varied. These residues project away from the coiled-coil interfaces, allowing them to promote higher order interactions between coiled-coil protofilaments, modulating nanofiber thicknesses and persistence length.163,202 More recent work by Hume et al. has shown that the coiled-coil heterodimer need not be considered the only structural unit capable of assembling into α-helical nanofibers. Instead, they showed that α-helical homopentamers can be engineered to present positive and negative charges in specific regions of the pentamer surface.203 The pentamers can undergo further assembly in to nanofibers through a charge-driven staggered pentamer alignment that resembles that of sticky-end assembly of individual α-helices.
The ability to computationally optimize inter-helix association and packing has enabled the design of nanoparticles and nanosheets composed of α-helical designer peptides. For example, Zaccai et al. demonstrated that a parallel 6-helix bundle can be designed through strategic optimization of helix-helix interactions.157 In addition, researchers have shown that it is possible to control nanoscale geometries by designing peptides having more than one α-helical domain. Boyle et al. designed peptides composed of two α-helical domains connected by flexible linkers, and demonstrated the ability of these peptides to assemble into nanoparticles.156 Demonstrating additional control over assembly properties, Zhang et al. designed α-helical 29-residue peptides that changed their assembly topology from nanoparticles to nanosheets in response to computationally-optimized variations in sequence.133 Tian et al. rationally designed a single peptide sequence having the same length, but which formed a lattice-like structure in response to solution conditions. Their 29-residue sequence, generated by computer-aided design, was found to be disordered in acidic solution, but assembles to form nanosheets at neutral or basic pH.136 The design of peptides that form bundles of multiple α-helices has even made it possible to produce nanofibers having well-controlled thicknesses, bundles can also be designed to assemble end-to-end with little lateral association.204
3.4. Rational Design of β-Sheet Peptide Assemblies
When multiple β-strands assemble into β-sheets, the resultant assembled structure is determined by interplay between the energetics of three types of surfaces. One type of surface is created by the amino acid sidechains. For a single β-sheet, the sidechain surface has the highest surface area and consequently significantly impacts the energetics of solvent interactions and β-sheet stacking (see Figures 9b and 9c).69,71,72 A second surface is defined by hydrogen bond donors (N-H) and acceptors (C=O) along the peptide backbone. The arrangement of the amino acid side chains within β-sheets (Figure 9b) defines strand registry via side chain cross-strand packing.205 The third surface is created by the peptide termini or turn signatures. The area of this type of surface increases with numbers of β-sheets stacks and strands in each sheet. For a parallel β-sheet structure, the N-termini and C-termini would be sequestered on opposite surfaces and both termini would be equally distributed on each surface for antiparallel β-sheets. It is from these energetic determinants that design rules for cross-β assemblies begin to emerge.
3.4.1. Controlling β-Strand Conformations Through Primary Structure.
Although the relationship between amino acid sequence and molecular structure is not comprehensively understood for proteins and peptides, it is clear that specific patterns of hydrophobic, charged, and polar sidechains can be correlated with specific secondary structures. Table 2 lists the amino acid sequences of selected β-strand forming peptides. In early work, Zhang et al. observed that a 35 residue segment of the yeast protein zuotin contains (HP)n repeats of alanine with hydrophilic (charged or polar) sidechains.206,207 Observation of this pattern inspired the design of a series of peptides having (HP)n sequences with varying amino acid identities, which led to discovery of the designer self-assembling peptide RADA16-I (Figure 11b). Researchers have since discovered other self-assembling peptides having similar patterning (Table 2).132,208–212
Table 2.
Selected β-sheet-forming self-assembling designer peptides or segments of proteins and binary co-assembling peptide systems, color-coded to show amino acid sidechain patterning. Black, green, blue, and red letters indicate amino acids having hydrophobic, polar, positively charged, and negatively charged sidechains, respectively. Indicated charges correspond to neutral pH. Acetylated N-termini are indicated by the characters CH3CO−. Amidated C-termini are indicated by –NH2. The symbol DP represents a proline having non-natural D-chirality at the α-carbon.
| Peptide or Protein Segment | Amino Acid Sequence |
|---|---|
| zuotin(306–339) | EGARAEAEAKAKAEAEAKAKAESEAKANASAKAD |
| RADA16-I | CH3CO-RADARADARADARADA-NH2 |
| DN1 | QQRFQWQFEQQ |
| Q11 | CH3CO- QQKFQFQFEQQ-NH2 |
| MAX1 | VKVKVKVKVDPPTKVEVKVKV-NH2 |
| p1/p2 | EEFKWKFKEE / KKFEWEFEKK |
| CATCH+/CATCH− | CH3CO-QQKFKFKFKQQ-NH2 CH3CO-EQEFEFEFEQE-NH2 |
An alternative approach to creating non-natural β-strand forming peptides is to modify the amino acid sequences associated with β-strand forming amino acid sequences. Many naturally occurring β-strand peptides do not exhibit (HP)n patterning. Fragments of Alzheimer’s β-amyloid peptide are popular choices for designer β-strand peptides.213,214 Although these peptides were not derived from de novo design of primary structure, sidechain patterning is sometimes considered in modification of the naturally-occurring sequences. For example, aromatic residues can be substituted with residues having larger aromatic sidechains to influence steric effects, and charged residues could be substituted with different charged residues to affect intermolecular Columbic interactions213,214 (termini could also be modified for similar reasons). This control has now expanded to chimeric biopolymer assemblies, including the incorporation of nucleic acid bases215 and lipids,216 further increasing the range of novel functional assemblies that can be designed and used.98
3.4.2. Controlling β-Strand Arrangement Within β-Sheets.
Recent structural investigations of naturally occurring amyloid fibrils have revealed that amyloids can be composed of either parallel or antiparallel β-sheets.150 Both configurations are stabilized by intermolecular backbone hydrogen bonding, and the parallel β-sheet often has the advantage of maximizing overlap between equivalent amino acid sidechains, promoting favorable hydrophobic and polar zipper interactions. While the antiparallel β-sheet has less overlap between identical sidechains, this can provide the advantage of avoiding potentially unfavorable electrostatic interactions between like-charged sidechains. The rational design of peptides capable of adopting specific β-strand arrangements within β-sheets would benefit from understanding the interplay between different types of energetically similar interactions. One aspect of the design challenge that may be unique to designer peptides concerns the co-assembly of distinct β-strand peptide molecules into β-sheets. For these binary systems of co-assembling β-strand peptides, rational design requires the engineering of cross-strand pairing interactions between complementary peptides such that neighboring peptides alternate within the β-sheet.69,71,72,152,213,217
The MAX1 peptide (Table 2), along with related peptides inspired by this design, utilize a type-II’ β-turn or a double proline hinge -VDPPT-, which promotes a β-hairpin molecular conformation and assembly of antiparallel β-sheets.218 The DP in this sequence denotes a D-proline, having opposite chirality of naturally occurring L-proline. The MAX1 peptide was designed to undergo salt-triggered assembly – at low ionic strength and neutral pH, repulsion between the positively-charged lysine sidechains promotes solubility by preventing β-strand formation. When ionic strength is increased to near physiological levels, electrostatic screening reduces repulsion between these sidechains, allowing the MAX1 peptide to form a β-hairpin structure and self-assemble into nanofibers. The -VDPPT- hinge makes it possible for each peptide molecule to contribute two neighboring β-strands having a specific alignment within a β-sheet. Other peptides designed using -VDPPT- turns include the TSS1 peptide, which has three β-strand domains and two -VDPPT- turns, and the VEQ1 peptide, which is similar to MAX1 but has negatively charged amino acids in place of the lysine residues.166,219
Recent efforts in the rational design of binary systems of co-assembling β-strand peptides have primarily focused on engineering the arrangement of neighboring β-strands. The p1/p2 system (Table 2) was designed based upon the DN1 self-assembling peptide,152,209 with the p1 and p2 peptide sequences having similar sidechain patterning to DN1, but using positively charged lysine or negatively charged glutamic acid in place of glutamine. The differing placements of lysine and glutamic acid residues in p1 and p2 were chosen such that oppositely charged sidechains would form salt bridges when the p1 and p2 peptides were arranged into antiparallel β-sheets. Individually, the p1 and p2 peptides were each found to be highly soluble in water, but mixing of the two peptides induced co-assembly, which was detected and characterized using Fourier transform infrared spectroscopy, small-angle X-ray scattering, and electron microscopy.152 The CATCH+/CATCH- co-assembling system was created using similar reasoning,217 but was inspired by the Q11 peptide, which has a similar sequence to the DN1 peptide (Table 2).220 Compared to the p1/p2 peptides, the CATCH peptides have a larger number of charged sidechains at neutral pH, with CATCH+ having exclusively lysine as the charged sidechains and CATCH- having exclusively glutamic acid as the charged sidechains. As with the p1/p2 system, assembly of CATCH peptides was only observed when both CATCH+ and CATCH-peptides were co-dissolved in solution, and these assemblies were characterized using circular dichroism, thioflavin-T fluorescence, and electron microscopy.217
In principle, it should be possible to create a peptide nanosheet by engineering a brickwork-like intermolecular organization within a β-sheet. However, alignment of β-strands within β-sheets usually maximizes hydrogen bonds between adjacent pairs of β-strands, creating β-strand alignments that lend themselves to nanofiber, and not nanosheet structures. In very early work, Zhang et. al. reported assembly of a peptide nanosheet in solution and proposed a brickwork-like intermolecular organization within β-sheets that can extend indefinitely in 2-dimensions.206 To our knowledge, this proposed brickwork-like configuration does not occur in any extant peptide assembly, but there is experimental and computational evidence for a brickwork-like molecular configuration within peptoid nanosheets.221–223 Although not free-floating or well-characterized at the molecular level, Rapaport et. al. reported formation of what could be a nanosheet-like self-assembled peptide at an air-water interface.224
3.4.3. Higher Order β-Sheet Assembly.
The canonical amino acid sequence of β-strands having alternating hydrophobic and hydrophilic sidechains generates an amphipathic folded structure. Subsequent assembly into β-sheets thus creates a hydrophobic surface which can stack with the hydrophobic surface of another β-sheet, the result of which is often a hydrophobic nanofiber core. Sathaye et al. demonstrated that the relative orientations of stacked β-sheets can be controlled through the choice of hydrophobic residues within the hydrophobic nanofiber core.225 Starting with the MAX1 self-assembling peptide sequence, selected valine residues were replaced by 2-naphthylalanine or alanine residues. The 2-napthylalanine sidechain is bulky and hydrophobic, while the alanine sidechain is hydrophobic, but less bulky than valine. The resultant LNK1 peptide assembles into a nanofiber structure in which the 2-napthylalanine sidechains on one β-sheet interact with alanine sidechains on the adjacent β-sheet.
The problem of creating β-strand peptide nanotube and nanosheet assemblies could be considered a challenge of β-sheet organization. Li et. al. produced a nanotube by modifying the termini of the nucleating core of the Alzheimer’s β-amyloid peptide (Aβ(16–22) to promote leaflet stabilization of cross-β membranes (see Figure 10c).98,226 The interface between leaflets is the surface associated with the peptide termini. There is now evidence that this interface can be cooperatively stabilized with other polymers to provide multi-lamellar assemblies having much larger molecular dimensions and co-assembled nanostructure on the 100 nm length scale. Furthermore, the nature of this interface can be modified through control of β-strand organization. Since the backbone amide is a dipole, one shift in registry of one strand requires a 180° rotation and places the side chains on the opposite surface; such a change can greatly impact the resulting morphology. For example, a simple change in pH stabilizes a shift in registry that alters the facial complementarity and drives sheet lamination to create peptide nanotubes of homogeneous diameter.227 This energetic constraint has now been used to design and construct peptide nanotubes having controlled internal diameters and surfaces.205 The leaflet interface has been used to construct the first self-assembling asymmetric membrane,98 and as a site to create novel functions including photochemical energy transfer228 and as specific catalysts.229 Since the β-sheet surface sidechains define axial growth via sheet stacking,214 it may now be possible to use and further develop these interactions for assembly design.
3.5. Structural Transformations Between α-Helices and β-Strands
While some designer peptides exclusively form α-helices or β-sheets, peptides have also been designed that are capable of switching between these structures in response to changes in their environment. In this sense, conformational switching increases the accessible structural and functional diversity in assembling peptide systems, and enables the design of stimuli-responsive assemblies. For example, structural transformations between α-helical and β-strand states of a peptide could serve as the basis for triggered or reversible assembly.129,230 An early example of such a system is the DAR16-IV peptide, which has β-strand-promoting (HP)n patterning and assembles into β-sheet nanofibers at room temperature. Zhang et al. discovered that upon heating above 70°C, the DAR16-IV nanofibers dissociate and the individual peptides convert to an α-helical secondary structure.142 This process is reversible, as cooling below 70 °C causes the DAR16-IV α-helices to reassemble into β-sheet nanofibers. Conformation-switching behavior has also been observed for peptides designed with α-helix-promoting (HPPHPPP)n sidechain patterning. Dong et al. demonstrated that soluble heterodimeric coiled-coil peptide systems could exhibit temperature-dependent or pH-dependent conversion into β-sheet structures, followed by irreversible assembly into β-sheet nanofibers.143,172 Kammerer et al. rationally designed the ccβ peptides to have amino acid patterning that is compatible with both α-helix and β-strand structures, and showed that water-soluble α-helical trimers of this peptide could be driven through heating to convert into β-strands and assemble further into amyloid-like fibrils in solution.231,232 Using solid-state NMR, Verel et al. further demonstrated that for the ccβ-p variant, alignments of β-strands within β-sheet fibrils was dependent upon pH during assembly.233,234
3.6. Remaining Challenges and Future Directions
Recent effort toward the rational design of peptide assemblies follows the story arc described in Chapter 1. Initially, the designs of α-helical and β-strand peptides relied on cues from the sequences and structures of natural peptides and proteins, including folded proteins, coiled-coil assemblies, and amyloid fibrils. There is now considerable experimental support for the paradigm that α-helical and β-strand secondary structures can be promoted by patterning of molecular recognition elements within the sequence of a peptide strand. The ability to rationally design peptide folds that display these elements at specific orientations in turn enables the engineering of intermolecular organization through careful placement of molecular complementarity.
A toolkit is emerging for predictive design coupled with experimental realization of the desired peptide assembly structures. Nanoparticles composed of α-helices can be created from assemblies of small numbers of peptide molecules (e.g., blunt-end dimers through hexamers), and further assembly into nanofibers can be achieved by a rationally designed change in intermolecular organization to generate sticky-ended geometries.143,185,187,190,195 Larger α-helical nanoparticles as well as nanosheets can be formed when 3-dimensional interfaces are precisely engineered between helical bundles.133,196,197 Designer peptides in linear β-strand conformations are now appearing where parallel or antiparallel β-sheet organization, the strand registry, sheet facial complementarity, and leaflet interface associations can be controlled during assembly. The LNK1 peptide system has been used to rationally control the relative orientations of stacked β-sheets within peptide nanofibers.225 For the RADA16-I peptide, although predicted antiparallel β-strand organizations differed from the parallel β-sheet organizations, it is encouraging that this level of structural detail can be probed using solid-state NMR spectroscopy.145,227 Peptides such as MAX1 were designed to adopt β-hairpin conformations and form nanofibers with antiparallel β-sheets. While structural characteristics such as the relative orientations of MAX1 β-hairpins within these β-sheets were not predicted in the peptide design process, the monomorphic structure of this assembly indicates that it should be possible to control the relative orientation of molecules within each β-sheet and between β-sheets.178 Research on naturally occurring peptide aggregation indicates that β-sheet nanoparticles (oligomers) do exist and what is known about oligomer structure and dynamics are discussed in Chapter 2 as a critical pathway to access all of these assemblies. Given this two-step nucleation pathway, size-limited nanoparticle assembly could be engineered by design of peptides having more than one secondary structural domain, as was accomplished by King et al. for a designer folded protein,235 or even with other combined polymer scaffolds.98,216
We predict that structural control of peptide assemblies will improve as we gain understanding of assembly mechanisms. At this point, it is clear that biology has exploited the surfaces generated in both α-helical and β-sheet elements for both protein folding and larger scale aggregation, and sufficient information is now emerging to use the folding energetics of these motifs to design new materials across large length scales. The importance of understanding assembly mechanisms is especially apparent for β-sheet assemblies; while a single polypeptide is capable of adopting an α-helix, the β-fold requires intermolecular assembly with a critical concentration dependence for liquid-liquid phase transitions. Individual folded structural units can be even more readily predicted at this point for α-helical assemblies using computer-aided design tools, and the side chains on the surfaces of these units can be computationally optimized. The intermolecular association of α-helices also likely depends on 2-state nucleation, further extending the predictions of energetic potentials for structural design. Thus, while the initial folding of α-helical structures can be engineered by design approaches that consider only the final assembled structures, predictive design of assembly for both α-helical and β-sheet elements requires an understanding of the entire folding and assembly process as outlined in Chapter 2. As also outlined in Chapter 2 and shown for the β-hairpin peptides such as MAX1, many assemblies undergo specific structural transformations during the assembly process.218 Although the structures of binary co-assembled β-sheets such as CATCH+/CATCH- have not been analyzed in detail, we suggest that highly specific intermediates are on pathway for co-assembly,217 and it is likely that improved structural control will be important for optimization of future co-assembling designs if high molecular selectivity is to be achieved. While much remains to be defined as the range of folding elements expands beyond the α-helical and β-sheet elements, we appear at the early stage of developing polymer co-assembly strategies for more sophisticated predictive design algorithms in protein assembly. As described in Chapter 1, experimental observations can be iterated to refine models, and thus improve predictive design.
As a final note, we observe that while many α-helices in nature do contain the heptad repeat pattern that has been adopted for helix design, naturally occurring β-strands do not necessarily exhibit the (HP)n patterning employed in designer β-strands. Dobson et al. have argued that the amyloid state may be a general free energy minimum that can be accessed by any polypeptide, and most polypeptides do not conform to (HP)n sidechain patterning.236 For assemblies composed solely of single β-strand peptides, Sawaya et al. defined a set of 8 symmetry classes for a basic “steric zipper” structural unit composed of two stacked β-sheets.237 Given that peptides having (HP)n patterning might be predicted to form β-sheets with hydrophobic residues organized on one face, and these β-sheets would be expected to stack along hydrophobic interfaces, we would expect peptides with (HP)n patterning to be compatible only with steric zipper symmetry classes that correspond to the “face-to-face” β-sheet stacking known as facial complementarity. Nevertheless, Sawaya et al. discovered several (HP)n peptides that assemble into structures having symmetries that are not consistent with “face-to-face” stacking, highlighting the complexity of predictive design for β-sheet assembly.237 We suggest that a remaining “grand challenge” in the field is the predictive design of β-strand peptides capable of controlled assembly into each of the 8 symmetry classes.
4. FUNCTIONAL PROTEINS ASSEMBLED IN STRUCTURED ENVIRONMENTS
4.1. Introduction to Functional Protein Assemblies
Proteins possess a diverse array of functions, including binding or affinity, catalysis, signal generation or propagation, and structural stabilization. While many biochemical studies and applications utilize pure, isolated proteins, especially in the case of enzymes and fluorescent proteins, this represents a departure from the natural environment of these molecules. In nature, proteins typically exist in assemblies, which can be formed with other proteins, and may also include other types of biomolecules.238 Even in prokaryotic cellular environments, which have relatively little sub-cellular organization, proteins are present at such exceptionally high concentrations (~300 g/L)239 that they naturally form interactions with other proteins. Additionally, the very early emergence of ribosomes, formed by assembly of proteins with RNA, suggests that functional protein assembly may be a characteristic element of life.240 Eukaryotic cells are capable of increasingly complex assemblies, which may be generated or stabilized by organelles, and serve as the inspiration for many designed assemblies.238 Further contributing to the diversity of functional protein assemblies, the cellular environment contains non-biological components, such as salts and buffers, which modulate protein folding and assembly via charge screening, electrostatic interactions, and steric attraction or repulsion.
4.2. Structure and Components of Functional Protein Assemblies
4.2.1. Components, Interactions, and Examples from Nature.
In the context of this Chapter, we define functional proteins as those whose function can be captured by a unique germane assay, such as catalytic activity or fluorescence, but not shape, oligomeric state, or folding status. Aside from the functional proteins themselves, the assemblies we describe here may include surfactants, lipids, nucleic acids, and other proteins. The role of these elements is typically to provide the 2- or 3-dimensional structure needed to assemble and constrain the functional proteins in their optimal spatial arrangement.
Assembly in lipid membranes is a motif frequently employed by cells, and in the case of eukaryotes, these assemblies may occur at either the outer cellular membrane or at sub-cellular membranes, such as the endoplasmic reticulum or the nuclear envelope. Importantly, proteins assembled in membranes experience a primarily lipophilic environment, which can be mimicked in the laboratory through the use of surfactants or synthetic lipids. Admittedly, the boundary between the definitions for surfactants and lipids is not clear. Both natural lipids, such as lecithin, as well as synthetic lipids designed to mimic natural lipids, such as dipalmitoylphosphatidylcholine (DPPC), have surfactant properties and can form structures to encapsulate proteins.241,242 These structures are generally vesicles, also termed “liposomes,” which can be uni- or multilamellar and have various sizes ranging from 100 nm to 100 µm. Beyond vesicles, the templating structures of designer protein-lipid assemblies can also include micelles or reversed micelles, microemulsions, or cubic or lamellar liquid crystals. Formation of these alternative structures can depend on environmental variables including temperature, pressure, degree of hydration, and salt type or concentration. Many proteins can even retain their structure and function in environments comprised almost entirely of non-aqueous, organic solvents, though the lack of templating structure in these media generally makes them less effective for functional protein assembly.243,244
Protein assembly can also be templated by nucleic acids. While the ribosome and other ribonucleoproteins serve as examples of RNA-driven assembly, most protein-nucleic acid assemblies involve DNA. 1-dimensional DNA-protein assemblies (“nanowires”), 2-dimensional DNA-protein arrays, and 3-dimensional DNA-protein structures (“nanohedra”) have all been constructed. In principle, the templating can originate either from the DNA or the protein, and multi-enzyme complexes or cascades can be optimized by varying the density of enzymes on the template or the distance between individual enzymes.245
Finally, functional protein assembly can be templated by other proteins, which are presumed to be “non-functional,” at least with respect to the target protein. The most prominent group of structural proteins is the virus-like particles (VLPs), which form highly symmetrical assemblies, often in regular geometric shapes such as icosahedrons. VLPs are designed to mimic the shell of viruses, and typically have sizes ranging between 10 and 100 nm. However, unlike viruses, VLPs do not carry DNA or RNA in their interior and thus are not infective. While this Chapter will focus on the use of VLPs for the design of functional protein assemblies, VLPs themselves are highly amenable to design and engineering. As comprehensively reviewed elsewhere, slight changes in the amino acid sequence of VLP proteins can result in changes to assembly size, structure, or function, and such changes can be generated through both rational design and laboratory evolution.3,110,246–249
4.2.2. Description of Interactions.
The interactions of proteins in ordered structural environments are guided by the nature of intermolecular forces that can be utilized and by the geometry of the interacting partners. In energetic terms, these interactions can range from covalent bonds (~120 kJ/mol) to electrostatic and hydrophobic interactions (~10–50 kJ/mol) or H-bonding interactions (~5–20 kJ/mol). Covalent interactions for protein-protein assemblies can be genetically encoded, utilizing constructs in which the genes for the structural and functional components are fused in tandem. In these cases, expression of the overall construct produces the assembled target functional protein. In contrast, surfactant-protein and lipid-protein assemblies rely on non-covalent interactions for construction of the lipid or surfactant structure and binding of the proteins to this structure and to one another within the structure.250 Micelles, reversed micelles, and microemulsions are thermodynamically stable phases that can be represented using specific coordinates of a phase diagram. In the case of micelles, the minimum concentration at which assembly occurs is referred to as the critical micelle concentration (CMC). These lipid or surfactant assemblies often display a characteristic diameter, which is related to the curvature of the surfactant layer.251,252 The curvature in turn depends primarily on the geometric shape of the surfactant or lipid molecule, and secondarily on the energy of interaction among the head groups and tail groups in the ensemble of molecules.
4.2.3. Focus and Scope.
This Chapter will focus on the co-localization and immobilization of functional proteins in or on organized media including surfactants or lipids, nucleic acids, and proteins using the variety of covalent and non-covalent interactions described above. We will specifically focus on volume-based immobilization, as encountered with micelles, water-in-oil microemulsions, liquid crystals, or uni- and multilamellar vesicles, as well as biological assemblies, such as virus-like particles (VLPs) or cellulosomes.253 While significant research effort has focused on surface-based immobilization using supports such as glass, non-porous silica, alumina, titania, gold, graphene, and metal-oxide frameworks (MOFs), these studies are considered to be generally outside of the scope of this Review. Readers interested in these topics are referred to recent reviews in this area.254–262 In these systems, the characteristic length of the surface often far exceeds that of the immobilized surface, and covalent immobilization produces random orientation, for example through non-specific cross-linking of lysine, aspartic acid, or glutamic acid sidechains on the protein with complementary functional groups on the inorganic surface. Frequently, a significant fraction of enzyme activity is also lost upon immobilization on solid surfaces, in part attributable to the random orientation of the immobilized proteins relative to the surface. However, in a few examples, this limitation has been overcome, and immobilization has even led to increased activity. For example, in an approach involving protein engineering, Candida antarctica lipase B (CALB), one of the most versatile industrial biocatalysts,263 was covalently immobilized on superparamagnetic beads via copper-catalyzed azide-alkyne cycloaddition (CuAAC) as a cysteine-free variant having a single non-canonical amino acid, para-propargyloxy-phenylalanine (pPA).264 This PRECISE (Protein Residue-Explicit Covalent Immobilization for Stability Enhancement) procedure resulted in a 1.2-fold increase in specific activity, albeit after tedious and low-yield expression and purification of the CALB-N98-pPA.265 The PRECISE protocol was extended to T4 lysozyme, and over time was demonstrated to result in 50–73% more active enzyme compared to the wild type.266
While the immobilization techniques described above enable exciting applications such as sensing or catalysis, these assemblies pose a challenge for specific structural design and engineering. In comparison, the assemblies that are the focus of this Chapter provide specific spatial arrangements of proteins, which either significantly enhance function or even enable new functionality. Moreover, the well-defined nature of these interactions makes them amenable to engineering and design. Many of the individual topics covered in this Chapter have been the subject of previous reviews. These include the functionality of enzymes in reversed micelles and microemulsions,267,268 assembly of functional proteins in both surface-based and volume-based scaffolds,253 supramolecular assemblies based on DNA templates,245 and the natural occurrence of supramolecular protein assemblies.269
4.3. Surfactant-Protein and Lipid-Protein Assemblies
4.3.1. Rational Design of Surfactant-Protein or Lipid-Protein Assemblies.
The basis for designing a surfactant-protein assembly system is the phase diagram between the hydrophilic solvent, typically water, the hydrophobic solvent, most often a water-immiscible solvent, such as an alkane or chloroform, and the surfactant. Subsequently, one can generate the assembly medium by mixing specific amounts of components according to the phase diagram. In most cases, several phases are possible, including micelles, reversed micelles, microemulsions, and cubic, hexagonal, or lamellar liquid crystals. These phases are all thermodynamically produced, with the exception of emulsions, which require stirring to form. The phase diagrams that describe these assemblies can currently be predicted qualitatively with reasonable accuracy, but quantitative prediction has yet to be fully realized.
Reverse micelles and microemulsions contain a water droplet that typically has a diameter between 1–20 nm. The size of this droplet can be predictably controlled, as it increases proportionally with the molar water:surfactant ratio w0, and the slope of this relationship correlates with a geometric factor that is derived from the headgroup area of the surfactant, the length of the tail, and the spread of the tail groups. Enzymes from almost all classes and structures have been solubilized in microemulsion systems and used to catalyze reactions.267 The activity and stability of enzymes in these assemblies are often comparable to their values in aqueous media. A key benefit to the use of microemulsions is the expansion of substrate scope, as many substrates which are not soluble in water or pure organic solvents such as hexane can be solubilized and reacted in microemulsions. Whereas enzyme structure and mechanism do not seem to change upon transition from water to organic or microemulsion phase,270–272 partitioning effects often play a key role in these reactions. For example, substrate binding and product release can be influenced by the surrounding medium and the Michaelis constant (KM) is known to depend on the partitioning coefficient (P = [A]organic/[A]water) of the organic medium surrounding the enzyme.273 While the key benefit of assembling enzymes in microemulsions is the enhanced concentration of substrate molecules, the use of charged surfactants to form these emulsions can also enable modulation of the effective pH value in the water pool of the droplets. Frequently, observed acceleration or deceleration effects on enzyme reactions can be explained by such partitioning effects.274
While microemulsions feature several advantages for controlling enzymatic reactions, a key disadvantage is the difficulty of separating the reaction product from the other components of the system. This disadvantage can be overcome by the use of highly viscous or solid liquid crystalline phases, which consist of the same components as microemulsions (water, surfactant, and organic solvent) (Figure 12). Several enzymes, such as peroxidases, lipoxygenase, and lipases, display activity across several phases of this three-component mixture.243 Cubic inverse phases are especially attractive due to their high mechanical stability, and a series of enzymatic reactions have been conducted in this liquid crystalline phase.275 Interestingly, the stability of enzymes has often been found to be significantly higher in liquid crystalline phases compared to pure isotropic continuous phases and media, such as buffer solutions.
Figure 12.

Ternary mixtures of water, organic solvent, and surfactant can give rise to diverse assembly types. The phase diagram for water:hexanol:cetyltrimethylammonium bromide (CTAB) indicates conditions for formation of (a) micelles, (b) reverse micelles, (c) lamellar aggregates, and (d) hexagonal aggregates. Adapted with permission from Martinek, K.; Levashov, A. V.; Klyachko, N.; Khmelnitski, Y. L.; Berezin, I. V. Micellar enzymology. FEBS J. 1986, 155, 453–468. Copyright 1986 John Wiley and Sons.
In contrast to surfactants, assemblies of lipids, such as phosphatidylcholine (PC), phosphatidylethanolamine (PE), or dipalmitoylphosphatidylcholine (DPPC), form in aqueous media alone and do not require a hydrophobic solvent. As a consequence, however, these assemblies are not thermodynamically stable and prediction of important length scales, such as the diameter of a cubic phase element or the distance between two lamellae, is not currently possible. However, various lipid-protein assemblies such as micelles, bicelles and nanodiscs can mitigate issues related to stability. Notably, BioNanoStacks are relatively stable materials that can be reversibly assembled and disassembled through thermal melting of the DNA duplex.276 The melting transition temperature of these novel materials can be tuned via engineering of the DNA sequence, salt concentration, and lipid composition within these superstructures. Recently, gas vesicles, i.e. up to micron-sized gas bubbles surrounded by a lipid or protein shell, have been discovered and characterized.277 Such vesicles, if filled with xenon gas, can be important tools for ultrasound spectroscopy and magnetic resonance imaging MRI).
4.3.2. Computational Methods for Design of Surfactant-Protein or Lipid-Protein Assemblies.
De novo computational design of surfactant- and lipid-protein assemblies has not yet been demonstrated at the levels presented above for other assembly types. However, looking to the future, the development of such methods is critically dependent upon the enhanced predictability of both length scales in the various phases and the locus of the transition between phases. As a first step toward achieving this goal, we propose that detailed understanding is needed for the preferred conformation of an ensemble of surfactant or lipid molecules as a function of molecular structure, ionic strength, temperature, pressure, and concentration of components. While the control of structure and function in assemblies of lipids and proteins remains a formidable challenge, some progress has been made. Bilgicer and Kuman reported a rational design strategy for creating self-assemblies of protein components and biological membranes.278 This design schema involved an incrementally staged assembly process that leverages the unique properties of fluorinated amino acids to drive transmembrane helix–helix interactions. At the outset, engineered hydrophobic peptides are partitioned into micellar lipids resulting in phase separation of hydrophobic and lipophobic fluorinated helical surfaces. This process drives the spontaneous self-assembly of higher order oligomers, and the ordered transmembrane protein ensembles were verified experimentally. This study represents a first step toward the development of first generation computational design tools. However, full realization of de novo design will require development of in silico force fields that are parameterized explicitly for both polar and non-polar milieu, in addition to improvements in complementary polarization metrics.
4.4. DNA-Protein Assemblies
4.4.1. Rational Design and Engineering of DNA-Protein Assemblies.
Nucleic acids offer an attractive scaffold for the design and engineering of protein assemblies, owing to the predictability of Watson-Crick base-pairing. While naturally occurring nucleic acid-protein assemblies may be comprised of either DNA or RNA, the higher stability of DNA has made it the overwhelming scaffold of choice for engineering applications. As described in Chapter 1, computational methods for the design and assembly of complex nucleic acid architectures are highly advanced, enabling the rapid generation of structures across diverse length scales, and having functional groups arrayed at specific locations and orientations.7–9
Given the high level of structural control that can be achieved using DNA, these scaffolds have emerged as exciting platforms for the assembly of enzyme cascades. In one example, single DNA chains, synthesized via rolling circle amplification,279 were used to assemble a cascade of glucose oxidase (GOx) and horseradish peroxidase (HRP).280 GOx catalyzes the oxidation of glucose to gluconic acid and hydrogen peroxide, which then serves as a substrate for HRP, resulting in oxidation of a dye molecule to produce an optical signal. Highlighting the efficacy of the assembly approach, neither the free enzymes nor the DNA template alone could produce the activity of the cascade. Moving into 2-dimensional assemblies, Willner et al. have demonstrated the construction of DNA-protein assemblies using hexagonal DNA tiles. Immobilization of different combinations of enzymes or enzyme and cofactor resulted again in activity that could not be realized using the individual components in homogeneous solution.281 In this design, an additional benefit is that the 2-dimensional DNA tiles enable precise control over the distance between components of the reaction, as shown in Figure 13.
Figure 13.
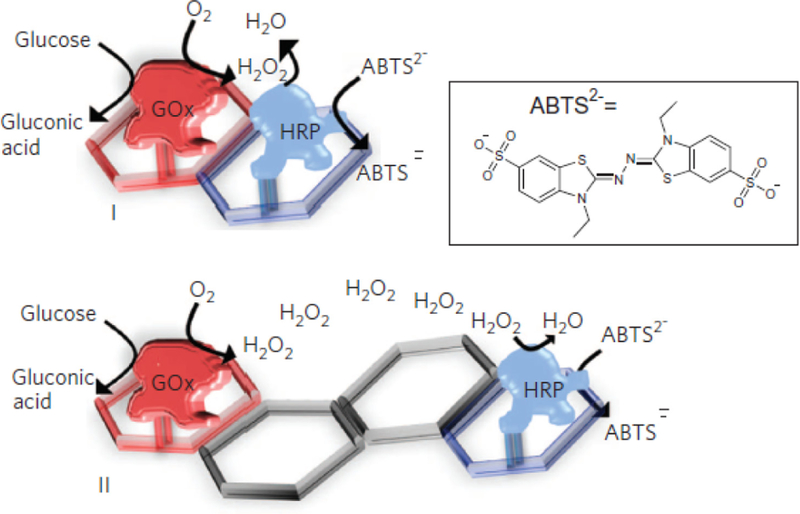
Assembly of GOx and HRP enzymes on hexagonal DNA tiles enables precise control over length scales between enzymes. Reprinted with permission from Wilner, O. I.; Weizmann, Y.; Gill, R.; Lioubashevski, O.; Freeman, R.; Willner, I. Enzyme cascades activated on topologically programmed DNA scaffolds. Nature Nanotechnol. 2009, 4, 249–254. Copyright 2009 Springer Nature.
4.4.2. Computational Methods for Design of DNA-Protein Assemblies.
The computational design of DNA sequences and structural elements, such as hairpins and turns, as well as protein secondary and tertiary structures, has undergone rapid progress owing to the increasing availability of comprehensive sequence and structure databases. However, the corresponding modeling and prediction of DNA-protein assemblies is far less advanced and mostly relies on empirical data from a limited number of systems. Of special interest are methods to predict overall geometric structure and to forecast the dependence of overall functional traits, such as global enzymatic activity of a cascade of proteins, on the distance between individual members of the cascade. Baker et al. recently presented the current state of protein-DNA interface design via the Rosetta design suite.282 The main testing ground for these in silico protocols has been the LAGLIDADG endonuclease family of DNA-cleaving enzymes. Computational methods (Rosetta) applied to this system are limited to designing endonuclease variants that can accommodate small numbers of target site substitutions. In a recent study, Mayo et al. leveraged iterative computational protein design and docking models to create a protein-DNA co-assembling nanomaterial.283 Namely, a homodimerization interface was engineered onto the Drosophila Engrailed homeodomain, allowing the dimerized protein complex to bind to two double-stranded DNA molecules. Varying the arrangement of protein-binding sites on the DNA strand (followed by the mixing of protein and engineered DNA building blocks), a nanowire with single-molecule width can spontaneously form. While the fundamental method for designing preliminary DNA-protein assemblies has been demonstrated, more advanced procedures that incorporate DNA flexibility and other properties are necessary for reliable modeling of more extensive target site changes and complementarity.
4.5. Protein-Protein Assemblies
4.5.1. Engineering of Functional Protein Assemblies Using VLPs.
VLPs are generated by self-assembly of capsid proteins into geometrically well-defined structures, and can present a near-spherical interior space suitable for encapsulation of functional proteins. The dimensions of these spaces coupled with the ability of small-molecule substrates to diffuse through small pores in the VLP structure makes these scaffolds especially well-suited for the encapsulation of multiple enzymes involved in a cascade reaction. Given the close proximity of the enzymes when encapsulated, reaction intermediates are only required to travel short distances, often protecting them from unwanted side reactions.
