

Abstract
Background
Evolutionary patterns of scleractinian (stony) corals are difficult to infer given the existence of few diagnostic characters and pervasive phenotypic plasticity. A previous study of Hawaiian Montipora (Scleractinia: Acroporidae) based on five partial mitochondrial and two nuclear genes revealed the existence of a species complex, grouping one of the rarest known species (M. dilatata, which is listed as Endangered by the International Union for Conservation of Nature - IUCN) with widespread corals of very different colony growth forms (M. flabellata and M. cf. turgescens). These previous results could result from a lack of resolution due to a limited number of markers, compositional heterogeneity or reflect biological processes such as incomplete lineage sorting (ILS) or introgression.
Results
All 13 mitochondrial protein-coding genes from 55 scleractinians (14 lineages from this study) were used to evaluate if a recent origin of the M. dilatata species complex or rate heterogeneity could be compromising phylogenetic inference. Rate heterogeneity detected in the mitochondrial data set seems to have no significant impacts on the phylogenies but clearly affects age estimates. Dating analyses show different estimations for the speciation of M. dilatata species complex depending on whether taking compositional heterogeneity into account (0.8 [0.05–2.6] Myr) or assuming rate homogeneity (0.4 [0.14–0.75] Myr). Genomic data also provided evidence of introgression among all analysed samples of the complex. RADseq data indicated that M. capitata colour morphs may have a genetic basis.
Conclusions
Despite the volume of data (over 60,000 SNPs), phylogenetic relationships within the M. dilatata species complex remain unresolved most likely due to a recent origin and ongoing introgression. Species delimitation with genomic data is not concordant with the current taxonomy, which does not reflect the true diversity of this group. Nominal species within the complex are either undergoing a speciation process or represent ecomorphs exhibiting phenotypic polymorphisms.
Electronic supplementary material
The online version of this article (10.1186/s12862-019-1476-2) contains supplementary material, which is available to authorized users.
Keywords: Across-branch compositional heterogeneity, Introgressive hybridization, Species complex, RADseq, Corals
Background
Reef-building corals are a complex ecosystem involving biotic interactions within the holobiont (the cnidarian host and its associated microorganisms). Multiple sources of genetic variation involving e.g. parallel evolution [1], and the observed ubiquitous phenotypic plasticity [2] have recognizably obscured phylogenetic inference and species delimitation within the order Scleractinia (stony corals). Defining species boundaries plays a central role in the establishment of conservation policies [3] and biodiversity assessment [4].
The use of a small number of loci usually provides suitable results for distantly related organisms but often produce gene tree discordance when shallower divergences are involved due to population-level effects such as allele frequency changes [5]. However, the concatenation of multiple independent loci can also produce misleading results [6], when, for example, heterogeneity among gene trees is not modeled appropriately [7]. Incomplete lineage sorting (ILS) and interspecific hybridization are among the most common causes of genome-wide heterogeneity in closely related species [8]. Across-branch compositional heterogeneity (i.e., nonstationarity) may also have strong effects on the phylogenetic inference because it groups unrelated taxa that share similar base compositional biases [9–11].
The multispecies coalescent approach [12, 13] and Bayesian species delimitation models using genome-wide data [14] may represent an alternative when single locus are insufficient to solve species boundaries (African beetles, [15]; green algae, [16]). Restriction-site associated DNA sequencing (RADseq) along with the production of thousands of single nucleotide polymorphisms (SNPs) is being increasingly used in shallow systematics, particularly in species delimitation of recent radiations [17, 18] or to evaluate the existence ancestral hybridization and introgression [19].
Phylogenetic inference within stony corals of the order Scleractinia has been hindered by a suit of factors including hybridization [20] and historical introgression [21]. Nuclear internal transcribed spacers (ITS) and a suite of mitochondrial markers have been widely used in coral phylogenies [22–24], but the existence of multiple ITS copies [25, 26] or the reduced utility of mitochondrial markers to delimit species [27, 28] has hindered our understanding of the evolution and molecular ecology of scleractinian corals.
The coral genus Montipora (Scleractinia, Acroporidae) is widely distributed across the Indian and the Pacific Oceans [29]. While putative reticulate evolution and hybridization were detected in some Australian Montipora species [30], it is not clear to what extent this phenomenon is widespread. A study on the Hawaiian congeners based on several mitochondrial (cytochrome oxidase subunit I, control region, cytochrome b, 16S rRNA, and ATP6) and nuclear (ITS and ATPsβ) markers, revealed the existence of a species complex (M. dilatata/M. flabellata/M. cf. turgescens), and phylogenetic patterns consistent with ILS, hybridization, polymorphism or phenotypic plasticity but the available data could not resolve among these alternate hypotheses [31].
Our objective was to determine if sampling all 13 nearly complete protein-coding genes of the mitochondrial genome and phylogenomic (RADseq) data would allow the resolution of species boundaries within the Montipora dilatata/M. flabellata/M. cf. turgescens complex. Conservation actions should be based on evolutionary distinctiveness metrics (e.g. full-coverage species-level phylogenies diversity over time; [32, 33]). This approach requires a significant amount of data, not always available for the target clade. Other approaches, including expert opinions or phylogenies with lower taxonomic coverage, may also perform well in the establishment of species boundaries [34].
We used the mitogenomic data set to analyse across-branch compositional biases and perform a dating analysis. We included a larger number of scleractinian representatives (six families and 15 genera) with an extensive fossil record required for date estimation. We intended to evaluate if a recent origin of the M. dilatata species complex could be hindering phylogenetic inference. RADseq data from 16 samples representing seven nominal Montipora species was used to investigate if ILS and/ or introgressive hybridization could be compromising species delimitation within the M. dilatata complex. We also evaluate whether the morphological variants of the highly polymorphic congener M. capitata can be distinguished at genomic level and how the range of variation within this undisputed species can be compared to their endangered congeners. In addition, a range of phenotypes (laminar, encrusting, plating, and branching) of M. capitata and included pooled libraries of distinct colour morphs (red and yellow/orange) previously shown to have different symbiont communities [35] were sampled to characterize within-species variation.
Results
Phylogenetic relationships within Scleractinian based on mitogenomic data
Mapping paired reads in Geneious v.8.1.4 to the mitochondrial genome of Montipora cactus (GenBank # NC_006902) resulted in a mean of 4583 reads per sample covering 97% of the reference sequence at a mean depth of 61 ± 60 (mean ± standard deviation) per library (Additional file 1: S1B). The concatenated nucleotide dataset of the 55 scleractinians (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups (57 taxa in total) comprised 11,484 characters. The net uncorrected p-distance between M. capitata and M. dilatata species complex was 0.004 ± 0.001. Uncorrected p-distances within the M. capitata clade and the M. dilatata species complex clade (M. dilatata/M. flabellata/M. cf. turgescens) were both zero.
P4 analysis of the concatenated mitochondrial dataset rejected among-lineage composition homogeneity (p-value = 0.00). The analysis under the node-discrete composition heterogeneity model (NDCH2; p-value = 0.85) indicated the existence four main clades including genera from the following families: clade 1 - Rhizangiidae + Merulinidae; clade 2 - Pocilloporidae; clade 3 - Poritidae; clade 4 - Acroporidae (Fig. 1). Similar to previous studies [36, 37], the clade corresponding to the family Acroporidae also included the genus Alveopora (Poritidae). Phylogenetic relationships within the M. dilatata species complex were unresolved. Montipora capitata was retrieved as the sister lineage to the remaining species of the genus. Montipora patula and M. verrilli grouped together to the exclusion of M. cactus. BI analyses performed with MrBayes under the assumption of a homogeneous model of rate change yielded a topology (Additional file 2: S2) identical to P4.
Fig. 1.

a Majority-rule consensus tree of a composition-heterogeneous Bayesian analysis of the concatenated dataset of the 13 protein-coding genes of the mitochondrial genome of 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile. The NDCH2 analysis was performed in P4 using duplicate runs each consisting of 2 million generations using a GTR + I + Γ model. Bayesian posterior probability values are shown in black circles for values of maximal probability (1.00) and grey circles for values between 0.95 and 0.97. b Inset highlighted in dark grey showing a detail of the inferred phylogenetic relationships within Montipora. Families are highlighted in light grey. Specimens in bold were sequenced in this study
In RAxML, we used the concatenated mitochondrial nucleotide dataset with eight partition schemes as determined by PartitionFinder, and corresponding evolutionary models, all accommodating rate heterogeneity: (1) nad5; (2) nad1; (3) cyt b and cox3; (4) nad2, nad3 and nad4; (5) cox2, nad6 and nad4L; (6) atp6; (7) atp8 and (8) cox1. The best substitution model was GTR + I + Γ for all partitions. This analysis (Additional file 3: S3) yielded a similar topology to the P4 tree with the two following exceptions: (1) phylogenetic relationships within the Montipora complex (M. dilatata/M. flabellata/M. cf. turgescens) were unresolved in P4 and Mrbayes whereas in RAxML M. dilatata (sample Mdil12) from Kane‘ohe bay was retrieved as the sister lineage of all remaining species, and (2) phylogenetic relationships between M. capitata morphs were unresolved in MrBayes, while samples 1 and 2 (red plate/branch and red branch) clustered together with high statistical support in RAxML.
Dating analyses
Multidivtime (Fig. 2) estimate for the crown group age of Montipora is 10.5 [5.3–16.6] Myr, while Beast (Additional file 4: S4) shows a more recent origin for the genus at 4.3 [3.6–5.1] Myr. According to Multidivtime, the crown group age of M. capitata is 0.6 [0.02–2.4] Myr, whereas Beast estimates it at 0.2 [0.06–0.4] Myr. The crown group age of the complex Montipora dilatata/M. flabellata/M. cf. turgescens was estimated at 0.8 [0.05–2.6] Myr or 0.4 [0.1–0.8], as estimated by Multidivtime and Beast, respectively.
Fig. 2.

Bayesian divergence dating analysis obtained with Multidivtime. Divergence dates were estimated on the Bayesian topology inferred by P4 and based on the concatenated dataset of the nearly complete 13 mitochondrial protein-coding genes of 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile (which are automatically removed from the resulting topology). Numbers at the nodes represent age estimates for the main groups in million years. Asteriks at the nodes represent minimum age constraints obtained from the fossil record, and 95% confidence intervals are represented by the blue bars
SNP-based analyses
The ‘coral’ data set averaged ~ 946,887 reads passing filtering per library with a coverage depth of 15.63 ± 8.77 (mean ± stdev) (Additional file 1: S1B).
