

Abstract
The surge of public disease and drug-related data availability has facilitated the application of computational methodologies to transform drug discovery. In the current chapter, we outline and detail the various resources and tools one can leverage in order to perform such analyses. We further describe in depth the in silico workflows of two recent studies that have identified possible novel indications of existing drugs. Lastly, we delve into the caveats and considerations of this process to enable other researchers to perform rigorous computational drug discovery experiments of their own.
Keywords: Systems pharmacology, Drug discovery, Big data, Electronic medical records, Clinical informatics, Bioinformatics, Drug repurposing, Drug repositioning, Gene expression data, Pharmacogenomics
1. Introduction
Preclinical drug discovery efforts are typically led by target-based or systems (phenotypic)-based strategies. In target-based screenings, the underlying goal is development around certain target or pathway with a priori evidence for its role in a disease or phenotype. Systems-based screenings are typically performed in a high-throughput fashion without an initial hypothesis of the target. In these models, many drugs, with and without known pharmacology, are tested against an assay evaluating properties of the phenotype of interest. From 1998 to 2006, 70% of first-in-class drugs discovered have been target-based, with only 30% directed by systems-based approaches [1]. This traditional drug discovery framework is both costly and timely, with a relatively low overall average success rate of 9.6% across all diseases [2]. While the max average overall success rate is 26.1% for hematology, the lowest is a mere 5.1% for oncology.
Other issues with traditional clinical trial design revolve around biased trials and selective publishing [3] that affect both internal and external validity [4] and therefore the implications of studies. One study evaluated the diversity of race, ethnicity, age, and sex in participants of cancer trials [5]. They found large differences in representation in all these realms: for instance, lower enrollment fractions in Hispanic and African-American participants compared to Caucasian participants (p < 0.001 for both comparisons). They also found an inverse relationship between age and enrollment fraction across all racial and ethnic groups and significant differences for men and women depending on the disease (e.g., men had higher enrollment fractions for lung cancer; p < 0.001). These differences in representation also have serious implications in practice when, for instance, a treatment studied in one population is given to another. Making clinical decisions based on studies with these issues can lead to expensive, sub-optimal treatment rates or missed opportunities at best and harmful events at worst. There are plenty of examples of randomized controlled trials that were judged to be beneficial but shown to be harmful (e.g., fluoride treatment for osteoporosis) [6]. Another weakness of clinical trial design is the relaxing of inclusion criteria for disease group in order to bolster study numbers, which is especially problematic in heterogeneous diseases.
These issues, along with the vast resources required for these studies and overall low success rates, necessitate complementary approaches. The recent surge of biomedical information pertaining to molecular characterizations of diseases and chemical compounds has facilitated a new age of drug discovery through computational predictions [7, 8]. By analyzing FDA-approved compounds to discover novel indications, one can leverage the massive amount of research and effort that have already been completed and bypass many steps in the traditional drug development framework. While there are many innovative strategies to reduce cost and improve success rates during the traditional drug discovery process [9, 10], drug repurposing is a viable and growing discipline with documented advantages: for instance, traditional drug discovery pipelines take on average from 12 to 16 years from inception to market and cost on average one to two billion dollars, while successful drug repurposing can be done in less than half the time (6 years on average) at a quarter of the cost (~$300 million) [11,12]. Incorporating drug repurposing strategies into workflows can drastically increase productivity for biopharmaceutical companies [13], and there are growing numbers of successful examples that prove its worth [14–17].
Thalidomide, for instance, was originally developed in Germany and England as a sedative and was prescribed to treat morning sickness in pregnant women. It soon became apparent that this treatment caused severe and devastating skeletal birth defects in thousands of babies born to mothers taking it during the first trimester of pregnancy. Years later, after the drug was banned for this purpose, it was serendipitously found to be effective for the treatment of erythema nodosum leprosum, a very serious complication ofleprosy. In a subsequent double-blind study of over 4500 patients with this condition, thalidomide treatment led to full remission in 2 weeks for 99% patients [18]. Not only is thalidomide the current (and only) standard of care for erythema nodosum leprosum, it has also proven beneficial in other disorders. Soon after, in fact, it was found to significantly improve survival in patients with multiple myeloma [19]. Celgene, the biopharmaceutical company responsible for driving the resurgence of thalidomide through these repurposed indications, derived 75% or more of its revenue in 2016 from this one drug, primarily for treating multiple myeloma [20].
This case, although extreme, illustrates the value and possibilities of drug repurposing even in the face of documented failure. There are countless avenues to explore for new indications of old drugs, which are not necessarily surveyed in traditional clinical trial design strategies. In the current chapter, we will outline and describe the tools and best-practice methodologies that can be used to successfully leverage big data for drug discovery by detailing the pipelines of two recent studies as templates. We further discuss limitations and important considerations of this process in Subheading 4. We expect that one can apply the approach to discover new therapeutics for other diseases ofinterest after reading this chapter.
2. Materials
In this section, we will cover an overview of the materials and tools that can be used to perform an in silico drug discovery experiment, along with how to go about accessing them. We also provide a visual guide for this process in Fig. 1. First, we will discuss recommended software and computational resources that can be used for this purpose. Next we describe the types of data that are typically used in these experiments along with ontological resources on how they can be effectively integrated. We then go into the specific databases that house disease and chemogenomic-related gene expression data. We conclude with specialized software and packages that can enhance figure making capabilities to visualize ensuing results.
Fig. 1.
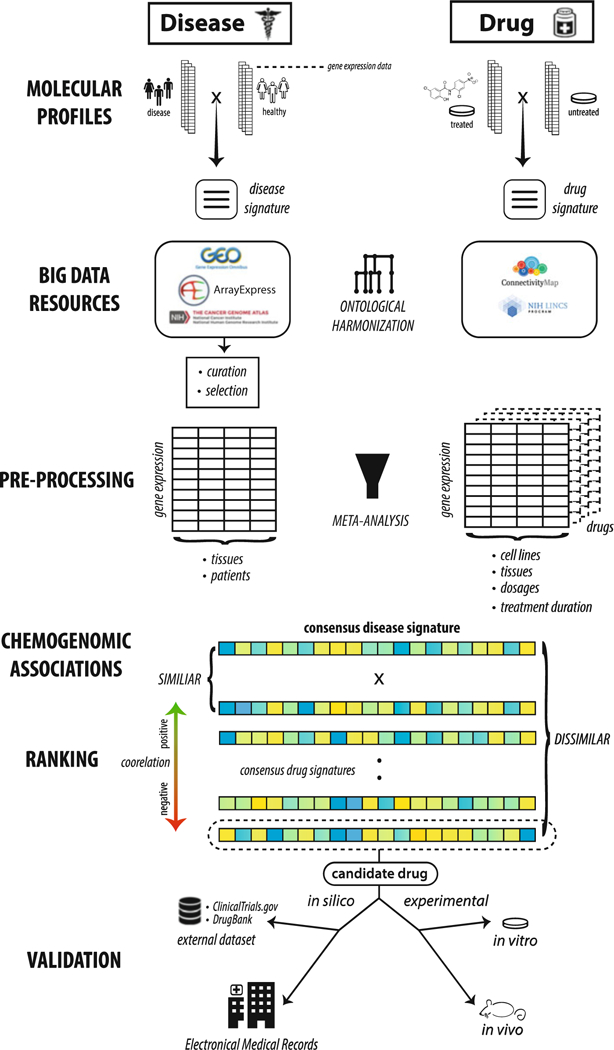
Full generic workflow for enabling drug discovery through big data resources. We start by illustrating the mechanism behind disease and drug gene expression signatures and highlighting a few public repositories where they can be found. Based on research focus, it is often recommended to harmonize these disparate sources of data through use of ontologies. Multiple signatures per disease or drug can be integrated through rigorous meta-analysis procedures. Chemogenomic association testing assesses similarity between drug and disease signatures and can be performed using procedures like the Kolmogorov-Smirnov (KS) test. These drug signatures can then be ranked according to their correlation, or anticorrelation, to the disease signature of interest. Drug signatures that are highly anticorrelated to the disease signature are potential treatment candidates. Drug candidates that are selected for follow-up need further validation, in the form of in silico (e.g., other external datasets or electronic medical records), in vitro, and/or in vivo experiments
2.1. Software and ComputationalResources
Drug discovery using big data resources is accomplished computationally. For the individual researcher, a modern computer is really the only hardware requirement. For software, essentially any programming language whether open-source (e.g., R [21], Python [22]) or closed and commercially licensed (e.g., SAS [SAS Institute Inc., Cary, NC]) can perform data organization, statistical analyses, and figure generation with the inclusion of freely available packages (e.g., SciPy [23] for Python) when needed. There exist numerous resources (e.g., edX; https://www.edx.org) that provide an introduction on using these programming languages for data science. For infrastructure, cloud computing (e.g., Amazon Web Services Cloud) enables managing large datasets and performing computationally intensive tasks without the need of building in-house clusters.