A synthetic metabolon was constructed by co-expressing the icosahedral VLP from bacteriophage P22 along with three enzymes: tetrameric β-glucosidase CelB to hydrolyze lactose to glucose and galactose, and both the dimeric ADP-dependent glucose kinase (GluK) and the monomeric ATP-dependent galactose kinase (GalK) to catalyze the formation of glucose-6-phophate or galactose-1-phosphate, respectively, in parallel reactions.284 Light-scattering coupled HPLC-SEC demonstrated that, on average, 61 CelB-GluK-SP units (SP = scaffolding protein) were packaged within one capsid, corresponding to a local protein concentration of 218 g/L, close to the calculated intracellular value of 300 g/L.239 Under optimum conditions for the activity of both enzymes, no channeling effect was observed, as the overall kinetic performance of packaged CelB-GluK-SP units was equal, but not superior to, that of CelB and GluK packaged separately, or of the two free enzymes in solution. Thus, proximity alone is not sufficient for enhanced activity. However, the authors showed that if the activity of the first enzyme, in this case CelB, were to be lowered by inhibition, an increase in KM value due to specific conditions, or a decrease in concentration relative to the subsequent enzyme, then a mathematical model predicts that channeling would be observed, with up to several-fold advantage of the VLP-enzyme assemblies compared to enzymes in solution.284
Another VLP system which has been highly amenable to engineering is the bacteriophage Qβ VLP, self-assembled from 180 copies of a 14.2-kD truncated capsid protein to give a 28.5 nm diameter spherical capsid (Figure 14). A variety of point mutations can be made to the Qβ capsid at solvent-exposed residues to give variants that are still expressed and assemble in high yields, and form particles of exactly the same size, shape, and quaternary structure as the starting (“wild-type”) particle.285 These simple mutations can generate large changes in the overall charge of the capsid, for example, the K16M mutation removes 180 positive charges from the external surface of the capsid, whereas K13E reverses the charge from positive to negative at 180 solvent-exposed sites. Modulating the charge state of these particles can in turn impact their ability to bind other biomolecules such as heparin.286 One key challenge to the use of VLPs for the construction of enzyme cascades is that significant amounts of bacterial and plasmid RNA are packaged during VLP expression, limiting available space for enzymes. However, in the case of Qβ, this RNA can either be biased in its composition287 or removed almost entirely by a hydrolytic procedure.288 Further increasing the flexibility of functionalization, Qβ VLPs can be expressed as “hybrid” particles containing extensions on the exterior surface at a randomly-distributed subset of subunits that form the particle.289–291 This is accomplished by co-expressing in the same E. coli cell a mixture of wild-type and extended coat proteins. When the extension is added to either the N- or C-terminus, the larger protein self-assembles with the co-expressed regular (truncated) protein in amounts that are proportional to their relative expression levels. These particles are not homogeneous, but rather exist as an ensemble having a range of ratios between wild-type and extended monomers. However, considerable flexibility is afforded to vary the size, charge, and conformation of the extensions to enable attachment of functional proteins. For example, Qβ VLPs have been produced in high yields having multiple copies of encapsulated fluorescent proteins or enzymes.287,292 The enzymes have been shown to retain activity, and may be significantly more stable toward thermal denaturation compared to the corresponding free enzymes in solution.
Figure 14.

Ribbon diagram representation of the Qβ capsid protein (pdb: 1 qbe). The C74-C80 disulfide bonds are depicted in yellow. Reprinted with permission from Fiedler, J. D.; Higginson, C.; Hovlid, M. L.; Kislukhin, A. A.; Castillejos, A.; Manzenrieder, F.; Campbell, M. G.; Voss, N. R.; Potter, C. S.; Carragher, B.; Finn, M. G. Engineered mutations change the structure and stability of a virus-like particle. Biomacromolecules 2012, 13, 2339–2348. Copyright 2012 American Chemical Society.
4.5.2. Computational Methods for Design of VLP-Protein Assemblies.
As genetically encodable proteins, the building blocks of VLPs are amenable to the same computational treatment as other proteins to predict their folding and assembly properties. However, for assemblies such as VLPs from Qβ, properties including the overall structure or size distribution of the assembled VLPs cannot yet be precisely designed using computational approaches. Nonetheless, some progress has been made toward this end. In a collection of studies, computational design was leveraged to produce nascent VLP systems.293–295 In an initial study, computational design enabled the generation of non-natural co-assembling, two-component, 120-subunit icosahedral protein nanostructures.295 Experimental characterization of the synthetic icosahedral systems revealed molecular weights and dimensions comparable to those of small viral capsids – i.e., 1.8–2.8 megadaltons and 24–40 nanometers in diameter, respectively. In a companion study, 25-nanometre icosahedral nanocages that self-assemble from trimeric protein building blocks were developed via rational (computational) protein design.294 A unique feature of these designed systems is the ability to intercalate engineered protein pentamers in the center of each of the 20 pentameric faces to modulate the size of the entrance and exit channels of these synthetic nanocages. Finally, the question of whether synthetic icosahedral protein nanostructures can be evolved to acquire additional functions was investigated in a follow-up study.293 The purpose of this study was to investigate whether putative (non-natural) VLP-like systems can be tuned by way of laboratory evolution to acquire virus-like properties, and this was accomplished by generating diversified populations using E. coli as an expression host. Taken together, this collection of studies lays an important foundation that can be used toward the de novo design of VLP systems having prescribed properties.
4.6. Remaining Challenges and Future Directions
While significant amounts of empirical knowledge have been gathered for functional proteins in structured environments, their rational design has not yet advanced in corresponding fashion. Towards the rational design of surfactant- and lipid-protein assemblies, reliable information about phase diagrams of more surface-active agents is required, with the goal of rapidly identifying regions of the phase diagram containing surfactant- or lipid-containing assembled phases. Geometric and physicochemical models, currently available only in rudimentary forms, would significantly accelerate mapping of these phase diagrams and ultimately enable prediction of such phases for molecularly well-characterized surfactants or lipids.
For assemblies based on DNA-protein or protein-protein interactions, the design rules for constructing synthetic DNA or genes and expressing the corresponding proteins are comparatively well-developed and protocols for the generation of variants are now almost routine. While many examples of the engineering of VLPs resulting in altered size, geometry, or interactions have been described, the correspondence between amino acid sequence of the building block assembling into a VLP and its geometric properties cannot be described systematically. In the case of loading functional proteins onto or into VLPs, we have barely scratched the surface: determining optimal linker length and sequences between the coat protein and functional protein or controlling functional protein density within a VLP represent just two of the future goals for moving this field forward. Generation and maintenance of a database analogous to the Protein Data Bank for assemblies of functional proteins with DNA or structural proteins would open the field to the power of sequence-based correlation between the structure and function of the corresponding assemblies, enabling the fully predictive design of assemblies having new functions.
5. TRANSCRIPTION FACTOR ASSEMBLY
5.1. Introduction to Transcription Factor Assemblies
Transcription factors (TFs) are macromolecular assemblies that control gene output by modulating the rate of mRNA production at specific sites in the genome. TFs are integral to the normal development of an organism, guiding routine cellular functions including metabolism and cell growth, and thus TFs can also play a role in the cause of, or response to, disease. Transcription factors are a diverse family of proteins that interact with specific regions of DNA, termed enhancers or promotors, to initiate transcription of RNA. Accordingly, transcription factors can be classified based on their requisite DNA binding domain. TFs can work alone or with other proteins such as activators, which also enhance transcription. An exhaustive review of all transcription factors is beyond the scope of this article, but several reviews and data bases have been developed that provide a broad assessment of TFs, and these are excellent resources for information regarding the general features of TFs and the scope of different transcription factors and TF families.12 In this Review, we will focus on the transcription factor model system lactose repressor (LacI), with the goal of exploring the intermolecular interactions that lead to TF assembly, and how an understanding of these interactions can enable TF engineering. LacI is a unique system, in that it has been extensively studied biochemically, biophysically, and genetically. Moreover, LacI has been shown to have broad biotechnological importance, and thematically (i.e., following the story arc outlined in Chapter 1) represents an assembly system that has progressed from observation to predictive design, at least in part. LacI is a native transcription factor to E. coli and has served as the model system for understanding transcriptional controls. In addition, LacI has been used as the workhorse for developing our extensive understanding of the role of allosteric communication during the process of gene regulation.
5.2. The Lac Operon as a Model for Transcription Factor Assembly
5.2.1. Introduction to the Lactose Repressor.
The LacI transcription factor is the master controller for the lac operon, and several reviews are available that specifically focus on the elements of the lac operon and the LacI transcription factor.296–299 In this chapter, we will address the fundamental aspects of the lac operon in relation to the elucidation of the structure and function of the principal transcription factor, LacI. In 1961, Jacob and Monod proposed a scheme for the utilization of alternate carbon sources in E. coli, implicating the transcription factor LacI as the primary regulatory protein of the lac operon (Figure 15).300 Jacob and Monod postulated that the ratios of glucose (primary carbon source) to lactose (secondary carbon source) influence the expression of three genes: beta-galactosidase (lacZ), lactose permease (lacY), and thiogalactoside transacetylase (lacA), all of which aid in the metabolism of lactose. Experimental studies subsequently identified a putative transcription factor (LacI) that regulates transcription of the lacZ, lacA and lacY genes301,302 via a specific interaction between LacI and a region of DNA referred to as the operator.303,304 A critical binding event was also elucidated between LacI and the natural inducer ligand allolactose, an isomer of lactose generated via the product of lacZ.300,305,306 Functionally, the lac operon has three modes in which it exerts control over the transcription of the polycistronic lac ZYA mRNA: (i) glucose-depleted environment in the presence of relatively high levels of the alternate carbon source lactose; (ii) an environment having both glucose and lactose present in high concentration; (iii) lactose-depleted environment having an abundance of glucose (Figure 15).
Figure 15.

Function of the lactose operon is dependent upon available carbohydrate sources. Changes in carbohydrate binding impact assembly, and thus function.
In the absence of lactose, the LacI transcription factor is expressed in high abundance, and binds to the operator DNA with high affinity, upstream of the genes lacZ, lacY, and lacA307,308 and downstream of the promotor. This binding event physically prevents RNA polymerase from initiating transcription,309–311 dramatically reducing mRNA production from lacZ, lacY, and lacA, which encode three proteins involved in lactose metabolism — β-galactosidase, lac permease, and thiogalactoside transacetylase, respectively (Figure 15).296,300,312,313 Mechanistically, lac permease transports the alternate carbon source lactose into the cytosol. Lactose is subsequently catabolized into galactose and glucose via β-galactosidase. To maintain cell viability, thiogalactoside transacetylase transfers an acetyl group from coenzyme A to the hydroxyl group of galactosides.313 Basal levels of β-galactosidase and lac permease expression facilitate the transport of lactose. β-galactosidase also catalyzes the isomerization of cytosolic lactose to the inducer ligand 1,6-allolactose.314,315 The alternate sugar 1,6-allolactose binds specifically to LacI, which reduces the affinity of the transcription factor for the operator DNA, initiating transcription of the polycistronic lac ZYA mRNA.306
When both glucose and lactose are present, low levels of polycistronic lac ZYA mRNA are still produced, as the ligand-bound LacI transcription factor is dislocated from the operator DNA. However, E. coli will preferentially metabolize glucose, eventually leading to depletion.300 Once the primary carbon source is depleted, adenylyl cyclase produces high levels of cAMP from ATP,316 which subsequently binds to cyclic AMP-dependent catabolite activator/repressor protein (CAP). The CAP-cAMP complex binds to a DNA sequence located upstream of the promoter region, increasing the affinity of RNA polymerase for the promoter and resulting in a 50-fold increase in the expression of the lac ZYA genes.317 Thus, the process originally observed by Jacob and Monod for the selective utilization of available metabolites300 can be reconciled mechanistically by our understanding of the assembly of the transcription factor LacI and corresponding interactions with ligands and operator DNA developed over the past four decades.
A variety of allolactose analogs can induce the wild-type LacI transcription factor,300,318 with the most commonly used example being isopropyl-β,D-thiogalactoside (IPTG). LacI transcription factor coupled with IPTG (and corresponding pseudo–palindromic DNA operator) has served as a key component in the development of a broad range of synthetic genetic circuits,319–321 and is commonly leveraged in biotechnology to regulate bespoke gene expression by substituting lacZYA DNA with any gene of choice. Accordingly, the LacI transcription factor and corresponding operator DNA have become a technological juggernaut, finding wide use in mass protein production, synthetic biology, and related fields. At a molecular level, the lactose repressor requires protein-protein, protein-DNA, and protein-ligand assembly to function. In the following sections, we will describe in detail each type of assembly and the role of these interactions with respect to the overall function and engineering of the transcription factor system.
5.2.2. Elucidating the Structure of the LacI Transcription Factor.
Structurally, LacI is a homo-tetrameric protein comprised of identical monomers (Figure 16). The structure of LacI was elucidated via several crystallographic and NMR studies, developed over a decade by numerous structural biology laboratories.299,322–324 Each LacI monomer is 360 amino acids in length, with a molecular weight of approximately 37.5 kDa, and is composed of a DNA binding domain (half-site), a regulatory core domain that encompasses the ligand binding site, and a C-terminal tetramerization domain (Figure 16).322,325 The regulatory core forms the monomer-monomer interface used for protein dimerization, and contains the allosteric region, and the tetramerization domain facilitates the dimerization of minimal functional LacI dimer units. Thus, the overall assembly of the LacI transcription factor can be described as the dimerization of dimers, with a total molecular weight of 150 kDa.322,325 Extensive experimental studies have shown that upon ligand binding, LacI undergoes a conformational change that reduces the affinity of the transcription factor for the operator DNA. These ligand-induced global conformational changes for LacI were initially observed via spectroscopic and hydrodynamic studies, and have been attributed to an allosteric response.326,327
Figure 16.
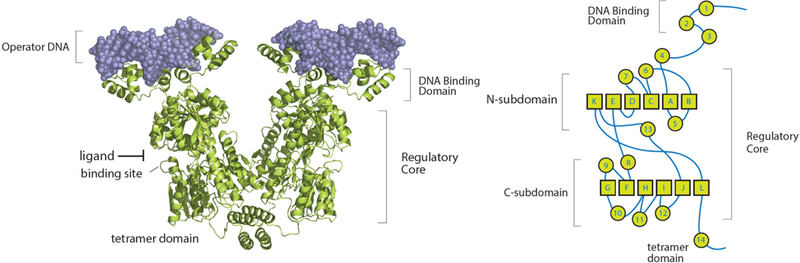
Structure of tetrameric LacI-Osym assembly with domains labeled (left). LacI monomer topology and domain structure (right). Note that the topology of folding involves three cross-overs between the N- and C-subdomains.
Residues 1–60 encompass the DNA binding domain, which includes the hinge region (residues 50–60). From a functional vantage point, the LacI dimer is regarded as the minimal functional unit capable of interacting specifically with operator DNA. Dimerization is required for LacI function because each monomer only possesses a DNA binding domain half-site and thus two copies are required to form a complete DNA binding domain capable of interacting with the full operator sequence. Once the functional unit is bound to operator DNA, the DNA binding domain of the transcription factor folds into a classic helix-turn-helix motif328 that fits into the major groove of operator DNA, burying more than 3300 Å2 of solvent accessible surface area.322,329 The protein-DNA interaction is facilitated by an intricate network of electrostatic interactions between amino acid sidechains and the phosphate backbone, which are consistent with the physicochemical properties identified for protein-DNA assemblies discussed in Chapter 4 (Figure 16).
Subsequent to dimerization of the DNA binding domain, the regulatory core is formed. The LacI core domain (residues 60–340) is composed of two subdomains having similar structures.322,325 Specifically, the N- and C-subdomains are interwoven with three crossover points between the two subdomains, thus the subdomains are not autonomous. The cleft between the subdomains forms the ligand binding site. The N-subdomain is regarded as the allosteric region, and upon ligand binding, the signal is propagated through this domain to the DNA binding domain. Distal to the N-subdomain, the C-subdomain is formed, and provides the requisite protein-protein interactions for dimerization of monomers, producing the minimal functional unit.
The C-terminal region from residues 340–360 represents the tetramerization domain. Once a pair of minimal functional units are formed via the C-subdomain,330,331 the tetramerization domain forms a highly stable interface composed of a four-helix bundle, arranged as anti-parallel coiled-coils.322 In addition to facilitating the dimerization of functional units, the tetramerization domain confers 10 kcal/mol of additional stability to the LacI structure. Functionally, the LacI tetramer can interact with two independent DNA operators, facilitating higher-order gene suppression.
5.2.3. LacI Function – Assembly Required for Cooperative Communication.
Allosteric communication is critical to the function of the LacI transcription factor. Upon ligand binding, the transcription factor must propagate this signal to the DNA binding domain to initiate transcription. X-Ray crystallographic studies comparing LacI with and without the IPTG inducer bound reveal two distinct protein conformations.322,325 The protein-ligand complex, relative to the un-liganded transcription factor, shows that the C-subdomain remains fixed, while the N-subdomain undergoes a conformational change. Structurally, ligand binding results in a 6° rotation and 4 Å translation of the N-subdomain. This conformational change decreases the affinity of LacI for the operator DNA, allowing RNA polymerase to transcribe genes downstream of the operator. In addition to the conformational change to the N-subdomain, ligand binding also results in the un-structuring of the DNA binding domain, making this structure undetectable by X-ray crystallographic analysis. Phenotypically, the concerted conformational rearrangement of the N-subdomain in tandem with the un-structuring of the DNA binding domain is observed as a cue to initiate transcription of the genes downstream of the operator for the metabolism of alternate carbon sources (Figure 15). It is this induction method that has become invaluable for large-scale protein production in biotechnological, biomedical, and industrial applications.
The lac operon is an ideal model for studying negative and positive control over gene expression. As presented, the LacI transcription factor principally functions as a dimer, and thus the fully assembled tetrameric LacI can interact with two independent DNA operators. Studies have shown that in E. coli, LacI can interact with three native DNA operators – the primary pseudo-palindromic operator O1 and two auxiliary operators (O2 and O3),307,332,333 (Figure 17). The natural auxiliary operators of the lac operon exhibit different affinities for LacI. Moreover, the auxiliary operators O2 and O3 are positioned just downstream and far upstream, respectively, of the O1 regulatory operator that is proximal to the promoter.332–338 In vivo, O1 works in collaboration with auxiliary operators to form extensive repression loops in the presence of LacI. Specifically, In E. coli, LacI tetramer binds simultaneously to a promoter-proximal DNA binding site (O1) and an auxiliary operator, resulting in a DNA loop, which increases repression efficiency. Functionally, when a repression loop is formed, gene suppression is increased 50-fold.339–344 Goodson et al. propose that these obstinate DNA loops near the operator can be used to accelerate re-repression upon exhaustion of inducer.345
Figure 17.
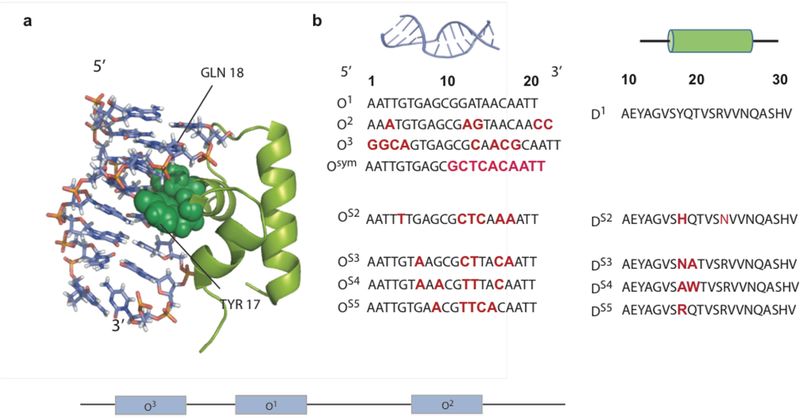
Protein DNA Assemblies. (a) Structure of DNA binding domain and operator DNA (Osym). (b) Wild-type operator DNA (O1) and auxiliary operators O2 and O3. O1, O2, O3, and Osym all assemble with wild-type the DNA binding domain D1. Variations of operator DNA (OS2, OS3, OS4, and OS5) assemble with orthogonal DNA binding domains (DS2, DS3, DS4, and DS5).
5.3. Modeling the Assembly and Function of LacI
5.3.1. Modeling Allosteric Communication in the Functional Assembly.
Biochemical and X-ray crystallographic studies have been essential to gaining a general understanding of lac repressor function. Specifically, the crystal structures corresponding to the induced and repressed states of LacI reveal two important protein conformations.322,325 However, these static structures do not reveal any information regarding the mechanism by which allosteric binding of a ligand induces conformation change between the repressed and induced states. An in silico study conducted by Ma et al. utilized a molecular mechanics simulation to elucidate important details of the putative allosteric pathway between the repressed and induced states of the LacI transcription factor.346 In this study, targeted molecular dynamics (TMD) was used to calculate allosteric trajectories with atomic level detail. Canonical molecular dynamics (MD) simulations are valuable tools used to investigate protein dynamics at discrete temperatures, however, all-atom MD simulations are typically computationally expensive, and thus are restricted to very modest time and length scales. Accordingly, an unrestricted MD simulation for a process such as the propagation of an allosteric signal is not practical. TMD overcomes this limitation by restricting calculations through the use of a significantly reduced conformational search space, thereby allowing for an assessment of processes that have considerably longer time and length scales. Specifically, TMD uses anchoring structures of the initial and final assembly states being investigated. In the study by Ma et al., the two anchoring structures were those of the repressed state (protein-protein-DNA assembly) and induced state (protein-protein-ligand assembly) of the functional unit. The anchoring structures were acquired from existing high-resolution crystallographic structures, highlighting the complementary use of experimental data and computational modeling.
TMD simulation of the allosteric transition between the repressed and induced states did not reveal any significant conformational rearrangement within the C-subdomains of the dimer. Given the existing structural data, this result was expected and served as a control to validate the accuracy of the simulation. In contrast, the N-subdomains undergo significant interpolated motions during the transition between states, and the TMD simulation suggested that the allosteric signal originates asymmetrically within the LacI N-subdomains. The trajectory was followed starting from the inducer-binding site of one monomer and propagating to the other monomer via three interconnected pathways. All three pathways propagated the allosteric signal by way of a series of noncovalent interactions. In the first pathway, conformational changes were found to be restricted to one monomer, and move from the IPTG-binding pocket, through the N-subdomain β-sheet, to a hydrophobic cluster at the top of this region proximal to the DNA binding domain. A similar transition subsequently occurred to the same areas on the opposing monomer. The observed motions resulted in changes at the sidechains that form the interface with the DNA-binding domains and residues that reside at the monomer–monomer interface. The second pathway produced a reorganization across this subunit interface, forming a putative intermediate state. Pathway three extends from the rear of the inducer-binding pocket, and transverses the monomer–monomer interface. It is important to note that none of the putative intermediate states detected in silico have been observed experimentally. However, the results from the simulated TMD trajectories agree overall with experimental biochemical and genetic studies. This TMD study reveals an important spatial and temporal edifice for allosteric communication within the LacI functional unit that could not be achieved via experimental assessment alone. In addition, this study has established a valuable platform that can be used to generate new and exciting hypotheses, which in turn can drive the development of novel experimental studies.
5.3.2. Modeling and Elucidating the Folding Mechanism of monomeric LacI.
Given the complex topology of the LacI monomer, folding (or partial folding) is a likely requirement preceding protein assembly. In a study conducted by Wilson et al., the experimental folding mechanism for the LacI monomer was elucidated.347 Many large proteins (>100 residues) fold by way of one or more populated intermediate states, i.e. in both the kinetic and equilibrium folding reactions.348,349 Moreover, as outlined in Chapter 2, protein assembly can significantly complicate the protein folding mechanism, rendering simple equilibrium studies insufficient to capture all of the relevant mechanistic details required to fully understand the folding and assembly processes. Accordingly, to conduct the initial kinetic folding study of LacI, Wilson et al. reduced the tetrameric LacI to a monomer through a Leu251Ala perturbation to the monomer-monomer interface of the C-subdomain, combined with elimination of the last eleven residues of the teteramerization domain. The rationale behind these two perturbations is that modification to position 251 disrupts the protein assembly without affecting the topology, but the monomer cannot be fully liberated without partial elimination of the teteramerization domain.
The resulting monomeric variant of LacI (MLAc) was purified and characterized using hydrodynamic methods including molecular sieve chromatography and analytical ultra-centrifugation. These experiments revealed a molecular weight and shape factor consistent with a folded LacI monomer. MLAc was then assessed for function using isothermal titration calorimetry (ITC) to probe the thermodynamics of IPTG binding. ITC thermograms showed that MLAc retains the same capacity and enthalpy profile for ligand binding as the wild-type transcription factor. Taken together, these data suggest that MLAc is structurally well-ordered and functional, making it an excellent model for studying the LacI monomer in solution. After characterization of the monomeric variant, stopped-flow kinetic experiments were used to elucidate both the unfolding and re-folding reactions for MLAc. These experiments utilized the chemical denaturant urea to ensure that all processes were reversible. Global assessment of MLAc refolding using circular dichroism revealed a three-state refolding reaction. Interestingly, the local assessment of tryptophan florescence revealed a four-state refolding reaction. Both the florescence and circular dichroism refolding assessments presented burst-phase reactions that occurred within the dead time of each instrument (~ 3 milliseconds), thus this phase of the reaction could not be fully resolved experimentally.
Given the limitations of the experimental study of MLAc folding, a complementary study was conducted in silico. Unlike the experimental assessment of the folding reaction, computational studies are well suited for processes that occur on sub-millisecond timescales. However, an a priori prediction of a folding reaction of this scale is not practical given the uncertainty regarding the accuracy of model parametrization, in addition to the need for proper selection of reaction order parameters. However, a comprehensive assessment of the folding mechanism of MLAc can be achieved by pairing experimental folding kinetics data with a theoretical assessment of the MLAc folding reaction via a course-grain molecular mechanics model. This strategy has two advantages: (i) the computational assessment can be achieved on a timescale that is on par with the experimental study; (ii) the assessment of theoretical landscapes can be guided by experimental results. As mentioned above, all-atom MD is far too computationally expensive to cover the time and length scales of a protein folding reaction for a larger protein. Thus, in this study the authors leveraged a course-grained model350 that reduced each sidechain to a unified bead. Hence, MLAc was modelled as beads on a string with information regarding the final topology remaining intact.347,351 In this landmark study, theoretical predictions of both the detailed folding mechanism and global and local structural changes were found to be nearly identical to experimental results (Figure 18). Moreover, computational analysis revealed detailed information regarding the burst-phase, which had proven to be too fast for experimental observation. In conclusion, simulation combined with experimental results produced a folding mechanism for MLAc at a level of confidence that could not be achieved by simulation or experiments alone, and the resulting insight into the intermediate folding states provides important structural details that can be leveraged to address the corresponding assembly mechanism. Notably, the LacI protein system builds on many of the fundamental peptide structural assembly motifs discussed in Chapter 3 (Figure 9).
Figure 18.
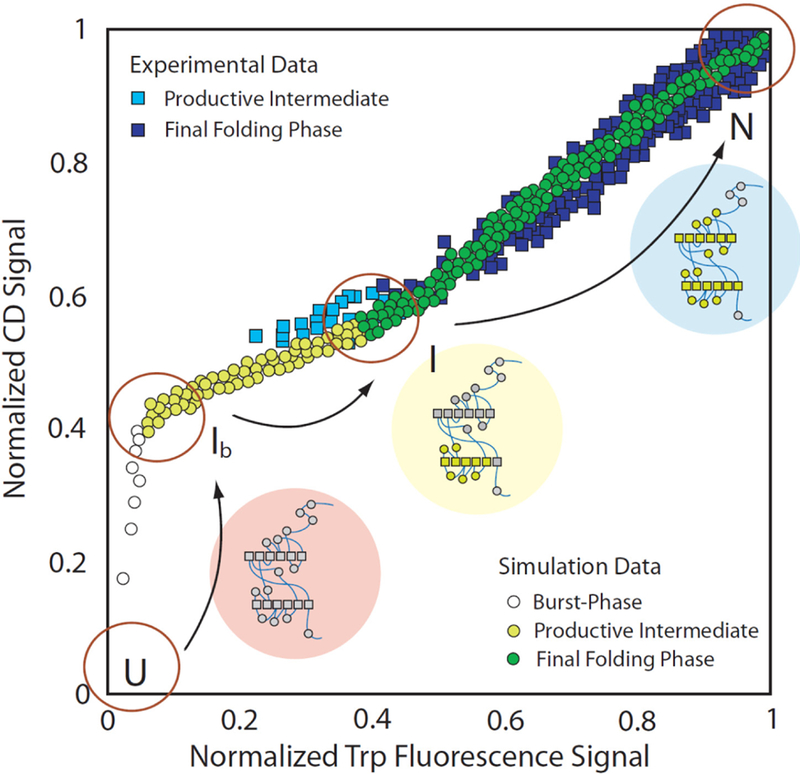
Comparing theoretical prediction to experimental data for LacI folding. Normalized CD signals are plotted as a function of fluorescence signals and color coded by phase (squares = experimental; circles = theoretical).
5.3.3. Elucidating the Folding and Assembly Mechanism of LacI.
In a follow up study, Wilson et al. leveraged the resulting MLAc folding landscape to evaluate the folding and assembly mechanism for dimeric and tetrameric LacI transcription factors.352 Conceptually, the energy landscapes and corresponding reaction coordinates that govern the protein folding reaction are similar to the physical interactions and mechanisms required in protein assembly mechanisms. However, protein assembly mechanisms that occur concurrently with protein folding are considerably more challenging to reconcile and deconvolute, as stated in Chapter 2.353 Guided by the theoretical-experimental mechanism of MLAc folding, Wilson et al. proposed new kinetic experiments to assess and deconvolute folding and assembly mechanisms of oligomeric LacI. In this study, the folding of dimeric LacI was monitored via circular dichroism (CD), intrinsic fluorescence, and Förster resonance energy transfer (FRET), with variation in protein concentration. Using stopped-flow kinetic experiments similar to those in the MLAc study, the experimental dimer folding-assembly mechanism was elucidated as a four-phase folding reaction in which the first three transitions are tantamount to monomer folding. The final reaction phase was attributed to protein-protein dimer assembly. Including the resolved burst phase from the theoretical-experimental mechanism of MLAc folding, the dimeric transcription factor folds and assembles via a five-state mechanism. The assembly reaction phase of the dimer is influenced by protein concentration and was independently observed using inter-molecular FRET experiments. Interestingly, unlike dimer formation, the folding and assembly reaction for the tetrameric LacI transcription factor is not influenced by protein concentration. This lack of concentration dependence is hypothesized to result from strong tethering of the monomers via the tetramerization domain. From this study, the authors concluded that the folding-assembly mechanism of the LacI tetramer is initiated by the rapid assembly of the unfolded monomers via the tetramerization domain, which simplifies the folding and assembly reactions by reducing the search space, trapping folded proteins as dimers once the folding reaction is completed.
5.4. Engineering and Designing the LacI Transcription Factor
5.4.1. Engineering Alternate Assembly by Modifying Allosteric Communication.
Allosteric communication is a hallmark of many transcription factors used to control gene expression and has enabled synthetic biologists to reprogram cells to function as sensors,354,355 toggle356 and memory switches,357 and biological clocks.358,359 The lactose repressor (LacI) is the standard model system for studying allostery and understanding how transcription is controlled by using small metabolites to modulate protein association with specific DNA sites.360 In a recent study, Wilson et al. used random mutagenesis laboratory evolution to re-route allosteric communication in the LacI scaffold to confer anti-lac functionality.361,362 (Figure 19). Anti-lac function is the inverse of wild-type LacI function, where a given anti-lac cannot bind to the operator DNA in the absence of IPTG, but binds specifically to the operator DNA when in complex with IPTG. Conferring anti-lac function required an initial block in allosteric communication, which was accomplished via an Is mutation. This single perturbation blocks transmission of the allosteric signal allowing a given Is variant to remain bound to DNA in the presence or absence of IPTG. With the allosteric block in place, error-prone PCR (EP-PCR) was used to randomly introduce compensatory mutations to the regulatory core in the N- and C-subdomains. The resulting DNA sequence space was evaluated using a screen in which the gene for green florescent protein (GFP) is placed downstream of the O1 operator DNA, such that transcription of the reporter gene is under the control of the LacI variants. Using this engineering approach, the authors produced more than a dozen anti-lac variants having re-routed allosteric communication. For these anti-lac variants, GFP is expressed in the absence of inducer, and the presence of IPTG triggers formation of a LacI-ligand-DNA assembly, which suppresses gene expression.
Figure 19.

Selected examples of engineering of the lac repressor.
While many of the mutations that confer anti-lac function occur within the allosteric N-subdomain, several of the anti-lac variants produced by random mutagenesis have mutations exclusively within the C-subdomain. The authors suggest that C-subdomain perturbations influence the orientation of the monomer-monomer assembly in a given anti-lac. Considering that the C-subdomain remains fixed during the allosteric transition, it is hypothesized that in these mutants, re-orientation of monomer assembly may be responsible for abrogated protein-DNA assembly, for example by perturbing the orientation of the N-subdomains such that they cannot properly position the DNA-binding domains to form an assembly with operator DNA. However, upon ligand binding, the N-subdomains could undergo a conformational change that aligns the system to a repressed configuration, rather than the canonical induced state, resulting in assembly and DNA binding to suppress gene expression. With variations in allosteric routes (and in some cases subdomain orientation), this collection of anti-lac variants displays a variable dynamic range and alternate ligand sensitivity, which is anticipated to be of use in a broad range of biotechnological applications.
5.4.2. Engineering Alternate Protein-DNA Assemblies.
The native DNA-operator site O1 is a 20 base pair pseudo-palindrome composed of two asymmetric half-sites (Figure 17). The left half-site of O1 is believed to contribute more to TF binding, as evidenced by its low mutational tolerance relative to the right half-site of O1. Based upon these observations, in 1983 Sadler et al. designed a perfectly symmetric palindromic DNA operator (Osym) variant fashioned after the left half-site of the natural lac O1 operator.363 By repairing the “flaws” to the right half-site, the wild-type LacI transcription factor bound to Osym with 10-fold higher affinity compared to the native operator O1.
In a subsequent study, Sartorius et al. simultaneously re-designed the DNA-binding domain and the corresponding DNA operator sequence to create a LacI-DNA assembly that is orthogonal to the wild-type assembly.364 Briefly, amino acid positions 17 (tyrosine) and 22 (arginine) were mutated to histidine and asparagine, respectively to create LacIS2. In addition, the operator DNA Osym was modified at positions 5 and 16, representing symmetric changes to each half-site, to create OS2 (Figure 17). Modification of the operator DNA to OS2 disrupts wild-type LacI protein-DNA assembly, and a novel assembly between LacIS2 and OS2 was observed. Using a similar engineering strategy, Zhan and coworkers created three additional orthogonal LacI-DNA assemblies.365
Inspired by the fact that protein dimer assembly is required for formation of the minimal functional unit, Daber and Lewis rationalized that the creation of a heterodimeric monomer-monomer interaction could result in a heteromeric LacI variant capable of interacting with operator DNA composed of two different half-sites (Figure 19)366 Accomplishing this first required the development of a non-symmetric operator sequence that could interact specifically with the heterodimeric complex. By virtue of the asymmetry of the new operator, no assembly with wild-type LacI was possible, rendering it orthogonal to the native transcription factor. The two monomers that compose the heterodimer were then engineered such that each monomer was comprised of a unique DNA binding domain that recognized a distinctive operator DNA half-site. The DNA binding domains were engineered by construction of a DNA sequence library of approximately 8,000 mutant repressor proteins having mutations at positions 17, 18, and 22, all within the DNA binding domain.322,367 This sequence library was screened for heterodimer transcription factor activity by placing GFP under the control of the non-symmetric DNA operator. If a mutant heterodimer was functional, then GFP production would be suppressed by protein-DNA assembly in the absence of IPTG, but protein-ligand assembly would induce the production of GFP in high yield upon addition of IPTG. In a subsequent study, Daber and Lewis leveraged this heterodimeric LacI transcription factor to demonstrate that induction requires two signals,368 which is consistent with the putative allosteric mechanism projected from the molecular dynamics study described above.
5.4.3. Engineering Alternate Protein-Ligand Assemblies Using Computer-Aided Protein Design.
In 1975, Barkley and coworkers demonstrated that LacI could bind and respond to a broad range of ligands.318 The benefits of alternate ligand binding in vivo are self-evident, as this is a hallmark of the lac-operon function. Additionally, researchers recognized that this characteristic could be powerfully harnessed for biotechnological applications in which the LacI transcription factor exclusively recognizes and responds to unique ligands that are orthogonal to IPTG. Toward this end, Ramand and coworkers leveraged computer-aided protein design (CaPD) combined with directed evolution to generate LacI variants capable of binding gentiobiose, fucose, lactitol, and sucralose. (Figure 19). None of these ligands interact with native LacI, and thus the engineered systems could offer orthogonal control over gene expression. The engineering workflow initially leveraged CaPD to create new ligand binding pockets in silico within the LacI fold. CaPD was specifically used to search a sequence space comprised of variation to residues 79–125, 148–197, and 245–296, which represents all of the relevant regions of the inducer binding pocket. In silico evaluation of a given target inducer molecule in situ was conducted, such that the computational procedure was accomplished via a series of iterations of rigid body docking, rotamer repacking, and backbone refinement as part of the optimization procedure to accommodate a given non-natural ligand. Initial candidate sequences generated by CaPD were moderately functional. A critical issue with CaPD is the reliance on predictions of protein-substrate (or in this case, protein-ligand) transition state assemblies, which are often inaccurate. One solution to this problem is to achieve “weak” function via CaPD, then carry out one or more rounds of laboratory evolution using error-prone PCR to introduce modifications and screen for improved function. The rationale behind this two-step engineering strategy is that the initial sequence space is far too large to search via directed evolution alone, and the use of CaPD reduces the search space to a more manageable level of diversity.
5.4.4. Modular Design of New Transcription Factors based on the LacI Topology.
Structural analysis coupled with large scale genetic studies has allowed for the identification of LacI functional domains, setting the stage for the modular design of transcription factors fashioned after the LacI topology. Our understanding of the LacI structure–function relationship has been expanded to the identification and study of more than 1000 homologous proteins. Curating of the LacI/GalR family has primarily been undertaken by Swint-Kruse et al..369–371 The common function of this protein family features allosteric regulation of DNA binding to modulate transcription, and each LacI homologue has evolved a unique variation in ligand specificity and DNA target sequence. To achieve this, each protein system shares analogous regulatory domains and DNA binding function. Specifically, achieving high-affinity binding to target DNA operator sequences requires the formation of homodimers. DNA binding function is paired with the biosensing function of the regulatory domain, having a similar topology and function to that of LacI. The regulatory domains are responsible for mediating homodimer formation and sensing and communicating the binding of effector molecules through allosteric communication, in turn modulating DNA binding. In principle, this collection of homologues provides a rich array of parts that can be used interchangeably in modular design to create new unnatural regulatory proteins (Figure 19).
