

Abstract
The field of environmental public health is at an important crossroad. Our current biomonitoring efforts document widespread exposure to a host of chemicals for which toxicity information is lacking. At the same time, advances in the fields of genomics, proteomics, metabolomics, genetics and epigenetics are yielding volumes of data at a rapid pace. Our ability to detect chemicals in biological and environmental media has far outpaced our ability to interpret their health relevance, and as a result, the environmental risk paradigm, in its current state, is antiquated and ill-equipped to make the best use of these new data. In light of new scientific developments and the pressing need to characterize the public health burdens of chemicals, it is imperative to reinvigorate the use of environmental epidemiology in chemical risk assessment. Two case studies of chemical assessments from the Environmental Protection Agency Integrated Risk Information System database are presented to illustrate opportunities where epidemiologic data could have been used in place of experimental animal data in dose-response assessment, or where different approaches, techniques, or studies could have been employed to better utilize existing epidemiologic evidence. Based on the case studies and what can be learned from recent scientific advances and improved approaches to utilizing human data for dose-response estimation, recommendations are provided for the disciplines of epidemiology and risk assessment for enhancing the role of epidemiologic data in hazard identification and dose-response assessment.
Keywords: Risk assessment, epidemiology, methylmercury, phthalates, di-butyl phthalate, dose-response assessment, developmental toxicity, reproductive toxicity, EPA, Integrated Risk Information System (IRIS), cardiovascular disease
INTRODUCTION
The field of environmental public health science is at an important crossroad. Advances in the interrelated disciplines it encompasses hold promise for our most comprehensive understanding of the influence of environmental stressors on human health to date. Estimates of the number of chemicals in commerce range from the tens of thousands to over 140,000 [1]; the majority of these are still lacking any sort of toxicological evaluation necessary to set regulatory standards aimed at protecting public health [2, 3]. At the same time, advances in the fields of genomics, proteomics, metabolomics, genetics and epigenetics are yielding volumes of data at a rapid pace [4]. Our ability to detect chemicals in biological and environmental media has far outpaced our ability to interpret their health relevance [5]. Our increasing recognition of the joint behavior of multiple stressors (often those of varying nature) offers challenges to the traditional Hill Criteria-based approach and overemphasis on causation in toxicological assessment [6]. The environmental risk paradigm, in its current state, is antiquated and ill-equipped to make the best use of these new data that are being generated in a high-throughput fashion [7]. In light of these new scientific developments and the ongoing and pressing need to characterize the public health impacts and burdens of chemicals, we need to reinvigorate the use of environmental epidemiology in chemical risk assessment.
The purpose of this paper is to highlight opportunities and approaches that allow for the enhanced incorporation of epidemiologic data in risk assessment. We present case studies of chemical assessments from the Environmental Protection Agency (EPA) Integrated Risk Information System (IRIS) database to illustrate opportunities where epidemiologic data could have been used in place of experimental animal data in dose-response assessment, or where different approaches, techniques, or studies could have been employed to better utilize existing epidemiologic evidence. Based on the case studies and what can be learned from recent scientific advances and improved approaches to utilizing human data for dose-response estimation, we make recommendations for the disciplines of epidemiology and risk assessment for enhancing the role of epidemiologic data in hazard identification and dose-response assessment.
EPIDEMIOLOGY IN RISK ASSESSMENT
Environmental risk assessment, as a policy-driven framework established to facilitate use and consideration of scientific information in the decision-making process, is reliant on information from studies of the adverse effects of chemicals in living organisms. The process of quantifying the relationship between chemical exposures and health consequences in humans, known as dose-response assessment, is informed by data from studies of human exposures to chemicals and studies in experimental animals, and can be supplemented by in vitro assays.
Historically, epidemiology has played the leading role in characterizing risks from agents with limited numbers of sources that are attributable to relatively greater morbidity and mortality, such as smoking. In contrast, risks resulting from multi-pathway lower level exposures to environmental chemicals have often relied on experimental animal data for toxicity characterization. Experimental animal data, as a function of laboratory-controlled settings, facilitate estimation of quantitative dose-response relationships and allow for the examination of the full range of toxic effects. As compared to epidemiologic studies, which usually are forced to rely on observational data [8, 9], animal studies are often considered to be better suited to evaluate effects for susceptible lifestages. Further, associations between exposure and effect estimated in human observational studies are subject to exposure and outcome misclassification due to frequent absence of direct exposure and outcome measurements and subsequent reliance on surrogate data; as a result of these often non-differential biases, estimations of the strength of these associations likely result in underestimation [9].
The utility of animal studies in estimating dose-response relationships in humans has limitations. In particular, animal studies often require extrapolations (often from high to environmentally-relevant doses, and from shorter- to longer-term exposure durations) and applications of assumptions regarding similarities in toxicokinetics and dynamics across species [10]. In addition, while the controlled environment of animal studies may be useful in moving towards identifying causal relationships, studies of exposures in humans are better suited for evaluating exposures to mixtures and interactions among different classes of agents, direct evaluation of chemical exposures at environmentally-relevant levels, and characterization of actual human disease states.
Numerous advantages afforded by epidemiologic studies relative to those conducted in experimental animals have been identified (see Table 1) [11]. Use of human data in dose-response estimation obviates or minimizes the impact of uncertainties that arise from certain necessary extrapolations inherent in using animal data, and it has been argued that uncertainties derived from exposure misclassification in epidemiologic studies are dwarfed by those stemming from potential interspecies differences [12]. Further, while high-to-low dose extrapolation may still be necessary in utilization of epidemiologic data (when extrapolating from occupational exposures to those in the general population, for example), the magnitude of such an extrapolation is often substantially reduced relative to experimental animal studies, coupled with a limiting of the associated uncertainty.
Table 1.
Comparison of Strengths and Weaknesses in Epidemiologic and Experimental Animal Studies
| Strengths | Weaknesses | |
|---|---|---|
| Epidemiologic Studies | Examination of exposure/effect relationships in species of interest | Increased potential for exposure and outcome misclassification and confounding |
| Often able to study effects in heterogeneous population | Limited measures of exposure and dose lead to challenges in characterizing quantitative dose-response relationships | |
| Better suited to consider true range of population exposures | Can take longer time to observe effects from exposure | |
| Exposures better reflect temporal nature and delivery mechanism | Relatively more expensive | |
| Evaluate cumulative exposures and health impacts | Confounding exposures/risk factors | |
| Experimental Animal Studies | Allow for examination of full spectrum of toxic effects | Extrapolations required (interspecies, dose, temporal) |
| Facilitates estimation of quantitative dose-response relationships | Dose delivery is artificial and poorly approximates true patterns of exposure | |
| Reduced misclassification of exposures and outcomes | Homogeneity of experimental animals in genetics, lifestage, diet, and initial health state | |
| Study times can be shorter and allow for early indication of harm |
While subject to its own concerns, exposure assessment as conducted in epidemiologic studies may more accurately reflect the temporal nature (and delivery mechanism) of exposure as compared to experimental animal studies, and epidemiologic studies incorporate the true range of population exposures. Exposures to environmental contaminants rarely occur at a consistent rate and dose magnitude; in reality, exposures to environmental chemicals can be intermittent and at inconsistent doses. As a result, the careful, controlled administration of chemicals in laboratory animal studies creates an artificial dose regime that may poorly approximate the true nature of exposure. Further, studies in experimental animals rely on a genetically similar strain, often of a single gender and lifestage, providing a limited context in which inter-individual variability or heterogeneity in susceptibility can be assessed [11, 13]. In addition, efforts to maintain an unchanging diet and consistent state of good health for experimental animals (in the absence of the substance being evaluated) results in a scenario that bears little semblance to that induced by real-world complexities that accompany human exposure [13]. Molecular epidemiologic methods, which employ biologic markers of early biological effect and susceptibility (such as genetic polymorphism data), allow for enhanced sensitivity and human relevance as compared to findings from studies in experimental animals [14, 15]. For these reasons, epidemiologic data may present opportunities not afforded by the experimental setting.
Despite potential advantages afforded by epidemiologic data in assessing dose-response relationships, numerous challenges have repeatedly been highlighted that argue against its use [11, 16], and a reluctance on the part of epidemiologists to participate in the risk assessment process [17] or tailor the reporting of their results for use in risk assessment has been noted [16, 18]. Prominent among these criticisms are issues regarding the sensitivity of epidemiologic methods, limitations of exposure measurements and the potential for confounding and other biases [11]. Further, challenges inherent in interpretation of the results of epidemiologic research may further inhibit its incorporation. Inconclusive results, poor documentation of methods and results, study design flaws, or positive findings in the face of considerable uncertainty may limit the utility of these data in quantification of relationships.
Additional challenges to incorporating epidemiologic data into risk assessment may arise from inherent differences in needed outputs between epidemiologists and risk assessors. In epidemiologic studies, information on dose is sparse, and in many cases, individual-level exposure data may be unavailable and estimation may rely on area-level or categorical assignments. It is often the case that these exposure and dose surrogates, as employed in epidemiologic studies, cannot be used in efforts to characterize quantitative dose-response relationships for individual chemicals, a critical component of risk assessment [16].
Risk assessments are frequently conducted and communicated to support a decision-making process, often occurring in the regulatory or standard-setting context and on a shorter time scale. On the other hand, the conduct of epidemiologic research is typically not driven by immediate regulatory or management need [16]. To this end, there exists inconsistency in the metrics utilized by epidemiologists and risk assessors to characterize dose-response relationships, particularly for noncancer health endpoints. The results of observational studies are typically presented in the form of odds ratios (ORs) or relative risks (RRs) associated with some increase in exposure, whereas reference values utilized in noncancer risk assessments have historically attempted to identify a dose threshold below which exposures are not likely to elicit adverse health consequences.
SCIENTIFIC ADVANCES AND DEVELOPMENTS
Much attention has been lent to scientific advances that have occurred over the last decade and hold promise for improving the value of epidemiologic studies in the elucidation of dose-response relationships. Among these are the improved understanding and use of biological markers, wider acceptance of the concept of toxicity pathways and the significance of early biological perturbations, and potential applications of genomics, proteomics and metabolomics in support of chemical prioritization and dose-response assessment. In addition to these advances, evolving perspectives on issues critical to dose-response assessment, such as variability in human susceptibility to chemical exposures as influenced by genetic and epigenetic factors, provide additional insight into the value of epidemiologic data in risk assessment. These advances, and their promise for enhancing the role of epidemiology in risk assessment, are discussed below.
Biomonitoring
The NRC defines biomonitoring as “one method for assessing human exposures to chemicals by measuring chemicals or their metabolites in human tissues or specimens” [5]. While biomarkers exist that serve as essential measures of chemical exposure, the same concept can be applied to evaluate chemical compounds in tissues as indicators of effect or susceptibility [19].
The rapidly expanding volume of biomonitoring data is changing the environmental epidemiologic landscape by improving our ability to evaluate exposures and outcomes and is becoming a critical component of modern epidemiologic investigations [20], involving measurement of chemicals or their metabolites in biological matrices at lower concentrations than those typically employed in experimental animal toxicological or human clinical studies [21]. This affords epidemiologists the ability to investigate changes that may occur at environmentally-relevant exposure concentrations [22] and, in many cases, allows for improved precision in characterization of exposure. In addition, biological markers of outcome and susceptibility allow for the ascertainment of frequency of subtle, subclinical toxicodynamic processes or the occurrence of unique vulnerabilities. Attention to these early changes and special populations will bolster the effectiveness of epidemiologic studies in steering the risk assessment process to provide the best possible characterization of the true nature of dose-response relationships, based on available data. Biomonitoring evaluations also hold promise for prioritizing epidemiologic studies [23], by highlighting gaps in toxicological knowledge corresponding to ubiquitous population exposures, as has been demonstrated in the case of polybrominated and polyfluoroalkyl compounds [24, 25]. Additionally, data collected in large-scale biomonitoring investigations can be useful in setting reference ranges to assist in exposure classification in epidemiologic studies [26, 27]. While the ongoing development of biomarkers holds promise for enhancing environmental epidemiology, biomarker development can be challenging [28]; to this end, efforts to identify the ideal characteristics for new biomarkers have been made [5, 29].
A number of domestic and international large-scale biomonitoring initiatives are either proposed, currently underway, or completed [30]. In the US, the National Health and Nutrition Examination Survey (NHANES), as conducted by the CDC and the National Human Exposure Assessment Survey (NHEXAS), as conducted by EPA constitute the largest nationally-representative single-agency biomonitoring efforts, and a number of state-based biomonitoring initiatives are underway [31]. The National Children’s Study (NCS), a longitudinal study aiming to evaluate the influence of environmental factors on health and development, will recruit 100,000 participants and encompass fourteen data collection events from parental pre-conception to the child between sixteen and twenty years of age. A number of biological specimens will be collected from the parents and the children [32]. Internationally, the European Commission, as part of the European Environment and Health Action Plan 2004–2010, has called for a Unionwide coordinated approach to biomonitoring in member states [33].
In its 2006 report, the NRC Committee on Human Biomonitoring for Environmental Toxicants noted that while population-based biomonitoring initiatives such as NHANES and NHEXAS are in place, and while advances in the laboratory have afforded us the ability to measure chemicals in tissues and other biological samples, there are relatively few chemicals for which methods for evaluating and interpreting the public health significance of these biological markers of exposure are in place [5]. The Committee recommended enhancing the use of biomonitoring data as it relates to risk assessment by calling for a coordinated strategy for biomarker development and continued population biomonitoring, prioritized on the basis of potential for exposure and public health concerns. The Committee also highlighted the current shortcoming of most large-scale biomonitoring efforts to collect data on important uniquely susceptible subpopulations; infants and children are often omitted from biomonitoring studies due to challenges related to sample collection. Further, a number of unique ethical issues pertaining to conducting biomonitoring studies in children have been identified [34]. While these difficulties in data collection may be viewed as prohibitory, information regarding exposure and susceptibility during earlier lifestages is essential in determination of critical exposure windows for young children. The development of new biomonitoring-based investigations as well as the enhancement of existing studies with biomonitoring components, both in experimental animals and humans, were recommended by the Committee [5].
Susceptibility
Inter-individual variability in susceptibility in response to chemical exposure, as mediated by age, disease status and a variety of other factors, has recently been highlighted as a critical consideration in dose-response assessment, particularly for elucidating the shape of the lower portion of the dose response curve. There has been increasing scrutiny of the adequacy of a threshold-based approach for noncancer effects; one perspective that has been presented is that individuals may exhibit a true dose threshold below which a response will not occur, though these individuals may differ in a variety of factors that influence where such a threshold may occur. Population-based variability in these influences, including genetic, epigenetic and behavioral factors, as well as existing morbidities and co-exposures, may obscure the true shape of the dose response relationship at low, environmentally-relevant doses, even in the case of chemicals with identified MOAs that are inconsistent with stochastic processes for key events [35]. Consequently, the use of a threshold-based toxicity value as a means of risk management at the population level is often not an accurate reflection of our scientific understanding of the dose-response relationship within the population.
Hattis has suggested that improvements in the precision of exposure assessment in environmental epidemiologic studies, coupled with evaluation of appropriately selected biological markers, will provide critical insight into the degree and nature of interindividual variability in susceptibility to chemical insult [36]. Hattis argues that epidemiologists can play a key role in discerning the relevance of interindividual variability in dose-response relationships by moving towards acknowledging mechanistically-relevant factors in their study designs. In particular, for evaluating variation in susceptibility to noncancer quantal endpoints, Hattis advocates for categorization of variability parameters into human factors that influence chemical uptake, pharmacokinetics and pharmacodynamics [13]. Discrete consideration of variability within these categories across the human population, while adding complexity to dose-response characterization, may improve the ability of risk assessments to provide adequate protection to those uniquely susceptible while improving overall precision in specification of the dose-response relationship.
The NRC has recently reaffirmed the importance of addressing susceptibility in dose-response assessment and provided recommendations for formalizing its consideration in the risk assessment process [7]. The enhanced susceptibility at early ages has begun to be addressed in risk assessment, with EPA adopting new guidelines to address increased risk during childhood from exposure to mutagenic compounds [37], and the California Environmental Protection Agency adopting guidelines to incorporate an extra factor to account for increased sensitivity to all carcinogens prenatally and during childhood [38]. While these actions incorporate some features of early-life sensitivity, the NRC notes that there are still further steps that can be taken, particularly for EPA, including development of methods to account for prenatal and other noncarcinogen exposures. The NRC also cautioned against defaulting to an assumption of zero variability in susceptibility in the absence of evidence to the contrary [7]. Evaluation of variation in population susceptibility plays a prominent role in the unified approach for dose-response assessment proposed by the NRC, assisting both in dose-response model selection and in identifying susceptible subpopulations for further assessment [7].
Advances in ‘Omics
The NRC committee on Toxicity Testing and Assessment of Environmental Agents released a report in 2007, stressing the inadequate capacity of current toxicity testing methodologies, which primarily rely on in vivo studies in experimental animals, and calling for a strategy that enables the scientific community to generate comprehensive toxicological information at an increasing rate [39]. As part of the Committee’s vision, attention was placed on identification and mapping of toxicity pathways, or cellular response pathways that result in adverse health effects when perturbed [4]. The Committee emphasized focus on preventing these perturbations, rather than clinically-observable endpoints, as a more appropriate goal for risk assessment. The view of the Committee was that the most effective means of achieving this goal is the eventual replacement (or near-replacement) of animal studies with the use of in vitro and in silico models of cellular and molecular systems to evaluate biologically-relevant perturbations, which would allow for higher throughput chemical evaluation.
