

Abstract
Nucleosomes represent the elementary units of chromatin packing and hubs in epigenetic signaling pathways. Across the chromatin and over the lifetime of the eukaryotic cell, nucleosomes experience a broad repertoire of alterations that affect their structure and binding with various chromatin factors. Dynamics of the histone core, nucleosomal and linker DNA, and intrinsic disorder of histone tails add further complexity to the nucleosome interaction landscape. In light of our understanding through the growing number of experimental and computational studies, we review the emerging patterns of molecular recognition of nucleosomes by their binding partners and assess the basic mechanisms of its regulation.
Introduction
Epigenetic signaling complexity can be achieved by the modularity and hierarchical organization of its components. Nucleosomes are the elementary building blocks of chromatin and represent points of coordination between DNA, histones and factors participating in epigenetic regulation. The nucleosome core particle (NCP, for simplicity called “nucleosome”) consists of two copies of four types of histones (H3, H4, H2A, H2B), and ~147 DNA base pairs wrapped around them in ~1.7 negative superhelical turns [1]. Nucleosomes can be affected by changes in histone and DNA sequences, by introducing covalent modifications in histones and DNA, or by the deposition of histone variants. This diverse set of nucleosome alterations may result in differences in nucleosomal structure and dynamics, and lead to the recruitment of specific chromatin components with spatiotemporal precision.
Our understanding of how the specific features of nucleosomes are recognized at the molecular level has been greatly enhanced due to the progress in experimental approaches, such as X-Ray, NMR, Cryo-EM and high-throughput and high-precision techniques, such as ChiP-Exo, DNA footprinting, and SILAC [2–11]. In addition, the hybrid approaches integrating experimental data with molecular modeling and molecular dynamics simulations may also provide clues about novel binding interfaces and the dynamic nature of interactions with nucleosomes [12–16]. Some of these experimental and hybrid methods can map binding sites on the nucleosome up to single-nucleotide and single-residue resolution, allowing one to start deciphering the main principles of molecular recognition of nucleosomes by epigenetic factors.
In this brief review, we assess the most recent advances in the field and provide a conceptual summary of up-to-date knowledge about binding modes and recognition motifs which are employed by the nucleosome to elicit highly specific responses upon binding to its partners. We end the review with a remark on the implications of these interactions for higher order chromatin structure.
Binding modes and motifs of the nucleosome core
The rate of deposition of complexes of nucleosomes with different binding partners into the Protein Data Bank (PDB) [17] shows an exponential increase within the last few years (Figure 1). As of writing this review, there are 36 structures of the NCP in complex with other proteins. Nucleosome-binding partners include chromatin factors, pathogen peptides, linker histones and other nucleosomes. Binding of proteins to individual NCP components may be spatially restricted by the nucleosome presence. Nevertheless many proteins can be recruited to the nucleosome by direct or indirect readout of nucleosomal DNA, by features of histone sequence and geometry [11, 18], by histone and DNA covalent modifications, or by a combination thereof (Figure 2). Pioneering transcription factors may bind and recognize distinct nucleosomal features including but not limited to: major and minor DNA grooves, the dyad region, two DNA gyres, nucleosomal DNA ends and periodic patterns on nucleosomal DNA [19, 20]. A typical nucleosome partner can contact multiple NCP components or can read more than one covalent modification to ensure the selectivity of recognition. For example, it was recently shown that the binding of kinetochore protein Mif2/CENP-C to centromeric nucleosomes involves a complex interface composed of DNA- and histone-binding domains harboring the CENP-C motif, an AT-hook, and clusters enriched in arginine and lysine residues [21].
Figure 1. Growth trends in the number of nucleosome structures.
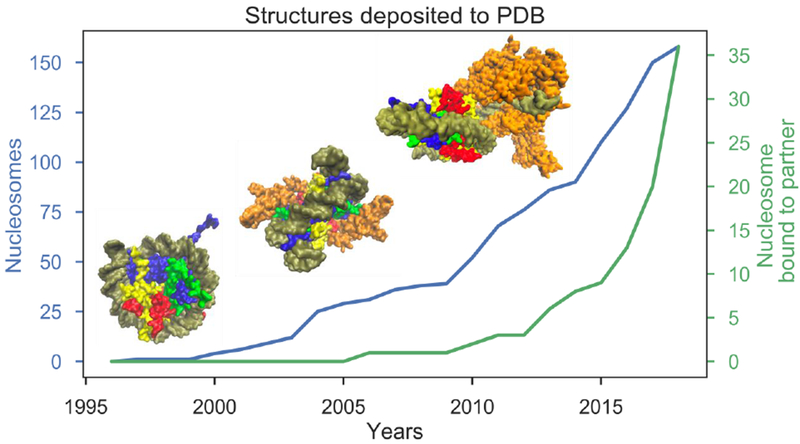
Blue graph shows the cumulative number of NCP structures in the PDB while the green graph shows the structures of the NCP in complex with a protein or peptide. From left to right, insets illustrate the first nucleosome crystal structure (1AOI, [1]), Sir3 BAH domain bound to yeast nucleosome (3TU4, [64]) and RNA Polymerase II elongation complex stalled at nucleosomal DNA SHL-2 (6A5R, [65]).