Swint-Kruse et al. were the first to demonstrate that the modular design of homologous LacI parts could be used to create new transcription factor assemblies.372 In their initial study, the regulatory domain of purine repressor protein (PurR) was combined with the DNA binding domain of LacI. While structurally similar, PurR differs from LacI in that it binds its respective PurR-operator DNA sequence in the presence of the effector molecule hypoxanthine or guanine.373 In addition, PurR has a similar allosteric response to that of anti-lacs, with ligand binding resulting in protein-DNA assembly.374 Experimental characterization of the LacI-PurR construct demonstrated that the designed protein is capable of repressing the native LacI operator O1 and has a two-fold increase in affinity for O1 in the presence of the ligand hypoxanthine. In a subsequent study, the authors modularly designed TFs capable of assembling with O1 but using GalR, ribose repressor, fructose repressor, galactose isorepressor, trehalose repressor, and cryptoic asc operon repressor biosensing functions (Figure 19).375
5.5. Remaining Challenges and Future Directions
The lactose repressor (LacI) is among the most utilized regulatory protein assemblies in biotechnology. Our extensive knowledge of the LacI structure-function relationship has been leveraged to expand our understanding of cooperative assemblies that utilize protein-protein, protein-DNA, and protein-ligand interactions. LacI represents a complex functional assembly, in that dimerization is required for assembly with DNA, and the functional unit can only achieve allosteric communication as an assembly. Moreover, the folding and assembly of LacI are inextricably linked, where preliminary tetramer assembly dramatically simplifies the folding mechanism. Ultimately our understanding of the mechanochemical properties of the LacI structure has provided us with a viable path to move from observation to predictive design, at least in part. Looking ahead, a key challenge will be the a priori design of cooperative function.
The control of gene expression is an important tool for metabolic engineering, the design of synthetic gene networks, gene-function analysis, and protein manufacturing.319–321 The most successful approaches to date are based on modulating mRNA synthesis via an inducible coupling to transcriptional effectors, which requires a biosensing function. A hallmark of biological sensing is the conversion of an exogenous signal, usually a small molecule or environmental cue such as a protein-ligand interaction into a useful output or response. Over the past 17 years, biologists and engineers have designed many genetic parts and gene circuits, such as sensors,376 switches,356,377 and oscillators,358,378–380 that can be combined to modify existing cell functions381 and generate new ones. All of the examples above utilize the LacI transcription factor assembly (in conjunction with complementary gene regulators - e.g., tetR, araC, luxR) to control these sophisticated genetic networks. In turn, newly engineered assembly systems based on the LacI architecture promise to revolutionize the development of synthetic biology systems by conferring bespoke gene controls that are orthogonal to the native cellular machinery. This ability will expand our biological “computing power,” facilitating the development of biological programs at the scale and complexity of living systems.
6. SELF-ASSEMBLED PROTEIN SCAFFOLDS
6.1. Introduction to Self-Assembled Protein Scaffolds
The term “self-assembled protein scaffolds” covers a wide range of structures and materials that can be found in nature or created in the laboratory. Broadly defined, these assemblies are constructed from folded proteins in a manner that generates repeated quaternary structure. They can be particulate and have discrete perimeters, or be gel-like with shapes that are dependent on their container. The quaternary structure linking separate proteins can be formed by a variety of interactions including hydrophobic interactions, hydrogen bonding, electrostatic interactions, disulfide bonds, van der Waals forces, and depletion interactions (Figure 1). Additionally, hierarchical structures can be formed from many copies of quaternary structures that further assemble with a higher level of order. The number of proteins in a scaffold can range from three, in simple trimers, to more than a trillion in bulk hydrogels. As such, the size of protein scaffolds varies widely from the nanometer to centimeter range, and beyond. Scaffolds may be homo-assemblies, comprised of a single protein, or hetero- or co-assemblies comprised of several protein constituents. The self-assembled protein scaffolds found in nature illustrate the diversity of this category of materials. For example, ferritin-like cages are nanoscale quaternary scaffolds arising from assembly of both homologous or heterologous protein subunits.382 These cages can have different shapes, including spherical or octahedral, as dictated by different types of symmetry at the molecular level. Ferritin-like cages can also exist in different sizes, as dictated by the number and size of individual protein subunits, but these are typically on the nanometer scale. Collagen serves as an example of a hierarchical self-assembled protein scaffold, as three collagen type I (homo)monomers assemble into the 300 nm long triple helix collagen molecule, which can further assemble into fibrils having lengths on the micron scale.383 These fibrils can undergo an additional level of assembly, which includes the participation of other molecules and enzymatic processes, to ultimately produce the bulk material known as extracellular matrix. The breadth of naturally occurring self-assembled protein scaffolds has inspired the creation of even more diversity in the laboratory with synthetic scaffolds comprised of either modified versions of natural proteins or rationally designed proteins.
Nanoscale self-assembled protein scaffolds have been recently and extensively reviewed by Luo et al.3 Thus, this chapter will focus primarily on scaffolds having sizes on the micron scale or larger. Though the difference in size can be dramatic between nano- and micro- or macro-scale scaffolds, the mechanisms for assembly are similar. More important to mechanism is the monomer size, as scaffolds such as those described in Chapter 3 that are assembled from small peptides are characterized by different mechanisms and features compared to those assembled from full-length, folded proteins, as described in this Chapter. The scaffolds discussed in this Chapter are primarily the products of rational, modular design, and are typically constructed from protein domains that are modified versions of naturally occurring structures. This contrasts with the designer peptides described in Chapter 3, which are primarily designed and synthesized de novo.
6.2. Early History of Designing Assemblies
Though the first designers of self-assembled protein scaffolds were undoubtedly inspired by the structure and function of naturally occurring protein scaffolds, these assemblies also reflect significant influence from abiotic polymeric self-assembled “scaffolds” or materials.384 This influence can be observed in the design of the proteins and mechanisms of assembly, as many early examples of engineered protein scaffolds resemble organic block co-polymers or utilize triggered conformational changes to induce assembly.
6.2.1. Elastin-Like Polypeptide Coacervates.
Coacervates formed from elastin-like polypeptides (ELPs) represent one of the earliest synthetic self-assembled protein scaffolds. Coacervates are a liquid phase comprised of condensed hydrophobic proteins that separates from the aqueous phase. ELPs are characterized by a pentapeptide repeat, (Val-Pro-Gly-Val-Gly)n derived from tropoelastin. In early work, these proteins were relatively small with 10 ≤ n ≤ 15. At a specific transition temperature, ELP undergoes a temperature-responsive inverse phase transition from soluble to coacervate phase, as illustrated schematically in Figure 20a.385 The soluble phase is disordered, whereas in the coacervate phase, the ELP scaffold contains both filamentous and amorphous regions and exhibits circular dichroism spectra for Type II β turns.386
Figure 20.
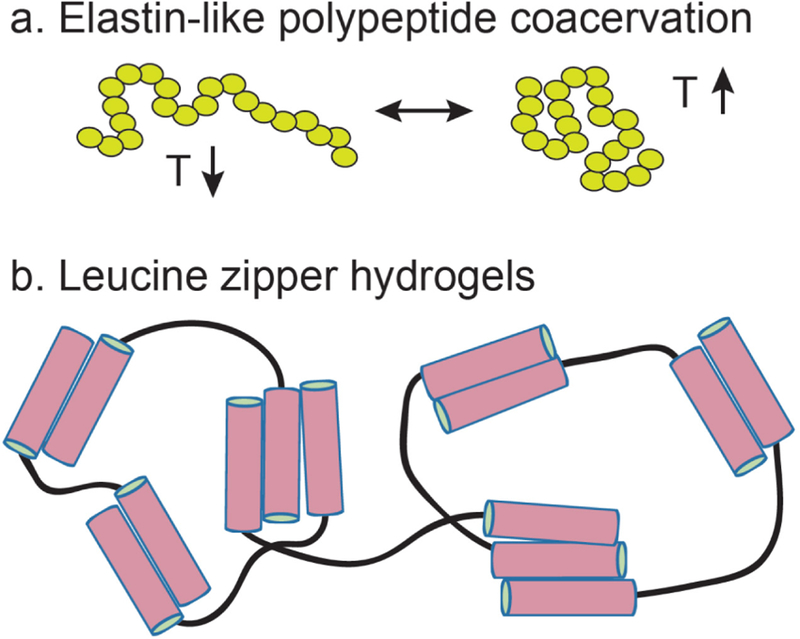
Early examples of assembled protein scaffolds: (a) elastin-like polypeptide coacervates and (b) leucine zipper hydrogels.
6.2.2. Leucine Zipper Hydrogels.
Another early example of self-assembled protein scaffolds arose from rational design of a tri-block fusion protein consisting of identical coiled-coil leucine zippers flanking a central alanine-glycine–rich sequence [(AlaGly)3ProGluGly]10 (known as C10 in this chapter) that is water soluble and unstructured.387 As shown in Figure 20b, association of the leucine zippers via coiled-coil interactions produces primarily dimers and some high ordered multimers, which in turn promote protein gelation. These self-assembled protein hydrogels can be reversibly solubilized by dissociating the coiled-coils, either through deprotonation of Glu sidechains at high pH or by heating above the melting temperature of the protein structures.
6.3. Engineering of Protein Scaffolds
The initial discovery of ELP scaffolds and design of leucine zipper scaffolds has been followed by a significant body of work aimed at modifying these systems and creating entirely new self-assembled protein scaffolds. These examples span a wide variety of different protein building blocks, assembly mechanisms, and bulk properties of the resulting produced scaffolds. A summary of the driving forces that can be harnessed to induce self-assembly of protein scaffolds is illustrated in Figure 21, and this section of the Chapter is organized according to these motifs. Many of these motifs, including templating, chemical modification, and supramolecular assembly, are primarily observed in nanoscale assemblies,3 and thus will not be covered here. Other motifs, such as coiled-coil interactions and secondary structure transitions, have already been introduced for the designer peptides covered in Chapter 3, but here will be applied to scaffolds comprised of folded proteins. The secondary structure transitions observed in some scaffolds also drive the assembly of naturally-occurring amyloids, which are discussed in Chapters 2 and 7. The assemblies described in this Chapter can be described as designed scaffolds, in that rational decisions were made to modify existing proteins or create de novo protein sequences that were hypothesized to self-assemble. Typically, these rational designs are based on one or more of the following: modifications of previously reported scaffolds, known protein-protein interactions from the literature, crystal structures from the Protein Data Bank, and experience or intuition, using similar strategies to those outlined in Chapter 5. Figure 22 provides a summary of the scaffolds described in this chapter.
Figure 21.
Common motifs utilized in assembly of protein scaffolds.
Figure 22.

Selected examples of engineered protein scaffolds.
6.3.1. Protein Scaffolds Formed by Secondary Structure Transition
6.3.1.1. Elastin-Like Polypeptide Scaffolds.
ELPs have been extensively explored beyond the early work of Urry et al., resulting in self-assembled scaffolds capable of forming a wide range of structures including micelles, vesicles, and hydrogels. The section will focus on the microscale and larger self-assembled ELP scaffolds, as opposed to the nanoscale or covalently crosslinked scaffolds, which have been extensively reviewed elsewhere.388,389 Modified ELPs are characterized by a (Val-Pro-Gly-Xaa-Gly)n sequence, but within this constraint, a number of opportunities for engineering exist. Parameters that can be explored include the identity of the 4th position guest residue, Xaa, and the number of repeats n of the pentapeptide. Additionally, ELP blocks having different Xaa and n can be fused to one another, or to other non-ELP proteins.
Wright et al. designed triblock ELPs in which the flanking ELP blocks have more hydrophobic character than the central ELP block, owing to the choice of amino acids at the Xaa position.390 Upon warming above the transition temperature, these ELPs undergo microscopic phase separation from aqueous solution to form a thermoplastic elastomer hydrogel. Triblock ELPs have also been rationally designed to specifically mimic the full sequence of tropoelastin, which consists of the typical (Val-Pro-Gly-Xaa-Gly)n sequence flanked on both ends by glycine-rich hydrophobic domains, (Val-Gly-Gly-Val-Gly)5.391 The glycine domains form β-sheet structures when warmed, distinct from the β-turn structures formed by the typical ELP domain. Just as described for model protein assemblies in Chapter 2, the structural transitions of the distinct domains do not occur simultaneously. Rather, the proteins initially assemble into nanoparticles that are rich in β-turn structures, and over time the β-sheet content increases and the nanoparticles connect to form long (>10 μm) beaded nanofibers.
Fusion of ELPs to high affinity coiled-coil leucine zipper partners has further expanded the type of self-assembled scaffolds that can be formed. For example, a basic leucine zipper was fused to ELP and mixed with minor amount of an mCherry or eGFP fluorescent protein fused to the partner acidic leucine zipper. The zippers exhibit femtomolar affinity heterodimer interactions and more moderate micromolar affinity homodimer interaction, both of which occur since the molar ratio of basic and acidic leucine zippers is not balanced.392 Upon warming above the transition temperature, the proteins assembled into hollow micron sized single-layer vesicles having a hydrophobic interior.161 Increasing the protein concentration or increasing the difference between the vesicle formation temperature and the transition temperature led to the formation of bilayer vesicles with a hydrophilic interior. Tuning these parameters also enables control of vesicle size over a range from hundreds of nanometers to a few microns.393 These same proteins can self-assemble into dynamic coacervate microparticles when introduced to model extracellular matrix under physiological conditions in a two-step process.394 First, introduction of cold, soluble ELP-zipper fusion induces simultaneous diffusion and phase transition into micro-coacervates trapped in the matrix. Subsequent addition of the mCherry-zipper partner fusion induces high affinity heterodimeric zipper binding, which changes the transition temperature and “dilutes” the ELP character. This leads to slow disassembly of the coacervates, upon which the released proteins continue to diffuse into the matrix.
Microscale ELP scaffolds can also be fused to calmodulin, enabling them to undergo calcium-triggered assembly.395 Upon calcium binding, calmodulin undergoes a conformational change that decreases the surface exposed charged residues and increases the surface exposed hydrophobic residues. This change is sufficient to decrease the transition temperature of the ELP domain to below room temperature, thus triggering the assembly of micron-sized coacervate particles. These results demonstrate the capacity of ELP assembly to be modulated by an array of triggers beyond temperature, salt, and monomer concentration.
6.3.1.2. Beta Roll Scaffolds.
The assembly mechanism of ELP can be generalized as a stimuli-induced change in secondary structure that enables quaternary interactions between proteins. This concept has been extended to other proteins having very different structures and utilizing different stimuli than ELP. Calcium-dependent hydrogel scaffolds were designed utilizing a beta roll domain from Bordetella pertussis that switches from an unstructured state to a β-helix upon addition of calcium.396 The beta roll domains were mutated to introduce leucines on one face, and were fused to leucine zippers via the unstructured C10 linker. Upon folding in response to calcium, the leucine residues are exposed and hydrophobic interactions between leucines induces dimerization of the beta rolls. The combination of beta roll dimerization and leucine zipper oligomerization forms hydrogels only in the presence of calcium. In subsequent work, the beta roll was mutated with additional leucine residues, such that both faces displayed exposed leucines in the presence of calcium.397 This doubled the hydrophobic crosslinking interface between beta roll domains, promoting gelation without the need for leucine zippers.
6.3.1.3. Silk Scaffolds.
Inspired by the incredible mechanical properties of spider dragline silk and centuries of use of silkworm silk as biomedical sutures, recombinant silk proteins have been engineered to produce self-assembled scaffolds.398 Dragline silk proteins are highly repetitive sequences having alanine- and glycine-rich motifs, and it is thought that the poly(alanine) patches adopt β-sheet structures to form crystalline-like particles, which provide strength and are embedded in an amorphous matrix of the glycine-rich motifs. It is proposed that these domains further assemble into 31-helical conformations and β-turns that form right-handed β-spirals, giving silk its elasticity. Comparison of Sup35p-NM amyloid-like fibrils, discussed in Chapter 7, with silk fibrils shows that they share structural characteristics, but their overall structures have distinct differences.399 Dragline silk proteins ADF-4 and ADF-3 have been engineered from consensus domains of the natural repeats, which are multimerized and produced in E. coli.400 Upon addition of methanol or potassium phosphate, the silk protein assembles into nanofibers, which undergo further slow assembly into a weak hydrogel.401 In the case of natural silk, precise processing modulated by the spider gland is critical to achieving the correct assembly. This can be mimicked using microfluidics to control potassium phosphate introduction and pH, and provide shear and elongation. The resulting engineered ADF-4 proteins assemble into spheres, but only form fibers when co-assembled with engineered spider silk protein ADF-3, which contains the 31-helices.402
An alternative approach to generating silk scaffolds is the block-copolymer design approach explored by Kaplan et al.. In this scaffold, the blocks consist of an alanine-rich hydrophobic A block and a glycine-rich hydrophilic B block, which mimic the natural silk sequence, and a histidine tag for purification.403 Increasing the portion of block A increased β-sheet content, as expected, but it was also observed that the histidine tag reduced β-sheet content. The morphology of the self-assembled scaffolds was dependent on both the block A content and the solvent. In water, an increase in the number of A blocks resulted in transition from thin films to bowl-shaped micelles to large compound micelles. In isopropanol, increasing A block content resulted in transition from thin films to nanofibers to large compound micelles. Continuing the parallel between engineered proteins and block copolymers, the high A content silk proteins were further studied to generate a phase diagram in water with ammonium sulfate at acidic pH that shows the transition between sheet-like morphologies and fibrils.404
Fusion of silk with other protein domains can introduce additional assembly properties. Silk-ELP fusion proteins having different ratios of silk to ELP content also self-assemble in response to temperature changes.405 Nanoscale micellar structures were formed below room temperature, but warming above room temperature produced larger spherical nanoparticles for proteins having low silk to ELP ratios. The highest silk to ELP ratio produced a gel scaffold, likely due to physical β-sheet “crosslinking” between silk groups.
6.3.2. Coiled-Coil Scaffolds.
The original leucine zipper hydrogel scaffold reported by Petka et al. has been adapted extensively to alter its properties, enabling use in a wide variety of applications.406 In one example, the zippers on either end of the C10 midblock were modified by asymmetric insertion of cysteine residues.407 Close association of the leucine zippers upon oligomerization enabled formation of intermolecular disulfide bonds between the zippers, which stabilized the gel upon placement in open buffer.
The leucine zipper scaffold has also been modified to generate assemblies having bioelectrocatalytic properties.408 A polyphenol oxidase, small laccase, was fused in place of one leucine zipper, preserving one zipper and the midblock C10 unstructured domain. The remaining zipper participated in coil-coil formation, leading to incorporation of the enzyme in the hydrogel scaffold and production of a current in the presence of oxygen. Dimerization of the laccase, which is necessary for activity, also contributed to physical crosslinking within the hydrogel. This scaffold was further elaborated through the incorporation of a different dimeric enzyme and recombination of the individual components. In this design, the enzyme used was organophosphate hydrolase, which is capable of neutralizing organophosphate neurotoxins, and the leucine zippers were placed on both termini of the hydrolase monomer.409 The N-terminal zipper was linked to the enzyme via the C10 unstructured midblock while the C-terminal zipper had no linker. Interestingly, addition of 6xHis tags further stabilized the assembled gels, potentially due to interactions with the divalent metal bound to the hydrolase. Thus, even in these examples of modular rational design using known domains, new assembly-stabilizing interactions can be discovered. Also interesting to note is the difference in the net effect of these interactions, as the histidine tag stabilizes the hydrolase assembly, but disrupts β-sheet assembly in the silk scaffold example.403
The above examples all derived from the same parent leucine zipper, but a variety of different zippers exist, some of which are characterized by higher oligomerization numbers.158 The scaffold reported by Petka et al. was modified by replacing the dimeric zippers with a pentameric leucine zipper having the same unstructured C10 midblock.410 Another variation on this assembly extended the midblock length by three-fold (C30). Together, the assemblies that resulted from these proteins demonstrated that the effect of the zipper and midblock properties on the scaffold are not entirely independent. Hydrogels assembled from these proteins exhibited extreme shear thinning behavior, which is fortuitous, as this property is useful for cell injection applications. Further modification of the pentameric fusion protein was accomplished by inserting nucleoporin-like peptides into the midblock, giving rise to self-assembling hydrogels having the capacity to selectively transport biomolecules.411
ELP domains can also be used to modify the nanoscale structure of self-assembled protein hydrogels, for example when incorporated at the ends of the pentameric leucine zipper-midblock fusions.412 Upon warming, micelle-like structures were generated from assembly of ELP domains and created nanostructure within the coiled-coil gel, which increased the moduli of the gels and enabled control over viscoelastic behavior. When ELP domains are fused to one or both sides of a single pentameric zipper, the proteins self-assemble into nanoparticles at low concentration, regardless of the order of the zipper and ELP.413 However, at high protein concentrations, self-assembly into a gel only occurs when the ELP is fused to the N-terminus of the zipper, demonstrating that the orientation of self-assembly motifs can significantly impact scaffold morphology. These differences can be overcome through replacement of phenylalanine with para-fluorophenylalanine, which leads to all three scaffolds showing the ability to form gels.414 Though non-natural amino acids have been incorporated into a number of protein scaffolds, this is typically for the purpose of chemical crosslinking, and this example demonstrates how incorporation of amino acids having unique chemical properties can alter self-assembly properties.
6.3.3. Co-Assembly of Scaffolds Using Affinity Domains.
While the leucine zipper-based scaffolds owe their assembly to homo-oligomerization processes in which coiled-coils in the same gel may contain different numbers of zippers, scaffolds formed by specific affinity interactions form co-assemblies having well-defined binding stoichiometry. Wong Po Foo et al. designed a two-component self-assembling hydrogel scaffold that relies on the specific molecular interaction of WW domains, which form anti-parallel β-sheet structures, and proline-rich peptides.415 In this co-assembly, each component consists of seven WW domains or nine proline-rich peptides, which are separated by shorter hydrophilic, unstructured C2 or C4 linkers. Upon mixing the two components, gelation occurs without the requirement for an external stimulus or environmental change, though reducing the number of domains does prevent gel formation. The physical properties of the gels can be further modulated through the use of different WW domains, which have varying affinities for the proline-rich peptide. These assemblies exhibited shear-thinning, injectable, and self-healing properties, allowing their use as scaffolds for the growth and differentiation of neural stem cells.
Protein hydrogels have also been generated using the SpyTag and SpyCatcher affinity system, which enabled comparison of the roles of non-covalent and covalent interactions in the assembly process.416 Component A consisted of three SpyTag domains separated by ELP linkers, and a mutated A’ component was generated having a point mutation in the center SpyTag, which retained the μM affinity toward SpyCatcher, but eliminated the ability to react covalently. Component B consisted of two SpyCatcher domains separated by ELP linkers. The ELP linkers were designed to have a transition temperature higher than the temperature used to make or use the gels, preventing coacervation. Upon mixing of A and B or A’ and B, gels formed. However, while the AB gels exhibited properties of covalently crosslinked hydrogels, the A’B gels eroded quickly in water as they swelled, indicating that the non-covalent interaction was not strong enough to support a stable gel. Furthermore, the covalent gels had sufficient stability to allow for incorporation of globular proteins or cell binding peptides via genetic fusion, and supported encapsulation and preservation of pluripotent mouse embryonic stem cells.
6.3.4. Other Self-Assembled Protein Scaffolds.
The natural sunflower surfactant-like protein, oleosin, was modified to create mutants having different lengths of hydrophilic arms flanking two central, hairpinned hydrophobic blocks.417 These proteins are insoluble in water, but adopt secondary structure in organic solvent. When injected into buffer, the protein self-assembled into a variety of structures including fibers, vesicles, and sheets, depending on the relative hydrophilic content of the protein and the ionic strength of the buffer. The fibers and sheets formed with dimensions ranging across the nano- to micro-scale, while the vesicles were either nano-scale or macro-scale, depending on the emulsion conditions.
6.4. Process of Modeling Data and Progress Toward Predictive Design
There has yet to be a wide-scale modeling effort focused on micro- to macro-scale self-assembled protein scaffolds, likely due to the large size and complex interactions of these assemblies. Among the modeling experiments that have been undertaken, the bulk of the effort has focused on ELP assemblies. As evidenced in the above descriptions of various ELP scaffolds, the transition temperature (Tt) and the assembly temperature relative to Tt are critical properties in assembly. Meyer and Chilkoti characterized a number of ELP libraries having blocks of different lengths n and containing different Xaa guest residues. These data were used to develop quantitative correlations between Tt, chain length, and concentration for a given ELP sequence.362 This work was later extended to include the effect of pH for ELPs having ionizable sidechains.418 A model was next developed to specifically incorporate a term to account for the effect of sequence composition on Tt.419 This model enables de novo design of ELP sequences and molecular weight combinations that exhibit the desired Tt in a specified concentration range. While this work predicts Tt, and thus whether assembly of a particular ELP will occur at a given temperature, it does not predict the nature of the structures that form, which can range from coacervates to micelles, vesicles, and gels. Recently, molecular dynamics simulations have been applied to predict the structural transitions that occur upon warming of a small virtual library of ELP sequences varying in length and Xaa identity.420 Replica Exchange Molecular Dynamics was performed at 60 different temperatures and for different salt concentrations, producing predictive data for radius of gyration, hydrogen bonding, solvent accessible surface area, solvation water, and secondary structure. The structural predictions were validated by experiment, and give information about the transition itself and the structure of the proteins in the coacervate phase. For example, doubling the length of an ELP does not significantly affect the Tt, but it increases the magnitude of the transition by increasing the degree of molecular collapse. This prediction of protein structure within coacervates is an important step toward prediction of the coacervate assembly process as a whole. The systems simulated here are small, and coarse graining would be required for simulations in the size range relevant to coacervate nano- or micro-particles.
Recently, exciting progress has been made toward the ability to predict and program complex coacervate structures formed by ELPs in water-in-oil emulsion droplets.421 Using a small group of rationally designed ELP sequences, Simon et al. demonstrated formation of multiphase, hierarchical coacervates from single or binary ELP mixtures in water. Further, a ternary mixture of a hydrophobic ELP, less hydrophobic ELP, and an amphiphilic di-block ELP in water arrested coacervate formation and produced punctae with sizes ranging from nano- to microscale. A framework was developed based on Flory-Huggins theory, which was used to build phase diagrams for these ELP systems, enabling prediction of assembled coacervate phases. Additionally, interpretation of structures formed across all combinations of ELP types enabled the establishment of design rules, which can be used to guide sequence and molecular weight selection with the goal of producing coacervates or punctae having desired morphologies. This work is possible due to the unstructured, disordered nature of ELPs in solution, which enables them to be treated analogously to organic polymers, indicating that this approach may be applicable to other intrinsically disordered proteins.
The Silk Integrative Theory Experiment Project is aimed at improving the design of engineered silk fibers having desired mechanical properties, and has established a workflow of modeling, silk protein synthesis, and fiber spinning.422 The modeling step of this workflow utilizes dissipative particle dynamics, a computational method used to simulate dynamic and rheological properties of fluids. Silk block co-polypeptides similar to those described earlier in this chapter are modeled as beads of 3 amino acids each and secondary structure information from experimental data is incorporated into the model. The structure of the protein is initially random, then shear flow is applied, and simulated mechanical measurements are performed. The proteins are then produced in the laboratory and extruded, and their fiber properties measured experimentally. These experimental data can be incorporated back into the model, enabling iteration of the design cycle. The modeling process is not yet fully predictive, but does enable more rational design, and the authors suggest that this approach should be applicable to other protein scaffolds beyond silk.
6.5. Remaining Challenges and Future Directions
Creation of novel and useful self-assembled protein scaffolds is largely driven by rational design based upon literature precedent, past experience, intuition, and creativity. This process is relatively slow, as each protein must be designed and produced, then the resulting assemblies characterized for size, morphology, and material properties. This is in stark contrast to the process of laboratory evolution, in which ~106–1015 variants of a protein are produced in parallel using cellular or acellular display methods and screened in a high-throughput manner to identify those having the desired properties.423 This cycle can be repeated multiple times to rapidly produce new proteins having unique structure or function. However, a key obstacle to the use of laboratory evolution is the need for a screening or selection step capable of separating the large library of variants into functional and non-functional pools. In the case of protein nanocapsids, laboratory evolution has been successfully applied, as the function of the capsid was to encapsulate a protease that is toxic to E. coli. In this case, cell viability could be used to identify functional variants, as only the cells expressing capsid protein mutants capable of both self-assembly and protease sequestration would survive and replicate.424 Using this process, a mutant capsid was identified having 5- to 10-fold greater protease encapsulation capability than the wild type. Looking ahead, increased use of laboratory evolution could rapidly move the field of self-assembled protein scaffolds. However, in addition to the need for a screening or selection step that can be applied at high throughput, a number of other challenges exist. These include the ability to produce sufficient numbers of identical proteins to enable self-assembly, physically separating different variant proteins, and preserving the genotype-phenotype link so that the sequences of the successful variants can be elucidated and replicated. It is possible that recent technological advances in laboratory evolution, such as cell-free protein synthesis and encapsulation, could be combined with recent advances in screening methods for high-throughput, combinatorial materials synthesis in order to broadly enable laboratory evolution of self-assembled protein scaffolds.425–428
In the meantime, computer-aided protein design (CaPD) approaches may provide viable options for in silico screening of assembly. The advantage of in silico screening is that much larger sequence spaces can be evaluated, several orders of magnitude greater than experimental screening or selection. CaPD platforms, such as Rosetta, are now being harnessed to predict and identify self-assembling nanoscale polyhedral protein scaffolds made from two different protein monomers.429 However, limitations to current CaPD approaches do exist, in that these methods require high-resolution structural input, which may not be available for the protein systems of interest. Moreover, given that many of the models used in CaPD have inaccuracies in parametrization, a priori predictions are unlikely to be successful. Accordingly, it will be challenging to adapt this approach to micro- or macroscale scaffolds that are not symmetric or highly ordered. However, CaPD platforms could potentially be used in combination with small scale simulations used to model or predict block co-polymer or surfactant self-assembly, provided the monomers used for assembly are relatively simple.430,431
The majority of self-assembled protein scaffolds are created using rational design and are modular in nature. However, the number of protein domains or “modules” used to create the currently reported scaffolds remains relatively small. Increasing the diversity of scaffolds and their functional properties requires a corresponding increase in the number of protein modules used for self-assembly. This in turn requires extensive characterization of protein domains, including both domains new to the self-assembled scaffolds community and those that have been previously used in scaffolds, with the aim of identifying new ways to combine these modules to create new assemblies. The family of leucine zippers is an obvious group of modules that could integrate well into this approach, yet highlight the challenges that exist. While though these domains are extremely well-characterized, their behavior can change significantly when they are fused to other protein domains, as is necessary to assemble scaffolds. This was demonstrated in a study of the oligomerization state of coiled-coils fused to GFP, which found that very few modules retained their original oligomerization number upon fusion, but that some oligomerization states could be restored through sequence modifications.158 This example further highlights that growing the protein assembly toolbox not only requires expansion of the current “parts list,” but also a greater understanding of each part. Given the recent pace of progress in the field and the growing number of researchers working with proteins as building blocks for self-assembly, it is guaranteed that the number of parts will grow. However, curation and accessibility of the assembly conditions and characterization data for each modular component remain essential to moving the field forward. The Protein Data Bank is an excellent example of accessible, high quality protein data used by many for protein engineering and design, and has grown consistently over the years. An analogous bank for protein assemblies and their modular components would be quite valuable to the field.
7. AMYLOIDS AND PRIONS: PATHOGENIC OR HERITABLE PROTEIN AGGREGATES
7.1. Introduction to Amyloids
The term “amyloid” was initially introduced by R. Virchow to describe iodine-stained depositions in human tissues,432 which were later discovered to be accumulations of protein aggregates.433 The propensity of amyloid to undergo this iodine staining is explained by the presence of glycosylated proteins in some of these depositions. Elucidating the composition of amyloid led to a more modern definition, referring to highly ordered fibrous protein (or peptide) aggregates, typically held together by intermolecular cross-β interactions. These “classic” amyloids possess a number of characteristic features, including: binding of specific dyes (e.g. Congo Red and thioflavine), optical anisotropy (birefringence) in the Congo Red bound state, and regular patterns of X-ray diffraction.150,434–436 Recently, an even broader definition of the term “amyloid” has emerged, as it has become evident that a variety of fibrillar cross-β polymers share the same basic structural features, despite the fact that some of these do not exhibit “classic” amyloid staining patterns.
As introduced with the example in Chapter 2, amyloid fibrils grow and spread via the process of nucleated polymerization (Figure 23), which involves immobilization of the monomeric peptide or protein into a fibril, followed by conformational conversion into an amyloid fold via the formation of hydrogen bonds with the β-strands in the existing fibril. This process is highly specific, as it requires a large degree of similarity between the interacting amino acid sequences; typically, only polypeptides having identical cross-β regions will assemble into an amyloid fibril. However, “cross-seeding” interactions between different amyloidogenic proteins have been reported, especially in cases when such proteins possess significant sequence similarities,437,438 though assembly via cross-seeding is typically much less efficient than proliferation of a homogenous amyloid. As discussed in Chapter 3, the amyloid fold relies on intermolecular interactions, and thus it can exist only within assemblies; monomeric units of the same peptide or protein would assume a different fold. From this protein folding perspective, long fibrils and so-called “on-pathway” short oligomers or “protofibrils” all possess the same amyloid fold and thus should be equally termed “amyloids.” We argue that application of the term “amyloid” exclusively to long fibrils or large deposits of fibrils, as is occasionally found in the literature, is misleading. For the purposes of this Review, we will utilize the term “amyloid” to describe all peptide or protein polymers possessing an amyloid fold, independent of their size or assembly morphology.
Figure 23.
Nucleated polymerization of amyloids or prions (squares) from non-amyloid isoform (circle).
Many peptides and proteins have been shown to form amyloids in vitro under specific solvent conditions such as pH, ionic strength, temperature, or shaking.439,440 Some of these amyloids form reversibly and can be depolymerized if the conditions are changed. Other amyloids form irreversibly and remain assembled and reproducible in a “seeded” fashion even if placed into conditions which are different from those in which they were initially formed. The ubiquity of the amyloid fold in nature has led to the proposal that it existed as an ancient protein fold, potentially to protect proteins from harsh prebiotic conditions.439–441 Moreover, amyloid fibrils tend to form higher order assemblies via lateral bonds, which may have contributed to the initial steps of biological compartmentalization.206,442 For most proteins, the ability to form an amyloid under physiological conditions has been suppressed over time via evolution, as the need for protection decreased and demand for more complex cellular functions increased. However, a subset of proteins has retained this ability, with amyloid formation playing a role in either pathogenic processes or biological functions.
While amyloids are ubiquitous in nature, a detailed picture of their molecular structure is only beginning to emerge, and the challenges associated with structural studies on amyloid have prevented the emergence of a complete understanding of the forces that drive these assembly processes. As a result, the field of amyloid engineering remains in its infancy. This Chapter will thus focus on what is known about amyloids in nature, and will present the limited examples where amyloid formation has yielded to external control or engineering. While this field is less developed than the engineering of other protein systems, the future potential appears to be significant, and we argue that understanding amyloid assembly is a critical step towards successful future engineering attempts.
7.2. Pathological Effects of Amyloids and Prions
To date, approximately 50 human diseases have been linked to the formation of amyloids or similar types of ordered protein aggregates (see examples in Table 3).443,444 The most widespread and devastating of these is Alzheimer’s disease (AD), a fatal and incurable pathology typically affecting aged people and rapidly growing in abundance due to the extermination of other diseases and corresponding increase of human lifespan.445,446 Studies of both heritable (rare) and sporadic (frequent) cases clearly point to amyloid formation by a peptide termed amyloid β (Aβ) as a triggering factor of AD. Aβ is processed from amyloid precursor protein (APP) and exists in several length variants, of which the 42 amino acid variant (Aβ42) is the most aggregation-prone and pathogenic, while Aβ40, lacking the last two amino acids, is less aggregation-prone and pathogenic.447 Notably, the progression of AD is associated with accumulation of a second amyloid, formed by the microtubule-stabilizing intracellular protein tau.448,449 Amyloid formation by tau has also been implicated in diseases other than AD, and these diseases are referred to as tauopathies.449,450 In the case of AD, it remains unclear whether Aβ amyloids directly cross-seed amyloid formation by tau, or whether they change the physiological status of the cell in a way that indirectly induces tau aggregation.
Table 3.
Examples of diseases associated with amyloids and prions
| Disease | Heritability | Cells affected | Outcome | Protein(s) involved | Protein localization |
|---|---|---|---|---|---|
| Alzheimer disease | Sporadic, rarely heritable | Neurons | Fatal | Aβ, tau | Extra-(Aβ) or intracellular (tau) |
| Parkinson disease | Sporadic, sometimes heritable | Neurons | Motor disfunction | α-synuclein | Intracellular |
| Huntington disease | Heritable | Neurons | Fatal | Huntingtin | Intracellular |
| TSE (prion diseases) | Infectious, sporadic or heritable | Neurons | Fatal | Prion protein (PrP) | Extracellular |
| Type II diabetes | Usually sporadic | β-cells (pancreas) | Defect in glucose signaling | Amylin (IAPP) | Extracellular |
| ATTR amyloidosis | Heritable or sporadic | Myocytes, neurons | Cardiomyopathy, polyneuropathy | Transthyretin (TTR) | Extracellular |
Other examples of prominent amyloid diseases in humans include Parkinson’s disease (PD), related to amyloid formation by the intracellular protein α-synuclein,447 and ATTR amyloidosis, associated with the amyloid formation by transthyretin protein, a transporter of the retinol-binding protein-vitamin A complex and thyroxin.451 Huntington’s disease (HD) is also associated with the formation of fibrillar aggregates from the protein called huntingtin,452 although some researchers still refrain from equating these aggregates with “classic” amyloids. PD and ATTR amyloidosis include both sporadic and heritable forms, while HD is always heritable and is caused by an expansion of the polyglutamine (polyQ) region in huntingtin, which in turn leads to protein aggregation. PD and HD are incurable, with existing treatments only ameliorating some consequences of disease,447,452 while ATTR amyloidosis can be treated by therapeutics that stabilize the normal tetrameric structure of transthyretin, preventing its conversion into an amyloid.453 Recent data also point to links between amyloids and type II diabetes,454 severe preeclampsia,455 and some forms of cancer,456 although the causative relationships between amyloids and the diseases have not yet been fully established. Type II diabetes is associated with amyloids formed by amylin or IAPP, a blood borne peptide hormone having noticeable sequence similarities to Aβ454 and capable of cross-seeding Aβ aggregation in experimental models.457
An extreme case of amyloid disease is represented by transmissible spongiform encephalopathies (TSEs), or prion diseases. These incurable diseases include sheep scrapie, cervide chronic wasting disease, and human disorders such as kuru and Creutzfeldt-Jakob diseases.458,459 Likley the best known example of TSEs is bovine spongiform encephalopathy (BSE) or “mad cow” disease, which devastated the European cattle industry in the 1990s due to the possibility of its transmission to humans. These infectious neurodegenerative diseases are transmitted by prion protein (PrP), a normally extracellular protein that can adopt an unusual (prion) isoform that possesses amyloidogenic properties. The amyloidogenic isoform of PrP can seed aggregation of normal PrP protein in vitro, via a process termed Protein Misfolding Cyclic Amplification (PMCA),460 and these in vitro produced PrP fibrils can infect laboratory animals, confirming the “protein only” model of prion transmission.461–463 Interestingly, some other disease-related amyloid proteins, including Aβ, tau, α-synuclein and amyllin, which are normally not infectious at the organismal level, can spread between cells or areas of brain (and in experimental conditions, even between organisms) using a mechanism that is essentially identical to the mechanism of prion infection.464–469 Therefore, a recent tendency in the field is to broaden the term “prion” by defining it as any transmissible protein isoform, allowing this term to be applied to a variety of amyloid diseases.470 Moreover, the ability of prions to switch between conformational states and to reproduce some of these states in a templated fashion allows prions to serve as carriers of biological information that can be used not only in infection, but also in inheritance. Indeed, experiments with fungal models have identified the prion-based heritable elements which are described below.
