

Abstract
Meiotic recombination comprises crossovers and noncrossovers. Recombination, crossover in particular, shuffles mutations and impacts both the level of genetic polymorphism and the speed of adaptation. In many species, the recombination rate varies across the genome with hot and cold spots. The hotspot paradox hypothesis asserts that recombination hotspots are evolutionarily unstable due to self-destruction. However, the genomic landscape of double-strand breaks (DSBs), which initiate recombination, is evolutionarily conserved among divergent yeast species, casting doubt on the hotspot paradox hypothesis. Nonetheless, because only a subset of DSBs are associated with crossovers, the evolutionary conservation of the crossover landscape could differ from that of DSBs. Here, we investigate this possibility by generating a high-resolution recombination map of the budding yeast Saccharomyces paradoxus through whole-genome sequencing of 50 meiotic tetrads and by comparing this recombination map with that of S. cerevisiae. We observe a 40% lower recombination rate in S. paradoxus than in S. cerevisiae. Compared with the DSB landscape, the crossover landscape is even more conserved. Further analyses indicate that the elevated conservation of the crossover landscape is explained by a near-subtelomeric crossover preference in both yeasts, which we find to be attributable at least in part to crossover interference. We conclude that the yeast crossover landscape is highly conserved and that the evolutionary conservation of this landscape can differ from that of the DSB landscape.
Keywords: crossover, double-strand break, hotspot paradox, recombination rate, Saccharomyces cerevisiae, Saccharomyces paradoxus
Introduction
Meiotic recombination is initiated by a double-strand break (DSB) in a DNA molecule, created by a suite of proteins including the transesterase Spo11 that cuts DNA (Keeney et al. 1997). DSBs are followed by single strand invasions, leading to either DSB repair (DSBR) or synthesis-dependent strand annealing (SDSA) (Baudat et al. 2013). DSBR predominately leads to crossovers (COs), whereas SDSA leads to noncrossovers (NCOs) (Allers and Lichten 2001) (fig. 1). COs involve a reciprocal exchange of genetic material between two homologous chromosomes accompanied by a tract subject to gene conversion, which is a nonreciprocal change of alleles that typically leads to a 3:1 segregation. NCOs are not associated with a reciprocal genetic exchange but involve a gene conversion. Although recombination formally includes both COs and NCOs, in practice it frequently refers to COs only, because NCOs seldom have phenotypic consequences.
Fig. 1.
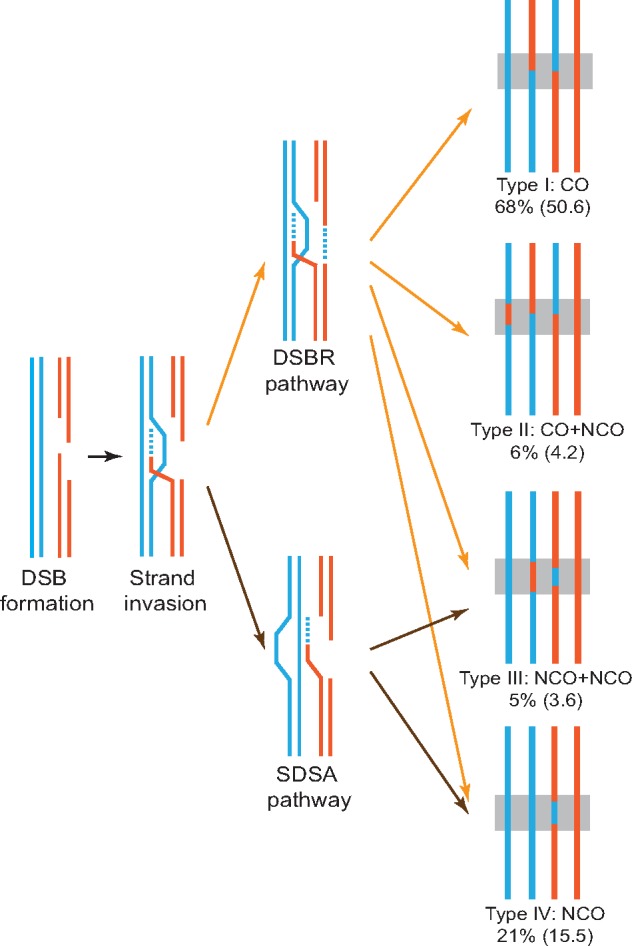
Schematic illustration of recombination processes. Recombination is initiated by a DSB, followed by single strand invasion, leading to either DSBR or SDSA. In this study, four different types of recombined tetrads are observed: a single CO event (Type I); a CO event and an NCO event at the same locus (Type II), which could be caused by two CO chromatids invading a third chromatid during recombination or two DSBs occurring in two different chromatids at the same locus; two NCO events at the same locus (Type III), which could be explained by resolution of a double Holiday junction in an NCO fashion or breakage of two chromatids at the same locus; and a single NCO event (Type IV). Frequencies of events are shown along with numbers of events per meiosis in the parentheses. The figure design follows figure 2 in Liu et al. (2018).
Recombination, CO in particular, is of fundamental importance to evolution for at least three reasons. First, recombination shuffles genetic variations, alleviating clonal interference and allowing natural selection to more efficiently sort beneficial from deleterious mutations (McDonald et al. 2016). Second, the rate of recombination is a key determinant of the level of intraspecific genetic polymorphism (Coop and Przeworski 2007). For instance, studies in flies and humans found that recombination rate explains more than 50% of the variation in nucleotide diversity across the genome (Nachman 2002). This is because recombination can be mutagenic (Yang et al. 2015; Liu et al. 2017) and because it reduces the size of the genomic region subject to the purge of polymorphism by positive or negative selection at linked sites (Charlesworth and Charlesworth 2018). Third, recombination may shape genomic features such as the nucleotide composition and codon usage through a process known as GC-biased gene conversion (Duret and Galtier 2009).
In many species, recombination events are not evenly distributed across the genome; instead, they are often concentrated in narrow ranges known as recombination hotspots (Kauppi et al. 2004). Previous theoretical work led to a prevailing hypothesis on the evolution of recombination hotspots known as the “hotspot paradox” (Boulton et al. 1997; Calabrese 2007; Coop and Myers 2007). In this hypothesis, the occurrence of DSB is mainly determined by a specific nucleotide sequence. During recombination, the active allele with DSB is often repaired and replaced with the inactive unbroken allele. Thus, recombination hotspots should quickly go extinct due to self-destruction. Paradoxically, hotspots still exist despite the predicted rapid extinction. The hotspot paradox hypothesis coincides with the discovery of PRDM9 and the rapid change of recombination hotspots in apes and mice (Baudat et al. 2010; Myers et al. 2010; Parvanov et al. 2010). PRDM9 is a histone methyltransferase that targets specific sequence motifs to direct DSB formation. PRDM9 evolves quickly under positive selection, resulting in a rapid turnover of its target sequence and hence recombination hotspots (Oliver et al. 2009; Hinch et al. 2011; Auton et al. 2012; Baudat et al. 2013). Although the selective agent behind PRDM9’s rapid evolution remains unclear despite the presence of several candidates (Coop and Myers 2007; Myers et al. 2010; Ubeda and Wilkins 2011; Davies et al. 2016; Smagulova et al. 2016), the above findings support the hotspot paradox hypothesis and explain why recombination hotspots still exist in the face of self-destruction.