Figure 2. Features of the nucleosomal recognition.
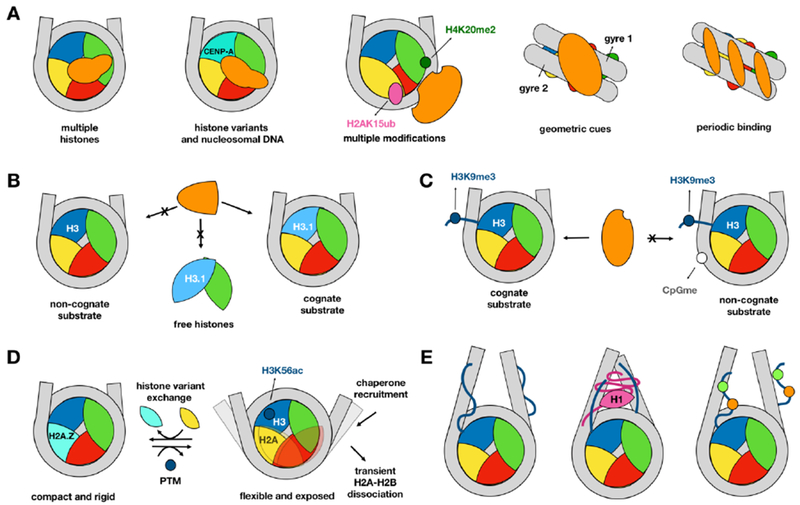
Nucleosome binding partners are shown in orange and specific examples are given in parentheses. A. Multivalent recognition of the nucleosome (from left to right): through multiple histones (Sir3 BAH domain, [64]), via specific histone variants and nucleosomal DNA (CENP-N bound to centromeric nucleosome [66]), through multiple modifications (53BP1, [27]), multiple geometric cues or periodic patterns (transcription factors [19]). B. A binding partner recognizes variant nucleosome and does not recognize canonical nucleosome or free histones (ATXR5, [3]). C. Multiple modifications can antagonistically influence the recognition by binding partner (Fdxl11/KDM2A, [11]). D. Multiple modifications and histone variants can have opposing effects on nucleosome dynamics with consequences for recognition ([28]). E. Competitive binding between core histone tails, H1, linker DNA and histone modifications ([45]).
Proteins can exclusively recognize nucleosomes while not interacting with the free DNA, free histones or histone octamers (Figure 2A). ATXR5 monomethyltransferase, for example, preferentially methylates NCPs over histone octamers (i.e., NCP without nucleosomal DNA) ensuring that H3K27me1 is not introduced on free histones and is not erroneously incorporated into active chromatin [3]. For some nucleosome binding partners, on the other hand, nucleosomal context can be preferential, but not an exclusive target. One such interactor is Spt-Ada-Gcn5 acetyltransferase (SAGA) coactivator that binds to monoubiquitinated histone H2B in nucleosomes and at the same time it binds to H2A/H2B dimers that are in complex with the histone chaperone, FACT [22]. This can be explained by the possible involvement of SAGA in the intermediate steps of nucleosomal assembly and disassembly.
Although the importance of non-random nucleosome positioning and its sequence preference has long been debated, the sequence and geometry of nucleosomal DNA can be vital for modulating the recognition for some protein partners. Shifting the position of DNA in the nucleosome by only one base pair leads to changes in its rotational positioning of around 36°, which can affect the binding of proteins that are recruited by specific DNA motifs. Examples include A-tracts and CpG methylation of nucleosomal DNA. The location of A-tracts with respect to the nucleosomal dyad has been shown to be crucial for directing the binding of proteins of the centromere machinery because these DNA elements are known for their specific properties, such as conformational rigidity and narrow minor grooves [12, 23, 24]. Covalent modifications of nucleosomal DNA can also affect its geometry, affinity and selectivity of NCP binding to other proteins. The methyl-CpG-binding protein MeCP2 and many other proteins may bind to DNA-methylated nucleosomes more favorably than to the free methylated DNA [11]. The affinity of these interactions depends on the locations of methylated CpG dinucleotides on the nucleosomal DNA. In case of methyl-binding domain MBD1, evidence suggests its preference for the methylated CpG of the dyad region facing the histone octamers [25]. It is known that many chromatin and transcription factors may recognize specific DNA sequence motifs on the free DNA. As to their binding to the nucleosomal DNA, it was recently shown that transcription factors can recognize partial or degenerate DNA sequence motifs and this ability correlates with the flexibility of their DNA binding domains [20].
