

Autophagy-dependent cell death may hold the key to understanding the molecular basis of ferroptosis.
Abstract
Ferroptosis is a form of nonapoptotic regulated cell death driven by iron-dependent lipid peroxidation. Autophagy involves a lysosomal degradation pathway that can either promote or impede cell death. A high level of autophagy has been associated with ferroptosis, but the mechanisms underpinning this relationship are largely elusive. We characterize the contribution of autophagy to ferroptosis in human cancer cell lines and mouse tumor models. We show that “clockophagy,” the selective degradation of the core circadian clock protein ARNTL by autophagy, is critical for ferroptosis. We identify SQSTM1 as a cargo receptor responsible for autophagic ARNTL degradation. ARNTL inhibits ferroptosis by repressing the transcription of Egln2, thus activating the prosurvival transcription factor HIF1A. Genetic or pharmacological interventions blocking ARNTL degradation or inhibiting EGLN2 activation diminished, whereas destabilizing HIF1A facilitated, ferroptotic tumor cell death. Thus, our findings reveal a new pathway, initiated by the autophagic removal of ARNTL, that facilitates ferroptosis induction.
INTRODUCTION
Cell death plays a major role in physiological and pathological processes, meaning that derangements in its molecular control contribute to the pathogenesis of human diseases. Different stimuli can trigger different types of cell death, which are categorized into accidental cell death (ACD) and regulated cell death (RCD) (1). Unlike ACD, which is an uncontrolled process, RCD is a tightly fine-tuned process that is accompanied by stereotyped biochemical and morphological alterations that reflect the activation of distinct RCD subroutines (1). Apart from apoptosis, defined in 1972, a number of new forms of RCD have been identified over the past decade. Among them, ferroptosis was described in cancer cells in 2012. This nonapoptotic form of RCD is driven by the iron-induced production of reactive oxygen species (ROS) and subsequent lipid peroxidation (2). Small-molecule compounds such as erastin or RSL3 have been established as pharmacological stimulators of ferroptosis that act by inhibiting the cystine-glutamate exchanger system xc− or the lipid hydroperoxidase GPX4 (glutathione peroxidase 4) (2, 3). Although GPX4 plays an important role in the suppression of ferroptosis, Gpx4 depletion may also be involved in the induction of apoptosis (4), necroptosis (5), and pyroptosis (6) under some conditions. The therapeutic induction or inhibition of ferroptosis is becoming an attractive strategy for intervening in human diseases including cancer, neurodegeneration, and ischemic disease (7, 8).
Macroautophagy (hereafter referred to as autophagy) is a phylogenetically ancient degradation process relying on the formation of specialized membrane structures including phagophores, autophagosomes, and autolysosomes (9). Autophagy plays a complex role in human health and disease (10). At the molecular level, autophagy is executed by autophagy-related (ATG) proteins that can undergo multiple posttranslational modifications (11). Autophagy often precedes cell death that it may either postpone or accelerate, depending on the precise circumstances. Autophagy can either act nonspecifically to remove cytoplasmic structures or selectively degrade substrates such as aggregated proteins, damaged organelles, and invading pathogens. This selective autophagy requires a cargo receptor for the recognition of its substrate (12). Recently, accumulating evidence indicates that autophagy contributes to ferroptosis (13). However, the precise molecular mechanisms that link ferroptosis to autophagy remain poorly understood.
The circadian rhythm is the endogenous oscillating mechanism that controls various cellular processes, including iron metabolism, oxidative stress, and cell death (14). The transcription factor ARNTL/BMAL1 (aryl hydrocarbon receptor nuclear translocator-like protein 1/brain and muscle ARNT-like 1) is a central component of the mammalian circadian clock because it regulates the expression of other clock-controlled genes such as those coding for the PER (period circadian regulator) and CRY (cryptochrome circadian regulator) families (15). Although the disruption of circadian clock signaling is involved in apoptosis (14), the expression and function of ARNTL in ferroptosis remain unknown.
In this study, we provide the first evidence that a novel mode of selective autophagy, “clockophagy,” i.e., the autophagic degradation of the key circadian clock protein ARNTL depending on the cargo receptor SQSTM1/p62 (sequestosome 1), markedly promotes ferroptosis through EGLN2/PHD1 (egl nine homolog 2/hypoxia-inducible factor prolyl hydroxylase 1)–mediated oxidative injury. The disruption of the ARNTL pathway improves the anticancer activity of ferroptosis activators in vitro and in vivo. These findings shed new light on the molecular mechanism of autophagy-dependent cell death.
RESULTS
Selective degradation of ARNTL in ferroptosis
We investigated the effects of type 1 and type 2 ferroptosis inducers on the expression of ARNTL in human cancer cell lines. Type 1 ferroptosis activators are system xc− inhibitors such as erastin, sulfasalazine, and sorafenib. Type 2 ferropoptosis inducers are GPX4 inhibitors including RSL3 and FIN56. Consistent with previous studies (2, 16), Calu-1 (a human non–small cell lung cancer cell line) cells were sensitive to these ferroptosis activators, whereas THP1 (a human acute monocytic leukemia cell line) cells were resistant to them (Fig. 1A). Immunoblot analyses revealed that the protein expression of ARNTL was suppressed by type 2 activators (RSL3 and FIN56) but not by type 1 activators (erastin, sulfasalazine, and sorafenib) in Calu-1 cells (Fig. 1B). RSL3 and FIN56, but not erastin, also suppressed ARNTL protein expression in other ferroptosis-sensitive cell lines including HT1080 (a human fibrosarcoma cell line) and HL-60 (a human promyelocytic leukemia cell line) (Fig. 1C). In contrast, THP1 cells were refractory to alterations in the protein expression of ARNTL following treatment with type 1 and type 2 activators (Fig. 1B).
Fig. 1. Selective degradation of ARNTL in ferroptosis.
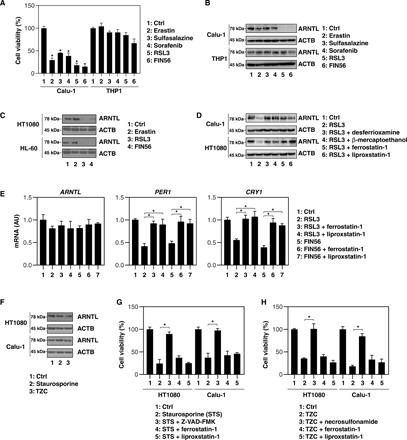
(A) Cell viability of Calu-1 and THP1 cells following treatment with erastin (10 μM), sulfasalazine (500 μM), sorafenib (10 μM), RSL3 (0.5 μM), or FIN56 (5 μM) for 12 hours (n = 3, *P < 0.05 versus control group). (B) In parallel, Western blot analyses were conducted to assess the expression of the indicated proteins in Calu-1 and THP1 cells. (C) Immunoblot analysis of the indicated proteins in HT1080 and HL-60 cells following treatment with erastin (10 μM), RSL3 (0.5 μM), or FIN56 (5 μM) for 12 hours. (D) Western blot analysis of the indicated proteins in HT1080 and Calu-1 cells following treatment with RSL3 (0.5 μM) for 12 hours in the absence or presence of desferrioxamine (10 μM), β-mercaptoethanol (5 μM), ferrostatin-1 (0.5 μM), or liproxstatin-1 (0.5 μM). (E) Quantitative polymerase chain reaction (qPCR) analysis of the indicated mRNAs in HT1080 cells following treatment with RSL3 (0.5 μM) or FIN56 (5 μM) for 12 hours in the absence or presence of ferrostatin-1 (0.5 μM) or liproxstatin-1 (0.5 μM) (n = 3, *P < 0.05). (F) Western blot analysis of the indicated proteins in HT1080 and Calu-1 cells following treatment with staurosporine (1 μM) or TZC [TNF (50 nM), ZVAD-FMK (20 μM), and cycloheximide (10 μg/ml)] for 12 hours. (G) Viability of HT1080 cells following treatment with staurosporine (1 μM) for 12 hours in the absence or presence of Z-VAD-FMK (20 μM), ferrostatin-1 (0.5 μM), or liproxstatin-1 (0.5 μM) (n = 3, *P < 0.05). (H) Viability of HT1080 cells after treatment with TZC [TNF (50 nM), ZVAD-FMK (20 μM), and cycloheximide (10 μg/ml)] for 12 hours in the absence or presence of necrosulfonamide (1 μM), ferrostatin-1 (0.5 μM), or liproxstatin-1 (0.5 μM) (n = 3, *P < 0.05). AU, arbitrary units.
We next investigated whether pharmacological blockade of ferroptosis inhibits ARNTL down-regulation in ferroptosis-suscpetible Calu-1 and HT1080 cells. Ferroptosis inhibitors, such as desferrioxamine, β-mercaptoethanol, ferrostatin-1, and liproxstatin-1, reversed RSL3-induced ARNTL protein down-regulation in these cell lines (Fig. 1D). The mRNA level of ARNTL was not remarkably changed by RSL3 and FIN56 in the absence or presence of ferrostatin-1 or liproxstatin-1 (Fig. 1E). In contrast, the mRNA of ARNTL-targeted clock genes such as PER1 and CRY1 was down-regulated by RSL3 and FIN56, and this effect was reversed by ferrostatin-1 or liproxstatin-1 (Fig. 1E). In addition, typical inducers of apoptosis—e.g., staurosporine—or necroptosis—e.g., TCZ [TNF (tumor necrosis factor), Z-VAD-FMK, and cycloheximide]—failed to induce ARNTL degradation (Fig. 1F). As a positive control, Z-VAD-FMK (a pan caspase inhibitor) and necrosulfonamide [a necroptosis inhibitor targeting MLKL (mixed lineage kinase domain–like pseudokinase)], but not ferrostatin-1 or liproxstatin-1, inhibited staurosporine- and TZC-induced cell death, respectively (Fig. 1, G and H). Collectively, these findings suggest that type 2 ferroptosis activators selectively induce ARNTL protein degradation.
SQSTM1 is a receptor for autophagic ARNTL degradation
Mammalian cells have two intracellular protein degradation pathways, namely the ubiquitin-proteasome system and autophagy. MG-132, a proteasome inhibitor, failed to block RSL3-induced ARNTL protein degradation in Calu-1 and HT1080 cells (Fig. 2A). As a positive control, MG-132 inhibited TNF-induced NFKBIA/IκBα (nuclear factor κB inhibitor α) degradation in THP1 cells (Fig. 2B), which is consistent with previous findings that TNF-induced NFKBIA degradation is proteasome dependent (17). Unlike MG-132, spautin-1 (an early-stage inhibitor of autophagy) and chloroquine (a late-stage inhibitor of autophagy) protected against RSL3-induced ARNTL protein degradation in Calu-1 and HT1080 cells (Fig. 2A). These findings indicate that autophagy, but not proteasomes, may contribute to ARNTL protein degradation during ferroptosis.
Fig. 2. Contribution of SQSTM1 to the autophagic degradation of ARNTL.
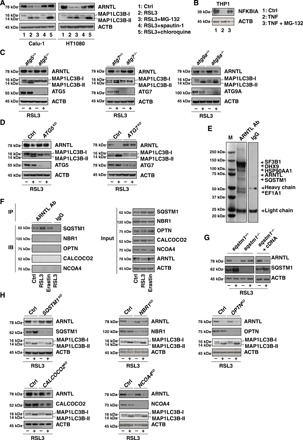
(A) Western blot analysis of the indicated proteins in HT1080 and Calu-1 cells following treatment with RSL3 (0.5 μM) in the absence or presence of MG-132 (2.5 μM), spautin-1 (5 μM), or chloroquine (50 μM) for 12 hours. (B) Western blot analysis of the indicated proteins in THP1 cells after treatment with TNF (10 ng/ml) in the absence or presence of MG-132 (2.5 μM) for 1 hour. (C) Western blot analysis of the indicated proteins in wild-type, atg5−/−, atg7−/−, and atg9a−/− mouse embryonic fibroblasts (MEFs) after treatment with RSL3 (0.5 μM) for 12 hours. (D) Western blot analysis of the indicated protein expression in control, ATG5 knockdown (ATG5KD), and ATG7 knockdown (ATG7KD) HT1080 cells following treatment with RSL3 (0.5 μM) for 12 hours. (E) Mass spectrometry analysis identified SQSTM1 as a direct binding protein of ARNTL in HT1080 cells. These are gels of the proteins that bind to ARNTL, stained with Coomassie Brilliant Blue. Ab, antibody; IgG, immunoglobulin G. (F) Immunoprecipitation (IP) analysis of ARNTL-binding proteins in HT1080 cells following treatment with erastin (10 μM) or RSL3 (0.5 μM) for 6 hours. IB, immunoblot. (G) Western blot analysis of the indicated proteins in wild-type cells, sqstm1−/− MEFs, or sqstm1−/− cells transfected with Sqstm1 complementary DNA (cDNA) (sqstm1−/− + cDNA) following treatment with RSL3 (0.5 μM) for 12 hours. (H) Western blot analysis of the indicated proteins in control and the indicated gene knockdown HT1080 cells following treatment with RSL3 (0.5 μM) for 12 hours. ACTB, actin beta.
We next addressed which autophagy pathway is involved in the regulation of ARNTL degradation. ATG5 and ATG7 are essential for starvation-induced autophagosome formation. The knockout of Atg5 or Atg7 inhibited the conversion of MAP1LC3B (microtubule-associated protein 1 light chain 3β)–I to MAP1LC3B-II (a marker of autophagosome formation), as well as ARNTL degradation in mouse embryonic fibroblasts (MEFs) responding to RSL3 (Fig. 2C). Similarly, the knockdown of Atg5 or Atg7 by specific short hairpin RNAs (shRNAs) suppressed RSL3-induced MAP1LC3B-II production and ARNTL degradation in HT1080 cells (Fig. 2D). However, the knockout of Atg9a, the transmembrane core ATG protein, which supplies membrane from vesicles to autophagosomes, failed to block these processes (Fig. 2C). Thus, ATG5 and ATG7, but not ATG9A, contribute to autophagosome formation and subsequent autophagic degradation of ARNTL during RSL3-induced ferroptosis.