However, PRDM9 is absent in plants, fungi, and many animals, and its role in recombination has not been found outside placental mammals (Baker et al. 2017). Whether recombination hotspots are evolutionarily conserved or short lived in the absence of PRDM9 is thus of great interest. PRDM9 has been lost in birds, where recombination hotspots inferred from population genomic data (and thus largely reflecting COs) appear conserved between zebra finch and long-tailed finch, two species with ∼1.5% genomic sequence divergence (Singhal et al. 2015). Similarly, an early yeast population genomic study of one chromosome showed that recombination hotspots are conserved between sister species Saccharomyces cerevisiae (Scer) and S. paradoxus (Spar) (Tsai et al. 2010), which diverged from each other ∼5 Ma and exhibit 17% genomic sequence divergence (Liti et al. 2009). However, population genomic inferences of recombination rates (i.e., CO rates) are subject to error due to assumptions about effective population size, demography, and natural selection that may not be realistic as well as a puzzling phenomenon that yeast cells can exit the meiosis program and return to mitotic growth after prophase I (Slatkin 2008; Laureau et al. 2016). More recently, based on genome-wide Spo11 footprints, Lam and Keeney examined the DSBs in yeast species that diverged ∼15 Ma and found the DSB landscape evolutionarily conserved (Lam and Keeney 2015). Nevertheless, a conserved DSB landscape does not necessarily mean a conserved CO landscape, because DSBs created by Spo11 are resolved as COs or NCOs in a fashion that is not entirely random at least in the nematode worm Caenorhabditis elegans and the mustard plant Arabidopsis thaliana (Rosu et al. 2011; Libuda et al. 2013; Jahns et al. 2014). Hence, it is possible that CO and NCO landscapes show different conservation levels from that of DSBs.
In this study, we attempt to directly quantify the evolutionary conservation of the yeast recombination landscape at CO and NCO levels. We produce a recombination map of Spar at the resolution of 100 nucleotides via genomic sequencing of parents and offspring and show that the CO landscape is more conserved than the DSB landscape. We explain the elevated CO landscape conservation by a near-subtelomeric CO preference in both yeasts, which is attributable at least in part to CO interference.
Results
Genome-Wide High-Resolution Mapping of Recombination Events in Spar
The resolution of a recombination map is determined by the mean distance between adjacent markers. To produce a recombination map at the resolution of 100 nucleotides in Spar, we crossed two divergent strains of the species, N17 and N44, whose genomes differ at 1.19% of nucleotide positions. We then performed high-coverage whole-genome resequencing of the two parental strains as well as 200 spores from 50 meiotic tetrads of the N17/N44 hybrid strain. The average sequencing depth is 12× and the average genome coverage is 99% (supplementary table S1, Supplementary Material online). Single-nucleotide polymorphisms (SNPs) in the hybrid strain were used as genetic markers to identify CO and NCO events (see Materials and Methods). In total, 140,294 SNPs were used as markers, with a mean marker density of 11.9 per kb, more than twice that previously used in similar studies of Scer (supplementary fig. S1, Supplementary Material online) (Mancera et al. 2008; Martini et al. 2011; Liu et al. 2018).
Based on these markers, we observed 54.8 CO and 26.9 NCO events per meiosis in Spar (table 1). After accounting for unobserved NCO events that fall between consecutive markers using a simulation method (Wijnker et al. 2013; Liu et al. 2018) (supplementary fig. S2, Supplementary Material online; see Materials and Methods), we inferred that the corrected NCO number is 27.3 (table 1). While most recombination events are classical CO or NCO (Type I and Type IV in fig. 1), two kinds of nonclassical events are also present. The first kind is where a CO and an NCO take place at the same locus (Type II in fig. 1), indicating that the two CO chromatids invade a third chromatid during recombination (Oh et al. 2007, 2008) or two DSBs occur in two different chromatids at the same locus. The second kind is where two NCOs take place at the same locus (Type III in fig. 1), which could be explained by resolution of a double Holiday junction in an NCO fashion (Oh et al. 2007, 2008) or breakage of two chromatids at the same locus. Of all CO events, 94% are associated with gene conversions (CO-GeneConv); this percentage exceeds all previously reported values from species including Scer, Neurospora crassa, Chlamydomonas reinhardtii, and A. thaliana (Mancera et al. 2008; Wijnker et al. 2013; Liu et al. 2018), probably in part due to the high SNP density of the Spar hybrid used here that yields a high detectability of gene conversion. CO-GeneConv events convert ∼1% of all markers per meiosis, and the median length of CO-GeneConv is 2,425 nucleotides. By contrast, NCO events convert ∼0.34% markers per meiosis, and the median conversion length of NCO is 1,320 nucleotides, much shorter than CO-GeneConv (P < 0.001, Wilcoxon rank-sum test; supplementary fig. S3, Supplementary Material online). We found a significant GC bias associated with CO-GeneConv, but no significant GC bias in NCO (table 2). When CO-GeneConv and NCO are combined, a weak but significant GC bias is detected (table 2).
Table 1.
Comparison of per Meiosis Rates of CO and NCO Events between Spar and Scer.
| Organism | Parental Strains | CO |
NCO |
||||
|---|---|---|---|---|---|---|---|
| Events | CO-GeneConv |
Observed Events | Corrected Events | % Markers Converteda | |||
| Events | % Markers Converteda | ||||||
| Spar | N44 × N17 | 54.8 | 51.6 | 1.0 | 26.9 | 27.3 | 0.34 |
| Scerb | YJM789 × S96 | 91.5 | 63.9 | 1.1 | 46.6 | 53.9 | 0.86 |
| YJM789 × YPS128 | 76.5 | 63.2 | 0.98 | 46.4 | 49.9 | 0.74 | |
| SK1 × S288C | 73 | — | — | 27 | — | — | |
Number of markers converted per meiosis by the mechanism indicated, relative to the total number of markers.
Data from Mancera et al. (2008), Liu et al. (2018), and Martini et al. (2011), respectively.
Table 2.
Nucleotide Composition Changes in Spar Gene Conversion Events.
| CO-GeneConv | NCO | Total | |
|---|---|---|---|
| AT → GC | 30,622 (50.56%) | 10,318 (49.76%) | 40,940 (50.36%) |
| GC → AT | 29,943 (49.44%) | 10,416 (50.24%) | 40,359 (49.64%) |
| P value | 0.00587 | 0.5005 | 0.0419 |
Note.—Numbers of nucleotide conversions (and percentages) in each direction are shown, along with two-tailed binomial P values.
Differences in Meiotic Recombination Rates between Spar and Scer
We compared our recombination rate estimate for Spar with that previously obtained in the hybrid of Scer strains YJM789 and S96 and the hybrid of Scer strains YJM789 and YPS128 (Mancera et al. 2008; Liu et al. 2018). The CO rate showed a 34.8% reduction and the corrected NCO rate showed a 47.4% reduction in Spar when compared with the corresponding rates in Scer (both P < 0.001, t-test; table 1). After dividing chromosomes to centromeres, subtelomeres, chromosome ends, and interstitial regions as in a previous study (Yue et al. 2017), we found that CO and NCO rates are suppressed in centromeres, subtelomeres, and chromosome ends in both species and that the rate differences between the two species are concentrated in interstitial regions (supplementary fig. S4, Supplementary Material online). In addition, detailed characteristics of gene conversions differ. The median tract length of CO-GeneConv is greater in Spar than in Scer (2,425 vs. 1,841 nucleotides; P < 0.001; Wilcoxon rank-sum test), whereas the median tract length of NCO is greater in Scer than in Spar (1,681 vs. 1,320 nucleotides; P < 0.001). The latter difference is at least in part due to a higher marker density in the Spar strains than in the Scer strains used. In both species, the numbers of CO and NCO events on a chromosome are respectively positively correlated with the chromosome length (supplementary fig. S5A–D, Supplementary Material online). In Scer, the CO rate (no. of CO events per Mb) and NCO rate (no. of NCO events per Mb) in a chromosome are respectively negatively correlated with the chromosome length (supplementary fig. S5E and F, Supplementary Material online), a pattern believed to have resulted from a network of intersecting negative regulatory circuits that control DSB formation (Thacker et al. 2014). In Spar, however, this negative correlation is observed between the CO rate and chromosome length (r = −0.67, P = 0.0046; supplementary fig. S5G, Supplementary Material online) but not between the NCO rate and chromosome length (r = 0.12, P = 0.65) (supplementary fig. S5H, Supplementary Material online), despite that the DSB rate is negatively correlated with chromosome length (Lam and Keeney 2015). Thus, patterns of recombination are not solely determined by those of DSBs.