Covalent modifications, especially charge altering modifications, in histones and nucleosomal DNA influence the stability, dynamics, accessibility of DNA, and may mediate or disrupt binding of other proteins [26]. Multiple modifications can be recognized simultaneously such as in the case of 53BP1 that is recruited by both methylation of H4 and ubiquitylation of H2A in response to the DNA double-strand breaks [27]. Computational studies predict that the effects of multiple modifications can be amplified as in the case of the simultaneous acetylation of H4K79 and H3K122 [14]. Modifications can assist the recognition of another modification by altering its accessibility, as in the case for the modifications on H3 tail [15] or can act antagonistically as shown in the following examples. Lysine demethylase Fbxl11/KDM2A binds to H3K9me3-modified nucleosomes via HP1 but this interaction is disrupted by CpG methylation of nucleosomal DNA [11]. In another case, H3K56ac and the exchange of H2A with the H2A.Z variant can antagonistically influence the lifetime of “open” nucleosome conformations that could be implicated in the recruitment of nucleosome-binding proteins [28]. Namely, H3K56 acetylation makes the interface between the (H3-H4)2 tetramer and the (H2A-H2B) dimer more flexible and shifts the equilibrium to a more open nucleosome state. On the other hand, H2A.Z deposition makes the dimer-tetramer interface less flexible and therefore the closed state dominates [28].
Intrinsic nucleosome dynamics and effects on binding
Nucleosomes are not static and represent the dynamic ensemble of interconverting structural substates. The intrinsic motions of nucleosomes can have implications for their geometry, stability, and solvent exposure, with further consequences for the binding of proteins to nucleosome. These motions include breathing and unwrapping of nucleosomal DNA [28], gaping of gyres [29] and conformational changes within the histone core [30]. The chromatin remodeler Snf2h, for example, illustrates how dynamic properties of the interface between histones H3 and H4 can be important for its function. Snf2h cannot slide DNA efficiently when these two histones are covalently crosslinked to each other and their dynamics is restrained [30]. Rearrangements in the H2A-H2B dimer within a nucleosome are also necessary to maintain interactions with the unwrapping DNA upon DNA breathing motions [31]. The analysis of intrinsic nucleosome dynamics may help to understand the mechanisms by which nucleosomes partially or completely disassemble. It is especially important since partially (dis)assembled nucleosome states, observed in several experimental and computational studies [13], may represent the means to regulate the nucleosomal DNA accessibility for DNA binding partners [32].
Binding of proteins to the nucleosome can be accompanied by substantial conformational changes in the histone octamer and DNA; it can unlock novel interfaces that are otherwise buried in the nucleosome in its canonical form. Dot1L, for example, binds to nucleosomes ubiquitinated at H2B K120, and with the help of H4 tail induces an H3 conformational change such that the core residue H3 K79 becomes accessible for methylation [33]. In another case, ANP32E-ZID binding drives a conformational change in the H2A.Z αC helix such that it becomes extended twice as long compared to its counterpart in the canonical nucleosome [34]. Nucleosome binding of the Mid domain of histone chaperone FACT induces conformational changes that lead to the disruption of the H3 αN helix holding the nucleosomal DNA ends together and cause DNA unwrapping from histones [35]. Several recent Cryo-EM structures reveal that interactions between nucleosomes and chromatin remodelers are accompanied by distortions in the nucleosomal DNA, such as formation of bulges [5] and widening of the gap between the two gyres [36].