Specific cargo receptors are involved in selective autophagy (12). SQSTM1 is a multifunctional cargo receptor implicated in the autophagic degradation of ubiquitinated substrates, including proteins and organelles. Mass spectrometry analysis identified SQSTM1 as an interactor of ARNTL under normal conditions (Fig. 2E). Immunoprecipitation analysis revealed that the SQSTM1-ARNTL interaction increased in RSL3-induced but not erastin-induced ferroptosis (Fig. 2F). In contrast, ARNTL failed to bind to other cargo receptors such as NBR1 (NBR1, autophagy cargo receptor), OPTN (optineurin), CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), and NCOA4 (nuclear receptor coactivator 4) be it in the absence or presence of RSL3 (Fig. 2F). Sqstm1 deletion diminished RSL3-induced ARNTL degradation in MEFs (Fig. 2G). Conversely, the expression of Sqstm1 complementary DNA (cDNA) in sqstm1−/− MEFs restored RSL3-induced ARNTL degradation (Fig. 2G). The knockdown of Sqstm1 (but not Nbr1, Optn, Calcoco2, or Ncoa4) by shRNA also blocked RSL3-induced ARNTL protein degradation (but not MAP1LC3B-II expression) in HT1080 cells (Fig. 2H). Immunoprecipitation analysis further found that the ubiquitin-associated (UBA) domain of SQSTM1 was required for SQSTM1-ARNTL interaction in the absence or presence of RSL3 (fig. S1). Unlike the down-regulation of SQSTM1 that occurs in starvation-induced bulk autophagy (18), SQSTM1 up-regulation was observed in RSL3-induced selective autophagy (Fig. 2, F to H), indicating that SQSTM1 changes can be cell type and context specific.
Confocal microscopy analysis further found that the colocalization between MAP1LC3B, SQSTM1, LAMP2 (lysosomal-associated membrane protein 2), and ARNTL was enhanced by RSL3 but not by erastin (fig. S2, A to C). Moreover, RSL3-induced colocalization between ARNTL, MAP1LC3B, and SQSTM1 (but not LAMP2) was enhanced by chloroquine (fig. S2, A to C). Western blot analysis of lysosomal fractions confirmed an increased expression of ARNTL in response to RSL3 but not erastin (fig. S2D). Collectively, these findings suggest that SQSTM1 is a direct receptor for the autophagic degradation of ARNTL in lysosome during RSL3-induced ferroptosis.
Autophagy-mediated ARNTL degradation promotes ferroptosis
We next investigated the impact of autophagy-mediated ARNTL degradation on ferroptosis. First, ARNTL was overexpressed by gene transfection in ferroptosis-sensitive cell lines (Calu-1 and HT1080) (Fig. 3A). The overexpression of ARNTL reduced RSL3- and FIN56-induced malondialdehyde (MDA; an end-product of lipid peroxidation) production (Fig. 3B) and cell death (Fig. 3C). Although the basic expression of ARNTL was not affected by type 1 activators (erastin, sulfasalazine, and sorafenib) (Fig. 1B), the overexpression of ARNTL inhibited type 1 activator–induced cell death and MDA production in Calu-1 and HT1080 cells (Fig. 3, B and C), indicating an ARNTL expression threshold effect on type 1 activator–induced ferroptosis. In contrast, knockdown of Arntl by two different shRNAs (Fig. 3D) restored MDA production (Fig. 3E) and cell death induction by type 1 and type 2 ferroptosis activators (Fig. 3F) in THP1 cells, indicating that ARNTL depletion can overcome resistance to ferroptosis.
Fig. 3. Autophagy-mediated ARNTL degradation promotes ferroptosis.
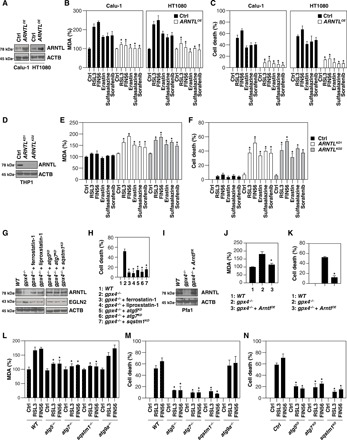
(A) Western blot analysis of the indicated proteins in control and ARNTL-overexpressing (ARNTLOE) HT1080 and Calu-1 cells. (B and C) Analysis of MDA levels (B) and cell death (C) in control and ARNTL-overexpressing (ARNTLOE) HT1080 and Calu-1 cells following treatment with erastin (10 μM), sulfasalazine (500 μM), sorafenib (10 μM), RSL3 (0.5 μM), or FIN56 (5 μM) for 12 hours (n = 3, *P < 0.05 versus control group). (D) Western blot analysis of the indicated proteins in control and ARNTL knockdown (ARNTLKD) THP1 cells. (E and F) Analysis of MDA levels (E) and cell death (F) in control and ARNTL knockdown (ARNTLKD) THP1 cells following treatment with erastin (10 μM), sulfasalazine (500 μM), sorafenib (10 μM), RSL3 (0.5 μM), or FIN56 (5 μM) for 12 hours (n = 3, *P < 0.05 versus control group). (G) Western blot analysis of the indicated proteins in gpx4−/− Pfa1 cells cultured in the absence or presence of ferroptosis inhibitors [e.g., ferrostatin-1 (0.5 μM, 24 hours) and liproxstatin-1 (0.5 μM, 24 hours)] or the knockdown of Atg5, Atg7, or Sqstm1. (H) Cell death in the setting of (G) (n = 3, *P < 0.05 versus single gpx4−/− group). (I) Western blot analysis of the indicated proteins in gpx4−/− Pfa1 cells with or without ARNTL overexpression. (J and K) MDA levels (J) and cell death (K) in the setting of (I) (n = 3, *P < 0.05 versus single gpx4−/− group). (L and M) MDA levels (L) and cell death (M) in MEFs with the indicated genotypes following treatment with RSL3 (0.5 μM) or FIN56 (5 μM) for 12 hours [n = 3, *P < 0.05 versus control wild-type (WT) group]. (N) Cell death in a panel of gene knockdown HT1080 cells following treatment with RSL3 (0.5 μM) or FIN56 (5 μM) for 12 hours (n = 3, *P < 0.05 versus control group).
GPX4 is a central negative regulator of lipid peroxidation under various stress conditions. A previous study has shown that the inducible knockout of Gpx4 leads to ferroptotic cell death in Pfa1 cells (19). Similar to RSL3 treatment (Fig. 2A), the knockout of Gpx4 increased MAP1LC3B-II production (fig. S3). We observed that ARNTL down-regulation (Fig. 3G) and cell death (Fig. 3H) in gpx4−/− Pfa1 cells were reversed by ferroptosis inhibitors (e.g., ferrostatin-1 and liproxstatin-1) or the knockdown of Atg5, Atg7, or Sqstm1. Transfection-enforced overexpression of ARNTL (Fig. 3I) decreased MDA production (Fig. 3J) and cell death in gpx4−/− Pfa1 cells (Fig. 3K), indicating that Gpx4 depletion–mediated ARNTL protein degradation is required for ferroptosis. Moreover, the knockout of Atg5, Atg7, or Sqstm1 (but not Atg9a) reduced RSL3- and FIN56-induced MDA production (Fig. 3L) and cell death in MEFs (Fig. 3M). The knockout of Atg5, Atg7, or Sqstm1 also reduced RSL3- and FIN56-induced cell death in HT1080 cells (Fig. 3N). Collectively, these findings suggest that autophagy-mediated ARNTL degradation promotes ferroptosis by the activation of lipid peroxidation.
ARNTL-mediated EGLN2 down-regulation blocks ferroptosis
ARNTL is a circadian transcription factor that regulates gene expression mainly via the binding of E-box motifs (CAGCTG or CACGTG) in their promoters. The Eukaryotic Promoter Database (https://epd.vital-it.ch/index.php) lists 1666 human genes with E-box motifs. The gene ontology analysis of gene clusters by DAVID (Database for Annotation, Visualization and Integrated Discovery; https://david.ncifcrf.gov/) online tool further revealed that 12 E-box–containing genes—EGLN2, NNT (nicotinamide nucleotide transhydrogenase), DNAJC16 [DnaJ heat shock protein family (Hsp40) n member C16], CUL5 (cullin 5), TXNRD1 (thioredoxin reductase 1), MLYCD [malonyl–CoA (coenzyme A) decarboxylase], GLRX5 (glutaredoxin 5), SH3BGR (SH3 domain binding glutamate rich protein), PPARG (peroxisome proliferator–activated receptor γ), PDIA5 (protein disulfide isomerase family A member 5), DIO2 (iodothyronine deiodinase 2), and ERO1B (endoplasmic reticulum oxidoreductase 1β)—may be involved in the regulation of oxidative stress. We used quantitative polymerase chain reactions (qPCRs) to determine whether these genes are directly controlled by ARNTL as part of ferroptosis. EGLN2 mRNA was up-regulated in both Calu-1 and HT1080 cells responding to RSL3 (Fig. 4A). In contrast, the overexpression of ARNTL blocked RSL3-induced EGLN2 up-regulation in both Calu-1 and HT1080 cells (Fig. 4A). mRNAs coding for other EGLN family members, including EGLN1 and EGLN3, did not undergo any major changes in their abundance after treatment with RSL3 and/or following ARNTL overexpression (Fig. 4A). Accordingly, a chromatin immunoprecipitation (ChIP) assay also found that ARNTL bonded to the promoter of EGLN2 (but not EGLN1 and EGLN3) in Calu-1 and HT1080 cells (Fig. 4B). Reporter gene (Fig. 4C) and Western blot (Fig. 4D) analyses confirmed that EGLN2 was transrepressed by ARNTL in RSL3-induced ferroptosis. Thus, the knockdown of ARNTL by shRNA increased EGLN2 expression in Calu-1 and HT1080 cells (Fig. 4E). In addition to ARNTL down-regulation (Fig. 3G), EGLN2 up-regulation (Fig. 3G) in gpx4−/− Pfa1 cells was also reversed by ferroptosis inhibitors (e.g., ferrostatin-1 and liproxstatin-1) or the knockdown of Atg5, Atg7, or Sqstm1, supporting that autophagy regulates ARNTL and EGLN2 expression in response to Gpx4 depletion–induced lipid peroxidation.
Fig. 4. ARNTL-mediated EGLN2 down-regulation blocks ferroptosis.
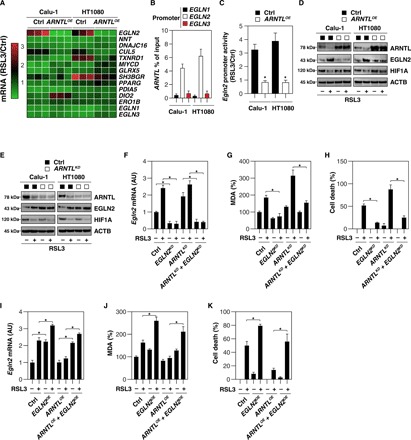
(A) Heat map of mRNA expression levels in control or ARNTL-overexpressing (ARNTLOE) HT1080 and Calu-1 cells following treatment with RSL3 (0.5 μM) for 12 hours. (B) Binding of ARNTL to EGLN1, EGLN2, or EGLN3 promoter was analyzed by ChIP-qPCR in HT1080 or Calu-1 cells (n = 3). (C) EGLN2 promoter activity in control or ARNTL-overexpressing (ARNTLOE) HT1080 and Calu-1 cells after treatment with RSL3 (0.5 μM) for 12 hours (n = 3, *P < 0.05 versus control group). (D) Western blot analysis of the indicated protein expression in control and ARNTL-overexpressing (ARNTLOE) HT1080 and Calu-1 cells upon treatment with RSL3 (0.5 μM) for 12 hours. (E) Western blot analysis of the indicated proteins in control and ARNTL knockdown (ARNTLKD) HT1080 and Calu-1 cells following treatment with RSL3 (0.5 μM) for 12 hours. (F to H) Analysis of EGLN2 mRNA (F), MDA levels (G), and cell death (H) in the indicated gene knockdown HT1080 cells after treatment with RSL3 (0.5 μM) for 12 hours (n = 3, *P < 0.05). (I to K) Analysis of EGLN2 mRNA (I), MDA level (J), and cell death (K) in the indicated gene-overexpressing HT1080 cells following treatment with RSL3 (0.5 μM) for 12 hours (n = 3, *P < 0.05).
To determine whether the up-regulation of EGLN2 contributes to ferroptosis, we knocked down EGLN2 by shRNA in Calu-1 and HT1080 cells. The suppression of EGLN2 expression (Fig. 4F) limited RSL3-induced MDA production (Fig. 4G) and cell death (Fig. 4H) in control and ARNTL knockdown HT1080 cells. In contrast, transfection-enforced EGLN2 overexpression (Fig. 4I) increased RSL3-induced MDA production (Fig. 4J) and cell death (Fig. 4K) both in control and in ARNTL-overexpressing HT1080 cells. Collectively, these findings support the hypothesis that ARNTL suppresses ferroptosis through the down-regulation of EGLN2 expression.