7.3. Fungal Prions as Heritable Elements
7.3.1. Discovery and Diversity of Fungal Prions.
Fungal prions are heritable protein isoforms, which are transmissible between cell generations or by cytoplasmic infection, and may control detectable phenotypic traits.437,471 The extension of a prion concept to heritable elements was first established by Wickner472 on the basis of data obtained by his and other labs from studies of yeast non-Mendelian heritable factors [PSI+] and [URE3], which were initially identified via their phenotypic manifestations and an unusual mode of inheritance.473,474 Subsequent data from various research groups confirmed this concept, including a direct demonstration of the transmission of a prion-based trait via transfection of the yeast cells with a purified protein in an amyloid form.475,476 Heritable prions are most commonly studied in the yeast model organism Saccharomyces cerevisiae, although examples have been reported from other species as well, including the filamentous fungus Podospora anserina.477 To date, at least 10 endogenous proteins are proven to form heritable prions in the cells of yeast and other fungi (Table 4).437,471 These proteins typically contain regions termed “prion domains” (PrDs), which are responsible for prion formation and propagation, and at least in some cases, are distinct from the regions responsible for the major cellular function of the respective protein (Figure 24). In most cases, fungal PrDs are enriched in N and/or Q residues, although exceptions to this rule have been reported.477,478 PrDs are transferable to other proteins via artificial constructions, enabling the engineering of new synthetic prions. In addition to yeast proteins capable of forming a prion in their native state, at least 10 additional PrD-like regions have been shown to do so when fused to a reporter,479 and at least 100 yeast proteins are suspected of having prion-like capabilities based on the patterns of their amino acid composition.479,480 Arguably, even this number is an underestimate, as these searches employed features such as the presence of a QN-rich domain that are characteristic of most, but not all fungal prions.
Table 4.
Fungal amyloid-based prions known to date
| Prion | Protein | Function |
|---|---|---|
| Saccharomyces cerevisiae | ||
| [PSI+] | Sup35 (eRF3) | Translation termination factor |
| [URE3] | Ure2 | Regulator in nitrogen metabolism |
| [RNQ+], or [PIN+] | Rnq1 | ? |
| [SWI+] | Swi1 | Chromatin remodeling factor |
| [OCT+] | Cyc8 | Transcriptional corepressor |
| [MOT3+] | Mot3 | Transcriptional repressor |
| [MOD+] | Mod5 | Transfer RNA isopentenyltransferase |
| [NUP100+] | Nup100 | FG-nucleoporin |
| [LSB+] | Lsb2 | Cytoskeletal assembly protein |
| Podospora anserina | ||
| [HetS] | Het-s | Cytoplasmic incompatibility |
Figure 24.

Structural and functional organization of fungal prion proteins.
Fungal prion proteins are homologous to neither each other, nor to known human disease-related amyloidogenic proteins, although some similarities in amino acid composition can be found (for example, the presence of QN-rich aggregation-prone domains in many yeast prion proteins resembles polyQ proteins, such as huntingtin). For the majority of known yeast prions, the molecular foundation of inheritance is through nucleated polymerization of amyloid fibrils. Other mechanisms for the formation of heritable protein isoforms in yeast, such as a self-activating proteolytic activity, have also been described for specific cases, but are not considered in this Review. In the case of amyloid-based yeast prions, only the PrDs are assembled into a cross-β structure, leaving the remainder of the protein exposed on the side of the fibril, where it can assume its normal fold.481 The phenotypic effects of prion formation are typically manifested as a decrease of protein function in the amyloid state, however prion formation could in principle be phenotypically undetectable for enzymes working on easily diffusible substrates.
7.3.2. De novo Formation of Fungal Prions in vivo and in vitro.
Transient overproduction of a prion protein or its PrD can nucleate de novo prion formation in yeast (Figure 25),482–484 after which point, prion assemblies can be propagated at normal expression levels of the prion-forming protein. This process of prion induction by protein overproduction is greatly facilitated by the presence of other proteins in an aggregated state, suggesting the possibility of cross-seeding interactions.485–487 Some yeast prion assemblies can also be induced by environmental stresses, for example by heat shock, long term storage at low temperature, or genetic alterations that increase protein oxidation.488–490 Notably, a yeast ribosome-associated member of the Hsp70 family, Ssb, counteracts spontaneous and overproduction-induced de novo prion formation,491 apparently via antagonizing the initial misfolding of the nascent polypeptide. A similar effect is observed in the case of disruption of the ribosome-associated chaperone complex, RAC. The RAC complex is composed of Zuo1, the ribosome-associated member of the Hsp40 co-chaperone family, and Ssz1, a non-conventional Hsp70 protein. RAC facilitates association of Ssb with the translating ribosome, and if either component of RAC is deleted, Ssb is released from the ribosome to the cytosol, leading to an increase in de novo prion formation.492,493 Ssb release may also occur in wild type cells under unfavorable growth conditions, implicating Ssb as a potential modulator of prion induction by environmental stresses.493,494
Figure 25.
De novo prion formation, cross-seeding and propagation in yeast. (a) Induction of the formation of a prion isoform ([PSI+]) of Sup35 protein by transient overproduction of Sup35 protein or its prion domain (PrD) is facilitated in the presence of a prion isoform ([PIN+]) of another protein, Rnq1, presumably due to a cross-seeding. Misfolded intermediate is indicated by a green ellipse, other designations are as on Figure 23. (b) Prion fragmentation and propagation by a chaperone machinery. Chaperone proteins are as designated (Hsp104 is a hexamer). Green rectangles indicate units of an amyloid fibril.
Yeast prion proteins or their fragments containing PrDs are also known to form amyloids in vitro.495–497 The NM fragment of the yeast translation termination factor, Sup35 (termed Sup35NM) is most frequently employed in the in vitro formation of yeast prions,498 as this protein is comprised of a QN-rich PrD (Sup35N) and a middle region (Sup35M) having clusters of charged residues. Electrostatic repulsion between Sup35M units slows aggregation of the PrD, making the process of in vitro amyloid formation more likely to produce the thermodynamically favored assembly. Given the role of electrostatic interactions, it is not surprising that in vitro amyloid formation by Sup35NM is modulated by the anions of the Hofmeister series.499,500 These ions are arranged according to their ability to salt out (precipitate) and salt in (solubilize) most proteins.501 In this series, smaller, strongly hydrated ions are termed “kosmotropes” and are known to decrease protein solubility, while larger, weakly hydrated ions that interfere with the hydrogen-bonding of water are termed “chaotropes” and are known to increase protein solubility. In the case of Sup35NM, kosmotropes promote amyloid formation, while chaotropes antagonize it, consistent with the increased formation of hydrogen bonds in the presence of kosmotropes and the “screening” effect of chaotropes.499,500 Similar effects of kosmotropes on amyloid formation have been detected for some other proteins such as PrP502 and amylin.503
7.3.3. Chaperone-Based Prion Propagation in Yeast.
Propagation and inheritance of yeast prions is typically achieved via concerted action of the same chaperone machinery that is involved in disaggregation of stress-damaged proteins (Figure 25).471,489 This machinery fragments prion fibrils into oligomers, thus multiplying them and initiating new rounds of fibril growth. Understanding the molecular composition of this chaperone machinery began with identification of its crucial component, Hsp104, as a protein required for the propagation of [PSI+], a prion form of Sup35 protein.504 Other components of the prion fragmentation machinery include a major cytosolic member of the Hsp70 family, Ssa,505–508 and co-chaperones of the Hsp40 family, typically Sis1 or Ydj1, which exhibit differential effects on different prions.509 In the mechanism of prion propagation, the Hsp70/40 complex is thought to bind to amyloid fibrils first, followed by Hsp104.510 Interestingly, the same chaperone machinery is involved in disaggregation of cellular proteins that have been damaged by environmental proteotoxic stresses.511 Thus, yeast prions are “hijacking” the cellular stress defense machinery for the purpose of prion propagation, in the same way as some viruses hijack the cellular DNA replication apparatus for the purpose of viral replication.
While Hsp104 plays a role in prion propagation, its overproduction can destabilize or “cure” some yeast prions,471,489,512 potentially due to “non-productive” binding of Hsp104 alone to amyloid fibrils.510 This has been hypothesized to solubilize fibrils by “chopping” monomers from the termini,513 although more recent data point to prion malpartition during cell divisions as a mechanism of prion loss in the presence of excess Hsp104.514
Although most components of the anti-stress chaperone machinery are evolutionarily conserved, Hsp104 orthologs are absent from the cytosol of multicellular animals.515 It remains unclear which proteins might play similar roles to Hsp104 in regard to disaggregation of mammalian amyloids, but recent data516 point to the possibility of that some Hsp104 functions could be assumed by its distant mammalian paralogs, RuvbL1 and RuvbL2, also found in yeast under the names of Rvb1 and Rvb2, respectively. However, further research is needed to elucidate whether RuvbL1/2 proteins have the same impact on propagation of mammalian amyloids as Hsp104 does in yeast. In the meantime, attempts are ongoing to engineer variants of Hsp104 as anti-amyloid tools for the treatment of human amyloid diseases.515,517
7.4. Non-Pathological Roles of Amyloids and Prions
7.4.1. Biological Roles of Fungal Prions.
The biological impact of prion formation in fungi remains a matter of debate.437,471 Prion isoforms of some yeast proteins, including Sup35 are cytotoxic,518 however other yeast or fungal prions appear to be associated with adaptive functions, such as control of cytoplasmic incompatibility477 or ethanol resistance.519 While Sup35 and Ure2 prions are extremely rare in the natural or industrial isolates of Saccharomyces yeast, some other prions are found in the wild-type strains.520–522 If prion isoforms are considered as protein “mutants” capable of generating heritable changes without mutating DNA,523 then by analogy, the effects of protein “mutations” could be either detrimental or adaptive, depending on the altered protein, type of alteration, and its functional consequences under specific conditions.
7.4.2. Prion-Like Oligomerization and Memory.
Given their thermodynamic stability, prions can be considered as molecular memory devices, “remembering” and reproducing a change that has occurred to a protein structure. Indeed, a synthetic “memory” device has been built on the basis of a yeast prion,524 and endogenous yeast non-heritable prion-like complexes (“mnemons”) have been identified.525,526 The metastable prion formed by a yeast cytoskeleton-associated protein Lsb2 is induced by heat stress and the prion assembly is maintained after the stress is reversed. Thus, the prion generates a cellular memory of stress, which can be inherited over an indefinite number of cell generations, although the prion fraction is constantly diminishing due to the metastable nature of the Lsb2 prion transmission.527 It is proposed that the prion form of Lsb2 promotes stress resistance via facilitation of the assembly of stress-damaged protein into cytoprotective cytoskeleton-associated deposits.527,528 If this is the case, the prion-based stress memory may play an adaptive role in case of repeated stress events.
Prion-like oligomers of the actin-binding protein CPEB, a regulator in mRNA translation, have also been implicated in long-term memory in animals, including the shellfish Aplysia, fruit fly Drosophila melanogaster, and mouse.529–531 CPEB oligomers are formed in response to physiological stimuli and maintained in the oligomeric state, modulating translation of other proteins in the synapse. Members of the CPEB family involved in this process contain a QN-rich region similar to yeast PrDs, and form amyloids in vitro and in yeast cells.532–534 Understanding the structure and assembly of these proteins in their native systems remains an exciting challenge.
7.4.3. Other Biological and Technological Applications of Amyloids and Prions.
Amyloids have been found to play a number of roles in biology beyond those described above, including: attachment to surfaces or other cells (e.g. biofilm formation) in bacteria and fungi,535,536 storage of peptide hormones in mammalian cells,537 and scaffolding the formation of covalent polymers (such as melanin) in animals.538 Several plant proteins have been shown to form amyloids in vitro or in heterologous systems, leading to hypotheses postulating functional importance of these amyloids.539,540 Known functional amyloids are typically either constitutively present in an amyloid state or induced (and sometimes reversed) by a change in environmental or physiological conditions, rather than spontaneously switching from the soluble to an amyloid form, as is case with pathogenic amyloids and prions. An important class of functional amyloids or amyloid-like proteins is represented by silks/fibroins. Spider silk is in fact a variation of an amyloid formed by a very long protein with the inclusion of the elastin domains.541,542 These proteins are employed for macroscale construction purposes both in nature (e.g. spider webs) and by humans. As described in Chapter 6, higher order macroscale scaffolds have been engineered from proteins having silk domains that retain their β-sheet folding, though replicating the assembly process that is utilized in nature has proven challenging. In addition to technological applications inspired by nature, modified Sup35NM fragment capable of binding gold particles has been used for the construction of self-assembled nanowires.543 Table 5 summarizes known biological roles of amyloids and prions known to date, whereas detailed descriptions of non-natural amyloid-like assemblies can be found in Chapters 3 and 6.
Table 5.
Examples of proven or proposed positive biological roles for amyloids and amyloid-like oligomers
| Protein(s) | Organism(s) | Biological role in polymeric form | Amyloid type |
|---|---|---|---|
| Het-s | Fungus Podospora | Vegetative incompatibility | Switchable |
| Mot3 | Yeast | Multicellularity, ethanol resistance | Switchable |
| Lsb2 | Yeast | Stress memory | Switchable |
| Adhesins | Fungi | Substrate attachment, biofilm | Inducible |
| CsgA,B (curli) | Bacteria | Substrate attachment, biofilm | Inducible |
| Peptide hormones | Mammals | Storage | Reversible |
| Pmel17 | Animals | Melanin synthesis | Reversible |
| CPEB (Orb) | Shellfish, flies, mice | Long term memory | Switchable |
| Silk/fibroin | Spiders, insects | Structure formation | Constitutive |
7.4.4. Amyloid Assembly with Other Proteins.
In addition to amyloids, living cells form a variety of other multi-protein and protein-RNA deposits, and the relationship of these with amyloid and prion proteins remain a matter of investigation. Of greatest interest are liquid droplets, hydrogels, and protein or protein-RNA assemblies generated via phase separation process. These assemblies include cytoprotective deposits formed in response to unfavorable conditions, such as stress granules (SGs) found in both yeast and mammalian cells,544–547 yeast Q-bodies,548 and JUxtaNuclear (JUNQ) and INtraNuclear (INQ) quality control deposits.549,550 Functional assemblies such as P-bodies, which serve sites of mRNA degradation, can also be formed via phase separation processes.551 While phase separation based assemblies are clearly different from amyloids, they may involve proteins capable of amyloid formation. Moreover, the PrD-like region of the protein TIA-1/Pub1 is important for SG assembly,552 and the same region of the FUS19 protein is involved in both amyloid formation and phase separation process.553 Human Tau protein is another SG component, and appears to play a role in SG assembly. Moreover, extensive SG formation in turn promotes amyloid formation by tau.554 These observations suggest that deposits generated by phase separation could serve as intermediates in the amyloid formation pathway,555,556 according to the two-step nucleation mechanism described in Chapter 2 and illustrated in Chapter 5.
Other self-assembled complexes such as cytoskeletal networks and virus-like particles may also involve proteins having amyloidogenic domains, however a relationship between such structures and amyloids has not yet been systematically addressed. As described above, some cytoskeleton-associated proteins such as tau in mammals and Lsb2 in yeast527 are shown to form amyloids. Cytoskeletal networks are involved in the formation of cytoprotective deposits which are, in contrast to those formed by phase separation, composed of amyloid-like highly ordered solid fibrillar materials. Examples of such deposits include the aggresome, a perinuclear structure formed with participation of the microtubular cytoskeleton in mammalian cells,557 and Intracelluar peripheral PrOtein Deposits (IPOD),549 formed in yeast cells and asymmetrically inherited through a process involving the actin cytoskeleton. Yeast cells also contain an analog of the aggresome,558 which may represent a perinuclear version of IPOD. These cytoprotective protein deposits are induced by proteotoxic stresses or through accumulation of aggregation-prone proteins (such as polyQs), and may represent an attempt by the cell to remove potentially toxic misfolded amyloidogenic proteins from the cytosol and confine them to specific locations, allowing them to be eliminated via autophagy or asymmetric inheritance in cell divisions. In the case of overproduction of Sup35 PrD, some Sup35 assemblies colocalize with IPOD markers,559 providing evidence for the hypothesis that prion generation occurs as a byproduct of processes aimed at assembly of cytoprotective deposits of misfolded proteins.560
7.5. Structure and Polymorphism of Amyloids and Prions
7.5.1. Structural Studies of Amyloids.
Structural characterization of amyloid fibrils is complicated by the fact that amyloidogenic proteins do not form crystals. In fact, amyloids could be considered as linear 2-dimensional crystals, and as such, represent an alternative to classical 3-dimensional crystal formation. While amyloid fibrils do show characteristic patterns of X-ray diffraction, indicative of a regular high-order structure, it is impossible to deduce atomic resolution or even near atomic structures from such images due to lack of a solution to the phase problem. Only very small amyloids, formed by short peptides such as a 6–7 amino acid segment of Sup35 PrD have been grown into microcrystals and resolved by X-ray crystallography.561,562 These studies revealed parallel in-registered cross-β sheets, stacked through a dry interface formed by sidechains. However, these structures cannot be directly extrapolated to the amyloids formed by larger peptides or whole proteins without further structural investigations.
Given the challenges of X-ray crystallography, solid state NMR has been used to obtain high resolution structures for some amyloids.563 Recently, significant advances in solving amyloid structures have also been made by using cryo-electron microscopy (cryo-EM).564,565 Some information regarding the proportion of β-structures and location of cross-β regions can also be drawn from low-resolution techniques, such as circular dichroism and hydrogen-deuterium (H-D) exchange.
Two major types of motifs have been detected in the structures solved to date for amyloids formed by relatively large proteins or peptides (Figure 26):
The superpleated parallel in-register β-sheet, in which the units of an amyloid fibril are linked to each other via intermolecular hydrogen bonds between identical and identically located residues within β-strands, while resulting intermolecular β-sheets are stacked into a structure sometimes termed “β-arcade”.566
The β-helix or β-solenoid, in which different molecules are linked to each other via cross-β interactions between short identical regions, while the remaining amyloid core region is packed into an intramolecular helical β-β structure. In the simplest version of a β-solenoid, units of each fibril are linked to each in a “head-to-head, tail-to-tail” fashion, although in reality, more complex assemblies are typically observed, for example the four intermolecular cross-β regions observed in the Het-s amyloid.567
Figure 26.
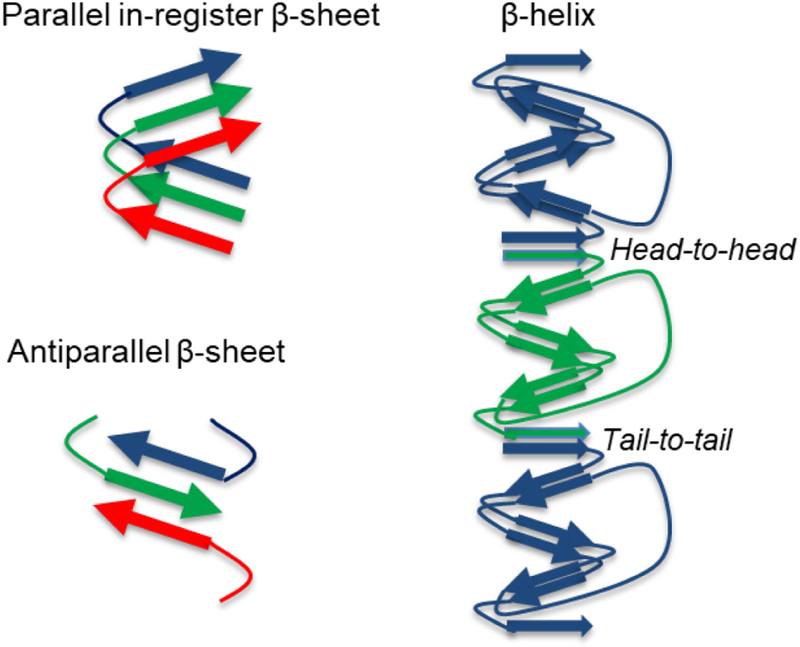
Types of amyloid structures. Arrows indicate β-strands, different polypeptides are shown by different colors.
In addition to these two predominant motifs, antiparallel cross-β structures have also been observed, for example in the case of an Aβ40 mutant.568
To date, relatively high resolution structures exist for the fungal prion Het-s (NMR),567 Aβ40 (NMR),569,570 Aβ42 (both NMR and cryo-EM),565,571,572 and tau (cryo-EM).564 Some examples of these are shown in Figure 27. Aβ structures typically adopt parallel in-register β-sheets, while Het-s and tau form complex β-solenoids. Partial structural information has also been obtained for several other amyloids, including yeast Sup35NM,573,574 bacterial surface amyloid (curli),575 and transthyretin.576 While not complete, these partial structures can enable researchers to formulate models and draw some conclusions about overall organization of fibrils. Interestingly, while β-strands in amyloids are typically produced from unstructured regions during the process of amyloid formation, the transthyretin amyloid core preserves two β-strands that are involved in the non-amyloid fold of the same protein.576 Another unique characteristic of amyloids is that, in some cases, the basic unit of a fibril is not a monomer. For example, a structural model of the Aβ42 fibril suggests that it exists as a cross-β polymer of a non-cross-β dimer.572,577 Multiple structural models are proposed and supported by data for the PrP prion, among which there is a structure having a β-solenoid fibril that forms as a polymer of a trimer.578,579
Figure 27.
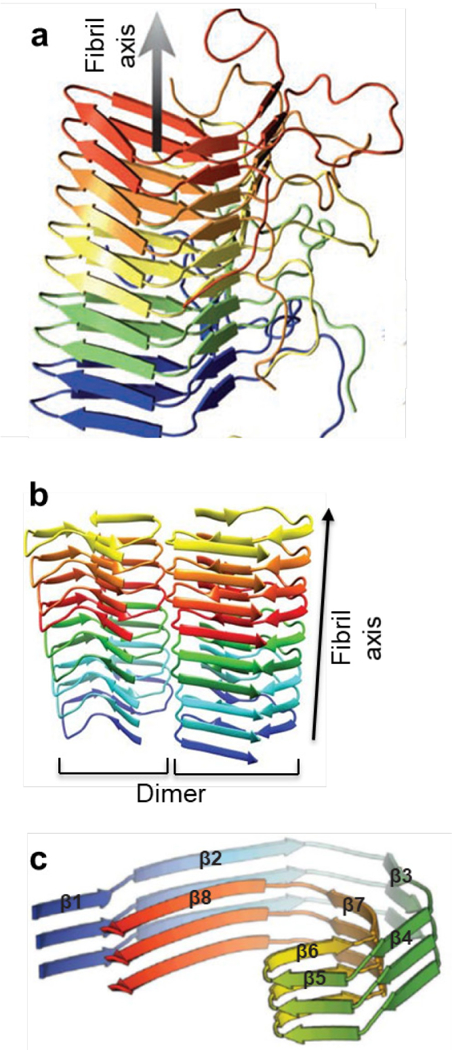
Examples of amyloid structural models. (a) Het-s473. (b) Aβ42 478. (c) Amyloid core of a tau fibril470. Arrows indicate β-strands. Reprinted with permission from Wasmer, C.; Lange, A.; Van Melckebeke, H.; Siemer, A. B.; Riek, R.; Meier, B. H. Amyloid fibrils of the HET-s (218–289) prion form a β solenoid with a triangular hydrophobic core. Science 2008, 319, 1523–1526. Copyright 2008 American Association for the Advancement of Science. Reprinted with permission from Wälti, M. A.; Ravotti, F.; Arai, H.; Glabe, C. G.; Wall, J. S.; Böckmann, A.; Güntert, P.; Meier, B. H.; Riek, R. Atomic-resolution structure of a disease-relevant Aβ (1–42) amyloid fibril. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E4976-E4984. Copyright 2016 National Academy of Sciences. Reprinted with permission from Fitzpatrick, A. W. P.; Falcon, B.; He, S.; Murzin, A. G.; Murshudov, G.; Garringer, H. J.; Crowther, R. A.; Ghetti, B.; Goedert, M.; Scheres, S. H. W. Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 2017, 547, 185–190. Copyright 2017 Springer Nature.
7.5.2. Structural Variants of Amyloids and Prions.
The major obstacle to structural studies of amyloids and prions is that a single amino acid sequence may form multiple amyloid conformations, referred to as polymorphs, or “strains.” The presence of different strains was initially identified in vivo for mammalian PrP on the basis of different disease characteristics, such as incubation period and host specificity of infection, which were stably reproduced in subsequent rounds of infection.580–582 Differences between PrP strains were then confirmed at a molecular level by identifying different protease-resistant cores in PrP protein isoforms originating from different strains. The notion of a single protein sequence not only forming transient alternative conformations but a variety of stable and reproducible conformations has long been considered an obstacle to the “protein only” concept of prion infection. However, yeast prions also form multiple strains, typically referred to as “variants” to distinguish them from microbial strains. For example, Sup35 protein can generate a variety of prion variants, which differ from each other by phenotypic stringency (“strong” or “weak”), stability of transmission in cell divisions, proportion of aggregated versus soluble protein, and range distribution of protein aggregate sizes.484,583,584 In the case of Sup35, it has been proven that variants can be generated in vitro and reproduced by transfection into the yeast cells,476 thus there is no doubt that variant characteristics are controlled by the Sup35 protein itself. Overproduction of Sup35 generates multiple prion variants.484,585 However, once formed, a given variant is faithfully reproduced. In vitro, Sup35NM preferentially forms different variants under different conditions. For example, “strong” variants are preferentially formed at low temperature or in the presence of kosmotropic anions, while “weak” variants are preferentially formed at high temperature or in the presence of chaotropic anions.476,500,586 Among mammalian amyloids, different strains have also been identified for Aβ582,587,588 and α-synuclein.589
7.5.3. Molecular Basis of Amyloid Strains.
The underlying molecular mechanisms controlling strain properties are most easily explained for the parallel in-register cross-β structures.590 In this structural model, multiple variants of β-strand formations could exist for a single sequence. However, once a specific variant is generated, it is reproduced in a template-like fashion, because each newly immobilized monomer is aligned to the preexisting structure and assumes the same conformation upon forming hydrogen bonds between identical residues. It is not yet clear how strain patterns are determined for amyloids in a β-solenoid structure.
Notably, analysis of the yeast prion Sup35 using H-D exchange shows that the “weak” variants contain a longer cross-β core region, while the “strong” variants contain a shorter cross-β region (Figure 28).591 This agrees with the observation that fibrils of “strong” variants are more readily fragmented by the chaperone machinery, and as a result, have lower average molecular weight, while fibrils of “weak” variants are less efficiently fragmented and have higher average molecular weight.88 At first glance, it appears somewhat paradoxical that biologically “weaker” strains are physically stronger, while biologically “stronger” strains are physically weaker. However, such an inverse correlation between the biological stringency and physical stability of an amyloid is in fact quite logical. As explained in Chapter 2, lower physical stability and more efficient fragmentation leads to formation of a larger number of oligomeric proliferating units for a set amount of protein, translating into a larger number of monomer immobilization sites and more efficient conversion of the monomeric protein into an amyloid.
Figure 28.
Molecular basis of prion/amyloid strains. (a) Phenotypic stringency of the strong and weak strains of yeast prion protein Sup35, as indicated by color on complete medium (stronger prion phenotype is associated with less accumulation of a red pigment, leading to a lighter color). (b) Differences in mitotic stability between strong and weak prion strains of Sup35 protein (mitotic loss of a prion leads to generation of red colonies). (c) Differences in proportion of aggregated (P, pellet) and non-aggregated (S, supernatant) protein between extracts of yeast cells bearing the strong and weak strains of the Sup35 prion, as demonstrated by differential centrifugation, followed by SDS-PAGE and reaction to Sup35 antibodies. (d) Differences in the length of amyloid core between the strong and weak strains of the Sup35 prion. Presumable β-strands are schematically indicated by boxes, and hydrogen bonds by dashes.
Strain reproduction is not error-proof – occasional strain “mutations” have been observed both in vivo and in vitro, and in the latter case are referred to as “deformed templating.”463,592–594 In these cases, an amyloid “population” typically represents a “cloud of substrains” composed of the predominant species and the derivatives generated via deformed templating.581,592,595 The efficiency of deformed templating may vary depending on the amyloid protein sequence, strain, and conditions. Indeed, changes in conditions may lead to a switch in strain patterns due to a change in the relative stability of strains or substrains, enabling these to proliferate better and therefore become predominant in new conditions. Such a switch has been described as “Darwinian evolution” in the case of prion strains.592 The existence of strains, as well as phenomena of deformed templating and strain evolution further complicate the interpretation of structural data for amyloids. Indeed, a structure of amyloid fibrils formed from a purified protein may only represent the strain which is predominant in the given conditions, and may not reflect the strain or strains that are predominant in vivo. Even if the in vitro aggregated amyloid is seeded using in vivo generated extracts, deformed templating or changes in environment may lead to predominance of a different strain. This issue could be addressed for yeast prions, where it is possible to confirm that the amyloid seeded by cell extracts reproduces the original phenotype after the transfection into the yeast cells,476 however it remains a serious obstacle for the structural studies of other amyloids, for which transfection and phenotypic characterization procedures are not as readily available.
7.5.4. Impact of Strains on Sequence-Specificity of Amyloid Transmission.
The strain issue is closely related to the issue of sequence specificity of amyloid propagation. Although rare cross-seeding events between distantly related or unrelated amyloidogenic proteins have been observed, efficient amyloid propagation requires a high level of identity between the interacting protein sequences. Indeed, transmission of the prion state even to a closely related protein (e.g. an ortholog from a different species) is impaired in both mammals and yeast, resulting in a so-called species barrier.580,596 However, the species barrier is not absolute – for example, “mad cow” disease can be transmitted to humans,597 and different strains of an amyloid or prion protein can differ from one other in transmission specificity.586,595,598–600 This likely occurs due to the fact that different regions having different levels of sequence divergence are involved in the cross-β core and into the intermolecular interactions in the various strains. Interestingly, conditions of amyloid formation can influence transmission specificity, primarily via influencing the spectrum of amyloid strains present. For example, kosmotropic and chaotropic anions alter the patterns of prion transmission among the closely related proteins from different species of Saccharomyces yeast by favoring the formation of different prion variants, but these ions do not have a significant impact on transmission specificity of the given variant.601
7.6. Computational Approaches to Prediction of Amyloid and Prion Potential
Considering the large number of proteins that can adopt an amyloid fold and the fact that there is little to no homology between different groups of amyloidogenic proteins, it is very difficult to predict the amyloidogenic potential of a given amino acid sequence. As a result, engineering and design of new amyloidogenic proteins is a very difficult task. Several prediction algorithms have been proposed (Table 6), and some of these work with some reliability in vitro, especially for relatively short peptides. However, none of the available algorithms appear to be capable of predicting the majority of amyloidogenic sequences, and most algorithms are highly ineffective for predictions of in vivo amyloidogenic properties or in the case of longer proteins.602 A key challenge may be that some of these approaches are based solely on the analysis of amyloid aggregation in vitro. For example, a vague consensus hexapeptide “amyloid stretch” motif has been found in most proteins that form amyloids in vitro at neutral pH, and a variation of this motive acting at acidic pH has also been described.603,604 Many proteins that form amyloid in vivo also contain this motif. However, some known amyloidogenic proteins do not contain “amyloid stretch” hexapeptides.
Table 6.
Examples of algorithms for amyloid prediction
| Algorithm | Principle of analysis | Website/server |
|---|---|---|
| Waltz | Presence of hexapeptides with amyloidogenic structures | http://waltz.switchlab.org |
| AGGRESCAN | Search for 5–11 aa amyloidogenic stretches, based on aa composition | http://bioinf.uab.es/aggrescan/ |
| FoldAmyloid | Scanning for 5 aa windows, based on certain chemical properties | http://bioinfo.protres.ru/fold-amyloid/oga.cgi |
| AmylPred | Lack of stable secondary structure, combined aa composition patterns | http://biophysics.biol.uoa.gr/AMYLPRED/ |
| TANGO | β-structure forming propensity and charge | http://tango.crg.es/ |
| SARP | Aa sequences with strong compositional biases | N/A |
| ArchCandy | High propensity of β-arc formation | https://omictools.com/archcandy-tool |
Several computational approaches are based on studies of the NNQQNY hexapeptide, derived from the prion domain of the yeast Sup35 protein, which forms amyloids that generate microcrystals having one of the few solvable structures described above. Based on these studies and computational calculations of probabilities of the formation of similar structures, a dataset of hexapeptides termed AmylHex was composed, containing the predicted capacity of each sequence to form an amyloid.195 This dataset was employed to generate a Waltz algorithm for the prediction of amyloidogenic hexapeptides.605 Similar to the “amyloid stretch” approach, the assumption of this algorithm is that a short amyloidogenic sequence can drive amyloid formation for a whole protein, which may not always be true.
Other computational approaches are aimed at the analysis of aggregation properties for known amyloidogenic proteins having specific amino acid alterations. For example, AGGRESCAN predicts the amyloidogenic properties of individual amino acid stretches of 5–11 amino acids based on the impact of individual residues on amyloidogenecity when substituted for the F residue at position 19 of Aβ.606 The FoldAmyloid program scans across tiled windows of 5 amino acids, employing specific characteristics shown to correlate with amyloidogenicity, such as the mean number of atom–atom contacts per residue, and the mean number of backbone hydrogen bonds per residue.607 This principle, in combination with several approaches for the prediction of secondary structures, is also included in the internet-based AmylPred tool, which predicts amyloids among sequences having ambivalent secondary structures.608 The TANGO algorithm employs the propensity of amino acids to be included in β-strands and searches for oligopeptide sequences having at least 5 residues in row that possess a high β-structure propensity, in a combination with an overall charge that is close to neutral, which promotes assembly by decreasing repulsive electrostatic interactions.609 Other computational approaches use properties of β-structures, estimates of cross-β pairing, or the probability of unfolding of the structured regions in an attempt to predict the propensity for amyloid formation.602 Several algorithms, including Waltz, AGGRESCAN, FoldAmyloid, AmylPred, and TANGO, were compared for their ability to predict amyloidogenicity using a set of proteins having known amyloidogenic properties (as well as a set of control non-amyloidogenic proteins). The approaches performed at varying levels for predicting known amyloids, and all exhibited a very high rate (35–75%) of false positive results.602 The major challenges observed with these approaches were an overprediction of amyloids among hydrophobic sequences, and a poor ability to predict amyloids in the sequences enriched by polar residues such as Q or N.
As mentioned above, some yeast PrDs are QN-rich. Moreover, “scrambled” versions of Sup35 and Ure2 PrDs, that retain amino acid composition but have entirely different sequences, typically retain an ability to form an amyloid-based prion in yeast.610,611 This has led to the suggestion that amino acid composition is more important to prion-forming potential than is the actual amino acid sequence. A list of potential amyloid/prion candidates in the yeast proteome has been composed based on amino acid composition patterns of QN-rich PrDs,480 and further refined and extended employing the hidden Markov Model based approach.479 Some PrDs from this list have been confirmed in experimental assays. Furthermore, at least 1% of proteins in the human proteome possesses domains with patterns of amino acid composition that are similar to known yeast PrDs.480 Subsequent research employing mutational redesigning of a sequence of the small piece of “scrambled” Sup35 PrD helped to define amino acid residues that are more likely to support prion propagation in yeast.612 However, even some yeast or fungal prions are not QN-rich, and prion propensity of amino acid residues identified by the mutagenesis approach applies specifically to prion propagation in the yeast cell rather than to amyloid formation in general.
Recently, a handful of promising new algorithms for amyloid prediction have been proposed. Sequence Analysis based on the Ranking of Probabilities (SARP)613,614 represents a further development of the principal approach that has been used by the Lowest Probability Subsequences (LPS)615 algorithm, and identifies amino acid sequences having strong compositional biases, including but not restricted to amyloidogenic domains. In contrast, the ArchCandy algorithm566 predicts the ability of a given sequence to generate stacked parallel in-register β-sheet structures (β-arcs). ArchCandy predicted known amyloids with a success rate of 85%, placing it close to the best performing computational tools that are based on other principles. Excitingly, ArchCandy produced only ~6% false positives, placing it well ahead of other algorithms. However, one limitation of ArchCandy is that it is unlikely to be capable of efficiently predicting amyloid structures other than β-arcs, such as β-solenoids or antiparallel β-sheets.
7.7. Remaining Challenges and Future Directions
Significant progress has been made recently in understanding the fundamental basis of amyloid formation and propagation, not only in vitro but also in vivo, in particular in yeast models. In addition to their role in diseases and inheritance, amyloids possess significant potential for use in technological applications that require self-assembly. However, there remain only a few amyloid structures that have been solved at atomic or near-atomic resolution. In this sense, amyloids are in the opposite situation of the transcriptional factors described in Chapter 5, for which structural information is readily available, and therefore designing of new structures is possible. In contrast, amyloid studies suffer from poor predictability, making the design of new structures extremely difficult, though some progress has been made in the case of amyloids formed by short peptides, as described in Chapter 3. Further development and refining of amyloid prediction tools, and their verification by experimental approaches, are necessary as a first step toward engineering of longer amyloid proteins.
As described in the latter portion of this Chapter, the other major obstacle to technological applications of amyloids is the issue of amyloid strains. Given that the same amino acid sequence can form different amyloid structures, formation of amyloids having specific properties is difficult or nearly impossible to predict. The widely-known challenges associated with producing high-quality artificial spider silks might potentially be explained, at least in part, in this way. Spiders employ an extrusion apparatus that modulates folding and assembly of silk fibrils through a combination of pressure and ionic strength. As explained in Chapter 6, these conditions are difficult to reproduce in recombinant systems, and therefore structural variants having different properties are likely to be formed in these artificial environments. Understanding the driving forces responsible for the structural properties of amyloid strains, as well as the mechanisms by which conditions influence these strain patterns, will undoubtedly serve as a key to engineering amyloids for human use. This is important, as the self-assembly capabilities of amyloids generate tremendous potential for technological applications including production of nanofibrils, nanowires and scaffolds, or development of new therapies based on sequestration of potentially toxic disease agents by amyloids. Prediction of the amyloid-forming potential of newly produced protein- or peptide-based drugs or hormones is also crucial because generation of amyloid aggregates may reduce their biological activities.