The CO Landscape Is More Conserved Than the DSB Landscape
To compare the recombination landscape between Spar and Scer, we first examined the numbers of CO and NCO events in the two species in 20-kb syntenic blocks across the genome excluding subtelomeric and centromeric regions, as in the previous analysis of DSBs (Lam and Keeney 2015). A clear covariation of CO rates (fig. 2A) between the two species exists, so does that of NCO rates (fig. 2B). These patterns are similar to what was previously reported for DSBs (Lam and Keeney 2015) reanalyzed here (fig. 2C).
Fig. 2.

Comparative distributions of (A) CO rate, (B) NCO rate, and (C) DSB rate across all 16 chromosomes (I–XVI) in Scer (red) and Spar (blue). CO and NCO rates are shown by numbers of events in a 20-kb window per meiosis, whereas the DSB rate is the fraction of all DSB reads in a 20-kb window. X-axis shows genomic positions on each chromosome (×100 kb).
To quantify the evolutionary conservations of the CO and NCO landscapes, we plotted the rate of CO (or NCO) in each syntenic block in Spar against that in Scer. Significant correlations are observed for both CO and NCO landscapes between the two species, but the correlation is substantially stronger for CO (linear correlation r = 0.52, rank correlation ρ = 0.48; fig. 3A) than NCO (r = 0.16, ρ = 0.17; fig. 3B) landscapes. Comparing interspecific correlations in CO, NCO, and DSB landscapes requires considering equal numbers of events in the three landscapes. Because there are more DSB than CO and NCO events in the data analyzed, we down-sampled DSBs reported in the previous study (Lam and Keeney 2015) to the same number as CO (or NCO) events in each species and then computed the linear correlation using the down-sampled DSBs. In only 14,052 of 1 million down-samplings do DSBs show higher between-species correlations than CO rates. Thus, the interspecific CO correlation is significantly higher than the DSB correlation (two-tailed P = 0.028; fig. 3C). Using the same approach, we found the interspecific NCO correlation to be significantly lower than the DSB correlation (two-tailed P = 0.00034; fig. 3D). Similar results were obtained when the rank instead of linear correlation was examined (two-tailed P = 0.0067 for CO and two-tailed P = 0.010 for NCO). The NCO result may be in part due to unobserved NCOs, including NCOs that fall between consecutive genetic markers and NCOs fixed by sister chromatids (i.e., genetically silent NCOs). The fraction of unobserved NCOs can be roughly estimated by the fraction of COs that are not associated with gene conversion. This fraction is only 5.8% in the present study but is 17.4–30.7% in the previous Scer studies (Mancera et al. 2008; Liu et al. 2018). Similar results regarding the conservation of CO and NCO landscapes were obtained from analyses of 5-, 10-, and 40-kb syntenic blocks. Because DSBs are resolved as either COs or NCOs, the evolutionary conservation of the combined landscape of COs and NCOs should not differ significantly from that of the DSB landscape. This is indeed the case (two-tailed P = 0.72).
Fig. 3.
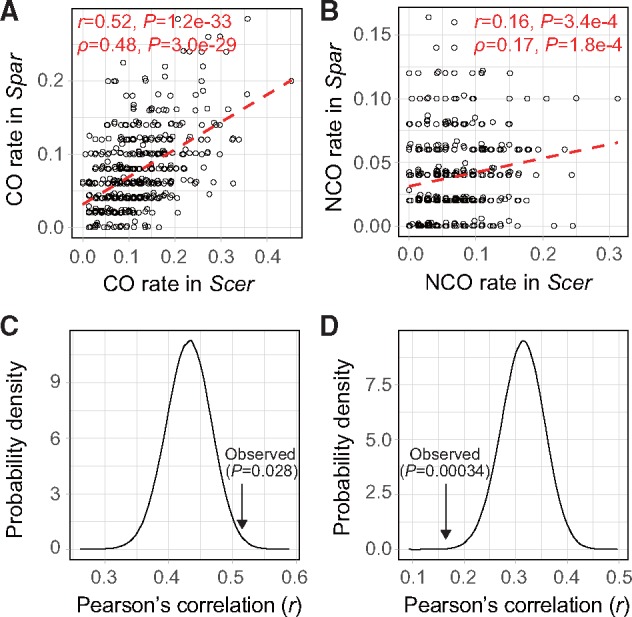
Between-species correlations in CO and NCO rates, compared with the between-species correlation in DSB rates. (A) Correlation in CO rates between Scer and Spar. (B) Correlation in NCO rates between Scer and Spar. In (A) and (B), each dot represents one 20 kb window and the red dashed line shows the linear regression. Pearson’s correlation (r) and associated P value are indicated, followed by Spearman’s correlation (ρ) and associated P value. (C) Probability density of the distribution of Pearson’s correlation in DSB rates between Scer and Spar, based on 1 million randomly down-sampled data sets with the size of the CO data. The arrow indicates the observed between-species correlation in CO rates, and the P value shows the two-tailed probability under the null hypothesis that the DSB and CO landscapes have equal conservations. Use of Spearman’s correlation yields similar results. (D) Probability density of the distribution of Pearson’s correlation in DSB rates between Scer and Spar, based on 1 million randomly down-sampled data sets with the size of the NCO data. The arrow indicates the observed between-species correlation in NCO rates, and the P value shows the two-tailed probability under the null hypothesis that the DSB and NCO landscapes have equal conservations. Use of Spearman’s correlation yields similar results.
A previous study revealed a variation in the ratio of the CO rate to NCO rate across the Scer genome (Mancera et al. 2008). Interestingly, we found no significant correlation in this ratio between Spar and Scer (r = −0.020, P = 0.67), indicating that the genomic variation of the CO/NCO rate ratio is not evolutionarily conserved.
Impacts of Heterozygosity on CO and NCO Landscapes
Because the yeast DSB landscapes were measured in homozygous strains whereas the CO and NCO landscapes were measured in heterozygous strains and because the heterozygosity of a strain impacts its overall recombination rate (Hunter et al. 1996; Greig et al. 2003), it is important to ask whether it is meaningful to compare the conservation level between such DSB landscapes and CO (or NCO) landscapes. To this end, we examined the impact of heterozygosity on CO and NCO rates by correlating local heterozygosity with CO (or NCO) rate at various scales (5, 10, 20, and 40 kb window sizes) (supplementary table S2, Supplementary Material online). In the analysis of Scer, we examined both the YJM789 × S96 hybrid and the YJM789 × YPS128 hybrid. Because the latter data (from 15 tetrads) are much smaller than the former (60 tetrads), the following inference is based on the former although the results from the two data sets are consistent with each other (supplementary table S2, Supplementary Material online).