EGLN2-mediated HIF1A down-regulation promotes ferroptosis
HIF1A (hypoxia-inducible factor 1 subunit α) is a transcription factor that mediates homeostatic responses to reduced oxygen availability in the microenvironment. Given that the major function of EGLN2 is to suppress HIF1A activation (20), we sought to determine whether the expression of HIF1A is regulated by ARNTL-mediated EGLN2 down-regulation. ARNTL overexpression inhibited EGLN2 expression, which in turn sustained HIF1A expression in Calu-1 and HT1080 cells following RSL3 treatment (Fig. 4D). In contrast, ARNTL knockdown promoted EGLN2 expression, correlating with reduced HIF1A expression in Calu-1 and HT1080 cells following RSL3 treatment (Fig. 4E). The knockdown of ARNTL partly reduced HIF1A expression under baseline conditions (Fig. 4E), indicating that other non-ARNTL pathways may contribute to basic HIF1A expression. Genetic or pharmacological inhibition of EGLN by means of a specific shRNA (Fig. 5A) or the administration of adaptaquin (Fig. 5B) increased HIF1A expression in RSL3-treated Calu-1 and HT1080 cells. Together, these findings indicate that ARNTL promotes HIF1A expression through the down-regulation of EGLN2 in RSL3-induced ferroptosis.
Fig. 5. EGLN2-mediated HIF1A down-regulation promotes ferroptosis.
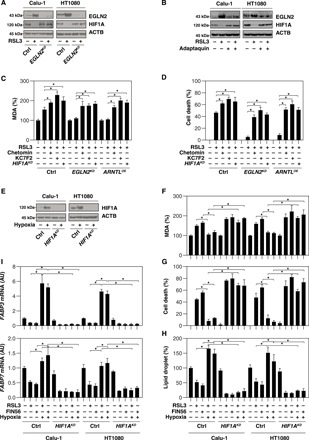
(A) Western blot analysis of the indicated proteins in control and EGLN2 knockdown (EGLN2KD) HT1080 and Calu-1 cells upon treatment with RSL3 (0.5 μM) for 12 hours. (B) Western blot analysis of the indicated protein expression in HT1080 and Calu-1 cells after treatment with RSL3 (0.5 μM) in the absence or presence of adaptaquin (4 μM) for 12 hours. (C and D) Analysis of MDA level (C) and cell death (D) in the indicated HT1080 cells subsequent to treatment with RSL3 (0.5 μM) in the absence or presence of chetomin (0.25 μM) and KC7F2 (25 μM) for 12 hours (n = 3, *P < 0.05). (E) Western blot analysis of the indicated proteins in control and HIF1A knockdown (HIF1AKD) HT1080 and Calu-1 cells following treatment with hypoxia (1% O2) for 24 hours. (F to I) Analysis of MDA level (F), cell death (G), lipid droplet (H), and gene mRNA (I) in the indicated hypoxia (1% O2, 24 hours)–pretreated HT1080 and Calu-1 cells after being cultured with RSL3 (0.5 μM) and FIN56 (5 μM) for 12 hours (n = 3, *P < 0.05).
HIF1A degradation is mediated by the ubiquitin-proteasome pathway (21). As expected, MG-132 inhibited RSL3-induced HIF1A down-regulation but not ARNTL down-regulation and EGLN2 up-regulation in HT1080 cells (fig. S4A). HIF1A can also be induced by iron-chelator desferrioxamine (22). As expected, desferrioxamine restored both ARNTL and HIF1A protein levels with decreased EGLN2 expression in HT1080 cells following RSL3 treatment (fig. S4B). Unlike RSL3 treatment (fig. S4, A and B), the expression of EGLN2 and HIF1A was not changed by erastin treatment (fig. S4C).
We next sought to investigate the function of HIF1A in ferroptosis. The administration of HIF1A inhibitors (e.g., chetomin and KC7F2) or knockdown of HIF1A restored RSL3-induced MDA production (Fig. 5C) and cell death (Fig. 5D) in EGLN2 knockdown or ARNTL-overexpressing HT1080 cells. Furthermore, hypoxia pretreatment induced HIF1A expression (Fig. 5E), as it limited RSL3- and FIN56-induced MDA production (Fig. 5F) and cell death (Fig. 5G) in Calu-1 and HT1080 cells. In contrast, the formation of lipid droplets, the intracellular sites for neutral lipid storage, was restored by hypoxia-induced HIF1A activation in response to RSL3 and FIN56 (Fig. 5H). Moreover, the mRNA expression of FABP3 (fatty acid binding protein 3) and FABP7 (fatty acid binding protein 7), two key HIF1A target genes responsible for fatty acid uptake and lipid storage (23), was restored by HIF1A activation in Calu-1 and HT1080 cells following RSL3 and FIN56 treatment (Fig. 5I). Collectively, these findings confirmed that HIF1A is a prosurvival factor in ferroptosis, whereas EGLN2-mediated HIF1A down-regulation promotes ferroptosis.
The ARNTL pathway regulates ferroptosis in vivo
To determine whether the ARNTL pathway also regulates tumor sensitivity to ferroptosis activators in vivo, we subcutaneously inoculated ARNTL, EGLN2, and HIF1A knockdown HT1080 cells into the right flank of immunodeficient mice. Beginning at day 7, these mice were systemically treated with (1S,3R)-RSL3, a metabolically stable RSL3 derivative that is suitable for in vivo experiments (3), for 2 weeks. Compared with the control shRNA group, RSL3 treatment effectively reduced the size of tumors formed in mice carrying ARNTL or HIF1A knockdown cells (Fig. 6A). In contrast, mice with EGLN2 knockdown cells were resistant to RSL3 treatment. qPCR analyses of the expression of PTGS2 (prostaglandin-endoperoxide synthase 2), a marker for the assessment of ferroptosis in vivo (Fig. 6B) (3) and for the quantification of MDA (Fig. 6C), indicated that the ARNTL and HIF1A knockdown increased while EGLN2 knockdown inhibited ferroptosis in vivo. In contrast, CASP3 (caspase 3; a marker of apoptosis) activity (a marker of apoptosis) was not altered by RSL3 in these gene-deficient tumors (Fig. 6D). Notably, the formation of lipid droplets (Fig. 6E) and the mRNA expression of FABP3 (Fig. 6F) and FABP7 (Fig. 6G) decreased in ARNTL and HIF1A knockdown HT1080 tumors following RSL3 treatment. In contrast, they remained largely unaffected in EGLN2 knockdown HT1080 cells (Fig. 6, E to G).
Fig. 6. Effects of genetic inhibition of ARNTL, EGLN2, and HIF1A on ferroptosis in vivo.
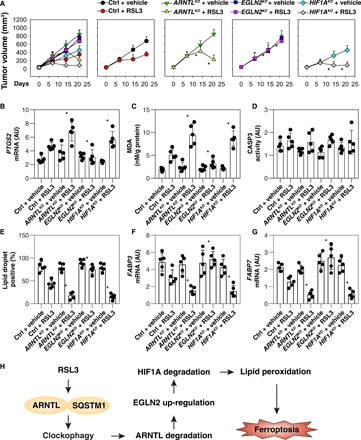
(A) Athymic nude mice were injected subcutaneously with the indicated HT1080 cells for 7 days and then treated with RSL3 (30 mg/kg; intraperitoneally, once every other day) at day 7 for 2 weeks. Tumor volumes were calculated weekly (n = 5 mice per group, *P < 0.05 versus ctrl + RSL3 group). (B to G) In parallel, PTGS2 mRNA (B), MDA level (C), CASP3 activity (D), lipid droplets (E), FABP3 mRNA (F), and FABP7 mRNA (G) in isolated tumors at day 14 after treatment were assayed (n = 5 mice per group, *P < 0.05 versus ctrl + RSL3 group). (H) Schematic summary of the role of clockophagy in the regulation of lipid peroxidation and ferroptosis.
Spautin-1 (an autophagy inhibitor), adaptaquin (an EGLN2 inhibitor), and liproxstatin-1 (a ferroptosis inhibitor) blocked the RSL3-mediated tumor growth reduction (fig. S5A), an effect that was associated with decreased PTGS2 mRNA expression (fig. S5B) and MDA production (fig. S5C). In contrast, the HIF1A inhibitor chetomin enhanced the anticancer activity of RSL3 (fig. S5A), PTGS2 mRNA expression (fig. S5B), and MDA production (fig. S5C), without affecting CASP3 activity (fig. S5D). In parallel, lipid droplet formation (fig. S5E) and the abundance of FABP3 (fig. S5F) and FABP7 (fig. S5G) mRNAs were decreased by chetomin, contrasting with the observation that these parameters increased in response to spautin-1, adaptaquin, and liproxstatin-1. Collectively, these findings indicate that ARNTL antagonizes the anticancer activity of RSL3-mediated ferroptosis in vivo.
DISCUSSION
Excessive ROS generated from oxidative stress has long been implicated in cell death and tissue injury. Regulating oxidative stress by controlled ROS-generating and ROS-scavenging mechanisms represents a promising therapeutic approach in various human diseases, including cancer and aging-associated diseases. Although ferroptosis is a recently identified form of ROS-dependent RCD (2), its molecular mechanism and biochemical functions still remain poorly understood. In this study, we uncovered a novel role for ARNTL in the blockade of ferroptotic cancer cell death through control of the EGLN2-HIF1A pathway (Fig. 6H). Thus, targeting the ARNTL-dependent pathway may represent a potential therapeutic avenue for enhancing ferroptosis-based anticancer therapy.
Lipid peroxidation plays a cardinal role in executing ferroptosis (7, 8). In contrast, antioxidant regulators such as GPX4 (3), system xc− (2), and NFE2L2/NRF2 (nuclear factor, erythroid 2 like 2) (24) act at different levels to limit oxidative injury leading to ferroptosis. We observed that ARNTL is selectively degraded in response to type 2 ferroptosis activators (RSL3 and FIN56). Others have found that RSL3 binds and inactivates GPX4 (3), whereas FIN56 induces GPX4 degradation (25). Although the mechanism of GPX4 degradation remains unclear, the molecular chaperone HSPA5 [heat shock protein family A (Hsp70) member 5] can bind and prevent GPX4 degradation in pancreatic cancer cells (26). The NFE2L2-mediated transactivation of MTIG (metallothionein 1G; a metal-binding protein) and SLC7A11 (solute carrier family 7 member 11) blocks ferroptosis (27, 28). In contrast to type 2 activators, type 1 ferroptosis activator (e.g., erastin, sulfasalazine, and sorafenib), which targets system xc− activity, failed to cause ARNTL degradation, indicating that these two types of stimuli induce different downstream signaling pathways, only one of which culminates in ARNTL degradation (for type 2 activators).
Historically, ferroptosis was described as nonautophagic cell death (2) until it was recently found that ferritinophagy-mediated ferritin degradation can promote ferroptosis via the release of free iron and subsequent Fenton reaction–induced oxidative injury (29, 30). Notably, autophagy plays a dual role in cell survival and cell death depending on the context. On the one hand, free amino acids and fatty acids resulting from the autophagic degradation of unused protein aggregates or damaged organelles can be used for protein synthesis and energy production in adaptive response to environmental stresses (31). On the other hand, autophagy can lead to the degradation of prosurvival proteins, thereby promoting cell death (32). In addition to ferritinophagy (29, 30), the autophagy regulator BECN1 (beclin 1) promotes ferroptosis through the binding and inhibition of system xc−, and this process requires the activation of an energy sensor, adenosine monophosphate–activated protein kinase (33). The inhibition of lysosomal activity and acid hydrolases released into the cytosol after lysosomal membrane permeabilization reportedly prevents ferroptosis (34, 35). Moreover, the degradation of lipid droplets by autophagy promotes RSL3-induced ferroptosis in hepatocytes (36). Our current findings indicate that the degradation of ARNTL (“clockophagy”) by ATG5-, ATG7-, and SQSTM1-dependent selective autophagy promotes ferroptosis through the activation of lipid peroxidation. NCOA4 and SQSTM1 are cargo receptors required for ferritinophagy and clockophagy, respectively, thus favoring ferroptosis through distinct molecular routes. These findings support the notion that ferroptosis is a form of autophagy-dependent cell death that can be driven by free iron accumulation (downstream of ferritinophagy) and transcriptional regulation (downstream of clockophagy).
Our findings raise the possibility that ARNTL serves a previously unknown prosurvival function by inhibiting the expression of EGLN2 during ferroptosis. EGLN2, also known as PHD1 and HPH3, is a member of the EGLN family of proline hydroxylases. Under normal conditions, EGLN2 is an oxygen sensor and hydroxylates proline residues of HIF1A, favoring HIF1A degradation (20). Under hypoxic conditions, EGLN2 activity is decreased, thus increasing HIF1A protein stability (21). HIF1A is a master transcriptional regulator of the hypoxic response and favors cell survival regulation in response to various stresses. Our results indicate that ARNTL increases HIF1A levels and subsequent ferroptosis resistance through the direct down-regulation of EGLN1 expression. HIF1A inhibition restored ferroptosis sensitivity in EGLN1 knockdown or ARNTL-overexpressing cells. Lipid droplets are dynamic storage organelles that are found in most eukaryotic cells. HIF1A promotes lipid storage and reduces fatty acid β-oxidation, which contributes to tumor cell survival (23, 37). Our current data suggest that HIF1A limits ferroptosis, presumably by influencing lipid metabolism and storing lipids in droplets, thus minimizing peroxidation-mediated endomembrane damage (36).
In summary, we report a previously unappreciated mechanism by which selective autophagy promotes ferroptosis. The autophagy-mediated degradation of ARNTL facilitates EGLN2 expression, thus destabilizing the prosurvival factor HIF1A, ultimately favoring lipid peroxidation and cell death. Our results also suggest that targeting this novel ARNTL-EGLN1-HIF1A pathway may enhance the anticancer activity of ferroptosis activators.
MATERIALS AND METHODS
Reagent
Reagent is as described in table S1.