Nucleosome recognition involving intrinsic protein disorder
Nearly a quarter of NCP histone sequence and more than half of linker histone H1 sequence are intrinsically disordered [37]. Perhaps not surprisingly, disorder is equally prevalent among nucleosome interactors. The disordered regions can explore a broad ensemble of conformations according to the “fly-casting” model, speeding up the search process in a manner reminiscent of the dimensionality reduction mechanism [38]. The intrinsic disorder allows for high specificity and low affinity interactions as is the case in many signaling processes [39, 40]. Binding can either maintain disorder or it can be accompanied by a disorder-to-order transition depending on whether the stable, short-range interactions can energetically compensate for the loss in conformational entropy. For example, high mobility group nucleosome (HMGN) binding proteins maintain their disordered state even when bound to nucleosomes. These proteins regulate chromatin structure through either directly interfering with the binding of linker histone H1 to the nucleosome [4] or by altering the condensation of the disordered H1 C-terminal domain and the interactions of the histone tails with nucleosomal DNA [41]. Figure 3 illustrates examples of recent structures of nucleosome-remodeler complexes and shows how histone tails may recognize acidic patches of binding partners. The disordered histone tails do not only provide sites for binding and PTMs, but also regulate the amount of surface area on the nucleosome accessible for binding other partners. For example, H3 N-terminal tails play especially crucial roles in this process because they can bind to linker DNA and modulate its accessibility for binding of H1, HMGN and effector proteins [15, 42–45].
Figure 3. Interactions between the basic patch within the histone H4 tail and acidic binding pockets of chromatin remodelers.

A. Snf2 (5X0Y [5]), B. Chd1 (5O9G [6]). Molecular surfaces are rendered in orange (Snf2 or Chd1), blue (H3), green (H4), yellow (H2A), red (H2B), and brown (DNA), respectively. Insets show the electrostatic potential mapped of the remodelers’ molecular surfaces. H4 sidechains on the binding interface are labeled.
Mechanisms of formation of higher order chromatin structure
The molecular basis for the transformation of open arrangements of neighboring nucleosomes into compact chromatin fiber structures continues to be an active area of research. The acidic patch, spanned by H2A and H2B residues exposed at the histone core, is a binding region for many binding partners including those responsible for the formation of higher order chromatin structure [4, 46, 47]. The acidic patch is commonly recognized by the “arginine anchors” of binding partners and can be modified by introducing histone variants. H2A.Z variants have additional negatively charged residues in the acidic patch that increase the compaction of nucleosome arrays whereas the H2A.B variant lacks some of the acidic patch residues [48, 49], resulting in a decrease in chromatin compaction. These variations in the acidic patch are specifically recognized by several binding partners including ISWI-family chromatin remodeling enzymes [50]. Another important regulator of chromatin compaction is the H4 N-terminal tail, in particular a small basic patch, whose deletion or modifications disrupt the condensation of chromatin [51]. Crystal-packing interactions between the acidic patch and the H4 tail of the neighboring nucleosomes were observed in early nucleosome studies [1], later also in a twisted tetranucleosomal structure [52], and more recently in the structure of a hexanucleosomal array forming a low-packing density chromatin fiber [53]. Interestingly, not only tail-core interactions, but also histone tail-tail interactions are important in chromatin fiber folding, and this process is highly regulated by tail acetylation [16, 54].
Linker histone H1 facilitates the formation of a compact arrangement of linker DNA strands [55, 56] and is an important hub in maintaining an extensive interaction network with many different partners [57]. Recent structures of linker histones bound to nucleosomes resolved the central, folded globular domain in contact with the dyad as well as with both DNA linker strands [58, 59]. These structures differ from prior, off-dyad models [52, 60], possibly due to a small number of key interfacial residues that vary between different species and histone variants as well as differences in the experimental setups and the heterogeneous nature of linker histone binding. Computational studies suggested that the alternative binding modes of the globular domain could be due to its conformational plasticity [61]. Even more puzzling remains the disordered C-terminal tail of H1 histone which leaves a distinct, ordered protection pattern on linker DNA when treated with hydroxyl radical footprinting [55]. In solution, the H1 C-terminal tail can form complexes with DNA, and the properties of its condensate depend on the phosphorylation level of the tail, a cell-cycle-dependent modification that correlates with the degree of chromatin compaction [62]. In addition, several studies pointed out the importance of salt concentration in regulating chromatin compaction [53, 63], however the molecular details of the transitions between different chromatin states remain to be elucidated.
Conclusions and Outlook
Chromatin is highly dynamic and interactions between its components occur on different time scales spanning several orders of magnitude. How the nucleosome is recognized by a multitude of diverse interactors and how this binding is controlled in time and space still remains ambiguous. However, the rapid development of experimental and computational techniques of high temporal and spatial precision permits a gain of more data about structure, binding kinetics and thermodynamics of different proteins to the nucleosome, including the dynamic crosstalk between histone and DNA modifications, and histone variants. The critical analysis and interpretation of this data should ultimately lead to a better conceptual understanding of how epigenetic signals can be integrated at the level of nucleosomes leading to signal amplification, enhanced response sensitivity and specificity.