While numerous challenges exist in facilitating such a paradigm shift, including the considerable burden of adequately characterizing the toxicodynamics of these toxicity pathways, improved in vitro and in silico methods may hold promise for identification of biomarkers of exposure, effect and susceptibility that will be useful in epidemiologic investigations [39]. To this end, numerous ‘Omics technologies have been introduced, focusing on various levels of biological molecules and configurations ranging from the sequence of DNA and alterations of expression of DNA, RNA transcription, and the synthesis of proteins and other small molecules; however, the use of these technologies in risk assessment to date has been limited [40]. The output of these assays also holds promise for identifying the potential for chemical interactions or target organ potentiation, as elucidation of chemicals capable of eliciting the same initial biological perturbation can aid in targeting evaluation of commonly occurring mixtures where risk assessment conducted using standard dose-response assumptions may not reflect our best understanding of the underlying science.
Interest in characterizing the influence of epigenetic factors on disease susceptibility has become increasingly prominent in recent years. The term epigenetics refers to changes in gene expression that occur in the absence of alteration of DNA sequence [41]. It is increasingly recognized that environmental stressors are capable of eliciting epigenetic alterations; three targets of susceptibility for epigenetic insult, transposable elements, promoter regions of housekeeping genes, and imprinting genes, have been identified as alterable by changes in methylation patterns [42]. These targets are believed to be most sensitive to insult during certain lifestages, such as gestation, neonatal development, puberty, and old age [42]. The consequences of alterations to the epigenome and subsequent modifications of gene expression can translate to phenotypic changes that may impact disease susceptibility, behavior, and survival [41]. Recognition of the epigenome’s influence on gene expression offers a new perspective on interactions between genetic and environmental factors in predicting susceptibility and likelihood of disease development [43]. In particular, the heritable nature of some environmentally-induced epigenetic alterations highlights the concern that early-life exposures to environmental chemicals may not only be concerning for the exposed later in life, but also for subsequent generations. Epigenetic alterations induced by some endocrine disrupting chemicals, such as vinclozolin (as demonstrated by Jirtle and Skinner), have been demonstrated to elicit transgenerational disease phenotypes leading to adverse health consequences [41]. A recent workshop held by the British Committee on Toxicology addressed the issue of use of transgenerational epigenetics in regulatory toxicity testing [44]. The Committee outlined suggestions and key questions for the regulatory community with regard to incorporation of epigenetics data in risk assessment. Prominent among these were a call for alternative methods to test the potential for transgenerational epigenetic effects that would obviate the need for testing as far as the F3 and F4 generations [44].
Increased recognition of the potential for chemicals to elicit toxicity through an epigenetic MOA will improve the ability of toxicity assessments to realize the role of epigenetic changes in the context of disease development [45]. Consideration of epigenetic alterations as markers of early biological perturbations along an effect continuum concluding with frank disease has the potential to improve the sensitivity of epidemiologic evaluations and may allow for identification of uniquely vulnerable subpopulations. Characterization of such markers may allow for the design of biomonitoring strategies that will generate data on epigenetic alterations in support of epidemiologic investigations.
Adversity and Recognition of Upstream Effects
Historically, risk assessments have relied largely on clinically-overt diseases or defects as measures of effect in dose-response estimation. In the context of derivation of noncancer toxicity values at EPA, these effects are referred to as “critical effects”, and are defined as “the first adverse effect, or its known precursor, that occurs to the most sensitive species as the dose rate of an agent increases” [46]. In contrast to its tendency to identify overt outcomes as critical effects, EPA defines an adverse effect as a “biochemical change, functional impairment, or pathologic lesion that affects the performance of the whole organism, or reduces an organism’s ability to respond to an additional environmental challenge” [47]. In an increasing number of cases, recent toxicological assays characterize early biological perturbations along the toxicodynamic continuum that precede frank effects; it is increasingly recognized that many of these initial perturbations, or upstream effects, are consistent with EPA’s current view of adversity. This will enhance the usefulness of epidemiology, because early biological markers can be more sensitive endpoints than more overt outcomes and are often measured on a continuous scale, so fewer observations are required to achieve sufficient statistical power.
The selection of critical endpoints in EPA risk assessments has been subject to criticism stemming from inconsistency with its definition of an adverse event. Debates about the significance of neurotoxicological endpoints highlight this confusion. Subtle perturbations that may influence deficits have frequently been dismissed for a number of reasons, including statistical imprecision in endpoint measurement instruments or population variability in a given neurological outcome masking a meaningful shift in a neurological parameter [48]. These shifts, while potentially viewed as insignificant for an individual, may have greater implications at the population level, where a slight shift in the population median value may result in a considerably larger number of persons within the range of adversity [49].
As provision of data regarding the doses at which these upstream effects occur becomes more routine, and as understanding of these early biological effects as adverse increases, there is mounting potential for these effects to be used as outcomes in risk assessments [50]. A scientific workshop was convened in 2007 to investigate the relationship between early biological changes and adversity. Participants identified three classes of early effects (thyroid perturbation, anti-androgen activity and immunological changes) where sufficient evidence exists to demonstrate linkage between these changes and observable disease, facilitating the direct use of these early biological changes in risk assessments. The recommendations of the workgroup also address consideration of factors that may influence the relationship between early biological markers and downstream effects, including biological background, possible dose additivity or interaction, windows of susceptibility, the importance of multiple or complex modes of action, and impact of co-morbidities, unique susceptibilities and background chemical exposures [50]. Workshop participants emphasized that assessing risks for noncancer effects should move towards a non-threshold model.
Unified Approach to Dose-Response Assessment
The approach to noncancer dose-response assessment has been repeatedly criticized [7, 51–53]. Noting the current dichotomy in methods for cancer and noncancer dose-response assessment, the NRC identified a goal of greater balance and coherence between the two by recommending use of information understood to influence individual and population dose-response curves (such as mechanism of action (MOA), susceptibility, background exposures and others) to inform the model selection for characterization of dose-response relationship. This approach recommends the use of low-dose linear non-threshold model unless the basis for a threshold or non-linear relationship in the low dose range is clearly established. The NRC proposes a dose-response framework that redefines the RfD as a risk-specific dose, or a dose corresponding to a particular risk considered to be de minimis at a defined confidence interval, rather than as a “dichotomous risk - unappreciable risk descriptor” [7]. Such an approach could be used not only to derive the dose equated with a particular risk level but also to characterize risk at any dose. The NRC recommends that risk assessments for all endpoints be quantitatively-based and that a threshold should not be assumed for effects, instead recognizing the potential for continuous dose-response relationships in the population. [7].
The outputs from epidemiologic studies, when presented as ORs and RRs, provide the means to move towards non-threshold models and risk characterization metrics for non-cancer endpoints because these measured differences in health outcomes can be readily converted to dose-response relationships. As opposed to being used to identify PODs from which bright lines can be identified, these epidemiologic outputs lend themselves to development of slope-based dose-response metrics. Bright line approaches to characterizing noncancer hazards are problematic, since a ‘safe/unsafe’ dichotomy does not speak to the probability or severity of health effects that occur at various points in the dose-response relationship [54].
EPA’S INTEGRATED RISK INFORMATION SYSTEM
The IRIS database is an online repository of toxicological evaluations of chemicals maintained by the National Center for Environmental Assessment at the EPA. It currently serves as the primary repository of risk assessments conducted by the EPA and its toxicity values are employed by entities at the international, national, state and local levels. Given its widespread use and recognition, toxicity assessments from the IRIS database will serve as case studies in this manuscript.
IRIS toxicological assessments encompass the hazard identification and dose-response assessment steps of the risk paradigm, as outlined by the National Research Council [7, 55]. Database entries for each chemical, depending on available evidence to support a relationship between exposure and effect, include the qualitative conclusions of a weight of evidence discussion regarding a chemical’s carcinogenicity, quantitative estimates of carcinogenic potency (cancer slope factors [CSF] and inhalation and drinking water unit risks [IURs and DWURs]) and noncancer reference values (reference doses [RfDs] and concentrations [RfCs]). When attempting to characterize dose-response relationships at low, environmentally-relevant doses, IRIS assumes carcinogens to operate in a linear, non-threshold fashion (except in the case of chloroform), whereas noncancer adverse effects are assumed to have dose thresholds below which these effects are unlikely. These toxicity values can be employed alongside site- or scenario-specific exposure data to estimate carcinogenic risk or potential noncancer hazard in exposed persons.
As of November 2009, the IRIS database contains evaluations of 553 chemicals. A recently published evaluation of the full IRIS database by EPA/NCEA scientists through June 2007 found that human data was employed in the derivation of at least one chronic toxicity value for 44 chemicals (< 8% of all assessed) [56]. Of these, epidemiologic data directly supported the derivation of 27 RfDs (<8% of all RfDs), 12 RfCs (<16%), 3 CSFs (<4%) and 12 IURs (<22%). Based on this, while RfDs and CSF comprise more than 75% of all IRIS toxicity values, human data is more frequently employed in derivation of inhalation toxicity values. Potential explanations for the relatively higher use of human data in derivation of inhalation toxicity values could be that human observational studies employed in risk assessment often evaluate occupational (and non-dietary) exposures to air contaminants, and that the costs and ease of conducting oral exposure studies in experimental animals as compared to inhalation studies inadvertently result in the tendency for non-human toxicological evaluations to examine the oral route.
A subsequent inspection of assessments conducted since this evaluation did not reveal additional assessments using human data; the last assessment or update to employ epidemiologic data in the derivation of toxicity values was that for toluene, most recently revised in 2005. Further evaluation of use of human data in support of IRIS toxicological assessments over time reveals its limited application (<10%) prior to 1995, with an increase to its peak usage of 44% between 1995 and 1999, and again declining to support only 11% of critical effects in toxicological assessments conducted between 2005–2009 (see Fig. 1). Despite this, Persad and Cooper have noted that epidemiologic data are expected to play a more prominent role in assessments in preparation [56]. It should be noted that this limited inspection did not include an evaluation of the frequency of cases where epidemiologic data were available but were not used to support selection of a critical effect. Such an effort would be instructive in understanding the role of and identifying opportunities for epidemiologic data to play a role in future IRIS assessments.
Fig. (1).
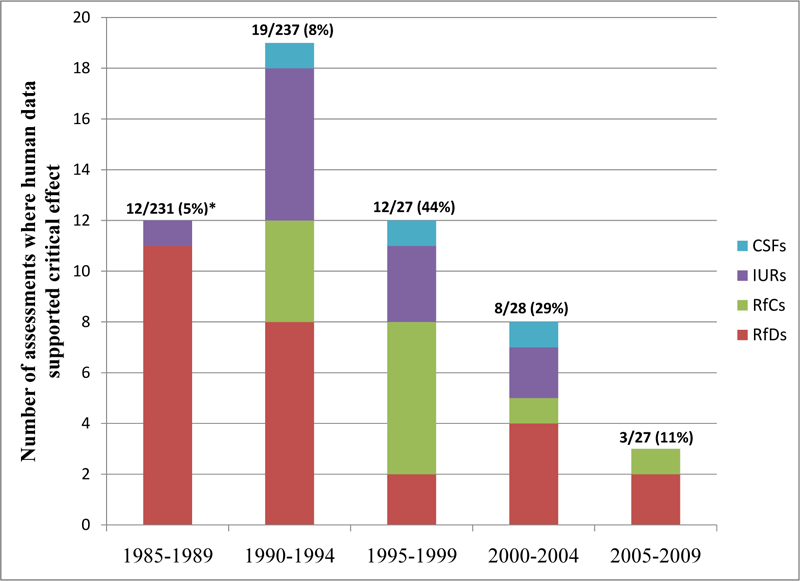
IRIS toxicity assessments (or updates) using human data for toxicity value derivation, 5-year increments.
*Displayed numbers represent number of assessments/updates where human data supported critical effect during 5-year increment, the total number of assessments/updates completed during 5-year increment, and in parentheses, the percentage of assessments/updates where human data supported the critical effect.
METHODOLOGY/CASE STUDY APPROACH
To illustrate how epidemiologic data could be better integrated and utilized in risk assessment, we discuss two case studies of chemical assessments from the IRIS database. The IRIS Toxicological Review of Di-butyl Phthalate (External Review Draft) [57] was selected as an example of a case where toxicological data were used in derivation of toxicity values despite the availability of epidemiologic data. The IRIS Toxicological Review of Methylmercury [58] will be used as the second case study to demonstrate alternate approaches for incorporation of epidemiologic data to facilitate toxicity value derivation. Within case studies, opportunities for incorporation of newer perspectives and the aforementioned shifts and advances in environmental public health science will be discussed. Following the case studies, recommendations will be provided for epidemiologists and risk assessors in pursuit of a more seamless translation of human data into guidance for decisionmakers.
CASE STUDIES – DI-BUTYL PHTHALATE
Background on Di-Butyl Phthalate
Di-butyl phthalate (DBP) is one of a number of phthalate esters used in the manufacture of consumer products such as cosmetics, drugs, personal care products and building materials. Phthalates are used as solvents, plasticizers, and emollients in these products. As constituents of consumer products and our physical environments, population exposure is common, as illustrated in recent biomonitoring surveys and research that demonstrates exposure to multiple phthalates in the US population [27, 59].
IRIS History and Health Effects of Concern
An oral RfD for DBP was first available in 1987, with the critical effect established as increased mortality based on a one-year feeding study of rats [60]. A contractor for EPA reviewed and updated the assessment in 1990 (adding references and minor text edits), but no changes were made to the RfD [57, 61]. In 2006, a revised draft toxicological review (TR) [57] was released and underwent external peer review. The 2006 Draft TR has not been finalized, and the IRISTrack system indicates that DBP is currently under re-assessment [62]. The next milestone listed is “Draft Development” – the first step of the IRIS review process - with an estimated due date of 4th quarter FY2010. Other phthalate esters are on the same timeline for draft development, suggesting a mixture approach is being considered, as recommended by the NRC Committee on the Health Effects of Phthalates [61]. It appears that EPA has moved quickly to undertake a new risk assessment that will incorporate multiple phthalates on the basis of population exposure to phthalate mixtures and common adverse outcomes observed in animals and humans. While the 2006 Draft TR is likely to differ substantially from the impending phthalates mixture assessment, it provides an opportunity to illustrate alternative approaches for using epidemiological data to develop risk-based toxicity values.
The noncancer health effects of primary concern identified in the TR (External Peer Review Draft) include reproductive performance, male reproductive development, histopathological changes in adult testis, and hepatic effects [57]. Male reproductive development was identified as the critical effect in the 2006 Draft TR.
The male reproductive malformations reported in toxicological evaluations of DBP include hypospadias; decreased anogenital distance (AGD); delayed preputial separation; agenesis of the prostate, epididymis, and vas deferens; degeneration of the seminiferous epithelium; nipple retention; gubernacular malformations and cryptorchidism [57]. The mechanism for many of the reproductive malformations has been identified as perturbation of androgen concentrations or androgen-receptor signaling [61]. Gubernacular malformations and cryptorchidism are thought be to related to reduced insulin-like growth factor 3 (insl3) and reduced testosterone [61].
The constellation of fetal and adult male reproductive effects of in utero phthalate (DBP and others) exposures in animals described above has been termed “phthalate syndrome” [61, 63]. The NRC [61] noted “excellent concordance” between the health effect data in animals and human “testicular dysgenesis syndrome” posited by Skakkebaek et al. [64]. Reproductive effects thought to have in utero origins in humans (e.g., infertility, decreased sperm count, cryptochordism, reproductive tract malformations, hypospadias, and testicular tumors) have been shown in rats exposed to phthalates in utero. Androgen insufficiency or disruption of androgen action underlies or contributes to all of these effects [61, 64]. The biological basis for the parallels between phthalate syndrome in animals and the hypothesized testicular dysgenesis syndrome in humans lies in our understanding of the specific requirement for androgen in normal male reproductive development in all mammals [61].
Overview of the 2006 Draft TR and RfD Derivation
Highlights of the DBP 2006 Draft TR are summarized in Table 2. Notable are the presence of both epidemiological and toxicological data, the large size of the toxicological database including multiple test species and multiple health effects; a good understanding of toxicokinetics in animals and some data for humans; and evidence in animals indicate that monobutyl phthalate is the toxic metabolite.
Table 2.
Key Features of the 2006 DBP Toxicological Review (External Peer Review Draft)
| Features | Summary |
|---|---|
| Epidemiology Data | Six observational studies reviewed Phthalate metabolites in ejaculate, urine or breast milk were associated with semen and sperm quality in adults, anogenital distance in male infants, and levels of reproductive hormones in infants |
| Toxicology Data | 40 studies covering acute, sub-chronic and chronic exposure durations and multiple test species (rat, mouse, rabbit) Outcomes observed in categories of reproductive performance, male reproductive development, histopathological changes in adult testis and liver |
| Toxicokinetics | Toxicokinetics are well understood in animals and several models exist; MBP identified as the proposed toxic metabolite |
| Sensitive Populations/Life Stages | Defined as developing (male) fetus |
| RfD Derivation | Animal study (Lehmann et al. 2004) showing decreased testosterone in fetal testis with maternal exposure to DBP 30 mg/kg-d defined as NOAEL UFs at 100, no database deficiency UF RfD proposed at 0.3 mg/kg-d |
Abbreviations: DBP, di-butyl phthalate; MBP, monobutyl phthalate; mg/kg-d, milligram per kilogram per day; NOAEL, no-observed adverse effect level; RfD, reference dose; UF, uncertainty factor.