Cell culture
All human tumor cell lines (Calu-1, THP1, HT1080, and HL-60) were obtained from the American Type Culture Collection. atg5−/−, atg7−/−, atg9a−/−, sqstm1−/−, and gpx4−/− cells were gifts from N. Mizushima, M. Komatsu, T. Saitoh, T. Yanagawa, and M. Conrad, respectively. These cell lines were grown in Dulbecco’s modified Eagle’s medium or RPMI-1640 medium with 10% fetal bovine serum, 2 mM l-glutamine, and penicillin and streptomycin (100 U/ml). For hypoxia treatment, Petri dishes containing cells were incubated in a hypoxic chamber (Forma Scientific) with a 94:5:1 mixture of N2/CO2/O2. All cells were mycoplasma free and authenticated using short tandem repeat DNA profiling analysis.
Animal study
To generate murine subcutaneous tumors, 5 × 106 HT1080 cells in 100 μl of phosphate-buffered saline (PBS) were injected subcutaneously at the right of the dorsal midline in 6- to 8-week-old female athymic nude mice (no. 490, Charles River Laboratories). We conducted all animal care and experiments in accordance with the Association for Assessment and Accreditation of Laboratory Animal Care guidelines (www.aaalac.org) and with approval from our institutional animal care and use committee. All mice were housed under a 12-hour light-dark diurnal cycle with controlled temperature (21° to 23°C) and provided a standard rodent diet (no. 5001, LabDiet) and water ad libitum throughout all experiments.
Cytotoxicity assays
Cells were seeded into 96-well plates and incubated with the indicated treatments. Subsequently, 100 μl of fresh medium was added to cells containing 10 μl of Cell Counting Kit-8 solutions (no. CK04, Dojindo Laboratories) and incubated for 1 hour (37°C, 5% CO2). Absorbance at 450 nm was measured using a microplate reader (Tecan). Trypan blue staining was used to assay cell death.
Lysosome isolation
Lysosomes were isolated using a lysosome isolation kit (ab234047, Abcam) according to the manufacturer’s protocol. Briefly, cells were isolated in ice-cold lysosome isolation buffer for 2 min and homogenized using a precooled glass Dounce homogenizer (Sigma-Aldrich). The supernatant was collected by centrifugation (500g, 10 min) at 4°C and layered onto discontinuous density gradient. The lysosomes were further isolated using an ultracentrifuge for 2 hours at 145,000g at 4°C.
Western blot analysis
Cells or lysosomes were lysed three times with 1× cell lysis buffer (no. 9803, Cell Signaling Technology) containing protease inhibitor on ice for 10 min. Protein was quantified using the bicinchoninic acid (BCA) assay (no. 23225, Thermo Fisher Scientific), and 20 to 40 μg of each sample was resolved on 4 to 12% Criterion XT Bis-Tris gels (no. 3450124, Bio-Rad) in XT MES running buffer (no. 1610789, Bio-Rad) and transferred to polyvinylidene difluoride membranes (no. 1620233, Bio-Rad) using the Trans-Blot Turbo Transfer Pack and System. Membranes were blocked with tris-buffered saline with Tween 20 (TBST) containing 5% skim milk for 1 hour and incubated overnight at 4°C with various primary antibodies. Following three washes in TBST, membranes were incubated with goat anti-rabbit or anti-mouse immunoglobulin G (IgG) horseradish peroxidase secondary antibody (no. 7074 or no. 7076, Cell Signaling Technology) at room temperature for 1 hour and washed. Chemiluminescence substrate was applied using the SuperSignal West Pico Chemiluminescent Substrate (no. 34080, Thermo Fisher Scientific) or the SuperSignal West Femto Maximum Sensitivity Substrate (no. 34095, Thermo Fisher Scientific), and blots were analyzed using the ChemiDoc Touch Imaging System (Bio-Rad) and Image Lab Software (Bio-Rad) (38).
Immunoprecipitation analysis
Cells were lysed at 4°C in ice-cold radioimmunoprecipitation assay buffer (no. 9806, Cell Signaling Technology), and cell lysates were cleared by brief centrifugation (13,000g, 15 min). Concentrations of proteins in the supernatant were determined using the BCA assay (no. 23225, Thermo Fisher Scientific). Before immunoprecipitation, samples containing equal amounts of proteins were precleared with protein A agarose beads (4°C, 3 hours; no. sc-2027, Santa Cruz Biotechnology) and subsequently incubated with various irrelevant IgG or specific antibodies (5 μg/ml) in the presence of protein A agarose beads for 2 hours or overnight at 4°C with gentle shaking. Following incubation, agarose beads were washed extensively with PBS, and proteins were eluted by boiling in 2× sodium dodecyl sulfate sample buffer before SDS–polyacrylamide gel electrophoresis.
RNA interference and gene transfection
The transfection of shRNA or cDNA was performed with Lipofectamine 3000 (no. L3000-015, Thermo Fisher Scientific) or the Neon Transfection System (no. MPK5000, Thermo Fisher Scientific) according to the manufacturer’s protocol.
qPCR analysis
Total RNA was extracted and purified from cultured cells using the RNeasy Plus Mini Kit (no. 74136, QIAGEN). First-strand cDNA was synthesized from 1 μg of RNA using the iScript cDNA Synthesis Kit (no. 1708890, Bio-Rad). Briefly, 20-μl reactions were prepared by combining 4 μl of iScript Select reaction mix, 2 μl of gene-specific enhancer solution, 1 μl of reverse transcriptase, 1 μl of gene-specific assay pool (20×, 2 μM), and 12 μl of RNA diluted in ribonuclease-free water. cDNA from various cell samples were then amplified by real-time qPCR with specific primers using the CFX96 Touch Real-Time PCR Detection System (Bio-Rad) with the CFX Manager Software (Bio-Rad).
MDA assay
The relative MDA concentration in cell or tumor lysates was assessed using a Lipid Peroxidation (MDA) Assay Kit (no. ab118970, Abcam) according to the manufacturer’s instructions. Briefly, MDA in the sample reacts with thiobarbituric acid (TBA) to generate an MDA-TBA adduct. The MDA-TBA adduct can be easily quantified colorimetrically (optical density = 532 nm).
CASP3 activity assay
The activity of CASP3 was assayed by the CASP3 Activity Assay Kit (no. ab39383, Abcam) according to the manufacturer’s protocol. Briefly, the assay was based on the detection of cleavage of the fluorogenic substrate DEVD–AFC (7-amino-4-trifluoromethyl coumarin). DEVD-AFC emitted blue light (λ maximum = 400 nm); upon cleavage of the substrate by CASP3 or related caspases, free AFC emitted a yellow-green fluorescence (excitation/emission = 400/505 nm), which can be quantified using a microplate reader (Tecan).
Lipid droplets assay
The level of lipid droplets was assayed using boron-dipyrromethene (BODIPY) 493/503 (no. D3922, Thermo Fisher Scientific) according to the manufacturer’s protocol. Briefly, cells were fixed with 4% paraformaldehyde for 15 min at room temperature and then stained with 2 μM BODIPY 493/503 working solution for 15 min at 37°C. After being washed with PBS, coverslips were mounted and imaged on a laser scanning confocal microscope (ZEISS LSM 800).
Secrete-pair luminescence assay
Calu-1 and HT1080 cells were transfected with pEZX-PG04-Egln2-promoter–Gaussia luciferase/secreted alkaline phosphatase (no. HPRM51834-PG04, GeneCopoeia). After 48 hours, these cells were treated with RSL3 (0.5 μM) for 12 hours. The EGLN2 promoter luciferase activity was measured with a secrete-pair dual luminescence assay kit (no. SPDA-D010, GeneCopoeia) in accordance with the manufacturer’s guidelines.
In-gel trypsin digestion
In-gel trypsin digestion was carried out as previously described (39). Excised gel bands were washed with high-performance liquid chromatography (HPLC) water and destained with 50% acetonitrile (ACN)/25 mM ammonium bicarbonate until there was no visible staining. Gel pieces were dehydrated with 100% ACN, reduced with 10 mM dithiothreitol (DTT) at 56°C for 1 hour, followed by alkylation with 55 mM iodoacetamide (IAA) at room temperature for 45 min in the dark. Gel pieces were then again dehydrated with 100% ACN to remove excess DTT and IAA, rehydrated with trypsin (20 ng/μl)/25 mM ammonium bicarbonate, and digested overnight at 37°C. The resultant tryptic peptides were extracted with 70% ACN/5% formic acid, vacuum dried, and reconstituted in 18 μl of 0.1% formic acid.
Tandem mass spectrometry
Proteolytic peptides from in-gel trypsin digestion were analyzed using nanoflow reverse-phased liquid chromatography tandem mass spectrometry (LC-MS/MS). Tryptic peptides were loaded onto a C18 column [PicoChip column packed with 10.5-cm ReproSil C18 (3 μm and 120 Å) chromatography media with a column with an internal diameter of 75 μm and a tip of 15 μm; New Objective Inc., Woburn, MA, USA] using a Dionex HPLC system (Dionex UltiMate 3000, Thermo Fisher Scientific, San Jose, CA, USA) operated with a double-split system to provide an in-column nanoflow rate (~300 nl/min). Mobile phases used were 0.1% formic acid for A and 0.1% formic acid in ACN for B. Peptides were eluted off the column using a 52-min gradient (2 to 40% B in 42 min, 40 to 95% B in 1 min, 95% B for 1 min, and 2% B for 8 min) and injected into a linear ion trap MS (LTQ XL, Thermo Fisher Scientific) through electrospray.
The LTQ XL was operated in a date-dependent MS/MS mode in which each full MS spectrum [acquired at 30,000 automatic gain control (AGC) targets, 50-ms maximum ion accumulation time, and precursor ion selection range of mass/charge ratio 375 to 1800] was followed by MS/MS scans of the eight most abundant molecular ions determined from full MS scan (acquired on the basis of the setting of 1000 signal thresholds, 10,000 AGC targets, 100-ms maximum accumulation time, 2.0-Da isolation width, 30-ms activation time, and 35% normalized collision energy). Dynamic exclusion was enabled to minimize redundant selection of peptides previously selected for collision-induced dissociation.
Peptide identification by database search
MS/MS spectra were searched using Mascot search engine (version 2.4.0, Matrix Science Ltd.) against the UniProt human proteome database. The modifications used were the following: static modification of cysteine (carboxyamidomethylation, +57.05 Da), variable modification of methionine (oxidation, +15.99 Da), and protein N-terminal acetylation. The mass tolerance was set to 1.4 Da for the precursor ions and 0.8 Da for the fragment ions. Peptide identifications were filtered using PeptideProphet and ProteinProphet algorithms with a protein threshold cutoff of 99% and a peptide threshold cutoff of 90% implemented in Scaffold (Proteome Software, Portland, OR, USA).
Immunofluorescence assay
The cells were fixed with 2% paraformaldehyde and incubated with primary antibodies in PBS with 1% bovine serum albumin overnight at 4°C, followed by washing and the application of secondary antibodies (40). After final washing, sections were protected with coverslips with an anti-fading mounting medium sealed with nail polish and stored at 4°C for preservation. Immunofluorescence images were acquired using a confocal laser scanning microscope (ZEISS LSM 800).
Chromatin immunoprecipitation
A ChIP assay was performed using the Pierce ChIP Kit (no. 26156, Thermo Fisher Scientific) according to the manufacturer’s guidelines. One-twentieth of the immunoprecipitated DNA was used in qPCR. Results were shown as a percentage of input. ARNTL antibody (no. 14020) used for ChIP was acquired from Cell Signaling Technology.
Statistical analysis
Data are presented as means ± SEM. Unpaired Student’s t tests were used to compare the means of two groups. One-way analysis of variance (ANOVA) was used for comparison among the different groups. When the ANOVA was significant, post hoc testing of differences between groups was performed using the least significant difference test. The Kaplan-Meier method was used to compare differences in mortality rates between groups. A P value of <0.05 was considered statistically significant. We did not exclude samples or animals. For every figure, statistical tests are justified as appropriate. All data meet the assumptions of the tests (e.g., normal distribution). No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those generally used in the field.
Supplementary Material
Acknowledgments
We thank D. Primm (Department of Surgery, University of Texas Southwestern Medical Center) for critical reading of the manuscript. We thank N. Yates (University of Pittsburgh) and X. Zeng (University of Pittsburgh) for help in the MS/MS assay (P30CA047904). We also thank S. Gygi (Department of Cell Biology, Harvard Medical School) for suggestion in the use of Dionex HPLC system. Funding: D.T. was supported by grants from the American Cancer Society (Research Scholar Grant RSG-16-014-01-CDD). R.K., D.J.K., and M.T.L. were supported by grants R01CA211070, GM131919, and R01CA181450 and R01CA206012, respectively, from the U.S. National Institutes of Health. J.L. was supported by grants from the National Natural Science Foundation of China (81830048, 81772508, and 31671435). M.Y. was supported by grants from the National Natural Science Foundation of China (81570154 and 81100359) and the Natural Science Foundation of Hunan Province (2016JJ3171). G.K. was supported by the Ligue contre le Cancer Comité de Charente-Maritime (équipe labellisée), the Agence National de la Recherche (ANR)—Programme Blanc, the ANR under the frame of E-Rare-2 (the ERA-Net for Research on Rare Diseases), the Association pour la recherche sur le cancer (ARC), the Cancéropôle Ile-de-France, the Chancellerie des universités de Paris (Legs Poix), the Fondation pour la Recherche Médicale (FRM), the European Commission (ArtForce), the European Research Council (ERC), the Fondation Carrefour, the Institut National du Cancer (INCa), INSERM [Heterogeneity of Tumors & Ecosystem (HTE) Program], the Institut Universitaire de France, the Leducq Foundation, the LabEx Immuno-Oncology, the RHU Torino Lumière, the Seerave Foundation, the SIRIC Stratified Oncology Cell DNA Repair and Tumor Immune Elimination (SOCRATE), the SIRIC Cancer Research and Personalized Medicine (CARPEM), and the Paris Alliance of Cancer Research Institutes (PACRI). Author contributions: D.T. and M.Y. designed the experiments. M.Y., P.C., J.L., S.Z., R.K., and D.T. conducted the experiments. D.T. wrote the paper. M.T.L. and H.J.Z. provided the reagents. G.K., D.J.K., M.T.L., and H.J.Z. assisted in the data interpretation and edited the manuscript. Competing interests: The authors declare that they have no competing or financial interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw2238/DC1
Fig. S1. SQSTM1’s UBA domain is required for the formation of the SQSTM1-ARNTL complex.