2.2. Ontologies and Reference Databases
The landscape of big data in biomedicine is expansive yet unsystematic: encompassing a tremendous amount of data points across a multitude of modalities. As such, there are clear hurdles in precise characterization and proper integration procedures. To address these challenges, researchers have created tools and ontologies to map and normalize these disparate data types in order to facilitate data harmonization and reproducible methodologies. This is especially important for the purposes of leveraging big data for drug discovery, as many different data types have to be seamlessly and reproducibly integrated along the entire drug discovery pipeline. A more comprehensive list of these various resources can be found in other related reviews [24].
The exponential growth of data entities with heterogeneous data types from multiple resources calls for developing ontologies to define entities and centralized reference databases. Meta-thesauruses, like the Unified Medical Language System (UMLS) [25], have been developed to organize, classify, standardize, and distribute key terminology in biomedical information systems and are invaluable for computational biology research. Essentially, each medical term (e.g., disease) has a Concept Unique Identifier (CUI) code that then can be referenced from other related ontologies. The Systematized Nomenclature of Medicine-Clinical Terms (SNOMED-CT; http://www.ihtsdo.org/snomed-ct/), for instance, is one of the largest healthcare-related ontologies and has over 300,000 medical concepts ranging from body structure to clinical findings. There are specific ontologies that aim to characterize the continually evolving representations of the phenotypic space. Many clinical datasets encode diseases using International Classification of Diseases (ICD) codes, for instance. The Disease Ontology (http://disease-ontology.org/) organizes and standardizes various aspects (e.g., synonyms, relationships, ICD codes) of disease-oriented clinical topics into representative terms. The Human Phenotype Ontology (http://human-phenotype-ontology.github.io/) has a similar goal but expands to more broad aspects of human phenotypes, including abnormalities and side effects. There are many resources that organize information pertaining to various properties of genetic data, such as dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP/), dbVar (https://www.ncbi.nlm.nih.gov/dbvar), RegulomeDB (information regarding regulatory elements; http://www.regulomedb.org/), and UniProt (protein sequence and functional information; http://www.uniprot.org/). The 1000 Genomes Project (www.internationalgenome.org/) is a collection of thousands of whole genomes from populations across the globe. The Exome Aggregation Consortium (ExAC) is an accumulation of whole-exome data for over 60,000 unrelated individuals from multiple contributing projects at the time of this publication. There are also resources that are specifically focused on compiling information related to the associations of genotypes and phenotypes. The Online Mendelian Inheritance in Man (OMIM; https://www.omim.org/) is an online compendium of genotype/phenotype information focusing on Mendelian disorders. As of June 15, 2017, this resource has information on over 6000 phenotypes for which the molecular basis is known and over 3700 genes with gene-causing mutations. Other related resources include the Human Gene Mutation Database (HGMD; http://www.hgmd.cf.ac.uk/) and ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/). The Mouse Genome Informatics (MGI; http://www.informatics.jax.org/) is a collection of data pertaining to various experiments performed on mice, including disease models with phenotype and genotype information.
In the drug space, medication and prescription data are often unsystematically organized due to issues like conflicting nomenclature or spellings (i.e., brand name vs. generic name, American vs. British terminology). Additionally, the same medication may be prescribed with different dosages, minor formulation differences, and routes of administration. All the information is presented as free text in the medical records, requiring additional text mining effort to connect them to other modalities. RxNorm [26] was developed as an UMLS-based, standardized medication vocabulary that intersects with known clinical knowledge repositories like Micromedex (Micromedex Solutions, Truven Health Analytics, Inc. Ann Arbor, MI). Essentially, related complex drug name strings can be mapped to a common identifier. Linking medications to an RxNorm identifier facilities easy connection to other related resources that include other modalities of data, such as Sider [27] and Offsides/Two sides [28], which document known and predicted drug-drug interactions and connect medications to known clinical side effects. Through public databases like DrugBank [29], one can cross-reference drug targets, pharmacological properties, and clinical indications. Using RepurposeDB (http://repurposedb.dudleylab.org/) [30], one can explore the known drug repurposing space and explore various factors (e.g., chemical properties) that might underlie these successes. The US National Institutes of Health clinical trial repository (https://ClinicalTrials.gov) is a collection of clinical trial data from various diseases and treatments along with study outcomes and can open the door for accessing and reanalyzing clinical trial data [31], such as research into medication efficacy at various stages of clinical trials. PubChem (https://pubchem.ncbi.nlm.nih.gov/) is a reference database for information on the biological activities of small molecules, including compound structure and substance information.
2.3. Data Resources that Can Be Leveraged for Drug Discovery
A growing priority of increasing data accessibility through open-access efforts has considerably boosted computational-based drug discovery capabilities. In fact, Greene et al. created the Research Parasite Award, which honors researchers who perform rigorous secondary analyses on existing, open-access data to make novel insights independent from the original investigators [32]. These data exist in many forms, including clinical (i.e., disease comorbidities), genomic (e.g., genetic, transcriptomic), and proteomics.
Drug discovery is multifaceted at its core, at the very least involving some combination of chemical data in addition to those mentioned above. This is even more relevant for computational-based drug discovery, where complex intersections of many disparate data types are required. Fortunately, there are many public repositories that contain a plethora of data around these spheres, which can be connected utilizing the aforementioned ontologies and reference databases. We outline the various data sources in the current section and present two examples ofsuccessful drug discovery utilizations of these datasets in detail in Subheading 3.
2.3.1. Hospitals and Academic Health Centers
Due to regulatory requirements, almost all American medical centers and health systems store patient data collected during outpatient and hospital visits using software platforms such as those provided by EPIC (Epic Systems Corporation, Madison, WI) and Cerner (Cerner Corporation, Kansas City, MO). These electronic medical records (EMRs), or alternately electronic health records (EHRs), are composed of a number of data types such as disease diagnoses, medication prescriptions, lab test results, surgical procedures, and physician notes. Generally, data warehouse administration takes responsibility for housing and creating an anonymized or de-identified version of the EMR. Affiliated faculty and researchers then apply for access to this resource through their Institutional Review Board (IRB). While the primary role of EMR is for institutional or administrative purposes, one important benefit of the digitization of health records is that they can be more easily adapted for powerful healthcare research purposes. For drug discovery, these data can be used for genotype-phenotype relationship discovery that could drive target selection or to analyze medication efficacy and side effects in a real-world context [33]. The implications and impact of precise electronic phenotyping procedures for these types of analyses will be discussed in Subheading 4.
There are many hospital systems and affiliated medical centers that have successfully used their in-house EMR systems for important scientific and clinical discoveries [34–38]. The Mount Sinai Hospital and the Icahn School of Medicine at Mount Sinai organize and protect their EMR data (beginning in 2003) within the Mount Sinai Data Warehouse, which is comprised of over 7.5 million patients and 2 billion points of data, including disease diagnoses and lab test results. The University of California, San Francisco (UCSF) is leading a massive effort to coordinate EMR data from five UC medical centers. The University of California Research eXchange (UC ReX; https://myresearch.ucsf.edu/uc-rex) provides a framework for UC-affiliated researchers to query de-identified clinical and demographic data, providing a natural cross-validation opportunity to compare findings across these sites.
Data analyses using EMRs can be enhanced with the inclusion of genetic data from biobanks or repositories of biological samples (e.g., blood) of recruited participants generally from a hospital setting. Many institutions have frameworks set up that facilitate this type of research, such as The Charles Bronfman Institute of Personalized Medicine BioMe biobank within the Mount Sinai Hospital system, BioVU from the Vanderbilt University Medical Center, and DiscovEHR, which is a collaboration between Regen-eron Genetics Center and the Geisinger Health System. Coupling clinical and genetic data has led to important findings in the area of drug and target discovery. Using the DiscovEHR resource, for instance, researchers recently characterized the distribution and clinical impact of rare, functional variants (i.e., deleterious) in whole-exome sequences for over 50,000 individuals [39]. The associations they identified add insight to the current understanding of therapeutic targets and clinically actionable genes.
A limitation of these biobanks and EMR systems is that they are often restricted to researchers affiliated with the associated institution. There are a number of initiatives and resources that allow researchers to apply for access of these types of data. For instance, the Centers for Medicare & Medicaid Services offer relevant healthcare-related data dumps (https://data.medicare.gov/) for sites that accept Medicare including hospitals, nursing home, and hospices with information on various factors and outcomes (e.g., infection rates). The DREAM challenges (http/www.dreamchallenges.org), for instance, consist of various “open science” prediction challenges, some of which are disease-centric that incorporate real clinical datasets often coupled with other modalities of data (e.g., genetics). In the Alzheimer’s Disease Big Data DREAM Challenge #1 (Synapse ID: syn2290704; June–-October 2014), challengers were tasked with developing the best performing machine learning model to predict disease progression, using actual genetic data (e.g., genotypes) and clinical data (e.g., cognitive assessments) from patients with mild cognitive impairment, early Alzheimer’s disease, and elderly controls. The UK Biobank (http://www.ukbiobank.ac.uk/) is an unprecedented health resource of over 500,000 recruited participants aged 40–69 with genetic (genotyping) and a wide variety of clinical data, including longitudinal follow-ups (original data collected from 2006 to 2010). These data include online questionnaires (e.g., about diet, cognitive function), EMR, blood biochemistry (e.g., hormone levels), and urinalysis, among others. Further, more specialized data is available for a subset of patients, including a 24-h activity monitoring for a week (n = 100,000) and image scans (e.g., brain, heart, abdomen; n = 100,000).