Critical evaluation of available data and scientific judgments on data quality and usability are inherent in the risk assessment process. Selecting the Lehmann et al. [65] rat study as basis for the point-of-departure was a significant scientific judgment made in the 2006 Draft TR.
The Lehmann et al. 2004 study was designed to investigate upstream or early biological effects, changes in gene and protein expression, and fetal testicular testosterone levels. Dams were exposed to DBP doses of ranging from 0.1, to 500 mg/kg-d in corn oil (by gavage) on gestational days 12–19. There were seven control animals and five animals in each dose group. Dose-dependent reductions in gene and protein expression and reduced testosterone in fetal testis were observed at dose levels below those shown to affect the developing reproductive tract. Lehmann et al. concluded that altered gene and protein expression and testosterone synthesis may be sensitive indicators of the testicular response to DBP [65].
In justifying the choice of an animal study, several limitations of the epidemiological literature were identified. Despite this, epidemiological data were cited as evidence supporting the main outcomes of concern (male reproductive development and function) but were not quantitatively applied in RfD derivation.
Concerns Related to the 2006 Draft TR
The RfD Derivation
One of the controversial decisions in the Draft TR was the definition of the no-observed-adverse-effect level (NOAEL) at the 30 mg/kg-d dose in Lehmann et al. 2004. This issue was raised by the National Resources Defense Council (NRDC) in a presentation to the National Academy Committee on Health Risks of Phthalates in December 2007 [66]. The Draft TR authors cited difficulty in determination of the magnitude of hormone reduction that can be considered adverse (and were unsure of the biological significance of 26% hormone decrease at 30 mg/kg-d), and elected to rely on the statistical differences observed in hormone levels at the 50 mg/kg-d dose as the lowest-observed-adverse-effect level (LOAEL). The lack of statistical significance observed in the 30 mg/kg-d dose in the study was likely due to the small sample sizes employed in Lehmann et al., and a larger sample size would likely have yielded a significant difference. Accordingly, NRDC presented a position that the 26% decrease in fetal testicular testosterone at 30 mg/kg-d should instead be considered biologically significant and defined as a LOAEL. In this case, an additional LOAEL to NOAEL conversion factor would have been applied in RfD derivation, reducing the RfD by a factor of 10 (Table 3).
Table 3.
Alternative RfD Calculations Using Toxicological and Epidemiological Studies
| Dataset or Source(s) | Potential RfDs (mg/kg-day) |
|---|---|
| Draft Toxicological Review (Lehmann et al. 2004) | 0.3 |
| Alt 1) Applying a Database Deficiency adjustment of 3 | 0.1 |
| Alt 2) Applying LOAEL to NOAEL adjustment (additional factor of 10) | 0.03 |
| Alt 3) Combining 1 and 2 | 0.01 |
| Swan et al. 2005 with Marsee et al. 2006 | 0.001 |
| Using UF of 1 for human variability | |
| Swan et al. 2005 with Marsee et al. 2006 | 0.00001 |
| Using UF of 110* for human variability |
Upper end of range of human susceptibility from various sources as reported in Science and Decisions: Advancing Risk Assessment (NRC 2009).
Epidemiological Data
Exposure Issues
The provision of information on phthalate mixture exposures is cited by the Draft TR as an impediment to use of those data for risk assessment. It can be argued that the epidemiologic data are more relevant for policy and preferred over the animal studies for the same reason.
The Draft TR reviewed four studies that found relationships between MBP (in urine or breast milk) and reduced sperm quality in adult men and AGI and reproductive hormones in infant boys (all of these studies examined and report exposures to multiple phthalate metabolites). MBP levels were reported differently in these studies. Duty et al. (2003a and 2004) reported selected urinary percentiles including 25th%iles at 9.6 and 10.1 ng/ml and maximums at 488 ng/ml and 3169.9 ng/ml (not adjusted for specific gravity). The Main et al. (2005) study of reproductive hormones reported breast milk MBP minimums and maximums of 0.6 to 10,900 µg/L in the Denmark cohort and 2.4 to 123 µg/L in the Finnish cohort. Swan et al. (2005) provides observations similar to those found in the animal studies and reported 25th%ile urinary MBP at 7.4 ng/ml and a maximum of 337 ng/ml [67].
Marsee et al. used available pharmacokinetic models to back-calculate DBP exposure from urinary MBP levels presented in Swan et al. [67, 68]. They calculated daily median and 95%ile DBP exposures of 1.00 µg/kg-d and 2.68 µg/kg-d, respectively. (The odds ratio for shorter than expected AGI at MBP levels above the 75th%ile was 10.2.) Using this information an epidemiology-based alternative RfD can be derived (Table 3). With no uncertainty factors (UFs) or adjustment for human variability applied, the Swan study data could result in an RfD of 0.001 mg-kg-d. Because effects on male infant development were observed in the Swan et al. study, it is likely that an additional UF adjustment for interindividual differences would be applied. As an example, considering various sources of human susceptibility as highlighted by the NRC [7] (up to 110 times more susceptible) an RfD of 0.00001 mg/kg-d could be calculated. If the Swan et al. study were used as the principal study for deriving an RfD, the likely value would fall within the range of values presented.
Alternatives 1 to 3 presented in Table 3 show RfD derivations (based on Lehmann et al. 2004) that could result from differing scientific judgments regarding the data available at the time of the Draft TR. These alternatives range from 0.1 mg/kg-d to 0.01 mg/kg-d. Using the Swan et al. and the Marsee et al. work, an epidemiology-based RfD can be derived that is one or more orders of magnitude less than what was proposed in the Draft TR or developed as examples here (ranging from 0.001 to 0.00001 mg/kg-d). The difference between the toxicology-based and epidemiologically-based RfDs could be because the epidemiologic data includes multiple exposures, which could lower the risk level, and/or due to increased biological sensitivity in humans compared to rats. For DBP, a risk assessment approach using epidemiologic data results in a substantially lower RfD than can be derived from animal data.
Study Design, Outcome Measures and Analyses
The Draft TR noted a particular concern about Swan et al. (2005), questioning the validity of AGI as an outcome measure for humans and raising doubt about the suitability of the study as a basis for risk assessment. At the time of the Draft TR there was only one other study of human males using this outcome [69], though human studies noting effects on sperm characteristics and postnatal reproductive hormone production added to the weight of evidence [70, 71]. The reliability of AGI was also questioned due to the challenge of controlling for differing age and growth at time of measurement.
Other limitations of the epidemiological studies were cited to justify the choice of the animal data, including the lack of causal data and methodological limitations; in particular, the Draft TR cited a lack of adjustment for possible confounders. Despite this claim, statistical modeling procedures in each of the recent studies did address selected confounders and known important covariates, including ethnicity, age (and gestational age), smoking status, and body mass index and other demographic factors [68, 70–74]. Citing the lack of causal data as justification for the animal data raises questions regarding evidence standards for regulatory risk assessment. What standard of evidence is appropriate? What are EPA’s data evaluation standards? The standards of evidence for policy making and specific examples of EPA guidance on data sufficiency are explored further below.
Standards of Data Sufficiency for Risk Assessment
EPA designs risk assessments to avoid underestimation and gross overestimation of risk and to ensure protection of public health and environment [75]. This approach to evaluation of scientific information differs from the higher bar set for standards of scientific proof.
Establishing proof within a scientific process has a very high standard of evidence to avoid false positives [6, 76]. In this way, scientific knowledge grows by an accumulation of the strongest evidence. In contrast, for risk assessments used in the regulatory policy arena the standard of evidence shifts. Regulatory processes are designed to establish health protective policies and a wide range of scientific data are evaluated in an effort to avoid false negatives [77]. It also allows for decision-making even when uncertainties remain in the science. As science is an evolving process, complete certainty is elusive, and requirements for certainty are not compatible with decision-making needs, which are often on a shorter time scale. Thus, having a likely concern is sufficient for taking preventive action in some areas (e.g., cancer).
Data Sufficiency: The Case of DBP
The health effects stemming from DBP exposure can be categorized as reproductive and developmental, adding complexity to the selection of critical endpoint; in addition, the observed effects are linked by the overarching biological framework of phthalate syndrome/testicular dysgenesis. Further complicating matters are EPA’s guidance documents for risk assessments for developmental and reproductive toxins, which differ regarding evaluation of epidemiological data to establish “sufficient human evidence” in support of quantitative risk assessment.
The guidelines for risk assessment of reproductive toxins [78] have very stringent standards for “sufficient human evidence” (emphasis added):
“This category includes agents for which there is convincing evidence from epidemiologic studies (e.g., case control and cohort) to judge whether exposure is causally related to reproductive toxicity. A case series in conjunction with other supporting evidence also may be judged as Sufficient Evidence. An evaluation of epidemiologic and clinical case studies should discuss whether the observed effects can be considered biologically plausible in relation to chemical exposure. (EPA 1996, page 72)”
In comparison, the guidelines for risk assessment of developmental toxins [79] have a less-stringent data sufficiency standard for epidemiological data (emphasis added):
“Sufficient Human Evidence: This category includes data from epidemiologic studies (e.g., case control and cohort) that provide convincing evidence for the scientific community to judge that a causal relationship is or is not supported. A case series in conjunction with strong supporting evidence may also be used. Supporting animal data may or may not be available. (EPA 1991, page 40)”
The EPA 2005 Guidelines for Carcinogen Risk Assessment present an additional approach to data evaluation and applications of epidemiological data in risk assessment [80]. The cancer guidelines adopt data sufficiency standards for each descriptor used to characterize cancer hazards. The guidelines anticipate that combinations of animal, human and in vitro data and studies needed to support any particular descriptor may vary and can be used in concert to support the different weight of evidence descriptors. For example, the descriptor “carcinogenic to humans” can be based on convincing epidemiological evidence of a causal association, or in exceptional cases, for cases where a less robust epidemiological dataset is corroborated other lines of evidence from nonhuman studies. For the descriptor “likely to be carcinogenic in humans”, supporting evidence can include a wide range of study types and data, including plausible associations between human exposure and cancer; definitive causal data in humans is not required. “Likely to be carcinogenic in humans” is the most common descriptor and quantitative risk analyses are conducted for these agents. For dose-response assessment of carcinogens, the cancer risk guidelines state a preference for epidemiological data. Common limitations of epidemiological data are acknowledged as part of this process, and strategies for adjusting the data to derive epidemiologically-based CSFs or unit risks are provided.
Applying the Data Sufficiency Guidance to DBP
At the time of the Draft TR, an argument for using epidemiological data for the DBP RfD derivation could have been built upon the guidelines for risk assessment for developmental toxics [79]. Two studies found developmental effects of reduced AGD and hormone changes [68, 71] and two others in adults found male reproductive effects [72, 73]. While the adult studies are technically considered reproductive rather than developmental, they still lend support to the developmental findings, as DBP is thought to affect a continuum of male reproductive health endpoints by interfering with androgens and other points along that pathway. This dataset includes case-control, cohort and cross-sectional study designs, strengthening the weight of this evidence. Given this diversity of studies, the epidemiologic dataset could stand alone as “convincing” evidence. Further, the toxicological evidence for DBP’s antiandrogenic activity and effects on AGD is also strong [81, 82]. Considering the epidemiological findings and their consistency with results from animal studies and the hypothesized toxic mechanism, the available data at the time of the Draft TR provided convincing evidence in favor of a causal relationship.
Under the guidelines for risk assessment of reproductive toxins, the research of Duty et al. [72–74] and Main et al. [71] would have been the foundations of the case for using the epidemiological data in derivation of the DBP RfD. In the work of Duty et al. there was one statistically significant study [73] and one study suggestive of a relationship between urinary MBP levels and reduced sperm motility [72]. Main et al. found statistically significant associations between MBP in breast milk and higher levels of sex-hormone binding globulin (p=0.01) and lower levels of free testosterone (p=0.03) in 3-month old infant boys [71]. The strengths of the epidemiological dataset regarding reproductive effects were the statistically-significant findings in multiple studies of varied design showing effects on adult sperm parameters and infant hormone levels. The reproductive toxicity findings in humans were consistent with the toxicological findings and the proposed MOA. In sum, the totality of the epidemiological data in conjunction with the animal evidence provides a strong case that the human evidence was sufficient for the quantitative risk assessment of DBP.
The cancer guidelines present a more graded and contemporary discussion of data sufficiency that incorporates a pragmatic approach to data evaluation. They maximize the utility of epidemiological data with adjustments that account for common limitations, and as in previous guidance, advocate the consideration of the whole of the available literature (including human, animal and in vitro evidence). An argument for deriving the RfD on the basis of epidemiological data on male development or reproductive effects could be developed with the frameworks laid out in the cancer guidelines.
Epidemiological Data then and Now
Table 4 summarizes the epidemiological studies reviewed in the Draft TR as well as several important studies released after the publication of the Draft TR that have strengthened the evidence base for male reproductive effects in infants and adults [70, 83, 84]. These include further findings of an association between MBP and reduced sperm count and motility from continued evaluations of patients from male fertility clinics [72–74]; further expansion of the findings of the association with AGD from the Study for Future Families cohort [68], and a new finding of an association with behavioral differences in boys. In addition to further follow-up and new statistical analyses to address the reliability of the AGD measure, Swan et al. [83] reviewed the evidence on AGD in humans to address concerns about the validity of the outcome.
Table 4.
Epidemiological Studies of DBP and Other Phthalates
| References | Discussed in Draft TR | Type of Study (Number of Participants) | Exposure Measures | Outcomes and Findings |
|---|---|---|---|---|
| Murature et al. 1987 | √ | Cross sectional College student volunteers (n=21) |
DBP concentration in cellular fraction of ejaculate | Strong negative correlation (r = −0.6 and −0.7) between sperm density and DBP concentration |
| Duty et al. 2003a | √ | Cross-sectional Men at fertility clinic (n=168) |
Urinary levels of several phthalate metabolites. | Dose response relation observed between increased urinary MBP and poor sperm motility (statistically significant trend) and sperm concentration. Similar findings for MBzP. |
| Duty et al. 2003b | √ | Cross-sectional Men at fertility clinic (n=168) |
Urinary levels of several phthalate metabolites | MBP was not found to be associated with sperm DNA integrity (using neutral comet assay). |
| Duty et al. 2004 | √ | Cross-sectional Men at fertility clinic (n=220) |
Urinary levels of several phthalate metabolites. | Suggestive negative dose-response relationships between MBzP, MBP, and MEHP with VCL and VSL; MEP negatively associated with LIN but positively associated with VCL and VSL. |
| Swan et al. 2005 | √ | Pregnancy cohort follow-up (n= 85, males only) | Phthalate metabolites in prenatal urine samples | Statistically significant increased odds of reduced AGI (AGD normalized by body weight) in male infants with increasing levels of maternal urinary MBP and several other phthalate metabolites (association with MBP levels strongest and of greatest magnitude) |
| Main et al. 2006 | √ | Case-control (n=130; 62 cases, 68 controls) | Phthalate metabolites in breast milk (1 −3 months postnatal) | No associations found with cryptochidism. MBP was negatively correlated with free testosterone. MBP and other metabolites were positively correlated with SHBG and LH: free testosterone ratio. MiNP was associated with LH. |
| Hauser et al. 2006 | Cross-sectional Men at fertility clinic (n=463) |
Urinary concentrations of several DBP and DEHP metabolites | Statistically significant dose response gradient observed for MBP and low sperm concentration and reduced motility | |
| Swan et al. 2008 | Reanalysis/expansion of 2005 study (n=106) Outcomes: AGD (adjusted for age and weight percentile), penile width and testicular descent |
Assessed individual and joint exposure to phthalate metabolites in prenatal urine samples | Higher metabolite concentrations (individual metabolites and combined exposures) associated with shorter AGD; higher metabolite concentrations associated with increased probability of incomplete testicular descent; MEHP and sum of DEHP metabolites associated with penile width | |
| Swan et al. 2009 | Pregnancy cohort follow-up Subset of boys (n=74) and girls (n=71) |
Phthalate metabolites in prenatal urine samples | Levels of prenatal MBP and MiBP associated with reduced masculine play in boys; no associations observed for girls |
Abbreviations: AGD, anogenital distance; AGI, anogenital index; DBP, dibutyl phthalate; DNA, deoxyribonucleic acid; LH, lutenizing hormone; LIN, sperm linearity; MBP, mono-butyl phthalate; MBzP, mono-benzyl phthalate; MEP, mono-ethyl phthalate; MiNP, mono-isononyl phthalate; MiBP, mono-isobutyl phthalate; PK, pharmacokinetic; SHBG, sex-hormone binding globulin; VCL, sperm curvilinear velocity; VSL, sperm straight-line velocity.
Alternative Approaches Using the Epidemiological Data
As demonstrated by this case example, development of the RfD using epidemiologic data could take several forms, including: the RfD based on the Lehmann et al. could have been adjusted to reflect the differences in levels of exposure between Lehmann et al. and in the Swan et al. and Hauser et al. papers (with back-calculation of DBP exposure as demonstrated by Marsee et al); Swan et al. or Hauser et al. could have served as the critical study for identifying the POD and deriving the RfD (again with back-calculation of DBP exposure); or use of a statistical approach to develop the POD combining information from multiple human studies integrating different populations and endpoints. Basing the POD on the human data is the preferred approach as it eliminates the need for animal to human extrapolation and ensures that the assessment reflects realistic population settings and co-exposures to other phthalates. An assessment using epidemiologic data would rely on the same foundations (strong toxicological dataset with described mechanisms of toxicity from animal studies, which are plausible in humans) as laid out in the Draft TR.