We found a significant positive correlation between the CO rate and heterozygosity in Scer for 5, 10, and 20 kb window sizes but there was no significant correlation in Spar at almost any scale (supplementary table S2, Supplementary Material online). If anything, a negative correlation was observed in Spar for the 10 kb window size (P = 0.039), but the correlation is no longer significant if one corrects the P value by considering that four tests are conducted for the four window sizes. Note that heterozygosity does not affect the detectability of COs. Given that heterozygosity impacts the CO rate differently in the two yeasts, the similarity in the CO landscape between homozygotes of the two yeasts would have been even greater than what is currently observed from the heterozygotes of the two yeasts. In other words, our conclusion that the CO landscape is more conserved than the DSB landscape cannot be an artifact of using heterozygotes in CO mapping but homozygotes in DSB mapping.
Similarly, we found a significant positive correlation between the local NCO rate and heterozygosity in Scer for all window sizes but no significant correlation in Spar at any scale (supplementary table S2, Supplementary Material online). Given the different impacts of heterozygosity on NCO rates in the two yeasts, using heterozygotes in NCO mapping but homozygotes in DSB mapping could make the NCO landscape look less conserved than the DSB landscape.
The Elevated CO Landscape Conservation Is Explained by a Near-Subtelomeric CO Preference in Both Yeasts
Because DSBs represent initiations of CO and NCO events, the different levels of evolutionary conservation of the CO, NCO, and DSB landscapes suggest the existence of nonrandom forces in determining CO and NCO events upon DSB formation. Specifically, a more conserved landscape of COs than DSBs is explainable if COs preferentially occur at the same subset of DSBs in the two species. Similarly, a less conserved landscape of NCOs than DSBs is explainable if different subsets of DSBs are favored by NCOs in the two species. We thus examined four factors known to affect the CO and NCO distributions: distance to subtelomere, distance to centromere, nucleotide GC content, and gene density (Barton et al. 2008; Liu et al. 2015). To this end, we first controlled the impact of the DSB distribution by computing the ratio of the number of CO events to that of DSBs in each 20-kb synthetic block. We then binned the blocks according to each of the four factors considered and tested if the CO/DSB ratio of a bin is significantly different from that of all other bins combined. For instance, in examining the distance to subtelomere, we found the ratio to be significantly higher in the bin of 0–20 kb and the bin of 20–40 kb for Scer (fig. 4A) as well as Spar (fig. 4B); these regions are referred to as near-subtelomeric regions because they are relatively close to subtelomeres. No significant variation in the CO/DSB ratio was observed in the investigation of the other three factors (supplementary fig. S6A–C, Supplementary Material online).
Fig. 4.
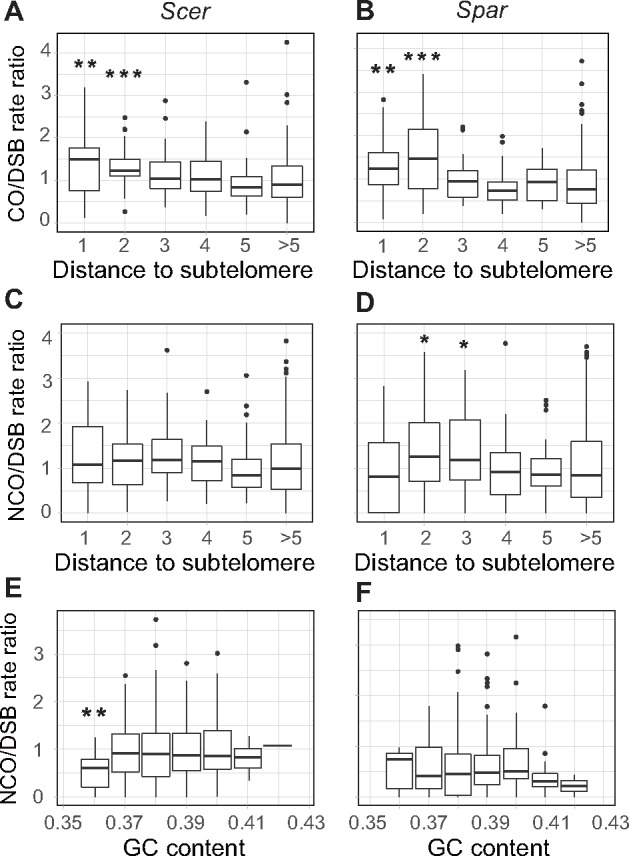
CO/DSB rate ratio and NCO/DSB rate ratio vary across genomic regions. (A and B) The CO/DSB rate ratio is significantly higher in near-subtelomeric regions than the rest of the genome in both Scer (A) and Spar (B). (C and D) The NCO/DSB rate ratio is not significantly variable in Scer (C) but is significantly variable in Spar (D) among regions with different distances to telomere. (E and F) The NCO/DSB rate ratio is significantly lower in the lowest GC content bin than other bins in Scer (E) but does not vary significantly among these bins in Spar (F). On the X-axis of panels (A)–(D), “1” indicates the 20-kb bin closest to subtelomeres (toward centromere), “2” is the next closest 20-kb bin, and so on. In (E) and (F), 20-kb windows are binned based on GC content, which ranges from 35.5% to 42.5%, and the bin width is 1%. In each bin, the data are presented as a bar plot; the lower and upper edges of a box represent the first (qu1) and third quartiles (qu3), respectively, the horizontal line inside the box indicates the median (md), the whiskers extend to the most extreme values inside inner fences, md ± 1.5(qu3 − qu1), and the dots represent values outside the inner fences (outliers). Wilcoxon rank-sum tests were performed between each bin and all other bins combined. *P < 0.05, **P < 0.01, and ***P < 0.001.
We conducted two tests to examine if the near-subtelomeric preference of CO events is necessary and sufficient to explain the elevated interspecific conservation of the CO landscape. In the first test, we removed near-subtelomeric regions in each species and found that the interspecific correlation in CO rates is no longer significantly different from that of randomly down-sampled DSBs (two-tailed P = 0.22). In the second test, we preferentially retained DSBs in near-subtelomeric regions when down-sampling the DSBs to the number of COs observed in each species. Specifically, in each species, we computed a CO/DSB ratio (α), dividing the total number of COs by the total number of DSBs in all near-subtelomeric regions combined. We similarly computed a CO/DSB ratio (β) for all other regions combined. In Spar, the probability that a near-subtelomeric DSB was retained was 1.498 (=α/β) times the probability that a non–near-subtelomeric DSB was retained. The corresponding value was 1.374 in Scer. Remarkably, such down-sampled DSBs show an interspecific correlation that is not significantly different from the corresponding CO rate correlation (two-tailed P = 0.26). Thus, the near-subtelomeric CO preference is necessary and sufficient to explain the higher evolutionary conservation of the CO landscape than the DSB landscape.
By contrast, NCOs apparently favor different subsets of DSBs in the two yeasts. In particular, when syntenic regions are binned according to the distance to subtelomeres, a significantly higher NCO/DSB ratio was found in the 20–40 and 40–60 kb bins in Spar but not any bins in Scer (fig. 4C and D). Similarly, when syntenic regions are binned according to the GC content, a significantly lower NCO/DSB ratio was found in the lowest GC bin in Scer but not in any bins of Spar (fig. 4E and F). No significant variation in the NCO/DSB ratio was observed in the investigation of the other two factors (supplementary fig. S6D and E, Supplementary Material online).