8. CONCLUSION AND OUTLOOK
This Review highlights the remarkable diversity of structural forms and functional capabilities of peptide and protein assemblies, illustrated through examples spanning wide length and complexity scales. While nucleic acid assembly is dominated by base pair specificity and the study of polysaccharides remains constrained synthetic accessibility, the intermolecular determinants that dictate stability in protein assemblies are beginning to define an arc of understanding that may now be used to design and construct new biopolymer assemblies of interest. The context dependence of peptide association energies, important to both the mechanism of the assembly process and the final structure, contributes to the remarkable polymorphic diversity of these assemblies. As our design process becomes more intuitive and our simulations of structural stability become more predictive, the construction of new functional materials will become increasingly robust. The examples herein demonstrate that this progress in engineering and design is directly proportional to our understanding of the structure and function of each assembly, motivating future studies aimed at increasing the depth of our knowledge in these realms.
The opportunities to extend these materials and their functions to the field of biomedical science already shows remarkable potential. In functional co-assemblies, as highlighted with transcriptional factors, in structural materials such as elastins, collagens, and silks, and at the interface of health and disease in the amyloids and prions, new frontiers for the treatment of disease to the creation of new evolvable materials are emerging. As the rules of assembly and co-assembly in proteins are translated into different biopolymer backbones, the designs of chimeric or hybrid biopolymers becomes possible, creating materials that may be orthogonal to, yet compatible with, the existing biopolymers.
On a more basic and fundamental level, the physical condensation and assembly of extant biopolymers are foundational to our grasp of all living systems. An understanding of how the laws of chemistry and physics may constrain the evolutionary history of biology on Earth and elsewhere may arguably be defined by the extent to which we are able to extend the structures and functions of biopolymers into new materials. Our hope is that this review will motivate and empower others to expand and exploit the emerging assembly codes for the design and creation of new functional materials through biopolymer assembly.
Acknowledgements
Y.O.C. thanks Dr. Tatiana Chernova and Dr. Kathryn Bruce for the help in preparing initial figure drafts. A.K.P. thanks Evan K. Roberts and Kong M. Wong for helpful discussions and guidance with the references. The authors gratefully acknowledge support from the National Science Foundation (MCB 1747439, CHE 1507385, CBET 1133834, and CBET 1844289 to C.J.W., DMR 1709428 to J.A.C., MCB 1516872 and MCB 1817976 to Y.O.C., CHE 1507932 and DMR BSF 1610377 to D.G.L., CBET 1743432 to A.K.P., CBET 1818476, CHE 1818781, and DMR 1822262 to J.M.H.), National Institutes of Health (P50AG025688 to D.G.L and Y.O.C., R01AG045703 to A.K.P., 1R01GM116991 to J.M.H.), Defense Threat Reduction Agency (CB10543 to J.M.H.), Howard Hughes Medical Institute (to D.G.L.), Russian Science Foundation (14–50-00069 to Y.O.C.), and St. Petersburg State University (15.61.2218.2013 to Y.O.C.)
Author Biographies
Corey J. Wilson is an Associate Professor in the School of Chemical & Biomolecular Engineering at the Georgia Institute of Technology. Dr. Wilson was an Assistant and Associate Professor of Chemical & Environmental Engineering at Yale University (2008–2016). He earned his B.S. at the University of Houston-Clear Lake in 2002, and earned his Ph.D. in Molecular Biophysics at Rice University in 2005. Dr. Wilson was a Gordon E. Moore Postdoctoral Scholar and National Science Foundation Fellow in the Division of Biology, Chemistry and Chemical Engineering at California Institute of Technology (2006–2008), where he focused on computational protein design. Prof. Wilson leverages protein and genetic engineering strategies to advance our understanding of protein structure and function, in the context of bespoke genetic architectures. His studies have significantly improved our understanding of enzyme temperature adaptation, enzyme structural and functional resistance to oxidative stress, energy transfer within a protein structure, and allostery. His current efforts are focused on biomolecular systems engineering.
Andreas (Andy) S. Bommarius obtained his education at the Technical University of Munich (Chemistry diploma 1984) and the Massachusetts Institute of Technology (BS 1982, PhD 1989 in Chemical Engineering). His doctoral thesis dealt with enzymatic reactions and transport phenomena in reversed micellar systems. Dr. Bommarius joined the Georgia Tech faculty in 2000 after 10 years with Degussa (now Evonik) in amino acid specialty chemicals where he headed the Biocatalysis laboratory and pilot plant. At Georgia Tech, he is a faculty member of the School of Chemical and Biomolecular Engineering as well as the School of Chemistry and Biochemistry as well as the interdisciplinary Bioengineering Program. Since February 2010, he is the Director of the NSF I/UCRC Center for Pharmaceutical Development (CPD). He also is co-director of the Center for Drug Design, Development, and Delivery (CD4) at Georgia Tech. His research interests are in biocatalysis, enzyme reaction engineering, Green Chemistry, protein engineering, and protein stability
Julie Champion is an Associate Professor at Georgia Institute of Technology in the School of Chemical & Biomolecular Engineering and a member of the Petit Institute for Bioengineering and Biosciences. Her lab develops functional protein materials, materials made directly from therapeutic proteins or enzymes by self-assembly or bioconjugation, for immunomodulatory, cancer and biocatalysis applications. She earned her Ph.D. in Chemical Engineering from University of California Santa Barbara and was a National Institutes of Health postdoctoral fellow at California Institute of Technology.
Yury O. Chernoff has received his B.Sc. and Ph.D. degrees in Biology from Leningrad (now St. Petersburg) State University in Russia, and performed postdoctoral studies at Okayama University (Japan) and University of Illinois (Chicago, USA). He is a faculty member (currently a full Professor of Biological Sciences) at Georgia Institute of Technology, Atlanta, USA since 1995, and is also supervising a research lab at St. Petersburg State University, Russia since 2013. His major areas of research include protein biosynthesis, folding, aggregation, protein-based epigenetic inheritance, and yeast models for amyloid and prion disorders. Dr. Chernoff has demonstrated induction of prion formation by transient protein overproduction, and established the crucial role of chaperones in prion propagation. He is a founding Editor-in-Chief of the international journal Prion, currently published by Taylor & Francis, Inc., and a Fellow of the American Association for the Advancement of Science (AAAS).
David G. Lynn has contributed broadly in the areas of systems chemistry, molecular recognition, synthetic biology and chemical evolution, and has developed chemical and physical methods for the analysis of supramolecular self-assemblies, in signal transduction in cellular development and pathogenesis, in molecular skeletons for storing and reading information, and for the evolution of biological order. He holds a Howard Hughes Medical Institute Professorship and is the Asa Griggs Candler Professor in Chemistry and Biology at Emory University.
Anant K. Paravastu is an Associate Professor at the Georgia Institute of Technology, affiliated with the School of Chemical & Biomolecular Engineering and the Petit Institute for Bioengineering and Biosciences. He has degrees in chemical engineering from the Massachusetts Institute of Technology (S.B.) and the University of California Berkeley (Ph.D.). At Berkeley, his Ph.D. research was on optically pumped NMR (advisor: Jeffrey A. Reimer), which is the use of laser excitation to enhance NMR signal strengths in semiconductors. Subsequently, Dr. Paravastu worked as a postdoctoral research fellow (under Robert Tycko) at the Laboratory of Chemical Physics at the National Institutes of Health, where he employed solid-state NMR to probe the molecular structures of amyloid fibrils of the Alzheimer’s amyloid-β peptide. His research group maintains an interest in understanding mechanisms of protein aggregation, expanding the capabilities of NMR techniques to address smaller “oligomeric” protein assemblies, and analyzing the structures formed by designer assembling peptides.
Chen Liang was born in Liaoning, China and received her BS degree from China Pharmaceutical University in 2011. She obtained her Ph.D. degree from Emory University Department of Chemistry in 2017 under the supervision of Dr. David G. Lynn. She has contributed broadly to the dynamics and structural changes associated with peptide condensation and assembly, most notably along the amyloid assembly pathway.
Ming-Chien Hsieh was born in Taipei, Taiwan and received his B.S. degree in Chemical Engineering from National Central University, his M.S. degree in Chemical Engineering from National Taiwan University under the supervision of Prof. Steven S.-S. Wang, and his Ph.D. degree in Georgia Institute of Technology under the supervision of Prof. Martha A. Grover. He joined Professor David G. Lynn’s group as a postdoctoral researcher in 2017. His research interests are investigating the kinetics of peptide assembly and functionalities of peptide-based materials.
Jennifer (Jen) Heemstra received her B.S. in Chemistry from the University of California, Irvine, in 2000. At Irvine, she performed undergraduate research with Prof. James Nowick investigating the folding of synthetic beta-sheet mimics, which instilled in her a love of supramolecular chemistry. She then moved to the University of Illinois, Urbana-Champaign, where she completed her Ph.D. with Prof. Jeffrey Moore in 2005 studying the reactivity of pyridine-functionalized phenylene ethynylene cavitands. After a brief stint in industry as a medicinal chemist, she moved to Harvard University to pursue postdoctoral research with Prof. David Liu exploring mechanisms for templated nucleic acid synthesis. Dr. Heemstra began her independent career in 2010, and is currently an Associate Professor in the Department of Chemistry at Emory University. Research in the Heemstra lab is focused on harnessing the molecular recognition and self-assembly properties of biomolecules for applications in biosensing and bioimaging.
References
- (1).Barabasi AL; Oltvai ZN Network Biology: Understanding the Cell’s Functional Organization. Nat. Rev. Genet 2004, 5, 101–113. [DOI] [PubMed] [Google Scholar]
- (2).Ross PD; Subramanian S Thermodynamics of Protein Association Reactions: Forces Contributing to Stability. Biochemistry 2002, 20, 3096–3102. [DOI] [PubMed] [Google Scholar]
- (3).Luo Q; Hou C; Bai Y; Wang R; Liu J Protein Assembly: Versatile Approaches to Construct Highly Ordered Nanostructures. Chem. Rev 2016, 116, 13571–13632. [DOI] [PubMed] [Google Scholar]
- (4).Yeates TO; Padilla JE Designing Supramolecular Protein Assemblies. Curr. Opin. Struct. Biol 2002, 12, 464–470. [DOI] [PubMed] [Google Scholar]
- (5).Jackel C; Kast P; Hilvert D Protein Design by Directed Evolution. Annu. Rev. Biophys 2008, 37, 153–173. [DOI] [PubMed] [Google Scholar]
- (6).Rothemund PW Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440, 297–302. [DOI] [PubMed] [Google Scholar]
- (7).Seeman NC An Overview of Structural DNA Nanotechnology. Mol. Biotechnol 2007, 37, 246–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Pinheiro AV; Han D; Shih WM; Yan H Challenges and Opportunities for Structural DNA Nanotechnology. Nat. Nanotechol 2011, 6, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Guo P The Emerging Field of Rna Nanotechnology. Nat. Nanotechol 2010, 5, 833–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Seeberger PH The Logic of Automated Glycan Assembly. Acc. Chem. Res 2015, 48, 1450–1463. [DOI] [PubMed] [Google Scholar]
- (11).Winder SJ; Ayscough KR Actin-Binding Proteins. J. Cell. Sci 2005, 118, 651–654. [DOI] [PubMed] [Google Scholar]
- (12).Yusuf D; Butland SL; Swanson MI; Bolotin E; Ticoll A; Cheung WA; Zhang XYC; Dickman CT; Fulton DL; Lim JS The Transcription Factor Encyclopedia. Genome Biol 2012, 13, R24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Harris BZ; Lim WA Mechanism and Role of Pdz Domains in Signaling Complex Assembly. J. Cell Sci 2001, 114, 3219–3231. [DOI] [PubMed] [Google Scholar]
- (14).Lad C; Williams NH; Wolfenden R The Rate of Hydrolysis of Phosphomonoester Dianions and the Exceptional Catalytic Proficiencies of Protein and Inositol Phosphatases. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 5607–5610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Breslow R; Dong SD Biomimetic Reactions Catalyzed by Cyclodextrins and Their Derivatives. Chem. Rev 1998, 98, 1997–2012. [DOI] [PubMed] [Google Scholar]
- (16).Qi D; Tann C-M; Haring D; Distefano MD Generation of New Enzymes Via Covalent Modification of Existing Proteins. Chem. Rev 2001, 101, 3081–3112. [DOI] [PubMed] [Google Scholar]
- (17).Dunker AK; Lawson JD; Brown CJ; Williams RM; Romero P; Oh JS; Oldfield CJ; Campen AM; Ratliff CM; Hipps KW Intrinsically Disordered Protein. J. Mol. Graph. Model 2001, 19, 26–59. [DOI] [PubMed] [Google Scholar]
- (18).Dunker AK; Oldfield CJ; Meng J; Romero P; Yang JY; Chen JW; Vacic V; Obradovic Z; Uversky VN The Unfoldomics Decade: An Update on Intrinsically Disordered Proteins. BMC Genomics 2008, 9, S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Tompa P Intrinsically Disordered Proteins: A 10-Year Recap. Trends. Biochem. Sci 2012, 37, 509–516. [DOI] [PubMed] [Google Scholar]
- (20).Wright PE; Dyson HJ Intrinsically Disordered Proteins in Cellular Signalling and Regulation. Nat. Rev. Mol. Cell Biol 2015, 16, 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Eichner T; Radford SE A Diversity of Assembly Mechanisms of a Generic Amyloid Fold. Mol. Cell 2011, 43, 8–18. [DOI] [PubMed] [Google Scholar]
- (22).Kay LE New Views of Functionally Dynamic Proteins by Solution Nmr Spectroscopy. J. Mol. Biol 2016, 428, 323–331. [DOI] [PubMed] [Google Scholar]
- (23).Kendrew JC; Bodo G; Dintzis HM; Parrish R; Wyckoff H; Phillips DC A Three-Dimensional Model of the Myoglobin Molecule Obtained by X-Ray Analysis. Nature 1958, 181, 662–666. [DOI] [PubMed] [Google Scholar]
- (24).Onuchic JN; Wolynes PG Theory of Protein Folding. Curr. Opin. Struct. Biol 2004, 14, 70–75. [DOI] [PubMed] [Google Scholar]
- (25).Bartesaghi A; Merk A; Banerjee S; Matthies D; Wu X; Milne JL; Subramaniam S 2.2 Å Resolution Cryo-Em Structure of Β-Galactosidase in Complex with a Cell-Permeant Inhibitor. Science 2015, 348, 1147–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Liu H; Jin L; Koh SBS; Atanasov I; Schein S; Wu L; Zhou ZH Atomic Structure of Human Adenovirus by Cryo-Em Reveals Interactions among Protein Networks. Science 2010, 329, 1038–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Chou PY; Fasman GD Prediction of Protein Conformation. Biochemistry 1974, 13, 222–245. [DOI] [PubMed] [Google Scholar]
- (28).Dill KA; Ozkan SB; Shell MS; Weikl TR The Protein Folding Problem. Annu. Rev. Biophys 2008, 37, 289–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Buell AK The Nucleation of Protein Aggregates - from Crystals to Amyloid Fibrils. Int. Rev. Cell. Mol. Biol 2017, 329, 187–226. [DOI] [PubMed] [Google Scholar]
- (30).Lindenberg C; Mazzotti M Effect of Temperature on the Nucleation Kinetics of Α L-Glutamic Acid. J. Cryst. Growth 2009, 311, 1178–1184. [Google Scholar]
- (31).Sauter A; Roosen-Runge F; Zhang F; Lotze G; Jacobs RM; Schreiber F Real-Time Observation of Nonclassical Protein Crystallization Kinetics. J. Am. Chem. Soc 2015, 137, 1485–1491. [DOI] [PubMed] [Google Scholar]
- (32).Sauter A; Roosen-Runge F; Zhang F; Lotze G; Feoktystov A; Jacobs RM; Schreiber F On the Question of Two-Step Nucleation in Protein Crystallization. Faraday Discuss 2015, 179, 41–58. [DOI] [PubMed] [Google Scholar]
- (33).Vorontsova MA; Maes D; Vekilov PG Recent Advances in the Understanding of Two-Step Nucleation of Protein Crystals. Faraday Discuss 2015, 179, 27–40. [DOI] [PubMed] [Google Scholar]
- (34).Erdemir D; Lee AY; Myerson AS Nucleation of Crystals from Solution: Classical and Two-Step Models. Acc. Chem. Res 2009, 42, 621–629. [DOI] [PubMed] [Google Scholar]
- (35).Tauer K; Kühn I Modeling Particle Formation in Emulsion Polymerization: An Approach by Means of the Classical Nucleation Theory. Macromolecules 1995, 28, 2236–2239. [Google Scholar]
- (36).Girshick SL; Chiu CP Kinetic Nucleation Theory: A New Expression for the Rate of Homogeneous Nucleation from an Ideal Supersaturated Vapor. J. Chem. Phys 1990, 93, 1273–1277. [Google Scholar]
- (37).Oxtoby DW Homogeneous Nucleation: Theory and Experiment. J. Phys.: Condens. Matter 1992, 4, 7627. [Google Scholar]
- (38).Auer S; Ricchiuto P; Kashchiev D Two-Step Nucleation of Amyloid Fibrils: Omnipresent or Not? J. Mol. Biol 2012, 422, 723–730. [DOI] [PubMed] [Google Scholar]
- (39).Hills RD; Brooks CL Hydrophobic Cooperativity as a Mechanism for Amyloid Nucleation. J. Mol. Biol 2007, 368, 894–901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Hurshman AR; White JT; Powers ET; Kelly JW Transthyretin Aggregation under Partially Denaturing Conditions Is a Downhill Polymerization. Biochemistry 2004, 43, 7365–7381. [DOI] [PubMed] [Google Scholar]
- (41).Massi F; Straub JE Energy Landscape Theory for Alzheimer’s Amyloid Β-Peptide Fibril Elongation. Proteins 2001, 42, 217–229. [DOI] [PubMed] [Google Scholar]
- (42).Powers ET; Powers DL The Kinetics of Nucleated Polymerizations at High Concentrations: Amyloid Fibril Formation near and above the “Supercritical Concentration”. Biophys. J 2006, 91, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Bieler NS; Knowles TP; Frenkel D; Vacha R Connecting Macroscopic Observables and Microscopic Assembly Events in Amyloid Formation Using Coarse Grained Simulations. PLoS Comput. Biol 2012, 8, e1002692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Fawzi NL; Okabe Y; Yap EH; Head-Gordon T Determining the Critical Nucleus and Mechanism of Fibril Elongation of the Alzheimer’s Abeta(1–40) Peptide. J. Mol. Biol 2007, 365, 535–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Nasica-Labouze J; Mousseau N Kinetics of Amyloid Aggregation: A Study of the Gnnqqny Prion Sequence. PLoS Comput. Biol 2012, 8, e1002782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Nguyen PH; Li MS; Stock G; Straub JE; Thirumalai D Monomer Adds to Preformed Structured Oligomers of Abeta-Peptides by a Two-Stage Dock-Lock Mechanism. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Nguyen P; Derreumaux P Understanding Amyloid Fibril Nucleation and Aβ Oligomer/Drug Interactions from Computer Simulations. Acc. Chem. Res 2013, 47, 603–611. [DOI] [PubMed] [Google Scholar]
- (48).Xue WF; Homans SW; Radford SE Systematic Analysis of Nucleation-Dependent Polymerization Reveals New Insights into the Mechanism of Amyloid Self-Assembly. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 8926–8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Arnaudov LN; de Vries R Strong Impact of Ionic Strength on the Kinetics of Fibrilar Aggregation of Bovine Β-Lactoglobulin. Biomacromolecules 2006, 7, 3490–3498. [DOI] [PubMed] [Google Scholar]
- (50).Chen S; Ferrone FA; Wetzel R Huntington’s Disease Age-of-Onset Linked to Polyglutamine Aggregation Nucleation. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 11884–11889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Kar K; Jayaraman M; Sahoo B; Kodali R; Wetzel R Critical Nucleus Size for Disease-Related Polyglutamine Aggregation Is Repeat-Length Dependent. Nat. Struct. Mol. Biol 2011, 18, 328–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).van der Linden E; Venema P Self-Assembly and Aggregation of Proteins. Curr. Opin. Coll. Interf. Sci 2007, 12, 158–165. [Google Scholar]
- (53).Klimov DK; Thirumalai D Dissecting the Assembly of Aβ 16–22 Amyloid Peptides into Antiparallel Β Sheets. Structure 2003, 11, 295–307. [DOI] [PubMed] [Google Scholar]
- (54).Xie L; Luo Y; Wei G Abeta(16–22) Peptides Can Assemble into Ordered Beta-Barrels and Bilayer Beta-Sheets, While Substitution of Phenylalanine 19 by Tryptophan Increases the Population of Disordered Aggregates. J. Phys. Chem. B 2013, 117, 10149–10160. [DOI] [PubMed] [Google Scholar]
- (55).Barz B; Wales DJ; Strodel B A Kinetic Approach to the Sequence-Aggregation Relationship in Disease-Related Protein Assembly. J. Phys. Chem. B 2014, 118, 1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Hwang W; Zhang S; Kamm RD; Karplus M Kinetic Control of Dimer Structure Formation in Amyloid Fibrillogenesis. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 12916–12921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Santini S; Mousseau N; Derreumaux P In Silico Assembly of Alzheimer’s Aβ16–22 Peptide into Β-Sheets. J. Am. Chem. Soc 2004, 126, 11509–11516. [DOI] [PubMed] [Google Scholar]
- (58).Arosio P; Beeg M; Nicoud L; Morbidelli M Time Evolution of Amyloid Fibril Length Distribution Described by a Population Balance Model. Chem. Eng. Sci 2012, 78, 21–32. [Google Scholar]
- (59).Cohen SI; Vendruscolo M; Welland ME; Dobson CM; Terentjev EM; Knowles TP Nucleated Polymerization with Secondary Pathways. I. Time Evolution of the Principal Moments. J. Chem. Phys 2011, 135, 065105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Knowles TP; Waudby CA; Devlin GL; Cohen SI; Aguzzi A; Vendruscolo M; Terentjev EM; Welland ME; Dobson CM An Analytical Solution to the Kinetics of Breakable Filament Assembly. Science 2009, 326, 1533–1537. [DOI] [PubMed] [Google Scholar]
- (61).Morris AM; Watzky MA; Finke RG Protein Aggregation Kinetics, Mechanism, and Curve-Fitting: A Review of the Literature. Biochim. Biophys. Acta 2009, 1794, 375–397. [DOI] [PubMed] [Google Scholar]
- (62).Morris AM; Watzky MA; Agar JN; Finke RG Fitting Neurological Protein Aggregation Kinetic Data Via a 2-Step, Minimal/”Ockham’s Razor” Model: The Finke− Watzky Mechanism of Nucleation Followed by Autocatalytic Surface Growth. Biochemistry 2008, 47, 2413–2427. [DOI] [PubMed] [Google Scholar]
- (63).ten Wolde PR; Frenkel D Enhancement of Protein Crystal Nucleation by Critical Density Fluctuations. Science 1997, 277, 1975–1978. [DOI] [PubMed] [Google Scholar]
- (64).Vekilov PG The Two-Step Mechanism of Nucleation of Crystals in Solution. Nanoscale 2010, 2, 2346–2357. [DOI] [PubMed] [Google Scholar]
- (65).Levin A; Mason TO; Adler-Abramovich L; Buell AK; Meisl G; Galvagnion C; Bram Y; Stratford SA; Dobson CM; Knowles TP, et al. Ostwald’s Rule of Stages Governs Structural Transitions and Morphology of Dipeptide Supramolecular Polymers. Nat. Commun 2014, 5, 5219. [DOI] [PubMed] [Google Scholar]
- (66).Liu Y; Wang X; Ching CB Toward Further Understanding of Lysozyme Crystallization: Phase Diagram, Protein−Protein Interaction, Nucleation Kinetics, and Growth Kinetics. Cryst. Growth Des 2010, 10, 548–558. [Google Scholar]
- (67).Pouget EM; Bomans PH; Goos JA; Frederik PM; Sommerdijk NA The Initial Stages of Template-Controlled Caco 3 Formation Revealed by Cryo-Tem. Science 2009, 323, 1455–1458. [DOI] [PubMed] [Google Scholar]
- (68).Savage JR; Dinsmore AD Experimental Evidence for Two-Step Nucleation in Colloidal Crystallization. Phys. Rev. Lett 2009, 102, 198302. [DOI] [PubMed] [Google Scholar]
- (69).Hsieh MC; Liang C; Mehta AK; Lynn DG; Grover MA Multistep Conformation Selection in Amyloid Assembly. J. Am. Chem. Soc 2017, 139, 17007–17010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Hsieh MC; Lynn DG; Grover MA Kinetic Model for Two-Step Nucleation of Peptide Assembly. J. Phys. Chem. B 2017, 121, 7401–7411. [DOI] [PubMed] [Google Scholar]
- (71).Liang C; Ni R; Smith JE; Childers WS; Mehta AK; Lynn DG Kinetic Intermediates in Amyloid Assembly. J. Am. Chem. Soc 2014, 136, 15146–15149. [DOI] [PubMed] [Google Scholar]
- (72).Smith JE; Liang C; Tseng M; Li N; Li S; Mowles AK; Mehta AK; Lynn DG Defining the Dynamic Conformational Networks of Cross-Β Peptide Assembly. Isr. J. Chem 2015, 55, 763–769. [Google Scholar]
- (73).Luiken JA; Bolhuis PG Primary Nucleation Kinetics of Short Fibril-Forming Amyloidogenic Peptides. J. Phys. Chem. B 2015, 119, 12568–12579. [DOI] [PubMed] [Google Scholar]
- (74).Pan W; Vekilov PG; Lubchenko V Origin of Anomalous Mesoscopic Phases in Protein Solutions. J. Phys. Chem. B 2010, 114, 7620–7630. [DOI] [PubMed] [Google Scholar]
- (75).Krishnan R; Lindquist SL Structural Insights into a Yeast Prion Illuminate Nucleation and Strain Diversity. Nature 2005, 435, 765–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Guo Q; Mehta AK; Grover MA; Chen W; Lynn DG; Chen Z Shape Selection and Multi-Stability in Helical Ribbons. Appl. Phys. Lett 2014, 104, 211901. [Google Scholar]
- (77).Ilie IM; den Otter WK; Briels WJ A Coarse Grained Protein Model with Internal Degrees of Freedom. Application to Alpha-Synuclein Aggregation. J. Chem. Phys 2016, 144, 085103. [DOI] [PubMed] [Google Scholar]
- (78).Kashchiev D; Vekilov PG; Kolomeisky AB Kinetics of Two-Step Nucleation of Crystals. J. Chem. Phys 2005, 122, 244706. [DOI] [PubMed] [Google Scholar]
- (79).Pan W; Kolomeisky AB; Vekilov PG Nucleation of Ordered Solid Phases of Proteins Via a Disordered High-Density State: Phenomenological Approach. J. Chem. Phys 2005, 122, 174905. [DOI] [PubMed] [Google Scholar]
- (80).Aich A; Pan W; Vekilov PG Thermodynamic Mechanism of Free Heme Action on Sickle Cell Hemoglobin Polymerization. AIChE J 2015, 61, 2861–2870. [Google Scholar]
- (81).Kusumoto Y; Lomakin A; Teplow DB; Benedek GB Temperature Dependence of Amyloid - Protein Fibrillization. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 12277–12282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Childers WS; Anthony NR; Mehta AK; Berland KM; Lynn DG Phase Networks of Cross-Beta Peptide Assemblies. Langmuir 2012, 28, 6386–6395. [DOI] [PubMed] [Google Scholar]
- (83).Collins SR; Douglass A; Vale RD; Weissman JS Mechanism of Prion Propagation: Amyloid Growth Occurs by Monomer Addition. PLoS Biol 2004, 2, e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Lomakin A; Chung DS; Benedek GB; Kirschner DA; Teplow DB On the Nucleation and Growth of Amyloid Beta-Protein Fibrils: Detection of Nuclei and Quantitation of Rate Constants. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 1125–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Carulla N; Caddy GL; Hall DR; Zurdo J; Gairi M; Feliz M; Giralt E; Robinson CV; Dobson CM Molecular Recycling within Amyloid Fibrils. Nature 2005, 436, 554–558. [DOI] [PubMed] [Google Scholar]
- (86).O’Nuallain B; Shivaprasad S; Kheterpal I; Wetzel R Thermodynamics of Aβ (1− 40) Amyloid Fibril Elongation. Biochemistry 2005, 44, 12709–12718. [DOI] [PubMed] [Google Scholar]
- (87).Carnall JM; Waudby CA; Belenguer AM; Stuart MC; Peyralans JJ-P; Otto S Mechanosensitive Self-Replication Driven by Self-Organization. Science 2010, 327, 1502–1506. [DOI] [PubMed] [Google Scholar]
- (88).Tanaka M; Collins SR; Toyama BH; Weissman JS The Physical Basis of How Prion Conformations Determine Strain Phenotypes. Nature 2006, 442, 585–589. [DOI] [PubMed] [Google Scholar]
- (89).Wang SSS; Chen Y-T; Chen P-H; Liu K-N A Kinetic Study on the Aggregation Behavior of Β-Amyloid Peptides in Different Initial Solvent Environments. Biochem. Eng. J 2006, 29, 129–138. [Google Scholar]
- (90).Lomakin A; Teplow DB; Kirschner DA; Benedek GB Kinetic Theory of Fibrillogenesis of Amyloid Β-Protein. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 7942–7947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Debeljuh N; Barrow CJ; Byrne N The Impact of Ionic Liquids on Amyloid Fibrilization of Abeta16–22: Tuning the Rate of Fibrilization Using a Reverse Hofmeister Strategy. Phys. Chem. Chem. Phys 2011, 13, 16534–16536. [DOI] [PubMed] [Google Scholar]
- (92).Lomakin A; Asherie N; Benedek GB Liquid-Solid Transition in Nuclei of Protein Crystals. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 10254–10257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (93).Lee J; Culyba EK; Powers ET; Kelly JW Amyloid-Beta Forms Fibrils by Nucleated Conformational Conversion of Oligomers. Nat. Chem. Biol 2011, 7, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Tomiyama T; Matsuyama S; Iso H; Umeda T; Takuma H; Ohnishi K; Ishibashi K; Teraoka R; Sakama N; Yamashita T, et al. A Mouse Model of Amyloid Beta Oligomers: Their Contribution to Synaptic Alteration, Abnormal Tau Phosphorylation, Glial Activation, and Neuronal Loss in Vivo. J. Neurosci 2010, 30, 4845–4856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (95).Jang Y; Champion JA Self-Assembled Materials Made from Functional Recombinant Proteins. Acc. Chem. Res 2016, 49, 2188–2198. [DOI] [PubMed] [Google Scholar]
- (96).Laganowsky A; Liu C; Sawaya MR; Whitelegge JP; Park J; Zhao M; Pensalfini A; Soriaga AB; Landau M; Teng PK Atomic View of a Toxic Amyloid Small Oligomer. Science 2012, 335, 1228–1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (97).Yu L; Edalji R; Harlan JE; Holzman TF; Lopez AP; Labkovsky B; Hillen H; Barghorn S; Ebert U; Richardson PL Structural Characterization of a Soluble Amyloid Β-Peptide Oligomer. Biochemistry 2009, 48, 1870–1877. [DOI] [PubMed] [Google Scholar]
- (98).Li S; Mehta AK; Sidorov AN; Orlando TM; Jiang Z; Anthony NR; Lynn DG Design of Asymmetric Peptide Bilayer Membranes. J. Am. Chem. Soc 2016, 138, 3579–3586. [DOI] [PubMed] [Google Scholar]
- (99).Watanabe-Nakayama T; Ono K; Itami M; Takahashi R; Teplow DB; Yamada M High-Speed Atomic Force Microscopy Reveals Structural Dynamics of Amyloid Beta1–42 Aggregates. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 5835–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Goodwin JT; Mehta AK; Lynn DG Digital and Analog Chemical Evolution. Acc. Chem. Res 2012, 45, 2189–2199. [DOI] [PubMed] [Google Scholar]
- (101).Childers WS; Mehta AK; Bui TQ; Liang Y; Lynn DG, Toward Intelligent Materials. In Molecular Self-Assembly: Advances and Applications, Li A, Ed. Pan Stanford Publishing: Singapore, 2013. [Google Scholar]
- (102).Hartgerink JD; Zubarev ER; Stupp SI Supramolecular One-Dimensional Objects. Curr. Opin. Solid State Mater. Sci 2001, 5, 355–361. [Google Scholar]
- (103).Woolfson DN; Bartlett GJ; Bruning M; Thomson AR New Currency for Old Rope: From Coiled-Coil Assemblies to Alpha-Helical Barrels. Curr. Opin. Struct. Biol 2012, 22, 432–441. [DOI] [PubMed] [Google Scholar]
- (104).MacPhee CE; Woolfson DN Engineered and Designed Peptide-Based Fibrous Biomaterials. Curr. Opin. Solid State Mater. Sci 2004, 8, 141–149. [Google Scholar]
- (105).Huang PS; Boyken SE; Baker D The Coming of Age of De Novo Protein Design. Nature 2016, 537, 320–327. [DOI] [PubMed] [Google Scholar]
- (106).Regan L; Woolfson DN Protein Folding and Design: From Simple Models to Complex Systems. Curr. Opin. Struct. Biol 2008, 18, 475–476. [DOI] [PubMed] [Google Scholar]
- (107).Ulijn RV; Woolfson DN Peptide and Protein Based Materials in 2010: From Design and Structure to Function and Application. Chem. Soc. Rev 2010, 39, 3349–3350. [DOI] [PubMed] [Google Scholar]
- (108).Woolfson DN The Design of Coiled-Coil Structures and Assemblies. Adv. Prot. Chem 2005, 70, 79–112. [DOI] [PubMed] [Google Scholar]
- (109).Bowerman CJ; Nilsson BL Self-Assembly of Amphipathic Beta-Sheet Peptides: Insights and Applications. Biopolymers 2012, 98, 169–184. [DOI] [PubMed] [Google Scholar]
- (110).Boyle AL; Woolfson DN De Novo Designed Peptides for Biological Applications. Chem. Soc. Rev 2011, 40, 4295–4306. [DOI] [PubMed] [Google Scholar]
- (111).Bromley EH; Channon K; Moutevelis E; Woolfson DN Peptide and Protein Building Blocks for Synthetic Biology: From Programming Biomolecules to Self-Organized Biomolecular Systems. ACS Chem. Biol 2008, 3, 38–50. [DOI] [PubMed] [Google Scholar]
- (112).Channon K; Bromley EH; Woolfson DN Synthetic Biology through Biomolecular Design and Engineering. Curr. Opin. Struct. Biol 2008, 18, 491–498. [DOI] [PubMed] [Google Scholar]