Considering the syndromic morbidity associated with DBP exposure, a third approach that allows combining data from multiple studies could be explored. With the addition of Swan et al. 2009 [84], the epidemiologic dataset includes four different outcomes, alternations in adult male sperm parameters related to the men’s urinary MBP levels, AGD in infant boys and less-masculine play behaviors in young boys related to prenatal DBP exposure as assessed by MBP in maternal urine samples, and postnatal reproductive hormone differences in boys associated with MBP in breast milk. It may be premature to pursue a data combination approach based on the existing DBP epidemiology; however, as the dataset grows, opportunities to use statistical methods to integrate findings from multiple studies may arise. Several methodologies are available including Bayesian approaches as described by Sutton and Abrams [85] or a composite score method as described by Coffey et al. [86] and illustrated by the NRC Committee on the Health Risks of Phthalates [61]. As with any new method, the utility and technical feasibility of a data combination approach would need to be evaluated carefully. Some approaches to data combination (e.g., Bayesian) will foster advances in dose-response modeling such as those recommended by NRC (2009) in the unified approach to dose-response.
Epidemiological Data Have Advantages But Many Challenges Persist
Use of epidemiologic data will eliminate the significant uncertainty stemming from interspecies extrapolation but will not eliminate numerous other uncertainties or obviate scientific judgments from risk assessment. For example, one of the challenges is characterizing the population-level implications of changes in continuous biological parameters, such as testosterone. Recent workshops have identified any shift in such parameters as adverse, but further work is needed to translate this into quantitative terms. Evaluation of certainty regarding important sources of human variability would need to be conducted to ensure accurate, science-based characterization of dose-response relationships that account for uniquely-susceptible subpopulations. Uncertainties related to exposure, dose, and dose-response modeling must also be addressed.
Di-Butyl Phthalate Case Study Conclusions
The DBP example illustrates challenges and complexities encountered in cumulative risk analyses, including evaluation and incorporation of realistic population exposures to multiple chemicals, analyses of multiple endpoints and evaluation of multiple populations of concern. The use of DBP epidemiological data would have better addressed these challenges by improving accuracy in determination of the dose-response relationship, thus providing a stronger basis for risk assessment.
CASE STUDY - METHYLMERCURY
An extensive toxicological and epidemiological literature base for methylmercury (MeHg) enabled the EPA in 2001 to establish an RfD incorporating a number of risk assessment best practices [58]. Methylmercury is among the few chemicals for which the RfD is derived from studies of general population exposure, obviating the need for high- to low-dose extrapolations from occupational studies or cross-species extrapolations from animal data. Biomarkers of internal dose were employed to characterize MeHg exposure, likely reducing misclassification bias. The MeHg RfD was derived based on the benchmark dose (BMD) method, which is superior to the NOAEL/LOAEL approach as it uses all dose-response information [87]. The case study also illustrates the potential for the MeHg risk assessment to continue to evolve towards the unified dose-response and risk-specific dose framework set out in the new Science and Decisions risk assessment paradigm [7] and to incorporate additional, potentially more sensitive endpoints such as cardiovascular disease (CVD). It also highlights the fact that, despite this potential, no re-assessment of MeHg is currently planned by EPA [62].
Background
Mercury (Hg) is a ubiquitous heavy metal contaminating the global seafood supply. Elemental mercury vapor is released into the atmosphere by anthropogenic and natural events, deposited onto waterways, and converted to MeHg by aquatic microorganisms. Methylmercury bioaccumulates efficiently in fish tissue and biomagnifies up the food chain. About one-third of inorganic mercury (iHg) emissions is anthropogenically-generated, in large part through combustion of fossil fuels and industrial and waste management processes; another third is re-emission of human and natural emissions; and the remaining one-third is from natural processes such as volcanic eruptions; [88]. Elemental mercury vapor has a long half-life (up to a year) in the atmosphere, and therefore travels with meteorological systems and deposits globally [89]. The dominant pathway of seafood [90]. Mercury has been found in the tissues of all human populations, including those in the remotest parts of the world [91]. Detectable levels of Hg are found in the blood of over 90% of reproductive age women in the US [92].
IRIS History and Health Effect of Concern
Mercury affects multiple organ systems, though its effects on the central nervous system (CNS) are best studied. While Hg’s neurotoxicity has been known for centuries, major accidental poisoning events in Minamata, Japan in the 1950s (as a result of industrial-effluent contamination of seafood) and in Iraq in the 1970s (due to fungicidal contamination of wheat used in bread) demonstrated MeHg’s differential neurotoxic effects in adults and children. Minamata was the first demonstration of infants with brain damage -- ranging from subtle development delays to mental retardation and cerebral palsy -- born to mothers who themselves suffered few or no adverse effects. Similar results were seen in Iraq, where dose-response relationships suggested delayed walking could occur as at concentrations as low as 10 ppm in maternal hair [93]. This suggested a much greater sensitivity of the developing fetus to MeHg’s neurotoxic effects.
The mechansim for MeHg neurotoxicity is complex and incompletely understood. MeHg crosses the blood-brain and placental barriers, providing one key explanation for enhanced fetal vulnerability. Autopsy studies have found that while adult exposures result in limited lesions in the brain, prenatal exposures produce widespread changes in the brain’s cytoarchitecture [93]. Several main MOAs have been suggested for CNS damage: (i) disrupted protein synthesis; (ii) cell structure damage; (iii) induction of lipid peroxidation and generation of reactive oxygen species (ROS); (iv) and interference with calcium homeostasis [93, 94].
The EPA’s chronic oral RfD for MeHg of 0.1 ug/kg/d was set in 1995 based on the poisoning episode in Iraq [58, 95]. The NAS, at the request of Congress, reviewed substantial then-available experimental and epidemiological evidence, concluded that “neurodevelopmental deficits are the most sensitive, well-documented effects”, and provided a recommendation for an RfD [90].
The epidemiological data included three large population-based cohort studies focusing on developmental neurotoxicity resulting from in utero exposure to MeHg-contaminated seafood in high fish-consuming communities in the Seychelles [96, 97], the Faroes Islands [98], and New Zealand [90]. Statistically-significant associations of mercury exposure biomarkers and a range of subtle neurological effects in children born to MeHg-exposed mothers were found in the Faroes and New Zealand studies, while no such clear relationships were found in the Seychelles study. The NRC committee chose the Faroes as the principal study due to its strengths. The lowest benchmark dose (BMD) considered reliable was for the Boston Naming Test from which it derived a lower confidence limit (BMDL05) of 58 ppb in cord blood, equivalent to about 12 ppm in hair (Table 5). The benchmark dose is the dose corresponding to a pre-selected change (often 5 or 10%) in response [99]. The committee identified biological variability in estimating dose and database insufficiency as the two major areas of uncertainty, and recommended an UF of 2 to 3 for variability and an additional factor “given the data indicating possible long-term neurological effects not evident at childhood, immunotoxicity, and cardiovascular effects,” for a total of “at least 10” [90].
Table 5.
Key Features of NRC Recommendation and EPA RfD
| Scientific Bases for Risk Assessment | ||
|---|---|---|
| NRC Recommendation and EPA RfD | ||
| Toxicological studies | 29 toxicological studies covering acute, sub-chronic and chronic exposures and in young animals of 3 species (monkey, rat and mouse); no NOAEL detected | |
| Epidemiological studies | 16 published reports on 9 observational studies of developmental neurotoxicity in infants and children in 9 countries; studies in the Faroes Islands, Seychelles and New Zealand considered strongest basis for quantitative risk assessment because they were large, well-conducted and in the dose-range of exposed US populations | |
| Toxicokinetics | One-compartment (IPCS 1990; EPA 1997) and PBPK (Clewell et al. 1999) models | |
| Sensitive population/lifestage | Developing fetus to in utero exposure (reproductive age women) | |
| Principal study | Faroes 7-year old cohort (Grandjean et al. 1997) | |
| RfD Derivation | ||
| NRC Recommendation | EPA RfD | |
| Critical health effect | Developmental neurotoxicity - score on Boston Naming Test | Developmental neurotoxicity -scores on several neuropsychological test scores |
| BDML05 | 58 ppb blood | 46–79 ppb blood |
| Uncertainty factor (biological variability and database insufficiency) and rationale | Factor of 2–3 for intra-species variability, another factor for database uncertainty with a total of “at least 10” | 3-fold for toxicokinetic variability (estimating ingested MeHg dose from cord blood) and 3-fold for a total of 10 |
| RfD | 0.1 ug/kg/d | 0.1 ug/kg/d |
Sources: NRC 2000; EPA 2001.
Overview of the RfD
Broadly following the NRC’s recommendation, the EPA’s RfD was based on the Faroes study. While the RfD itself remained the same, the basis was adjusted to reflect newer epidemiological evidence in a chronic exposure setting more comparable with real life exposures in the general population. The EPA chose BMDLs of 46–79 ppb in maternal blood for several neurological test scores in the Faroes cohort, supported by an integrative analysis of the three studies [90], rather than relying on one test score [58]. An UF of ten was applied (three for toxicokinetic variability and three for toxicodynamic variability) to account for the inability to quantify long-term sequelae, the lack of a two-generation reproductive-effects assay, and issues related to selection of critical effect -- specifically, concern about the possibility of MeHg effects lower than the chosen BMDL [58]. Table 5 shows a comparison of the NRC recommendations and the EPA RfD.
Evidence of effects on the cardiovascular system (at lower doses than those associated with neurological effects), delayed and lifespan neurotoxic effects, and immune and reproductive system effects was available at the time the RfD was established. However, as the NRC Committee concluded “on the basis of the body of evidence from human and animal studies … neurodevelopmental deficits are the most sensitive, well-documented effects and currently the most appropriate for derivation of the RfD.” Lack of completeness in the epidemiological knowledge base was explicitly built into UFs.
Moving Toward Unified Dose-Response Assessment
Bright line approaches to characterizing noncancer hazards are problematic (as noted above), since a ‘safe/unsafe’ dichotomy does not speak to the probability or severity of health effects that occur at various points in the dose-response relationship [54]. Using MeHg to illustrate, Clewell and Crump note that the difference in distributions of mean neurological test scores modeled in the NRC risk assessment between children of mothers who were exposed at the RfD compared with unexposed was a 0.25%, a change the authors describe as “barely discernable”. To characterize risk of developmental neurotoxicity at higher-end exposures, biomonitoring results from the NHANES 1999–2000 cross-sectional survey were employed, choosing the 99.8th percentile (second highest observed value) blood mercury level. Based on the modeled mean test score difference they found a decrease in mean test score of 1.6% in children born to mothers exposed at the 99.8th percentile compared with those born to unexposed mothers. Based on NHANES sampling, they report an estimated 8,000 births at risk annually (Table 6).
Table 6.
Dose-Response Relationships from Epidemiology Used to Improve Risk Estimates
| Number of Annual Births at Risk | Clewell & Crump (2005) | Axelrad et al. (2007) | |
|---|---|---|---|
| Predicted Difference, Mean Test Score | Predicted Difference, Mean IQ | ||
| Maternal Hg, unexposed vs exposed at RfD | 60,000 (NRC 2000) | 0.25 % | 0.13 points |
| Maternal Hg, unexposed vs exposed at 99.8th percentile | 8,000 (Clewell & Crump 2005) | 1.6% | 0.82 points |
Axelrad et al. estimated a dose-response relationship by standardizing neurological test scores to the IQ scale and using a Bayesian model to perform an integrative re-assessment of the data from the three key MeHg epidemiologic studies [100]. For exposed children, they estimated a loss of 0.18 IQ point for each part per million (ppm) of mercury in maternal hair. Adapting the Clewell and Crump approach using the Axelrad et al. dose-response relationship suggests risk of loss of 0.82 IQ point at the 99.8th percentile of maternal exposure for the 8,000 annual births (Table 6). With a quantified dose-response relationship and distribution of internal dose for a representative sample of the population, similar estimates are possible for any percentile of the exposure distribution. In improving the precision of estimated severity and frequency of risk, this type of characterization is likely to be more informative for risk management and communication, as well as benefit-cost assessment [54]. Stating that 8,000 children born annually to mothers at the highest percentile of MeHg exposure dose may be at risk of an approximately 2% lower performance on a standardized neurodevelopmental test, or a nearly 1-point loss in IQ, is likely to be more informative for decision-makers than stating that 60,000 births annually to mothers with intake over the RfD are at some unspecified level of risk [90]. These examples from recent publications offer insight into methodological refinements and are presented for illustration. The examples presented address only two of the many neurological effects of MeHg; the effects estimates above do not capture (and therefore underestimate) the full impact of MeHg exposure.
Building on such approaches, the NRC set out three conceptual models for calculation of a risk-specific dose, based on individual and population dose-response relationships [7]. Examination of MOA, background exposure and variability would guide choice of a dose-response conceptual model. Epidemiological research will provide much of the basis for these assessments. Given observed individual low-dose linearity [90] and sufficient background exposure and variability to presume low-dose population linearity [7], MeHg is likely to fall into the category of the third conceptual model which extrapolates from the human point of departure linearly to low dose and then estimates population and individual risk and uncertainty based on inter-individual variability distributions [7].
To this end, research in the last ten years has contributed to reduced uncertainty in the relationship between mercury measured in newborn cord blood to that in maternal blood. In determination of the RfD, maternal intake doses were modeled from measured MeHg in fetal cord blood. While aware of data reflecting cord to maternal blood ratios substantially above one [101–103], EPA chose to use a ratio of one for this purpose representing the central tendency of this ratio. Interindividual variability of this ratio was included as a component of the toxicokinetic UF [58]. A meta-analysis of ten published studies found a ratio of 1.7 for cord to maternal blood mercury [104]; some researchers have interpreted this finding to mean the estimated internal dose equivalent of the RfD (5.8 ug/L in blood) should be adjusted downward by this ratio, and have reported blood levels of mercury using 3.5 ug/L as a reference value [105, 106].
In a subsequent probabilistic assessment incorporating the 1.7 cord-to-maternal blood ratio and reconsidering key parameters of the NRC MeHg model, Stern derived a distribution of the intake needed to reach 58 ppb MeHg in hair, NRC’s POD [107]. For example, an estimated intake of 0.2 ug/kg/day would be sufficient to reach this level at the 1st (99th) percentile. The author argues that reducing uncertainty in cord-to-maternal blood and other model parameters substantially reduces the need for toxicokinetic UFs, although factors for database sufficiency and toxicodynamic variability would still be applied [107]. To illustrate, a factor 3-fold for toxicodynamic variability applied to the 1st (99th) percentile suggests an intake of 0.067 ug/kg/d, or two-thirds of the current RfD. This analysis moves in the direction of a risk-specific dose by reducing uncertainty in an important measure of inter-individual variability and evaluating risk at a specific percentile of MeHg intake.
Susceptibility factors are “broadly considered to include any factor that increases (or decreases) the response of an individual to a dose relative to a typical individual in the population” [7]. The NRC concluded that variability in human susceptibility had not been adequately considered in EPA risk assessments and recommended that more explicit evaluation of population variability should be incorporated in both exposure and dose-response assessment. The RfD for MeHg is unusual in that it targets a specific group that is susceptible due to gender and lifestage; however, the population of reproductive-age women is large and heterogeneous, and substantial regional differences have been documented. While based on NHANES 1999–2004, on average 4.7% of reproductive age women were over the RfD, only 1.2% were over the level in the Midwest compared with 9.0% in the Northeast [106]. Similarly, 8.1% of the coastal population was over this limit compared with 2.1% of non-coastal. A cross-sectional survey in New York City found 25% of women over the RfD equivalent, and nearly two-thirds of those of Asian origin over that level [108]. Blood mercury levels are strongly associated with seafood intake in these studies [106]. Even among NHANES seafood consumers in the same monthly frequency category substantial variability in internal dose exists, ranging from 4 to 8-fold [92].
Not all individuals in the population will respond the same way to a given exposure dose; genetics, lifestyle, co-exposures and other factors are likely to also play a role in chemical metabolism and elimination. In the case of MeHg, Castoldi et al. have raised the importance of diet (essential fatty acids, selenium, vitamin E), gender, temporal patterns of exposure, and co-exposure with other toxicants in potentially modifying the effects of MeHg on neurotoxicity throughout the lifespan [109]. Essential fatty acids in seafood, in particular eicosapentaenoic and docosahexaenoic acids, are important for brain function [110]. Studies have suggested essential fatty acids and MeHg may interact antagonistically with respect to neurotoxicity. Caution in epidemiological studies is warranted because unadjusted confounding from a beneficial moderating factor may lead to an underestimate of adverse effects [111]. Similarly, the need for examining nutrients, chemicals (particularly persistent organic pollutants), and the social environment for possible modification of MeHg neurotoxicity has been identified [112]. A recent re-analysis of the 9 year follow-up of the Seychelles cohort -- a study that found no consistent adverse effect associated with seafood-related MeHg exposure -- concluded that motor function decreased significantly with MeHg exposure in children with a below-average home environment [113]. The NRC has identified that “the data gap represented by interhuman pharmacodynamic (PD) variability presents a critical research need that can be approached by mining the existing epidemiology literature and by designing new studies in which biomarkers of exposure and effect are used to describe variability in sensitivity to health outcomes in similarly exposed people.” Further development of such research will be important in determining risk-specific doses [7].