What distinguishes the UK Biobank from other large-scale initiatives is that it is a “fair access” biobank [40], one that has infrastructure to facilitate collection, storage, protection, and distribution (with data update releases) to allow academic and industrial researchers to apply for access. As of July 2016, there have been around 100 publications featuring or involving data from the UK Biobank (http://www.ukbiobank.ac.uk/published-papers/; latest update), although this number is rapidly increasing. While these studies include major findings in the realm of genetics and phenotypes (e.g., diseases and health outcomes), very few are directed at drug discovery. Wain et al. have demonstrated the value of these data for the identification of potential drug targets, specifically for lung function and chronic obstructive pulmonary disease (COPD) [41]. The authors leveraged the large cohort size to select ~50,000 individuals at the extreme ends of lung function (e.g., forced expired volume in 1 s) for their analyses. From a GWAS, they identified 97 signals (43 reported as novel) and created a polygenic risk score from around six alleles that confers a 3.7-fold change in COPD risk between high- and low-risk scored individuals. The authors then analyzed these signals in the context of variant function (i.e., deleteriousness) that cause expression changes in other genes (eQTLs), resulting in 234 genes with potentially causal effects on lung function. Seven out of these 234 genes are already targets of approved or drugs currently in development. The remaining genes, along with others they identified by expanding their network via protein-protein interactions, represent potential novel targets for drug development.
2.3.2. Genomic Experimental Data Repositories
Each and every research study is crucial for furthering scientific knowledge, but sharing of the experimental data collected can additionally benefit other researchers in the field directly. The pooling of several sources of data can allow for meta-analyses and other types of research not possible in isolation, thereby facilitating further discoveries. There have been outstanding efforts to provide a framework for researchers to deposit and share data, with many high-impact journals even requiring it for publication. There are other reviews that go into detail about these resources, but we will describe a few here.
The Gene Expression Omnibus (GEO) is a public repository from NCBI that allows researchers to upload high-quality functional genomics data (e.g., microarray) that meet their guidelines [42]. GEO organizes, stores, and freely distributes these data to the research community. As of 2017, GEO already surpasses over two million samples. ArrayExpress (http://www.ebi.ac.uk/arrayexpress/) [43] is an archive of functional genomics data (some overlap with GEO) comprised of over 70,000 experiments, 2.2. million assays, and 45 terabytes of data as of July 2017. The Immunology Database and Analysis Portal (ImmPort; www.immport.org) is a related resource focused on data from immunology studies of various types, focuses, and species that provides a framework to share and use genomic data of clinical samples.
The Cancer Genome Atlas (TCGA; https://cancergenome.nih.gov/) from the NIH is a comprehensive resource that collects data relating to the genomic aspects in over 33 types of cancer. TCGA harmonizes clinical, sequencing, transcriptomic, and other data types from various studies in a user-friendly Genomic Data Commons Data Portal (https://portal.gdc.cancer.gov/). As of Data Release 6.0 (May 9, 2017), TCGA encompasses around 250,000 files from almost 15,000 patients with cancer in 29 primary sites (e.g., kidney). As the volume of the data from TCGA is both immense and complex, Firehose GDAC (https://gdac.broadinstitute.org/) was developed to systematize analyses and pipelines using TCGA in order to facilitate smooth research efforts. The Cancer Cell Line Encyclopedia (CCLE; http://portals.broadinstitute.org/ccle/) is a public compendium of over 1000 cell lines that aims to characterize the genetic and pharmacologic aspects of human cancer models. The Genome-Tissue Expression (GTEx; https://gtexportal.org/home/) portal is a collection of genetic and gene expression data from the Broad Institute, with data for 544 healthy individuals in 53 tissues as of the V6p release. Using this resource, researchers have been able to delineate cis and trans expression quantitative trait loci (eQTL), a unique capability from having access to both genotype and gene expression data.
2.3.3. Chemogenomic Data Resources
The last crucial component to computational-based drug discovery is a resource that relates effects of chemical exposure on biological systems. The Connectivity Map (CMap; https://portals.broadinstitute.org/cmap/) is a landmark repository that seeks to provide a systematic representation of the transcriptomic effects of cell lines being treated with various drugs. The current version (build 02) of CMap contains over 7000 drug-induced gene expression profiles for 1309 compounds across five cell lines.
A spiritual successor of CMap, the Library of Integrated Cellular Signatures (LINCS; http://www.lincsproject.org/) project is a large-scale collaboration from 12 institutions. As of the writing of this chapter, LINCS encompasses 350 datasets using 14 different methods (e.g., KINOMEscan) across six subject areas (e.g., binding, proteomics) and 11 biological processes (e.g., gene expression, cell proliferation). Alarge component of LINCS is L1000 Connectivity Map - a collection of assays that measure transcriptomic profiles for a variety of pharmacological and genetic (knockdown and overexpression) perturbations across different cell lines. L1000 contains data for roughly 20,000 compounds across over 50 different cell lines - a substantial increase from CMap. One large difference between these two resources, however, is that the L1000 assay directly measures only 978 “landmark” genes and uses imputation methods to gain information for others. Researchers can obtain these datasets through a convenient data portal (http://lincsportal.ccs.miami.edu/dcic-portal/). As referenced above, there are countless successful research applications using these resources in the fields oftarget discovery, drug discovery, and drug repurposing along with many others.
2.4. Visualization Tools and Software
Effective data visualization is pertinent for network-based target (e.g., key driver) discovery or data exploration in drug discovery. The programming languages mentioned before can produce high-quality figures of results. While the base graphical output style is often adequate, there are a number of packages that can be used to create superior and publication-ready graphics, including ggplot2 [44] for R, matplotlib [45], PyX (http://pyx.sourceforge.net/), and seaborn (http://seaborn.pydata.org/) for Python. D3 (https://d3js.org/), a JavaScript library, can also create impressive static and interactive figures for feature on web sites. While these languages can generate network figures, such as NetworkX(http://networkx.github.io/) for Python, specialized software such as Cytoscape [46], Gephi [47], and igraph (http://http://igraph.org/) or online tools such as Plot.ly (http://plot.ly/) may be preferred for this use case.
3. Methods
In the current section, we will describe a general workflow that leverages public data for drug discovery using, exemplified in two of our recent studies. We provide the general step-by-step framework for performing such experiments in Fig. 2. While both studies are methodologically similar, they have disparate focuses, illustrating the wide variety of possibilities of this framework. In the first study, Li and Greene et al. sought to identify novel therapeutic options for chronic allograft damage, specifically to limit the progression of interstitial fibrosis and tubular atrophy (IF/TA) [48]. In the next study, Chen and Wei et al. discovered a novel, potentially therapeutic drug to treat hepatocellular carcinoma (HCC) revealing reduced growth of cancer cells in both in vitro and in vivo models [49].
Fig. 2.