Using the same two tests described above, we investigated whether the preference of NCO in regions that are 20–60 kb from subtelomeres in Spar and the disfavor of NCO in low-GC bins in Scer together explain the lower interspecific correlation in NCO rates than that in DSB rates. In the first test, we found that removing the NCO-preferred region in Spar and NCO-disfavored region in Scer renders the interspecific correlations in NCO rates and down-sampled DSBs similar (two-tailed P = 0.063). In the second test, the interspecific correlations in NCOs and DSBs remain significantly different (two-tailed P = 0.0014) even after we down-sampled the DSBs differentially according to the NCO preference and disfavor discovered. Thus, the identified preference and disfavor are necessary but not sufficient to explain the reduced correlation in NCO rates between the two yeasts, and additional factors including unobserved NCOs and effects of heterozygosity may be involved.
The Near-Subtelomeric Preference of CO Is At Least in Part Attributable to CO Interference
That CO is enriched near chromosome ends exclusive of subtelomeres has been reported in yeast, C. elegans, and mammals (McKim et al. 1988; Villeneuve 1994; Barlow and Hultén 1998; Lander et al. 2001; Jensen-Seaman et al. 2004; Barton et al. 2008), and two potential mechanistic explanations have been proposed (Barton et al. 2008). The first explanation is based on the fact that telomere clustering at the leptotene/zygotene transition during meiosis brings the endmost homologous sequences to close proximity; this kind of spatial positioning may increase the probability of CO. The second explanation is CO interference, which refers to the phenomenon that the occurrence of a CO reduces the probability of its nearby COs (Hillers 2004); CO interference may push CO events toward near-subtelomeric regions when multiple COs occur on the same chromosome. Our data allow a test of the second hypothesis. Specifically, for all COs in Spar and Scer, we divided them into two categories: single COs, which do not co-occur with other COs on a chromosome, and multiple COs, which co-occur with at least another CO on the same chromosome. Single COs should not be affected by CO interference, whereas multiple COs may. We found that multiple COs show significant near-subtelomeric preferences when compared with DSBs or a random distribution (all P < 10−18, chi-squared test; table 3), whereas single COs do not share these preferences (all P > 0.05, chi-squared test; table 3), suggesting that the near-subtelomeric CO preference is at least in part attributable to CO interference. Notwithstanding, the above finding should be verified in the future because the numbers of single COs are relatively small here. Surprisingly, the fraction of DSBs in near-subtelomeric regions is only 95% (Spar) to 97% (Scer) of the expected value under the random distribution (P < 10−10 in both species, chi-squared test), which contrasts the recent report that Hop1, a DSB-promoting protein, tends to persist in near-subtelomeric regions (Subramanian et al. 2017). Future studies are required to resolve this apparent contradiction.
Table 3.
Numbers of Single COs and Multiple COs in Near-Subtelomeric Regions and Other Regions.
| Categories | Near-Subtelomeric Regionsa | Other Regions | Near-Subtelomeric/Other Ratio | P Value (compared with random 1) | P Value (compared with random 2) |
|---|---|---|---|---|---|
| Spar | |||||
| Single COs | |||||
| Random expectation 1b | 28.27% | 71.73% | 0.394 | ||
| Random expectation 2b | 28.64% | 71.36% | 0.401 | ||
| Observed | 34 | 58 | 0.586 | 0.064 | 0.24 |
| Multiple COs | |||||
| Random expectation 1 | 10.66% | 89.34% | 0.119 | ||
| Random expectation 2 | 9.63% | 90.37% | 0.107 | ||
| Observed | 388 | 1,974 | 0.197 | 1.1 × 10−19 | 4.2 × 10−29 |
| Scer | |||||
| Single COs | |||||
| Random expectation 1 | 40.63% | 59.37% | 0.684 | ||
| Random expectation 2 | 42.96% | 57.04% | 0.753 | ||
| Observedc | 8 | 19 | 0.421 | 0.078 | 0.16 |
| Multiple Cos | |||||
| Random expectation 1 | 10.81% | 89.19% | 0.121 | ||
| Random expectation 2 | 10.23% | 89.77% | 0.114 | ||
| Observedc | 875 | 4,896 | 0.179 | 1.8 × 10−26 | 4.1 × 10−35 |
Note.—P values in the last two columns are from chi-squared tests.
Near-subtelomeric regions are defined as 0–40 kb from subtelomere toward centromere.
Random expectation 1 is based on the relative lengths of near-subtelomeric regions and other regions. Random expectation 2 is based on relative numbers of DSBs in near-subtelomeric regions and other regions, according to DSB data from Lam and Keeney (2015). Single and multiple COs are not evenly distributed among chromosomes. When calculating random expectations, we weight each chromosome by the number of single (or multiple) COs observed on the chromosome.
Data from Mancera et al. (2008) and Liu et al. (2018).
Discussion
In this work, we generated the first high-resolution recombination map of Spar, the sister species of the model organism Scer. The map provides detailed information on the genomic distributions of CO and NCO rates and characteristics of gene conversions including tract length and GC bias. More importantly, comparing this map with that of Scer offers insights on the evolution of the recombination landscape in yeasts. Several findings are worth discussion. First, we found that the recombination rate is much lower in Spar than in Scer in the hybrids examined here. It is known that the rate of recombination is under strict controls and also responds to selection (Martini et al. 2006; Otto and Barton 1997; Séguéla-Arnaud et al. 2015; Ritz et al. 2017). Theoretical analysis showed that, during rapid adaptations, modifiers increasing recombination rates tend to spread by hitchhiking beneficial mutations brought together by recombination (Otto and Barton 1997). In fact, domestication tends to lead to increased recombination rates (Otto and Barton 2001; Ross-Ibarra 2004; Groenen et al. 2009). It is thus possible that the higher recombination rate in Scer than Spar is a result of Scer domestication. Another possibility is that high rates of recombination are selected against in natural environments while this selective constraint is relieved during the domestication of laboratory yeast strains. The rate of CO is limited to 1–2 per chromosome per meiosis in most species, possibly because of the recombination load (Charlesworth and Barton 1996), which refers to the effect that recombination breaks up coadapted clusters of alleles established in evolution. Indeed, multiple pathways limiting CO rates have been identified in Arabidopsis (Crismani et al. 2012; Séguéla-Arnaud et al. 2015). Under this logic, because laboratory strains of yeast overwhelmingly divide mitotically, the selective constraint on the recombination rate is relaxed and the pathways limiting the recombination rate erode over time, resulting in a higher rate of recombination today. The above explanations are consistent with the observation that a wild/laboratory hybrid Scer exhibits a significantly lower CO rate when compared with a laboratory/laboratory hybrid Scer (Liu et al. 2018) and can be further tested by measuring the recombination rate in a wild/wild hybrid Scer. Alternatively, the lower CO rate in Spar could be a result of the relatively high sequence divergence between the two parental strains used in the current study (1.19%), which is much greater than that of previously used Scer parental strains (∼0.5%). It has been shown in Scer that high sequence divergence decreases the probability of homologous recombination at the whole genome scale (Hunter et al. 1996; Greig et al. 2003). This explanation is also consistent with the CO rate difference between the wild/laboratory hybrid and laboratory/laboratory hybrid of Scer above mentioned, because the parental sequence divergence is higher in the former than the latter (Liu et al. 2018). To test the effect of heterozygosity on the rate of recombination, we examined the heterozygosity in 1 kb regions around CO sites, NCO sites, and 1 million random sites. Compared with random sites, COs are associated with significantly lower heterozygosity in Scer but not Spar (supplementary fig. S7, Supplementary Material online). The Scer finding is consistent with the previous findings at the whole genome scale (Hunter et al. 1996; Greig et al. 2003), but the different observations in Scer and Spar suggest that the potential impact of sequence divergence on recombination varies among species or exists within a limited range of sequence divergence. Furthermore, it is worth noting that, the CO rate is positively correlated with local heterozygosity in Scer at the scale of 5–40 kb (supplementary table S2, Supplementary Material online). It is possible that the potential impact of sequence divergence on recombination varies depending on the scale of analysis. Compared with random sites, NCOs are associated with significantly higher heterozygosity in both Scer (the YJM789 × S96 hybrid) and Spar (supplementary fig. S7, Supplementary Material online), possibly because a higher marker density improves the detection of NCOs. No significant difference in heterozygosity was observed between NCOs and random sites in the YJM789 × YPS128 hybrid (supplementary fig. S7, Supplementary Material online), possibly due to the relatively small data size in this hybrid.