Multiple Endpoints and Data Sufficiency
Since the NRC mercury report, the scientific basis for MeHg’s adverse effects on multiple health endpoints has strengthened. In particular, substantial epidemiologic evidence of risk to the cardiovascular system at doses well below the BMD for developmental neurotoxicity has emerged.
At the time of the NRC review, while the committee observed that “the cardiovascular system appears to be a target for MeHg toxicity… at very low Hg exposures,” only two large epidemiological studies had been conducted (Table 7). One found an association between MeHg exposure and acute coronary events in Finnish seafood-consuming men [114] and the second an association between MeHg and blood pressure and heart rate variability in Faroes Island children [115]. Over the last ten years at least 11 additional reports have been published (Table 7), summarizing findings from 10 primarily observational studies of MeHg exposure from seafood consumption in general populations of mostly men covering 16 countries. These studies examine three sets of CVD-related outcomes: (i) acute coronary events (myocardial infarction, MI) and other heart disease events and risk markers, (ii) hypertension and (iii) heart rate variability. Most found positive associations between MeHg exposure and risk of CVD outcomes, particularly those examining adult male populations. The mean mercury level in the four published reports from the large Finnish KIHD cohort study (about 1.9 ppm in hair) is about six-fold lower than the BMD for developmental neurotoxicity for the EPA RfD. Thus, as early as 15 years ago, epidemiological evidence was available suggesting population health effects at levels substantially lower than the RfD. Given this, the issue of data sufficiency needed to spur action once again becomes critically important.
Table 7.
Epidemiological Studies of MeHg and Cardiovascular Risks
| Population | Study Type | Exposure Measure (Mean) | Outcome Measure and Findings | References |
|---|---|---|---|---|
| Finland, men aged 42–60, n=1,833 | KIHD prospective cohort (6 yr follow up) | Hair mercury (1.9 ug/g) | AMI and BP: RR 1.96 acute coronary events (MI, CHD/CVD death); Hg second strongest predictor of IMT (carotid atherosclerosis) after systolic BP | Salonen et al. 1995 |
| Faroes Islands children (birth and 7 yrs) n=1,000 | Prospective cohort study (7 yr follow up) | Cord blood mercury | BP and HRV: BP increase and HRV decrease (boys) with Hg level; prenatal Hg may affect CVD homeostasis | Sorensen et al. 1999 |
| Finland, men n=1,084 | KIHD prospective cohort, (4 yr follow up) | Hair mercury (1.8 ug/g) | BP: Hg associated with IMT (progression of carotid atherosclerosis) | Salonen et al. 2000 |
| Finland, men n=1,871 | KIHD prospective cohort (10 yr follow up) | Hair mercury (1.9 ug/g) | AMI: High hair Hg attenuated benefits of PUFAs on CVD risk | Rissanen et al. 2000 |
| Sweden, men and women, n=78 cases and 156 controls | Nested case control (1.7 yrs) | Erythrocyte mercury (4.4–5.4 ng/g) | AMI: Did not find association with first myocardial infarction | Hallgren et al. 2001 |
| Eight EU countries, Israel, men n=684 cases, 724 controls | EURAMIC case control | Toenail mercury (0.25 ug/g controls; 0.29 ug/g cases) | AMI: 2.16 OR of first myocardial infarction associated with highest compared to lowest quintile Hg | Guallar et al. 2002 |
| United States, men n=470 cases and 474 controls (63% dentists) | US Health Professionals Follow up nested case control study | Toenail Hg (0.72–0.74 ug/g; 0.45 ug/g non-dentists, 0.91 ug/g dentists) | AMI: No increased risks comparing cases to control when dentists included in controls; 1.27 RR when dentists excluded, 1.7 when adjusted for PUFAs | Yoshizawa et al. 2002 |
| Finland, men n=1,871 | KIHD cohort (13.9 yr follow up) | Hair mercury (1.9 ug/g) | AMI: Highest one-third (over 2.0 ppm hair Hg) ~1.6-fold risk lowest one-third acute coronary event | Virtanen et al. 2005 |
| United States, reproductive age (16–49) women n=1,245 | NHANES cross-sectional study | Blood mercury (1 ug/g), 30-day fish consumption (avg 2.9 meals) | CVD risk markers: 30-day fish negatively associated with C-reactive protein and positively with HDL cholesterol | Smith et al. 2008 |
| Faroes Islands whaling men aged 30–70 yrs) n=42 | Clinical study | Toenail and blood mercury (toenail 2 ug/g; hair 6 ug/g) | BP: Increased BP and IMT associated with mercury level; toenail Hg highly correlated with hair Hg | Choi et al. 2009 |
| Japan men and women n=54 (experiment and control) | Intervention study | Hair mercury (control 2.1 ug/g; experiment 2.3–8.8 ug/g) | HRV: Significantly (reversibly) altered after 14 wk exposure at UN reference rate; PTWI not CVD protective | Yaginuma-Sakurai et al. 2009 |
| Canada, Nunavik Inuit men and women n=732 | Qanuippitaa population cross sectional | Blood mercury (50.2 nmol/L) | BP: Hg associated with increasing systolic and diastolic BP | Valera et al. 2009 |
| Japan, Minamata (n=833), Goshonoura (n=1,450), Araike (n=755) men and women | Retrospective (1971 population based survey) | Residence area and hair mercury | BP: Prevalence of hypertension higher in Minamata than Araike, OR 1.4–1.6 for hypertension, increasing dose-response trend | Yorifuji et al. 2010 |
A 2005 review of the MeHg-CVD epidemiology found convincing evidence in support of an association between dietary MeHg exposure rates from fish and CVD endpoints in adult men, including MI [107]. This review further concluded that the Finnish KIDH cohort study provided the strongest basis for quantitative risk assessment of the cardiovascular effects of MeHg. Despite this, several concerns with the epidemiological and experimental bases for risk assessment were raised. First, while large and well-conducted case-control studies on men in the US [116], the EU and Israel [117] examining MI and other CVD risk parameters could provide the basis for risk assessment, these studies are based on a biomarker (toenail mercury) with a poorly studied relationship with MeHg intake. Second, mechanistic evidence for MeHg’s role in CVD remained weak, though oxidative stress was noted as a plausible mechanism. Finally, the evidence for hypertension was less conclusive than for CVD outcomes, and while heart rate variability was clearly a nervous system effect, its link to CVD risk was unclear [107].
Additional research conducted since the 2005 review has strengthened the case for cardiovascular endpoints in adult males. Over the last several years, associations between toenail mercury, hair and blood mercury and MeHg intake have been quantified [118], and toenail Hg has been significantly correlated with finfish and shellfish intake [119], facilitating analysis of the toenail-based studies in quantitative risk assessment. Recent experimental research has also strengthened the basis for MeHg’s pro-oxidant MOA on the cardiovascular system, increasing ROS and depressing nitric oxide availability [120]. Three additional studies have been published since the Stern review supporting an association between MeHg and measures of increased blood pressure [121–123]. The implications for cardiovascular risk of MeHg’s observed effects on heart rate variability, which seem reversible, remain unclear.
Numerous epidemiologic investigations have shown essential omega-3 fatty acids in seafood to be protective of cardiovascular function in both healthy and high-risk populations [110]. Mozaffarian and Rimm reviewed the published evidence for both beneficial and adverse effects of seafood, and concluded that modest seafood and/or fish oil consumption decreased the relative risk of mortality from coronary heart disease by 25% or more [124]. These authors also pooled five large studies examining association of MeHg with coronary heart disease and found results were inconclusive. However, it should be noted that the two studies in this pooled analysis which did not find associations included women. The authors conclude that the key public health question of interest may be whether intake of MeHg decreases the heart-protective effects of seafood. Seafood species vary in their essential fatty acid and MeHg content (e.g., most salmon is high in essential fatty acids and low in MeHg, while most swordfish is low in essential fatty acids and high MeHg). Several computer models have been developed that can help both policy-makers and seafood consumers evaluate the net risks and benefits of seafood consumption on a fish-species specific basis, considering omega-3 fatty acid and MeHg content [125–127].
In sum, the epidemiologic evidence base for MeHg’s CVD effects in men has strengthened substantially since the NRC report and establishment of the RfD. The epidemiologic base (Table 7) is beginning to approach the strength of the base for developmental neurotoxicity at the time of the NRC review in 2000 (Table 5). Mechanistic support has also strengthened, and measurement concerns related to biomarkers may be more readily addressed. The EPA defines as the basis for the critical effect for establishing RfDs “the first adverse effect, or its known precursor that occurs to the most sensitive species as the dose rate of an agent increases” [46]. The range of 2 ppm mercury in hair may be approximately the 90th percentile for men in the US [107], and is near the median for men in high seafood-consuming countries like Japan [128]. This suggests a large population worldwide may be at risk, or at least may not be fully reaping the heart-protective benefits of seafood. It is now worth considering whether the Finnish, EU and American studies of MI and other CVD outcome risks in men provide sufficient basis for quantitative risk assessment of the cardiovascular health effects of MeHg for adult males. Risk characterization should employ dose-response modeling and risk-based characterization.
Methylmercury Case Study Conclusions
The MeHg case study reiterates that the “acceptable” level of risk targeted for a chemical and health endpoint is a matter of environmental public health policy. Given the erroneous assumption that the RfD represents negligible lifetime risk (similar to a 1 in 1,000,000 risk for carcinogens), and the estimation that risk level for MeHg at the current RfD is 5 in 1,000 [52], outputs of NRC-recommended unified dose-response modeling approaches and use of risk-specific doses will require explicit policy decisions to determine acceptable levels of risk for non-cancer endpoints such as developmental neurotoxicity and cardiovascular effects from MeHg.
The use of RfDs as “bright lines” can be misleading for risk management. This case study demonstrates how the use of epidemiologic studies can be used to forego assumptions of thresholds in order to establish dose-response relationships and how cross-sectional data can inform probabilistic characterizations of population risk, contributing to this goal. A better understanding of inter-individual variability will be important in both confirming the modeling approach as well as estimating population risk under the new framework. With a strong epidemiologic base, ubiquitous exposures and irreversible effects, MeHg will be a key candidate for improved understanding of interindividual variability that leads into risk-specific dose characterizations.
The epidemiologic base has now evolved on cardiovascular disease endpoints in men, and suggests risk at doses well below the current point of departure for the developmental neurotoxicity. This demonstrates the importance of following up on early epidemiologic signals.
COMMON ISSUES AND LESSONS LEARNED FROM CASE STUDIES
In the DBP case study, we examined the rationale and various methods for using epidemiologic data in RfD derivation. In the case of MeHg, we assessed the developments in the epidemiologic and other scientific literature that show a way forward towards the unified dose-response assessment process described in the Science and Decisions report [7]. The case studies point to several overarching risk assessment issues: how to evaluate and incorporate multiple endpoints in risk assessment; the need for consistent guidelines for data sufficiency; and the implications of the unified dose-response and development of risk-specific doses for non-cancer endpoints. The case studies also present a common lesson about the incremental nature of epidemiologic research and its utility for risk assessment.
Multiple Endpoints
The existence of multiple health effects from a particular chemical exposure has been addressed in risk assessment by the selection of a critical effect. Critical effects are defined as “the first effect, or its known precursor, that occurs in the most sensitive species as the dose rate of an agent “increases” [46]. Risk management decisions based on critical effects are assumed to be protective of the other effects. In the case of DBP, syndromes -- and multiple effects within same syndrome -- affect different populations. If various effects are observed at roughly the same dose, the RfD (or RfC) set for the critical effect should be protective of all populations. MeHg illustrates another case, that of multiple endpoints and different susceptible populations. In this situation, additional analysis may be necessary to ensure that the original critical effect (i.e., developmental neurotoxicity) and subsequent risk management is adequately protective of populations susceptible to other effects observed (e.g., cardiovascular outcomes in adult males).
Weight of Evidence Evaluations, Data Sufficiency, and Data Combination
The case study examples explored questions related to utility of existing data for the purpose of hazard identification and dose-response assessment. The DBP case illustrated the need for consistent guidelines across all health effects, endpoints and outcomes. Because DBP is capable of eliciting developmental and reproductive effects, different risk assessment guidelines were applicable to evaluation of these data. Within those guidelines were inconsistencies in data sufficiency criteria, particularly as it relates to the certainty of the epidemiologic evidence. In the case of MeHg, at issue is evaluating the quality of evidence for a new health endpoint -- in this case cardiovascular outcomes such as increased risk of myocardial infarction -- and in determining when there is enough evidence of sufficiently high quality to act on a new endpoint.
Use of Epidemiologic Data to Determine Quantitative Dose-Response Relationships
Epidemiologic data reported as RRs or ORs provide opportunities to move toward a noncancer quantitative dose-response as recommended by the NAS. The DBP case study illustrated how different study results could be interpreted and demonstrated how epidemiologic data provided a more scientifically accurate POD. As exemplified in the MeHg case study, epidemiologic data provide dose-response information that can be used to establish such relationships, which in turn can be used in developing risk-specific doses as well as in improving risk communication and in undertaking cost-benefit analysis.
Defining Acceptable Risk for Noncancer Effects
The RfD is currently defined as the dose unlikely to result in appreciable adverse health effects over a lifetime [46]. A challenge for developing risk-specific doses within the unified dose-response process is defining acceptable levels of increased risk for noncancer outcomes. The target range of acceptable increased risk for cancer is 1 in 10,000 to 1 in 1,000,000 [80]. No such guideline exists for noncancer health effects. Such a guideline will be needed to provide a context for risk-specific doses.
It should be noted that use of epidemiologic data in dose-response estimation usually obviates the need for interspecies and subchronic-to-chronic adjustment factors (more commonly and somewhat misleadingly referred to as uncertainty factors). Accordingly, a POD derived using benchmark dose modeling will correspond to the predetermined benchmark response, placing it in the range of 1% to 10% response. In the case of human data, use of the remaining adjustment factors (database deficiency, interindividual variability, and LOAEL-to-NOAEL), typically result in an adjustment downward by a factor of 10 to 100 (though rarely more than 10), ultimately yielding a level of risk between 1 in 10,000 and 1 in 100. Such a risk range is in contrast with the range considered to be de minimis used to guide risk management and decision-making in the context of carcinogen risk assessment. Efforts to develop guidelines for setting risk management objectives for noncancer endpoints would be first step towards reconciling these differences in target risks.
Use of Epidemiologic Data to Improve Estimates of Interhuman Variability
Quantification of variability in sensitivity to specific health outcomes among similarly exposed people will be needed to contribute to development of risk-specific doses. While epidemiologic studies have begun to test for effect modification that may shed light on this variability [112], additional work is needed both to mine existing data sources and develop new research.
Lesson Learned: Why Epidemiologic Data Provide Critical Information for Risk Assessment
The development of the epidemiologic datasets for DBP and MeHg highlights facets of epidemiologic research that argue for its use in risk assessment. Health effect signals from epidemiologic research are important because there are many factors that can obscure results by biasing observed associations towards the null; therefore, when associations are found in the face of these challenges, attention is warranted. Further, in both of the cases reviewed, a pattern was evident -- early studies of health effect signals were replicated later, particularly for effects which were concordant with animal studies. Therefore, early use of epidemiologic studies is both prudent and scientifically justified.
DISCUSSION AND RECOMMENDATIONS
As efforts to expand the characterization of the impacts of chemical exposures on human health continue [129], it is increasingly apparent that reliance on our current decision-making process, especially as supported by historic methods of dose-response estimation, is untenable. We present two case studies that examine how animal and human evidence were incorporated into risk assessments. In both cases, we propose that epidemiologic data could be used in place of animal data, or used differently, to take full advantage of the state of knowledge. Based on case studies results and the discussion of new directions in environmental public health science, we provide recommendations for enhancing the use of epidemiology in risk assessment (Table 8).
Table 8.
Summary of Recommendations
| Recommendations for Epidemiology |
|---|
| Epidemiologic investigations should strive to employ markers of exposure and upstream effect to enhance study sensitivity |
| Develop a consistent format for epidemiologic results reporting to facilitate cross-study comparisons and data combination methods |
| Susceptibility factors should be analyzed as effect modifiers |
| Conduct joint analyses when data on multiple related exposures or outcomes are available |
| Recommendations for Risk Assessment |
| Use the dose-response information from epidemiologic studies of noncancer effects as the basis for developing quantitative risk estimates |
| Risk assessments should move toward a more systematic and transparent evaluation of the existing evidence |
| EPA should develop a set of data sufficiency and evaluation criteria for epidemiologic data to be applied uniformly across different types of endpoints |
| EPA’s definition of “critical effect” used in developing toxicity values for risk assessment should be consistent with its definition of “adverse effect” and include early upstream events |
| Risk assessors should reframe the concept of critical effect to move beyond determination of a single adverse endpoint to consider syndromes and groups of adverse endpoints that stem from the same initial biological perturbation |
| Trans-Disciplinary Recommendation |
| Create opportunities for cross-disciplinary training and collaboration between epidemiologists and toxicologists. Epidemiologists should be provided with training in regulatory risk assessment |
Recommendations for Epidemiology
Recommendation: Epidemiologic investigations should strive to employ markers of exposure and upstream effect to enhance study sensitivity.
Epidemiologists can improve the utility of their research through lessons from mechanistic toxicology. Detection of rare effects and subtle (but meaningful) changes in continuous biological parameters is a persistent challenge in the field of epidemiology, compounded by difficulties in ascertaining an adequate sample size. In an example provided in a minority report accompanying an EPA Science Advisory Board comment on human subjects testing, Needleman and Reigert assert that in the case of pesticides, detection of a small effect, such as a 1% increase in the rate of a rare neurobehavioral toxicity with a base rate of deficit of 1%, would require a sample size exceeding 3,000 subjects per group [130], highlighting the burden epidemiologists face in ensuring the ability to discern a true effect, as many overt diseases are in the 1% or less range of observations (e.g. cancer, birth defects).