The Venn diagram (Additional file 6: S6) plotted using the program BioVenn [38] shows minimal overlap between putative coral and symbiont loci. 64% (43,423) of the overall loci (67,598) had significant hits to the M. capitata draft genome, [39], whereas only 6% (4,286) were significant against the Symbiodinium minutum genome [40] and 7% (4,727) against the Fugacium kawagutii genome [41]. As such, we used the ‘coral’ data set in all analyses.
The net uncorrected p-distance based on the ‘coral’ data set between Montipora capitata and Montipora dilatata species complex was 0.301 ± 0.043. Uncorrected p-distances within the M. capitata clade and M. dilatata species complex clade (M. dilatata/M. flabellata/M. cf. turgescens) were 0.056 ± 0.011 and 0.140 ± 0.022, respectively.
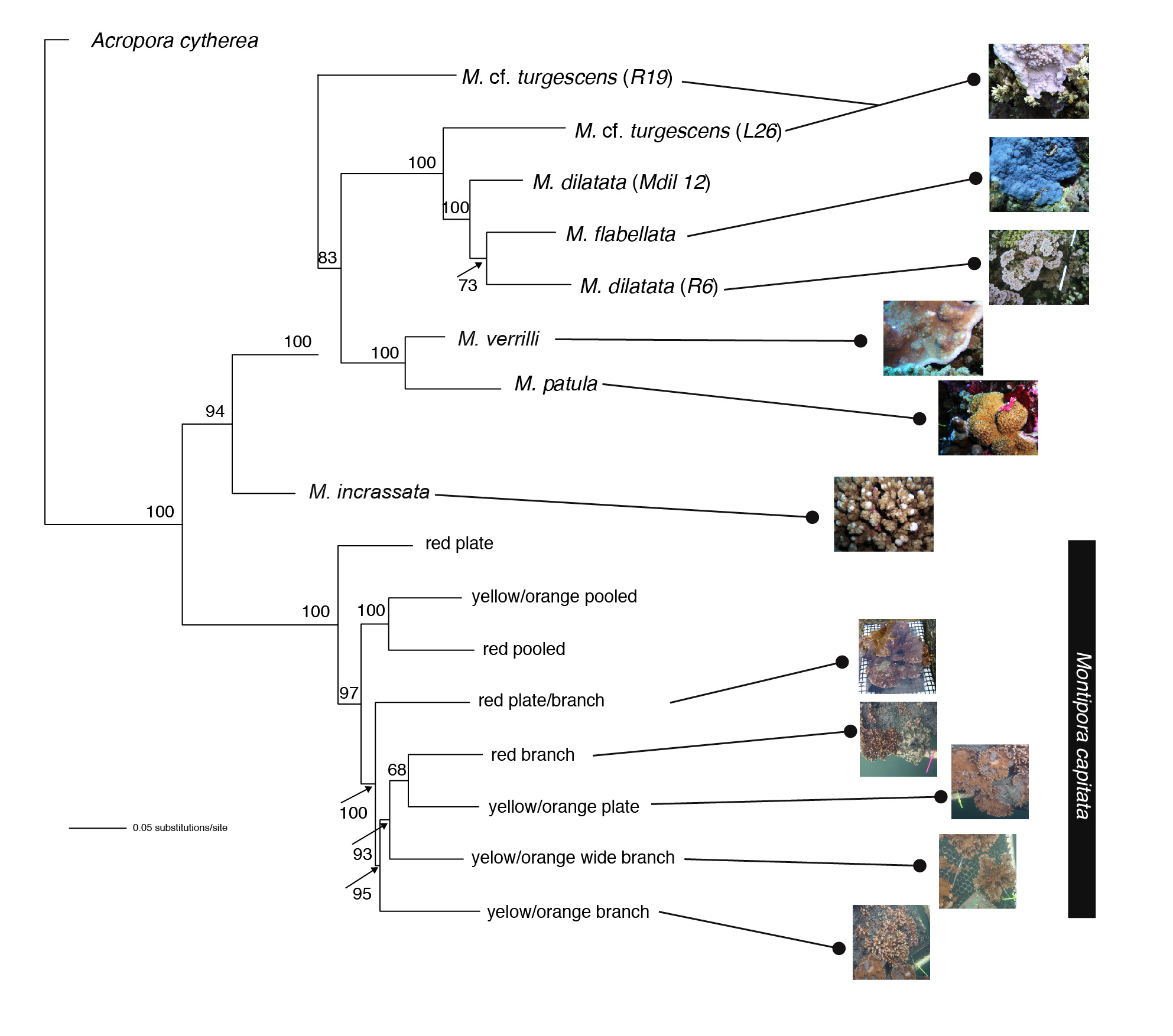
ML analysis based on the ‘coral’ dataset (Fig. 3), recovered the two M. dilatata samples in a clade that received high statistical support. Montipora cf. turgescens (sample L26) did not cluster with its conspecifics (sample R6). Montipora patula and M. verrilli were retrieved as the sister clade of the species. The ML analysis (Fig. 3) showed some level of correspondence between phenotypic variability and genetic clustering within Montipora capitata. Yellow/orange colonies 3 and 5 and red colonies 1 and 2 grouped together but with relatively low statistical support. Images of each species and Montipora morphotypes used in the analyses are shown in Additional file 7: S7. We found no genetic association between samples with similar shapes (e.g., colonies 3 and 5 grouped together and show plate and branch shapes, respectively) thus we performed no further analyses on this relationship. More stringent options regarding missing data (−-max-missing 0.75; 16,379 SNPs) yielded an identical ML topology but with less statistical support for the recovered clades (not shown); therefore, we used the file with a larger number of SNPs (60,602 SNPs) in the analyses.
Fig. 3.

Maximum likelihood phylogram of Hawaiian Montipora corals inferred with RAxML based on the ‘coral’ dataset (draft genome-based assembly of RADseq data from 16 samples; 60,602 SNPs). Numbers at the nodes represent Bayesian Posterior Probabilities. Acropora cytherea and A. hyacinthus are the selected outgroup. Numbers in the black circles, both on the panel images and branches of the phylogenetic tree, correspond to five distinct morphotypes within Montipora capitata. Specimen labels are indicated within parentheses
Bayes factor comparisons for species delimitation using MLE (Table 1) clearly reject current taxonomy, favouring the hypothesis that groups M. capitata by colour morphs and splits the remaining samples into different species. The second-best hypothesis is the one that considers M. dilatata and M. flabellata as a single species and splits the remaining samples into different species. The hypothesis that considers the M. dilatata complex as a single species received low support.
Table 1.
Species delimitation using BFD* with SNP data from the Hawaiian Montipora based on the ‘coral’ data set. The six analysed models are ranked according to their Bays Factor (BF) value
| Model | MLE | Rank | BF |
|---|---|---|---|
| A - current taxonomy | − 572.42 | 5 | N/A |
| B - splits M. dilatata and M. cf. turgescens samples into different species | −318.88 | 2 | −507.1 |
| C - M. dilatata complex as a single species | − 721.67 | 5 | 298.5 |
| D - groups M. capitata by colour morphs (Yellow/orange; Red) | − 288.39 | 1 | − 568.1 |
| E - Lumps M. dilatata into a single species; splits all remaining samples of the complex as different species | −338.36 | 4 | − 468.1 |
| F - Groups M. dilatata and M. flabellata into a single species; splits M. cf. turgescens | −328.74 | 3 | − 487.4 |
MLE marginal likelihood estimate, BF Bayes factor.
Values in bold correspond to the best estimated model
The species tree based on the ‘coral’ data set estimated by SVDQuartets yielded a topology (Additional file 5: S5) in which M. dilatata (samples Mdil12 and R6) groups with M. flabellata in the same clade, and the two samples of M. cf. turgescens do not cluster together. The species tree marginally supports a genetic basis for the observed polymorphisms; only yellow/orange colonies 4 and 5 grouped together (Additional file 5: S5).
Genomic admixture
Given the conflicting results we obtained regarding phylogenetic relationships within the M. dilatata complex depending on the methods used (RAxML: M. dilatata monophyletic; SVDQuartets: M. dilatata paraphyletic) we tested for the existence of ILS/introgression. We used the D-statistic calculation [42] to distinguish between both processes. Considering the dubious taxonomic status of the species belonging to the complex, we tested for all 30 possible combinations of donor/admixed lineages between the five samples (Table 2).
Table 2.