- (113).Dasgupta A; Mondal JH; Das D Peptide Hydrogels. RSC Advances 2013, 3, 9117. [Google Scholar]
- (114).Edwards-Gayle CJC; Hamley IW Self-Assembly of Bioactive Peptides, Peptide Conjugates, and Peptide Mimetic Materials. Org. Biomol. Chem 2017, 15, 5867–5876. [DOI] [PubMed] [Google Scholar]
- (115).Gelain F; Horii A; Zhang S Designer Self-Assembling Peptide Scaffolds for 3-D Tissue Cell Cultures and Regenerative Medicine. Macromol. Biosci 2007, 7, 544–551. [DOI] [PubMed] [Google Scholar]
- (116).Liu J; Zhao X Design of Self-Assembling Peptides and Their Biomedical Applications. Nanomedicine 2011, 6, 1621–1643. [DOI] [PubMed] [Google Scholar]
- (117).Matsuurua K Rational Design of Self-Assembled Proteins and Peptides for Nano- and Micro-Sized Architectures. RSC Adv 2014, 4, 2942–2953. [Google Scholar]
- (118).Rad-Malekshahi M; Lempsink L; Amidi M; Hennink WE; Mastrobattista E Biomedical Applications of Self-Assembling Peptides. Bioconjug. Chem 2016, 27, 3–18. [DOI] [PubMed] [Google Scholar]
- (119).Hosseinkhani H; Hong PD; Yu DS Self-Assembled Proteins and Peptides for Regenerative Medicine. Chem. Rev 2013, 113, 4837–4861. [DOI] [PubMed] [Google Scholar]
- (120).Ulijn RV; Smith AM Designing Peptide Based Nanomaterials. Chem. Soc. Rev 2008, 37, 664–675. [DOI] [PubMed] [Google Scholar]
- (121).Woolfson DN; Ryadnov MG Peptide-Based Fibrous Biomaterials: Some Things Old, New and Borrowed. Curr. Opin. Chem. Biol 2006, 10, 559–567. [DOI] [PubMed] [Google Scholar]
- (122).Yu Z; Cai Z; Chen Q; Liu M; Ye L; Ren J; Liao W; Liu S Engineering Beta-Sheet Peptide Assemblies for Biomedical Applications. Biomater. Sci 2016, 4, 365–374. [DOI] [PubMed] [Google Scholar]
- (123).Zhang S Fabrication of Novel Biomaterials through Molecular Self-Assembly. Nat. Biotechnol 2003, 21, 1171–1178. [DOI] [PubMed] [Google Scholar]
- (124).Rajagopal K; Schneider JP Self-Assembling Peptides and Proteins for Nanotechnological Applications. Curr. Opin. Struct. Biol 2004, 14, 480–486. [DOI] [PubMed] [Google Scholar]
- (125).Zhang S; Marini DM; Hwang W; Santoso S Design of Nanostructured Biological Materials through Self-Assembly of Peptides and Proteins. Curr. Opin. Chem. Biol 2002, 6, 865–871. [DOI] [PubMed] [Google Scholar]
- (126).Zhao X; Zhang S Fabrication of Molecular Materials Using Peptide Construction Motifs. Trends Biotechnol 2004, 22, 470–476. [DOI] [PubMed] [Google Scholar]
- (127).Zhao X; Zhang S Molecular Designer Self-Assembling Peptides. Chem. Soc. Rev 2006, 35, 1105–1110. [DOI] [PubMed] [Google Scholar]
- (128).Yanlian Y; Ulung K; Xiumei W; Horii A; Yokoi H; Shuguang Z Designer Self-Assembling Peptide Nanomaterials. Nano Today 2009, 4, 193–210. [Google Scholar]
- (129).Lowik DW; Leunissen EH; van den Heuvel M; Hansen MB; van Hest JC Stimulus Responsive Peptide Based Materials. Chem. Soc. Rev 2010, 39, 3394–3412. [DOI] [PubMed] [Google Scholar]
- (130).Apostolovic B; Danial M; Klok HA Coiled Coils: Attractive Protein Folding Motifs for the Fabrication of Self-Assembled, Responsive and Bioactive Materials. Chem. Soc. Rev 2010, 39, 3541–3575. [DOI] [PubMed] [Google Scholar]
- (131).Jung JP; Nagaraj AK; Fox EK; Rudra JS; Devgun JM; Collier JH Co-Assembling Peptides as Defined Matrices for Endothelial Cells. Biomaterials 2009, 30, 2400–2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Geisler IM; Schneider JP Evolution-Based Design of an Injectable Hydrogel. Adv. Funct. Mater 2012, 22, 529–537. [Google Scholar]
- (133).Zhang HV; Polzer F; Haider MJ; Tian Y; Villegas JA; Kiick KL; Pochan DJ; Saven JG Computationally Designed Peptides for Self-Assembly of Nanostructured Lattices. Sci. Adv 2016, 2, e1600307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Smith AM; Banwell EF; Edwards WR; Pandya MJ; Woolfson DN Engineering Increased Stability into Self-Assembled Protein Fibers. Adv. Funct. Mater 2006, 16, 1022–1030. [Google Scholar]
- (135).Ryadnov MG; Woolfson DN Map Peptides: Programming the Self-Assembly of Peptide-Based Mesoscopic Matrices. J. Am. Chem. Soc 2005, 127, 12407–12415. [DOI] [PubMed] [Google Scholar]
- (136).Tian Y; Zhang HV; Kiick KL; Saven JG; Pochan DJ Transition from Disordered Aggregates to Ordered Lattices: Kinetic Control of the Assembly of a Computationally Designed Peptide. Org. Biomol. Chem 2017, 15, 6109–6118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Fletcher JM; Harniman RL; Barnes FR; Boyle AL; Collins A; Mantell J; Sharp TH; Antognozzi M; Booth PJ; Linden N Self-Assembling Cages from Coiled-Coil Peptide Modules. Science 2013, 340, 595–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Li IC; Moore AN; Hartgerink JD “Missing Tooth” Multidomain Peptide Nanofibers for Delivery of Small Molecule Drugs. Biomacromolecules 2016, 17, 2087–2095. [DOI] [PubMed] [Google Scholar]
- (139).Sharp TH; Bruning M; Mantell J; Sessions RB; Thomson AR; Zaccai NR; Brady RL; Verkade P; Woolfson DN Cryo-Transmission Electron Microscopy Structure of a Gigadalton Peptide Fiber of De Novo Design. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 13266–13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Yucel T; Micklitsch CM; Schneider JP; Pochan DJ Direct Observation of Early-Time Hydrogelation in Β-Hairpin Peptide Self-Assembly. Macromolecules 2008, 41, 5763–5772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Egelman EH; Xu C; DiMaio F; Magnotti E; Modlin C; Yu X; Wright E; Baker D; Conticello VP Structural Plasticity of Helical Nanotubes Based on Coiled-Coil Assemblies. Structure 2015, 23, 280–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Zhang S; Rich A Direct Conversion of an Oligopeptide from a Β-Sheet to an Α-Helix: A Model for Amyloid Formation. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 23–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Dong H; Hartgerink JD Short Homodimeric and Heterodimeric Coiled Coils. Biomacromolecules 2006, 7, 691–695. [DOI] [PubMed] [Google Scholar]
- (144).Baker EG; Bartlett GJ; Crump MP; Sessions RB; Linden N; Faul CF; Woolfson DN Local and Macroscopic Electrostatic Interactions in Single Alpha-Helices. Nat. Chem. Biol 2015, 11, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Cormier AR; Pang X; Zimmerman MI; Zhou H-X; Paravastu AK Molecular Structure of Rada16-I Designer Self-Assembling Peptide Nanofibers. ACS Nano 2013, 7, 7562–7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Papapostolou D; Smith AM; Atkins ED; Oliver SJ; Ryadnov MG; Serpell LC; Woolfson DN Engineering Nanoscale Order into a Designed Protein Fiber. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 10853–10858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Breukels V; Konijnenberg A; Nabuurs SM; Doreleijers JF; Kovalevskaya NV; Vuister GW Overview on the Use of Nmr to Examine Protein Structure. Curr. Protoc. Prot. Sci 2011, 64, 17.15. 11–17.15. 44. [DOI] [PubMed] [Google Scholar]
- (148).Wider G Nmr Techniques Used with Very Large Biological Macromolecules in Solution. Methods Enzymol 2005, 394, 382–398. [DOI] [PubMed] [Google Scholar]
- (149).Tugarinov V; Kanelis V; Kay LE Isotope Labeling Strategies for the Study of High-Molecular-Weight Proteins by Solution Nmr Spectroscopy. Nat. Protoc 2006, 1, 749–754. [DOI] [PubMed] [Google Scholar]
- (150).Tycko R; Wickner RB Molecular Structures of Amyloid and Prion Fibrils: Consensus Versus Controversy. Acc. Chem. Res 2013, 46, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Burgess NC; Sharp TH; Thomas F; Wood CW; Thomson AR; Zaccai NR; Brady RL; Serpell LC; Woolfson DN Modular Design of Self-Assembling Peptide-Based Nanotubes. J. Am. Chem. Soc 2015, 137, 10554–10562. [DOI] [PubMed] [Google Scholar]
- (152).King PJ; Giovanna Lizio M; Booth A; Collins RF; Gough JE; Miller AF; Webb SJ A Modular Self-Assembly Approach to Functionalised Beta-Sheet Peptide Hydrogel Biomaterials. Soft Matter 2016, 12, 1915–1923. [DOI] [PubMed] [Google Scholar]
- (153).Bagrov D; Gazizova Y; Podgorsky V; Udovichenko I; Danilkovich A; Prusakov K; Klinov D Morphology and Aggregation of Rada-16-I Peptide Studied by Afm, Nmr and Molecular Dynamics Simulations. Biopolymers 2016, 106, 72–81. [DOI] [PubMed] [Google Scholar]
- (154).Meng Q; Kou Y; Ma X; Liang Y; Guo L; Ni C; Liu K Tunable Self-Assembled Peptide Amphiphile Nanostructures. Langmuir 2012, 28, 5017–5022. [DOI] [PubMed] [Google Scholar]
- (155).Ho SP; DeGrado WF Design of a 4-Helix Bundle Protein: Synthesis of Peptides Which Self-Associate into a Helical Protein. J. Am. Chem. Soc 1987, 109, 6751–6758. [Google Scholar]
- (156).Boyle AL; Bromley EH; Bartlett GJ; Sessions RB; Sharp TH; Williams CL; Curmi PM; Forde NR; Linke H; Woolfson DN Squaring the Circle in Peptide Assembly: From Fibers to Discrete Nanostructures by De Novo Design. J. Am. Chem. Soc 2012, 134, 15457–15467. [DOI] [PubMed] [Google Scholar]
- (157).Zaccai NR; Chi B; Thomson AR; Boyle AL; Bartlett GJ; Bruning M; Linden N; Sessions RB; Booth PJ; Brady RL, et al. A De Novo Peptide Hexamer with a Mutable Channel. Nat. Chem. Biol 2011, 7, 935–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (158).Cristie-David AS; Sciore A; Badieyan S; Escheweiler JD; Koldewey P; Bardwell JCA; Ruotolo BT; Marsh ENG Evaluation of De Novo-Designed Coiled Coils as Off-the-Shelf Components for Protein Assembly. Mol. Sys. Design Engineer 2017, 2, 140–148. [Google Scholar]
- (159).Lopez De La Paz M; Goldie K; Zurdo J; Lacroix E; Dobson CM; Hoenger A; Serrano L De Novo Designed Peptide-Based Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16052–16057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Bromley EH; Channon KJ; King PJ; Mahmoud ZN; Banwell EF; Butler MF; Crump MP; Dafforn TR; Hicks MR; Hirst JD, et al. Assembly Pathway of a Designed Alpha-Helical Protein Fiber. Biophys. J 2010, 98, 1668–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Park WM; Champion JA Thermally Triggered Self-Assembly of Folded Proteins into Vesicles. J. Am. Chem. Soc 2014, 136, 17906–17909. [DOI] [PubMed] [Google Scholar]
- (162).Ozbas B; Kretsinger J; Rajagopal K; Schneider JP; Pochan DJ Salt-Triggered Peptide Folding and Consequent Self-Assembly into Hydrogels with Tunable Modulus. Macromolecules 2004, 37, 7331–7337. [Google Scholar]
- (163).Banwell EF; Abelardo ES; Adams DJ; Birchall MA; Corrigan A; Donald AM; Kirkland M; Serpell LC; Butler MF; Woolfson DN Rational Design and Application of Responsive Alpha-Helical Peptide Hydrogels. Nat. Materials 2009, 8, 596–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Dooling LJ; Tirrell DA Engineering the Dynamic Properties of Protein Networks through Sequence Variation. ACS Cent. Sci 2016, 2, 812–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Nagy-Smith K; Beltramo PJ; Moore E; Tycko R; Furst EM; Schneider JP Molecular, Local, and Network-Level Basis for the Enhanced Stiffness of Hydrogel Networks Formed from Coassembled Racemic Peptides: Predictions from Pauling and Corey. ACS Cent. Sci 2017, 3, 586–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Rughani RV; Salick DA; Lamm MS; Yucel T; Pochan DJ; Schneider JP Folding, Self-Assembly, and Bulk Material Properties of a De Novo Designed Three-Stranded Beta-Sheet Hydrogel. Biomacromolecules 2009, 10, 1295–1304. [DOI] [PubMed] [Google Scholar]
- (167).Micklitsch CM; Medina SH; Yucel T; Nagy-Smith KJ; Pochan DJ; Schneider JP Influence of Hydrophobic Face Amino Acids on the Hydrogelation of Β-Hairpin Peptide Amphiphiles. Macromolecules 2015, 48, 1281–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Hule RA; Nagarkar RP; Hammouda B; Schneider JP; Pochan DJ Dependence of Self-Assembled Peptide Hydrogel Network Structure on Local Fibril Nanostructure. Macromolecules 2009, 42, 7137–7145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Cerf E; Sarroukh R; Tamamizu-Kato S; Breydo L; Derclaye S; Dufrene YF; Narayanaswami V; Goormaghtigh E; Ruysschaert JM; Raussens V Antiparallel Beta-Sheet: A Signature Structure of the Oligomeric Amyloid Beta-Peptide. Biochem. J 2009, 421, 415–423. [DOI] [PubMed] [Google Scholar]
- (170).Sarroukh R; Goormaghtigh E; Ruysschaert JM; Raussens V Atr-Ftir: A “Rejuvenated” Tool to Investigate Amyloid Proteins. Biochim. Biophys. Acta 2013, 1828, 2328–2338. [DOI] [PubMed] [Google Scholar]
- (171).Frederix PW; Scott GG; Abul-Haija YM; Kalafatovic D; Pappas CG; Javid N; Hunt NT; Ulijn RV; Tuttle T Exploring the Sequence Space for (Tri-)Peptide Self-Assembly to Design and Discover New Hydrogels. Nat. Chem 2015, 7, 30–37. [DOI] [PubMed] [Google Scholar]
- (172).Dong H; Hartgerink JD Role of Hydrophobic Clusters in the Stability of Α-Helical Coiled Coils and Their Conversion to Amyloid-Like Β-Sheets. Biomacromolecules 2007, 8, 617–623. [DOI] [PubMed] [Google Scholar]
- (173).Ridgley DM; Claunch EC; Barone JR Characterization of Large Amyloid Fibers and Tapes with Fourier Transform Infrared (Ft-Ir) and Raman Spectroscopy. Appl. Spectrosc 2013, 67, 1417–1426. [DOI] [PubMed] [Google Scholar]
- (174).Eisenberg D; Wilcox W; Eshita SM; Pryciak PM; Ho SP; Degrado WF The Design, Synthesis, and Crystallization of an Alpha-Helical Peptide. Proteins 1986, 1, 16–22. [DOI] [PubMed] [Google Scholar]
- (175).Apostolovic B; Klok H-A Ph-Sensitivity of the E3/K3 Heterodimeric Coiled Coil. Biomacromolecules 2008, 9, 3173–3180. [DOI] [PubMed] [Google Scholar]
- (176).Naiki H; Higuchi K; Hosokawa M; Takeda T Fluorometric Determination of Amyloid Fibrils in Vitro Using the Fluorescent Dye, Thioflavine T. Anal. Biochem 1989, 177, 244–249. [DOI] [PubMed] [Google Scholar]
- (177).Nilsson MR Techniques to Study Amyloid Fibril Formation in Vitro. Methods 2004, 34, 151–160. [DOI] [PubMed] [Google Scholar]
- (178).Nagy-Smith K; Moore E; Schneider J; Tycko R Molecular Structure of Monomorphic Peptide Fibrils within a Kinetically Trapped Hydrogel Network. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 9816–9821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Leonard SR; Cormier AR; Pang X; Zimmerman MI; Zhou HX; Paravastu AK Solid-State Nmr Evidence for Beta-Hairpin Structure within Max8 Designer Peptide Nanofibers. Biophys. J 2013, 105, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (180).Nilsson BL; Doran TM, Peptide Self-Assembly: Methods and Protocols Humana Press: 2018. [Google Scholar]
- (181).Takegoshi K; Nakamura S; Terao T 13c–1h Dipolar-Assisted Rotational Resonance in Magic-Angle Spinning Nmr. Chem. Phys. Lett 2001, 344, 631–637. [Google Scholar]
- (182).Tycko R Symmetry-Based Constant-Time Homonuclear Dipolar Recoupling in Solid State Nmr. J. Chem. Phys 2007, 126, 064506. [DOI] [PubMed] [Google Scholar]
- (183).Jaroniec CP; Tounge BA; Rienstra CM; Herzfeld J; Griffin RG Recoupling of Heteronuclear Dipolar Interactions with Rotational-Echo Double-Resonance at High Magic-Angle Spinning Frequencies. J. Mag. Reson 2000, 146, 132–139. [DOI] [PubMed] [Google Scholar]
- (184).Ciani B; Hutchinson EG; Sessions RB; Woolfson DN A Designed System for Assessing How Sequence Affects Alpha to Beta Conformational Transitions in Proteins. J. Biol. Chem 2002, 277, 10150–10155. [DOI] [PubMed] [Google Scholar]
- (185).Moutevelis E; Woolfson DN A Periodic Table of Coiled-Coil Protein Structures. J. Mol. Biol 2009, 385, 726–732. [DOI] [PubMed] [Google Scholar]
- (186).Gribbon C; Channon KJ; Zhang W; Banwell EF; Bromley EH; Chaudhuri JB; Oreffo RO; Woolfson DN Magicwand: A Single, Designed Peptide That Assembles to Stable, Ordered Alpha-Helical Fibers. Biochemistry 2008, 47, 10365–10371. [DOI] [PubMed] [Google Scholar]
- (187).Pandya MJ; Spooner GM; Sunde M; Thorpe JR; Rodger A; Woolfson DN Sticky-End Assembly of a Designed Peptide Fiber Provides Insight into Protein Fibrillogenesis. Biochemistry 2000, 39, 8728–8734. [DOI] [PubMed] [Google Scholar]
- (188).Ryadnov MG; Bella A; Timson S; Woolfson DN Modular Design of Peptide Fibrillar Nano-to Microstructures. J. Am. Chem. Soc 2009, 131, 13240–13241. [DOI] [PubMed] [Google Scholar]
- (189).Hadley EB; Testa OD; Woolfson DN; Gellman SH Preferred Side-Chain Constellations at Antiparallel Coiled-Coil Interfaces. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Smith AM; Acquah SF; Bone N; Kroto HW; Ryadnov MG; Stevens MS; Walton DR; Woolfson DN Polar Assembly in a Designed Protein Fiber. Angew. Chem. Int. Ed. Engl 2004, 44, 325–328. [DOI] [PubMed] [Google Scholar]
- (191).Zhou NE; Kay CM; Hodges R Synthetic Model Proteins. Positional Effects of Interchain Hydrophobic Interactions on Stability of Two-Stranded Alpha-Helical Coiled-Coils. J. Biol. Chem 1992, 267, 2664–2670. [PubMed] [Google Scholar]
- (192).Hartmann MD; Ridderbusch O; Zeth K; Albrecht R; Testa O; Woolfson DN; Sauer G; Dunin-Horkawicz S; Lupas AN; Alvarez BH A Coiled-Coil Motif That Sequesters Ions to the Hydrophobic Core. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 16950–16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (193).Fletcher JM; Bartlett GJ; Boyle AL; Danon JJ; Rush LE; Lupas AN; Woolfson DN N@a and N@D: Oligomer and Partner Specification by Asparagine in Coiled-Coil Interfaces. ACS Chem. Biol 2017, 12, 528–538. [DOI] [PubMed] [Google Scholar]
- (194).Steinkruger JD; Woolfson DN; Gellman SH Side-Chain Pairing Preferences in the Parallel Coiled-Coil Dimer Motif: Insight on Ion Pairing between Core and Flanking Sites. J. Am. Chem. Soc 2010, 132, 7586–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Thompson MJ; Sievers SA; Karanicolas J; Ivanova MI; Baker D; Eisenberg D The 3d Profile Method for Identifying Fibril-Forming Segments of Proteins. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 4074–4078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).Jiang T; Xu C; Liu Y; Liu Z; Wall JS; Zuo X; Lian T; Salaita K; Ni C; Pochan D, et al. Structurally Defined Nanoscale Sheets from Self-Assembly of Collagen-Mimetic Peptides. J. Am. Chem. Soc 2014, 136, 4300–4308. [DOI] [PubMed] [Google Scholar]
- (197).Jiang T; Xu C; Zuo X; Conticello VP Structurally Homogeneous Nanosheets from Self-Assembly of a Collagen-Mimetic Peptide. Angew. Chem. Int. Ed. Engl 2014, 53, 8367–8371. [DOI] [PubMed] [Google Scholar]
- (198).Magnotti EL; Hughes SA; Dillard RS; Wang S; Hough L; Karumbamkandathil A; Lian T; Wall JS; Zuo X; Wright ER Self-Assembly of an Α-Helical Peptide into a Crystalline Two-Dimensional Nanoporous Framework. J. Am. Chem. Soc 2016, 138, 16274–16282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Cortajarena AL; Grove TZ, Protein-Based Engineered Nanostructures Springer: Switzerland, 2016. [Google Scholar]
- (200).Wood CW; Bruning M; Ibarra AA; Bartlett GJ; Thomson AR; Sessions RB; Brady RL; Woolfson DN Ccbuilder: An Interactive Web-Based Tool for Building, Designing and Assessing Coiled-Coil Protein Assemblies. Bioinformatics 2014, 30, 3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Thomson AR; Wood CW; Burton AJ; Bartlett GJ; Sessions RB; Brady RL; Woolfson DN Computational Design of Water-Soluble Α-Helical Barrels. Science 2014, 346, 485–488. [DOI] [PubMed] [Google Scholar]
- (202).Papapostolou D; Bromley EH; Bano C; Woolfson DN Electrostatic Control of Thickness and Stiffness in a Designed Protein Fiber. J. Am. Chem. Soc 2008, 130, 5124–5130. [DOI] [PubMed] [Google Scholar]
- (203).Hume J; Sun J; Jacquet R; Renfrew PD; Martin JA; Bonneau R; Gilchrist ML; Montclare JK Engineered Coiled-Coil Protein Microfibers. Biomacromolecules 2014, 15, 3503–3510. [DOI] [PubMed] [Google Scholar]
- (204).Xu C; Liu R; Mehta AK; Guerrero-Ferreira RC; Wright ER; Dunin-Horkawicz S; Morris K; Serpell LC; Zuo X; Wall JS Rational Design of Helical Nanotubes from Self-Assembly of Coiled-Coil Lock Washers. J. Am. Chem. Soc 2013, 135, 15565–15578. [DOI] [PubMed] [Google Scholar]
- (205).Liang Y; Pingali SV; Jogalekar AS; Snyder JP; Thiyagarajan P; Lynn DG Cross-Strand Pairing and Amyloid Assembly. Biochemistry 2008, 47, 10018–10026. [DOI] [PubMed] [Google Scholar]
- (206).Zhang S; Holmes T; Lockshin C; Rich A Spontaneous Assembly of a Self-Complementary Oligopeptide to Form a Stable Macroscopic Membrane. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 3334–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Zhang S; Lockshin C; Herbert A; Winter E; Rich A Zuotin, a Putative Z-DNA Binding Protein in Saccharomyces Cerevisiae. EMBO J 1992, 11, 3787–3796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Danilkovich AV; Lipkin VM; Udovichenko IP Classification of Self-Organizing Peptides. Russ. J. Bioorg. Chem 2011, 37, 707–712. [DOI] [PubMed] [Google Scholar]
- (209).Aggeli A; Nyrkova IA; Bell M; Harding R; Carrick L; McLeish TC; Semenov AN; Boden N Hierarchical Self-Assembly of Chiral Rod-Like Molecules as a Model for Peptide Beta -Sheet Tapes, Ribbons, Fibrils, and Fibers. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (210).Caplan MR; Schwartzfarb EM; Zhang S; Kamm RD; Lauffenburger DA Control of Self-Assembling Oligopeptide Matrix Formation through Systematic Variation of Amino Acid Sequence. Biomaterials 2002, 23, 219–227. [DOI] [PubMed] [Google Scholar]
- (211).Qin X; Xie W; Tian S; Cai J; Yuan H; Yu Z; Butterfoss GL; Khuong AC; Gross RA Enzyme-Triggered Hydrogelation Via Self-Assembly of Alternating Peptides. Chem. Commun 2013, 49, 4839–4841. [DOI] [PubMed] [Google Scholar]
- (212).Dong H; Paramonov SE; Aulisa L; Bakota EL; Hartgerink JD Self-Assembly of Multidomain Peptides: Balancing Molecular Frustration Controls Conformation and Nanostructure. J. Am. Chem. Soc 2007, 129, 12468–12472. [DOI] [PubMed] [Google Scholar]
- (213).Candreva J; Chau E; Aoraha E; Nanda V; Kim J Hetero-Assembly of a Dual Β-Amyloid Variant Peptide System. Chem. Commun 2018, DOI: 10.1039/C1038CC02724B. [DOI] [PubMed] [Google Scholar]
- (214).Childers WS; Mehta AK; Ni R; Taylor JV; Lynn DG Peptides Organized as Bilayer Membranes. Angew. Chem. Int. Ed 2010, 49, 4104–4107. [DOI] [PubMed] [Google Scholar]
- (215).Liu P; Ni R; Mehta AK; Childers WS; Lakdawala A; Pingali SV; Thiyagarajan P; Lynn DG Nucleobase-Directed Amyloid Nanotube Assembly. J. Am. Chem. Soc 2008, 130, 16867–16869. [DOI] [PubMed] [Google Scholar]
- (216).Ni R; Childers WS; Hardcastle KI; Mehta AK; Lynn DG Remodeling Cross-Β Nanotube Surfaces with Peptide/Lipid Chimeras. Angew. Chem. Int. Ed 2012, 51, 6635–6638. [DOI] [PubMed] [Google Scholar]
- (217).Seroski DT; Restuccia A; Sorrentino AD; Knox KR; Hagen SJ; Hudalla GA Co-Assembly Tags Based on Charge Complementarity (Catch) for Installing Functional Protein Ligands into Supramolecular Biomaterials. Cell. Mol. Bioeng 2016, 9, 335–350. [Google Scholar]
- (218).Haines-Butterick L; Rajagopal K; Branco M; Salick D; Rughani R; Pilarz M; Lamm MS; Pochan DJ; Schneider JP Controlling Hydrogelation Kinetics by Peptide Design for Three-Dimensional Encapsulation and Injectable Delivery of Cells. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 7791–7796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (219).Sinthuvanich C; Nagy-Smith KJ; Walsh STR; Schneider JP Triggered Formation of Anionic Hydrogels from Self-Assembling Acidic Peptide Amphiphiles. Macromolecules 2017, 50, 5643–5651. [Google Scholar]
- (220).Collier JH; Messersmith PB Enzymatic Modification of Self-Assembled Peptide Structures with Tissue Transglutaminase. Bioconjugate Chem 2003, 14, 748–755. [DOI] [PubMed] [Google Scholar]
- (221).Mannige RV; Haxton TK; Proulx C; Robertson EJ; Battigelli A; Butterfoss GL; Zuckermann RN; Whitelam S Peptoid Nanosheets Exhibit a New Secondary-Structure Motif. Nature 2015, 526, 415–420. [DOI] [PubMed] [Google Scholar]
- (222).Hudson BC; Battigelli A; Connolly MD; Edison J; Spencer RK; Whitelam S; Zuckermann RN; Paravastu AK Evidence for Cis Amide Bonds in Peptoid Nanosheets. J. Phys. Chem. Lett 2018, 9, 2574–2578. [DOI] [PubMed] [Google Scholar]
- (223).Edison JR; Spencer RK; Butterfoss GL; Hudson BC; Hochbaum AI; Paravastu AK; Zuckermann RN; Whitelam S Conformations of Peptoids in Nanosheets Result from the Interplay of Backbone Energetics and Intermolecular Interactions. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 5647–5651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (224).Rapaport H; Möller G; Knobler CM; Jensen TR; Kjaer K; Leiserowitz L; Tirrell DA Assembly of Triple-Stranded Β-Sheet Peptides at Interfaces. J. Am. Chem. Soc 2002, 124, 9342–9343. [DOI] [PubMed] [Google Scholar]
- (225).Sathaye S; Zhang H; Sonmez C; Schneider JP; MacDermaid CM; Von Bargen CD; Saven JG; Pochan DJ Engineering Complementary Hydrophobic Interactions to Control Beta-Hairpin Peptide Self-Assembly, Network Branching, and Hydrogel Properties. Biomacromolecules 2014, 15, 3891–3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Li S; Sidorov AN; Mehta AK; Bisignano AJ; Das D; Childers WS; Schuler E; Jiang Z; Orlando TM; Berland K Neurofibrillar Tangle Surrogates: Histone H1 Binding to Patterned Phosphotyrosine Peptide Nanotubes. Biochemistry 2014, 53, 4225–4227. [DOI] [PubMed] [Google Scholar]
- (227).Mehta AK; Lu K; Childers WS; Liang Y; Dublin SN; Dong J; Snyder JP; Pingali SV; Thiyagarajan P; Lynn DG Facial Symmetry in Protein Self-Assembly. J. Am. Chem. Soc 2008, 130, 9829–9835. [DOI] [PubMed] [Google Scholar]
- (228).Rengifo RF; Li NX; Sementilli A; Lynn DG Amyloid Scaffolds as Alternative Chlorosomes. Org. Biomol. Chem 2017, 15, 7063–7071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Omosun TO; Hsieh M-C; Childers WS; Das D; Mehta AK; Anthony NR; Pan T; Grover MA; Berland KM; Lynn DG Catalytic Diversity in Self-Propagating Peptide Assemblies. Nat. Chem 2017, 9, 805. [DOI] [PubMed] [Google Scholar]
- (230).Pagel K; Koksch B Following Polypeptide Folding and Assembly with Conformational Switches. Curr. Opin. Chem. Biol 2008, 12, 730–739. [DOI] [PubMed] [Google Scholar]
- (231).Kammerer RA; Kostrewa D; Zurdo J; Detken A; Garcia-Echeverria C; Green JD; Muller SA; Meier BH; Winkler FK; Dobson CM, et al. Exploring Amyloid Formation by a De Novo Design. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 4435–4440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Kammerer RA; Steinmetz MO De Novo Design of a Two-Stranded Coiled-Coil Switch Peptide. J. Struct. Biol 2006, 155, 146–153. [DOI] [PubMed] [Google Scholar]
- (233).Verel R; Tomka IT; Bertozzi C; Cadalbert R; Kammerer RA; Steinmetz MO; Meier BH Polymorphism in an Amyloid-Like Fibril-Forming Model Peptide. Angew. Chem. Int. Ed. Engl 2008, 47, 5842–5845. [DOI] [PubMed] [Google Scholar]
- (234).Steinmetz MO; Gattin Z; Verel R; Ciani B; Stromer T; Green JM; Tittmann P; Schulze-Briese C; Gross H; van Gunsteren WF, et al. Atomic Models of De Novo Designed Cc Beta-Met Amyloid-Like Fibrils. J. Mol. Biol 2008, 376, 898–912. [DOI] [PubMed] [Google Scholar]
- (235).King IC; Gleixner J; Doyle L; Kuzin A; Hunt JF; Xiao R; Montelione GT; Stoddard BL; DiMaio F; Baker D Precise Assembly of Complex Beta Sheet Topologies from De Novo Designed Building Blocks. Elife 2015, 4, e11012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Dobson CM Protein Folding and Misfolding. Nature 2003, 426, 884–890. [DOI] [PubMed] [Google Scholar]
- (237).Sawaya MR; Sambashivan S; Nelson R; Ivanova MI; Sievers SA; Apostol MI; Thompson MJ; Balbirnie M; Wiltzius JJ; McFarlane HT, et al. Atomic Structures of Amyloid Cross-Beta Spines Reveal Varied Steric Zippers. Nature 2007, 447, 453–457. [DOI] [PubMed] [Google Scholar]
- (238).Alberts B The Cell as a Collection of Protein Machines: Preparing the Next Generation of Molecular Biologists. Cell 1998, 92, 291–294. [DOI] [PubMed] [Google Scholar]
- (239).Ellis RJ Macromolecular Crowding: An Important but Neglected Aspect of the Intracellular Environment. Curr. Opin. Struct. Biol 2001, 11, 114–119. [DOI] [PubMed] [Google Scholar]
- (240).Petrov AS; Bernier CR; Hsiao C; Norris AM; Kovacs NA; Waterbury CC; Stepanov VG; Harvey SC; Fox GE; Wartell RM Evolution of the Ribosome at Atomic Resolution. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 10251–10256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Koide K; Karel M Encapsulation and Stimulated Release of Enzymes Using Lecithin Vesicles. Int. J. Food Sci. Technol 1987, 22, 707–723. [Google Scholar]
- (242).Grenha A; Remuñán-López C; Carvalho EL; Seijo B Microspheres Containing Lipid/Chitosan Nanoparticles Complexes for Pulmonary Delivery of Therapeutic Proteins. Eur. J. Pharm. Biopharm 2008, 69, 83–93. [DOI] [PubMed] [Google Scholar]
- (243).Martinek K; Levashov AV; Klyachko N; Khmelnitski YL; Berezin IV Micellar Enzymology. FEBS J 1986, 155, 453–468. [DOI] [PubMed] [Google Scholar]
- (244).Ruckenstein E; Karpe P Enzymatic Super-and Subactivity in Nonionic Reverse Micelles. J. Phys. Chem 1991, 95, 4869–4882. [Google Scholar]
- (245).Hou C; Guan S; Wang R; Zhang W; Meng F; Zhao L; Xu J; Liu J Supramolecular Protein Assemblies Based on DNA Templates. J. Phys. Chem. Lett 2017, 8, 3970–3979. [DOI] [PubMed] [Google Scholar]
- (246).Zeltins A Construction and Characterization of Virus-Like Particles: A Review. Mol. Biotechnol 2013, 53, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Ludwig C; Wagner R Virus-Like Particles—Universal Molecular Toolboxes. Curr. Opin. Biotechnol 2007, 18, 537–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Wang Q; Lin T; Tang L; Johnson JE; Finn M Icosahedral Virus Particles as Addressable Nanoscale Building Blocks. Angew. Chem. Int. Ed 2002, 41, 459–462. [DOI] [PubMed] [Google Scholar]
- (249).Zhang L; Lua LH; Middelberg AP; Sun Y; Connors NK Biomolecular Engineering of Virus-Like Particles Aided by Computational Chemistry Methods. Chem. Soc. Rev 2015, 44, 8608–8618. [DOI] [PubMed] [Google Scholar]
- (250).Kahya N Protein–Protein and Protein–Lipid Interactions in Domain-Assembly: Lessons from Giant Unilamellar Vesicles. Biochim. Biophys. Acta 2010, 1798, 1392–1398. [DOI] [PubMed] [Google Scholar]
- (251).Acosta EJ; Yuan JS; Bhakta AS The Characteristic Curvature of Ionic Surfactants. J. Surfactant. Detergent 2008, 11, 145–158. [Google Scholar]
- (252).Hensel JK; Carpenter AP; Ciszewski RK; Schabes BK; Kittredge CT; Moore FG; Richmond GL Molecular Characterization of Water and Surfactant Aot at Nanoemulsion Surfaces. Proc. Natl. Acad. Sci. U.S.A 2017, 114, 13351–13356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Kuchler A; Yoshimoto M; Luginbuhl S; Mavelli F; Walde P Enzymatic Reactions in Confined Environments. Nat. Nanotechol 2016, 11, 409–420. [DOI] [PubMed] [Google Scholar]
- (254).Chen Y-X; Triola G; Waldmann H Bioorthogonal Chemistry for Site-Specific Labeling and Surface Immobilization of Proteins. Acc. Chem. Res 2011, 44, 762–773. [DOI] [PubMed] [Google Scholar]
- (255).Hernandez K; Fernandez-Lafuente R Control of Protein Immobilization: Coupling Immobilization and Site-Directed Mutagenesis to Improve Biocatalyst or Biosensor Performance. Enzyme Microb. Technol 2011, 48, 107–122. [DOI] [PubMed] [Google Scholar]
- (256).Liese A; Hilterhaus L Evaluation of Immobilized Enzymes for Industrial Applications. Chem. Soc. Rev 2013, 42, 6236–6249. [DOI] [PubMed] [Google Scholar]
- (257).Talbert JN; Goddard JM Enzymes on Material Surfaces. Coll. Surf. B 2012, 93, 8–19. [DOI] [PubMed] [Google Scholar]
- (258).Sassolas A; Blum LJ; Leca-Bouvier BD Immobilization Strategies to Develop Enzymatic Biosensors. Biotechnol. Adv 2012, 30, 489–511. [DOI] [PubMed] [Google Scholar]
- (259).Saha K; Agasti SS; Kim C; Li X; Rotello VM Gold Nanoparticles in Chemical and Biological Sensing. Chem. Rev 2012, 112, 2739–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (260).Mohamad NR; Marzuki NHC; Buang NA; Huyop F; Wahab RA An Overview of Technologies for Immobilization of Enzymes and Surface Analysis Techniques for Immobilized Enzymes. Biotechnol. Biotechnol. Equip 2015, 29, 205–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Kim D; Herr AE Protein Immobilization Techniques for Microfluidic Assays. Biomicrofluidics 2013, 7, 041501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Ansari SA; Husain Q Potential Applications of Enzymes Immobilized on/in Nano Materials: A Review. Biotechnol. Adv 2012, 30, 512–523. [DOI] [PubMed] [Google Scholar]
- (263).Anderson EM; Larsson KM; Kirk O One Biocatalyst–Many Applications: The Use of Candida Antarctica B-Lipase in Organic Synthesis. Biocatal. Biotransform 2009, 16, 181–204. [Google Scholar]
- (264).Hein JE; Fokin VV Copper-Catalyzed Azide–Alkyne Cycloaddition (Cuaac) and Beyond: New Reactivity of Copper (I) Acetylides. Chem. Soc. Rev 2010, 39, 1302–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (265).Wu JC Enhancing Protein and Enzyme Stability through Rationally Engineered Site-Specific Immobilization Utilizing Non-Canonical Amino Acids. Brigham Young University, 2014.
