

Abstract
The hemochromatosis gene HFE was discovered in 1996, more than a century after clinical and pathologic manifestations of hemochromatosis were reported. Linked to the major histocompatibility complex (MHC) on chromosome 6p, HFE encodes the MHC class I-like protein HFE that binds beta-2 microglobulin. HFE influences iron absorption by modulating the expression of hepcidin, the main controller of iron metabolism. Common HFE mutations account for ~90% of hemochromatosis phenotypes in whites of western European descent. We review HFE mapping and cloning, structure, promoters and controllers, and coding region mutations, HFE protein structure, cell and tissue expression and function, mouse Hfe knockouts and knockins, and HFE mutations in other mammals with iron overload. We describe the pertinence of HFE and HFE to mechanisms of iron homeostasis, the origin and fixation of HFE polymorphisms in European and other populations, and the genetic and biochemical basis of HFE hemochromatosis and iron overload.
Keywords: hemochromatosis, iron, major histocompatibility complex, transferrin receptor
1. Introduction
The French clinicians Trousseau and Troisier described the rare triad of darkening of the skin, diabetes mellitus, and cirrhosis in the latter half of the 19th C [1,2]. Three decades later, von Recklinghausen, a German pathologist, named the condition hämochromatose [3]. In a 1935 monograph, English gerontologist Joseph Sheldon reported his analysis of 311 cases from the literature and speculated that hemochromatosis is heritable [4]. In the 1970s, Simon and colleagues reported that hemochromatosis is relatively common, is linked to human leukocyte antigen (HLA) markers, and is transmitted as an autosomal recessive trait [5–8]. In 1996, Rothenberg and Voland hypothesized that non-classical class I MHC genes that bind β2M also control iron absorption and that β2M knockout mice (beta2m−/−) would develop iron overload [9]. They and others confirmed this hypothesis [9–11], but humans with iron overload phenotypes did not have explanatory mutations in B2M [12]. In 1996, Feder and colleagues used positional cloning to identify HFE, the hemochromatosis gene, linked to the major histocompatibility complex (MHC) on chromosome 6p [13].
The membrane protein HFE is similar to MHC class I-type proteins and binds beta-2 microglobulin (β2M) [13]. HFE binds transferrin receptor (TFRC) in its extracellular α1-α2 domain [14,15]. HFE is required for normal regulation of hepatic synthesis of hepcidin, the main controller of iron metabolism [16]. Common HFE mutations account for ~90% of hemochromatosis phenotypes in whites of western European descent.
2. HFE mapping and cloning
In studies of French subjects, Simon et al. first reported that hemochromatosis is a heritable condition linked to MHC alleles HLA-A*03, B*07, and B*14 on the short arm of chromosome 6 (chromosome 6p) [5–8]. There is strong linkage disequilibrium within the MHC over a physical distance of 6 Mb in which there is a lack of recombination in most hemochromatosis patients [17]. Consequently, estimates of the position of the hemochromatosis locus relative to HLA-A and HLA-B [7,18–20] and HLA-F [21,22] were inconsistent. Polymorphic short tandem repeat markers within chromosome 6p enhanced the ability to conduct positional cloning [23–26]. Multipoint mapping within hemochromatosis families indicated that the gene was <1 cM proximal and ~0.5 cM distal to HLA-A [27]. D6S105(8), significantly associated with hemochromatosis, was the closest marker to the gene known at the time [27–31]. The gene was located telomeric to D6S105 [32]. D6S105(8) occurred in 30% of hemochromatosis-associated haplotypes in Italians [33]. Hemochromatosis ancestral haplotypes in Australians extended telomeric of HLA-A as far as D6S105 [34]. Yeast artificial chromosome LD5–1 hybridized with the hemochromatosis gene [35].
In 1996, Feder et. al. identified a 250 kb region between D6S2238 and D6S2241 that contained the hemochromatosis gene [13]. Within this region, they identified a MHC class I-like gene telomeric to the classical MHC that contained two missense mutations. Homozygosity for a c.845G→A mutation (cysteine→tyrosine at amino acid 282, p.C282Y) was found in 83% of hemochromatosis patients. This mutation was detected in 3.2% of control chromosomes. The remaining patients had c.187C→G (histidine→aspartic acid at amino acid 63; p.H63D) either in compound heterozygosity with p.C282Y or as p.H63D homozygosity [13].
Feder and colleagues named the gene HLA-H [13], although the name had been published earlier to designate a presumed pseudogene in the HLA class I region [36]. Bodmer and Mercier appealed for a more appropriate designation [37,38]. Both the WHO Nomenclature Committee for Factors of the HLA System and the HuGO Genome Nomenclature Committee approved the symbol HFE (H = high; FE = iron) (OMIM *613609). The cytogenetic location of HFE is 6p22.2 (genomic coordinates (GRCh37): 6:26,087,421–26,096,437).