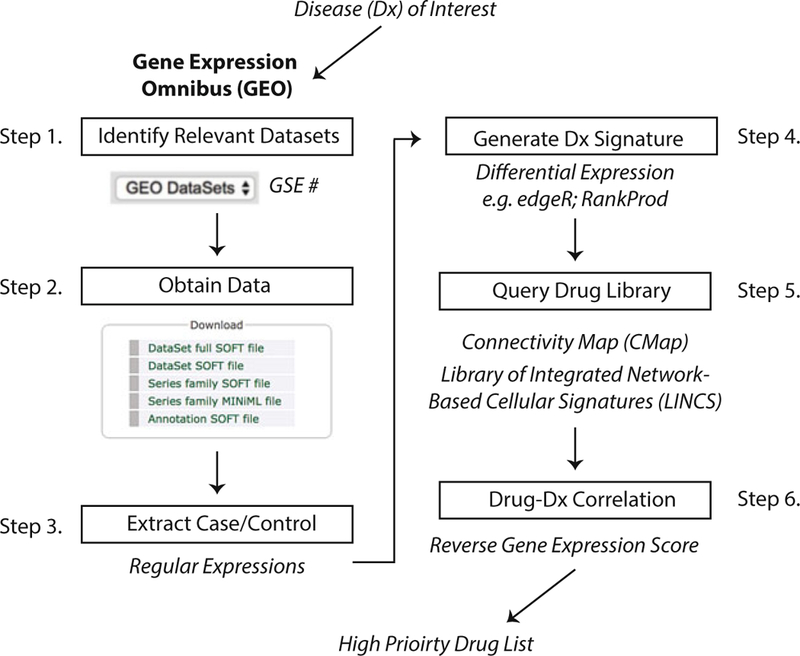
Step-by-step process for performing in silico drug discovery starting from a disease of interest. Researchers can go to the GEO online portal and search for their disease of interest, which will result in experiments and microarray datasets that other scientists have generated and uploaded. Having identified all datasets of interest, the data can be downloaded individually or via a tool, such as GEOquery [80] for R. Case and control data will have to be identified either manually or through regular expressions from the name strings. Next, disease signatures can be derived through various tools that perform differential expression analysis, such as edgeR [81] for RNA-Seq and RankProd and SAM for microarray data [82]. Drug libraries, such as CMap and LINCS, provide RNA expression data for a large number of compounds across different cell lines. Next, a correlation analysis can be performed comparing the disease signature and all drug signatures producing a reversal score for each drug. Significant scores can be ranked from the top most correlated signatures to the most anticorrelated signatures. These hits can then be used to select a candidate drug based on study goals, such as prioritizing candidates that would reverse disease signature (i.e., top anticorrelated hits)
3.1. Disease Signatures
A disease signature is the unique molecular state (i.e., RNA expression profile) of a phenotype (e.g., type 2 diabetes) that is altered from “wild-type” or healthy. Typically, these signatures can be characterized by multimodal biological data, including gene expression in tissues and protein composition in the microbiome. In the Li and Greene et al. study, they obtained kidney transplant microarray datasets from GEO in addition to an in-house dataset. They first restricted the possible datasets to those in humans with biopsy or peripheral blood samples, leaving five that were eligible. As is typical in these types of analyses, the phenotypic descriptors from the datasets did not directly coincide. As such, for each study, they selected data from specific biopsies that met their criteria for suitable comparison (i.e., “moderate” and “severe” IF/TA from one study, IF/TA “II” and “III” from another, etc.) including both case and control conditions. To rectify any possible conflicting probe annotations due to platform or versioning, they re-annotated all probes to the latest gene identifiers using AILUN [50]. In addition to ensuring consistent annotations, it is also important to account for potential discrepancies in expression measurements across studies and platforms. Accordingly, they standardized each dataset using quantile-quantile normalization to allow for more precise integration. The integrated dataset comprised 275 samples in two tissues from three different microarray platforms.
With the datasets normalized and integrated, a meta-analysis can be performed to create a consensus disease signature across multiple studies. There are many differing methodologies to perform meta-analysis on microarray data that we discuss in Subheading 4. Li and Greene et al. performed two meta-analysis techniques and included genes that had robust expression profiles significant in both, thus maximizing the methodological strengths of each. The first method involved evaluating the differential expression effect size for each. Specifically, these effect sizes were combined using a fixed-effect inverse-variance model by which the effect size from each study is weighted by the inverse of the intra-study variance resulting in a meta-effect size. From this approach, they identified 996 FDR-corrected significant genes measured in at least four studies and were concordantly expressed in the same direction (FDR < 0.05).
For the second method, they then utilized results from the significance analysis of microarrays (SAM) [51] of each study, classifying significant differentially expressed genes between IF/TA and non-IF/TA groups using q < 0.1 threshold. For the metaanalysis, they performed a Fisher’s exact test comparing the number of studies in which each gene was significantly differentially expressed by the hypergeometric distribution (p < 0.05). From this method, they identified 510 genes that were significant in at least three of the studies. With the two results from the meta-analyses in hand, they restricted their disease signature to genes that were significantly differentially expressed using both methods, resulting in 85 genes used for drug signature comparison.
Chen and Wei et al. also leveraged public, external datasets to generate a signature for the disease of interest: HCC. The authors constructed a multitiered pipeline that utilized various open-source databases to build a high-confidence HCC disease signature that was evaluated at multiple stages. To build the HCC disease signature, they first obtained RNA sequence profiles for 200 HCC and 50 adjacent non-tumor samples from GDAC and their corresponding clinical labels from TCGA. For subsequent evaluation of this disease signature, they downloaded data from GEO by querying “hepatocellular carcinoma,” resulting in seven potential independent datasets that had at least three samples in both disease and control (i.e., non-tumor liver sample) groups. Like the other study, they converted all probes to the most recent build and collapsed them to the gene level by mean value and then performed quantile normalization. As mentioned above, datasets from public resources may not coincide with the current research question or focus. Like before, different methodologies have to be employed to make best use of the exploitable aspects of interest (see Subheading 4). For the current study, in order to ensure that gene expression data from these HCC samples were of high quality, robust enough, and related to the cell lines that were later used for experimental validation, they performed an initial assessment of the similarity of each expression profile to cancer cell lines that are already characterized in detail. For the background set, they downloaded gene expression data for 1019 cancer cell lines, including 25 that were HCC-specific, from CCLE, and represented each by their expression profiles of 5000 genes that varied the most across all samples. For the similarity assessment, they performed a ranked-based Spearman correlation of the CCLE set to both the TCGA and GEO samples of interest. They then performed a Mann-Whitney test to differentiate the correlation outcomes of the samples of interest to the HCC-specific cell lines against the non-specific ones. The resulting p value outcomes of the sets of interest were compared to outcomes of 1000 random tissue samples (assessed via the same method) obtained from the Expression Project for Oncology (expO; GSE2109). From the original datasets of interest, samples were removed from consideration if their association was lower than 95% of the random samples based on the null distribution (p < 0.05). From this thorough assessment, eight tumor samples from TCGA and one dataset from GEO were removed from consideration. After restricting to high-quality TCGA data-sets (n = 192 tumor and 50 non-tumor samples), the authors built the initial HCC disease signature model using DESeq (version 1) [52] to perform differential expression analysis across various log-two fold change (FC) and significance thresholds.
To fine-tune and identify the best disease signatures, the authors evaluated them on the 1736 patient samples from the six curated datasets from GEO. For each study, they removed non-HCC genes from the gene expression data and then performed principal component analysis to build classifiers, with the first principal component representing the variation between tumor and non-tumor samples. They found that FC > 2.0 and p < 1E-20 thresholds led to the best separation of tumor and non-tumor samples (median AUC = 0.995) across all studies. Based on this best threshold, the integrated HCC signature consisted of 163 up-and 111 downregulated genes. In order to use the LINCS L1000, a drug gene expression database with 978 genes profiled, they also created another reduced disease signature.
3.2. Drug Signatures
Drug signatures are similar in nature to disease signatures in that they represent the global perturbation of gene expression compared to vehicle control. In the case of drug signatures, the perturbation is treatment exposure instead of disease state. As mentioned in Subheading 2, there are many databases that contain gene expression data for a variety of drugs across many different cell lines, tissues and organisms. In the Li and Greene et al. study, they collected all drug-induced transcriptional profiles for all drugs in CMap (n = 1309) across all experiments (n = 6100). They then created a consensus, representative profile for each drug, merging data from the associated studies using the prototype ranked list (PRL) method [53]. The PRL method works through a hierarchical majority-voting scheme for all ranked gene expression lists within a single compound where consistently over- or down-regulated genes are weighted toward the top of affiliated extremes. The various merging methodologies and their considerations will be discussed in the Notes.
Chen and Wei et al. utilized both CMap and LINCS data to generate drug signatures. For each drug of all CMap, they performed an initial quality control step keeping only instances (i.e., cell line experiment data) where its profile correlated ( p < 0.05) with at least one other profile (of the same drug), leaving 1329 high-quality instances. They gathered a high-quality list of data from LINCS by only including landmark genes (n = 978), restricting instances to HepG2 and Huh7 cell lines, and removing poor quality perturbations, resulting in 2816 profiles. As the authors were interested in repurposing an already approved compound, they intersected drugs from both data sources to DrugBank, leaving 380 instances of 249 drugs. Out of these 249 drugs, 83 were common between the two sources.
3.3. Integrating Disease and Drug Signatures: In Silico Methodologies for Therapeutic Predictions
The general methodology used to correlate drug-induced and disease-state gene expression profiles is well established and has been used successfully in a wide variety of studies. How well, and in what direction, drug and biological profiles correlate can direct drug discovery and repurposing efforts. The disease-state profile represents gene expression patterns that diverge from the norm. Accordingly, identifying a compound with an opposing (i.e., anticorrelated) profile can push biological expression patterns back to an unperturbed state. Typically, the signature matching is performed using a KS test [54]. As this type of analysis can produce many candidate predictions, we recommend additional refinement and filtration steps to prioritize drugs or targets with the highest confidence of success to move forward with testing.
In the Li and Greene et al. study, they compared the correlation for 1309 consensus compound signatures from CMap to their IF/TA disease signature using a modified KS test. The significance of these scores was calculated by comparing specific KS scores to those from a random permutation of 1000 drug signatures, producing a ranked list of potential treatments that were significantly anticorrelated to the disease signature (p ≤ 0.05). To further refine this list of candidates, they performed a literature review of the top hits and excluded drugs from their list of possibilities that would impede IF/TA improvement due to their side effect profile (i.e., those with negative neurological side effects). From this step, they decided to further pursue kaempferol and esculetin as candidate therapeutic drugs to treat renal fibrosis. To add further evidence to their findings, the authors performed a separate, additional in silico experiment to try to characterize potential immune-related effects of these two compounds through evaluating specific immune cell they may potentially influence during anti-fibrosis activity. As such, they first matched the 1309 drug expression profiles to 221 immune cell state profiles procured from immune-cell pharmacology map [55] to look for enrichments in specific immune cell subsets. This analysis predicted that esculetin and kaempferol would inhibit both active and innate immune cells (e.g., CD4 T cells) in IF/TA.