The challenges of detecting rare and difficult-to-measure effects may be in part addressed by development of toxicity pathways and establishment of markers of early biological perturbation. Utilization of these upstream effect measures in epidemiologic investigation increases sensitivity in outcome measurement; such refinements in outcome assessment techniques are likely to reduce the number of subjects needed to achieve sufficient power to see an effect. Changes in testosterone levels resulting from DBP exposure would be an example of a more sensitive and upstream event.
Markers of upstream effect also hold promise for reducing misclassification. Epidemiologists are often forced to rely upon non-objective and surrogate exposure measures, which suffer from numerous biases that may influence estimation of dose-response relationships. These misclassifications, when not differential, tend to dampen estimates of association, whereas when differential, may bias effect estimates in manner that is unpredictable in magnitude and direction. The potential impacts of biases stemming from exposure measurement errors are rarely investigated [131]. Reliance on frank effects as outcomes can intensify the likelihood that persons who have progressed along the toxicodynamic continuum but have not yet reached a clinically apparent disease state will be misclassified as not having the disease. Such misclassification can be differential and may result in the weakening of effect estimates, ultimately resulting in the inability to observe an effect.
Numerous changes in the landscape of environmental public health science may serve to enhance the ability of epidemiologists to build the case for relationships between exposures and subtle or rare events. As the knowledge base regarding toxicity pathways expands, it will become increasingly possible to examine upstream biological changes as outcomes in epidemiologic studies. The elucidation of these toxicity pathways may help identify new and previously unrecognized early biomarkers of toxicodynamic progression towards overt disease. The growing recognition of the value of biomonitoring data in epidemiologic investigations also holds promise for reducing misclassification by providing objective measures of exposure and outcome. As biomarker data for exposure, outcome and susceptibility are increasingly generated, epidemiology will play a prominent role in developing an understanding of toxicodynamic progression as a function of chemical exposure.
Recommendation: Develop a consistent format for epidemiologic results reporting to facilitate cross-study comparisons and data combination methods (such as meta-analysis).
Measures of exposure and effects estimates with associated variability (preferably in the form of standard errors) are the essential data elements to be evaluated when assembling a data set for risk assessment. In rare instances, access to primary data may be requested. Access to primary data allows evaluation of various types of measurement error in exposure assessment (and subsequent misclassification or bias) or may be combined in a dose or exposure response analysis using a meta-analysis or other data combination approach [131].
Recommendation: Susceptibility factors should be analyzed as effect modifiers.
Beyond the advantages afforded by improvement in exposure characterization, recognition of host factors that influence toxicity are also of key importance. More accurate quantification of susceptibility due to genetic differences, age and gender, diet, health status and socioeconomic status will inform probability of distributions of uncertainty and variability. One important strategy, where possible, is testing whether these specific susceptibility factors modify the relationship between intake of a chemical and its internal dose as measured in some body tissue (i.e., effect modification in toxicokinetic pathways), and between intake or internal dose and measures of early or later effect (i.e., modifications in toxicodynamic pathways). Often, these factors are simply adjusted for as potential confounders [112]. For chemicals with validated biomarkers of dose and early effect, NHANES and other large cross-sectional surveys can be used to estimate variability distributions for factors known to influence susceptibility.
Recommendation: Conduct joint analyses when data on multiple related exposures or outcomes are available.
Although epidemiologic data have the potential to inform pressing health risk questions related to multiple exposures and outcomes, most analytical epidemiologic studies try to isolate a specific causal or explanatory factor for a well-defined outcome. For outcomes thought to originate from a common biological perturbation, epidemiologic studies that examine associations between exposures and grouped outcomes may be informative in the risk assessment process. Similarly, for exposure to multiple agents thought to act on common targets, efforts to assess the joint action of the combined exposures would be highly relevant for risk assessments. As evidence of MeHg’s ability to elicit effects in multiple organ systems continues to accrue, it becomes increasingly clear that the current risk practice of selecting a single critical effect will underestimate MeHg’s total impact. Joint or combined analyses will provide better characterizations of risk.
Recommendations for Risk Assessment
Recommendation: Use the dose-response information from epidemiologic studies of noncancer effects as the basis for developing quantitative risk estimates.
Critical changes in how risk assessors use epidemiologic data, coupled with reconsideration of some of the fundamental methodologies in how dose-response data are employed in assessments are warranted. The unified approach for cancer and noncancer dose response assessment presented by the NRC [7] promotes the movement away from the default threshold-based hazard metric embodied by the RfD and RfC and allows for a spectrum of considerations, including factors that predict host susceptibility, among others, to influence the selection of dose-response model. This evidence and data-driven approach to model selection allows for a more holistic incorporation of available data, extending beyond the current method of selecting a single dataset and setting an artificial bright-line and moving towards a risk-specific dose that is better suited for evaluations of risk-tradeoffs and benefit-cost analyses [7]. Such an approach will allow risk assessors to employ estimates of slope for chemicals as the default approach on both individual and population bases and attempts to provide a quantitative description of risk across numerous doses with consideration of changes in slope across dose when mechanistic information suggests differences from the observed slope at high dose to the slope at low dose (in the range of environmental exposures). The risk-specific dose as proposed by NRC provides a risk metric for noncancer effects consistent with the existing approach for cancer risk estimates [132–134]. It is recognized that some estimates of slope derived from the observed range (especially those derived from evaluations of occupational exposures) may not reflect those in the lower, environmentally-relevant range; accordingly, steps should be taken to account for heterogeneity in response in the low dose range across the population [7]. Characterization of risks corresponding to a range of doses, combined with the NRC recommendation to combine risks for chemicals that elicit similar health effects or act on the same organ system, allows risk assessors to more fully consider the relevance of multiple exposures in the decision-making process.
Well-researched examples from the epidemiologic literature (particulate matter, ozone, lead, secondhand or environmental tobacco smoke and radon) reinforce the case and provide examples for updating dose-response assessment and procedures for extrapolation of dose-response to low doses. Population studies have found no threshold for particulate matter and all-cause and cardio-respiratory mortality [135], ozone and mortality [136], lead and cognitive function [137], second hand tobacco smoke and radon and lung cancer [138–140], consistent with the NRC recommendations.
Recommendation: Risk assessments should move toward a more systematic and transparent evaluation of the existing evidence.
Evidence-based medicine and public health rely on systematic literature review and evidence synthesis methods [141, 142]. A valuable attribute of these methods is that a transparent literature search strategy assures that all relevant research has been gathered for review. Sometimes proprietary data are used in risk assessments. Incorporating data from a systematic literature review would expand the dataset for the risk assessment and also balance proprietary data with information from the broader literature.
Systematic reviews use well-defined literature search and statistical evaluation techniques to appraise study validity and summarize research on specific health effects. Systematic review has been defined as “the application of scientific strategies, in ways that limit bias, to the assembly, critical appraisal, and synthesis of all relevant studies” [143]. The statistical tools of meta-analysis, which capture an effect size from various studies, are often but not always applied to a systematic review. Systematic reviews for intervention-based clinical medicine review randomized clinical trials (RCTs) as the “gold standard.” However, environmental epidemiology relies mainly on observational studies such as cohort and case-control studies, due to ethical and other issues related to designing experimental studies to examine chemical health effects in populations. The Cochrane Collaboration and other organizations have developed transparent guidelines for systematic reviews of intervention-based RCTs in developing the underpinnings of evidence-based medicine [142]. However, these organizations are now beginning to also look at observational studies. Key concerns with performing systematic review of observational studies are heterogeneity of design and inherent biases and confounding, need to search extensively and demonstrate reproducibility due to tendency of observational studies to overestimate exposure effect [144]. One new method that may be particularly useful for risk assessment is the Grading of Recommendations Assessment, Development and Evaluation (GRADE) approach, which is a system for grading, or rating, evidence in the health field. In using a weight of evidence approach, risk analysts and policy makers are influenced by both the conclusion of an assessment but also their confidence in the estimates. The GRADE system is a systematic method to rank confidence in the summarized evidence [145]. An approach that values the supporting toxicological evidence is important for this approach, as animal evidence is not currently part of the GRADE system, and toxicological evidence has clearly been identified (e.g. cancer guidelines) as critical supporting information. Work is ongoing in this area (Sutton, personal communication).
Recommendation: EPA should develop a set of data sufficiency and evaluation criteria for epidemiologic data to be applied uniformly across different types of endpoints (cancer and noncancer).
The 2005 Guidelines for Carcinogen Risk Assessment discussion of data sufficiency for each descriptor provides a useful foundation for this effort; a summary of this is provided in Table 9. The review of the EPA’s developmental, reproductive toxicity and carcinogen risk assessment guidance documents in the DBP case example found that differing standards of data sufficiency apply to epidemiologic data evaluations for risk assessments of these endpoints. Differing guidance presented a challenge for assessing DBP and will be a challenge for other compounds where there is evidence for multiple effects stemming from a single biological perturbation or exposure. Similarly, as methods for cumulative risk assessments develop and move towards addressing multiple endpoints in addition to multiple stressors, the existing endpoint-specific guidance will impede rather than foster those more comprehensive assessments.
Table 9.
Guidance for Consideration of Evidence in Support of Cancer Descriptors (Adapted from EPA 2005 Guidelines for Carcinogen Risk Assessment)
| Cancer Descriptor | Sufficient Evidence |
|---|---|
| Carcinogenic to humans | Convincing evidence of causal association between human exposure and cancer (e.g., from well conducted cohort study[ies]). |
| OR | |
| Strong evidence of association between human exposure and cancer or key precursor events AND extensive evidence of carcinogenicity in animals AND mode(s) of action identified in animals AND strong evidence that mode(s) of action identified in animals occur in humans. | |
| Likely to be carcinogenic to humans | Plausible (but not definitively causal) association between human exposure and cancer in most cases with some supporting biological and experimental evidence (not necessarily carcinogenicity data from animal experiments). |
| OR | |
| An agent that has tested positive in animal experiments in more than one species, sex, strain, or exposure route, with or without evidence of carcinogenicity in humans. | |
| OR | |
| A positive tumor study that raises additional biological concerns beyond that of a statistically significant result, e.g., a high degree of malignancy, or an early age at onset. | |
| OR | |
| A rare animal tumor response in a single experiment that is assumed to be relevant to humans. | |
| OR | |
| A positive tumor study that is strengthened by other lines of evidence, e.g., either plausible association between human exposure and cancer or evidence that the agent or an important metabolite causes events generally known to be associated with tumor formation or likely to be related to the tumor response in the this particular case. | |
| Suggestive evidence of carcinogenic potential | A small and possible not statistically significant increase in tumor incidence observed in a single animal or human study that does not reach the weight of evidence for “Likely to be carcinogenic to humans”. The study findings would generally not be contradicted by other similar studies of equal quality. |
| OR | |
| A small increase in tumor with a high background rate in that sex and strain when there is some evidence that the observed tumors may be due to intrinsic factors that cause background tumors and not due to the agent being assessed. | |
| OR | |
| Evidence of a positive response in a study whose power, design, or conduct limits the ability to draw a confident conclusion, but where the carcinogenic potential is strengthened by other lines of evidence (e.g., structure activity relationships). | |
| OR | |
| A statistically significant increase at one dose only, but no significant response at other doses and no overall trend. | |
| Inadequate information to assess carcinogenic potential | Little or no pertinent information |
| OR | |
| Conflicting evidence in studies of equal quality | |
| OR | |
| Negative results that are not sufficiently robust to support “Not likely to be carcinogenic to humans” | |
| Not likely to be carcinogenic to humans | Animal evidence that demonstrates lack of carcinogenic effect in both sexes in at least two appropriate animal species (in absence of other animal or human data suggesting a potential for cancer effects) |
| OR | |
| Convincing and extensive experimental evidence showing that the only carcinogenic effects observed in animals are not relevant to humans | |
| OR | |
| Convincing evidence that carcinogenic effects are not likely by a particular exposure route | |
| OR | |
| Convincing evidence that carcinogenic effects are not likely below a defined dose range | |
Recommendation: EPA’s definition of “critical effect” used in developing toxicity values for risk assessment should be consistent with its definition of “adverse effect” and include early upstream events.
There is a growing consensus in the risk assessment community that the definition of critical endpoint needs to be refined to reflect our rapidly advancing understanding of adversity [50]. Further, we have identified clear inconsistencies between the EPA IRIS definitions for critical and adverse effects. As more attention is focused on elucidation of toxicity pathways [4] and our ability to detect early biological perturbations improves through advances in biomonitoring and in ‘omics technologies, it is incumbent upon risk assessors to acknowledge subtle changes early along the toxicodynamic pathway that are indicative of increased potential for downstream effects [50]. As such, the selection of clinically-overt health effects as critical endpoints upon which dose-response assessments are based is an outdated practice in need of modernizing. A move towards recognition of upstream, subclinical effects in risk assessments shows promise for recognition of the potential for shared toxicity pathways across chemicals. As stated in the EPA IRIS definition of “adverse effect”, such an acknowledgement that these subtle biochemical changes may themselves affect the performance of an organism or reduce the capacity for response to additional environmental challenges would well serve risk assessors to meet newer types of dose-response data generated as the field of environmental epidemiology moves forward.
Recommendation: Risk assessors should reframe the concept of critical effect to move beyond determination of a single adverse endpoint to consider syndromes and groups of adverse endpoints that stem from the same initial biological perturbation.
In regards to selection of critical endpoints, it is relevant that risk assessors evaluate the evidence in support of multiple adverse outcomes stemming from the same initial biological perturbation. Given interindividual variability in numerous host factors, including metabolic profiles, co-exposures and co-morbidities, it is possible that variation may exist in the manifestation of adverse outcomes in response to a chemical challenge; phthalate exposures and testicular dysgenesis syndrome and the collection of motor skill, cognitive and behavioral outcomes resulting from lead exposures are two prominent examples of this phenomenon [61, 64, 146]. Ideally, the relationships between exposures and combinations of related endpoints would be addressed in epidemiologic studies, though this is frequently not the case. Accordingly, the field of risk assessment would be well served by developing procedures for evaluating evidence for co-occurrence of multiple adverse outcomes as a critical endpoint for toxicity value derivation.
Historically, when faced with data from multiple epidemiologic and experimental animal studies that evaluate the relationship between exposures and different health outcomes, risk assessors seek to identify the most sensitive adverse endpoint (derived from a single study) for quantitative estimation of a point of departure. The shortcoming of this approach is that, in the instance of multiple, exposure-related outcomes, characterization of risk based on a POD derived from one of many outcomes may neglect the true population risk of developing any number of adverse conditions relevant to the exposure of interest. Increased recognition of syndromic morbidity in the epidemiologic community and improvements in endpoint selection in epidemiologic studies will aid risk assessors in critical endpoint selection. However, as risk assessors are often reliant on data from studies not recently conducted, awareness of the potential for related sequelae from exposures to the same agent should weigh heavily when attempting POD derivation.
Trans-Disciplinary Recommendation
Recommendation: Create opportunities for cross-disciplinary training and collaboration between epidemiologists and toxicologists. Epidemiologists should be provided with training in regulatory risk assessment.
Risk assessment aims to inform the decision-making process in the environmental policy arena. An understanding of the risk assessment process will equip epidemiologists with the basis for ensuring that the results of their work are translatable into a format that is amenable into the hazard identification and dose-response characterization steps of risk assessments.
Studies in experimental animals have largely provided the basis for derivation of toxicity values in IRIS toxicological assessments [56]. While the explanation for this may rely on available evidence in the literature, it may be partially explained by the possibility that the expertise of the IRIS team is more weighted towards toxicology. An increased recognition of the role of epidemiologists in the development of toxicity assessments could enhance the ability of IRIS to take full advantage of a chemical’s available health effect database. This must be combined with a concerted effort by EPA to bring in more epidemiologists, as the comparatively fewer epidemiologists at the agency contribute to epidemiologic data being undervalued.
CONCLUSIONS
Epidemiology is essential to our understanding of the role of environmental exposures in human disease. After all, it is only by studying the human population that we will understand the complex interactions of the environment, social factors, heredity, and behavior that determine individual and population health. This paper has explored the role of epidemiologic data in supporting and shaping chemical risk assessment and related risk management policies. Our examination of current chemical risk guidelines indicates the dominance of animal toxicology studies in chemical risk assessments and a disconnect between available epidemiologic data and the needs of risk decision makers. However, advances in exposure science, biomarkers of internal dose and early effect, understanding of population variability in susceptibility, and population health surveillance combine to provide an unprecedented opportunity to revitalize the role of epidemiology.
Our current biomonitoring efforts document widespread exposure to a host of chemicals for which toxicity information is lacking. Development of timely traditional chemical-specific risk assessments for the tens of thousands of potential environmental pollutants has proven to be an exercise in futility. In addition, it remains difficult if not impossible to demonstrate the public health benefits of many chemical risk management efforts. We can make advances now by better integrating existing epidemiologic studies with our current understanding of toxicodynamic processes from in vitro, in vivo, and ultimately in silico experiments. As biomonitoring advances our understanding of population exposures, and biomarkers expand our understanding of susceptibility and early biological effects, it is time to develop a new, trans-disciplinary lens through which we can examine environment and health. Improving risk assessment will require multidisciplinary involvement from the earliest problem formulation stage through the characterization and management of risks. Epidemiologists and other environmental health disciplines with cross-training in epidemiology can shape new approaches to addressing cumulative population risks, prioritizing chemicals for evaluation, and addressing the scientific needs of decision makers.