Acknowledgements
This work was supported by the Intramural Research Programs of the National Heart, Lung and Blood Institute (S. K.) and the National Library of Medicine (S. K., A. G., Y. M., D. L., and A. R. P.) at the National Institutes of Health. We thank Yawen Bai, Bing-Rui Zhou, Stefan Dimitrov, Thomas Madej, and Alexey Onufriev for critical reading of the manuscript.
Abbreviations
- NCP
nucleosome core particle
- PTM
posttranslational modification
- PDB
Protein Data Bank
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Luger K, Mader AW, Richmond RK, Sargent DF, and Richmond TJ (1997). Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature 389, 251–260. [DOI] [PubMed] [Google Scholar]
- 2.Fang Q, Chen P, Wang M, Fang J, Yang N, Li G, and Xu RM (2016). Human cytomegalovirus IE1 protein alters the higher-order chromatin structure by targeting the acidic patch of the nucleosome. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bergamin E, Sarvan S, Malette J, Eram MS, Yeung S, Mongeon V, Joshi M, Brunzelle JS, Michaels SD, Blais A, et al. (2017). Molecular basis for the methylation specificity of ATXR5 for histone H3. Nucleic Acids Res 45, 6375–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kato H, van Ingen H, Zhou BR, Feng HQ, Bustin M, Kay LE, and Bai YW (2011). Architecture of the high mobility group nucleosomal protein 2-nucleosome complex as revealed by methyl-based NMR. P Natl Acad Sci USA 108, 12283–12288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu X, Li M, Xia X, Li X, and Chen Z (2017). Mechanism of chromatin remodelling revealed by the Snf2-nucleosome structure. Nature 544, 440–445. [DOI] [PubMed] [Google Scholar]
- 6.Farnung L, Vos SM, Wigge C, and Cramer P (2017). Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature 550, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ayala R, Willhoft O, Aramayo RJ, Wilkinson M, McCormack EA, Ocloo L, Wigley DB, and Zhang XD (2018). Structure and regulation of the human INO80-nucleosome complex. Nature 556, 391–395. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) Cryo-EM structure of the INO80 chromatin remodeling complex bound to partially flanking nucleosome revealing a novel binding mode where the motor domain binds to DNA at the entry point to the nucleosome Two similar structures by independent efforts were reported in the same journal issue (also see highlight below). These structures provide insight into the processive nucleosome sliding and versatile editing functions of this remodeler complex.
- 8.Eustermann S, Schall K, Kostrewa D, Lakomek K, Strauss M, Moldt M, and Hopfner KP (2018). Structural basis for ATP-dependent chromatin remodelling by the INO80 complex. Nature 556, 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) Cryo-EM structure of the INO80 chromatin remodeling complex bound to partially flanking nucleosome. Also see highlight of Ayala et al., above
- 9.Rhee HS, and Pugh BF (2011). Comprehensive genome-wide protein-DNA interactions detected at single-nucleotide resolution. Cell 147, 1408–1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shaytan AK, Xiao H, Armeev GA, Gaykalova DA, Komarova GA, Wu C, Studitsky VM, Landsman D, and Panchenko AR (2018). Structural interpretation of DNA-protein hydroxyl-radical footprinting experiments with high resolution using HYDROID. Nat Protoc 13, 2535–2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartke T, Vermeulen M, Xhemalce B, Robson SC, Mann M, and Kouzarides T (2010). Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 143, 470–484. [DOI] [PMC free article] [PubMed] [Google Scholar]; (**) Comprehensive study to identify chromatin interactors that can recognize DNA- and histone H3 methylation patterns. The authors employ a SILAC-based nucleosome affinity purification (SNAP) technique to reveal several different forms of the crosstalk that can occur between these two different classes of modifications.