3. HFE gene structure
HFE contains 7 exons spanning 12 kb [13]. HFE encompasses 9,609 bp of DNA on chromosome 6p within the extended HLA class I region. Histone genes are present on both sides of HFE [39]. Exon 1 corresponds to the signal peptide and exons 2–4 to the α1, α2, and α3 domains, respectively. Exon 5 accounts for the transmembrane domain. The cytoplasmic tail is encoded by the 5’ portion of exon 6 that includes a native stop codon. Thus, the full-length transcript represents 6 exons [39].
3.1. HFE promoters and controllers
Alignment of the promoters of the human, rat, and mouse HFE genes reveals highly conserved elements, including binding sites for the transcription factors GATA, NF-IL6, AP1, AP2, CREB, PEA3, gamma-IRE, GFI1, HNF-3beta, and HFH2 [40].
The 5’ end of HFE mRNA includes two major initiation sites directed by TATA-like sequences and a window of initiation upstream of the first coding nucleotide [41]. HFE is activated by liver-enriched C/EBPalpha, erythropoiesis-specific GATA-1, and Sp1 transcription factors [41]. An inverted repeat sequence near the HFE promoter can bind poly (ADP-ribose) polymerase 1 (PARP1) and repress HFE promoter. Knockdown of PARP1 or treatments with either hemin or FeCl3 increase HFE mRNA and protein, leading to up-regulation of hepcidin mRNA [42]. An antisense transcript originating from HFE spans exon 1, exon 2, part of intron 1, and 1 kb upstream of it. The transcript was polyadenylated, had no open reading frame, and was expressed at low levels in all tissues and cell lines tested. This antisense RNA decreased HFE expression [43].
3.2. Alternative splicing
HFE expression is subjected to alternative splicing [44–47], like other MHC class I proteins [48,49]. Nine HFE splicing variants have been reported [39]. The predominant HFE full-length transcript is ~4.2 kb [47,50]. Other transcripts lack exon 2 or exon 3, or exons 2–3, 2–4, or 2–5 [50].
The full length and most of the alternatively spliced HFE transcripts were detected in ovary, testis, duodenum, heart, kidney, spleen, and liver [50]. Ovary and liver have the highest levels of total HFE mRNA. Duodenum has smallest amount. Exon 2–5 skipping was detected only in the gonads, duodenum and heart [50].
In HepG2 cells, full-length HFE appears in a perinuclear and cell membrane distribution and co-localizes with β2M and TFRC [50]. The intracellular distribution of HFE protein derived from a transcript lacking exon 2 is similar to that of p.C282Y. HFE protein transcribed from exons 4 and 5 with inclusion of intron 4 has a scattered intracellular distribution, is absent from the cell membrane, does not co-localize with either β2M or TFRC, and is present in the endoplasmic reticulum [50]. A soluble HFE isoform lacking transmembrane and intracellular domains (sHFE) was found predominantly in the duodenum, spleen, breast, skin, and testis [44], and was secreted into HepG2 cell culture medium in association with β2M [50]. Alternative HFE splice variants may play regulatory roles in specific cells or tissues [50].
3.3. Evolution of HFE
Proteins with partial orthology to human HFE occur in primitive animals and plants [51]. The earliest animals that express MHC class I genes or proteins are branchiostomes [52]. Six cosmids from amphioxus (Branchiostoma floridae) contained genes orthologous to those of human MHC-linked regions. The genes mapped to a single chromosome [52]. In sharks and bony fishes, there are some orthologs of human MHC class I proteins, especially α3 domains [53–56].
There is no positive evidence of HFE orthologs in seabass (Dicentrarchus labrax) [57], in zebrafish (Danio rerio) [58], or in other fish species for which sequence data are available [59], although fish MHC class I proteins with distant homology to HFE have been identified [59]. HFE has not been described in amphibians, reptiles, or birds, but has been characterized in numerous mammals. These observations suggest that HFE arose later than MHC genes.
Basic Local Alignment Search Tool (BLAST) comparisons demonstrate that human HFE is ~100% similar to chimpanzee HFE and 61–67% similar to HFE proteins of dogs, rats, cattle, and mice. HFE C282 is conserved because cysteine 282 is essential to β2M binding and extracellular presentation of HFE. H63 is also conserved. Histidine 63 forms a salt bridge in the α2 domain that binds TFRC, suggesting that the salt bridge is important for HFE function [60]. Histidines 116 and 145 and tyrosine 140 are widely conserved. A cluster of four histidine residues (H109, H111, H116, H145) is associated with Y140 in the α1 domain. This configuration resembles functional sites in other iron-binding proteins [61].
Proline 188 is highly conserved. The function of MHC class I molecules depends on the interaction of the α1–α2 ligand binding superdomain with nonameric peptides presented to αβ T-cell receptors [62]. P188 occurs in the α1 domain and is associated with a kink necessary for peptide binding by MHC molecules [61,63]. In human HFE, amino acids 307–329 in the transmembrane domain are partially conserved, suggesting that normal function of this domain may not depend on a high degree of amino acid conservation.