In the Chen and Wei et al. study, they were unable to use their best performing HCC signature profile of 274 genes as only 30 genes mapped to the landmark LINCS set. Accordingly, they relaxed the threshold (p < 0.001 and FC >2.0) to increase the power ofthe signature for this comparison task, producing a signature of 44 genes. The original signature was correlated to 1174 distinct drugs in CMap and the reduced signature was correlated to 249 distinct drugs in LINCS. Similar to the previous study, the authors utilized a nonparametric, rank-based pattern methodology based on the KS statistic to identify compounds curated from both CMap and LINCS that are anticorrelated with the HCC signature. They also assessed significance of these predictions through random permutations and multiple testing correction (FDR <0.05): there were 302 drugs from CMap and 39 drugs from LINCS that were anticorrelated at this threshold, with 16 overlapping. Out of this high-confidence intersected set, the top hit from ranking across both libraries was niclosamide. This drug had previously established antitumor properties in other cancers, but had not been assessed in HCC animal models. Therefore, it was selected as a candidate drug to undergo subsequent validation experiments.
Like the previous study, Chen and Wei et al. developed an innovative, additional in silico approach to evaluate the global performance of the predictions from their pipeline. They hypothesized that their top predictions (i.e., anticorrelated drugs) should be enriched for gold standard, or established, treatments of HCC. Accordingly, they extracted data from clinicaltrials.gov querying the terms “hepatocellular carcinoma” and “liver cancer” and filtering from the results trials that studied tumors and cancer in general. From this list of 960 trials, they extracted 76 drugs from the “interventions” column that appeared in more than one trial as their list of gold standard HCC drugs. When mapped to the che- mogenomic databases, there were 7 found in CMap and 16 in LINCS. Using ssGSEA from GSVA [56] with permutation testing (n = 10,000), a gene set enrichment package, they found that these gold standard drugs were more likely to reverse the HCC gene signature in both CMap (p = 0.012) and LINCS (p = 0.018), increasing the confidence of testing other hits in the lab.
3.4. Experimental Validation of Predictions
While the prediction methodology outlined above is powerful in its own right, it is often necessary to validate these findings in an experimental setting. Convincing validation of these predictions can be achieved in a multitude of ways in both in vitro and in vivo models. The considerations and criteria for this decision are discussed in Subheading 4. To investigate the utility of kaempferol and esculetin for fibrosis, Li and Greene et al. utilized human kidney 2 (KH2) in vitro cell line to determine perturbations in cellular pathways following drug exposure, specifically targeting biological aspects from their in silico-based hypotheses. As such, they showed kaempferol significantly reduced TGF-β1-mediated expression of SNAH (p = 0.014 for 15 μm exposure) and reversed CDH1 downregulation ( p = 0.045). They also found that esculetin treatment inhibits Wnt/β-catenin signaling in renal tubular cells: esculetin-treated cells caused a significant decrease in CCND1 protein levels after Wnt agonist stimulation ( p = 0.0054 for 60 μm exposure).
In addition to the encouraging in vitro results, they performed an additional experiment to assess the effects of kaempferol and esculetin on renal interstitial fibrosis in vivo. Specifically, they used a unilateral ureteric obstruction (UUO) mouse model to study gene expression, histological, immunohistochemistry (IHC) effects of treatment on renal fibrogenesis. Mice (Balb/c mice from Jackson Laboratory) were administered kaempferol (n = 5), esculetin (n = 5), or saline (n = 6) from 2 days prior to UUO surgery procedure until sample collection 7 days post-UUO. They found encouraging results supporting a beneficial role of these drugs in treating renal fibrosis: mice treated with both drugs had significantly lower amount of interstitial collagens and renal fibrosis in UUO kidneys by picrosirius red staining compared to controls (p = 0.0009 for kaempferol; p = 0.0011 for kaempferol). Their targeted analyses also allowed to assess their hypotheses of the mechanisms by which these drugs are effective in this system: for instance, kaempferol caused a significant reduction in the gene expression of Snail (p = 0.038), a key transcription factor in the TGF-β signaling pathway to confirm their in vitro analysis.
Like the first study, Chen and Wei et al. performed a series of both in vitro and in vivo experiments to systematically evaluate their prediction of niclosamide as a HCC drug candidate. Due to poor water solubility of niclosamide which could hamper its effectiveness, they also performed these experiments using niclosamide ethanolamine salt (NEN), which is known to have better systemic bioavailability and could have a better chance of reaching the tumor site. As an initial evaluation, the authors performed a cell viability assay of these two drugs on HCC cells to calculate inhibitory concentrations. They found that niclosamide and NEN both reduced HCC cell viability, being over sevenfold more cytotoxic to HCC cells than primary hepatocytes. With these findings, the authors felt confident to assess the antitumor effects ofniclosamide and NEN in a mouse primary HCC model. Mice with induced HCC were treated with food containing niclosamide or NEN (cases) or with autoclaved food (control). At 12 weeks, the livers were extracted and analyzed histologically. The authors found that niclosamide and NEN both reduced the number of tumor nodules compared to controls, but the effect of NEN was much more pronounced. Furthermore, the NEN-treated mice had lower over-all liver weights compared to both control and niclosamide groups (p < 0.001 for both), but there was no significant difference between niclosamide and control groups. This provided early evidence that NEN might be the superior treatment candidate.
Moving closer in relevance for human treatment, they evaluated the effect of these drugs on mice bearing orthotopic patient- derived xenografts (PDX) derived from HCC tissue of resected livers from three patients with the disease. After implantation, these mice were separated into groups like before, being fed either regular food or food with NEN or niclosamide. Like in the results for the previous experiment, the NEN group had the most pronounced effects in inhibiting PDX growth based on both bioluminescence and tumor volume (p < 0.05 for both) and did not significantly lower body weight. Niclosamide treatment, however, did not significantly reduce tumor growth. At the end of this experiment, they found that levels of niclosamide were over 15 × greater in xenografts in the NEN group compared to the niclosamide group providing further evidence for the limited bioavailability of the latter.
As an adjunct to this experiment, the authors assessed how niclosamide and NEN compared to sorafenib, one standard of care for HCC, in this PDX model. In the PDX model, the combination of NEN and sorafenib resulted in decreased tumor volume compared to control group (p = 0.013), NEN only (p = 0.030), and sorafenib only (p = 0.024). Treatment with niclosamide and sorafenib, however, did not result in reduced tumor volumes compared to the other groups. These experiments further verified that NEN was the preferred candidate over niclosamide for HCC treatment.
Relating to the original computation-based predictions, the authors experimentally assessed the capacity for these drugs to reverse the HCC gene signature they defined. As a first step, they treated HepG2 cells for 6 h with 10 μm niclosamide, 10 μm NEN, or 10 μm dimethyl sulfoxide (control). As hypothesized, they found that both niclosamide (p = 1.1 × 10−7; Spearman correlation of–0.32) and NEN (p = 9.8 × 10−6; Spearman correlation of –0.26) significantly reduced the 274 HCC gene expression profile. Furthermore, they observed similar gene expression changes for both drugs compared to control (p < 2.2 × 10−16; Spearman correlation of 0.87). As a complement to the in vitro assessment of gene expression changes, the authors additionally assessed effects on gene expression within the PDX model. They found that the differential gene expression profile between NEN-treated animals and controls was significantly anticorrelated with the HCC profile (p < 3.9 × 10−6; Spearman correlation of –0.25). Out of all the genes in the HCC profile, the authors observed that the anticorrelation signal was mostly driven by 20 upregulated and 29 suppressed genes from both the in vitro and in vivo experiments (FDR <0.25).
As a final piece to the puzzle, the authors sought to characterize by which biological mechanism NEN attenuates HCC. They focused their attention to chaperone proteins heat shock protein 90 (HSP90) and cell division cycle 37 (CDC37) that regulate kinases inhibited by NEN. They first confirmed that NEN inhibited the HSP90/CDC37 interaction and then assessed to which protein NEN binds. They found that NEN binds to CDC37 and also enhanced its thermal stability. To support this finding, they observed that CDC37 was overexpressed in 80% of HCC tissues compared to normal livers. To assess whether CDC37 is, in fact, mediating the effects of NEN in treating HCC, they performed a final experiment in which RNA interference was used to knockdown CDC37 in HCC cells treated with NEN. As hypothesized, they found that HCC cells lacking CDC37 expression were less sensitive to NEN, supporting the notion that the antiproliferative effects of NEN in HCC are partly dependent on CDC37. With these exhaustive in vitro and in vivo experiments, the authors successfully provided a molecular context of how their computationally predicted treatment for HCC worked at multiple biological levels.