- 12.Shaytan AK, Xiao H, Armeev GA, Wu C, Landsman D, and Panchenko AR (2017). Hydroxyl-radical footprinting combined with molecular modeling identifies unique features of DNA conformation and nucleosome positioning. Nucleic Acids Res 45, 9229–9243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rychkov GN, Ilatovskiy AV, Nazarov IB, Shvetsov AV, Lebedev DV, Konev AY, Isaev-Ivanov VV, and Onufriev AV (2017). Partially Assembled Nucleosome Structures at Atomic Detail. Biophys J 112, 460–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fenley AT, Anandakrishnan R, Kidane YH, and Onufriev AV (2018). Modulation of nucleosomal DNA accessibility via charge-altering post-translational modifications in histone core. Epigenetics Chromatin 11, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Morrison EA, Bowerman S, Sylvers KL, Wereszczynski J, and Musselman CA (2018). The conformation of the histone H3 tail inhibits association of the BPTF PHD finger with the nucleosome. Elife 7. [DOI] [PMC free article] [PubMed] [Google Scholar]; (*) An example of a hybrid approach integrating NMR spectroscopy and molecular dynamics simulations. The authors analyze the crosstalk between histone PTMs on H3 tail and binding of effector domains. They show that conformations of H3 tail inhibit the binding of PHD finger while certain modifications of H3 tail increase the accessibility of another tail modification, H3K4me3, to PHD finger domain.
- 16.Zhang R, Erler J, and Langowski J (2017). Histone Acetylation Regulates Chromatin Accessibility: Role of H4K16 in Inter-nucleosome Interaction. Biophys J 112, 450–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rose PW, Prlic A, Altunkaya A, Bi C, Bradley AR, Christie CH, Costanzo LD, Duarte JM, Dutta S, Feng Z, et al. (2017). The RCSB protein data bank: integrative view of protein, gene and 3D structural information. Nucleic Acids Res 45, D271–D281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu P, Li CM, Chen ZH, Jiang SY, Fan SL, Wang JW, Dai JB, Zhu P, and Chen ZC (2016). The NuA4 Core Complex Acetylates Nucleosomal Histone H4 through a Double Recognition Mechanism. Mol Cell 63, 965–975. [DOI] [PubMed] [Google Scholar]
- 19.Zhu FJ, Farnung L, Kaasinen E, Sahu B, Yin YM, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, et al. (2018). The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]; (**) 14 A comprehensive analysis of the binding motifs of transcription factors to the nucleosome. The authors show that while many transcription factors can not bind to DNA in the context of nucleosome, many others exploit a diverse set of binding modes and DNA motifs.
- 20.Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiao H, Wang F, Wisniewski J, Shaytan AK, Ghirlando R, FitzGerald PC, Huang Y, Wei D, Li S, Landsman D, et al. (2017). Molecular basis of CENP-C association with the CENP-A nucleosome at yeast centromeres. Genes Dev 31, 1958–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morgan MT, Haj-Yahya M, Ringel AE, Bandi P, Brik A, and Wolberger C (2016). Structural basis for histone H2B deubiquitination by the SAGA DUB module. Science 351, 725–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bao Y, White CL, and Luger K (2006). Nucleosome core particles containing a poly(dA.dT) sequence element exhibit a locally distorted DNA structure. J Mol Biol 361, 617–624. [DOI] [PubMed] [Google Scholar]
- 24.Buckwalter JM, Norouzi D, Harutyunyan A, Zhurkin VB, and Grigoryev SA (2017). Regulation of chromatin folding by conformational variations of nucleosome linker DNA. Nucleic Acids Res 45, 9372–9387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mendonca A, Sanchez OF, Liu WJ, Li Z, and Yuan CL (2017). CpG dinucleotide positioning patterns determine the binding affinity of methyl-binding domain to nucleosomes. Bba-Gene Regul Mech 1860, 713–720. [DOI] [PubMed] [Google Scholar]
- 26.Ruthenburg AJ, Li H, Patel DJ, and Allis CD (2007). Multivalent engagement of chromatin modifications by linked binding modules. Nat Rev Mol Cell Biol 8, 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson MD, Benlekbir S, Fradet-Turcotte A, Sherker A, Julien JP, McEwan A, Noordermeer SM, Sicheri F, Rubinstein JL, and Durocher D (2016). The structural basis of modified nucleosome recognition by 53BP1. Nature 536, 100–103. [DOI] [PubMed] [Google Scholar]