To be sure, many challenges remain in advancing the role of epidemiology in chemical risk assessment. Those in search of bright lines and thresholds will continue to be frustrated by the limits of human studies. However, bolstered by advanced tools and improved study designs, epidemiology may hold the key to shedding new light on the recognition, prevention, and management of chemical risks.
ABBREVIATIONS
- AGD
Anogenital distance
- AGI
Anogenital index
- BMD
Benchmark dose
- BMDL
Lower confidence limit on the BMD
- CDC
Centers for Disease Control and Prevention
- CNS
Central nervous system
- CSF
Cancer slope factor
- CVD
Cardiovascular disease
- DBP
Di-butyl phthalate
- DNA
Deoxyribonucleic acid
- DWUR
Drinking water unit risk
- EPA
United States Environmental Protection Agency
- GGT
Gamma glutamyltransferase
- GRADE
Grading of Recommendations Assessment, Development and Evaluation
- Hg
Mercury
- iHg
Inorganic mercury
- insl3
Insulin-like growth factor 3
- IRIS
EPA Integrated Risk Information System
- IUR
Inhalation unit risk
- LH
Lutenizing hormone
- LIN
Sperm linearity
- LOAEL
Lowest observed adverse effect level
- MBP
Mono-butyl phthalate
- MBzP
Mono-benzyl phthalate
- MeHg
Methylmercury
- MEP
Mono-ethyl phthalate
- MI
Myocardial infarction
- MiBP
Mono-isobutyl phthalate
- MiNP
Mono-isononyl phthalate
- MOA
Mode of action
- MR
Lymphatic cholinergic muscarinic receptor
- NCS
National Children’s Study
- NHANES
National Health and Nutrition Examination Survey
- NHEXAS
National Human Exposure Assessment Survey
- NOAEL
No observed adverse effect level
- NRC
National Research Council
- NRDC
National Resources Defense Council
- OR
Odds ratio
- PD
Pharmacodynamic
- PK
Pharmacokinetic
- POD
Point of departure
- PPARα
Peroxisome proliferator-activated receptor-α
- ppb
Parts per billion
- ppm
Parts per million
- RCT
Randomized clinical trial
- RfC
Reference concentration
- RfD
Reference dose
- ROS
Reactive oxygen species
- RR
Relative risk
- SES
Socioeconomic status
- SHBG
Sex-hormone binding globulin
- T4
Thyroxine
- TR
Toxicological Review
- UF
Uncertainty factor
- VCL
Sperm curvilinear velocity
- VSL
Sperm straight-line velocity
- AMI
Acute myocardial infarction
- BP
Blood pressure
- CHD
Chronic heart disease
- CVD
Cardiovascular disease
- EU
European Union
- EURAMIC
European Union Acute Myocardial Infarction and Cancer study
- g
Gram
- HRV
Heart rate variability
- IMT
Intima media thickness
- IPCS
International Programme on Chemical Safety
- KIHD
Kuopio Ischemic Heart Disease study
- L
Liter
- Nmol
Nanomol
- PTWI
Provisional tolerable weekly intake
- PUFA
Polyunsaturated fatty acid
- ug
Microgram
REFERENCES
- [1].Hartung T, Rovida C. Chemical regulators have overreached. Nature 2009; 460(7259): 1080–1. [DOI] [PubMed] [Google Scholar]
- [2].Roe D, Pease W, Florini K, Silbergeld E. Toxic Ignorance Washington, DC: Environmental Defense Fund; 1997. [December 15, 2009]; [Available from: http://www.edf.org/documents/243_toxicignorance.pdf] [Google Scholar]
- [3].Grandjean P Non-precautionary aspects of toxicology. Toxicol Appl Pharmacol 2005; 207(2 Suppl): 652–7. [DOI] [PubMed] [Google Scholar]
- [4].National Research Council. Toxicity Testing in the Twenty-first Century: A Vision and a Strategy Washington DC: National Academies Press; 2007. [Google Scholar]
- [5].National Research Council. Human Biomonitoring for Environmental Chemicals Washington DC: National Academies Press; 2006. [Google Scholar]
- [6].Gee D Late lessons from early warnings: Toward realism and precaution with endocrine-disrupting substances. Environ Health Perspect 2006; 114(Suppl) 1: 152–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].National Research Council. Science and Decisions: Advancing Risk Assessment Washington, DC National Academies Press; 2009. [PubMed] [Google Scholar]
- [8].Swaen GM. A framework for using epidemiological data for risk assessment. Hum Exp Toxicol 2006: 147–55. [DOI] [PubMed]
- [9].Vlaanderen J, Vermeulen R, Heederik D, Kromhout H. Guidelines to evaluate human observational studies for quantitative risk assessment. Environ Health Perspect 2008; 116(12): 1700–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].United States Environmental Protection Agency. A Review of the Reference Dose and Reference Concentration Properties Washington DC: Risk Assessment Forum; 2002; Contract No.: EPA/630/P-02/002F. [Google Scholar]
- [11].Hertz-Picciotto I. Epidemiology and quantitative risk assessment: a bridge from science to policy. Am J Public Health 1995; 85(4): 484–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Smith AH. Epidemiologic input to environmental risk assessment. Arch Environ Health 1988; 43(2): 124–9. [DOI] [PubMed] [Google Scholar]
- [13].Hattis D Human interindividual variability in susceptibility to toxic effects: from annoying detail to a central determinant of risk. Toxicology 1996; 111(1–3): 5–14. [DOI] [PubMed] [Google Scholar]
- [14].Perera F The potential usefulness of biological markers in risk assessment. Environ Health Perspect 1987; 76: 141–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Perera FP. Molecular epidemiology: insights into cancer susceptibility, risk assessment, and prevention. J Natl Cancer Inst 1996; 88(8): 496–509. [DOI] [PubMed] [Google Scholar]
- [16].Samet JM, Schnatter R, Gibb H. Epidemiology and risk assessment. Am J Epidemiol 1998; 148(10): 929–36. [DOI] [PubMed] [Google Scholar]
- [17].Rothman KJ, Greenland S. Modern Epidemiology 2nd ed. Philadelphia: Lipincott-Raven Publishers; 1998. [Google Scholar]
- [18].Pershagen G Environmental epidemiology in public health. Lancet 1998; 352(9126): 417. [DOI] [PubMed] [Google Scholar]
- [19].Committee on Biological Markers of the National Research Council. Biological markers in environmental health research. Environ Health Perspect 1987; 74: 3–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Barr DB, Wilder LC, Caudill SP, Gonzalez AJ, Needham LL, Pirkle JL. Urinary creatinine concentrations in the US. population: implications for urinary biologic monitoring measurements. Environ Health Perspect 2005; 113(2): 192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Albertini R, Bird M, Doerrer N, et al. The use of biomonitoring data in exposure and human health risk assessments. Environ Health Perspect 2006; 114(11): 1755–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Needham LL, Calafat AM, Barr DB. Uses and issues of biomonitoring. Int J Hyg Environ Health 2007; 210(3–4): 229–38. [DOI] [PubMed] [Google Scholar]
- [23].Robison SH, Barr DB. Use of biomonitoring data to evaluate methyl eugenol exposure. Environ Health Perspect 2006; 114(11): 1797–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Birnbaum LS, Cohen Hubal EA. Polybrominated diphenyl ethers: a case study for using biomonitoring data to address risk assessment questions. Environ Health Perspect 2006; 114(11): 1770–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Calafat AM, Wong LY, Kuklenyik Z, Reidy JA, Needham LL. Polyfluoroalkyl chemicals in the U.S. population: data from the National Health and Nutrition Examination Survey (NHANES) 2003–2004 and comparisons with NHANES 1999–2000. Environ Health Perspect 2007; 115(11): 1596–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Calafat AM, Ye X, Silva MJ, Kuklenyik Z, Needham LL. Human exposure assessment to environmental chemicals using biomonitoring. Int J Androl 2006; 29(1): 166–71; discussion 81–5. [DOI] [PubMed] [Google Scholar]
- [27].Centers for Disease Control and Prevention. Fourth National Report on Human Exposures To Environmental Chemicals - Executive Summary Atlanta, GA: 2009. [December 15, 2009]; [Available from: http://www.cdc.gov/exposurereport/pdf/FourthReport_ExecutiveSummary.pdf] [Google Scholar]
- [28].Weis BK, Balshaw D, Barr JR, et al. Personalized exposure assessment: promising approaches for human environmental health research. Environ Health Perspect 2005; 113(7): 840–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ryan PB, Burke TA, Cohen Hubal EA, Cura JJ, McKone TE. Using biomarkers to inform cumulative risk assessment. Environ Health Perspect 2007; 115(5): 833–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Angerer J, Bird MG, Burke TA, et al. Strategic biomonitoring initiatives: moving the science forward. Toxicol Sci 2006; 93(1): 3–10. [DOI] [PubMed] [Google Scholar]
- [31].Centers for Disease Control and Prevention. National Biomonitoring Program: State Grant Activities 2009. [updated September 9, 2009; cited 2009 December 3]; [Available from: http://www.cdc.gov/biomonitoring/state_grants.htm]
- [32].Lioy PJ, Isukapalli SS, Trasande L, et al. Using National and Local Extant Data to Characterize Environmental Exposures in the National Children’s Study: Queens County, New York. Environ Health Perspect 2009; 117(10): 1494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Viso AC, Casteleyn L, Biot P, Eilstein D. Human biomonitoring programmes and activities in the European Union. Epidemiol Commun Health 2009; 63(8): 623–4. [DOI] [PubMed] [Google Scholar]
- [34].Sly PD, Eskenazi B, Pronczuk J, et al. Ethical issues in measuring biomarkers in children’s environmental health. Environ Health Perspect 2009; 117(8): 1185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].White RH, Cote I, Zeise L, et al. State-of-the-science workshop report: issues and approaches in low-dose-response extrapolation for environmental health risk assessment. Environ Health Perspect 2009; 117(2): 283–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Hattis D The Use of Biological Markers in Risk Assessment. Stat Sci 1988; 3(3): 358–66. [Google Scholar]
- [37].United States Environmental Protection Agency. Supplemental Guidance for Assessing Susceptibility from Early-Life Exposure to Carcinogens Washington DC: 2005; Contract No.: EPA/630/R-03/003F. [Google Scholar]
- [38].California Environmental Protection Agency. Technical Support Document for Cancer Potency Factors: Methodologies for derivation, listing of available values, and adjustments to allow for early life stage exposures. In: Office of Environmental Health Hazard Assessment/Air Toxicol Epidemiol Branch 2009.
- [39].Krewski D, Andersen ME, Mantus E, Zeise L. Toxicity testing in the 21st century: implications for human health risk assessment. Risk Anal 2009; 29(4): 474–9. [DOI] [PubMed] [Google Scholar]
- [40].Vlaanderen J, Moore LE, Smith MT, et al. Application of OMICS technologies in occupational and environmental health research; current status and projections. Occup Environ Med 2010; 67(2): 136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Jirtle RL, Skinner MK. Environmental epigenomics and disease susceptibility. Nat Rev Genet 2007; 8(4): 253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Dolinoy DC, Jirtle RL. Environmental epigenomics in human health and disease. Environ Mol Mutagen 2008; 49(1): 4–8. [DOI] [PubMed] [Google Scholar]
- [43].Szyf M, McGowan P, Meaney MJ. The social environment and the epigenome. Environ Mol Mutagen 2008; 49(1): 46–60. [DOI] [PubMed] [Google Scholar]
- [44].Committee on Toxicology. COT statement on the COT workshop on transgenerational epigenetics London: 2008. [cited 2010 March 15]; [Available from: http://cot.food.gov.uk/cotstatements/cotstatementsyrs/cotstatements2008/cot200803] [Google Scholar]
- [45].Sutherland JE, Costa M. Epigenetics and the Environment. Ann NY Acad Sci 2003; 983: 151–60. [DOI] [PubMed] [Google Scholar]
- [46].United States Environmental Protection Agency. IRIS Glossary 2010. [cited 2010 March 10]; [Available from: http://www.epa.gov/iris/help_gloss.htm.
- [47].United States Environmental Protection Agency. IRIS Glossary 2009. [cited 2009 November 13]; [Available from: http://www.epa.gov/iris/help_gloss.htm]
- [48].Bellinger DC. Interpreting epidemiologic studies of developmental neurotoxicity: conceptual and analytic issues. Neurotoxicol Teratol 2009; 31(5): 267–74. [DOI] [PubMed] [Google Scholar]
- [49].Woodruff TJ, Wells EM, Holt EW, Burgin DE, Axelrad DA. Estimating risk from ambient concentrations of acrolein across the United States. Environ Health Perspect 2007; 115(3): 410–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Woodruff TJ, Zeise L, Axelrad DA, et al. Meeting report: moving upstream-evaluating adverse upstream end points for improved risk assessment and decision-making. Environ Health Perspect 2008; 116(11): 1568–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Hattis D, Baird S, Goble R. A straw man proposal for a quantitative definition of the RfD. Drug Chem Toxicol 2002; 25(4): 403–36. [DOI] [PubMed] [Google Scholar]
- [52].Castorina R, Woodruff TJ. Assessment of potential risk levels associated with U.S. Environmental Protection Agency reference values. Environ Health Perspect 2003; 111(10): 1318–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Axelrad DA, Baetcke K, Dockins C, et al. Risk assessment for benefits analysis: framework for analysis of a thyroid-disrupting chemical. J Toxicol Environ Health A 2005; 68(11–12): 837–55. [DOI] [PubMed] [Google Scholar]
- [54].Clewell HJ, Crump KS. Quantitative Estimates of Risk Noncancer Endpoints. Risk Anal 2005; 25: 285–9. [DOI] [PubMed] [Google Scholar]
- [55].National Research Council. Risk Assessment in the Federal Government Washington DC: National Academies Press; 1983. [PubMed] [Google Scholar]
- [56].Persad AS, Cooper GS. Use of epidemiologic data in Integrated Risk Information System (IRIS) assessments. Toxicol Appl Pharmacol 2008; 233(1): 137–45. [DOI] [PubMed] [Google Scholar]
- [57].United States Environmental Protection Agency. IRIS Toxicological Review of Dibutyl Phthalate (Di-N-Butyl Phthalate) (External Review Draft) Washington, DC: 2006. [Google Scholar]
- [58].United States Environmental Protection Agency. Methylmercury (MeHg) (CASRN 22967–92-6) Washington DC: 2001. [cited 2009 December 4]; [Available from: http://www.epa.gov/ncea/iris/subst/0073.htm] [Google Scholar]
- [59].Wolff MS, Engel SM, Berkowitz GS, et al. Prenatal phenol and phthalate exposures and birth outcomes. Environ Health Perspect 2008; 116(8): 1092–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Smith C Toxicity of butyl stearate, dibutyl sebacate, dibutyl phthalate, and methoxyethyl oleate. AMA Arch Ind Hyg Occup Med 1953; 7(4): 310–8. [PubMed] [Google Scholar]
- [61].National Research Council. Phthalates and Cumulative Risk Assessment: The Task Ahead Washington DC: National Academies Press; 2008. [PubMed] [Google Scholar]
- [62].IRISTrack [database on the Internet] 2010. [cited February 15, 2010]. [Available from: http://cfpub.epa.gov/ncea/iristrac/index.cfm]
- [63].Carruthers CM, Foster PM. Critical window of male reproductive tract development in rats following gestational exposure to di-n-butyl phthalate. Birth Defects Res B Dev Reprod Toxicol 2005; 74(3): 277–85. [DOI] [PubMed] [Google Scholar]
- [64].Skakkebak NE, Rajpert-De Meyts E, Main KM. Testicular dysgenesis syndrome: an increasingly common developmental disorder with environmental aspects: Opinion. Hum Reprod 2001; 16(5): 972–8. [DOI] [PubMed] [Google Scholar]
- [65].Lehmann KP, Phillips S, Sar M, Foster PM, Gaido KW. Dose-dependent alterations in gene expression and testosterone synthesis in the fetal testes of male rats exposed to di (n-butyl) phthalate. Toxicol Sci 2004; 81(1): 60–8. [DOI] [PubMed] [Google Scholar]
- [66].Sass J, Janssen S. Issues Concerning Phthalates and Cumulative Risk Assessments: NGO perspective: Comments from the National Resources Defense Council Washington DC: 2007. [Google Scholar]
- [67].Marsee K, Woodruff TJ, Axelrad DA, Calafat AM, Swan SH. Estimated daily phthalate exposures in a population of mothers of male infants exhibiting reduced anogenital distance. Environ Health Perspect 2006; 114(6): 805–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Swan SH, Main KM, Liu F, et al. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect 2005; 113(8): 1056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Salazar-Martinez E, Romano-Riquer P, Yanez-Marquez E, Longnecker MP, Hernandez-Avila M. Anogenital distance in human male and female newborns: a descriptive, cross-sectional study. Environ Health 2004; 3(1): 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Hauser R, Meeker JD, Duty S, Silva MJ, Calafat AM. Altered semen quality in relation to urinary concentrations of phthalate monoester and oxidative metabolites. Epidemiology 2006; 17(6): 682–91. [DOI] [PubMed] [Google Scholar]
- [71].Main KM, Mortensen GK, Kaleva MM, et al. Human breast milk contamination with phthalates and alterations of endogenous reproductive hormones in infants three months of age. Environ Health Perspect 2006; 114(2): 270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Duty SM, Calafat AM, Silva MJ, et al. The relationship between environmental exposure to phthalates and computer-aided sperm analysis motion parameters. J Androl 2004; 25(2): 293–302. [DOI] [PubMed] [Google Scholar]
- [73].Duty SM, Silva MJ, Barr DB, et al. Phthalate exposure and human semen parameters. Epidemiology 2003; 14(3): 269–77. [PubMed] [Google Scholar]
- [74].Duty SM, Singh NP, Silva MJ, et al. The relationship between environmental exposures to phthalates and DNA damage in human sperm using the neutral comet assay. Environ Health Perspect 2003; 111(9): 1164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].United States Environmental Protection Agency. An Examination of EPA Risk Assessment Principles and Practices Washington, DC: Office of the Science Advisor, United States Environmental Protection Agency; 2004. Report No.: EPA/100/B-04/001 [Google Scholar]
- [76].Woodruff T Policy Implications of Endocrine-Disrupting Compounds. In: Gore AC, ed Endocrine-Disrupting Chemicals: From Basic Research to Clinical Practice Totowa NJ: Humana Press; 2007. [Google Scholar]
- [77].Woodruff TJ, Janssen S. Interpreting Science in the Policy Context. In: Woodruff TJ, Guillette LJ Jr, Guidice LC, Eds Environ Impacts on ductive Health and Fertility NY: Cambridge University Press; 2010. [Google Scholar]
- [78].United States Environmental Protection Agency. Guidelines for Reproductive Toxicity Risk Assessment: Risk Assessment Forum. 1996. Contract No.: EPA/630/R-96/009.