Fig. S2. Analysis of ARNTL localization in ferroptosis.
Fig. S3. Effects of gpx4 depletion on MAP1LC3B-II expression in Pfa1 cells.
Fig. S4. Effects of MG-132 and desferrioxamine on protein expressions in HT1080 cells.
Fig. S5. Effects of spautin-1, adaptaquin, liproxstatin-1, and chetomin on ferroptosis in vivo.
Table S1. Reagent sources.
REFERENCES AND NOTES
- 1.Tang D., Kang R., Berghe T. V., Vandenabeele P., Kroemer G., The molecular machinery of regulated cell death. Cell Res. 29, 347–364 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dixon S. J., Lemberg K. M., Lamprecht M. R., Skouta R., Zaitsev E. M., Gleason C. E., Patel D. N., Bauer A. J., Cantley A. M., Yang W. S., Morrison B. III, Stockwell B. R., Ferroptosis: An iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yang W. S., SriRamaratnam R., Welsch M. E., Shimada K., Skouta R., Viswanathan V. S., Cheah J. H., Clemons P. A., Shamji A. F., Clish C. B., Brown L. M., Girotti A. W., Cornish V. W., Schreiber S. L., Stockwell B. R., Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ran Q., Van Remmen H., Gu M., Qi W., Roberts L. J. II, Prolla T., Richardson A., Embryonic fibroblasts from Gpx4+/− mice: A novel model for studying the role of membrane peroxidation in biological processes. Free Radic. Biol. Med. 35, 1101–1109 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Canli O., Alanku Y. B., Grootjans S., Vegi N., Hultner L., Hoppe P. S., Schroeder T., Vandenabeele P., Bornkamm G. W., Greten F. R., Glutathione peroxidase 4 prevents necroptosis in mouse erythroid precursors. Blood 127, 139–148 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kang R., Zeng L., Zhu S., Xie Y., Liu J., Wen Q., Cao L., Xie M., Ran Q., Kroemer G., Wang H., Billiar T. R., Jiang J., Tang D., Lipid peroxidation drives gasdermin d-mediated pyroptosis in lethal polymicrobial sepsis. Cell Host Microbe 24, 97–108.e4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stockwell B. R., Friedmann Angeli J. P., Bayir H., Bush A. I., Conrad M., Dixon S. J., Fulda S., Gascón S., Hatzios S. K., Kagan V. E., Noel K., Jiang X., Linkermann A., Murphy M. E., Overholtzer M., Oyagi A., Pagnussat G. C., Park J., Ran Q., Rosenfeld C. S., Salnikow K., Tang D., Torti F. M., Torti S. V., Toyokuni S., Woerpel K. A., Zhang D. D., Ferroptosis: A regulated cell death nexus linking metabolism, redox biology, and disease. Cell 171, 273–285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xie Y., Hou W., Song X., Yu Y., Huang J., Sun X., Kang R., Tang D., Ferroptosis: Process and function. Cell Death Differ. 23, 369–379 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Klionsky D. J., Emr S. D., Autophagy as a regulated pathway of cellular degradation. Science 290, 1717–1721 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Levine B., Kroemer G., Biological functions of autophagy genes: A disease perspective. Cell 176, 11–42 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xie Y., Kang R., Sun X., Zhong M., Huang J., Klionsky D. J., Tang D., Posttranslational modification of autophagy-related proteins in macroautophagy. Autophagy 11, 28–45 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gatica D., Lahiri V., Klionsky D. J., Cargo recognition and degradation by selective autophagy. Nat. Cell Biol. 20, 233–242 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou B., Liu J., Kang R., Klionsky D. J., Kroemer G., Tang D., Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2019, S1044-579X(19)30006-9 (2019). [DOI] [PubMed] [Google Scholar]
- 14.Magnone M. C., Langmesser S., Bezdek A. C., Tallone T., Rusconi S., Albrecht U., The mammalian circadian clock gene per2 modulates cell death in response to oxidative stress. Front. Neurol. 5, 289 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Partch C. L., Green C. B., Takahashi J. S., Molecular architecture of the mammalian circadian clock. Trends Cell Biol. 24, 90–99 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu Y., Xie Y., Cao L., Yang L., Yang M., Lotze M. T., Zeh H. J., Kang R., Tang D., The ferroptosis inducer erastin enhances sensitivity of acute myeloid leukemia cells to chemotherapeutic agents. Mol. Cell. Oncol. 2, e1054549 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fiedler M. A., Wernke-Dollries K., Stark J. M., Inhibition of TNF-α-induced NF-κB activation and IL-8 release in A549 cells with the proteasome inhibitor MG-132. Am. J. Respir. Cell Mol. Biol. 19, 259–268 (1998). [DOI] [PubMed] [Google Scholar]
- 18.Klionsky D. J., Abdelmohsen K., Abe A., Abedin M. J., Abeliovich H., Acevedo Arozena A., Adachi H., Adams C. M., Adams P. D., Adeli K., Adhihetty P. J., Adler S. G., Agam G., Agarwal R., Aghi M. K., Agnello M., Agostinis P., Aguilar P. V., Aguirre-Ghiso J., Airoldi E. M., Ait-Si-Ali S., Akematsu T., Akporiaye E. T., Al-Rubeai M., Albaiceta G. M., Albanese C., Albani D., Albert M. L., Aldudo J., Algül H., Alirezaei M., Alloza I., Almasan A., Almonte-Beceril M., Alnemri E. S., Alonso C., Altan-Bonnet N., Altieri D. C., Alvarez S., Alvarez-Erviti L., Alves S., Amadoro G., Amano A., Amantini C., Ambrosio S., Amelio I., Amer A. O., Amessou M., Amon A., An Z., Anania F. A., Andersen S. U., Andley U. P., Andreadi C. K., Andrieu-Abadie N., Anel A., Ann D. K., Anoopkumar-Dukie S., Antonioli M., Aoki H., Apostolova N., Aquila S., Aquilano K., Araki K., Arama E., Aranda A., Araya J., Arcaro A., Arias E., Arimoto H., Ariosa A. R., Armstrong J. L., Arnould T., Arsov I., Asanuma K., Askanas V., Asselin E., Atarashi R., Atherton S. S., Atkin J. D., Attardi L. D., Auberger P., Auburger G., Aurelian L., Autelli R., Avagliano L., Avantaggiati M. L., Avrahami L., Awale S., Azad N., Bachetti T., Backer J. M., Bae D. H., Bae J. S., Bae O. N., Bae S. H., Baehrecke E. H., Baek S. H., Baghdiguian S., Bagniewska-Zadworna A., Bai H., Bai J., Bai X. Y., Bailly Y., Balaji K. N., Balduini W., Ballabio A., Balzan R., Banerjee R., Bánhegyi G., Bao H., Barbeau B., Barrachina M. D., Barreiro E., Bartel B., Bartolomé A., Bassham D. C., Bassi M. T., Bast R. C. Jr., Basu A., Batista M. T., Batoko H., Battino M., Bauckman K., Baumgarner B. L., Bayer K. U., Beale R., Beaulieu J. F., Beck G. R. Jr., Becker C., Beckham J. D., Bédard P. A., Bednarski P. J., Begley T. J., Behl C., Behrends C., Behrens G. M., Behrns K. E., Bejarano E., Belaid A., Belleudi F., Bénard G., Berchem G., Bergamaschi D., Bergami M., Berkhout B., Berliocchi L., Bernard A., Bernard M., Bernassola F., Bertolotti A., Bess A. S., Besteiro S., Bettuzzi S., Bhalla S., Bhattacharyya S., Bhutia S. K., Biagosch C., Bianchi M. W., Biard-Piechaczyk M., Billes V., Bincoletto C., Bingol B., Bird S. W., Bitoun M., Bjedov I., Blackstone C., Blanc L., Blanco G. A., Blomhoff H. K., Boada-Romero E., Böckler S., Boes M., Boesze-Battaglia K., Boise L. H., Bolino A., Boman A., Bonaldo P., Bordi M., Bosch J., Botana L. M., Botti J., Bou G., Bouché M., Bouchecareilh M., Boucher M. J., Boulton M. E., Bouret S. G., Boya P., Boyer-Guittaut M., Bozhkov P. V., Brady N., Braga V. M., Brancolini C., Braus G. H., Bravo-San Pedro J. M., Brennan L. A., Bresnick E. H., Brest P., Bridges D., Bringer M. A., Brini M., Brito G. C., Brodin B., Brookes P. S., Brown E. J., Brown K., Broxmeyer H. E., Bruhat A., Brum P. C., Brumell J. H., Brunetti-Pierri N., Bryson-Richardson R. J., Buch S., Buchan A. M., Budak H., Bulavin D. V., Bultman S. J., Bultynck G., Bumbasirevic V., Burelle Y., Burke R. E., Burmeister M., Bütikofer P., Caberlotto L., Cadwell K., Cahova M., Cai D., Cai J., Cai Q., Calatayud S., Camougrand N., Campanella M., Campbell G. R., Campbell M., Campello S., Candau R., Caniggia I., Cantoni L., Cao L., Caplan A. B., Caraglia M., Cardinali C., Cardoso S. M., Carew J. S., Carleton L. A., Carlin C. R., Carloni S., Carlsson S. R., Carmona-Gutierrez D., Carneiro L. A., Carnevali O., Carra S., Carrier A., Carroll B., Casas C., Casas J., Cassinelli G., Castets P., Castro-Obregon S., Cavallini G., Ceccherini I., Cecconi F., Cederbaum A. I., Ceña V., Cenci S., Cerella C., Cervia D., Cetrullo S., Chaachouay H., Chae H. J., Chagin A. S., Chai C. Y., Chakrabarti G., Chamilos G., Chan E. Y., Chan M. T., Chandra D., Chandra P., Chang C. P., Chang R. C., Chang T. Y., Chatham J. C., Chatterjee S., Chauhan S., Che Y., Cheetham M. E., Cheluvappa R., Chen C. J., Chen G., Chen G. C., Chen G., Chen H., Chen J. W., Chen J. K., Chen M., Chen M., Chen P., Chen Q., Chen Q., Chen S. D., Chen S. Chen S. S., Chen W., Chen W. J., Chen W. Q., Chen W., Chen X., Chen Y. H., Chen Y. G., Chen Y., Chen Y., Chen Y., Chen Y. J., Chen Y. Q., Chen Y., Chen Z., Chen Z., Cheng A., Cheng C. H., Cheng H., Cheong H., Cherry S., Chesney J., Cheung C. H., Chevet E., Chi H. C., Chi S. G., Chiacchiera F., Chiang H. L., Chiarelli R., Chiariello M., Chieppa M., Chin L. S., Chiong M., Chiu G. N., Cho D. H., Cho S. G., Cho W. C., Cho Y. Y., Cho Y. S., Choi A. M., Choi E. J., Choi E. K., Choi J., Choi M. E., Choi S. I., Chou T. F., Chouaib S., Choubey D., Choubey V., Chow K. C., Chowdhury K., Chu C. T., Chuang T. H., Chun T., Chung H., Chung T., Chung Y. L., Chwae Y. J., Cianfanelli V., Ciarcia R., Ciechomska I. A., Ciriolo M. R., Cirone M., Claerhout S., Clague M. J., Clària J., Clarke P. G., Clarke R., Clementi E., Cleyrat C., Cnop M., Coccia E. M., Cocco T., Codogno P., Coers J., Cohen E. E., Colecchia D., Coletto L., Coll N. S., Colucci-Guyon E., Comincini S., Condello M., Cook K. L., Coombs G. H., Cooper C. D., Cooper J. M., Coppens I., Corasaniti M. T., Corazzari M., Corbalan R., Corcelle-Termeau E., Cordero M. D., Corral-Ramos C., Corti O., Cossarizza A., Costelli P., Costes S., Cotman S. L., Coto-Montes A., Cottet S., Couve E., Covey L. R., Cowart L. A., Cox J. S., Coxon F. P., Coyne C. B., Cragg M. S., Craven R. J., Crepaldi T., Crespo J. L., Criollo A., Crippa V., Cruz M. T., Cuervo A. M., Cuezva J. M., Cui T., Cutillas P. R., Czaja M. J., Czyzyk-Krzeska M. F., Dagda R. K., Dahmen U., Dai C., Dai W., Dai Y., Dalby K. N., Dalla Valle L., Dalmasso G., D’Amelio M., Damme M., Darfeuille-Michaud A., Dargemont C., Darley-Usmar V. M., Dasarathy S., Dasgupta B., Dash S., Dass C. R., Davey H. M., Davids L. M., Dávila D., Davis R. J., Dawson T. M., Dawson V. L., Daza P., de Belleroche J., de Figueiredo P., de Figueiredo R. C., de