4. Notes
In this chapter, we have briefly detailed the vast amount of public resources that can enable computational-based drug discovery. The methods used by Li and Greene et al. and Chen and Wei et al. can hopefully serve as a guide on how to leverage big data for drug discovery from prediction to validation. It is important, however, to note the limitations and caveats ofvarious aspects ofthese pipelines and the special considerations that should be accounted for.
4.1. Computational Considerations
As software is continually updated and new versions of tools and packages released, there is a large issue with reproducibility in research, often due to incompatibility of computing environments [57]. This often leads to inconsistent results, even when using the same computational protocols. Beaulieu-Jones and Greene have recently proposed a system that, if adopted, could avoid these potential incompatibility issues [58]. They outline a pipeline that combines Docker, a container technology that is distinct from the native operating system environment, with software that continually reruns the pipeline whenever new updates are released for the data or underlying packages. We generally recommend using open-source programming frameworks and distributable notebooks, such as Jupyter Notebook (http://jupyter.org/) and RMarkDown (http://rmarkdown.rstudio.com/), and following the emerging best practices for reproducible research whenever possible in order to most easily facilitate data sharing and research reproduction. With this said, the most important step toward enabling reproducible research is a willingness to share the data and code in a public repository. GitHub (http://github.com/) and Bit-bucket (http://bitbucket.org/) are two common code repos, and it is often encouraged to deposit large datasets in repositories like Synapse (http://www.synapse.org/).
Another issue plaguing biomedical research is the ever-changing nature of genome builds and identifiers, which can affect delineation of reference alleles, genomic coordinates, and probe annotations, among many others. As an early salutation to this phenomenon, Chen et al. built AILUN, a fully automated online tool that will re-annotate all microarray datasets to the latest annotations, allowing for compatible analyses across differing versions [50].
4.2. Robustness of Disease Signatures
The concept of a disease signature is continually evolving through the development of better methods to quantify health, the refinement of understanding roles of disease pathophysiology, and increase in both the availability and types of biological data collected. The extensive public repositories described above contain a wealth of data pertaining to a variety of diseases across many disease models, tissues, and conditions (i.e., exposures). To fully leverage these data and increase power, it is possible to collect and integrate multiple related datasets, but will require vigilant and methodological quality control for effective and accurate integration in both the phenotypic and genomic space. There are, however, potential critical issues to consider when performing a meta-analysis experiment of gene expression disease signatures, particularly in microarray experiments [59]. In addition to issues arising from experiments having mismatching platforms (described above), the disease characterization that is the basis of each experiment may differ, such as having different inclusion criteria or disease definitions. Population-level differences in racial, demographic, or environmental backgrounds of these studies can have a large effect on gene expression levels that are unrelated to disease and could lead to spurious associations if not properly controlled for [60]. One solution to deal with these and other potential unmodeled factors would be to utilize surrogate variable analysis to overcome gene expression heterogeneity within these studies [61].
One study assessed the robustness of disease signatures for over 8000 microarrays across over 400 experiments in GEO for a broad range of diseases and tissue types [62]. Fortunately, they concluded that the gene expression signatures within diseases are more concordant than tissue expression across diseases, lending further credibility to utilizing such data for meta-analysis studies. While this finding is encouraging, it may not always be true for new datasets or alternative public repositories. Another consideration is the type of statistical analysis performed to complete the meta-analysis. We have illustrated a few in experiments described within Subheading 3, but there are many other options. In fact, researchers have systematically compared eight different meta-analysis methods for combining multiple microarray experiments [63]. They found that these different approaches resulted in substantially different error rates in classifying the disease of interest when using the same datasets. As such, one must be conscientious of studies to be included and the method used in meta-analysis experiments.
4.3. Drug Profile Variation
Many related studies, including the highlighted one by Li and Greene et al., performed a meta-analysis to integrate multiple drug profiles into a consensus signature. Not surprisingly, there are caveats of this process pertaining to varying biological contexts that can affect its validity and utility [64]. Researchers integrated gene expression data for 11,000 drugs from LINCS with their chemical structure and bioactivity data from PubChem to assess correlation between structure and expression [65]. Specifically, they systematically evaluated the effect of various biological conditions, namely, cell line, dose, and treatment duration, on this relationship. They found that compounds that are more structurally similar tend to have similar transcriptomic profiles as well, but it is dependent on cell line. For instance, PC3 and VCAP, both prostate-related cancer cell lines, generally have significantly different patterns of similarity between structure and gene expression. Separately, they also found an interesting relationship between structure and gene expression in the context of dose: drugs with high structure similarity have stronger gene expression concordance at higher than lower doses. Another research group merged profiles of 1302 drugs from CMap across doses and cell lines to create a drug network with the goal ofusing it to predict drug effect and mechanism of action [53]. They noted that creating a consensus signature for a drug that has inconsistent effects on different cell lines could dilute its unique biological effects. That being said, they found even across heterogeneous cell types, a consensus drug signature could still be well classified in their drug network given a sufficiently large collection of data. In any case, further understanding and characterization of relationship between compound and biological context will undoubtedly improve accuracy of computational predictions and thereby bolster rates of successful translation into the clinic.
4.4. Selection of Validation Model
With a candidate drug selected for a disease ofinterest, it is imperative to perform a series of coordinated experiments (e.g., PK/PD/ toxicity studies), in addition to legal considerations (e.g., IP protection) to gauge its eligibility. Further, these decisions may be particularly critical due to time and financial limitations, where setbacks, mistakes, or poor choices could halt future progress. Drug candidates often fail to successfully translate into the clinic due to two main reasons: improper safety and efficacy assessments, both possibly a direct result of poor early target validation and unreliable preclinical models [66].
In terms of preclinical testing, the large number of avenues and models can be overwhelming, especially in light of variable reliability. While the accessibility to massive quantities of biological big data has been transformative in the field of drug discovery, not all experiments are of equal utility. In studying HCC, for example, researchers recently found that half of public HCC cell lines do not resemble actual HCC tumors in terms of gene expression patterns [67]. Interestingly, another group found that rarely used ovarian cancer cell lines actually have higher genetic similarity to ovarian tumors than more commonly used ones [68]. Due to phenomena like these, we recommend performing rigorous examinations like those above and leveraging knowledge from existing evaluations of these data when possible before usage. The nuances of subsequent steps of the validation process are beyond the scope of this chapter, but there are countless resources that delve into specifics regarding proper procedures for each stage [69–72].
4.5. Drug Discovery Using EMR Data
EMR systems contain a great deal of disease- and phenotype-related information that can be leveraged for drug discovery as “real-world data.” The multifaceted data that are collected in EMR facilitate flexibility in devising research questions that may be beyond the scope or focus of the original experiment. As such, unanticipated connections may be formed to biological aspects that would not be collected in traditional prospective study designs. Additionally, the large sample sizes and natural collection of longitudinal, follow-up information relating to patient outcomes from treatment are invaluable advantages over traditional randomized clinical trials [73].
As mentioned, EMR frameworks are primarily designed for infrastructural support and to facilitate billing. Like other research datasets, the raw data is often messy, incomplete, and subject to biases. Therefore, certain aspects may not be as reliable as unbiased collection measures. Despite better refined structuring, ICD codebased disease classification overall is often insufficient to accurately capture disease status [74]. In this notable example, clinicians manually reviewed the charts of 325 patients that had been recorded with the ICD-9 code for chronic kidney disease stage 3 (585.3). They found that 47% of these patients did not have any clinical indicators for the disease. Including other types of data, like medications, in phenotyping criteria often leads to better accuracy. For instance, researchers found that electronic phenotyping algorithms that require at least two ICD-9 diagnoses, prescription of antirheumatic medication, and participation of a rheumatologist resulted in the highest positive predictive value to identify rheumatoid arthritis [75]. In fact, a recent study analyzed ten common diseases (e.g., Parkinson’s disease) in an EMR system and compared the accuracy of classification through ICD codes, medications (i.e., those primarily prescribed for disease treatment), or clinician notes (i.e., if the disease was mentioned in the visit report) [76]. The “true” disease classification was determined by a physician’s manual chart review. It is clear that certain diseases are more often and better classified by certain modalities, such as clinician notes for Atrial Fibrillation or ICD codes for Parkinson’s disease, but combinations of all three lead generally to highest predictive power and lower rates of error. Fortunately, notable efforts by groups such as the electronic medical records and genomics (eMERGE) consortium (https://emerge.mc.vanderbilt.edu/) have led to rigorous, standardized electronic phenotyping algorithms that identify case and control cohorts utilizing multiple dimensions of EMR data to establish inclusion and exclusion criteria. The Phenotype KnowledgeBase (PheKB; https://phekb.org/) is a collaborative effort to collect, validate, and publish such algorithms for transportability and reproducibility for use in any medical system’s EMR With this in mind, researchers must understand these caveats and take much care as when incorporating public expression data.