Second, in contrast to the large difference in recombination rate between Scer and Spar, the CO landscape is conserved between the two species and is even more conserved than the DSB landscape. Thus, the hotspot paradox hypothesis is invalid in yeast at both the DSB level and CO level. Because yeast DSBs are enriched in promoters, especially in nucleosome-depleted regions (Lam and Keeney 2015), the conservation of the CO landscape could be a “side effect” of the functional constraint of promoters. Alternatively, the conservation of the CO landscape could be a result of direct selection on the landscape (Lam and Keeney 2015), although the potential benefit of this conserved landscape is currently unknown.
Third, we found that the elevated conservation of the yeast CO landscape relative to the DSB landscape is due to a near-subtelomeric CO preference. Two potential mechanisms have been proposed to explain this preference, and our analysis tested and confirmed one of them—CO interference. It is unknown whether the near-subtelomeric CO preference is also caused by the other potential mechanism—telomere clustering. Because CO interference is a general phenomenon in meiosis and because the enrichment of CO in chromosomal ends excluding subtelomeric regions is known in many organisms (McKim et al. 1988; Villeneuve 1994; Barlow and Hultén 1998; Trelles-Sticken et al. 1999; Lander et al. 2001; Jensen-Seaman et al. 2004), the stronger conservation of the CO landscape than DSB landscape may be more widespread than revealed in this study. Regardless, our results suggest that, while interspecific comparison of DSBs is a valuable approach to studying the evolutionary conservation of the DSB landscape, it does not provide an unbiased estimate of the conservation of the CO landscape.
Materials and Methods
Yeast Strains, Growth Conditions, Crossing, and Tetrad Dissection
Stable haploids derived from wild-type homothallic diploid strains of Spar N17 and N44 isolates were previously created (Cubillos et al. 2009) and were a gift from Gianni Liti. As the strains of both mating types carried the same auxotrophic and drug resistance markers (ura3::KanMX4, ho::HygMX4), marker switching was carried out in the N17 MATα strain to replace the ho::HygMX4 marker with the Nourseothricin resistance cassette NatMX4 to allow for easy hybrid detection. The two strains, N17 (MATα ura3::KanMX4, ho::NatMX4) and N44 (MATa ura3::KanMX4, ho::HygMX4), were mixed well on the surface of a YPD agar plate (1% yeast extract, 2% peptone, 2% glucose, and 2% agar) and incubated overnight at 25 °C. Diploids were then selected by replication of cells to an YPD agar plate containing 300 μg/ml Hygromycin B and 100 μg/ml Nourseothricin. After overnight incubation at 25 °C, single diploid genotypes were isolated by streaking to a fresh dual drug selection plate and a further ∼48 h growth at 25 °C for single colony formation. A single colony was picked and its ploidy confirmed by polymerase chain reaction of the mating type loci.
To induce meiosis, we cultured the N44/N17 hybrid overnight at 25 °C on an YPD plate and transferred the resulting patch of cells to a KAc sporulation plate (2% potassium acetate and 2% agar). The sporulation plate was incubated at 25 °C for 4 days and the formation of meiotic products (tetrads) was confirmed by light microscopy. Tetrads were treated with zymolyase and dissected onto YPD agar plates using an Axioskop 40 dissection microscope (Carl Zeiss Microscopy). Plates were incubated at 25 °C to allow for colony formation. One hundred tetrads were initially dissected and the N17/N44 hybrid was found to have a fertility level of ∼84%, in the range determined previously for similarly related Spar strains (Liti et al. 2006). Fifty complete tetrads (with all four spores viable) were randomly selected for further study following confirmation of the correct segregation of both Nourseothricin/Hygromycin drug resistance and mating type by replica plating and polymerase chain reaction, respectively. Each of the 200 meiotic products was frozen in 20% glycerol and stored at −80 °C.
DNA Extraction, Genome Sequencing, and Marker Filtering
The DNAs of haploid parental strains and each dissected meiotic spore were extracted and sequenced. DNA was extracted using the phenol/chloroform method, and whole-genome resequencing was performed at BGI-Shenzhen. In particular, paired-end sequencing libraries were constructed for each sample and 2× 100 bp paired-end reads were generated on Illumina’s Hiseq 2000 platform. In total, 202 samples were sequenced, including two parental strains and 200 meiotic spores from 50 tetrads. Parental strains were sequenced with higher depth (on average 52.6×) than offspring spores (on average 11.8×).
The Spar reference genome (strain N44) was downloaded from The Yeast Population Reference Panel (Yue et al. 2017) (https://yjx1217.github.io/Yeast_PacBio_2016/data/; last accessed December 30, 2018). The genomic coordinates of subtelomere and centromere were listed in the supplemental data of that paper (Yue et al. 2017) and the gff file on their website. For each sample, the Illumina reads were mapped onto the reference genome by Burrows–Wheeler Aligner (Li and Durbin 2009); duplicates marking and realignment around indels were carried out by Genome Analysis Toolkit (McKenna et al. 2010) and variants were called by Genome Analysis Toolkit HaplotypeCaller.
The sequenced samples in this study are all haploid. “Unambiguous” SNPs between the two parental strains (i.e., no shared state at the SNP site between the reads of the two alleles) with quality scores ≥30 were first called as marker candidates. For a tetrad, a marker candidate was used in identifying recombination events if it met the following criteria: 1) called “unambiguous” in all four spores, 2) genotyped with high quality (≥30) in all four spores, and 3) the genotypes of the four spores at the candidate site agree with their parental strains.
Identification of Recombination Events
We adopted a published method (Liu et al. 2018) to identify three types of recombination events: COs, crossovers associated gene conversions (CO-GeneConv), and NCOs. In each tetrad, the genotypes of all four spores were used collectively to identify these events in the following manner. Reciprocal changes of genotypes in two of the four spores are identified as a CO. If the genotype switch point is the same in these two spores, it is identified as a simple CO event. If the genotype switch point is not the same in these two spores, it is identified as a CO event associated with a continuous gene conversion. Sometimes, a CO-GeneConv emerges as a discontinuous conversion tract, where a short nonreciprocal genotype change appears near a CO event (distance < 10 kb). This is identified as a CO event associated with a discontinuous gene conversion. Nonreciprocal changes of genotypes that are not associated with CO events are identified as NCO events.
The tract length of a gene conversion event was determined using the published midpoint method (Mancera et al. 2008). In short, at each end of a gene conversion event, the midpoint between the end marker and the nearest flanking non-converted marker is considered the edge of the conversion tract.