- (266).Wu JC; Hutchings CH; Lindsay MJ; Werner CJ; Bundy BC Enhanced Enzyme Stability through Site-Directed Covalent Immobilization. J. Biotechnol 2015, 193, 83–90. [DOI] [PubMed] [Google Scholar]
- (267).Shield JW; Ferguson HD; Bommarius AS; Hatton TA Enzymes in Reversed Micelles as Catalysts for Organic-Phase Synthesis Reactions. Indust. Engineer. Chem. Fund 1986, 25, 603–612. [Google Scholar]
- (268).Orlich B; Schomäcker R Enzyme Catalysis in Reverse Micelles. Trends Bioprocess. Biotransform 2002, 185–208. [DOI] [PubMed]
- (269).Pieters BJ; van Eldijk MB; Nolte RJ; Mecinovic J Natural Supramolecular Protein Assemblies. Chem. Soc. Rev 2016, 45, 24–39. [DOI] [PubMed] [Google Scholar]
- (270).Kanerva LT; Klibanov AM Hammett Analysis of Enzyme Action in Organic Solvents. J. Am. Chem. Soc 1989, 111, 6864–6865. [Google Scholar]
- (271).Burke PA; Griffin RG; Klibanov A Solid-State Nmr Assessment of Enzyme Active Center Structure under Nonaqueous Conditions. J. Biol. Chem 1992, 267, 20057–20064. [PubMed] [Google Scholar]
- (272).Bommarius AS; Hatton TA; Wang DI Xanthine Oxidase Reactivity in Reversed Micellar Systems: A Contribution to the Prediction of Enzymic Activity in Organized Media. J. Am. Chem. Soc 1995, 117, 4515–4523. [Google Scholar]
- (273).Laane C; Hilhorst R; Veeger C [20] Design of Reversed Micellar Media for the Enzymatic Synthesis of Apolar Compounds. Methods Enzymol 1987, 136, 216–229. [DOI] [PubMed] [Google Scholar]
- (274).co/>Knoche W; Schomäcker R, Reactions in Compartmentalized Liquids: Proceedings of a Symposium Held at the Zentrum Fur Interdisziplinare Forschung, Bielefeld/Frg, September 11–14, 1988 Springer: 1989. [Google Scholar]
- (275).Klyachko NL; Levashov AV; Pshezhetsky AV; Bogdanova NG; Berezin IV; Martinek K Catalysis by Enzymes Entrapped into Hydrated Surfactant Aggregates Having Lamellar or Cylindrical (Hexagonal) or Ball-Shaped (Cubic) Structure in Organic Solvents. FEBS J 1986, 161, 149–154. [DOI] [PubMed] [Google Scholar]
- (276).Beales PA; Geerts N; Inampudi KK; Shigematsu H; Wilson CJ; Vanderlick TK Reversible Assembly of Stacked Membrane Nanodiscs with Reduced Dimensionality and Variable Periodicity. J. Am. Chem. Soc 2013, 135, 3335–3338. [DOI] [PubMed] [Google Scholar]
- (277).Lakshmanan A; Lu GJ; Farhadi A; Nety SP; Kunth M; Lee-Gosselin A; Maresca D; Bourdeau RW; Yin M; Yan J Preparation of Biogenic Gas Vesicle Nanostructures for Use as Contrast Agents for Ultrasound and Mri. Nat. Protoc 2017, 12, 2050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Bilgicer B; Kumar K De Novo Design of Defined Helical Bundles in Membrane Environments. Proc Natl Acad Sci U S A 2004, 101, 15324–15329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Cheglakov Z; Weizmann Y; Braunschweig AB; Wilner OI; Willner I Increasing the Complexity of Periodic Protein Nanostructures by the Rolling-Circle-Amplified Synthesis of Aptamers. Angew. Chem. Int. Ed. Engl 2008, 47, 126–130. [DOI] [PubMed] [Google Scholar]
- (280).Wilner OI; Shimron S; Weizmann Y; Wang Z-G; Willner I Self-Assembly of Enzymes on DNA Scaffolds: En Route to Biocatalytic Cascades and the Synthesis of Metallic Nanowires. Nano Lett 2009, 9, 2040–2043. [DOI] [PubMed] [Google Scholar]
- (281).Wilner OI; Weizmann Y; Gill R; Lioubashevski O; Freeman R; Willner I Enzyme Cascades Activated on Topologically Programmed DNA Scaffolds. Nature Nanotechnol 2009, 4, 249–254. [DOI] [PubMed] [Google Scholar]
- (282).Thyme S; Baker D Redesigning the Specificity of Protein-DNA Interactions with Rosetta. Methods Mol Biol 2014, 1123, 265–282. [DOI] [PubMed] [Google Scholar]
- (283).Mou Y; Yu JY; Wannier TM; Guo CL; Mayo SL Computational Design of Co-Assembling Protein-DNA Nanowires. Nature 2015, 525, 230–233. [DOI] [PubMed] [Google Scholar]
- (284).Patterson DP; Schwarz B; Waters RS; Gedeon T; Douglas T Encapsulation of an Enzyme Cascade within the Bacteriophage P22 Virus-Like Particle. ACS Chem. Biol 2014, 9, 359–365. [DOI] [PubMed] [Google Scholar]
- (285).Fiedler JD; Higginson C; Hovlid ML; Kislukhin AA; Castillejos A; Manzenrieder F; Campbell MG; Voss NR; Potter CS; Carragher B, et al. Engineered Mutations Change the Structure and Stability of a Virus-Like Particle. Biomacromolecules 2012, 13, 2339–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Udit AK; Everett C; Gale AJ; Reiber Kyle J; Ozkan M; Finn MG Heparin Antagonism by Polyvalent Display of Cationic Motifs on Virus-Like Particles. ChemBioChem 2009, 10, 503–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Fiedler JD; Brown SD; Lau JL; Finn MG Rna-Directed Packaging of Enzymes within Virus-Like Particles. Angew. Chem. Int. Ed. Engl 2010, 49, 9648–9651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Hovlid ML; Lau JL; Breitenkamp K; Higginson CJ; Laufer B; Manchester M; Finn M Encapsidated Atom-Transfer Radical Polymerization in Qβ Virus-Like Nanoparticles. ACS nano 2014, 8, 8003–8014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Brown SD; Fiedler JD; Finn MG Assembly of Hybrid Bacteriophage Qbeta Virus-Like Particles. Biochemistry 2009, 48, 11155–11157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Udit AK; Brown S; Baksh MM; Finn MG Immobilization of Bacteriophage Qbeta on Metal-Derivatized Surfaces Via Polyvalent Display of Hexahistidine Tags. J. Inorg. Biochem 2008, 102, 2142–2146. [DOI] [PubMed] [Google Scholar]
- (291).Pokorski JK; Hovlid ML; Finn MG Cell Targeting with Hybrid Qbeta Virus-Like Particles Displaying Epidermal Growth Factor. ChemBioChem 2011, 12, 2441–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Rhee JK; Hovlid M; Fiedler JD; Brown SD; Manzenrieder F; Kitagishi H; Nycholat C; Paulson JC; Finn MG Colorful Virus-Like Particles: Fluorescent Protein Packaging by the Qbeta Capsid. Biomacromolecules 2011, 12, 3977–3981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Butterfield GL; Lajoie MJ; Gustafson HH; Sellers DL; Nattermann U; Ellis D; Bale JB; Ke S; Lenz GH; Yehdego A, et al. Evolution of a Designed Protein Assembly Encapsulating Its Own Rna Genome. Nature 2017, 552, 415–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Hsia Y; Bale JB; Gonen S; Shi D; Sheffler W; Fong KK; Nattermann U; Xu C; Huang PS; Ravichandran R, et al. Corrigendum: Design of a Hyperstable 60-Subunit Protein Icosahedron. Nature 2016, 540, 150. [DOI] [PubMed] [Google Scholar]
- (295).Bale JB; Gonen S; Liu Y; Sheffler W; Ellis D; Thomas C; Cascio D; Yeates TO; Gonen T; King NP, et al. Accurate Design of Megadalton-Scale Two-Component Icosahedral Protein Complexes. Science 2016, 353, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Matthews KS; Nichols JC Lactose Repressor Protein: Functional Properties and Structure. Progress Nucleic Acid Res Mol. Biol 1997, 58, 127–164. [DOI] [PubMed] [Google Scholar]
- (297).Lewis M The Lac Repressor. C. R. Biol 2005, 328, 521–548. [DOI] [PubMed] [Google Scholar]
- (298).Miller JH; Reznikoff WS, The Operon 2nd ed.; Cold Spring Harbor Laboratory: 1980. [Google Scholar]
- (299).Bell CE; Lewis M The Lac Repressor: A Second Generation of Structural and Functional Studies. Curr. Opin. Struct. Biol 2001, 11, 19–25. [DOI] [PubMed] [Google Scholar]
- (300).Jacob F; Monod J Genetic Regulatory Mechanisms in the Synthesis of Proteins. J. Mol. Biol 1961, 3, 318–356. [DOI] [PubMed] [Google Scholar]
- (301).Gilbert W; Müller-Hill B Isolation of the Lac Repressor. Proc. Natl. Acad. Sci. U. S. A 1966, 56, 1891–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Riggs AD; Bourgeois S On the Assay, Isolation and Characterization of the Lac Repressor. J. Mol. Biol 1968, 34, 361–364. [DOI] [PubMed] [Google Scholar]
- (303).Gilbert W; Müller-Hill B The Lac Operator Is DNA. Proc. Natl. Acad. Sci. U. S. A 1967, 58, 2415–2421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Riggs AD; Bourgeois S; Newby RF; Cohn M DNA Binding of the Lac Repressor. J. Mol. Biol 1968, 34, 365–368. [DOI] [PubMed] [Google Scholar]
- (305).Riggs AD; Suzuki H; Bourgeois S Lac Repressor-Operator Interaction. J. Mol. Biol 1970, 48, 67–83. [DOI] [PubMed] [Google Scholar]
- (306).Jobe A; Bourgeois S Lac Repressor-Operator Interaction. J. Mol. Biol 1972, 69, 397–408. [DOI] [PubMed] [Google Scholar]
- (307).Gilbert W; Maxam A The Nucleotide Sequence of the Lac Operator. Proc. Natl. Acad. Sci. U. S. A 1973, 70, 3581–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Riggs AD; Newby RF; Bourgeois S Lac Repressor—Operator Interaction. J. Mol. Biol 1970, 51, 303–314. [DOI] [PubMed] [Google Scholar]
- (309).Schlax PJ; Capp MW; Record TM Jr Inhibition of Transcription Initiation Buiacrepressor. J. Mol. Biol 1995, 245, 331–350. [DOI] [PubMed] [Google Scholar]
- (310).Lee J; Goldfarb A Lac Repressor Acts by Modifying the Initial Transcribing Complex So That It Cannot Leave the Promoter. Cell 1991, 66, 793–798. [DOI] [PubMed] [Google Scholar]
- (311).Straney SB; Crothers DM Lac Repressor Is a Transient Gene-Activating Protein. Cell 1987, 51, 699–707. [DOI] [PubMed] [Google Scholar]
- (312).Miller JH; Calos MP; Galas D; Hofer M; Büchel DE; Benno M-H Genetic Analysis of Transpositions in the Lac Region of Escherichia Coli. J. Mol. Biol 1980, 144, 1–18. [DOI] [PubMed] [Google Scholar]
- (313).Roderick SL The Lac Operon Galactoside Acetyltransferase. C. R. Biol 2005, 328, 568–575. [DOI] [PubMed] [Google Scholar]
- (314).Juers DH; Heightman TD; Vasella A; McCarter JD; Mackenzie L; Withers SG; Matthews BW A Structural View of the Action of Escherichia Coli (Lac Z) Β-Galactosidase. Biochemistry 2001, 40, 14781–14794. [DOI] [PubMed] [Google Scholar]
- (315).Wheatley RW; Lo S; Jancewicz LJ; Dugdale ML; Huber RE Structural Explanation for Allolactose (Lac Operon Inducer) Synthesis by Lacz Beta-Galactosidase and the Evolutionary Relationship between Allolactose Synthesis and the Lac Repressor. J. Biol. Chem 2013, 288, 12993–13005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Emmer M; Pastan I; Perlman R Cyclic Amp Receptor Protein of E. Coli: Its Role in the Synthesis of Inducible Enzymes. Proc. Natl. Acad. Sci. U. S. A 1970, 66, 480–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (317).Kolb A; Busby S; Buc H; Garges S; Adhya S Transcriptional Regulation by Camp and Its Receptor Protein. Annu. Rev. Biochem 1993, 62, 749–797. [DOI] [PubMed] [Google Scholar]
- (318).Barkley MD; Riggs AD; Jobe A; Bourgeois S Interaction of Effecting Ligands with Lac Repressor and Repressor-Operator Complex. Biochemistry 1975, 14, 1700–1712. [DOI] [PubMed] [Google Scholar]
- (319).Nielsen AA; Segall-Shapiro TH; Voigt CA Advances in Genetic Circuit Design: Novel Biochemistries, Deep Part Mining, and Precision Gene Expression. Curr. Opin. Chem. Biol 2013, 17, 878–892. [DOI] [PubMed] [Google Scholar]
- (320).Clancy K; Voigt CA Programming Cells: Towards an Automated ‘Genetic Compiler’. Curr. Opin. Biotechnol 2010, 21, 572–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Voigt CA Genetic Parts to Program Bacteria. Curr. Opin. Biotechnol 2006, 17, 548–557. [DOI] [PubMed] [Google Scholar]
- (322).Lewis M; Chang G; Horton NC; Kercher MA; Pace HC; Schumacher MA; Brennan RG; Lu P Crystal Structure of the Lactose Operon Repressor and Its Complexes with DNA and Inducer. Science 1996, 271, 1247–1254. [DOI] [PubMed] [Google Scholar]
- (323).Bell CE; Lewis M A Closer View of the Conformation of the Lac Repressor Bound to Operator. Nat. Struct. Mol. Biol 2000, 7, 209–214. [DOI] [PubMed] [Google Scholar]
- (324).Bell CE; Barry J; Matthews KS; Lewis M Structure of a Variant of Lac Repressor with Increased Thermostability and Decreased Affinity for Operator. J. Mol. Biol 2001, 313, 99–109. [DOI] [PubMed] [Google Scholar]
- (325).Friedman AM; Fischmann TO; Steitz TA Crystal Structure of Lac Repressor Core Tetramer and Its Implications for DNA Looping. Science 1995, 268, 1721–1727. [DOI] [PubMed] [Google Scholar]
- (326).Ohshima Y; Matsuura M; Horiuchi T Conformational Change of the Lac Repressor Induced with the Inducer. Biochem. Biophys. Res. Commun 1972, 47, 1444–1450. [DOI] [PubMed] [Google Scholar]
- (327).Matthews KS Ultraviolet Difference Spectra of the Lactose Repressor Protein Ii. Trypsin Core Protein. Biochim. Biophys. Acta 1974, 359, 334–340. [DOI] [PubMed] [Google Scholar]
- (328).Slijper M; Bonvin A; Boelens R; Kaptein R Refined Structure Oflacrepressor Headpiece (1–56) Determined by Relaxation Matrix Calculations from 2d and 3d Noe Data: Change of Tertiary Structure Upon Binding to Thelacoperator. J. Mol. Biol 1996, 259, 761–773. [DOI] [PubMed] [Google Scholar]
- (329).Kaptein R; Zuiderweg E; Scheek R; Boelens R; Van Gunsteren W A Protein Structure from Nuclear Magnetic Resonance Data: Lac Repressor Headpiece. J. Mol. Biol 1985, 182, 179–182. [DOI] [PubMed] [Google Scholar]
- (330).Chen J; Matthews K Deletion of Lactose Repressor Carboxyl-Terminal Domain Affects Tetramer Formation. J. Biol. Chem 1992, 267, 13843–13850. [PubMed] [Google Scholar]
- (331).Alberti S; Oehler S; von Wilcken-Bergmann B; Muller-Hill B Genetic Analysis of the Leucine Heptad Repeats of Lac Repressor: Evidence for a 4-Helical Bundle. EMBO J 1993, 12, 3227–3236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Reznikoff WS; Winter RB; Hurley CK The Location of the Repressor Binding Sites in the Lac Operon. Proc. Natl. Acad. Sci. U. S. A 1974, 71, 2314–2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Pfahl M; Gulde V; Bourgeois S “Second” and “Third Operator” of the Lac Operon: An Investigation of Their Role in the Regulatory Mechanism. J. Mol. Biol 1979, 127, 339–344. [DOI] [PubMed] [Google Scholar]
- (334).Mossing MC; Record MT Jr Upstream Operators Enhance Repression of the Lac Promoter. Science 1986, 233, 889–893. [DOI] [PubMed] [Google Scholar]
- (335).Besse M; von Wilcken-Bergmann B; Müller-Hill B Synthetic Lac Operator Mediates Repression through Lac Repressor When Introduced Upstream and Downstream from Lac Promoter. EMBO J 1986, 5, 1377–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Brenowitz M; Pickar A; Jamison E Stability of a Lac Repressor Mediated” Looped Complex”. Biochemistry 1991, 30, 5986–5998. [DOI] [PubMed] [Google Scholar]
- (337).Borowiec JA; Zhang L; Sasse-Dwight S; Gralla JD DNA Supercoiling Promotes Formation of a Bent Repression Loop in Lac DNA. J. Mol. Biol 1987, 196, 101–111. [DOI] [PubMed] [Google Scholar]
- (338).Becker NA; Kahn JD; Maher LJ 3rd Bacterial Repression Loops Require Enhanced DNA Flexibility. J. Mol. Biol 2005, 349, 716–730. [DOI] [PubMed] [Google Scholar]
- (339).Whitson PA; Hsieh W; Wells R; Matthews K Influence of Supercoiling and Sequence Context on Operator DNA Binding with Lac Repressor. J. Biol. Chem 1987, 262, 14592–14599. [PubMed] [Google Scholar]
- (340).Hsieh W-T; Whitson PA; Matthews K; Wells RD Influence of Sequence and Distance between Two Operators on Interaction with the Lac Repressor. J. Biol. Chem 1987, 262, 14583–14591. [PubMed] [Google Scholar]
- (341).Krämer H; Amouyal M; Nordheim A; Müller-Hill B DNA Supercoiling Changes the Spacing Requirement of Two Lac Operators for DNA Loop Formation with Lac Repressor. EMBO J 1988, 7, 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Krämer H; Niemöller M; Amouyal M; Revet B; von Wilcken-Bergmann B; Müller-Hill B Lac Repressor Forms Loops with Linear DNA Carrying Two Suitably Spaced Lac Operators. EMBO J 1987, 6, 1481–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Levandoski MM; Tsodikov OV; Frank DE; Melcher SE; Saecker RM; Record TM Jr Cooperative and Anticooperative Effects in Binding of the First and Second Plasmid Osymoperators to a Laci Tetramer: Evidence for Contributions of Non-Operator DNA Binding by Wrapping and Looping. J. Mol. Biol 1996, 260, 697–717. [DOI] [PubMed] [Google Scholar]
- (344).Eismann ER; Müller-Hill B Lac Repressor Forms Stable Loops in Vitro with Supercoiled Wild-Type Lac DNA Containing All Three Natural Lac Operators. J. Mol. Biol 1990, 213, 763–775. [DOI] [PubMed] [Google Scholar]
- (345).Goodson KA; Wang Z; Haeusler AR; Kahn JD; English DS Laci-DNA-Iptg Loops: Equilibria among Conformations by Single-Molecule Fret. J. Phys. Chem. B 2013, 117, 4713–4722. [DOI] [PubMed] [Google Scholar]
- (346).Flynn TC; Swint-Kruse L; Kong Y; Booth C; Matthews KS; Ma J Allosteric Transition Pathways in the Lactose Repressor Protein Core Domains: Asymmetric Motions in a Homodimer. Protein Sci 2003, 12, 2523–2541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Wilson CJ; Das P; Clementi C; Matthews KS; Wittung-Stafshede P The Experimental Folding Landscape of Monomeric Lactose Repressor, a Large Two-Domain Protein, Involves Two Kinetic Intermediates. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 14563–14568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Gillespie B; Plaxco KW Using Protein Folding Rates to Test Protein Folding Theories. Annu. Rev. Biochem 2004, 73, 837–859. [DOI] [PubMed] [Google Scholar]
- (349).Matthews CR Pathways of Protein Folding. Annu. Rev. Biochem 1993, 62, 653–683. [DOI] [PubMed] [Google Scholar]
- (350).Kraus GA; Gottschalk P Direct Preparation of Bromoacetaldehyde. J. Org. Chem 1983, 48, 2111–2112. [Google Scholar]
- (351).Das P; Wilson CJ; Fossati G; Wittung-Stafshede P; Matthews KS; Clementi C Characterization of the Folding Landscape of Monomeric Lactose Repressor: Quantitative Comparison of Theory and Experiment. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 14569–14574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Ramot R; Kishore Inampudi K; Wilson CJ Lactose Repressor Experimental Folding Landscape: Fundamental Functional Unit and Tetramer Folding Mechanisms. Biochemistry 2012, 51, 7569–7579. [DOI] [PubMed] [Google Scholar]
- (353).Walters J; Milam SL; Clark AC Chapter 1 Practical Approaches to Protein Folding and Assembly. Methods Enzymol 2009, 455, 1–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (354).Angelici B; Mailand E; Haefliger B; Benenson Y Synthetic Biology Platform for Sensing and Integrating Endogenous Transcriptional Inputs in Mammalian Cells. Cell Rep 2016, 16, 2525–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Khalil AS; Collins JJ Synthetic Biology: Applications Come of Age. Nat. Rev. Genet 2010, 11, 367–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Gardner TS; Cantor CR; Collins JJ Construction of a Genetic Toggle Switch in Escherichia Coli. Nature 2000, 403, 339–342. [DOI] [PubMed] [Google Scholar]
- (357).Inniss MC; Silver PA Building Synthetic Memory. Curr. Biol 2013, 23, R812–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (358).Elowitz MB; Leibler S A Synthetic Oscillatory Network of Transcriptional Regulators. Nature 2000, 403, 335–338. [DOI] [PubMed] [Google Scholar]
- (359).Potvin-Trottier L; Lord ND; Vinnicombe G; Paulsson J Synchronous Long-Term Oscillations in a Synthetic Gene Circuit. Nature 2016, 538, 514–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Davey JA; Wilson CJ Deconstruction of Complex Protein Signaling Switches: A Roadmap toward Engineering Higher-Order Gene Regulators. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol 2017, 9, e1461. [DOI] [PubMed] [Google Scholar]
- (361).Richards DH; Meyer S; Wilson CJ Fourteen Ways to Reroute Cooperative Communication in the Lactose Repressor: Engineering Regulatory Proteins with Alternate Repressive Functions. ACS Synth. Biol 2017, 6, 6–12. [DOI] [PubMed] [Google Scholar]
- (362).Meyer DE; Chilkoti A Quantification of the Effects of Chain Length and Concentration on the Thermal Behavior of Elastin-Like Polypeptides. Biomacromolecules 2004, 5, 846–851. [DOI] [PubMed] [Google Scholar]
- (363).Sadler JR; Sasmor H; Betz JL A Perfectly Symmetric Lac Operator Binds the Lac Repressor Very Tightly. Proc. Natl. Acad. Sci. U. S. A 1983, 80, 6785–6789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (364).Sartorius J; Lehming N; Kisters B, v; von Wilcken-Bergmann, B.; Müller-Hill, B. Lac Repressor Mutants with Double or Triple Exchanges in the Recognition Helix Bind Specifically to Lac Operator Variants with Multiple Exchanges. EMBO J 1989, 8, 1265–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Zhan J; Ding B; Ma R; Ma X; Su X; Zhao Y; Liu Z; Wu J; Liu H Develop Reusable and Combinable Designs for Transcriptional Logic Gates. Mol. Syst. Biol 2010, 6, 388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (366).Daber R; Lewis M A Novel Molecular Switch. J. Mol. Biol 2009, 391, 661–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (367).Kaptein R; Boelens R; Chuprina VP; Rullmann JAC; Slijper M [20] Nmr and Nucleic Acid-Protein Interactions: The Lac Repressor-Operator System. Methods Enzymol 1995, 261, 513–524. [DOI] [PubMed] [Google Scholar]
- (368).Daber R; Sharp K; Lewis M One Is Not Enough. J. Mol. Biol 2009, 392, 1133–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (369).Sousa FL; Parente DJ; Shis DL; Hessman JA; Chazelle A; Bennett MR; Teichmann SA; Swint-Kruse L Allorep: A Repository of Sequence, Structural and Mutagenesis Data for the Laci/Galr Transcription Regulators. J. Mol. Biol 2016, 428, 671–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Meinhardt S; Manley MW Jr; Becker NA; Hessman JA; Maher III LJ; Swint-Kruse L Novel Insights from Hybrid Laci/Galr Proteins: Family-Wide Functional Attributes and Biologically Significant Variation in Transcription Repression. Nucleic Acids Res 2012, 40, 11139–11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Swint-Kruse L; Matthews KS Allostery in the Laci/Galr Family: Variations on a Theme. Curr. Opin. Microbiol 2009, 12, 129–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (372).Tungtur S; Egan SM; Swint-Kruse L Functional Consequences of Exchanging Domains between Laci and Purr Are Mediated by the Intervening Linker Sequence. Proteins 2007, 68, 375–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (373).Xu H; Moraitis M; Reedstrom RJ; Matthews KS Kinetic and Thermodynamic Studies of Purine Repressor Binding to Corepressor and Operator DNA. J. Biol. Chem 1998, 273, 8958–8964. [DOI] [PubMed] [Google Scholar]
- (374).Choi KY; Zalkin H Structural Characterization and Corepressor Binding of the Escherichia Coli Purine Repressor. J. Bacteriol 1992, 174, 6207–6214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (375).Meinhardt S; Manley MW Jr; Parente DJ; Swint-Kruse L Rheostats and Toggle Switches for Modulating Protein Function. PloS ONE 2013, 8, e83502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (376).Kobayashi H; Kaern M; Araki M; Chung K; Gardner TS; Cantor CR; Collins JJ Programmable Cells: Interfacing Natural and Engineered Gene Networks. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 8414–8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Kramer BP; Viretta AU; Daoud-El-Baba M; Aubel D; Weber W; Fussenegger M An Engineered Epigenetic Transgene Switch in Mammalian Cells. Nat. Biotechnol 2004, 22, 867–870. [DOI] [PubMed] [Google Scholar]
- (378).Fung E; Wong WW; Suen JK; Bulter T; Lee SG; Liao JC A Synthetic Gene-Metabolic Oscillator. Nature 2005, 435, 118–122. [DOI] [PubMed] [Google Scholar]
- (379).Stricker J; Cookson S; Bennett MR; Mather WH; Tsimring LS; Hasty J A Fast, Robust and Tunable Synthetic Gene Oscillator. Nature 2008, 456, 516–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (380).Tigges M; Marquez-Lago TT; Stelling J; Fussenegger M A Tunable Synthetic Mammalian Oscillator. Nature 2009, 457, 309–312. [DOI] [PubMed] [Google Scholar]
- (381).Lu TK; Khalil AS; Collins JJ Next-Generation Synthetic Gene Networks. Nat. Biotechnol 2009, 27, 1139–1150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (382).Zhang Y; Orner BP Self-Assembly in the Ferritin Nano-Cage Protein Superfamily. Int. J. Mol. Sci 2011, 12, 5406–5421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Gautieri A; Vesentini S; Redaelli A; Buehler MJ Hierarchical Structure and Nanomechanics of Collagen Microfibrils from the Atomistic Scale Up. Nano Lett 2011, 11, 757–766. [DOI] [PubMed] [Google Scholar]
- (384).Bates CM; Bates FS 50th Anniversary Perspective: Block Polymers Pure Potential. Macromolecules 2016, 50, 3–22. [Google Scholar]
- (385).Urry D; Long M; Cox B; Ohnishi T; Mitchell L; Jacobs M The Synthetic Polypentapeptide of Elastin Coacervates and Forms Filamentous Aggregates. Biochim. Biophys. Acta 1974, 371, 597–602. [DOI] [PubMed] [Google Scholar]
- (386).Urry DW; Trapane T; Prasad K Phase-Structure Transitions of the Elastin Polypentapeptide– Water System within the Framework of Composition–Temperature Studies. Biopolymers 1985, 24, 2345–2356. [DOI] [PubMed] [Google Scholar]
- (387).Petka WA; Harden JL; McGrath KP; Wirtz D; Tirrell DA Reversible Hydrogels from Self-Assembling Artificial Proteins. Science 1998, 281, 389–392. [DOI] [PubMed] [Google Scholar]
- (388).McDaniel JR; Callahan DJ; Chilkoti A Drug Delivery to Solid Tumors by Elastin-Like Polypeptides. Adv. Drug Deliv. Rev 2010, 62, 1456–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (389).Chilkoti A; Christensen T; MacKay JA Stimulus Responsive Elastin Biopolymers: Applications in Medicine and Biotechnology. Curr. Opin. Chem. Biol 2006, 10, 652–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Wright ER; McMillan RA; Cooper A; Apkarian RP; Conticello VP Thermoplastic Elastomer Hydrogels Via Self-Assembly of an Elastin-Mimetic Triblock Polypeptide. Adv. Funct. Mater 2002, 12, 149–154. [Google Scholar]
- (391).Le DH; Hanamura R; Pham DH; Kato M; Tirrell DA; Okubo T; Sugawara-Narutaki A Self-Assembly of Elastin-Mimetic Double Hydrophobic Polypeptides. Biomacromolecules 2013, 14, 1028–1034. [DOI] [PubMed] [Google Scholar]
- (392).Moll JR; Ruvinov SB; Pastan I; Vinson C Designed Heterodimerizing Leucine Zippers with a Ranger of Pis and Stabilities up to 10(−15) M. Protein Sci 2001, 10, 649–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (393).Jang Y; Choi WT; Heller WT; Ke Z; Wright ER; Champion JA Engineering Globular Protein Vesicles through Tunable Self-Assembly of Recombinant Fusion Proteins. Small 2017, 13, 1700399. [DOI] [PubMed] [Google Scholar]
- (394).Park WM; Champion JA Two-Step Protein Self-Assembly in the Extracellular Matrix. Angew. Chem. Int. Ed 2013, 52, 8098–8101. [DOI] [PubMed] [Google Scholar]
- (395).Kim B; Chilkoti A Allosteric Actuation of Inverse Phase Transition of a Stimulus-Responsive Fusion Polypeptide by Ligand Binding. J. Am. Chem. Soc 2008, 130, 17867–17873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (396).Dooley K; Kim YH; Lu HD; Tu R; Banta S Engineering of an Environmentally Responsive Beta Roll Peptide for Use as a Calcium-Dependent Cross-Linking Domain for Peptide Hydrogel Formation. Biomacromolecules 2012, 13, 1758–1764. [DOI] [PubMed] [Google Scholar]
- (397).Dooley K; Bulutoglu B; Banta S Doubling the Cross-Linking Interface of a Rationally Designed Beta Roll Peptide for Calcium-Dependent Proteinaceous Hydrogel Formation. Biomacromolecules 2014, 15, 3617–3624. [DOI] [PubMed] [Google Scholar]
- (398).Altman GH; Diaz F; Jakuba C; Calabro T; Horan RL; Chen J; Lu H; Richmond J; Kaplan DL Silk-Based Biomaterials. Biomaterials 2003, 24, 401–416. [DOI] [PubMed] [Google Scholar]
- (399).Slotta U; Hess S; Spieß K; Stromer T; Serpell L; Scheibel T Spider Silk and Amyloid Fibrils: A Structural Comparison. Macromol. Biosci 2007, 7, 183–188. [DOI] [PubMed] [Google Scholar]
- (400).Scheibel T Spider Silks: Recombinant Synthesis, Assembly, Spinning, and Engineering of Synthetic Proteins. Microb. Cell Fact 2004, 3, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Rammensee S; Huemmerich D; Hermanson K; Scheibel T; Bausch A Rheological Characterization of Hydrogels Formed by Recombinantly Produced Spider Silk. Appl. Phys. A 2006, 82, 261. [Google Scholar]
- (402).Rammensee S; Slotta U; Scheibel T; Bausch A Assembly Mechanism of Recombinant Spider Silk Proteins. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 6590–6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (403).Rabotyagova OS; Cebe P; Kaplan DL Self-Assembly of Genetically Engineered Spider Silk Block Copolymers. Biomacromolecules 2009, 10, 229–236. [DOI] [PubMed] [Google Scholar]
- (404).Krishnaji ST; Huang W; Cebe P; Kaplan DL Influence of Solution Parameters on Phase Diagram of Recombinant Spider Silk-Like Block Copolymers. Macromol. Chem. Phys 2014, 215, 1230–1238. [Google Scholar]
- (405).Xia XX; Xu Q; Hu X; Qin G; Kaplan DL Tunable Self-Assembly of Genetically Engineered Silk--Elastin-Like Protein Polymers. Biomacromolecules 2011, 12, 3844–3850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Banta S; Wheeldon IR; Blenner M Protein Engineering in the Development of Functional Hydrogels. Annu. Rev. Biomed. Eng 2010, 12, 167–186. [DOI] [PubMed] [Google Scholar]
- (407).Shen W; Lammertink RG; Sakata JK; Kornfield JA; Tirrell DA Assembly of an Artificial Protein Hydrogel through Leucine Zipper Aggregation and Disulfide Bond Formation. Macromolecules 2005, 38, 3909–3916. [Google Scholar]
- (408).Wheeldon IR; Gallaway JW; Barton SC; Banta S Bioelectrocatalytic Hydrogels from Electron-Conducting Metallopolypeptides Coassembled with Bifunctional Enzymatic Building Blocks. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 15275–15280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (409).Lu HD; Wheeldon IR; Banta S Catalytic Biomaterials: Engineering Organophosphate Hydrolase to Form Self-Assembling Enzymatic Hydrogels. Protein. Eng. Des. Sel 2010, 23, 559–566. [DOI] [PubMed] [Google Scholar]
- (410).Olsen BD; Kornfield JA; Tirrell DA Yielding Behavior in Injectable Hydrogels from Telechelic Proteins. Macromolecules 2010, 43, 9094–9099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Kim M; Chen WG; Kang JW; Glassman MJ; Ribbeck K; Olsen BD Artificially Engineered Protein Hydrogels Adapted from the Nucleoporin Nsp1 for Selective Biomolecular Transport. Adv. Mater 2015, 27, 4207–4212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (412).Glassman MJ; Olsen BD End Block Design Modulates the Assembly and Mechanics of Thermoresponsive, Dual-Associative Protein Hydrogels. Macromolecules 2015, 48, 1832–1842. [Google Scholar]
- (413).Haghpanah JS; Yuvienco C; Roth EW; Liang A; Tu RS; Montclare JK Supramolecular Assembly and Small Molecule Recognition by Genetically Engineered Protein Block Polymers Composed of Two Sads. Mol. Biosyst 2010, 6, 1662–1667. [DOI] [PubMed] [Google Scholar]
- (414).Yuvienco C; More HT; Haghpanah JS; Tu RS; Montclare JK Modulating Supramolecular Assemblies and Mechanical Properties of Engineered Protein Materials by Fluorinated Amino Acids. Biomacromolecules 2012, 13, 2273–2278. [DOI] [PubMed] [Google Scholar]
- (415).Wong Po Foo CT; Lee JS; Mulyasasmita W; Parisi-Amon A; Heilshorn SC Two-Component Protein-Engineered Physical Hydrogels for Cell Encapsulation. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 22067–22072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (416).Sun F; Zhang W-B; Mahdavi A; Arnold FH; Tirrell DA Synthesis of Bioactive Protein Hydrogels by Genetically Encoded Spytag-Spycatcher Chemistry. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 11269–11274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Vargo KB; Parthasarathy R; Hammer DA Self-Assembly of Tunable Protein Suprastructures from Recombinant Oleosin. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 11657–11662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).MacKay JA; Callahan DJ; FitzGerald KN; Chilkoti A Quantitative Model of the Phase Behavior of Recombinant Ph-Responsive Elastin-Like Polypeptides. Biomacromolecules 2010, 11, 2873–2879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (419).McDaniel JR; Radford DC; Chilkoti A A Unified Model for De Novo Design of Elastin-Like Polypeptides with Tunable Inverse Transition Temperatures. Biomacromolecules 2013, 14, 2866–2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (420).Tarakanova A; Huang W; Weiss AS; Kaplan DL; Buehler MJ Computational Smart Polymer Design Based on Elastin Protein Mutability. Biomaterials 2017, 127, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Simon JR; Carroll NJ; Rubinstein M; Chilkoti A; López GP Programming Molecular Self-Assembly of Intrinsically Disordered Proteins Containing Sequences of Low Complexity. Nat. Chem 2017, 9, 509–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (422).Rim NG; Roberts EG; Ebrahimi D; Dinjaski N; Jacobsen MM; Martin-Moldes Z; Buehler MJ; Kaplan DL; Wong JY Predicting Silk Fiber Mechanical Properties through Multiscale Simulation and Protein Design. ACS Biomater. Sci. Eng 2017, 3, 1542–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Park SJ; Cochran JR, Protein Engineering and Design CRC press: 2009; Vol. 75. [Google Scholar]
- (424).Wörsdörfer B; Woycechowsky KJ; Hilvert D Directed Evolution of a Protein Container. Science 2011, 331, 589–592. [DOI] [PubMed] [Google Scholar]
- (425).Carlson ED; Gan R; Hodgman CE; Jewett MC Cell-Free Protein Synthesis: Applications Come of Age. Biotechnol. Adv 2012, 30, 1185–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Zinchenko A; Devenish SR; Kintses B; Colin PY; Fischlechner M; Hollfelder F One in a Million: Flow Cytometric Sorting of Single Cell-Lysate Assays in Monodisperse Picolitre Double Emulsion Droplets for Directed Evolution. Anal. Chem 2014, 86, 2526–2533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (427).Kahn JS; Ruiz RC; Sureka S; Peng S; Derrien TL; An D; Luo D DNA Microgels as a Platform for Cell-Free Protein Expression and Display. Biomacromolecules 2016, 17, 2019–2026. [DOI] [PubMed] [Google Scholar]
- (428).Hook AL; Anderson DG; Langer R; Williams P; Davies MC; Alexander MR High Throughput Methods Applied in Biomaterial Development and Discovery. Biomaterials 2010, 31, 187–198. [DOI] [PubMed] [Google Scholar]
- (429).Bale JB; Gonen S; Liu Y; Sheffler W; Ellis D; Thomas C; Cascio D; Yeates TO; Gonen T; King NP Accurate Design of Megadalton-Scale Two-Component Icosahedral Protein Complexes. Science 2016, 353, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Zhang Q; Lin J; Wang L; Xu Z Theoretical Modeling and Simulations of Self-Assembly of Copolymers in Solution. Prog. Polym. Sci 2017, 75, 1–30. [Google Scholar]
- (431).Nagarajan R Constructing a Molecular Theory of Self-Assembly: Interplay of Ideas from Surfactants and Block Copolymers. Adv. Colloid Interface Sci 2017, 244, 113–123. [DOI] [PubMed] [Google Scholar]
- (432).Virchow R Ueber Eine Im Gehirn Und Rückenmark Des Menschen Aufgefundene Substanz Mit Der Chemischen Reaction Der Cellulose. Virchows Archiv 1854, 6, 135–138. [Google Scholar]
- (433).Friedreich N; Kekulé A Zur Amyloidfrage. Virchows Archiv 1859, 16, 50–65. [Google Scholar]
- (434).Eanes E; Glenner G X-Ray Diffraction Studies on Amyloid Filaments. J. Histochem. Cytochem 1968, 16, 673–677. [DOI] [PubMed] [Google Scholar]
- (435).LeVine H [18] Quantification of Β-Sheet Amyloid Fibril Structures with Thioflavin T. Methods Enzymol 1999, 309, 274–284. [DOI] [PubMed] [Google Scholar]
- (436).Sipe JD; Cohen AS Review: History of the Amyloid Fibril. J. Struct. Biol 2000, 130, 88–98. [DOI] [PubMed] [Google Scholar]
- (437).Liebman SW; Chernoff YO Prions in Yeast. Genetics 2012, 191, 1041–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Spires-Jones TL; Attems J; Thal DR Interactions of Pathological Proteins in Neurodegenerative Diseases. Acta Neuropathol 2017, 134, 187–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (439).Stefani M; Dobson CM Protein Aggregation and Aggregate Toxicity: New Insights into Protein Folding, Misfolding Diseases and Biological Evolution. J. Mol. Med 2003, 81, 678–699. [DOI] [PubMed] [Google Scholar]
- (440).Chiti F; Dobson CM Protein Misfolding, Functional Amyloid, and Human Disease. Annu. Rev. Biochem 2006, 75, 333–366. [DOI] [PubMed] [Google Scholar]
- (441).Chiti F; Dobson CM Amyloid Formation by Globular Proteins under Native Conditions. Nat. Chem. Biol 2009, 5, 15–22. [DOI] [PubMed] [Google Scholar]
- (442).Chernoff YO Amyloidogenic Domains, Prions and Structural Inheritance: Rudiments of Early Life or Recent Acquisition? Curr. Opin. Chem. Biol 2004, 8, 665–671. [DOI] [PubMed] [Google Scholar]
- (443).Sipe JD; Benson MD; Buxbaum JN; Ikeda S; Merlini G; Saraiva MJ; Westermark P Nomenclature 2014: Amyloid Fibril Proteins and Clinical Classification of the Amyloidosis. Amyloid 2014, 21, 221–224. [DOI] [PubMed] [Google Scholar]
- (444).Chiti F; Dobson CM Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress over the Last Decade. Annu. Rev. Biochem 2017, 27–68. [DOI] [PubMed]
- (445).Holtzman DM; Morris JC; Goate AM Alzheimer’s Disease: The Challenge of the Second Century. Sci. Transl. Med 2011, 3, 77sr71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Querfurth HW; LaFerla FM Alzheimer’s Disease. New. Engl. J. Med 2010, 362, 329–344. [DOI] [PubMed] [Google Scholar]