Measure of the phylogenetic admixture among the species of the Montipora dilatata complex. (M. dilatata/M. flabellata/M. cf. turgescens) based on the ‘coral’ data set
| Gene tree | Hypotheses | (((P1,P2),P3),O); | ABBA | BABA | D_stat | D_Pvalue | introgression |
|---|---|---|---|---|---|---|---|
| Donor = Montipora dilatata: specimen Mdil12 | H1 | P1 = R6Mdil; P2 = Mflab; P3 = Mdil12; Outgroup = L27incra | 247 | 286 | −0.073 | 0.091 | none |
| H2 | P1 = R6Mdil; P2 = L26turg; P3 = Mdil12; Outgroup = L27incra | 151 | 237 | −0.222 | 1.3E-05 | 13 | |
| H3 | P1 = R6Mdil; P2 = R19turg; P3 = Mdil12; Outgroup = L27incra | 102 | 195 | −0.313 | 6.8E-08 | 13 | |
| H4 | P1 = L26turg; P2 = R19turg; P3 = Mdil12; Outgroup = L27incra | 71 | 122 | −0.264 | 2.4E-04 | 13 | |
| H5 | P1 = Mflab; P2 = R19turg; P3 = Mdil12; Outgroup = L27incra | 99 | 204 | −0.347 | 1.6E-09 | 13 | |
| H6 | P1 = Mflab; P2 = L26turg; P3 = Mdil12; Outgroup = L27incra | 159 | 221 | −0.163 | 1.5E-03 | 13 | |
| Donor = M. dilatata: specimen R6Mdil | H7 | P1 = Mdil12; P2 = Mflab; P3 = R6Mdil; Outgroup = L27incra | 192 | 286 | −0.197 | 1.7E-05 | 13 |
| H8 | P1 = Mdil12; P2 = L26turg; P3 = R6Mdil; Outgroup = L27incra | 112 | 237 | −0.358 | 2.2E-11 | 13 | |
| H9 | P1 = Mdil12; P2 = R19turg; P3 = R6Mdil; Outgroup = L27incra | 82 | 195 | −0.408 | 1.1E-11 | 13 | |
| H10 | P1 = L26turg; P2 = Mflab; P3 = R6Mdil; Outgroup = L27incra | 182 | 166 | 0.046 | 0.391 | none | |
| H11 | P1 = L26turg; P2 = R19turg; P3 = R6Mdil; Outgroup = L27incra | 78 | 125 | −0.232 | 0.001 | 13 | |
| H12 | P1 = Mflab; P2 = R19turg; P3 = R6Mdil; Outgroup = L27incra | 109 | 202 | −0.299 | 0.000 | 13 | |
| Donor = M. flabellata: specimen Mflab | H13 | P1 = Mdil12; P2 = R6Mdil; P3 = Mflab; Outgroup = L27incra | 192 | 247 | −0.125 | 0.009 | 13 |
| H14 | P1 = Mdil12; P2 = R19turg; P3 = Mflab; Outgroup = L27incra | 86 | 204 | −0.407 | 0.000 | 13 | |
| H15 | P1 = Mdil12; P2 = L26turg; P3 = Mflab; Outgroup = L27incra | 134 | 221 | −0.245 | 0.000 | 13 | |
| H16 | P1 = R6Mdil; P2 = L26turg; P3 = Mflab; Outgroup = L27incra | 166 | 182 | −0.046 | 0.391 | none | |
| H17 | P1 = R6Mdil; P2 = R19turg; P3 = Mflab; Outgroup = L27incra | 112 | 202 | −0.287 | 0.000 | 13 | |
| H18 | P1 = L26turg; P2 = R19turg; P3 = Mflab; Outgroup = L27incra | 70 | 115 | −0.243 | 0.001 | 13 | |
| Donor = M. cf. turgescens: specimen R19turg | H19 | P1 = Mdil12; P2 = R6Mdil; P3 = R19turg; Outgroup = L27incra | 82 | 102 | −0.109 | 0.140 | none |
| H20 | P1 = Mdil12; P2 = L26turg; P3 = R19turg; Outgroup = L27incra | 84 | 71 | 0.084 | 0.296 | none | |
| H21 | P1 = Mdil12; P2 = Mflab; P3 = R19turg; Outgroup = L27incra | 86 | 99 | −0.070 | 0.339 | none | |
| H22 | P1 = R6Mdil; P2 = Mflab; P3 = R19turg; Outgroup = L27incra | 112 | 109 | 0.014 | 0.840 | none | |
| H23 | P1 = R6Mdil; P2 = L26turg; P3 = R19turg; Outgroup = L27incra | 92 | 78 | 0.082 | 0.283 | none | |
| H24 | P1 = L26turg; P2 = Mflab; P3 = R19turg; Outgroup = L27incra | 70 | 85 | −0.097 | 0.228 | none | |
| Donor = M. cf. turgescens: specimen L26turg | H25 | P1 = Mdil12; P2 = R6Mdil; P3 = L26turg; Outgroup = L27incra | 112 | 151 | −0.148 | 0.016 | none |
| H26 | P1 = Mdil12; P2 = R19turg; P3 = L26turg; Outgroup = L27incra | 84 | 122 | −0.184 | 0.008 | 13 | |
| H27 | P1 = Mdil12; P2 = Mflab; P3 = L26turg; Outgroup = L27incra | 134 | 159 | −0.085 | 0.144 | none | |
| H28 | P1 = R6Mdil; P2 = Mflab; P3 = L26turg; Outgroup = L27incra | 166 | 166 | 0.000 | 1.000 | none | |
| H29 | P1 = R6Mdil; P2 = R19turg; P3 = L26turg; Outgroup = L27incra | 92 | 125 | −0.152 | 0.025 | none | |
| H30 | P1 = R19turg; P2 = Mflab; P3 = L26turg; Outgroup = L27incra | 115 | 85 | 0.150 | 0.034 | none |
alpha = 0.01.
Significant P-values showing evidence of introgression are shown in bold
Results showed evidence of introgression because of the significant discordant ABBA/BABA site patterns (highlighted in bold in Table 2) between: (i) the donor population M. dilatata (sample Mdil12) vs. all samples but M. cf. turgescens (sample R19turg); (ii) the donor lineage M. dilatata (sample R6) vs. all samples but M. cf. turgescens (sample R19turg); (iii) the donor lineage M. flabellata vs. all samples but M. cf. turgescens (sample R19turg), and (iv) the donor lineage M. cf. turgescens (sample L26turg) vs. M. dilatata (sample Mdil12).
Discussion
Phylogenetic reconstructions and species delimitation of scleractinian corals have long been hampered by a disagreement between genetic data and colony-level morphology [43–47]. This challenge is particularly difficult among genera in which colony morphology is known to be highly variable, such as the Hawaiian Montipora. For example, the rare coral M. dilatata was listed as Endangered by IUCN following a 1996 bleaching event that reduced the population to just two known colonies in the Main Hawaiian Islands [48]. In 2009, M. dilatata, M. flabellata, M. turgescens, M. patula and M. verrilli were included in a petition to list 83 coral species for protection under the US Endangered Species Act [49]. Several genetic markers called into question the taxonomic validity of these species, however there was not enough data to test alternative hypotheses of possible introgression, incomplete lineage sorting or rapid speciation [31].
Here we used all 13 mitochondrial protein-coding genes and reduced representation genomic sequencing in excess of 60,000 SNPs to evaluate phylogenetic relationships within the Hawaiian M. dilatata species complex in a more rigorous hypotheses testing framework than the [31] study.
Montipora phylogenetic relationships sampling greater portions of the genome
Overall, our phylogenetic reconstructions based on mitogenomic data are consistent with previous work, particularly when examining deeper nodes but show some discrepancies, mostly at intra-generic level. BI and ML analyses (Fig. 1, Additional file 2: S2 and Additional file 3: S3) showed that Acropora and Isopora are sister genera to Montipora, as reported in previous studies using a more comprehensive taxon sampling of the Scleractinia [50, 51]. The inclusion of Anacropora matthai within the Montipora clade (Fig. 1, Additional file 2: S2 and Additional file 3: S3) was also already described in [36, 52, 53], suggesting that its taxonomic status needs a revision. Our mitochondrial-based analyses (Fig. 1) further confirmed lack of differentiation between Porites lobata and P. compressa as described in [54].
All analyses based on genomic data (with the single exception of the RAxML topology) revealed that M. dilatata and M. flabellata correspond to a single species, and the two samples of M. cf. turgescens represent different evolutionary significant units, which does not reflect current taxonomy. Additional population level sampling is needed to determine if the genetic structure within this clade is due to partitioning across geographic regions, habitats, or morphology. Results presented here show the existence of hidden diversity within the genus Montipora that was not detected by the mitochondrial markers, showing the importance of sampling a greater portion of the genome for species delimitation.
Impact of rate heterogeneity on phylogenetic inference and dating estimates
The use of models that do not consider lineage-based compositional heterogeneity may provide strong support for an incorrect topology (e.g., wrong placement of eukaryotes [55] or the placental mammals [56] in the tree of life). As such, we explored if there were topological differences between the analyses based on mitogenomic data performed under a homogeneous model of sequence evolution or accommodating among-lineage compositional heterogeneity.
Although our results showed the existence of heterogeneity in the concatenated mitochondrial dataset (the homogeneous composition vector was rejected, P = 0.000), its impact on the scleractinian phylogeny seems minimal given the similar topologies obtained with methods having different assumptions (RAxML: gamma model of rate heterogeneity, P4: across-branch compositional heterogeneity vs. MrBayes: rate homogeneity). On the other hand, our results show that heterogeneity in among-lineage base composition clearly affects dating analyses. Beast (rate homogeneity) showed more recent age estimates at the tips and an older root (Additional file 4: S4) when compared with Multidivtime (rate heterogeneity; Fig. 2). The crown group ages of the Hawaiian Montipora were quite different if estimated with Multidivtime (10.5 Myr) or Beast (4.3 Myr). Only a few studies are taking into account among-lineage compositional biases when dating divergences (e.g., cichlids [57]), and usually involve the removal of problematic genes and loss of significant phylogenetic information. In the methodology we used, age estimates are based on the complete data set and performed over a tree that was inferred with nonstationary models.
Genetic association with phenotypic variability
We used phenotypic variation among well-characterized colour variants of M. capitata to evaluate if there is a genetic basis for the observed polymorphisms. Bayes Factor Delimitation (BFD*) based on > 60,000 SNPs showed a significant correlation between colour variability and genetic clustering, as indicated by the highest BF value of the model that included the grouping of samples according to their colour (Hypothesis D; Table 1).
ML based on the ‘coral’ data set grouped M. capitata colonies by colour (Fig. 3), suggesting that there may be a genetic component to these variants, warranting further attention. Red and yellow/orange colonies of M. capitata growing under identical conditions revealed distinct fluorescent phenotypes [58], which may also support a genetic basis underlying the colour polymorphism. Furthermore, Innis and colleagues [35] found that M. capitata colour morphs contain distinct proportions of clades of photosynthetic dinoflagellates family Symbiodiniaceae, in the genera Cladocopium and Durusdinium (formerly clade C and D), and Shore-Maggio and colleagues showed the same was true of the microbial communities [59] and disease susceptibility [60] of these colour morphs. We found no genetic basis for the phenotypic differences associated to colony shape (Table 1).
Incomplete lineage sorting, introgressive hybridization or a recent origin for the Hawaiian Montipora?
The difficulty in establishing species boundaries within the M. dilatata complex has been attributed to either ILS [31] or interspecific hybridization [30], but these assumptions were not previously tested. D-statistics tests (Table 2) identified the existence of introgression between all samples of the complex. However, only M. dilatata, M. flabellata and sample L26turg (M. cf. turgescens) were identified as donor lineages. Genomic admixture was identified between sympatric lineages (e.g., M. dilatata - sample Mdil12 vs. M. flabellata both from Kāne‘ohe Bay, O‘ahu, Hawaii), but also among samples found in allopatry (e.g., M. cf. turgescens - sample L26turg, Kure Atoll, Northwestern Hawaiian Islands, NWHI vs. M. dilatata - sample Mdil12, Kāne‘ohe Bay).
Reproductive isolation between species is a gradual process allowing introgression of allopatric lineages upon secondary contact, long time after their divergence [61]. In broadcast spawners like Montipora corals, which have the ability to disperse over long distances and maintain gene flow among distant populations, divergence may take even longer and we estimated a very recent origin for the species complex (see next paragraph). Further, the geographic distribution of the nominal species of the complex largely overlaps, and introgression in parapatric lineages was recently described [62]. Introgressive hybridization was also detected in another group of scleractinian corals of the genus Pocillopora [63].