- 28.Kim J, Wei S, Lee J, Yue H, and Lee TH (2016). Single-Molecule Observation Reveals Spontaneous Protein Dynamics in the Nucleosome. J Phys Chem B 120, 8925–8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ngo TT, and Ha T (2015). Nucleosomes undergo slow spontaneous gaping. Nucleic Acids Res 43, 3964–3971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinha KK, Gross JD, and Narlikar GJ (2017). Distortion of histone octamer core promotes nucleosome mobilization by a chromatin remodeler. Science 355, eaaa3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bilokapic S, Strauss M, and Halic M (2018). Histone octamer rearranges to adapt to DNA unwrapping. Nat Struct Mol Biol 25, 101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bohm V, Hieb AR, Andrews AJ, Gansen A, Rocker A, Toth K, Luger K, and Langowski J (2011). Nucleosome accessibility governed by the dimer/tetramer interface. Nucleic Acids Res 39, 3093–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Worden EJ, Hoffmann NA, Hicks CW, and Wolberger C (2019). Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell 176, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Obri A, Ouararhni K, Papin C, Diebold ML, Padmanabhan K, Marek M, Stoll I, Roy L, Reilly PT, Mak TW, et al. (2014). ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature 505, 648–653. [DOI] [PubMed] [Google Scholar]
- 35.Tsunaka Y, Fujiwara Y, Oyama T, Hirose S, and Morikawa K (2016). Integrated molecular mechanism directing nucleosome reorganization by human FACT. Gene Dev 30, 673–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willhoft O, Ghoneim M, Lin CL, Chua EYD, Wilkinson M, Chaban Y, Ayala R, McCormack EA, Ocloo L, Rueda DS, et al. (2018). Structure and dynamics of the yeast SWR1-nucleosome complex. Science 362. [DOI] [PubMed] [Google Scholar]
- 37.Cutter AR, and Hayes JJ (2015). A brief review of nucleosome structure. FEBS Lett 589, 2914–2922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shoemaker BA, Portman JJ, and Wolynes PG (2000). Speeding molecular recognition by using the folding funnel: The fly-casting mechanism. P Natl Acad Sci USA 97, 8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuxreiter M (2018). Fold or not to fold upon binding - does it really matter? Curr Opin Struct Biol 54, 19–25. [DOI] [PubMed] [Google Scholar]
- 40.Darling AL, and Uversky VN (2018). Intrinsic Disorder and Posttranslational Modifications: The Darker Side of the Biological Dark Matter. Front Genet 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Murphy KJ, Cutter AR, Fang H, Postnikov YV, Bustin M, and Hayes JJ (2017). HMGN1 and 2 remodel core and linker histone tail domains within chromatin. Nucleic Acids Res 45, 9917–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Angelov D, Vitolo JM, Mutskov V, Dimitrov S, and Hayes JJ (2001). Preferential interaction of the core histone tail domains with linker DNA. Proc Natl Acad Sci U S A 98, 6599–6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arya G, and Schlick T (2006). Role of histone tails in chromatin folding revealed by a mesoscopic oligonucleosome model. Proc Natl Acad Sci U S A 103, 16236–16241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shaytan AK, Armeev GA, Goncearenco A, Zhurkin VB, Landsman D, and Panchenko AR (2016). Coupling between Histone Conformations and DNA Geometry in Nucleosomes on a Microsecond Timescale: Atomistic Insights into Nucleosome Functions. J Mol Biol 428, 221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stutzer A, Liokatis S, Kiesel A, Schwarzer D, Sprangers R, Soding J, Selenko P, and Fischle W (2016). Modulations of DNA Contacts by Linker Histones and Posttranslational Modifications Determine the Mobility and Modifiability of Nucleosomal H3 Tails. Mol Cell 61, 247–259. [DOI] [PubMed] [Google Scholar]; (*) The authors demonstrate that the dynamics of H3 tails are restricted in the presence of linker histones. They further show that modifications of H3 tails and the occupancies of linker histones are anti-correlated on a genome-wide level.
- 46.Lesbats P, Serrao E, Maskell DP, Pye VE, O’Reilly N, Lindemann D, Engelman AN, and Cherepanov P (2017). Structural basis for spumavirus GAG tethering to chromatin. Proc Natl Acad Sci U S A 114, 5509–5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, and Bai Y (2013). A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science 340, 1110–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sullivan SA, and Landsman D (2004). Mining core histone sequences from public protein databases. Methods Enzymol 375, 3–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Draizen EJ, Shaytan AK, Marino-Ramirez L, Talbert PB, Landsman D, and Panchenko AR (2016). HistoneDB 2.0: a histone database with variants--an integrated resource to explore histones and their variants. Database (Oxford) 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dann GP, Liszczak GP, Bagert JD, Muller MM, Nguyen UTT, Wojcik F, Brown ZZ, Bos J, Panchenko T, Pihl R, et al. (2017). ISWI chromatin remodellers sense nucleosome modifications to determine substrate preference. Nature 548, 607–611. [DOI] [PMC free article] [PubMed] [Google Scholar]; (**) High-throughput nucleosome remodeling assay using ISWI-family enzymes and a diverse library of posttranslationally modified nucleosomes. The authors characterize in detail which PTMs lead to elevated activity in comparison to unmodified nucleosomes.