- (447).Irvine GB; El-Agnaf OM; Shankar GM; Walsh DM Protein Aggregation in the Brain: The Molecular Basis for Alzheimer’s and Parkinson’s Diseases. Mol. Med 2008, 14, 451–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (448).de Calignon A; Polydoro M; Suarez-Calvet M; William C; Adamowicz DH; Kopeikina KJ; Pitstick R; Sahara N; Ashe KH; Carlson GA, et al. Propagation of Tau Pathology in a Model of Early Alzheimer’s Disease. Neuron 2012, 73, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (449).Spillantini MG; Goedert M Tau Pathology and Neurodegeneration. Lancet Neurol 2013, 12, 609–622. [DOI] [PubMed] [Google Scholar]
- (450).Lee VM; Goedert M; Trojanowski JQ Neurodegenerative Tauopathies. Annu. Rev. Neurosci 2001, 24, 1121–1159. [DOI] [PubMed] [Google Scholar]
- (451).Galant NJ; Westermark P; Higaki JN; Chakrabartty A Transthyretin Amyloidosis: An under-Recognized Neuropathy and Cardiomyopathy. Clin. Sci 2017, 131, 395–409. [DOI] [PubMed] [Google Scholar]
- (452).Shao J; Diamond MI Polyglutamine Diseases: Emerging Concepts in Pathogenesis and Therapy. Hum. Mol. Genet 2007, 16, R115–123. [DOI] [PubMed] [Google Scholar]
- (453).Bulawa CE; Connelly S; Devit M; Wang L; Weigel C; Fleming JA; Packman J; Powers ET; Wiseman RL; Foss TR, et al. Tafamidis, a Potent and Selective Transthyretin Kinetic Stabilizer That Inhibits the Amyloid Cascade. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 9629–9634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Westermark GT; Fandrich M; Lundmark K; Westermark P Noncerebral Amyloidoses: Aspects on Seeding, Cross-Seeding, and Transmission. Cold Spring Harb. Perspect. Med 2018, 8, advance article. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Buhimschi IA; Nayeri UA; Zhao G; Shook LL; Pensalfini A; Funai EF; Bernstein IM; Glabe CG; Buhimschi CS Protein Misfolding, Congophilia, Oligomerization, and Defective Amyloid Processing in Preeclampsia. Sci. Trans. Med 2014, 6, 245ra292. [DOI] [PubMed] [Google Scholar]
- (456).Antony H; Wiegmans AP; Wei MQ; Chernoff YO; Khanna KK; Munn AL Potential Roles for Prions and Protein-Only Inheritance in Cancer. Cancer Metastasis Rev 2012, 31, 1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (457).Moreno-Gonzalez I; Edwards Iii G; Salvadores N; Shahnawaz M; Diaz-Espinoza R; Soto C Molecular Interaction between Type 2 Diabetes and Alzheimer’s Disease through Cross-Seeding of Protein Misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (458).Prusiner SB Prions. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Colby DW; Prusiner SB Prions. Cold Spring Harb. Perspect. Biol 2011, 3, a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (460).Castilla J; Saá P; Morales R; Abid K; Maundrell K; Soto C Protein Misfolding Cyclic Amplification for Diagnosis and Prion Propagation Studies. Methods Enzymol 2006, 412, 3–21. [DOI] [PubMed] [Google Scholar]
- (461).Castilla J; Morales R; Saa P; Barria M; Gambetti P; Soto C Cell-Free Propagation of Prion Strains. EMBO J 2008, 27, 2557–2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Kim JI; Cali I; Surewicz K; Kong Q; Raymond GJ; Atarashi R; Race B; Qing L; Gambetti P; Caughey B, et al. Mammalian Prions Generated from Bacterially Expressed Prion Protein in the Absence of Any Mammalian Cofactors. J. Biol. Chem 2010, 285, 14083–14087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Makarava N; Kovacs GG; Bocharova O; Savtchenko R; Alexeeva I; Budka H; Rohwer RG; Baskakov IV Recombinant Prion Protein Induces a New Transmissible Prion Disease in Wild-Type Animals. Acta Neuropathol 2010, 119, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Kane MD; Lipinski WJ; Callahan MJ; Bian F; Durham RA; Schwarz RD; Roher AE; Walker LC Evidence for Seeding of Β-Amyloid by Intracerebral Infusion of Alzheimer Brain Extracts in Β-Amyloid Precursor Protein-Transgenic Mice. J. Neurosci 2000, 20, 3606–3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Clavaguera F; Bolmont T; Crowther RA; Abramowski D; Frank S; Probst A; Fraser G; Stalder AK; Beibel M; Staufenbiel M, et al. Transmission and Spreading of Tauopathy in Transgenic Mouse Brain. Nat. Cell Biol 2009, 11, 909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Desplats P; Lee HJ; Bae EJ; Patrick C; Rockenstein E; Crews L; Spencer B; Masliah E; Lee SJ Inclusion Formation and Neuronal Cell Death through Neuron-to-Neuron Transmission of Alpha-Synuclein. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (467).Stohr J; Watts JC; Mensinger ZL; Oehler A; Grillo SK; DeArmond SJ; Prusiner SB; Giles K Purified and Synthetic Alzheimer’s Amyloid Beta (Abeta) Prions. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 11025–11030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (468).Sanders DW; Kaufman SK; DeVos SL; Sharma AM; Mirbaha H; Li A; Barker SJ; Foley AC; Thorpe JR; Serpell LC, et al. Distinct Tau Prion Strains Propagate in Cells and Mice and Define Different Tauopathies. Neuron 2014, 82, 1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (469).Mukherjee A; Morales-Scheihing D; Salvadores N; Moreno-Gonzalez I; Gonzalez C; Taylor-Presse K; Mendez N; Shahnawaz M; Gaber AO; Sabek OM, et al. Induction of Iapp Amyloid Deposition and Associated Diabetic Abnormalities by a Prion-Like Mechanism. J. Exp. Med 2017, 214, 2591–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (470).Frost B; Diamond MI Prion-Like Mechanisms in Neurodegenerative Diseases. Nat. Rev. Neurosci 2010, 11, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Chernova TA; Wilkinson KD; Chernoff YO Prions, Chaperones, and Proteostasis in Yeast. Cold Spring Harb. Perspect. Biol 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Wickner RB [Ure3] as an Altered Ure2 Protein: Evidence for Prion Analog in Saccharomyces Cerevisiae. Science 1994, 264, 566–570. [DOI] [PubMed] [Google Scholar]
- (473).Cox BSΨ, a Cytoplasmic Suppressor of Super-Suppressor in Yeast. Heredity 1965, 20, 505–521. [Google Scholar]
- (474).Lacroute F Non-Mendelian Mutation Allowing Ureidosuccinic Acid Uptake in Yeast. J. Bacteriol 1971, 106, 519–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).King CY; Diaz-Avalos R Protein-Only Transmission of Three Yeast Prion Strains. Nature 2004, 428, 319–323. [DOI] [PubMed] [Google Scholar]
- (476).Tanaka M; Chien P; Naber N; Cooke R; Weissman JS Conformational Variations in an Infectious Protein Determine Prion Strain Differences. Nature 2004, 428, 323–328. [DOI] [PubMed] [Google Scholar]
- (477).Saupe SJ A Short History of Small S: A Prion of the Fungus Podospora Anserina. Prion 2007, 1, 110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (478).Suzuki G; Shimazu N; Tanaka M A Yeast Prion, Mod5, Promotes Acquired Drug Resistance and Cell Survival under Environmental Stress. Science 2012, 336, 355–359. [DOI] [PubMed] [Google Scholar]
- (479).Alberti S; Halfmann R; King O; Kapila A; Lindquist S A Systematic Survey Identifies Prions and Illuminates Sequence Features of Prionogenic Proteins. Cell 2009, 137, 146–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (480).Michelitsch MD; Weissman JS A Census of Glutamine/Asparagine-Rich Regions: Implications for Their Conserved Function and the Prediction of Novel Prions. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 11910–11915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Wickner RB; Shewmaker F; Kryndushkin D; Edskes HK Protein Inheritance (Prions) Based on Parallel in-Register Beta-Sheet Amyloid Structures. Bioessays 2008, 30, 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Chernoff YO; Derkach IL; Inge-Vechtomov SG Multicopy Sup35 Gene Induces De-Novo Appearance of Psi-Like Factors in the Yeast Saccharomyces Cerevisiae. Curr. Genet 1993, 24, 268–270. [DOI] [PubMed] [Google Scholar]
- (483).Masison DC; Wickner RB Prion-Inducing Domain of Yeast Ure2p and Protease Resistance of Ure2p in Prion-Containing Cells. Science 1995, 270, 93–95. [DOI] [PubMed] [Google Scholar]
- (484).Derkatch IL; Chernoff YO; Kushnirov VV; Inge-Vechtomov SG; Liebman SW Genesis and Variability of [Psi] Prion Factors in Saccharomyces Cerevisiae. Genetics 1996, 144, 1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (485).Derkatch IL; Bradley ME; Zhou P; Chernoff YO; Liebman SW Genetic and Environmental Factors Affecting the De Novo Appearance of the [Psi+] Prion in Saccharomyces Cerevisiae. Genetics 1997, 147, 507–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (486).Derkatch IL; Bradley ME; Hong JY; Liebman SW Prions Affect the Appearance of Other Prions: The Story of [Pin+]. Cell 2001, 106, 171–182. [DOI] [PubMed] [Google Scholar]
- (487).Osherovich LZ; Weissman JS Multiple Gln/Asn-Rich Prion Domains Confer Susceptibility to Induction of the Yeast [Psi+] Prion. Cell 2001, 106, 183–194. [DOI] [PubMed] [Google Scholar]
- (488).Tyedmers J; Madariaga ML; Lindquist S Prion Switching in Response to Environmental Stress. PLoS Biol 2008, 6, e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (489).Chernova TA; Wilkinson KD; Chernoff YO Physiological and Environmental Control of Yeast Prions. FEMS Microbiol. Rev 2014, 38, 326–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (490).Doronina VA; Staniforth GL; Speldewinde SH; Tuite MF; Grant CM Oxidative Stress Conditions Increase the Frequency of De Novo Formation of the Yeast [Psi+] Prion. Mol. Microbiol 2015, 96, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Chernoff YO; Newnam GP; Kumar J; Allen K; Zink AD Evidence for a Protein Mutator in Yeast: Role of the Hsp70-Related Chaperone Ssb in Formation, Stability, and Toxicity of the [Psi] Prion. Mol. Cell. Biol 1999, 19, 8103–8112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (492).Amor AJ; Castanzo DT; Delany SP; Selechnik DM; van Ooy A; Cameron DM The Ribosome-Associated Complex Antagonizes Prion Formation in Yeast. Prion 2015, 9, 144–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Kiktev DA; Melomed MM; Lu CD; Newnam GP; Chernoff YO Feedback Control of Prion Formation and Propagation by the Ribosome-Associated Chaperone Complex. Mol. Microbiol 2015, 96, 621–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Chernoff YO; Kiktev DA Dual Role of Ribosome-Associated Chaperones in Prion Formation and Propagation. Curr. Genet 2016, 62, 677–685. [DOI] [PubMed] [Google Scholar]
- (495).Glover JR; Kowal AS; Schirmer EC; Patino MM; Liu J-J; Lindquist S Self-Seeded Fibers Formed by Sup35, the Protein Determinant of [Psi+], a Heritable Prion-Like Factor of S. Cerevisiae. Cell 1997, 89, 811–819. [DOI] [PubMed] [Google Scholar]
- (496).King C-Y; Tittmann P; Gross H; Gebert R; Aebi M; Wüthrich K Prion-Inducing Domain 2– 114 of Yeast Sup35 Protein Transforms in Vitro into Amyloid-Like Filaments. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 6618–6622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Taylor KL; Cheng N; Williams RW; Steven AC; Wickner RB Prion Domain Initiation of Amyloid Formation in Vitro from Native Ure2p. Science 1999, 283, 1339–1343. [DOI] [PubMed] [Google Scholar]
- (498).Serio TR; Cashikar AG; Moslehi JJ; Kowal AS; Lindquist SL [41] Yeast Prion [Ψ+] and Its Determinant, Sup35p. Methods Enzymol 1999, 309, 649–673. [DOI] [PubMed] [Google Scholar]
- (499).Yeh V; Broering JM; Romanyuk A; Chen B; Chernoff YO; Bommarius AS The Hofmeister Effect on Amyloid Formation Using Yeast Prion Protein. Protein Sci 2010, 19, 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (500).Rubin J; Khosravi H; Bruce KL; Lydon ME; Behrens SH; Chernoff YO; Bommarius AS Ion-Specific Effects on Prion Nucleation and Strain Formation. J. Biol. Chem 2013, 288, 30300–30308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Hofmeister F Zur Lehre Von Der Wirkung Der Salze. Naunyn-Schmiedeberg’s Arch. Pharmacol 1888, 25, 1–30. [Google Scholar]
- (502).Diaz-Espinoza R; Mukherjee A; Soto C Kosmotropic Anions Promote Conversion of Recombinant Prion Protein into a Prpsc-Like Misfolded Form. PLoS One 2012, 7, e31678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (503).Marek PJ; Patsalo V; Green DF; Raleigh DP Ionic Strength Effects on Amyloid Formation by Amylin Are a Complicated Interplay among Debye Screening, Ion Selectivity, and Hofmeister Effects. Biochemistry 2012, 51, 8478–8490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Chernoff YO; Lindquist SL; Ono B.-i.; Inge-Vechtomov SG; Liebman SW Role of the Chaperone Protein Hsp104 in Propagation of the Yeast Prion-Like Factor [Psi+]. Science 1995, 268, 880–884. [DOI] [PubMed] [Google Scholar]
- (505).Newnam GP; Wegrzyn RD; Lindquist SL; Chernoff YO Antagonistic Interactions between Yeast Chaperones Hsp104 and Hsp70 in Prion Curing. Mol. Cell. Biol 1999, 19, 1325–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (506).Jung G; Jones G; Wegrzyn RD; Masison DC A Role for Cytosolic Hsp70 in Yeast [Psi+] Prion Propagation and [Psi+] as a Cellular Stress. Genetics 2000, 156, 559–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (507).Jones G; Song Y; Chung S; Masison DC Propagation of Saccharomyces Cerevisiae [Psi+] Prion Is Impaired by Factors That Regulate Hsp70 Substrate Binding. Mol. Cell. Biol 2004, 24, 3928–3937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (508).Hines JK; Li X; Du Z; Higurashi T; Li L; Craig EA [Swi], the Prion Formed by the Chromatin Remodeling Factor Swi1, Is Highly Sensitive to Alterations in Hsp70 Chaperone System Activity. PLoS Genet 2011, 7, e1001309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (509).Sporn ZA; Hines JK Hsp40 Function in Yeast Prion Propagation: Amyloid Diversity Necessitates Chaperone Functional Complexity. Prion 2015, 9, 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Winkler J; Tyedmers J; Bukau B; Mogk A Hsp70 Targets Hsp100 Chaperones to Substrates for Protein Disaggregation and Prion Fragmentation. J. Cell Biol 2012, 198, 387–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Glover JR; Lindquist S Hsp104, Hsp70, and Hsp40: A Novel Chaperone System That Rescues Previously Aggregated Proteins. Cell 1998, 94, 73–82. [DOI] [PubMed] [Google Scholar]
- (512).Matveenko AG; Barbitoff YA; Jay-Garcia LM; Chernoff YO; Zhouravleva GA Differential Effects of Chaperones on Yeast Prions: Current View. Curr. Genet 2017, 1–9. [DOI] [PubMed]
- (513).Park YN; Zhao X; Yim YI; Todor H; Ellerbrock R; Reidy M; Eisenberg E; Masison DC; Greene LE Hsp104 Overexpression Cures Saccharomyces Cerevisiae [Psi+] by Causing Dissolution of the Prion Seeds. Eukaryot. Cell 2014, 13, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Ness F; Cox BS; Wongwigkarn J; Naeimi WR; Tuite MF Over-Expression of the Molecular Chaperone Hsp104 in Saccharomyces Cerevisiae Results in the Malpartition of [Psi(+) ] Propagons. Mol. Microbiol 2017, 104, 125–143. [DOI] [PubMed] [Google Scholar]
- (515).Vashist S; Cushman M; Shorter J Applying Hsp104 to Protein-Misfolding Disorders. Biochem. Cell Biol 2010, 88, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (516).Zaarur N; Xu X; Lestienne P; Meriin AB; McComb M; Costello CE; Newnam GP; Ganti R; Romanova NV; Shanmugasundaram M, et al. Ruvbl1 and Ruvbl2 Enhance Aggresome Formation and Disaggregate Amyloid Fibrils. EMBO J 2015, 34, 2363–2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (517).Shorter J Engineering Therapeutic Protein Disaggregases. Mol. Biol. Cell 2016, 27, 1556–1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (518).McGlinchey RP; Kryndushkin D; Wickner RB Suicidal [Psi+] Is a Lethal Yeast Prion. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 5337–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Holmes DL; Lancaster AK; Lindquist S; Halfmann R Heritable Remodeling of Yeast Multicellularity by an Environmentally Responsive Prion. Cell 2013, 153, 153–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Resende CG; Outeiro TF; Sands L; Lindquist S; Tuite MF Prion Protein Gene Polymorphisms in Saccharomyces Cerevisiae. Mol. Microbiol 2003, 49, 1005–1017. [DOI] [PubMed] [Google Scholar]
- (521).Nakayashiki T; Kurtzman CP; Edskes HK; Wickner RB Yeast Prions [Ure3] and [Psi+] Are Diseases. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 10575–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Halfmann R; Jarosz DF; Jones SK; Chang A; Lancaster AK; Lindquist S Prions Are a Common Mechanism for Phenotypic Inheritance in Wild Yeasts. Nature 2012, 482, 363–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Chernoff YO Mutation Processes at the Protein Level: Is Lamarck Back? Mutation Res 2001, 488, 39–64. [DOI] [PubMed] [Google Scholar]
- (524).Newby GA; Kiriakov S; Hallacli E; Kayatekin C; Tsvetkov P; Mancuso CP; Bonner JM; Hesse WR; Chakrabortee S; Manogaran AL, et al. A Genetic Tool to Track Protein Aggregates and Control Prion Inheritance. Cell 2017, 171, 966–979 e918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Caudron F; Barral Y A Super-Assembly of Whi3 Encodes Memory of Deceptive Encounters by Single Cells During Yeast Courtship. Cell 2013, 155, 1244–1257. [DOI] [PubMed] [Google Scholar]
- (526).Caudron F; Barral Y Mnemons: Encoding Memory by Protein Super-Assembly. Microbial Cell 2014, 1, 100–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Chernova TA; Kiktev DA; Romanyuk AV; Shanks JR; Laur O; Ali M; Ghosh A; Kim D; Yang Z; Mang M, et al. Yeast Short-Lived Actin-Associated Protein Forms a Metastable Prion in Response to Thermal Stress. Cell Rep 2017, 18, 751–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (528).Chernova TA; Chernoff YO; Wilkinson KD Prion-Based Memory of Heat Stress in Yeast. Prion 2017, 11, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (529).Si K; Choi YB; White-Grindley E; Majumdar A; Kandel ER Aplysia Cpeb Can Form Prion-Like Multimers in Sensory Neurons That Contribute to Long-Term Facilitation. Cell 2010, 140, 421–435. [DOI] [PubMed] [Google Scholar]
- (530).Majumdar A; Cesario WC; White-Grindley E; Jiang H; Ren F; Khan MR; Li L; Choi EM; Kannan K; Guo F, et al. Critical Role of Amyloid-Like Oligomers of Drosophila Orb2 in the Persistence of Memory. Cell 2012, 148, 515–529. [DOI] [PubMed] [Google Scholar]
- (531).Fioriti L; Myers C; Huang YY; Li X; Stephan JS; Trifilieff P; Colnaghi L; Kosmidis S; Drisaldi B; Pavlopoulos E, et al. The Persistence of Hippocampal-Based Memory Requires Protein Synthesis Mediated by the Prion-Like Protein Cpeb3. Neuron 2015, 86, 1433–1448. [DOI] [PubMed] [Google Scholar]
- (532).Si K; Lindquist S; Kandel ER A Neuronal Isoform of the Aplysia Cpeb Has Prion-Like Properties. Cell 2003, 115, 879–891. [DOI] [PubMed] [Google Scholar]
- (533).Heinrich SU; Lindquist S Protein-Only Mechanism Induces Self-Perpetuating Changes in the Activity of Neuronal Aplysia Cytoplasmic Polyadenylation Element Binding Protein (Cpeb). Proc. Natl. Acad. Sci. U. S. A 2011, 108, 2999–3004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (534).Stephan JS; Fioriti L; Lamba N; Colnaghi L; Karl K; Derkatch IL; Kandel ER The Cpeb3 Protein Is a Functional Prion That Interacts with the Actin Cytoskeleton. Cell Rep 2015, 11, 1772–1785. [DOI] [PubMed] [Google Scholar]
- (535).Blanco LP; Evans ML; Smith DR; Badtke MP; Chapman MR Diversity, Biogenesis and Function of Microbial Amyloids. Trends Microbiol 2012, 20, 66–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (536).Lipke PN; Klotz SA; Dufrene YF; Jackson DN; Garcia-Sherman MC Amyloid-Like Β-Aggregates as Force-Sensitive Switches in Fungal Biofilms and Infections. Microbiol. Mol. Biol. Rev 2018, 82, e00035–00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (537).Maji SK; Perrin MH; Sawaya MR; Jessberger S; Vadodaria K; Rissman RA; Singru PS; Nilsson KP; Simon R; Schubert D, et al. Functional Amyloids as Natural Storage of Peptide Hormones in Pituitary Secretory Granules. Science 2009, 325, 328–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Fowler DM; Koulov AV; Balch WE; Kelly JW Functional Amyloid--from Bacteria to Humans. Trends Biochem. Sci 2007, 32, 217–224. [DOI] [PubMed] [Google Scholar]
- (539).Chakrabortee S; Kayatekin C; Newby GA; Mendillo ML; Lancaster A; Lindquist S Luminidependens (Ld) Is an Arabidopsis Protein with Prion Behavior. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 6065–6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (540).Antonets KS; Nizhnikov AA Amyloids and Prions in Plants: Facts and Perspectives. Prion 2017, 11, 300–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Heidebrecht A; Scheibel T Recombinant Production of Spider Silk Proteins. Adv. Appl. Microbiol 2013, 82, 115–153. [DOI] [PubMed] [Google Scholar]
- (542).Doblhofer E; Heidebrecht A; Scheibel T To Spin or Not to Spin: Spider Silk Fibers and More. Appl. Microbiol. Biotechnol 2015, 99, 9361–9380. [DOI] [PubMed] [Google Scholar]
- (543).Scheibel T; Parthasarathy R; Sawicki G; Lin XM; Jaeger H; Lindquist SL Conducting Nanowires Built by Controlled Self-Assembly of Amyloid Fibers and Selective Metal Deposition. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 4527–4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Anderson P; Kedersha N Stress Granules. Curr. Biol 2009, 19, R397–398. [DOI] [PubMed] [Google Scholar]
- (545).Buchan JR; Parker R Eukaryotic Stress Granules: The Ins and Outs of Translation. Mol. Cell 2009, 36, 932–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Cherkasov V; Hofmann S; Druffel-Augustin S; Mogk A; Tyedmers J; Stoecklin G; Bukau B Coordination of Translational Control and Protein Homeostasis During Severe Heat Stress. Curr. Biol 2013, 23, 2452–2462. [DOI] [PubMed] [Google Scholar]
- (547).Grousl T; Ivanov P; Malcova I; Pompach P; Frydlova I; Slaba R; Senohrabkova L; Novakova L; Hasek J Heat Shock-Induced Accumulation of Translation Elongation and Termination Factors Precedes Assembly of Stress Granules in S. Cerevisiae. PLoS One 2013, 8, e57083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Escusa-Toret S; Vonk WI; Frydman J Spatial Sequestration of Misfolded Proteins by a Dynamic Chaperone Pathway Enhances Cellular Fitness During Stress. Nat. Cell Biol 2013, 15, 1231–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (549).Kaganovich D; Kopito R; Frydman J Misfolded Proteins Partition between Two Distinct Quality Control Compartments. Nature 2008, 454, 1088–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (550).Miller SB; Ho CT; Winkler J; Khokhrina M; Neuner A; Mohamed MY; Guilbride DL; Richter K; Lisby M; Schiebel E, et al. Compartment-Specific Aggregases Direct Distinct Nuclear and Cytoplasmic Aggregate Deposition. EMBO J 2015, 34, 778–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (551).Sfakianos AP; Whitmarsh AJ; Ashe MP Ribonucleoprotein Bodies Are Phased In. Biochem. Soc. Trans 2016, 44, 1411–1416. [DOI] [PubMed] [Google Scholar]
- (552).Gilks N; Kedersha N; Ayodele M; Shen L; Stoecklin G; Dember LM; Anderson P Stress Granule Assembly Is Mediated by Prion-Like Aggregation of Tia-1. Mol. Biol. Cell 2004, 15, 5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (553).Murray DT; Kato M; Lin Y; Thurber KR; Hung I; McKnight SL; Tycko R Structure of Fus Protein Fibrils and Its Relevance to Self-Assembly and Phase Separation of Low-Complexity Domains. Cell 2017, 171, 615–627 e616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (554).Vanderweyde T; Apicco DJ; Youmans-Kidder K; Ash PEA; Cook C; Lummertz da Rocha E; Jansen-West K; Frame AA; Citro A; Leszyk JD, et al. Interaction of Tau with the Rna-Binding Protein Tia1 Regulates Tau Pathophysiology and Toxicity. Cell Rep 2016, 15, 1455–1466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Molliex A; Temirov J; Lee J; Coughlin M; Kanagaraj AP; Kim HJ; Mittag T; Taylor JP Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (556).Nizhnikov AA; Antonets KS; Bondarev SA; Inge-Vechtomov SG; Derkatch IL Prions, Amyloids, and Rna: Pieces of a Puzzle. Prion 2016, 10, 182–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (557).Johnston JA; Ward CL; Kopito RR Aggresomes: A Cellular Response to Misfolded Proteins. J. Cell Biol 1998, 143, 1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Wang Y; Meriin AB; Zaarur N; Romanova NV; Chernoff YO; Costello CE; Sherman MY Abnormal Proteins Can Form Aggresome in Yeast: Aggresome-Targeting Signals and Components of the Machinery. FASEB J 2009, 23, 451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).Tyedmers J; Treusch S; Dong J; McCaffery JM; Bevis B; Lindquist S Prion Induction Involves an Ancient System for the Sequestration of Aggregated Proteins and Heritable Changes in Prion Fragmentation. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 8633–8638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Chernoff YO Stress and Prions: Lessons from the Yeast Model. FEBS Lett 2007, 581, 3695–3701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (561).Balbirnie M; Grothe R; Eisenberg DS An Amyloid-Forming Peptide from the Yeast Prion Sup35 Reveals a Dehydrated Beta-Sheet Structure for Amyloid. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 2375–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (562).Nelson R; Sawaya MR; Balbirnie M; Madsen AØ; Riekel C; Grothe R; Eisenberg D Structure of the Cross-Β Spine of Amyloid-Like Fibrils. Nature 2005, 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Meier BH; Riek R; Bockmann A Emerging Structural Understanding of Amyloid Fibrils by Solid-State Nmr. Trends Biochem. Sci 2017, 42, 777–787. [DOI] [PubMed] [Google Scholar]
- (564).Fitzpatrick AWP; Falcon B; He S; Murzin AG; Murshudov G; Garringer HJ; Crowther RA; Ghetti B; Goedert M; Scheres SHW Cryo-Em Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (565).Gremer L; Schölzel D; Schenk C; Reinartz E; Labahn J; Ravelli RB; Tusche M; Lopez-Iglesias C; Hoyer W; Heise H Fibril Structure of Amyloid-Β (1–42) by Cryo–Electron Microscopy. Science 2017, 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (566).Ahmed AB; Znassi N; Chateau MT; Kajava AV A Structure-Based Approach to Predict Predisposition to Amyloidosis. Alzheimers Dement 2015, 11, 681–690. [DOI] [PubMed] [Google Scholar]
- (567).Wasmer C; Lange A; Van Melckebeke H; Siemer AB; Riek R; Meier BH Amyloid Fibrils of the Het-S (218–289) Prion Form a Β Solenoid with a Triangular Hydrophobic Core. Science 2008, 319, 1523–1526. [DOI] [PubMed] [Google Scholar]
- (568).Qiang W; Yau WM; Luo Y; Mattson MP; Tycko R Antiparallel Beta-Sheet Architecture in Iowa-Mutant Beta-Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 4443–4448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (569).Petkova AT; Ishii Y; Balbach JJ; Antzutkin ON; Leapman RD; Delaglio F; Tycko R A Structural Model for Alzheimer’s Beta -Amyloid Fibrils Based on Experimental Constraints from Solid State Nmr. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (570).Paravastu AK; Leapman RD; Yau WM; Tycko R Molecular Structural Basis for Polymorphism in Alzheimer’s Beta-Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Xiao Y; Ma B; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y Abeta(1–42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer’s Disease. Nat. Struct. Mol. Biol 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (572).Wälti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Böckmann A; Güntert P; Meier BH; Riek R Atomic-Resolution Structure of a Disease-Relevant Aβ (1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (573).Shewmaker F; Wickner RB; Tycko R Amyloid of the Prion Domain of Sup35p Has an in-Register Parallel Beta-Sheet Structure. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (574).Gorkovskiy A; Thurber KR; Tycko R; Wickner RB Locating Folds of the in-Register Parallel Beta-Sheet of the Sup35p Prion Domain Infectious Amyloid. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E4615–E4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (575).Shewmaker F; McGlinchey RP; Thurber KR; McPhie P; Dyda F; Tycko R; Wickner RB The Functional Curli Amyloid Is Not Based on in-Register Parallel Beta-Sheet Structure. J. Biol. Chem 2009, 284, 25065–25076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (576).Lim KH; Dasari AK; Hung I; Gan Z; Kelly JW; Wright PE; Wemmer DE Solid-State Nmr Studies Reveal Native-Like Beta-Sheet Structures in Transthyretin Amyloid. Biochemistry 2016, 55, 5272–5278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (577).Schmidt M; Rohou A; Lasker K; Yadav JK; Schiene-Fischer C; Fändrich M; Grigorieff N Peptide Dimer Structure in an Aβ (1–42) Fibril Visualized with Cryo-Em. Proc. Natl. Acad. Sci. U.S.A 2015, 112, 11858–11863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (578).Wille H; Govaerts C; Borovinskiy A; Latawiec D; Downing KH; Cohen FE; Prusiner SB Electron Crystallography of the Scrapie Prion Protein Complexed with Heavy Metals. Arch. Biochem. Biophys 2007, 467, 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Vazquez-Fernandez E; Vos MR; Afanasyev P; Cebey L; Sevillano AM; Vidal E; Rosa I; Renault L; Ramos A; Peters PJ, et al. The Structural Architecture of an Infectious Mammalian Prion Using Electron Cryomicroscopy. PLoS Pathog 2016, 12, e1005835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (580).Bruce M; Chree A; McConnell I; Foster J; Pearson G; Fraser H Transmission of Bovine Spongiform Encephalopathy and Scrapie to Mice: Strain Variation and the Species Barrier. Phil. Trans. Royal Soc. London B: Biol. Sci 1994, 343, 405–411. [DOI] [PubMed] [Google Scholar]
- (581).Collinge J; Clarke AR A General Model of Prion Strains and Their Pathogenicity. Science 2007, 318, 930–936. [DOI] [PubMed] [Google Scholar]
- (582).Walker LC Proteopathic Strains and the Heterogeneity of Neurodegenerative Diseases. Annu. Rev. Genet 2016, 50, 329–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Zhou P; Derkatch IL; Uptain SM; Patino MM; Lindquist S; Liebman SW The Yeast Non-Mendelian Factor [Eta+] Is a Variant of [Psi+], a Prion-Like Form of Release Factor Erf3. EMBO J 1999, 18, 1182–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (584).Kryndushkin DS; Alexandrov IM; Ter-Avanesyan MD; Kushnirov VV Yeast [Psi+] Prion Aggregates Are Formed by Small Sup35 Polymers Fragmented by Hsp104. J. Biol. Chem 2003, 278, 49636–49643. [DOI] [PubMed] [Google Scholar]
- (585).Sharma J; Liebman SW [Psi(+) ] Prion Variant Establishment in Yeast. Mol. Microbiol 2012, 86, 866–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (586).Chien P; Weissman JS Conformational Diversity in a Yeast Prion Dictates Its Seeding Specificity. Nature 2001, 410, 223–227. [DOI] [PubMed] [Google Scholar]
- (587).Cohen ML; Kim C; Haldiman T; ElHag M; Mehndiratta P; Pichet T; Lissemore F; Shea M; Cohen Y; Chen W, et al. Rapidly Progressive Alzheimer’s Disease Features Distinct Structures of Amyloid-Beta. Brain 2015, 138, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (588).Qiang W; Yau WM; Lu JX; Collinge J; Tycko R Structural Variation in Amyloid-Beta Fibrils from Alzheimer’s Disease Clinical Subtypes. Nature 2017, 541, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (589).Bousset L; Pieri L; Ruiz-Arlandis G; Gath J; Jensen PH; Habenstein B; Madiona K; Olieric V; Bockmann A; Meier BH, et al. Structural and Functional Characterization of Two Alpha-Synuclein Strains. Nat. Commun 2013, 4, 2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (590).Wickner RB; Shewmaker F; Edskes H; Kryndushkin D; Nemecek J; McGlinchey R; Bateman D; Winchester CL Prion Amyloid Structure Explains Templating: How Proteins Can Be Genes. FEMS Yeast Res 2010, 10, 980–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (591).Toyama BH; Kelly MJ; Gross JD; Weissman JS The Structural Basis of Yeast Prion Strain Variants. Nature 2007, 449, 233–237. [DOI] [PubMed] [Google Scholar]
- (592).Li J; Browning S; Mahal SP; Oelschlegel AM; Weissmann C Darwinian Evolution of Prions in Cell Culture. Science 2010, 327, 869–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (593).Ghaemmaghami S; Watts JC; Nguyen HO; Hayashi S; DeArmond SJ; Prusiner SB Conformational Transformation and Selection of Synthetic Prion Strains. J. Mol. Biol 2011, 413, 527–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (594).Makarava N; Baskakov IV Genesis of Tramsmissible Protein States Via Deformed Templating. Prion 2012, 6, 252–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Bateman DA; Wickner RB The [Psi+] Prion Exists as a Dynamic Cloud of Variants. PLoS Genet 2013, 9, e1003257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (596).Bruce KL; Chernoff YO Sequence Specificity and Fidelity of Prion Transmission in Yeast. Semin. Cell Dev. Biol 2011, 22, 444–451. [DOI] [PubMed] [Google Scholar]
- (597).Will RG Variant Creutzfeldt-Jakob Disease. Acta Neurobiol. Exp 2002, 62, 167–173. [DOI] [PubMed] [Google Scholar]
- (598).Beringue V; Vilotte JL; Laude H Prion Agent Diversity and Species Barrier. Vet. Res 2008, 39, 1–30. [DOI] [PubMed] [Google Scholar]
- (599).Chen B; Bruce KL; Newnam GP; Gyoneva S; Romanyuk AV; Chernoff YO Genetic and Epigenetic Control of the Efficiency and Fidelity of Cross-Species Prion Transmission. Mol. Microbiol 2010, 76, 1483–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Grizel AV; Rubel AA; Chernoff YO Strain Conformation Controls the Specificity of Cross-Species Prion Transmission in the Yeast Model. Prion 2016, 10, 269–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (601).Sharma A; Bruce KL; Chen B; Gyoneva S; Behrens SH; Bommarius AS; Chernoff YO Contributions of the Prion Protein Sequence, Strain, and Environment to the Species Barrier. J. Biol. Chem 2016, 291, 1277–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (602).Ahmed AB; Kajava AV Breaking the Amyloidogenicity Code: Methods to Predict Amyloids from Amino Acid Sequence. FEBS Lett 2013, 587, 1089–1095. [DOI] [PubMed] [Google Scholar]
- (603).Esteras-Chopo A; Serrano L; Lopez de la Paz M The Amyloid Stretch Hypothesis: Recruiting Proteins toward the Dark Side. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 16672–16677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (604).Pastor MT; Esteras-Chopo A; Serrano L Hacking the Code of Amyloid Formation: The Amyloid Stretch Hypothesis. Prion 2007, 1, 9–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (605).Maurer-Stroh S; Debulpaep M; Kuemmerer N; Lopez de la Paz M; Martins IC; Reumers J; Morris KL; Copland A; Serpell L; Serrano L, et al. Exploring the Sequence Determinants of Amyloid Structure Using Position-Specific Scoring Matrices. Nat. Methods 2010, 7, 237–242. [DOI] [PubMed] [Google Scholar]
- (606).Conchillo-Sole O; de Groot NS; Aviles FX; Vendrell J; Daura X; Ventura S Aggrescan: A Server for the Prediction and Evaluation of “Hot Spots” of Aggregation in Polypeptides. BMC Bioinformatics 2007, 8, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (607).Garbuzynskiy SO; Lobanov MY; Galzitskaya OV Foldamyloid: A Method of Prediction of Amyloidogenic Regions from Protein Sequence. Bioinformatics 2010, 26, 326–332. [DOI] [PubMed] [Google Scholar]
- (608).Hamodrakas SJ; Liappa C; Iconomidou VA Consensus Prediction of Amyloidogenic Determinants in Amyloid Fibril-Forming Proteins. Int. J. Biol. Macromol 2007, 41, 295–300. [DOI] [PubMed] [Google Scholar]
- (609).Fernandez-Escamilla A-M; Rousseau F; Schymkowitz J; Serrano L Prediction of Sequence-Dependent and Mutational Effects on the Aggregation of Peptides and Proteins. Nat. Biotechnol 2004, 22, 1302–1306. [DOI] [PubMed] [Google Scholar]
- (610).Ross ED; Baxa U; Wickner RB Scrambled Prion Domains Form Prions and Amyloid. Mol. Cell Biol 2004, 24, 7206–7213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (611).Ross ED; Edskes HK; Terry MJ; Wickner RB Primary Sequence Independence for Prion Formation. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 12825–12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (612).Toombs JA; McCarty BR; Ross ED Compositional Determinants of Prion Formation in Yeast. Mol. Cell Biol 2010, 30, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (613).Antonets KS; Nizhnikov AA Sarp: A Novel Algorithm to Assess Compositional Biases in Protein Sequences. Evol. Bioinform. Online 2013, 9, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (614).Antonets KS; Nizhnikov AA Predicting Amyloidogenic Proteins in the Proteomes of Plants. Int. J. Mol. Sci 2017, 18, 2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (615).Harrison PM; Gerstein M A Method to Assess Compositional Bias in Biological Sequences and Its Application to Prion-Like Glutamine/Asparagine-Rich Domains in Eukaryotic Proteomes. Genome Biol 2003, 4, R40. [DOI] [PMC free article] [PubMed] [Google Scholar]