Correction of Number of NCO Events
An NCO event is unobserved if there is no marker in the converted region. To account for such unobserved NCO events, we first applied a previously used simulation-based method (Wijnker et al. 2013; Liu et al. 2018) to estimate the average tract length of NCO events. For each tract length from 100 to 3,000 bp with a step size of 100 bp, 10,000 random NCO events were simulated and the mean number of markers converted among those tracts with at least one marker was calculated (supplementary fig. S2A, Supplementary Material online). This relationship between tract length and mean number of markers converted (among observable tracts) allowed us to infer the actual mean tract length from the observed mean number of markers converted in the real data. In Spar, an observed NCO event on average converted 18 markers, which correspond to a tract length of 1,500 bp in the simulation. Hence, our estimated mean tract length in NCO events is 1,500 bp. We used the above simulated NCOs to estimate the probability that a tract of a given length is detectable (i.e., covering at least one marker). We found that this probability exceeds 90% for tracts >300 bp (supplementary fig. S2B, Supplementary Material online), indicating that our observed number of NCOs should be close to the true number. For a tract of 1,500 bp, the probability of detection is 98.5%. Hence, the corrected number of NCO events is 26.9/98.5% = 27.3, where 26.9 is the observed number of NCO events.
Data Availability
The raw Illumina reads generated in this study were submitted to NCBI sequence read archive under accession number PRJNA473589. The genotypes of the 50 tetrads at all marker sites were submitted to figshare and can be accessed through the link https://figshare.com/articles/Genotypes_of_50_Spar_tetrads/6406475, last accessed December 30, 2018.
Supplementary Material
Supplementary data are available at Molecular Biology and Evolution online.
Supplementary Material
Acknowledgments
We thank Piaopiao Chen and Xinzhu Wei for valuable comments. This work was supported in part by a research grant from the U.S. National Institutes of Health (GM120093) to J.Z.
References
- Allers T, Lichten M.. 2001. Differential timing and control of noncrossover and crossover recombination during meiosis. Cell 1061: 47–57. [DOI] [PubMed] [Google Scholar]
- Auton A, Fledel-Alon A, Pfeifer S, Venn O, Ségurel L, Street T, Leffler EM, Bowden R, Aneas I, Broxholme J, et al. 2012. A fine-scale chimpanzee genetic map from population sequencing. Science 3366078: 193–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker Z, Schumer M, Haba Y, Bashkirova L, Holland C, Rosenthal GG, Przeworski M.. 2017. Repeated losses of PRDM9-directed recombination despite the conservation of PRDM9 across vertebrates. eLife 6: e24133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow AL, Hultén MA.. 1998. Crossing over analysis at pachytene in man. Eur J Hum Genet. 64: 350–358. [DOI] [PubMed] [Google Scholar]
- Barton AB, Pekosz MR, Kurvathi RS, Kaback DB.. 2008. Meiotic recombination at the ends of chromosomes in Saccharomyces cerevisiae. Genetics 1793: 1221–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G, de Massy B.. 2010. PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 3275967: 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Imai Y, de Massy B.. 2013. Meiotic recombination in mammals: localization and regulation. Nat Rev Genet. 1411: 794–806. [DOI] [PubMed] [Google Scholar]
- Boulton A, Myers RS, Redfield RJ.. 1997. The hotspot conversion paradox and the evolution of meiotic recombination. Proc Natl Acad Sci U S A. 9415: 8058–8063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese P. 2007. A population genetics model with recombination hotspots that are heterogeneous across the population. Proc Natl Acad Sci U S A. 10411: 4748–4752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B, Barton NH.. 1996. Recombination load associated with selection for increased recombination. Genet Res. 671: 27–41. [DOI] [PubMed] [Google Scholar]
- Charlesworth B, Charlesworth D.. 2018. Neutral variation in the context of selection. Mol Biol Evol. 356: 1359–1361. [DOI] [PubMed] [Google Scholar]
- Coop G, Myers SR.. 2007. Live hot, die young: transmission distortion in recombination hotspots. PLoS Genet. 33: e35.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coop G, Przeworski M.. 2007. An evolutionary view of human recombination. Nat Rev Genet. 81: 23–34. [DOI] [PubMed] [Google Scholar]
- Crismani W, Girard C, Froger N, Pradillo M, Santos JL, Chelysheva L, Copenhaver GP, Horlow C, Mercier R.. 2012. FANCM limits meiotic crossovers. Science 3366088: 1588–1590. [DOI] [PubMed] [Google Scholar]
- Cubillos FA, Louis EJ, Liti G.. 2009. Generation of a large set of genetically tractable haploid and diploid Saccharomyces strains. FEMS Yeast Res. 98: 1217–1225. [DOI] [PubMed] [Google Scholar]
- Davies B, Hatton E, Altemose N, Hussin JG, Pratto F, Zhang G, Hinch AG, Moralli D, Biggs D, Diaz R, et al. 2016. Re-engineering the zinc fingers of PRDM9 reverses hybrid sterility in mice. Nature 5307589: 171–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duret L, Galtier N.. 2009. Biased gene conversion and the evolution of mammalian genomic landscapes. Annu Rev Genomics Hum Genet. 10:285–311. [DOI] [PubMed] [Google Scholar]
- Greig D, Travisano M, Louis EJ, Borts RH.. 2003. A role for the mismatch repair system during incipient speciation in Saccharomyces. J Evol Biol. 163: 429–437. [DOI] [PubMed] [Google Scholar]
- Groenen MAM, Wahlberg P, Foglio M, Cheng HH, Megens H-J, Crooijmans RPMA, Besnier F, Lathrop M, Muir WM, Wong GK-S, et al. 2009. A high-density SNP-based linkage map of the chicken genome reveals sequence features correlated with recombination rate. Genome Res. 193: 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillers KJ. 2004. Crossover interference. Curr Biol. 1424: R1036–R1037. [DOI] [PubMed] [Google Scholar]
- Hinch AG, Tandon A, Patterson N, Song Y, Rohland N, Palmer CD, Chen GK, Wang K, Buxbaum SG, Akylbekova EL, et al. 2011. The landscape of recombination in African Americans. Nature 4767359: 170–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter N, Chambers SR, Louis EJ, Borts RH.. 1996. The mismatch repair system contributes to meiotic sterility in an interspecific yeast hybrid. EMBO J. 157: 1726–1733. [PMC free article] [PubMed] [Google Scholar]
- Jahns MT, Vezon D, Chambon A, Pereira L, Falque M, Martin OC, Chelysheva L, Grelon M.. 2014. Crossover localisation is regulated by the neddylation posttranslational regulatory pathway. PLoS Biol. 128: e1001930.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Seaman MI, Furey TS, Payseur BA, Lu Y, Roskin KM, Chen C-F, Thomas MA, Haussler D, Jacob HJ.. 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 144: 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppi L, Jeffreys AJ, Keeney S.. 2004. Where the crossovers are: recombination distributions in mammals. Nat Rev Genet. 56: 413.. [DOI] [PubMed] [Google Scholar]
- Keeney S, Giroux CN, Kleckner N.. 1997. Meiosis-specific DNA double-strand breaks are catalyzed by Spo11, a member of a widely conserved protein family. Cell 883: 375–384. [DOI] [PubMed] [Google Scholar]
- Lam I, Keeney S.. 2015. Nonparadoxical evolutionary stability of the recombination initiation landscape in yeast. Science 3506263: 932–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, et al. 2001. Initial sequencing and analysis of the human genome. Nature 4096822: 860–921. [DOI] [PubMed] [Google Scholar]