Phylogenetic inference between closely related taxa can be hindered by several factors such as lack of genetic variation in recently-derived taxa [64]. Our dating analysis estimated the crown group age of the complex Montipora dilatata/M. flabellata/M. cf. turgescens under the NDCH2 composition heterogeneity model at 0.8 Myr [0.04–2.62] (Fig. 2). This recent origin is most certainly hampering phylogenetic inference.
Conclusions
Delineating species is critical to our ability to address conservation and management goals, but the boundaries between species can be challenging in groups such as scleractinian corals because genetic data disagree with established taxonomy and gross morphology. Here, we included all 13 mitochondrial coding-genes and RADseq data (> 60,000 SNPs) to test how previous results could be impacted by lack of informative loci, across-branch compositional heterogeneity or biological processes such as introgressive hybridization. We generated the most comprehensive mitogenomic data set gathered to date for scleractinian corals. Despite the volume of data available, we still fail to clearly resolve relationships among nominal species within the M. dilatata species complex. Nevertheless, genomic data revealed new findings: (1) current taxonomy [65] does not reflect the true diversity within the genus; (2) species delimitation tests favoured the model that considered M. dilatata and M. flabellata as a single species and splits the two samples of M. cf. turgescens as different evolutionary significant units; (3) species delimitation tests and ML analysis supported a genetic basis for the observed colour polymorphisms in M. capitata, and (4) the existence of introgression among the species of the complex is confirmed. Dating analyses indicated a very recent origin for the complex. Age estimates varied depending on whether compositional heterogeneity was taken into account (0.8 Myr) or rate homogeneity was assumed (0.4 Myr). Genomic admixture was identified between sympatric lineages but also between samples found in allopatry; however, the geographic distribution of the nominal species belonging to the complex largely overlaps, allowing parapatric introgression. Phylogenomic data presented here questions the endangered status of M. dilatata and the taxonomic validity of the remaining species of the complex also showing the existence of cryptic genetic diversity within the genus warranting further study.
Methods
Taxon sampling
Tissue from 16 samples belonging to the genus Montipora were collected from Hawai’i and the Central Pacific between 2010 and 2013 and stored either in salt-saturated dimethyl sulfoxide (DMSO) buffer [66] or in 95% ethanol prior DNA extraction. All our samples had a mean sample size of 1–2 cm2 and were taken by non-lethal tissue biopsy of the corals as required by the State collection permits (SAP-HIMB2010, SAP-HIMB2011, SAP-HIMB2012, SAP-HIMB2013) that we obtained to sample these organisms. There were no colonies harvested (or permanently damaged) for this research (all heal within less than 30 days from the biopsy sampling), and all were sampled as approved by the State and Federal agencies responsible for their management.
The samples included six different morphological types (morphs) and two pooled samples (red and yellow/orange) of M. capitata, two samples of M. dilatata, two samples of M. turgescens, and one sample from each of the following nominal species: M. verrilli, M. patula, M. incrassata, and M. flabellata (Additional file 1: S1A). We sampled a range of variation within M. capitata including samples (five fragments from each colour morph) that were collected around the Hawai‘i Institute of Marine Biology and grown from ~2cm fragments in a ‘common garden’ floating rack, in a lagoon environment for six years.
For the mitogenomic analyses, we used all 13 mitochondrial protein-coding genes from 14 Montipora samples sequenced in this study and retrieved from the GeneBank 41 coral species of the order Scleractinia, representing a total of 6 families and 15 genera (GeneBank accession numbers in the Additional file 1: S1A, Genomic libraries were deposited in the NCBI short read archive with the bioproject ID PRJNA554733). Taxon sampling essentially focused on the family Acroporidae (genera Acropora, Montipora, Isopora and Anacropora). The remaining taxa were selected because of their fossil record, which is required for calibration in the dating analysis.
Laboratory procedures, sequence alignment and genetic distances
Genomic DNA was extracted from coral tissue using the OMEGA (BIO-TEK) E-Z 96 Tissue DNA Kit and eluted in 2 × 100 μl. Extractions were quantified using the AccuBlueTM (Biotium, Inc.). Libraries were generated using the ezRAD method [67, 68]. Genomic DNA was digested using the isoschizomer restriction enzymes MboI and Sau3AI (New England BioLab), which both cleave at GATC recognition sites. Details on library preparation are described in Johnston [63]. All libraries were size-selected between 300 and 500 bp and only after passing the quality control steps (bioanalyser and qPCR) were sequenced at the Hawai’i Institute of Marine Biology (HIMB) Genetics Core Facility following the Illumina TruSeq Sample Prep v2 Low Throughput protocol.
The 16 Illumina Montipora libraries were assembled to the mitochondrial genome of Montipora cactus (NC_006902) to get the consensus sequences using the default settings (high sensitivity iterated up to five times and the medium/read mapping settings) in GENEIOUS v.8.1.4 (Biomatters, Auckland, New Zealand; https://www.geneious.com). We failed to get reliable mitochondrial consensus sequences from two of the sequenced libraries corresponding to the samples MturgL26 (M. cf. turgescens) and mCAPsamp (M. capitata).
Following Medina et al. [69], the actinarians Nematostella sp. and Metridium senile (GenBank accession numbers: DQ643835 and HG423143, respectively) were selected as the outgroup of the Scleractinia used in the mitogenomic analyses. Each mitochondrial protein-coding gene from the 55 scleractinians (14 samples from this study) plus the two outgroups (57 taxa in total) was individually aligned with Mafft v7.245 [70]. The invertebrate mitochondrial genetic code was used to detect open reading frames (ORFs) and stop codons and deduce the amino acid sequences of each of the 13 partial mitochondrial protein-coding genes in Mesquite v3.2 [71]. Those alignments were concatenated into a single dataset (57 taxa, 11,484 bp) used in Bayesian inference (BI) and maximum likelihood (ML) analyses. All newly generated mitochondrial protein-coding sequences have been deposited in GenBank, for which accession numbers are provided in the Additional file 1: S1A.
Sequence distances of the mitochondrial concatenated data set with 500 bootstrap replicates were calculated in Mega 5 [72]. Uncorrected within p-distances were computed for the following groups: (1) all M. capitata samples representing a wide range of colony morphology, and (2) all samples within the M. dilatata complex (M. dilatata: Mdil12 + R6; M. cf. turgescens: R19 + L26 and M. flabellata). Net-sequence distances were estimated between M. capitata and M. dilatata complex.
Phylogenetic analyses of mitochondrial DNA based on standard homogeneous models
Bayesian Inference (BI) analyses based on the concatenated mitochondrial dataset (57 taxa, 11,484 bp) were performed with MrBayes v3.2.1 [73] under a homogeneous model of rate change (rates = equal). All analyses were run for 9 × 107 generations (four simultaneous Markov chains; 1 × 103 sample frequency) following a discarded burn-in of 10%. The convergence to the stationary distributions was confirmed by inspection of the MCMC samples using Tracer v. 1.6 [74].
Across-branch compositional heterogeneity
To analyse if there was across-branch compositional heterogeneity in the concatenated nucleotide data set from the 13 mitochondrial protein-coding genes representing 55 Scleractinian lineages, we first inferred a phylogeny under a model of composition homogeneity as implemented in P4 v1.2.0 [9]. As the results indicated a null probability for this model we used the node-discrete composition heterogeneity (NDCH2) model [9] with GTR + I + Γ, also implemented in P4 v1.2.0. This software performs a Bayesian MCMC analysis which allows composition to vary among lineages, with a distinct composition vector for each node [11].
We performed ML analysis of the concatenated mitochondrial data set with RAxML v8.2.10 [75] using the option –q that specifies the file name which contains the assignment of models to alignment partitions for multiple models of substitution under the GTR-CAT approximation (gamma model of rate heterogeneity). We used PartitionFinder2 v.2.1.1 [76] with the corrected Akaike Information Criterion (AICc) to select the best partitioning scheme and corresponding best-fit model of substitution. The best-scoring ML tree was determined from 100 randomized maximum-parsimony starting trees using the rapid hill-climbing algorithm, and 1000 bootstrap replicates were drawn on each best-scored ML tree using the exhaustive bootstrap algorithm.
P4, BI and ML analyses were performed on the CCMAR computational cluster facility (http://gyra.ualg.pt) and on the R2C2 computational cluster facility provided by the IT Department of the University of Algarve.
Dating analyses of mitogenomic data
To date major splitting events within Hawaiian Montipora and estimate the origin of the M. dilatata complex, we used 55 Scleractinian lineages that exhibit extensive fossil record and have mitogenomic data available. Two relaxed molecular-clock approaches were used to evaluate the impact of rate heterogeneity on date estimates. The software Multidivtime [77] includes an input topology, which in this case was set to the one inferred by P4 that accommodates across-branch compositional heterogeneity, and Beast v.1.8.4 [78] allows the use of rate homogeneity models.
Multidivtime: following Thorne and Kishino [79], we used PAML v.3.14 [80] to estimate ML parameters using a discrete gamma distribution with five rate categories and the F84 model of nucleotide substitution that was selected because of computational tractability [81]. Estbranches [82] was used to estimate branch lengths and subsequently Multidivtime was used to estimate divergence times. This method requires a prior assumption for the mean and standard deviation of the time of the ingroup root node (rttm) that represents the calibration of the root of the tree. This parameter was set to 24.5 time units, where in this analysis, one-time unit represents 10 million years (Myr). This value was based on the earliest record of the true scleractinians in during the Anisian (≈ 245) Myr [83, 84]. The standard deviation of the prior distribution was set to its maximum value (equal to the mean) to avoid violation of the definition of a prior. Calibrations are indicated in Table 3. The MCMC method was employed to approximate both prior and posterior distributions [82]. Initial parameter values were randomly selected to initialize the Markov chain and then, a burn-in period of 100,000 cycles was completed before parameters were sampled from the MCMC chain. Afterwards, the state of the Markov chain was sampled every 100 cycles until a total of 10,000 generations.
Table 3.