Strict data access stipulations and possible disparate EMR system frameworks oftentimes make cross-institution replication efforts difficult. To address this issue, there are resources that release public software and analytical tools for healthcare-related research, like i2b2 (https://www.i2b2.org/). OHDSI (https://www.ohdsi.org/) is a community of researchers who collaborate to solve biomedical problems across multiple disciplines, enabling reproducibility of research through a large-scaled, open-source workflow using observational health data [77]. Although EMR-based studies require stringent IRB approval, there is a growing concern for patient privacy and confidentiality, especially as this type of research, and those with access to these data, continues to expand [78]. On the other side of this issue, the stringency of data access makes reproducibility difficult. These types of multi-hospital EMR studies both facilitate cross validation for any findings and isolate any potential role of geographic environment.
Despite these challenges, EMR-based research will continue to evolve to produce even more outstanding insights that may direct drug discovery. Open platforms like the UK Biobank will be essential to allow more researchers to perform this type ofresearch. With nomenclature standardization practices improving and resources growing, integration with developing resources of other biological and environmental modalities (e.g., pollution data) and sensor- based data collection [79] will allow for a multi-scale understanding of findings. The integration of these types of data with state-of-the-art machine learning approaches, such as deep learning, can push predictive power well beyond the current success rates. Hopefully, we will continue to see findings from these works to continue to transform clinical care, leading to more cost-effective and efficient drug development along with better patient outcomes and satisfaction.
Acknowledgments
The research is supported by R21 TR001743, U24 DK116214, and K01 ES028047 (to BC). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Eder J, Sedrani R, Wiesmann C (2014) The discovery of first-in-class drugs: origins and evolution. Nat Rev Drug Discov 13 (8):577–587 [DOI] [PubMed] [Google Scholar]
- 2.Mullard A (2016) Parsing clinical success rates. Nat Rev Drug Discov 15(7):447. [DOI] [PubMed] [Google Scholar]
- 3.Every-Palmer S, Howick J (2014) How evidence-based medicine is failing due to biased trials and selective publication. J Eval Clin Pract 20(6):908–914 [DOI] [PubMed] [Google Scholar]
- 4.Rothwell PM (2006) Factors that can affect the external validity of randomised controlled trials. PLoS Clin Trials 1(1):e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Murthy VH, Krumholz HM, Gross CP (2004) Participation in cancer clinical trials: race-, sex-, and age-based disparities. JAMA 291 (22):2720–2726 [DOI] [PubMed] [Google Scholar]
- 6.Rothwell PM (2005) External validity of randomised controlled trials: “to whom do the results of this trial apply?”. Lancet 365 (9453):82–93 [DOI] [PubMed] [Google Scholar]
- 7.Hodos RA, Kidd BA, Shameer K, Readhead BP, Dudley JT (2016) In silico methods for drug repurposing and pharmacology. Wiley Interdiscip Rev Syst Biol Med 8(3):186–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Paik H, Chen B, Sirota M, Hadley D, Butte AJ (2016) Integrating clinical phenotype and gene expression data to prioritize novel drug uses. CPT Pharmacometrics Syst Pharmacol 5 (11):599–607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL (2010) How to improve R&D productivity: the pharmaceutical industry’s grand challenge. Nat Rev Drug Discov 9(3):203–214 [DOI] [PubMed] [Google Scholar]
- 10.Caskey CT (2007) The drug development crisis: efficiency and safety. Annu Rev Med 58:1–16 [DOI] [PubMed] [Google Scholar]
- 11.Nosengo N (2016) Can you teach old drugs new tricks? Nature 534(7607):314–316 [DOI] [PubMed] [Google Scholar]
- 12.Scannell JW, Blanckley A, Boldon H, Warrington B (2012) Diagnosing the decline in pharmaceutical R&D efficiency. Nat Rev Drug Discov 11(3):191–200 [DOI] [PubMed] [Google Scholar]
- 13.Ashburn TT, Thor KB (2004) Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov 3 (8):673–683 [DOI] [PubMed] [Google Scholar]
- 14.Jahchan NS, Dudley JT, Mazur PK, Flores N, Yang D, Palmerton A, Zmoos AF, Vaka D, Tran KQ, Zhou M et al. (2013)A drug repositioning approach identifies tricyclic antidepressants as inhibitors of small cell lung cancer and other neuroendocrine tumors. Cancer Discov 3(12):1364–1377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pessetto ZY, Chen B, Alturkmani H, Hyter S, Flynn CA, Baltezor M, Ma Y, Rosenthal HG, Neville KA, Weir SJ et al. (2017) In silico and in vitro drug screening identifies new therapeutic approaches for Ewing sarcoma. Oncotarget 8(3):4079–4095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dudley JT, Sirota M, Shenoy M, Pai RK, Roedder S, Chiang AP, Morgan AA, Sarwal MM, Pasricha PJ, Butte AJ (2011) Computational repositioning of the anticonvulsant topiramate for inflammatory bowel disease. Sci Transl Med 3(96):96ra76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sirota M, Dudley JT, Kim J, Chiang AP, Morgan AA, Sweet-Cordero A, Sage J, Butte AJ (2011) Discovery and preclinical validation of drug indications using compendia of public gene expression data. Sci Transl Med 3 (96):96ra77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stephens T, Brynner R (2009) Dark remedy: the impact of thalidomide and its revival as a vital medicine. Basic Books [Google Scholar]
- 19.Attal M, Harousseau JL, Leyvraz S, Doyen C, Hulin C, Benboubker L, Yakoub Agha I, Bour- his JH, Garderet L, Pegourie B et al. (2006) Maintenance therapy with thalidomide improves survival in patients with multiple myeloma. Blood 108(10):3289–3294 [DOI] [PubMed] [Google Scholar]
- 20.From nightmare drug to celgene blockbuster, thalidomide is back bloomberg. https:/www.bloomberg.com/news/articles/2016-08-22/from-nightmare-drug-to-celgene-blockbuster-thalidomide-is-back
- 21.R Core Team (2014) R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: In. 2014 [Google Scholar]
- 22.Van Rossum G, Drake FL: Python language reference manual: network theory; 2003 [Google Scholar]
- 23.Jones E, Oliphant T, Peterson P (2014) SciPy: open source scientific tools for Python [Google Scholar]
- 24.Chen B, Wang H, Ding Y, Wild D (2014) Semantic breakthrough in drug discovery. Synthesis Lectures on the Semantic Web: Theory and Technology 4(2):1–142 [Google Scholar]
- 25.Bodenreider O (2004) The unified medical language system (UMLS): integrating biomedical terminology. Nucleic Acids Res 32(Data-base issue):D267–D270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, Ma W, Moore R, Ganesan V, Nelson S (2005) RxNorm: prescription for electronic drug information exchange. IT professional 7 (5):17–23 [Google Scholar]
- 27.Kuhn M, Letunic I, Jensen LJ, Bork P (2016) The SIDER database of drugs and side effects. Nucleic Acids Res 44(D1):D1075–D1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tatonetti NP, Ye PP, Daneshjou R, Altman RB (2012) Data-driven prediction of drug effects and interactions. Sci Transl Med 4 (125):125ra131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P, Chang Z, Woolsey J (2006) DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res 34(Database issue): D668–D672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shameer K, Glicksberg BS, Hodos R, Johnson KW, Badgeley MA, Readhead B, Tomlinson MS, O’Connor T, Miotto R, Kidd BA et al. (2017) Systematic analyses of drugs and disease indications in RepurposeDB reveal pharmacological, biological and epidemiological factors influencing drug repositioning. Brief Bioinform [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geifman N, Bollyky J, Bhattacharya S, Butte AJ (2015) Opening clinical trial data: are the voluntary data-sharing portals enough? BMC Med 13:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Greene CS, Garmire LX, Gilbert JA, Ritchie MD, Hunter LE (2017) Celebrating parasites. Nat Genet 49(4):483–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yao L, Zhang Y, Li Y, Sanseau P, Agarwal P (2011) Electronic health records: implications for drug discovery. Drug Discov Today 16 (13–14):594–599 [DOI] [PubMed] [Google Scholar]
- 34.Wang G, Jung K, Winnenburg R, Shah NH (2015) A method for systematic discovery of adverse drug events from clinical notes. J Am Med Inform Assoc 22(6):1196–1204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Crosslin DR, Robertson PD, Carrell DS, Gordon AS, Hanna DS, Burt A, Fullerton SM, Scrol A, Ralston J, Leppig K et al. (2015) Prospective participant selection and ranking to maximize actionable pharmacogenetic variants and discovery in the eMERGE network. Genome Med 7(1):67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu H, Aldrich MC, Chen Q, Liu H, Peterson NB, Dai Q, Levy M, Shah A, Han X, Ruan X et al. (2015) Validating drug repurposing signals using electronic health records: a case study of metformin associated with reduced cancer mortality. J Am Med Inform Assoc 22 (1):179–191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kirkendall ES, Kouril M, Minich T, Spooner SA (2014) Analysis of electronic medication orders with large overdoses: opportunities for mitigating dosing errors. Appl Clin Inform 5 (1):25–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ramirez AH, Shi Y, Schildcrout JS, Delaney JT, Xu H, Oetjens MT, Zuvich RL, Basford MA, Bowton E, Jiang M et al. (2012) Predicting warfarin dosage in European-Americans and African-Americans using DNA samples linked to an electronic health record. Pharma- cogenomics 13(4):407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dewey FE, Murray MF, Overton JD, Habegger L, Leader JB, Fetterolf SN, O’Dushlaine C, Van Hout CV, Staples J, Gonzaga-Jauregui C et al. (2016) Distribution and clinical impact of functional variants in 50,726 whole-exome sequences from the DiscovEHR study. Science 354(6319) [DOI] [PubMed] [Google Scholar]