- Laureau R, Loeillet S, Salinas F, Bergström A, Legoix-Né P, Liti G, Nicolas A.. 2016. Extensive recombination of a yeast diploid hybrid through meiotic reversion. PLoS Genet. 122: e1005781.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R.. 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 2514: 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libuda DE, Uzawa S, Meyer BJ, Villeneuve AM.. 2013. Meiotic chromosome structures constrain and respond to designation of crossover sites. Nature 5027473: 703–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Barton DBH, Louis EJ.. 2006. Sequence diversity, reproductive isolation and species concepts in Saccharomyces. Genetics 1742: 839–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liti G, Carter DM, Moses AM, Warringer J, Parts L, James SA, Davey RP, Roberts IN, Burt A, Koufopanou V, et al. 2009. Population genomics of domestic and wild yeasts. Nature 4587236: 337–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Huang J, Sun X, Li J, Hu Y, Yu L, Liti G, Tian D, Hurst LD, Yang S.. 2018. Tetrad analysis in plants and fungi finds large differences in gene conversion rates but no GC bias. Nat Ecol Evol. 21: 164–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Jia Y, Sun X, Tian D, Hurst LD, Yang S.. 2017. Direct determination of the mutation rate in the bumblebee reveals evidence for weak recombination-associated mutation and an approximate rate constancy in insects. Mol Biol Evol. 341: 119–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Zhang X, Huang J, Chen J-Q, Tian D, Hurst LD, Yang S.. 2015. Causes and consequences of crossing-over evidenced via a high-resolution recombinational landscape of the honey bee. Genome Biol. 161: 15.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancera E, Bourgon R, Brozzi A, Huber W, Steinmetz LM.. 2008. High-resolution mapping of meiotic crossovers and non-crossovers in yeast. Nature 4547203: 479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini E, Borde V, Legendre M, Audic S, Regnault B, Soubigou G, Dujon B, Llorente B.. 2011. Genome-wide analysis of heteroduplex DNA in mismatch repair-deficient yeast cells reveals novel properties of meiotic recombination pathways. PLoS Genet. 79: e1002305.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini E, Diaz RL, Hunter N, Keeney S. 2006. Crossover homeostasis in yeast meiosis. Cell 1262: 285–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald MJ, Rice DP, Desai MM.. 2016. Sex speeds adaptation by altering the dynamics of molecular evolution. Nature 5317593: 233–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, et al. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 209: 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKim KS, Howell AM, Rose AM.. 1988. The effects of translocations on recombination frequency in Caenorhabditis elegans. Genetics 1204: 987–1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G, Donnelly P.. 2010. Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 3275967: 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman MW. 2002. Variation in recombination rate across the genome: evidence and implications. Curr Opin Genet Dev. 126: 657–663. [DOI] [PubMed] [Google Scholar]
- Oh SD, Lao JP, Hwang PY-H, Taylor AF, Smith GR, Hunter N.. 2007. BLM ortholog, Sgs1, prevents aberrant crossing-over by suppressing formation of multichromatid joint molecules. Cell 1302: 259–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh SD, Lao JP, Taylor AF, Smith GR, Hunter N.. 2008. RecQ helicase, Sgs1, and XPF family endonuclease, Mus81-Mms4, resolve aberrant joint molecules during meiotic recombination. Mol Cell. 313: 324–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N, Beatson SA, Lunter G, Malik HS, Ponting CP.. 2009. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 512: e1000753.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP, Barton NH.. 1997. The evolution of recombination: removing the limits to natural selection. Genetics 1472: 879–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SP, Barton NH.. 2001. Selection for recombination in small populations. Evolution 5510: 1921–1931. [DOI] [PubMed] [Google Scholar]
- Parvanov ED, Petkov PM, Paigen K.. 2010. Prdm9 controls activation of mammalian recombination hotspots. Science 3275967: 835.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritz KR, Noor MAF, Singh ND.. 2017. Variation in recombination rate: adaptive or not? Trends Genet. 335: 364–374. [DOI] [PubMed] [Google Scholar]
- Ross-Ibarra J. 2004. The evolution of recombination under domestication: a test of two hypotheses. Am Nat. 1631: 105–112. [DOI] [PubMed] [Google Scholar]
- Rosu S, Libuda DE, Villeneuve AM.. 2011. Robust crossover assurance and regulated interhomolog access maintain meiotic crossover number. Science 3346060: 1286–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Séguéla-Arnaud M, Crismani W, Larchevêque C, Mazel J, Froger N, Choinard S, Lemhemdi A, Macaisne N, Leene JV, Gevaert K, et al. 2015. Multiple mechanisms limit meiotic crossovers: TOP3α and two BLM homologs antagonize crossovers in parallel to FANCM. Proc Natl Acad Sci U S A. 11215: 4713–4718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal S, Leffler EM, Sannareddy K, Turner I, Venn O, Hooper DM, Strand AI, Li Q, Raney B, Balakrishnan CN, et al. 2015. Stable recombination hotspots in birds. Science 3506263: 928–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slatkin M. 2008. Linkage disequilibrium—understanding the evolutionary past and mapping the medical future. Nat Rev Genet. 96: 477–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagulova F, Brick K, Pu Y, Camerini-Otero RD, Petukhova GV.. 2016. The evolutionary turnover of recombination hot spots contributes to speciation in mice. Genes Dev. 303: 266–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian VV, Markowitz TE, Silva LAV, San-Segundo P, Hollingsworth NM, Hochwagen A.. 2017. Persistent DNA-break potential near telomeres increases initiation of meiotic recombination on small chromosomes. bioRxiv 201889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thacker D, Mohibullah N, Zhu X, Keeney S.. 2014. Homologue engagement controls meiotic DNA break number and distribution. Nature 5107504: 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trelles-Sticken E, Loidl J, Scherthan H.. 1999. Bouquet formation in budding yeast: initiation of recombination is not required for meiotic telomere clustering. J Cell Sci. 112:651–658. [DOI] [PubMed] [Google Scholar]
- Tsai IJ, Burt A, Koufopanou V.. 2010. Conservation of recombination hotspots in yeast. Proc Natl Acad Sci U S A. 10717: 7847–7852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubeda F, Wilkins JF.. 2011. The Red Queen theory of recombination hotspots. J Evol Biol. 243: 541–553. [DOI] [PubMed] [Google Scholar]
- Villeneuve AM. 1994. A cis-acting locus that promotes crossing over between X chromosomes in Caenorhabditis elegans. Genetics 1363: 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijnker E, James GV, Ding J, Becker F, Klasen JR, Rawat V, Rowan BA, de Jong DF, de Snoo CB, Zapata L, et al. 2013. The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. eLife 2:e01426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang L, Huang J, Zhang X, Yuan Y, Chen J-Q, Hurst LD, Tian D.. 2015. Parent-progeny sequencing indicates higher mutation rates in heterozygotes. Nature 5237561: 463–467. [DOI] [PubMed] [Google Scholar]
- Yue J-X, Li J, Aigrain L, Hallin J, Persson K, Oliver K, Bergström A, Coupland P, Warringer J, Lagomarsino MC, et al. 2017. Contrasting evolutionary genome dynamics between domesticated and wild yeasts. Nat Genet. 496: 913–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw Illumina reads generated in this study were submitted to NCBI sequence read archive under accession number PRJNA473589. The genotypes of the 50 tetrads at all marker sites were submitted to figshare and can be accessed through the link https://figshare.com/articles/Genotypes_of_50_Spar_tetrads/6406475, last accessed December 30, 2018.