la Fuente J., De Martino L., De Matteis A., De Meyer G. R., De Milito A., De Santi M., de Souza W., De Tata V., De Zio D., Debnath J., Dechant R., Decuypere J. P., Deegan S., Dehay B., Del Bello B., Del Re D. P., Delage-Mourroux R., Delbridge L. M., Deldicque L., Delorme-Axford E., Deng Y., Dengjel J., Denizot M., Dent P., Der C. J., Deretic V., Derrien B., Deutsch E., Devarenne T. P., Devenish R. J., Di Bartolomeo S., Di Daniele N., Di Domenico F., Di Nardo A., Di Paola S., Di Pietro A., Di Renzo L., DiAntonio A., Díaz-Araya G., Díaz-Laviada I., Diaz-Meco M. T., Diaz-Nido J., Dickey C. A., Dickson R. C., Diederich M., Digard P., Dikic I., Dinesh-Kumar S. P., Ding C., Ding W. X., Ding Z., Dini L., Distler J. H., Diwan A., Djavaheri-Mergny M., Dmytruk K., Dobson R. C., Doetsch V., Dokladny K., Dokudovskaya S., Donadelli M., Dong X. C., Dong X., Dong Z., Donohue T. M. Jr, Doran K. S., D’Orazi G., Dorn G. W. II, Dosenko V., Dridi S., Drucker L., Du J., Du L. L., Du L., du Toit A., Dua P., Duan L., Duann P., Dubey V. K., Duchen M. R., Duchosal M. A., Duez H., Dugail I., Dumit V. I., Duncan M. C., Dunlop E. A., Dunn W. A. Jr., Dupont N., Dupuis L., Durán R. V., Durcan T. M., Duvezin-Caubet S., Duvvuri U., Eapen V., Ebrahimi-Fakhari D., Echard A., Eckhart L., Edelstein C. L., Edinger A. L., Eichinger L., Eisenberg T., Eisenberg-Lerner A., Eissa N. T., El-Deiry W. S., El-Khoury V., Elazar Z., Eldar-Finkelman H., Elliott C. J., Emanuele E., Emmenegger U., Engedal N., Engelbrecht A. M., Engelender S., Enserink J. M., Erdmann R., Erenpreisa J., Eri R., Eriksen J. L., Erman A., Escalante R., Eskelinen E. L., Espert L., Esteban-Martínez L., Evans T. J., Fabri M., Fabrias G., Fabrizi C., Facchiano A., Færgeman N. J., Faggioni A., Fairlie W. D., Fan C., Fan D., Fan J., Fang S., Fanto M., Fanzani A., Farkas T., Faure M., Favier F. B., Fearnhead H., Federici M., Fei E., Felizardo T. C., Feng H., Feng Y., Feng Y., Ferguson T. A., Fernández Á. F., Fernandez-Barrena M. G., Fernandez-Checa J. C., Fernández-López A., Fernandez-Zapico M. E., Feron O., Ferraro E., Ferreira-Halder C. V., Fesus L., Feuer R., Fiesel F. C., Filippi-Chiela E. C., Filomeni G., Fimia G. M., Fingert J. H., Finkbeiner S., Finkel T., Fiorito F., Fisher P. B., Flajolet M., Flamigni F., Florey O., Florio S., Floto R. A., Folini M., Follo C., Fon E. A., Fornai F., Fortunato F., Fraldi A., Franco R., Francois A., François A., Frankel L. B., Fraser I. D., Frey N., Freyssenet D. G., Frezza C., Friedman S. L., Frigo D. E., Fu D., Fuentes J. M., Fueyo J., Fujitani Y., Fujiwara Y., Fujiya M., Fukuda M., Fulda S., Fusco C., Gabryel B., Gaestel M., Gailly P., Gajewska M., Galadari S., Galili G., Galindo I., Galindo M. F., Galliciotti G., Galluzzi L., Galluzzi L., Galy V., Gammoh N., Gandy S., Ganesan A. K., Ganesan S., Ganley I. G., Gannagé M., Gao F. B., Gao F., Gao J. X., García Nannig L., García Véscovi E., Garcia-Macía M., Garcia-Ruiz C., Garg A. D., Garg P. K., Gargini R., Gassen N. C., Gatica D., Gatti E., Gavard J., Gavathiotis E., Ge L., Ge P., Ge S., Gean P. W., Gelmetti V., Genazzani A. A., Geng J., Genschik P., Gerner L., Gestwicki J. E., Gewirtz D. A., Ghavami S., Ghigo E., Ghosh D., Giammarioli A. M., Giampieri F., Giampietri C., Giatromanolaki A., Gibbings D. J., Gibellini L., Gibson S. B., Ginet V., Giordano A., Giorgini F., Giovannetti E., Girardin S. E., Gispert S., Giuliano S., Gladson C. L., Glavic A., Gleave M., Godefroy N., Gogal R. M. Jr., Gokulan K., Goldman G. H., Goletti D., Goligorsky M. S., Gomes A. V., Gomes L. C., Gomez H., Gomez-Manzano C., Gómez-Sánchez R., Gonçalves D. A., Goncu E., Gong Q., Gongora C., Gonzalez C. B., Gonzalez-Alegre P., Gonzalez-Cabo P., González-Polo R. A., Goping I. S., Gorbea C., Gorbunov N. V., Goring D. R., Gorman A. M., Gorski S. M., Goruppi S., Goto-Yamada S., Gotor C., Gottlieb R. A., Gozes I., Gozuacik D., Graba Y., Graef M., Granato G. E., Grant G. D., Grant S., Gravina G. L., Green D. R., Greenhough A., Greenwood M. T., Grimaldi B., Gros F., Grose C., Groulx J. F., Gruber F., Grumati P., Grune T., Guan J. L., Guan K. L., Guerra B., Guillen C., Gulshan K., Gunst J., Guo C., Guo L., Guo M., Guo W., Guo X. G., Gust A. A., Gustafsson Å. B., Gutierrez E., Gutierrez M. G., Gwak H. S., Haas A., Haber J. E., Hadano S., Hagedorn M., Hahn D. R., Halayko A. J., Hamacher-Brady A., Hamada K., Hamai A., Hamann A., Hamasaki M., Hamer I., Hamid Q., Hammond E. M., Han F., Han W., Handa J. T., Hanover J. A., Hansen M., Harada M., Harhaji-Trajkovic L., Harper J. W., Harrath A. H., Harris A. L., Harris J., Hasler U., Hasselblatt P., Hasui K., Hawley R. G., Hawley T. S., He C., He C. Y., He F., He G., He R. R., He X. H., He Y. W., He Y. Y., Heath J. K., Hébert M. J., Heinzen R. A., Helgason G. V., Hensel M., Henske E. P., Her C., Herman P. K., Hernández A., Hernandez C., Hernández-Tiedra S., Hetz C., Hiesinger P. R., Higaki K., Hilfiker S., Hill B. G., Hill J. A., Hill W. D., Hino K., Hofius D., Hofman P., Höglinger G. U., Höhfeld J., Holz M. K., Hong Y., Hood D. A., Hoozemans J. J., Hoppe T., Hsu C., Hsu C. Y., Hsu L. C., Hu D., Hu G., Hu H. M., Hu H., Hu M. C., Hu Y. C., Hu Z. W., Hua F., Hua Y., Huang C., Huang H. L., Huang K. H., Huang K. Y., Huang S., Huang S., Huang W. P., Huang Y. R., Huang Y., Huang Y., Huber T. B., Huebbe P., Huh W. K., Hulmi J. J., Hur G. M., Hurley J. H., Husak Z., Hussain S. N., Hussain S., Hwang J. J., Hwang S., Hwang T. I., Ichihara A., Imai Y., Imbriano C., Inomata M., Into T., Iovane V., Iovanna J. L., Iozzo R. V., Ip N. Y., Irazoqui J. E., Iribarren P., Isaka Y., Isakovic A. J., Ischiropoulos H., Isenberg J. S., Ishaq M., Ishida H., Ishii I., Ishmael J. E., Isidoro C., Isobe K., Isono E., Issazadeh-Navikas S., Itahana K., Itakura E., Ivanov A. I., Iyer A. K., Izquierdo J. M., Izumi Y., Izzo V., Jäättelä M., Jaber N., Jackson D. J., Jackson W. T., Jacob T. G., Jacques T. S., Jagannath C., Jain A., Jana N. R., Jang B. K., Jani A., Janji B., Jannig P. R., Jansson P. J., Jean S., Jendrach M., Jeon J. H., Jessen N., Jeung E. B., Jia K., Jia L., Jiang H., Jiang H., Jiang L., Jiang T., Jiang X., Jiang X., Jiang X., Jiang Y., Jiang Y., Jiménez A., Jin C., Jin H., Jin L., Jin M., Jin S., Jinwal U. K., Jo E. K., Johansen T., Johnson D. E., Johnson G. V., Johnson J. D., Jonasch E., Jones C., Joosten L. A., Jordan J., Joseph A. M., Joseph B., Joubert A. M., Ju D., Ju J., Juan H. F., Juenemann K., Juhász G., Jung H. S., Jung J. U., Jung Y. K., Jungbluth H., Justice M. J., Jutten B., Kaakoush N. O., Kaarniranta K., Kaasik A., Kabuta T., Kaeffer B., Kågedal K., Kahana A., Kajimura S., Kakhlon O., Kalia M., Kalvakolanu D. V., Kamada Y., Kambas K., Kaminskyy V. O., Kampinga H. H., Kandouz M., Kang C., Kang R., Kang T. C., Kanki T., Kanneganti T. D., Kanno H., Kanthasamy A. G., Kantorow M., Kaparakis-Liaskos M., Kapuy O., Karantza V., Karim M. R., Karmakar P., Kaser A., Kaushik S., Kawula T., Kaynar A. M., Ke P. Y., Ke Z. J., Kehrl J. H., Keller K. E., Kemper J. K., Kenworthy A. K., Kepp O., Kern A., Kesari S., Kessel D., Ketteler R., Kettelhut Ido C., Khambu B., Khan M. M., Khandelwal V. K., Khare S., Kiang J. G., Kiger A. A., Kihara A., Kim A. L., Kim C. H., Kim D. R., Kim D. H., Kim E. K., Kim H. Y., Kim H. R., Kim J. S., Kim J. H., Kim J. C., Kim J. H., Kim K. W., Kim M. D., Kim M. M., Kim P. K., Kim S. W., Kim S. Y., Kim Y. S., Kim Y., Kimchi A., Kimmelman A. C., Kimura T., King J. S., Kirkegaard K., Kirkin V., Kirshenbaum L. A., Kishi S., Kitajima Y., Kitamoto K., Kitaoka Y., Kitazato K., Kley R. A., Klimecki W. T., Klinkenberg M., Klucken J., Knævelsrud H., Knecht E., Knuppertz L., Ko J. L., Kobayashi S., Koch J. C., Koechlin-Ramonatxo C., Koenig U., Koh Y. H., Köhler K., Kohlwein S. D., Koike M., Komatsu M., Kominami E., Kong D., Kong H. J., Konstantakou E. G., Kopp B. T., Korcsmaros T., Korhonen L., Korolchuk V. I., Koshkina N. V., Kou Y., Koukourakis M. I., Koumenis C., Kovács A. L., Kovács T., Kovacs W. J., Koya D., Kraft C., Krainc D., Kramer H., Kravic-Stevovic T., Krek W., Kretz-Remy C., Krick R., Krishnamurthy M., Kriston-Vizi J., Kroemer G., Kruer M. C., Kruger R., Ktistakis N. T., Kuchitsu K., Kuhn C., Kumar A. P., Kumar A., Kumar A., Kumar D., Kumar D., Kumar R., Kumar S., Kundu M., Kung H. J., Kuno A., Kuo S. H., Kuret J., Kurz T., Kwok T., Kwon T. K., Kwon Y. T., Kyrmizi I., La Spada A. R., Lafont F., Lahm T., Lakkaraju A., Lam T., Lamark T., Lancel S., Landowski T. H., Lane D. J., Lane J. D., Lanzi C., Lapaquette P., Lapierre L. R., Laporte J., Laukkarinen J., Laurie G. W., Lavandero S., Lavie L., LaVoie M. J., Law B. Y., Law H. K., Law K. B., Layfield R., Lazo P. A., Le Cam L., Le Roch K. G., Le Stunff H., Leardkamolkarn V., Lecuit M., Lee B. H., Lee C. H., Lee E. F., Lee G. M., Lee H. J., Lee H., Lee J. K., Lee J., Lee J. H., Lee J. H., Lee M., Lee M. S., Lee P. J., Lee S. W., Lee S. J., Lee S. J., Lee S. Y., Lee S. H., Lee S. S., Lee S. J., Lee S., Lee Y. R., Lee Y. J., Lee Y. H., Leeuwenburgh C., Lefort S., Legouis R., Lei J., Lei Q. Y., Leib D. A., Leibowitz G., Lekli I., Lemaire S. D., Lemasters J. J., Lemberg M. K., Lemoine A., Leng S., Lenz G., Lenzi P., Lerman L. O., Lettieri Barbato D., Leu J. I., Leung H. Y., Levine B., Lewis P. A., Lezoualc’h F., Li C., Li F., Li F. J., Li J., Li K., Li L., Li M., Li M., Li Q., Li R., Li S., Li W., Li W., Li X., Li Y., Lian J., Liang C., Liang Q., Liao Y., Liberal J., Liberski P. P., Lie P., Lieberman A. P., Lim H. J., Lim K. L., Lim K., Lima R. T., Lin C. S., Lin C. F., Lin F., Lin F., Lin F. C., Lin K., Lin K. H., Lin P. H., Lin T., Lin W. W., Lin Y. S., Lin Y., Linden R., Lindholm D., Lindqvist L. M., Lingor P., Linkermann A., Liotta L. A., Lipinski M. M., Lira V. A., Lisanti M. P., Liton P. B., Liu B., Liu C., Liu C. F., Liu F., Liu H. J., Liu J., Liu J. J., Liu J. L., Liu K., Liu L., Liu L., Liu Q., Liu R. Y., Liu S., Liu S., Liu W., Liu X. D., Liu X., Liu X. H., Liu X., Liu X., Liu X., Liu Y., Liu Y., Liu Z., Liu Z., Liuzzi J. P., Lizard G., Ljujic M., Lodhi I. J., Logue S. E., Lokeshwar B. L., Long Y. C., Lonial S., Loos B., López-Otín C., López-Vicario C., Lorente M., Lorenzi P. L., Lõrincz P., Los M., Lotze M. T., Lovat P. E., Lu B., Lu B., Lu J., Lu Q., Lu S. M., Lu S., Lu Y., Luciano F., Luckhart S., Lucocq J. M., Ludovico