- [79].United States Environmental Protection Agency. Guidelines for Developmental Toxicity Risk Assessment Washington DC: Risk Assessment Forum; 1991; Contract No.: EPA/600/FR-91/001. [Google Scholar]
- [80].United States Environmental Protection Agency. Guidelines for Carcinogen Risk Assessment Washington DC: 2005; Contract No.: EPA/630/P-03/001F [Google Scholar]
- [81].Barlow NJ, Foster PM. Pathogenesis of male reproductive tract lesions from gestation through adulthood following in utero exposure to Di(n-butyl) phthalate. Toxicol Pathol 2003; 31(4): 397–410. [DOI] [PubMed] [Google Scholar]
- [82].Foster PM, Cattley RC, Mylchreest E. Effects of di-n-butyl phthalate (DBP) on male reproductive development in the rat: implications for human risk assessment. Food Chem Toxicol 2000; 38(1 Suppl): S97–9. [DOI] [PubMed] [Google Scholar]
- [83].Swan SH. Environmental phthalate exposure in relation to reproductive outcomes and other health endpoints in humans. Environ Res 2008; 108(2): 177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Swan SH, Liu F, Hines M, et al. Prenatal phthalate exposure and reduced masculine play in boys. Int J Androl 2009; 33: 259–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Sutton AJ, Abrams KR. Bayesian methods in meta-analysis and evidence synthesis. Stat Method Med Res 2001; 10(4): 277–303. [DOI] [PubMed] [Google Scholar]
- [86].Coffey T, Gennings C, Moser VC. The simultaneous analysis of discrete and continuous outcomes in a dose-response study: Using desirability functions. Regul Toxicol Pharmacol 2007; 48(1): 51–8. [DOI] [PubMed] [Google Scholar]
- [87].Barnes DG, Daston GP, Evans JS, et al. Benchmark Dose Workshop: Criteria for Use of a Benchmark Dose to Estimate a Reference Dose. Regul Toxicol Pharmacol 1995; 21(2): 296–306. [DOI] [PubMed] [Google Scholar]
- [88].United Nations Environment Programme. Global Atmospheric Mercury Assessment: Sources, Emissions and Transport Geneva 2008.
- [89].Madison Ed. The Madison declaration on mercury pollution Ambio 2007. [PubMed]
- [90].National Research Council. Toxicological Effects of Methylmercury Washington DC: National Academies Press; 2000. [PubMed] [Google Scholar]
- [91].Pacyna EG, Pacyna JM, Steenhuisen F, Wilson S. Global anthropogenic mercury emission inventory for 2000. Atmospheric Environ 2006; 40(22): 4048–63. [Google Scholar]
- [92].Mahaffey KR, Clickner RP, Bodurow CC. Blood organic mercury and dietary mercury intake: National Health and Nutrition Examination Survey, 1999 and 2000. Environ Health Perspect 2004; 112(5): 562–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Clarkson TW, Magos L. The toxicology of mercury and its chemical compounds. Crit Rev Toxicol 2006; 36(8): 609–62. [DOI] [PubMed] [Google Scholar]
- [94].Taber KH, Hurley RA. Mercury exposure: effects across the lifespan. J Neuropsychiatry Clin Neurosci 2008; 20(4): iv–389. [DOI] [PubMed] [Google Scholar]
- [95].Marsh DO, Clarkson TW, Cox C, Myers GJ, Amin-Zaki L, Al-Tikriti Sa. (Fetal Methylmercury Poisoning: Relationship Between Concentration in Single Strands of Maternal Hair and Child Effects). Arch Neurol 1987; 44(10): 1017–22. [DOI] [PubMed] [Google Scholar]
- [96].Myers GJ, Davidson PW, Shamlaye CF, et al. Effects of prenatal methylmercury exposure from a high fish diet on developmental milestones in the Seychelles Child Development Study. Neurotoxicol 1997; 18(3): 819–29. [PubMed] [Google Scholar]
- [97].Davidson PW, Myers GJ, et al. (Effects of Prenatal and Postnatal Methylmercury Exposure From Fish Consumption on Neurodevelopment: Outcomes at 66 Months of Age in the Seychelles Child Development Study). JAMA 1998; 280(8): 701–7. [DOI] [PubMed] [Google Scholar]
- [98].Grandjean P, Weihe P, White RF, et al. (Cognitive Deficit in 7-Year-Old Children with Prenatal Exposure to Methylmercury). Neurotoxicol Teratol 1997; 19(6): 417–28. [DOI] [PubMed] [Google Scholar]
- [99].Crump KS. A new method for determining allowable daily intakes. Fundam Appl Toxicol 1984; 4(5): 854–71. [DOI] [PubMed] [Google Scholar]
- [100].Axelrad DA, Bellinger DC, Ryan LM, Woodruff TJ. Dose-response relationship of prenatal mercury exposure and IQ: an integrative analysis of epidemiologic data. Environ Health Perspect 2007; 115(4): 609–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Yang J, Jiang Z, Wang Y, Qureshi IA, Wu XD. Maternal-fetal transfer of metallic mercury via the placenta and milk. Ann Clin Lab Sci 1997; 27(2): 135–41. [PubMed] [Google Scholar]
- [102].Hansen JC, Tarp U, Bohm J. Prenatal exposure to methyl mercury among Greenlandic polar Inuits. Arch Environ Health 1990; 45(6): 355–8. [DOI] [PubMed] [Google Scholar]
- [103].Vahter M, Akesson A, Lind B, Bjors U, Schutz A, Berglund M. Longitudinal study of methylmercury and inorganic mercury in blood and urine of pregnant and lactating women, as well as in umbilical cord blood. Environ Res 2000; 84(2): 186–94. [DOI] [PubMed] [Google Scholar]
- [104].Stern AH, Smith AE. An assessment of the cord blood: maternal blood methylmercury ratio: implications for risk assessment. Environ Health Perspect 2003; 111(12): 1465–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Hightower JM, O’Hare A, Hernandez GT. Blood mercury reporting in NHANES: identifying Asian, Pacific Islander, Native American, and multiracial groups. Environ Health Perspect 2006; 114(2): 173–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Mahaffey KR, Clickner RP, Jeffries RA. Adult women’s blood mercury concentrations vary regionally in the United States: association with patterns of fish consumption (NHANES 1999–2004). Environ Health Perspect 2009; 117(1): 47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Stern AH. A revised probabilistic estimate of the maternal methyl mercury intake dose corresponding to a measured cord blood mercury concentration. Environ Health Perspect 2005; 113(2): 155–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].McKelvey W, Gwynn RC, Jeffery N, et al. A biomonitoring study of lead, cadmium, and mercury in the blood of New York city adults. Environ Health Perspect 2007; 115(10): 1435–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Castoldi AF, Johansson C, Onishchenko N, et al. Human developmental neurotoxicity of methylmercury: impact of variables and risk modifiers. Regul Toxicol Pharmacol 2008; 51(2): 201–14. [DOI] [PubMed] [Google Scholar]
- [110].He K. Fish, long-chain omega-3 polyunsaturated fatty acids and prevention of cardiovascular disease--eat fish or take fish oil supplement? Prog Cardiovasc Dis 2009; 52(2): 95–114. [DOI] [PubMed] [Google Scholar]
- [111].Budtz-Jorgensen E, Grandjean P, Weihe P. Separation of risks and benefits of seafood intake. Environ Health Perspect 2007; 115(3): 323–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Rice DC. Overview of modifiers of methylmercury neurotoxicity: chemicals, nutrients, and the social environment. Neurotoxicology 2008; 29(5): 761–6. [DOI] [PubMed] [Google Scholar]
- [113].Huang L-S, Myers GJ, Davidson PW, et al. Is susceptibility to prenatal methylmercury exposure from fish consumption non-homogeneous? Tree-structured analysis for the Seychelles Child Development Study. Neurotoxicology 2007; 28(6): 1237–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Salonen JT, Seppanen K, Nyyssonen K, et al. Intake of Mercury From Fish, Lipid Peroxidation, and the Risk of Myocardial Infarction and Coronary, Cardiovascular, and Any Death in Eastern Finnish Men. Circulation 1995; 91(3): 645–55. [DOI] [PubMed] [Google Scholar]
- [115].Sorensen N, Murata K, Budtz-Jorgensen E, Weihe P, Grandjean P. Prenatal Methylmercury Exposure as a Cardiovascular Risk Factor at Seven Years of Age. Epidemiology 1999; 10(4): 370–5. [PubMed] [Google Scholar]
- [116].Yoshizawa K, Rimm EB, Morris JS, et al. Mercury and the Risk of Coronary Heart Disease in Men. N Engl J Med 2002; 347(22): 1755–60. [DOI] [PubMed] [Google Scholar]
- [117].Guallar E, Sanz-Gallardo MI, Veer Pvt, et al. Mercury, Fish Oils, and the Risk of Myocardial Infarction. N Engl J Med 2002; 347(22): 1747–54. [DOI] [PubMed] [Google Scholar]
- [118].Ohno T, Sakamoto M, Kurosawa T, Dakeishi M, Iwata T, Murata K. Total mercury levels in hair, toenail, and urine among women free from occupational exposure and their relations to renal tubular function. Environ Res 2007; 103(2): 191–7. [DOI] [PubMed] [Google Scholar]
- [119].Rees JR, Sturup S, Chen C, Folt C, Karagas MR. Toenail mercury and dietary fish consumption. J Expos Sci Environ Epidemiol 2006; 17(1): 25–30. [DOI] [PubMed] [Google Scholar]
- [120].Virtanen JK, Rissanen TH, Voutilainen S, Tuomainen T-P. Mercury as a risk factor for cardiovascular diseases. Nutr Biochem 2007; 18(2): 75–85. [DOI] [PubMed] [Google Scholar]
- [121].Choi AL, Weihe P, Budtz-Jorgensen E, et al. Methylmercury exposure and adverse cardiovascular effects in Faroese whaling men. Environ Health Perspect 2009; 117(3): 367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Valera B, Dewailly E, Poirier P. Environmental Mercury Exposure and Blood Pressure Among Nunavik Inuit Adults Hypertension 2009; 54(5): 981–6. [DOI] [PubMed] [Google Scholar]
- [123].Yorifuji T, Tsuda T, Kashima S, Takao S, Harada M. Long-term exposure to methylmercury and its effects on hypertension in Minamata. Environ Res 2009; 110(1): 40–6. [DOI] [PubMed] [Google Scholar]
- [124].Mozaffarian D, Rimm EB. Fish intake, contaminants, and human health: evaluating the risks and the benefits. JAMA 2006; 296(15): 1885–99. [DOI] [PubMed] [Google Scholar]
- [125].Ginsberg GL, Toal BF. Quantitative approach for incorporating methylmercury risks and omega-3 fatty acid benefits in developing species-specific fish consumption advice. Environ Health Perspect 2009; 117(2): 267–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Mahaffey KR, Clickner RP, Jeffries RA. Methylmercury and omega-3 fatty acids: Co-occurrence of dietary sources with emphasis on fish and shellfish. Environ Res 2008; 107(1): 20–9. [DOI] [PubMed] [Google Scholar]
- [127].Domingo JL, Bocio A, Martí-Cid R, Llobet JM. Benefits and risks of fish consumption: Part II. RIBEPEIX, a computer program to optimize the balance between the intake of omega-3 fatty acids and chemical contaminants. Toxicology 2007; 230(2–3): 227–33. [DOI] [PubMed] [Google Scholar]
- [128].Yasutake A, Matsumoto M, Yamaguchi M, Hachiya N. Current Hair Mercury Levels in Japanese: Survey in Five Districts. Tohoku J Exp Med 2003; 199(3): 161–9. [DOI] [PubMed] [Google Scholar]
- [129].Hartung T A toxicology for the 21st century--mapping the road ahead. Toxicol Sci 2009; 109(1): 18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].United States Environmental Protection Agency. Comments on the Use of Data From the Testing of Human Subjects: A Report from the Science Advisory Board and the FIFRA Scientific Advisory Panel. Washington DC: 2000. Contract No.: EPA-SAB-EC-00–017. [Google Scholar]
- [131].Bateson TF, ed Current Practices for Characterizing Exposure Measurement Error in Epidemiologic Studies and Related Uncertainties in Health Assessments -Case Studies National Research Council Workshop: Quantitative Methods for Assessing Exposure Measurement Error in Epidemiologic Studies: Implications for Environmental Hazard Identification and Risk Analysis Washington DC: 2008. [Google Scholar]
- [132].United States Environmental Protection Agency. Formaldehyde (CASRN 50–00-0) Washington DC: 1998; [cited 2010 March 16]; Available from: http://www.epa.gov/ncea/iris/subst/0419.htm. [Google Scholar]
- [133].United States Environmental Protection Agency. Benzene (CASRN 71–43-2) Washington DC: 2000; [Cited 2010 March 16]; Available from: http://www.epa.gov/NCEA/iris/subst/0276.htm. [Google Scholar]
- [134].United States Environmental Protection Agency. 1,3-Butadiene (CASRN 106–99-0) Washington DC: 2002; [cited 2010 March 16]; Available from: http://www.epa.gov/NCEA/iris/subst/0139.htm. [Google Scholar]
- [135].Daniels MJ, Dominici F, Zeger SL, Samet JM. The National Morbidity, Mortality, and Air Pollution Study. Part III: PM10 concentration-response curves and thresholds for the 20 largest US cities. Res Rep Health Eff Inst 2004(94 Pt 3): 1–21; discussion 3–30. [PubMed] [Google Scholar]
- [136].Bell ML, Peng RD, Dominici F. The exposure-response curve for ozone and risk of mortality and the adequacy of current ozone regulations. Environ Health Perspect 2006; 114(4): 532–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Centers for Disease Control and Prevention. Preventing Lead Poisoning in Young Children Atlanta, Georgia: 2005. [Google Scholar]
- [138].United States Department of Health and Human Services. The Health Consequences of Involuntary Exposure to Tobacco Smoke: A Report of the Surgeon General. In: U.S. Department of Health and Human Services Centers for Disease Control & Prevntion, Coordinating Center for Health Promotion, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health Atlanta: Georgia: 2006. [PubMed] [Google Scholar]
- [139].Darby S, Hill D, Auvinen A, et al. Radon in homes and risk of lung cancer: collaborative analysis of individual data from 13 European case-control studies. BMJ 2005; 330(7485): 223-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Krewski D, Lubin JH, Zielinski JM, et al. Residential Radon and Risk of Lung Cancer: A Combined Analysis of 7 North American Case-Control Studies. Epidemiology 2005; 16(2): 137–45 10.1097/01.ede.0000152522.80261.e3. [DOI] [PubMed] [Google Scholar]
- [141].Allee N Supporting Decisions With Best Evidence 2005. [Public Health Information and Data: A Training Manual]; Available from: http://phpartners.org/pdf/BestEvidenceChapterPDF.pdf.
- [142].Cochrane Collaboration. Cochrane Reviews: What are Cochrane Reviews? 2010. [cited 2010 March 29]; Available from: http://www.cochrane.org/cochrane-reviews.
- [143].Cook DJ, Mulrow CD, Haynes RB. Systematic reviews: synthesis of best evidence for clinical decisions. Ann Intern Med 1997; 126(5): 376–80. [DOI] [PubMed] [Google Scholar]
- [144].Simunovic N, Sprague S, Bhandari M. Methodological Issues in Systematic Reviews and Meta-Analyses of Observational Studies in Orthopaedic Research. J Bone Joint Surg Am 2009; 91(Suppl 3): 87–94. [DOI] [PubMed] [Google Scholar]
- [145].Guyatt GH, Oxman AD, Vist GE, et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations BMJ 2008; 336(7650): 924–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Cory-Slechta DA. Bridging human and experimental animal studies of lead neurotoxicity: Moving beyond IQ. Neurotoxicol Teratol 1995; 17(3): 219–21. [DOI] [PubMed] [Google Scholar]