Calibration points used in the mitogenomic dating analysis
| Calibration points (in million years) | Description | References | |
|---|---|---|---|
| 1 | [70.1–69.9] | Divergence between Astrangia and Montastraea | Medina et al., 2006 [69]; Park et al., 2012 [100]; Veron, 1995 [101] |
| 2 | [56.0–33.9] | Divergence between Montipora and Acropora | Wells, 1956 [102] |
| 3 | [42.7–28.4] | Divergence between Pocillopora and Seriatopora | Simpson et al., 2011 [84] ; Strauss and Sadler, 1989 [103] |
| 4 | [5.0–0.0] | The origin of Porites porites | Budd and Jonhson, 1999 [104] |
| 5 | [15.0–5.3] | Divergence between Isopora and Acropora | Simpson et al., 2011 [84]; Strauss and Sadler, 1989 [103] |
| 6 | [0.99–2.99] | Estimated age for the genus Pocillopora | Jonhston et al., 2017 [63] |
Beast: we selected the Birth-Death Incomplete Sampling tree prior because we were only using a fraction of the extant Scleractinia. As we wanted to compare date estimates between models incorporating across-branch compositional heterogeneity as implemented in P4/Multidivtime and homogeneous models, we used Beast without the gamma model. We used the same calibrations as in the Multidivtime analysis described in Table 3 that were modeled with a normal distribution: (1) divergence between Astrangia and Montastraea (mean = 70, stdev = 0.1, 2) divergence between Montipora and Acropora (mean = 44.95, stdev = 11.05, 3) divergence between Pocillopora and Seriatopora (mean = 35.55, stdev = 7.15, 4) estimated origin of Porites porites (mean = 2.5; stdev = 2.5, 5) divergence between Isopora and Acropora (mean = 10.15, stdev = 4.85, 6) estimated age for the genus Pocillopora (mean = 1.99, stdev = 0.65). We ran two independent runs for 100,000,000 generations, sampling every 1000 generations. The final tree was produced by TreeAnnotator using the “maximum clade creditability” option and mean node height, after burn-in of 10% of the generations. The convergence to the stationary distribution was confirmed by inspection of the Markov Chain Monte Carlo (MCMC) samples and of effective sample sizes (ESS should be > 200) using Tracer v1.6.0 [74].
SNP-based analyses
We used 16 individuals representing a range of morphologies and two pooled samples representing common colour morphs (yellow/orange and red) of M. capitata, to characterize within species variability within this notoriously polymorphic species and determine possible relationships between genotype and phenotype (more details in Additional file 1: S1A). In addition, we included two samples of each nominal species M. dilatata and M. cf. turgescens, and one sample from M. flabellata to analyse phylogenetic relationships within the M. dilatata species complex. We also included a sample of M. incrassata and M. verrilli to further analyse phylogenetic relationships within Montipora.
FastQC was used for a preliminary quality control of the pair-end reads [85]. The raw reads from Illumina were cleaned with Trim Gallore! [86] and subsequently analysed with dDocent 2.2.25 [87, 88], a pipeline that combines several software packages (https://github.com/jpuritz/dDocent/). All libraries were mapped to the M. capitata draft genome [39] using BWA [89] with the following settings: -L 20,5 -t 32 -a -M -T 10 -A 1 -B 4 -O 6 -R. We used vcftools v0.1.15 [90] to filter the resulting variant call file (VCF) from dDocent analysis using the following options: --remove-indels --max-missing 0.50 --thin 300 --mac 3 --minQ 30. We performed an additional analysis using max-missing = 0.75 to evaluate the impact of missing data on phylogenomic reconstruction.
To evaluate the level of contamination from Symbiodiniaceae de-novo assembly of all data was performed with dDocent v2.2.25 [87, 88] with a clustering threshold of 0.85, default mapping and SNP calling parameters. Prior to filtering, only loci with at least 2x coverage within libraries and present in at least 3 libraries were retained. The reference sequences for all loci (n = 67,598) generated by dDocent were then compared at the amino acid-level using tBLASTx [91] to several available reference genomes in order to determine if the locus was of putative coral or Symbiodiniaceae origin.
To convert the filtered SNPs in VCF format to PHYLIP for phylogenetic analysis we used a python script (https://github.com/edgardomortiz/vcf2phylip) that also performs an additional filtering (a minimum of 10 samples are required to be present at a locus, where the default value is 4 to use in a phylogenomic analysis). The dataset hereafter called ‘coral’ consisted of 60,602 SNPs.
Uncorrected p-distances with 500 bootstrap replicates were calculated in Mega 5 [72] following the same procedure described for the mitochondrial data.
ML analyses based on the ‘coral’ data set were performed with RAxML v8.2.10 [75]. Following the manual recommendations, the analysis was run under the GTRGAMMA evolutionary model. The best-scoring ML tree was determined from 100 randomized maximum-parsimony starting trees using the rapid hill-climbing algorithm, and 100 bootstrap replicates were drawn on the best-scored ML tree using the exhaustive bootstrap algorithm. Acropora cytherea was selected as outgroup.
We used Bayes delimitation with genomic data (BFD*), a species delimitation method for analysis of SNP data [14] to establish the number of species within Montipora. BFD* combines the Beast v.2.4.8 [92] add-on SNAPP [93] with path sampling that estimates marginal likelihoods to use in Bayes factor model selection [14]. We used the VCF file from the dDocent run based on the M. capitata draft genome assembly. The filtered VCF (as described earlier) was converted to a binary nexus format using the script vcf2phylip (https://github.com/edgardomortiz/vcf2phylip; [94]). We conducted path sampling with 24 steps to estimate the marginal likelihood estimates (MLE). The MCMC chain for path sampling analysis was run in Beast v.2.4.8 for 1,000,000 generations and sampled every 1000 with a pre-burnin of 100,000. We used the framework of Kass and Raftery [95] to evaluate the strength of support of Bayes factor (BF) comparisons of competing models. A positive BF test statistic (2 x loge) reflects evidence in favor model 1, whereas negative BF values support model 2 [14]. A BF value between 2 and 6 represents positive evidence and BF > 10 is decisive. We tested six different hypotheses: (A) supports current taxonomy (M. verrilli, M. patula, M. capitata, M. incrassata, M. flabellata, M. cf. turgescens and M. dilatata) with all individuals of the same nominal species grouping together; (B) splits M. cf. turgescens and M. dilatata samples in different species; (C) groups all samples belonging to the Montipora dilatata complex into a single species: M. cf. turgescens (samples L26 + R19); M. flabellata; M. dilatata, (samples R6 + Mdil12); (D) groups M. capitata samples by colour morph (red and yellow/orange) as a single species, and the remaining Montipora samples as separate species; (E) lumps M. dilatata into a single species; splits all remaining samples of the complex as different species; (F) groups M. dilatata and M. flabellata into a single species; splits M. cf. turgescens. Acropora cytherea was used as outgroup in all models.
We used SVDQuartets (Singular Value Decomposition for quartets) [96] to estimate the species tree under the multi-species-coalescent model from SNP data (‘coral’ data set), as implemented in PAUP* v.4.0a (build 163) [97]. We assessed node support with 100 bootstraps, and an exhaustive search of all possible quartets (3060) were evaluated using the QFM quartet assembly algorithm.
Testing introgression
Phylogenetic discordances (differences in gene tree topologies) can result from either incomplete lineage sorting (ILS) or introgressive hybridization. Given the conflicting patterns of the ML SNP-based topology based and the species tree, we tested all 30 possible combinations of genomic admixture among the nominal species belonging to the species complex M. dilatata/M. flabellata/ M. cf. turgescens (see Table 2 for further details). We selected M. incrassata as outgroup based on the results we obtained in the phylogenetic analyses.
We estimated the Patterson’s D-statistic [98, 99] running the python script dfoil.py https://github.com/jbpease/dfoil with the “--mode dstat” option for a four-taxon D-statistic calculation [42]. In a four-taxon phylogenetic relationship represented by (((P1, P2), P3), O) where P represents the lineages of interest for hybridization and O the outgroup, the D-statistic compares two incongruent SNP patterns, ABBA and BABA, in which “B” is the derived allele and “A” the ancestral allele [98, 99]. In the absence of gene flow and random mating, frequencies of the two patterns are expected to be similar [99]. Under a ILS scenario, we would expect that both patterns (ABBA/BABA) would be sampled with equal frequency while a statistically significant imbalance would reflect introgression [42].
Additional files
S1. A. Species_list: Scleractinian corals used in the mitogenomic data set, accession numbers corresponding to the complete mitochondrial genomes retrieved from GenBank and to the newly sequenced 13 partial mitochondrial protein-coding genes of the 14 Montipora samples used in the mitogenomic analyses. List of Montipora samples used in the RADseq analyses. B. Sample_information: collected sample information regarding mitogenomic and genomic analyses (number of reads, reads length, mean depth coverage and standard deviation, percentage of the reference sequence). (XLSX 18 kb)
{kind=link}
S2. A. Bayesian analysis showing phylogenetic relationships among 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile based on the concatenated mitochondrial dataset (13 protein-coding genes). B. Inset showing a detail of the topology referring to the genus Montipora zoomed 20x. This analysis was produced by MrBayes under a homogeneous model of rate change. Bayesian posterior probability (BPP) values are shown in black circles for values of maximal probability (1.00) and a grey circle for a value of 0.57. (JPG 587 kb)
S3. A. Maximum likelihood phylogram of 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile based on the concatenated mitochondrial dataset (13 protein-coding genes) produced by RAxML under a gamma model of rate heterogeneity. B. Inset showing a detail of the topology referring to the genus Montipora zoomed 100x. Numbers at the nodes represent bootstrap proportions. (PDF 230 kb)
S4. Beast maximum clade credibility chronogram showing main cladogenetic events among 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile. The 95% highest posterior density (HPD) intervals are represented by the blue bars, and numbers at the nodes represent million years. (PDF 226 kb)
{kind=link}
S5. Species tree based on 60,602 SNPs from the ‘coral’ data set estimated with SVDquartets. Numbers at the nodes represent bootstrap proportions. Image plates represent in-situ photographs of Montipora species sampled for this study and of the morphotypes within Montipora capitata. (JPG 386 kb)
S6. Venn diagram showing the overlap between putative coral (Montipora capitata) and symbionts (Symbiodinium minutum and Fugacium kawagutii) loci. (PDF 109 kb)
{kind=link}
S7. In-situ photographs of Montipora species sampled for this study and of the morphotypes within Montipora capitata. (JPG 3488 kb)
Acknowledgements
We thank Cymon J. Cox and Filipe Sousa for all the help with P4. We are also very grateful to Edgardo M. Ortiz for sharing the python script to convert VCF files into PHYLIP and NEXUS binary formats. We thank Erika C. Johnston, Romina Henriques and Miguel Baltazar Soares for the help with RADseq data analyses. We wish to thank Ashley Smith and Christina Runyan for identifying and collecting the orange/yellow and red Montipora capitata phenotypes. Ashley Smith passed away before she was able to finish her dissertation research and we dedicate this work towards her research interests in her honour.