P., Lugea A., Lukacs N. W., Lum J. J., Lund A. H., Luo H., Luo J., Luo S., Luparello C., Lyons T., Ma J., Ma Y., Ma Y., Ma Z., Machado J., Machado-Santelli G. M., Macian F., MacIntosh G. C., MacKeigan J. P., Macleod K. F., MacMicking J. D., MacMillan-Crow L. A., Madeo F., Madesh M., Madrigal-Matute J., Maeda A., Maeda T., Maegawa G., Maellaro E., Maes H., Magariños M., Maiese K., Maiti T. K., Maiuri L., Maiuri M. C., Maki C. G., Malli R., Malorni W., Maloyan A., Mami-Chouaib F., Man N., Mancias J. D., Mandelkow E. M., Mandell M. A., Manfredi A. A., Manié S. N., Manzoni C., Mao K., Mao Z., Mao Z. W., Marambaud P., Marconi A. M., Marelja Z., Marfe G., Margeta M., Margittai E., Mari M., Mariani F. V., Marin C., Marinelli S., Mariño G., Markovic I., Marquez R., Martelli A. M., Martens S., Martin K. R., Martin S. J., Martin S., Martin-Acebes M. A., Martín-Sanz P., Martinand-Mari C., Martinet W., Martinez J., Martinez-Lopez N., Martinez-Outschoorn U., Martínez-Velázquez M., Martinez-Vicente M., Martins W. K., Mashima H., Mastrianni J. A., Matarese G., Matarrese P., Mateo R., Matoba S., Matsumoto N., Matsushita T., Matsuura A., Matsuzawa T., Mattson M. P., Matus S., Maugeri N., Mauvezin C., Mayer A., Maysinger D., Mazzolini G. D., McBrayer M. K., McCall K., McCormick C., McInerney G. M., McIver S. C., McKenna S., McMahon J. J., McNeish I. A., Mechta-Grigoriou F., Medema J. P., Medina D. L., Megyeri K., Mehrpour M., Mehta J. L., Mei Y., Meier U. C., Meijer A. J., Meléndez A., Melino G., Melino S., de Melo E. J., Mena M. A., Meneghini M. D., Menendez J. A., Menezes R., Meng L., Meng L. H., Meng S., Menghini R., Menko A. S., Menna-Barreto R. F., Menon M. B., Meraz-Ríos M. A., Merla G., Merlini L., Merlot A. M., Meryk A., Meschini S., Meyer J. N., Mi M. T., Miao C. Y., Micale L., Michaeli S., Michiels C., Migliaccio A. R., Mihailidou A. S., Mijaljica D., Mikoshiba K., Milan E., Miller-Fleming L., Mills G. B., Mills I. G., Minakaki G., Minassian B. A., Ming X. F., Minibayeva F., Minina E. A., Mintern J. D., Minucci S., Miranda-Vizuete A., Mitchell C. H., Miyamoto S., Miyazawa K., Mizushima N., Mnich K., Mograbi B., Mohseni S., Moita L. F., Molinari M., Molinari M., Møller A. B., Mollereau B., Mollinedo F., Mongillo M., Monick M. M., Montagnaro S., Montell C., Moore D. J., Moore M. N., Mora-Rodriguez R., Moreira P. I., Morel E., Morelli M. B., Moreno S., Morgan M. J., Moris A., Moriyasu Y., Morrison J. L., Morrison L. A., Morselli E., Moscat J., Moseley P. L., Mostowy S., Motori E., Mottet D., Mottram J. C., Moussa C. E., Mpakou V. E., Mukhtar H., Mulcahy Levy J. M., Muller S., Muñoz-Moreno R., Muñoz-Pinedo C., Münz C., Murphy M. E., Murray J. T., Murthy A., Mysorekar I. U., Nabi I. R., Nabissi M., Nader G. A., Nagahara Y., Nagai Y., Nagata K., Nagelkerke A., Nagy P., Naidu S. R., Nair S., Nakano H., Nakatogawa H., Nanjundan M., Napolitano G., Naqvi N. I., Nardacci R., Narendra D. P., Narita M., Nascimbeni A. C., Natarajan R., Navegantes L. C., Nawrocki S. T., Nazarko T. Y., Nazarko V. Y., Neill T., Neri L. M., Netea M. G., Netea-Maier R. T., Neves B. M., Ney P. A., Nezis I. P., Nguyen H. T., Nguyen H. P., Nicot A. S., Nilsen H., Nilsson P., Nishimura M., Nishino I., Niso-Santano M., Niu H., Nixon R. A., Njar V. C., Noda T., Noegel A. A., Nolte E. M., Norberg E., Norga K. K., Noureini S. K., Notomi S., Notterpek L., Nowikovsky K., Nukina N., Nürnberger T., O’Donnell V. B., O’Donovan T., O’Dwyer P. J., Oehme I., Oeste C. L., Ogawa M., Ogretmen B., Ogura Y., Oh Y. J., Ohmuraya M., Ohshima T., Ojha R., Okamoto K., Okazaki T., Oliver F. J., Ollinger K., Olsson S., Orban D. P., Ordonez P., Orhon I., Orosz L., O’Rourke E. J., Orozco H., Ortega A. L., Ortona E., Osellame L. D., Oshima J., Oshima S., Osiewacz H. D., Otomo T., Otsu K., Ou J. H., Outeiro T. F., Ouyang D. Y., Ouyang H., Overholtzer M., Ozbun M. A., Ozdinler P. H., Ozpolat B., Pacelli C., Paganetti P., Page G., Pages G., Pagnini U., Pajak B., Pak S. C., Pakos-Zebrucka K., Pakpour N., Palková Z., Palladino F., Pallauf K., Pallet N., Palmieri M., Paludan S. R., Palumbo C., Palumbo S., Pampliega O., Pan H., Pan W., Panaretakis T., Pandey A., Pantazopoulou A., Papackova Z., Papademetrio D. L., Papassideri I., Papini A., Parajuli N., Pardo J., Parekh V. V., Parenti G., Park J. I., Park J., Park O. K., Parker R., Parlato R., Parys J. B., Parzych K. R., Pasquet J. M., Pasquier B., Pasumarthi K. B., Patschan D., Patterson C., Pattingre S., Pattison S., Pause A., Pavenstädt H., Pavone F., Pedrozo Z., Peña F. J., Peñalva M. A., Pende M., Peng J., Penna F., Penninger J. M., Pensalfini A., Pepe S., Pereira G. J., Pereira P. C., Pérez-de la Cruz V., Pérez-Pérez M. E., Pérez-Rodríguez D., Pérez-Sala D., Perier C., Perl A., Perlmutter D. H., Perrotta I., Pervaiz S., Pesonen M., Pessin J. E., Peters G. J., Petersen M., Petrache I., Petrof B. J., Petrovski G., Phang J. M., Piacentini M., Pierdominici M., Pierre P., Pierrefite-Carle V., Pietrocola F., Pimentel-Muiños F. X., Pinar M., Pineda B., Pinkas-Kramarski R., Pinti M., Pinton P., Piperdi B., Piret J. M., Platanias L. C., Platta H. W., Plowey E. D., Pöggeler S., Poirot M., Polčic P., Poletti A., Poon A. H., Popelka H., Popova B., Poprawa I., Poulose S. M., Poulton J., Powers S. K., Powers T., Pozuelo-Rubio M., Prak K., Prange R., Prescott M., Priault M., Prince S., Proia R. L., Proikas-Cezanne T., Prokisch H., Promponas V. J., Przyklenk K., Puertollano R., Pugazhenthi S., Puglielli L., Pujol A., Puyal J., Pyeon D., Qi X., Qian W. B., Qin Z. H., Qiu Y., Qu Z., Quadrilatero J., Quinn F., Raben N., Rabinowich H., Radogna F., Ragusa M. J., Rahmani M., Raina K., Ramanadham S., Ramesh R., Rami A., Randall-Demllo S., Randow F., Rao H., Rao V. A., Rasmussen B. B., Rasse T. M., Ratovitski E. A., Rautou P. E., Ray S. K., Razani B., Reed B. H., Reggiori F., Rehm M., Reichert A. S., Rein T., Reiner D. J., Reits E., Ren J., Ren X., Renna M., Reusch J. E., Revuelta J. L., Reyes L., Rezaie A. R., Richards R. I., Richardson D. R., Richetta C., Riehle M. A., Rihn B. H., Rikihisa Y., Riley B. E., Rimbach G., Rippo M. R., Ritis K., Rizzi F., Rizzo E., Roach P. J., Robbins J., Roberge M., Roca G., Roccheri M. C., Rocha S., Rodrigues C. M., Rodríguez C. I., de Cordoba S. R., Rodriguez-Muela N., Roelofs J., Rogov V. V., Rohn T. T., Rohrer B., Romanelli D., Romani L., Romano P. S., Roncero M. I., Rosa J. L., Rosello A., Rosen K. V., Rosenstiel P., Rost-Roszkowska M., Roth K. A., Roué G., Rouis M., Rouschop K. M., Ruan D. T., Ruano D., Rubinsztein D. C., Rucker E. B. III, Rudich A., Rudolf E., Rudolf R., Ruegg M. A., Ruiz-Roldan C., Ruparelia A. A., Rusmini P., Russ D. W., Russo G. L., Russo G., Russo R., Rusten T. E., Ryabovol V., Ryan K. M., Ryter S. W., Sabatini D. M., Sacher M., Sachse C., Sack M. N., Sadoshima J., Saftig P., Sagi-Eisenberg R., Sahni S., Saikumar P., Saito T., Saitoh T., Sakakura K., Sakoh-Nakatogawa M., Sakuraba Y., Salazar-Roa M., Salomoni P., Saluja A. K., Salvaterra P. M., Salvioli R., Samali A., Sanchez A. M., Sánchez-Alcázar J. A., Sanchez-Prieto R., Sandri M., Sanjuan M. A., Santaguida S., Santambrogio L., Santoni G., Dos Santos C. N., Saran S., Sardiello M., Sargent G., Sarkar P., Sarkar S., Sarrias M. R., Sarwal M. M., Sasakawa C., Sasaki M., Sass M., Sato K., Sato M., Satriano J., Savaraj N., Saveljeva S., Schaefer L., Schaible U. E., Scharl M., Schatzl H. M., Schekman R., Scheper W., Schiavi A., Schipper H. M., Schmeisser H., Schmidt J., Schmitz I., Schneider B. E., Schneider E. M., Schneider J. L., Schon E. A., Schönenberger M. J., Schönthal A. H., Schorderet D. F., Schröder B., Schuck S., Schulze R. J., Schwarten M., Schwarz T. L., Sciarretta S., Scotto K., Scovassi A. I., Screaton R. A., Screen M., Seca H., Sedej S., Segatori L., Segev N., Seglen P. O., Seguí-Simarro J. M., Segura-Aguilar J., Seki E., Sell C., Seiliez I., Semenkovich C. F., Semenza G. L., Sen U., Serra A. L., Serrano-Puebla A., Sesaki H., Setoguchi T., Settembre C., Shacka J. J., Shajahan-Haq A. N., Shapiro I. M., Sharma S., She H., Shen C. K., Shen C. C., Shen H. M., Shen S., Shen W., Sheng R., Sheng X., Sheng Z. H., Shepherd T. G., Shi J., Shi Q., Shi Q., Shi Y., Shibutani S., Shibuya K., Shidoji Y., Shieh J. J., Shih C. M., Shimada Y., Shimizu S., Shin D. W., Shinohara M. L., Shintani M., Shintani T., Shioi T., Shirabe K., Shiri-Sverdlov R., Shirihai O., Shore G. C., Shu C. W., Shukla D., Sibirny A. A., Sica V., Sigurdson C. J., Sigurdsson E. M., Sijwali P. S., Sikorska B., Silveira W. A., Silvente-Poirot S., Silverman G. A., Simak J., Simmet T., Simon A. K., Simon H. U., Simone C., Simons M., Simonsen A., Singh R., Singh S. V., Singh S. K., Sinha D., Sinha S., Sinicrope F. A., Sirko A., Sirohi K., Sishi B. J., Sittler A., Siu P. M., Sivridis E., Skwarska A., Slack R., Slaninová I., Slavov N., Smaili S. S., Smalley K. S., Smith D. R., Soenen S. J., Soleimanpour S. A., Solhaug A., Somasundaram K., Son J. H., Sonawane A., Song C., Song F., Song H. K., Song J. X., Song W., Soo K. Y., Sood A. K., Soong T. W., Soontornniyomkij V., Sorice M., Sotgia F., Soto-Pantoja D. R., Sotthibundhu A., Sousa M. J., Spaink H. P., Span P. N., Spang A., Sparks J. D., Speck P. G., Spector S. A., Spies C. D., Springer W., Clair D. S., Stacchiotti A., Staels B., Stang M. T., Starczynowski D. T., Starokadomskyy P., Steegborn C., Steele J. W., Stefanis L., Steffan J., Stellrecht C. M., Stenmark H., Stepkowski T. M., Stern S. T., Stevens C., Stockwell B. R., Stoka V., Storchova Z., Stork B., Stratoulias V., Stravopodis D. J., Strnad P., Strohecker A. M., Ström A. L., Stromhaug P., Stulik J., Su Y. X., Su Z., Subauste C. S., Subramaniam S., Sue C. M., Suh S. W., Sui X., Sukseree S., Sulzer D., Sun F. L., Sun J., Sun J., Sun S. Y., Sun Y., Sun Y., Sun Y., Sundaramoorthy V., Sung J., Suzuki H., Suzuki K., Suzuki N., Suzuki T., Suzuki Y. J., Swanson M. S., Swanton C., Swärd K., Swarup G., Sweeney S. T., Sylvester P. W., Szatmari Z., Szegezdi E., Szlosarek P. W., Taegtmeyer H., Tafani M., Taillebourg E., Tait S. W., Takacs-Vellai K., Takahashi Y., Takáts S., Takemura G., Takigawa N., Talbot N. J., Tamagno E., Tamburini J., Tan C. P., Tan L., Tan M. L., Tan M., Tan Y. J., Tanaka K., Tanaka M., Tang D., Tang D., Tang G., Tanida I., Tanji K., Tannous B. A., Tapia J. A., Tasset-Cuevas