- 51.Dorigo B, Schalch T, Bystricky K, and Richmond TJ (2003). Chromatin fiber folding: Requirement for the histone H4 N-terminal tail. J Mol Biol 327, 85–96. [DOI] [PubMed] [Google Scholar]
- 52.Song F, Chen P, Sun DP, Wang MZ, Dong LP, Liang D, Xu RM, Zhu P, and Li GH (2014). Cryo-EM Study of the Chromatin Fiber Reveals a Double Helix Twisted by Tetranucleosomal Units. Science 344, 376–380. [DOI] [PubMed] [Google Scholar]
- 53.Garcia-Saez I, Menoni H, Boopathi R, Shukla MS, Soueidan L, Noirclerc-Savoye M, Le Roy A, Skoufias DA, Bednar J, Hamiche A, et al. (2018). Structure of an H1-Bound 6-Nucleosome Array Reveals an Untwisted Two-Start Chromatin Fiber Conformation. Mol Cell 72, 902–915. [DOI] [PubMed] [Google Scholar]
- 54.Bascom GD, and Schlick T (2018). Chromatin Fiber Folding Directed by Cooperative Histone Tail Acetylation and Linker Histone Binding. Biophysical Journal 114, 2376–2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Syed SH, Goutte-Gattat D, Becker N, Meyer S, Shukla MS, Hayes JJ, Everaers R, Angelov D, Bednar J, and Dimitrov S (2010). Single-base resolution mapping of H1-nucleosome interactions and 3D organization of the nucleosome. Proc Natl Acad Sci U S A 107, 9620–9625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fang H, Clark DJ, and Hayes JJ (2012). DNA and nucleosomes direct distinct folding of a linker histone H1 C-terminal domain. Nucleic Acids Res 40, 1475–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Szerlong HJ, Herman JA, Krause CM, DeLuca JG, Skoultchi A, Winger QA, Prenni JE, and Hansen JC (2015). Proteomic characterization of the nucleolar linker histone H1 interaction network. J Mol Biol 427, 2056–2071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou BR, Jiang J, Feng H, Ghirlando R, Xiao TS, and Bai Y (2015). Structural Mechanisms of Nucleosome Recognition by Linker Histones. Mol Cell 59, 628–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bednar J, Garcia-Saez I, Boopathi R, Cutter AR, Papai G, Reymer A, Syed SH, Lone IN, Tonchev O, Crucifix C, et al. (2017). Structure and Dynamics of a 197 bp Nucleosome in Complex with Linker Histone H1. Mol Cell 66, 384–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhou BR, Feng HQ, Kato H, Dai L, Yang YD, Zhou YQ, and Bai YW (2013). Structural insights into the histone H1-nucleosome complex. P Natl Acad Sci USA 110, 19390–19395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ozturk MA, Pachov GV, Wade RC, and Cojocaru V (2016). Conformational selection and dynamic adaptation upon linker histone binding to the nucleosome. Nucleic Acids Res 44, 6599–6613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Turner AL, Watson M, Wilkins OG, Cato L, Travers A, Thomas JO, and Stott K (2018). Highly disordered histone H1-DNA model complexes and their condensates. Proc Natl Acad Sci U S A 115, 11964–11969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Allahverdi A, Chen Q, Korolev N, and Nordenskiold L (2015). Chromatin compaction under mixed salt conditions: opposite effects of sodium and potassium ions on nucleosome array folding. Sci Rep 5, 8512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Armache KJ, Garlick JD, Canzio D, Narlikar GJ, and Kingston RE (2011). Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 A resolution. Science 334, 977–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kujirai T, Ehara H, Fujino Y, Shirouzu M, Sekine SI, and Kurumizaka H (2018). Structural basis of the nucleosome transition during RNA polymerase II passage. Science 362, 595–598. [DOI] [PubMed] [Google Scholar]; (**) Cryo-EM structures of transcribing RNA Polymerase II in complex with the nucleosome. The authors report multiple complexes stalled at different superhelical locations demonstrating that the elongation complex preserves the nucleosome intact.
- 66.Chittori S, Hong J, Saunders H, Feng H, Ghirlando R, Kelly AE, Bai Y, and Subramaniam S (2018). Structural mechanisms of centromeric nucleosome recognition by the kinetochore protein CENP-N. Science 359, 339–343. [DOI] [PMC free article] [PubMed] [Google Scholar]