Abbreviations
- BFD*
Bayes Factor Delimitation with genomic data
- DMSO
Salt-saturated dimethyl sulfoxide
- HIMB
Hawai’i Institute of Marine Biology
- ILS
Incomplete lineage sorting
- ITS
Nuclear internal transcribed spacers
- IUCN
International Union for Conservation of Nature
- NCBI
National Center for Biotechnology Information
- NDCH2
Node-discrete composition heterogeneity model
- NWHI
Northwestern Hawaiian Islands
- RADseq
Restriction-site associated DNA sequencing
- SNPs
Single nucleotide polymorphisms
- VCF
Variant call file
Authors’ contributions
RLC, ZHF and RJT conceived the study, ISSK and RB collected the data. RLC and ZHF analysed the data, RC provided financial support for the acquisition of computational facilities for genomic analyses and designed the figures. RLC and RJT wrote the manuscript and all authors provided critical feedback approving the final version of the manuscript.
Funding
RLC was supported by the Portuguese Foundation for Science & Technology (FCT) post-doctoral fellowship (SFRH/BPD/109685/2015). Sequencing for this project was supported by the Pauley Foundation and NSF-OA#1416889 to RJT. This is HIMB contribution # 1765 and SOEST # 10748. This study also received Portuguese national funds from FCT - Foundation for Science and Technology through project UID/Multi/04326/2019 and from the operational programs CRESC Algarve 2020 and COMPETE 2020 through projects EMBRC.PT ALG-01-0145-FEDER-022121 and BIODATA.PT ALG-01-0145-FEDER-022231.
Availability of data and materials
The datasets generated and/or analysed during the current study are available in the GeneBank (accession numbers in the Additional file 1: S1A and NCBI SRA bioproject ID PRJNA554733).
Ethics approval and consent to participate
Our research activities for this project were covered by the State of Hawai’i, Department of Land & natural Resources, Special Activity Permits: SAP-HIMB2010–1, SAP-HIMB2011–1, SAP-HIMB2012–63, SAP-HIMB2013–47 issued to the Hawai’i Institute of Marine Biology.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Regina L. Cunha, Email: rcunha@ualg.pt
Zac H. Forsman, Email: zac@hawaii.edu
Roy Belderok, Email: roy.belderok@gmail.com.
Ingrid S. S. Knapp, Email: iknapp@hawaii.edu
Rita Castilho, Email: rcastil@ualg.pt.
Robert J. Toonen, Email: rjtoonen@gmail.com
References
- 1.Fukami H, Budd AF, Paulay G, Solé-Cava A, Chen CA, Iwao K, Knowlton N. Conventional taxonomy obscures deep divergence between Pacific and Atlantic corals. Nature. 2004;427(6977):832–835. doi: 10.1038/nature02339. [DOI] [PubMed] [Google Scholar]
- 2.Sawall Y, Al-Sofyani A, Hohn S, Banguera-Hinestroza E, Voolstra CR, Wahl M. Extensive phenotypic plasticity of a Red Sea coral over a strong latitudinal temperature gradient suggests limited acclimatization potential to warming. Sci Rep. 2015;5:8940. [DOI] [PMC free article] [PubMed]
- 3.Vitecek S, Kučinić M, Previšić A, Živić I, Stojanović K, Keresztes L, Bálint M, Hoppeler F, Waringer J, Graf W. Integrative taxonomy by molecular species delimitation: multi-locus data corroborate a new species of Balkan Drusinae micro-endemics. BMC Evol Biol. 2017;17(1):129. doi: 10.1186/s12862-017-0972-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gómez-Zurita J, Cardoso A, De la Cadena G, Jurado-Rivera JA, Maes J-M, Montelongo T, Nguyen DT, Papadopoulou A. High-throughput biodiversity analysis: Rapid assessment of species richness and ecological interactions of Chrysomelidae (Coleoptera) in the tropics. ZooKeys. 2016;597:3. doi: 10.3897/zookeys.597.7065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Golding B, Felsenstein J. A maximum likelihood approach to the detection of selection from a phylogeny. J Mol Evol. 1990;31(6):511–523. doi: 10.1007/BF02102078. [DOI] [PubMed] [Google Scholar]
- 6.Kubatko LS, Degnan JH. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst Biol. 2007;56(1):17–24. doi: 10.1080/10635150601146041. [DOI] [PubMed] [Google Scholar]
- 7.Liu Y, Cox CJ, Wang W, Goffinet B. Mitochondrial phylogenomics of early land plants: mitigating the effects of saturation, compositional heterogeneity, and codon-usage bias. Syst Biol. 2014;63(6):862–878. doi: 10.1093/sysbio/syu049. [DOI] [PubMed] [Google Scholar]
- 8.Nater A, Burri R, Kawakami T, Smeds L, Ellegren H. Resolving evolutionary relationships in closely related species with whole-genome sequencing data. Syst Biol. 2015;64(6):1000–1017. doi: 10.1093/sysbio/syv045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foster PG. Modeling compositional heterogeneity. Syst Biol. 2004;53(3):485–495. doi: 10.1080/10635150490445779. [DOI] [PubMed] [Google Scholar]
- 10.Moran RJ, Morgan CC, O'Connell MJ. A guide to phylogenetic reconstruction using heterogeneous models—a case study from the root of the placental mammal tree. Computation. 2015;3(2):177–196. doi: 10.3390/computation3020177. [DOI] [Google Scholar]
- 11.Puttick MN, Morris JL, Williams TA, Cox CJ, Edwards D, Kenrick P, Pressel S, Wellman CH, Schneider H, Pisani D, et al. The Interrelationships of Land Plants and the Nature of the Ancestral Embryophyte. Curr Biol. 2018;28(5):733–745. doi: 10.1016/j.cub.2018.01.063. [DOI] [PubMed] [Google Scholar]
- 12.Rannala B, Yang Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 2003;164:1645–1656. doi: 10.1093/genetics/164.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang Z, Rannala B. Bayesian species identification under the multispecies coalescent provides significant improvements to DNA barcoding analyses. Mol Ecol. 2017;26(11):3028–3036. doi: 10.1111/mec.14093. [DOI] [PubMed] [Google Scholar]
- 14.Leaché AD, Fujita MK, Minin VN, Bouckaert RR. Species delimitation using genome-wide SNP data. Syst Biol. 2014;63(4):534–542. doi: 10.1093/sysbio/syu018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahrens D, Fujisawa T, Krammer HJ, Eberle J, Fabrizi S, Vogler AP. Rarity and incomplete sampling in DNA-based species delimitation. Syst Biol. 2016;65:478–494. doi: 10.1093/sysbio/syw002. [DOI] [PubMed] [Google Scholar]
- 16.Leliaert F, Verbruggen H, Vanormelingen P, Steen F, Lopez-Bautista J, Zuccarello G, Clerck O. DNA-based species delimitation in algae. Eur J Phycol. 2014;49(2):179–196. doi: 10.1080/09670262.2014.904524. [DOI] [Google Scholar]
- 17.Shaffer HB, Thomson RC. Delimiting Species in Recent Radiations. Syst Biol. 2007;56(6):896–906. doi: 10.1080/10635150701772563. [DOI] [PubMed] [Google Scholar]
- 18.Herrera S, Shank TM. RAD sequencing enables unprecedented phylogenetic resolution and objective species delimitation in recalcitrant divergent taxa. Mol Phylogenet Evol. 2016;100:70–79. doi: 10.1016/j.ympev.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 19.Eaton DA, Ree RH. Inferring phylogeny and introgression using RADseq data: an example from flowering plants (Pedicularis: Orobanchaceae) Syst Biol. 2013;62(5):689–706. doi: 10.1093/sysbio/syt032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Richards ZT, van Oppen MJH, Wallace CC, Willis BL, Miller DJ. Some Rare Indo-Pacific Coral Species Are Probable Hybrids. PloS One. 2008;3(9):e3240. doi: 10.1371/journal.pone.0003240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Putnam HM, Adams DK, Zelzion E, Wagner NE, Qiu H, Mass T, Falkowski PG, Gates RD, Bhattacharya D. Divergent evolutionary histories of DNA markers in a Hawaiian population of the coral Montipora capitata. PeerJ. 2017;5:e3319. doi: 10.7717/peerj.3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Forsman ZH, Guzman HM, Chen CA, Fox GE, Wellington GM. An ITS region phylogeny of Siderastrea (Cnidaria: Anthozoa): is S. glynni endangered or introduced? Coral Reefs. 2005;24:343–347. doi: 10.1007/s00338-005-0497-z. [DOI] [Google Scholar]
- 23.Forsman ZH, Wellington GM, Fox GE, Toonen RJ. Clues to unraveling the coral species problem: distinguishing species from geographic variation in Porites across the Pacific with molecular markers and microskeletal traits. PeerJ. 2015;3:e751. doi: 10.7717/peerj.7751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hellberg ME, Prada C, Tan MH, Forsman ZH, Baums IB. Getting a grip at the edge: recolonization and introgression in eastern Pacific Porites corals. J Biog. 2016;43(11):2147–2159. doi: 10.1111/jbi.12792. [DOI] [Google Scholar]
- 25.Vollmer SV, Palumbi SR. Testing the utility of internally transcribed spacer sequences in coral phylogenetics. Mol Ecol. 2004;13(9):2763–2772. doi: 10.1111/j.1365-294X.2004.02265.x. [DOI] [PubMed] [Google Scholar]
- 26.Chen CA, Chang C-C, Wei NV, Chen C, Lein Y-T, Lin H-E, Dai C-F, Wallace C. Secondary structure and phylogenetic utility of the ribosomal internal transcribed spacer 2 (ITS2) in scleractinian corals. Zool Stud. 2004;43(4):759–71.