I., Tatar M., Tavassoly I., Tavernarakis N., Taylor A., Taylor G. S., Taylor G. A., Taylor J. P., Taylor M. J., Tchetina E. V., Tee A. R., Teixeira-Clerc F., Telang S., Tencomnao T., Teng B. B., Teng R. J., Terro F., Tettamanti G., Theiss A. L., Theron A. E., Thomas K. J., Thomé M. P., Thomes P. G., Thorburn A., Thorner J., Thum T., Thumm M., Thurston T. L., Tian L., Till A., Ting J. P., Titorenko V. I., Toker L., Toldo S., Tooze S. A., Topisirovic I., Torgersen M. L., Torosantucci L., Torriglia A., Torrisi M. R., Tournier C., Towns R., Trajkovic V., Travassos L. H., Triola G., Tripathi D. N., Trisciuoglio D., Troncoso R., Trougakos I. P., Truttmann A. C., Tsai K. J., Tschan M. P., Tseng Y. H., Tsukuba T., Tsung A., Tsvetkov A. S., Tu S., Tuan H. Y., Tucci M., Tumbarello D. A., Turk B., Turk V., Turner R. F., Tveita A. A., Tyagi S. C., Ubukata M., Uchiyama Y., Udelnow A., Ueno T., Umekawa M., Umemiya-Shirafuji R., Underwood B. R., Ungermann C., Ureshino R. P., Ushioda R., Uversky V. N., Uzcátegui N. L., Vaccari T., Vaccaro M. I., Váchová L., Vakifahmetoglu-Norberg H., Valdor R., Valente E. M., Vallette F., Valverde A. M., Van den Berghe G., Van Den Bosch L., van den Brink G. R., van der Goot F. G., van der Klei I. J., van der Laan L. J., van Doorn W. G., van Egmond M., van Golen K. L., Van Kaer L., van Lookeren Campagne M., Vandenabeele P., Vandenberghe W., Vanhorebeek I., Varela-Nieto I., Vasconcelos M. H., Vasko R., Vavvas D. G., Vega-Naredo I., Velasco G., Velentzas A. D., Velentzas P. D., Vellai T., Vellenga E., Vendelbo M. H., Venkatachalam K., Ventura N., Ventura S., Veras P. S., Verdier M., Vertessy B. G., Viale A., Vidal M., Vieira H. L., Vierstra R. D., Vigneswaran N., Vij N., Vila M., Villar M., Villar V. H., Villarroya J., Vindis C., Viola G., Viscomi M. T., Vitale G., Vogl D. T., Voitsekhovskaja O. V., von Haefen C., von Schwarzenberg K., Voth D. E., Vouret-Craviari V., Vuori K., Vyas J. M., Waeber C., Walker C. L., Walker M. J., Walter J., Wan L., Wan X., Wang B., Wang C., Wang C. Y., Wang C., Wang C., Wang C., Wang D., Wang F., Wang F., Wang G., Wang H. J., Wang H., Wang H. G., Wang H., Wang H. D., Wang J., Wang J., Wang M., Wang M. Q., Wang P. Y., Wang P., Wang R. C., Wang S., Wang T. F., Wang X., Wang X. J., Wang X. W., Wang X., Wang X., Wang Y., Wang Y., Wang Y., Wang Y. J., Wang Y., Wang Y., Wang Y. T., Wang Y., Wang Z. N., Wappner P., Ward C., Ward D. M., Warnes G., Watada H., Watanabe Y., Watase K., Weaver T. E., Weekes C. D., Wei J., Weide T., Weihl C. C., Weindl G., Weis S. N., Wen L., Wen X., Wen Y., Westermann B., Weyand C. M., White A. R., White E., Whitton J. L., Whitworth A. J., Wiels J., Wild F., Wildenberg M. E., Wileman T., Wilkinson D. S., Wilkinson S., Willbold D., Williams C., Williams K., Williamson P. R., Winklhofer K. F., Witkin S. S., Wohlgemuth S. E., Wollert T., Wolvetang E. J., Wong E., Wong G. W., Wong R. W., Wong V. K., Woodcock E. A., Wright K. L., Wu C., Wu D., Wu G. S., Wu J., Wu J., Wu M., Wu M., Wu S., Wu W. K., Wu Y., Wu Z., Xavier C. P., Xavier R. J., Xia G. X., Xia T., Xia W., Xia Y., Xiao H., Xiao J., Xiao S., Xiao W., Xie C. M., Xie Z., Xie Z., Xilouri M., Xiong Y., Xu C., Xu C., Xu F., Xu H., Xu H., Xu J., Xu J., Xu J., Xu L., Xu X., Xu Y., Xu Y., Xu Z. X., Xu Z., Xue Y., Yamada T., Yamamoto A., Yamanaka K., Yamashina S., Yamashiro S., Yan B., Yan B., Yan X., Yan Z., Yanagi Y., Yang D. S., Yang J. M., Yang L., Yang M., Yang P. M., Yang P., Yang Q., Yang W., Yang W. Y., Yang X., Yang Y., Yang Y., Yang Z., Yang Z., Yao M. C., Yao P. J., Yao X., Yao Z., Yao Z., Yasui L. S., Ye M., Yedvobnick B., Yeganeh B., Yeh E. S., Yeyati P. L., Yi F., Yi L., Yin X. M., Yip C. K., Yoo Y. M., Yoo Y. H., Yoon S. Y., Yoshida K., Yoshimori T., Young K. H., Yu H., Yu J. J., Yu J. T., Yu J., Yu L., Yu W. H., Yu X. F., Yu Z., Yuan J., Yuan Z. M., Yue B. Y., Yue J., Yue Z., Zacks D. N., Zacksenhaus E., Zaffaroni N., Zaglia T., Zakeri Z., Zecchini V., Zeng J., Zeng M., Zeng Q., Zervos A. S., Zhang D. D., Zhang F., Zhang G., Zhang G. C., Zhang H., Zhang H., Zhang H., Zhang H., Zhang J., Zhang J., Zhang J., Zhang J., Zhang J. P., Zhang L., Zhang L., Zhang L., Zhang L., Zhang M. Y., Zhang X., Zhang X. D., Zhang Y., Zhang Y., Zhang Y., Zhang Y., Zhang Y., Zhao M., Zhao W. L., Zhao X., Zhao Y. G., Zhao Y., Zhao Y., Zhao Y. X., Zhao Z., Zhao Z. J., Zheng D., Zheng X. L., Zheng X., Zhivotovsky B., Zhong Q., Zhou G. Z., Zhou G., Zhou H., Zhou S. F., Zhou X. J., Zhu H., Zhu H., Zhu W. G., Zhu W., Zhu X. F., Zhu Y., Zhuang S. M., Zhuang X., Ziparo E., Zois C. E., Zoladek T., Zong W. X., Zorzano A., Zughaier S. M., Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Friedmann Angeli J. P., Schneider M., Proneth B., Tyurina Y. Y., Tyurin V. A., Hammond V. J., Herbach N., Aichler M., Walch A., Eggenhofer E., Basavarajappa D., Rådmark O., Kobayashi S., Seibt T., Beck H., Neff F., Esposito I., Wanke R., Förster H., Yefremova O., Heinrichmeyer M., Bornkamm G. W., Geissler E. K., Thomas S. B., Stockwell B. R., O’Donnell V. B., Kagan V. E., Schick J. A., Conrad M., Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat. Cell Biol. 16, 1180–1191 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Appelhoff R. J., Tian Y.-M., Raval R. R., Turley H., Harris A. L., Pugh C. W., Ratcliffe P. J., Gleadle J. M., Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 279, 38458–38465 (2004). [DOI] [PubMed] [Google Scholar]
- 21.Majmundar A. J., Wong W. J., Simon M. C., Hypoxia-inducible factors and the response to hypoxic stress. Mol. Cell 40, 294–309 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Woo K. J., Lee T.-J., Park J.-W., Kwon T. K., Desferrioxamine, an iron chelator, enhances HIF-1α accumulation via cyclooxygenase-2 signaling pathway. Biochem. Biophys. Res. Commun. 343, 8–14 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Bensaad K., Favaro E., Lewis C. A., Peck B., Lord S., Collins J. M., Pinnick K. E., Wigfield S., Buffa F. M., Li J.-L., Zhang Q., Wakelam M. J. O., Karpe F., Schulze A., Harris A. L., Fatty acid uptake and lipid storage induced by HIF-1α contribute to cell growth and survival after hypoxia-reoxygenation. Cell Rep. 9, 349–365 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Sun X., Ou Z., Chen R., Niu X., Chen D., Kang R., Tang D., Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shimada K., Skouta R., Kaplan A., Yang W. S., Hayano M., Dixon S. J., Brown L. M., Valenzuela C. A., Wolpaw A. J., Stockwell B. R., Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat. Chem. Biol. 12, 497–503 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu S., Zhang Q., Sun X., Zeh H. J. III, Lotze M. T., Kang R., Tang D., HSPA5 regulates ferroptotic cell death in cancer cells. Cancer Res. 77, 2064–2077 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun X., Niu X., Chen R., He W., Chen D., Kang R., Tang D., Metallothionein-1G facilitates sorafenib resistance through inhibition of ferroptosis. Hepatology 64, 488–500 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chen D., Tavana O., Chu B., Erber L., Chen Y., Baer R., Gu W., NRF2 is a major target of ARF in p53-independent tumor suppression. Mol. Cell 68, 224–232.e4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hou W., Xie Y., Song X., Sun X., Lotze M. T., Zeh H. J. III, Kang R., Tang D., Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gao M., Monian P., Pan Q., Zhang W., Xiang J., Jiang X., Ferroptosis is an autophagic cell death process. Cell Res. 26, 1021–1032 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kroemer G., Mariño G., Levine B., Autophagy and the integrated stress response. Mol. Cell 40, 280–293 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denton D., Kumar S., Autophagy-dependent cell death. Cell Death Differ. 26, 605–616 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Song X., Zhu S., Chen P., Hou W., Wen Q., Liu J., Xie Y., Liu J., Klionsky D. J., Kroemer G., Lotze M. T., Zeh H. J., Kang R., Tang D., AMPK-mediated BECN1 phosphorylation promotes ferroptosis by directly blocking system Xc− activity. Curr. Biol. 28, 2388–2399.e5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gao H., Bai Y., Jia Y., Zhao Y., Kang R., Tang D., Dai E., Ferroptosis is a lysosomal cell death process. Biochem. Biophys. Res. Commun. 503, 1550–1556 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Torii S., Shintoku R., Kubota C., Yaegashi M., Torii R., Sasaki M., Suzuki T., Mori M., Yoshimoto Y., Takeuchi T., Yamada K., An essential role for functional lysosomes in ferroptosis of cancer cells. Biochem. J. 473, 769–777 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Bai Y., Meng L., Han L., Jia Y., Zhao Y., Gao H., Kang R., Wang X., Tang D., Dai E., Lipid storage and lipophagy regulates ferroptosis. Biochem. Biophys. Res. Commun. 508, 997–1003 (2019). [DOI] [PubMed] [Google Scholar]
- 37.Huang D., Li T., Li X., Zhang L., Sun L., He X., Zhong X., Jia D., Song L., Semenza G. L., Gao P., Zhang H., HIF-1-mediated suppression of acyl-CoA dehydrogenases and fatty acid oxidation is critical for cancer progression. Cell Rep. 8, 1930–1942 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Yang M., Liu L., Xie M., Sun X., Yu Y., Kang R., Yang L., Zhu S., Cao L., Tang D., Poly-ADP-ribosylation of HMGB1 regulates TNFSF10/TRAIL resistance through autophagy. Autophagy 11, 214–224 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shevchenko A., Tomas H., Havlis J., Olsen J. V., Mann M., In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 1, 2856–2860 (2006). [DOI] [PubMed] [Google Scholar]
- 40.Tang D., Kang R., Livesey K. M., Cheh C.-W., Farkas A., Loughran P., Hoppe G., Bianchi M. E., Tracey K. J., Zeh H. J. III, Lotze M. T., Endogenous HMGB1 regulates autophagy. J. Cell Biol. 190, 881–892 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/7/eaaw2238/DC1
Fig. S1. SQSTM1’s UBA domain is required for the formation of the SQSTM1-ARNTL complex.
Fig. S2. Analysis of ARNTL localization in ferroptosis.
Fig. S3. Effects of gpx4 depletion on MAP1LC3B-II expression in Pfa1 cells.
Fig. S4. Effects of MG-132 and desferrioxamine on protein expressions in HT1080 cells.
Fig. S5. Effects of spautin-1, adaptaquin, liproxstatin-1, and chetomin on ferroptosis in vivo.
Table S1. Reagent sources.