- 40.Yuille M, Dixon K, Platt A, Pullum S, Lewis D, Hall A, Ollier W (2010) The UKDNA banking network: a “fair access” biobank. Cell Tissue Bank 11(3):241–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wain LV, Shrine N, Artigas MS, Erzurumluoglu AM, Noyvert B, Bossini-Castillo L, Obeidat M, Henry AP, Portelli MA, Hall RJ et al. (2017) Genome-wide association analyses for lung function and chronic obstructive pulmonary disease identify new loci and potential druggable targets. Nat Genet 49(3):416–425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30(1):207–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kolesnikov N, Hastings E, Keays M, Melnichuk O, Tang YA, Williams E, Dylag M, Kurbatova N, Brandizi M, Burdett T et al. (2015) ArrayExpress update--simplifying data submissions. Nucleic Acids Res 43(Database issue):D1113–D1116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wickham H (2016) ggplot2: elegant graphics for data analysis, 2nd edn Springer [Google Scholar]
- 45.Hunter JD (2007) Matplotlib: a 2D graphics environment. Comput Sci Eng 9(3):90–95 [Google Scholar]
- 46.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T (2003) Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13(11):2498–2504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bastian M, Heymann S, Jacomy M (2009) Gephi: an open source software for exploring and manipulating networks. Icwsm 8:361–362 [Google Scholar]
- 48.Li L, Greene I, Readhead B, Menon MC, Kidd BA, Uzilov AV, Wei C, Philippe N, Schroppel B, He JC et al. (2017) Novel therapeutics identification for fibrosis in renal allograft using integrative informatics approach. Sci Rep 7:39487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen B, Wei W, Ma L, Yang B, Gill RM, Chua MS, Butte AJ, So S (2017) Computational discovery of niclosamide ethanolamine, a repurposed drug candidate that reduces growth of hepatocellular carcinoma cells in vitro and in mice by inhibiting cell division cycle 37 signaling. Gastroenterology 152 (8):2022–2036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen R, Li L, Butte AJ (2007) AILUN: reannotating gene expression data automatically. Nat Methods 4(11):879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tusher VG, Tibshirani R, Chu G (2001) Significance analysis of microarrays applied to the ionizing radiation response. Proc Natl Acad Sci USA 98(9):5116–5121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11(10):R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Iorio F, Bosotti R, Scacheri E, Belcastro V, Mithbaokar P, Ferriero R, Murino L, Tagliaferri R, Brunetti-Pierri N, Isacchi A et al. (2010)Discovery of drug mode of action and drug repositioning from transcriptional responses. Proc Natl Acad Sci U S A 107 (33):14621–14626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lamb J, Crawford ED, Peck D, Modell JW, Blat IC, Wrobel MJ, Lerner J, Brunet JP, Subramanian A, Ross KN et al. (2006) The connectivity map: using gene-expression signatures to connect small molecules, genes, and disease. Science 313(5795):1929–1935 [DOI] [PubMed] [Google Scholar]
- 55.Kidd BA, Wroblewska A, Boland MR, Agudo J, Merad M, Tatonetti NP, Brown BD, Dudley JT (2016) Mapping the effects of drugs on the immune system. Nat Biotechnol 34(1):47–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanzelmann S, Castelo R, Guinney J (2013) GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dudley JT, Butte AJ (2010) In silico research in the era of cloud computing. Nat Biotechnol 28(11):1181–1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Beaulieu-Jones BK, Greene CS (2017) Reproducibility of computational workflows is automated using continuous analysis. Nat Biotechnol 35(4):342–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramasamy A, Mondry A, Holmes CC, Altman DG (2008) Key issues in conducting a meta-analysis of gene expression microarray datasets. PLoS Med 5(9):e184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Klebanov L, Yakovlev A (2006) Treating expression levels of different genes as a sample in microarray data analysis: is it worth a risk? Stat Appl Genet Molec Biol 5(1):1–9 [DOI] [PubMed] [Google Scholar]
- 61.Leek JT, Storey JD (2007) Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet 3(9):1724–1735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dudley JT, Tibshirani R, Deshpande T, Butte AJ (2009) Disease signatures are robust across tissues and experiments. Mol Syst Biol 5:307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Campain A, Yang YH (2010) Comparison study of microarray meta-analysis methods. BMC Bioinformatics 11:408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Chen B, Ma L, Paik H, Sirota M, Wei W, Chua MS, So S, Butte AJ (2017) Reversal of cancer gene expression correlates with drug efficacy and reveals therapeutic targets. Nat Commun (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen B, Greenside P, Paik H, Sirota M, Hadley D, Butte AJ (2015) Relating chemical structure to cellular response: an integrative analysis of gene expression, bioactivity, and structural data across 11,000 compounds. CPT Pharmacometrics Syst Pharmacol 4 (10):576–584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smith C (2003) Drug target validation: hitting the target. Nature 422(6929). 341, 343, 345 passim [DOI] [PubMed] [Google Scholar]
- 67.Chen B, Sirota M, Fan-Minogue H, Hadley D, Butte AJ (2015) Relating hepatocellular carcinoma tumor samples and cell lines using gene expression data in translational research. BMC Med Genet 8(Suppl 2):S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Domcke S, Sinha R, Levine DA, Sander C, Schultz N (2013) Evaluating cell lines as tumour models by comparison of genomic profiles. Nat Commun 4:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hefti FF (2008) Requirements for a lead compound to become a clinical candidate. BMC Neurosci 9(Suppl 3):S7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Empfield JR, Leeson PD (2010) Lessons learned from candidate drug attrition. IDrugs 13(12):869–873 [PubMed] [Google Scholar]
- 71.Hughes JP, Rees S, Kalindjian SB, Philpott KL (2011) Principles of early drug discovery. Br J Pharmacol 162(6):1239–1249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Meanwell NA (2011) Improving drug candidates by design: a focus on physicochemical properties as a means of improving compound disposition and safety. Chem Res Toxicol 24 (9):1420–1456 [DOI] [PubMed] [Google Scholar]
- 73.Bate A, Juniper J, Lawton AM, Thwaites RM (2016) Designing and incorporating a real world data approach to international drug development and use: what the UK offers. Drug Discov Today 21(3):400–405 [DOI] [PubMed] [Google Scholar]
- 74.Cipparone CW, Withiam-Leitch M, Kimminau KS, Fox CH, Singh R, Kahn L (2015) Inaccuracy of ICD-9 codes for chronic kidney disease: a study from two practice-based research networks (PBRNs). J Am Board Fam Med 28 (5):678–682 [DOI] [PubMed] [Google Scholar]
- 75.Chung CP, Rohan P, Krishnaswami S, McPheeters ML (2013) A systematic review of validated methods for identifying patients with rheumatoid arthritis using administrative or claims data. Vaccine 31(Suppl 10):K41–K61 [DOI] [PubMed] [Google Scholar]
- 76.Wei WQ, Teixeira PL, Mo H, Cronin RM, Warner JL, Denny JC (2016) Combining billing codes, clinical notes, and medications from electronic health records provides superior phenotyping performance. J Am Med Inform Assoc 23(e1):e20–e27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yoon D, Ahn EK, Park MY, Cho SY, Ryan P, Schuemie MJ, Shin D, Park H, Park RW (2016) Conversion and data quality assessment of electronic health record data at a Korean tertiary teaching hospital to a common data model for distributed network research. Healthc Inform Res 22(1):54–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Barrows RC Jr, Clayton PD (1996) Privacy, confidentiality, and electronic medical records. J Am Med Inform Assoc 3(2):139–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Shameer K, Badgeley MA, Miotto R, Glicksberg BS, Morgan JW, Dudley JT (2017) Translational bioinformatics in the era of real-time biomedical, health care and wellness data streams. Brief Bioinform 18(1):105–124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Davis S, Meltzer PS (2007) GEOquery: a bridge between the gene expression omnibus (GEO) and BioConductor. Bioinformatics 23 (14):1846–1847 [DOI] [PubMed] [Google Scholar]
- 81.Robinson MD, McCarthy DJ, Smyth GK (2010) edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26 (1):139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hong F, Breitling R, McEntee CW, Wittner BS, Nemhauser JL, Chory J (2006) RankProd: a bioconductor package for detecting differentially expressed genes in meta-analysis. Bioin formatics 22(22):2825–2827 [DOI] [PubMed] [Google Scholar]