- 27.Hellberg ME. No variation and low synonymous substitution rates in coral mtDNA despite high nuclear variation. BMC Evol Biol. 2006;6(1):24. doi: 10.1186/1471-2148-6-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Budd AF, Nunes FL, Weil E, Pandolfi JM. Polymorphism in a common Atlantic reef coral (Montastraea cavernosa) and its long-term evolutionary implications. Evol Ecol. 2012;26(2):265–290. doi: 10.1007/s10682-010-9460-8. [DOI] [Google Scholar]
- 29.Veron J, Stafford-Smith MG. Corals of the World. Townsville: Australian Institute of Marine Science; 2000. [Google Scholar]
- 30.Van Oppen M, Koolmees E, Veron J. Patterns of evolution in the scleractinian coral genus Montipora (Acroporidae) Mar Biol. 2004;144(1):9–18. doi: 10.1007/s00227-003-1188-3. [DOI] [Google Scholar]
- 31.Forsman ZH, Concepcion GT, Haverkort RD, Shaw RW, Maragos JE, Toonen RJ. Ecomorph or endangered coral? DNA and microstructure reveal Hawaiian species complexes: Montipora dilatata/flabellata/turgescens & M. patula/verrilli. PLoS One. 2010;5(12):e15021. doi: 10.1371/journal.pone.0015021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Redding DW, Hartmann K, Mimoto A, Bokal D, DeVos M, Mooers AØ. Evolutionarily distinctive species often capture more phylogenetic diversity than expected. J Theor Biol. 2008;251(4):606–615. doi: 10.1016/j.jtbi.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 33.Huang D. Threatened reef corals of the world. PLoS One. 2012;7(3):e34459. doi: 10.1371/journal.pone.0034459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Curnick D, Head C, Huang D, Crabbe MJC, Gollock M, Hoeksema B, Johnson K, Jones R, Koldewey H, Obura D. Setting evolutionary-based conservation priorities for a phylogenetically data-poor taxonomic group (Scleractinia) Anim Conserv. 2015;18(4):303–312. doi: 10.1111/acv.12185. [DOI] [Google Scholar]
- 35.Innis T, Cunning R, Ritson-Williams R, Wall CB, Gates RD. Coral color and depth drive symbiosis ecology of Montipora capitata in Kāne'ohe Bay, O'ahu, Hawai‘i. Coral Reefs. 2018;37(2):423–430. doi: 10.1007/s00338-018-1667-0. [DOI] [Google Scholar]
- 36.Fukami H, Chen CA, Budd AF, Collins A, Wallace C, Chuang Y-Y, Chen C, Dai C-F, Iwao K, Sheppard C. Mitochondrial and nuclear genes suggest that stony corals are monophyletic but most families of stony corals are not (Order Scleractinia, Class Anthozoa, Phylum Cnidaria) PloS one. 2008;3(9):e3222. doi: 10.1371/journal.pone.0003222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Budd AF, Romano SL, Smith ND, Barbeitos MS. Rethinking the phylogeny of scleractinian corals: a review of morphological and molecular data. Integr Comp Biol. 2010;50(3):411–427. doi: 10.1093/icb/icq062. [DOI] [PubMed] [Google Scholar]
- 38.Hulsen T, de Vlieg J, Alkema W. BioVenn–a web application for the comparison and visualization of biological lists using area-proportional Venn diagrams. BMC genomics. 2008;9(1):488. doi: 10.1186/1471-2164-9-488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shumaker A, Putnam H, Qiu H, Price DC, Zelzion E, Harel A, Wagner N, Gates RD, Yoon HS, Bhattacharya D. Genome analysis of the rice coral Montipora capitata. Sci Rep. 2019;9:2571. doi: 10.1038/s41598-019-39274-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Shoguchi E, Shinzato C, Kawashima T, Gyoja F, Mungpakdee S, Koyanagi R, Takeuchi T, Hisata K, Tanaka M, Fujiwara M. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr Biol. 2013;23(15):1399–1408. doi: 10.1016/j.cub.2013.05.062. [DOI] [PubMed] [Google Scholar]
- 41.Lin S, Cheng S, Song B, Zhong X, Lin X, Li W, Li L, Zhang Y, Zhang H, Ji Z. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science. 2015;350(6261):691–694. doi: 10.1126/science.aad0408. [DOI] [PubMed] [Google Scholar]
- 42.Pease JB, Hahn MW. Detection and Polarization of Introgression in a Five-taxon Phylogeny. Syst Biol. 2015;64(4):651–662. doi: 10.1093/sysbio/syv023. [DOI] [PubMed] [Google Scholar]
- 43.Gittenberger A, Reijnen BT, Hoeksema BW. A molecularly based phylogeny reconstruction of mushroom corals (Scleractinia Fungiidae) with taxonomic consequences and evolutionary implications for life history traits. Contrib Zool. 2011;80(2):107–132. doi: 10.1163/18759866-08002002. [DOI] [Google Scholar]
- 44.Huang D, Licuanan WY, Baird AH, Fukami H. Cleaning up the “Bigmessidae”: molecular phylogeny of scleractinian corals from Faviidae, Merulinidae, Pectiniidae and Trachyphylliidae. BMC Evol Biol. 2011;11:37. doi: 10.1186/1471-2148-11-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitahara MV, Stolarski J, Cairns SD, Benzoni F, Stake JL, Miller DJ. The first modern solitary Agariciidae (Anthozoa, Scleractinia) revealed by molecular and microstructural analysis. Inv Syst. 2012;26(3):303–315. doi: 10.1071/IS11053. [DOI] [Google Scholar]
- 46.Benzoni F, Arrigoni R, Stefani F, Reijnen B, Montano S, Hoeksema BW. Phylogenetic position and taxonomy of Cycloseris explanulata and C. wellsi (Scleractinia: Fungiidae): lost mushroom corals find their way home. Contrib Zool. 2012;81(3):125–46.
- 47.Arrigoni R, Kitano YF, Stolarski J, Hoeksema BW, Fukami H, Stefani F, Galli P, Montano S, Castoldi E, Benzoni F. A phylogeny reconstruction of the Dendrophylliidae (Cnidaria, Scleractinia) based on molecular and micromorphological criteria, and its ecological implications. Zool Scripta. 2014;43(6):661–688. doi: 10.1111/zsc.12072. [DOI] [Google Scholar]
- 48.DeVantier L, Hodgson G, Huang D, Johan O, Licuanan A, Obura D, Sheppard C, Syahrir M, Turak E. Montipora dilatata. . The IUCN Red List of Threatened Species 2008. 2008. [Google Scholar]
- 49.Brainard RE, Birkeland C, Eakin CM, Mcelhany P, Miller MW, Patterson M, Piniak GA. In: Status review report of 82 candidate coral species petitioned under the U.S. Endangered Species Act. Commer. D: NOAA Tech. Memo; NOAA-TM-NMFS-PIFSC-27, editor. 2011. [Google Scholar]
- 50.Wallace C, Chen C, Fukami H, Muir P. Recognition of separate genera within Acropora based on new morphological, reproductive and genetic evidence from Acropora togianensis, and elevation of the subgenus Isopora Studer, 1878 to genus (Scleractinia: Astrocoeniidae; Acroporidae) Coral Reefs. 2007;26(2):231–239. doi: 10.1007/s00338-007-0203-4. [DOI] [Google Scholar]
- 51.Kitahara MV, Lin M-F, Forêt S, Huttley G, Miller DJ, Chen CA. The “naked coral” hypothesis revisited–evidence for and against scleractinian monophyly. PLoS One. 2014;9(4):e94774. doi: 10.1371/journal.pone.0094774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fukami H, Omori M, Hatta M. Phylogenetic relationships in the coral family Acroporidae, reassessed by inference from mitochondrial genes. Zool Sci. 2000;17(5):689–696. doi: 10.2108/zsj.17.689. [DOI] [PubMed] [Google Scholar]
- 53.Kitahara MV, Cairns SD, Stolarski J, Blair D, Miller DJ. A comprehensive phylogenetic analysis of the Scleractinia (Cnidaria, Anthozoa) based on mitochondrial CO1 sequence data. PloS one. 2010;5(7):e11490. doi: 10.1371/journal.pone.0011490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Forsman ZH, Knapp ISS, Tisthammer K, Eaton DAR, Belcaid M, Toonen RJ. Coral hybridization or phenotypic variation? Genomic data reveal gene flow between Porites lobata and P. compressa. Mol Phylog Evol. 2017;111:132–148. doi: 10.1016/j.ympev.2017.03.023. [DOI] [PubMed] [Google Scholar]
- 55.Cox CJ, Foster PG, Hirt RP, Harris SR, Embley TM. The archaebacterial origin of eukaryotes. Proc Natl Acad Sci. 2008;105(51):20356–20361. doi: 10.1073/pnas.0810647105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgan CC, Foster PG, Webb AE, Pisani D, McInerney JO, O’Connell MJ. Heterogeneous Models Place the Root of the Placental Mammal Phylogeny. Mol Biol Evol. 2013;30(9):2145–2156. doi: 10.1093/molbev/mst117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Azuma Y, Kumazawa Y, Miya M, Mabuchi K, Nishida M. Mitogenomic evaluation of the historical biogeography of cichlids toward reliable dating of teleostean divergences. BMC Evol Biol. 2008;8(1):215. doi: 10.1186/1471-2148-8-215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Forsman ZH, Knapp ISS, Szabo Z, Toonen RJ. Intraspecific fluorescent phenotypes in Montipora capitata. Galaxea J Coral Reef Stud. 2014;16:17–18. doi: 10.3755/galaxea.16.17. [DOI] [Google Scholar]
- 59.Shore-Maggio A, Runyon CM, Ushijima B, Aeby GS, Callahan SM. Differences in bacterial community structure in two color morphs of the Hawaiian reef coral, Montipora capitata. Appl Env Microbiol. 2015;81:7312–7318. doi: 10.1128/AEM.01935-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shore-Maggio A, Callahan SM, Aeby GS. Trade-offs in disease and bleaching susceptibility among two color morphs of the Hawaiian reef coral, Montipora capitata. Coral Reefs. 2018;37(2):507–517. doi: 10.1007/s00338-018-1675-0. [DOI] [Google Scholar]
- 61.Huang JP. Parapatric genetic introgression and phenotypic assimilation: testing conditions for introgression between Hercules beetles (Dynastes, Dynastinae) Mol Ecol. 2016;25(21):5513–5526. doi: 10.1111/mec.13849. [DOI] [PubMed] [Google Scholar]
- 62.Souissi A, Bonhomme F, Manchado M, Bahri-Sfar L, Gagnaire P-A. Genomic and geographic footprints of differential introgression between two divergent fish species (Solea spp.) Heredity. 2018;121(6):579–593. doi: 10.1038/s41437-018-0079-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Johnston EC, Forsman ZH, Flot J-F, Schmidt-Roach S, Pinzón JH, Knapp ISS, Toonen RJ. A genomic glance through the fog of plasticity and diversification in Pocillopora. Sci Rep. 2017;7(1):5991. doi: 10.1038/s41598-017-06085-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cariou M, Duret L, Charlat S. Is RAD-seq suitable for phylogenetic inference? An in silico assessment and optimization. Ecol Evol. 2013;3(4):846–852. doi: 10.1002/ece3.512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hoeksema BW, Cairns S: World List of Scleractinia. 2019. http://www.marinespecies.org/scleractinia. Accessed 19 May 2019.
- 66.Gaither MR, Szabó Z, Crepeau MW, Bird CE, Toonen RJ. Preservation of corals in salt-saturated DMSO buffer is superior to ethanol for PCR experiments. Coral Reefs. 2011;30(2):329–333. doi: 10.1007/s00338-010-0687-1. [DOI] [Google Scholar]
- 67.Toonen RJ, Puritz JB, Forsman ZH, Whitney JL, Fernandez-Silva I, Andrews KR. Bird CE: ezRAD: a simplified method for genomic genotyping in non-model organisms. PeerJ. 2013;1:e203. doi: 10.7717/peerj.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Knapp ISS, Puritz JB, Bird CE, JL W, Sudek M, Forsman ZH, Toonen RJ. ezRAD- an accessible next-generation RAD sequencing protocol suitable for non-model organisms_v3.2. 2016. https://www.protocols.io/view/ezrad-an-accessible-next-generation-rad-sequencing-e9pbh5n. Accessed 15 May 2017.
- 69.Medina M, Collins A, Takaoka T, Kuehl J, Boore J. Naked corals: skeleton loss in Scleractinia. Proc Natl Acad Sci. 2006;103:9096–9100. doi: 10.1073/pnas.0602444103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Katoh K, Toh H. Parallelization of the MAFFT multiple sequence alignment program. Bioinformatics. 2010;26(15):1899–1900. doi: 10.1093/bioinformatics/btq224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Maddison WP, Maddison DR. Mesquite: a modular system for evolutionary analysis. Version 331. 2017. [Google Scholar]
- 72.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular Evolutionary Genetics Analysis Using Maximum Likelihood, Evolutionary Distance, and Maximum Parsimony Methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Hohna S, Larget B, Liu L, Suchard MA, Huelsenbeck JP. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rambaut A, Suchard MA, Xie D, Drummond AJ: Tracer version 1.7.1. Available from http://tree.bio.ed.ac.uk/software/tracer/. 2014.
- 75.Stamatakis A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014;30(9):1312–1313. doi: 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lanfear R, Frandsen PB, Wright AM, Senfeld T, Calcott B. PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol Biol Evol. 2017;34(3):772–773. doi: 10.1093/molbev/msw260. [DOI] [PubMed] [Google Scholar]
- 77.Thorne JL, Kishino H, Painter IS. Estimating the rate of evolution of the rate of molecular evolution. Mol Biol Evol. 1998;15:1647–1657. doi: 10.1093/oxfordjournals.molbev.a025892. [DOI] [PubMed] [Google Scholar]
- 78.Drummond AJ, Suchard MA, Xie D, Rambaut A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol Biol Evol. 2012;29(8):1969–1973. doi: 10.1093/molbev/mss075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thorne JL, Kishino H. Divergence time and evolutionary rate estimation with multilocus data. Syst Biol. 2002;51:689–702. doi: 10.1080/10635150290102456. [DOI] [PubMed] [Google Scholar]
- 80.Yang Z. PAML: a programme package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci. 1997;13:555–556. doi: 10.1093/bioinformatics/13.5.555. [DOI] [PubMed] [Google Scholar]
- 81.Wiegmann BM, Yeates DK, Thorne JL, Kishino H. Time flies, a new molecular time-scale for brachyceran fly evolution without a clock. Syst Biol. 2003;52(6):745–756. doi: 10.1093/sysbio/52.6.745. [DOI] [PubMed] [Google Scholar]
- 82.Kishino H, Thorne JL, Bruno WJ. Performance of a divergence time estimation method under a probabilistic model of rate evolution. Mol Biol Evol. 2001;18:352–361. doi: 10.1093/oxfordjournals.molbev.a003811. [DOI] [PubMed] [Google Scholar]
- 83.Stanley GD. The evolution of modern corals and their early history. Earth Sci Rev. 2003;60(3):195–225. doi: 10.1016/S0012-8252(02)00104-6. [DOI] [Google Scholar]
- 84.Simpson C, Kiessling W, Mewis H, Baron-Szabo RC, Müller J. Evolutionary diversification of reef corals: a comparison of the molecular and fossil records. Evolution. 2011;65(11):3274–3284. doi: 10.1111/j.1558-5646.2011.01365.x. [DOI] [PubMed] [Google Scholar]
- 85.Andrews S: FastQC: A quality control tool for high throughput sequence data. In: Available online at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/. Accessed 17 June 2018.
- 86.Krueger F, Andrews SR. Department of Medicine, Hematology and Oncology, Domagkstr. 3, 48149. Münster: Epigenesys; 2012. Quality control, trimming and alignment of Bisulfite-Seq data (Prot 57) pp. 1–13. [Google Scholar]
- 87.Puritz JB, Hollenbeck CM, Gold JR. dDocent: a RADseq, variant-calling pipeline designed for population genomics of non-model organisms. PeerJ. 2014;2:e431. doi: 10.7717/peerj.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Puritz JB, Matz MV, Toonen RJ, Weber JN, Bolnick DI, Bird CE. Demystifying the RAD fad. Mol Ecol. 2014;23(24):5937–5942. doi: 10.1111/mec.12965. [DOI] [PubMed] [Google Scholar]
- 89.Li H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv. 2013;1303.3997v2:1–3.
- 90.Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST. The variant call format and VCFtools. Bioinformatics. 2011;27(15):2156–2158. doi: 10.1093/bioinformatics/btr330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nuc Acids Res. 1997;25(17):3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bouckaert R, Heled J, Kühnert D, Vaughan T, Wu C-H, Xie D, Suchard MA, Rambaut A, Drummond AJ. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comp Biol. 2014;10(4):e1003537. doi: 10.1371/journal.pcbi.1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bryant D, Bouckaert R, Felsenstein J, Rosenberg NA, RoyChoudhury A. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol Biol Evol. 2012;29(8):1917–1932. doi: 10.1093/molbev/mss086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ortiz EM. vcf2phylip v1.5: convert a VCF matrix into several matrix formats for phylogenetic analysis. 2018. [Google Scholar]
- 95.Kass RE, Raftery AE. Bayes Factors. J Am Stat Assoc. 1995;90(430):773–795. doi: 10.1080/01621459.1995.10476572. [DOI] [Google Scholar]
- 96.Chifman J, Kubatko L. Quartet inference from SNP data under the coalescent model. Bioinformatics. 2014;30(23):3317–3324. doi: 10.1093/bioinformatics/btu530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Swofford DL. PAUP*: phylogenetic analysis using parsimony, version 4.0b10. 2003. [Google Scholar]
- 98.Green RE, Krause J, Briggs AW, Maricic T, Stenzel U, Kircher M, Patterson N, Li H, Zhai W, Fritz MH-Y. A draft sequence of the Neandertal genome. Science. 2010;328(5979):710–722. doi: 10.1126/science.1188021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Mol Biol Evol. 2011;28(8):2239–2252. doi: 10.1093/molbev/msr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Park Eunji, Hwang Dae-Sik, Lee Jae-Seong, Song Jun-Im, Seo Tae-Kun, Won Yong-Jin. Estimation of divergence times in cnidarian evolution based on mitochondrial protein-coding genes and the fossil record. Molecular Phylogenetics and Evolution. 2012;62(1):329–345. doi: 10.1016/j.ympev.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 101.Veron JEN. Corals in Space and Time: The Biogeography and Evolution of the Scleractinia. Ithaca: Cornell Univ. Press.; 1995.
- 102.Wells JW. Scleractinia. In: Treatise on Invertebrate Paleontology part F Coelenterata. Edited by Moore RC. Kansas, USA: Geol. Soc. Amer. & Univ. Kansas Press; 1956:328–478.
- 103.Strauss David, Sadler Peter M. Classical confidence intervals and Bayesian probability estimates for ends of local taxon ranges. Mathematical Geology. 1989;21(4):411–427. doi: 10.1007/BF00897326. [DOI] [Google Scholar]
- 104.Budd Ann F., Johnson Kenneth G. Origination preceding extinction during late Cenozoic turnover of Caribbean reefs. Paleobiology. 1999;25(2):188–200. doi: 10.1017/S009483730002649X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
S1. A. Species_list: Scleractinian corals used in the mitogenomic data set, accession numbers corresponding to the complete mitochondrial genomes retrieved from GenBank and to the newly sequenced 13 partial mitochondrial protein-coding genes of the 14 Montipora samples used in the mitogenomic analyses. List of Montipora samples used in the RADseq analyses. B. Sample_information: collected sample information regarding mitogenomic and genomic analyses (number of reads, reads length, mean depth coverage and standard deviation, percentage of the reference sequence). (XLSX 18 kb)
S2. A. Bayesian analysis showing phylogenetic relationships among 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile based on the concatenated mitochondrial dataset (13 protein-coding genes). B. Inset showing a detail of the topology referring to the genus Montipora zoomed 20x. This analysis was produced by MrBayes under a homogeneous model of rate change. Bayesian posterior probability (BPP) values are shown in black circles for values of maximal probability (1.00) and a grey circle for a value of 0.57. (JPG 587 kb)
S3. A. Maximum likelihood phylogram of 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile based on the concatenated mitochondrial dataset (13 protein-coding genes) produced by RAxML under a gamma model of rate heterogeneity. B. Inset showing a detail of the topology referring to the genus Montipora zoomed 100x. Numbers at the nodes represent bootstrap proportions. (PDF 230 kb)
S4. Beast maximum clade credibility chronogram showing main cladogenetic events among 55 scleractinian corals (49 species plus five morphotypes and pooled samples within M. capitata) representing 6 families and 15 genera plus the two outgroups Nematostella sp. and Metridium senile. The 95% highest posterior density (HPD) intervals are represented by the blue bars, and numbers at the nodes represent million years. (PDF 226 kb)
S5. Species tree based on 60,602 SNPs from the ‘coral’ data set estimated with SVDquartets. Numbers at the nodes represent bootstrap proportions. Image plates represent in-situ photographs of Montipora species sampled for this study and of the morphotypes within Montipora capitata. (JPG 386 kb)
S6. Venn diagram showing the overlap between putative coral (Montipora capitata) and symbionts (Symbiodinium minutum and Fugacium kawagutii) loci. (PDF 109 kb)
S7. In-situ photographs of Montipora species sampled for this study and of the morphotypes within Montipora capitata. (JPG 3488 kb)
Data Availability Statement
The datasets generated and/or analysed during the current study are available in the GeneBank (accession numbers in the Additional file 1: S1A and NCBI SRA bioproject ID PRJNA554733).