4. HFE coding region mutations
4.1. Common HFE mutations
The three most common coding-region mutations of HFE are: p.C282Y (exon 4; c.845G→A; rs1800562); p.H63D (exon 2; c.187C→G; rs1799945); and p.S65C (exon 2; c.193A→T; rs1800730) [64]. The p.C282Y mutation disrupts a critical disulfide bond in the α3 domain of HFE, abrogating its binding to β2M and limiting its localization mostly to the cytoplasm [65,66]. p.H63D and p.S65C affect the α1 binding groove but do not prevent HFE presentation on cell surfaces.
4.2. HFE mutations and iron phenotypes
Mean serum iron, transferrin saturation (TS), and serum ferritin levels are higher in adults with p.C282Y homozygosity than in adults with other common HFE genotypes [67]. Demonstration of small differences in mean levels of these blood iron measures in adults with HFE genotypes C282Y/wt, H63D/wt, C282Y/H63D and H63D/H63D usually requires large cohorts [64,67].
“Classical” hemochromatosis typically occurs in adults who are p.C282Y homozygotes [64]. The prevalence of this genotype in many western European [68] Caucasian control populations is 0.002–0.005 (2–5/1000) [64]. Lower prevalence estimates have been reported from control populations in northern Spain (Catalonia) [69], among Basques in Guipuzcoa, Spain [70], and in central Italy [71]. The combined prevalence of C282Y homozygosity in 404 control subjects in Northern Ireland [72] and 249 control subjects in the northwestern Republic of Ireland [73] was 0.012 [95% confidence interval 0.006, 0.025]. Penetrance of iron overload phenotypes is variable and often greater in men [64].
Liver is the predominant target organ of iron overload, although arthropathy, diabetes mellitus and other endocrinopathy, and additional manifestations consequent to severe iron overload occur in some p.C282Y homozygotes [64]. A small proportion of adults with p.C282Y/p.H63D or p.C282Y/p.S65C compound heterozygosity or p.H63D homozygosity develop mild iron overload, usually in the presence of concomitant liver disease [64,74–77]. Prevalences and characteristics of adults with HFE hemochromatosis are reviewed in detail elsewhere [64].
4.3. HFE alleles and hemochromatosis
Diverse mutations involving HFE introns and exons discovered in persons with hemochromatosis or their family members cause or probably cause high iron phenotypes. Other mutations are either synonymous or their effect on iron phenotypes, if any, has not been demonstrated. Most of these mutations are rare and many have been discovered in Caucasians, although interest in identifying HFE mutations is great in regions where large numbers of Caucasians reside. These mutations and their phenotypes are displayed in Table 1.
Table 1.
| Exon | cDNA alteration | Protein alteration | Phenotype3 | Reference |
|---|---|---|---|---|
| 5’UTR | c.−20G→A | fs | 1 | [78] |
| 2 | 88C→T | L30L | 0 | [79] |
| 2 | 4128G→A + 187C→G | G43D + H63D | 1 | [80] |
| 2 | 138T→G | L46W | 1 | [81] |
| 2 | c.del149–170 | L50fs | 0 | [82,83] |
| 2 | 128G→A | G43D | 1 | [84] |
| 2 | 157G→A | V53M | 0 | [85] |
| 2 | 175G→A | V59M | 0 | [85] |
| 2 | 187C→G | H63D | 1 | [13] |
| 2,4 | 4187C→G + 845G→C | H63D + C282Y | 1 | [86,87] |
| 2 | 189T→C | H63H | 0 | [85] |
| 2 | 193A→T | S65C | 1 | [88] |
| 2 | 196C→T | R66C | 1 | [79] |
| 2 | 199C→T | R67C | 1 | [83] |
| 2 | c.del203 | V68fs | 2 | [89] |
| 2 | 211C→T | R74X | 2 | [90] |
| 2 | 277G→C | G93R | 2 | [91] |
| 2 | 277del | G93fs | 2 | [92] |
| 2 | 314T→C | I105T | 1 | [91] |
| 2 | 340G→A | E114K | 1 | [83] |
| IVS2(+4)T→C | — | 0 | [90] | |
| 3 | 381A→C | Q127H | 1 | [85] |
| 3 | 385G→A | D129N | 0 | [81] |
| 3 | 414C→G | Y138X | 2 | [81] |
| 3 | 471del | A158fs | 2 | [93] |
| 3 | 478del | P160fs | 2 | [94] |
| 3 | 502G→C | E168Q | 1 | [95] |
| 3 | 502G→T | E168X | 2 | [96] |
| 3, 2 | 4502G→C + 187C→G | E168Q + H63D | 1 | [97] |
| 3 | 506G→A | W169X | 2 | [96] |
| 3 | 527C→T | A176V | 1 | [98] |
| 3 | 548T→C | L183P | 2 | [99] |
| IVS3(+1)G→T | (null allele) | 2 | [100] | |
| IVS3(+21)T→C | G43D | 1 | [79] | |
| IVS3(+21)T→C | — | 0 | [79] | |
| IVS3(−48)C→G | — | 0 | [101] | |
| 4 | c.del616–48C→T | — | 0 | [102] |
| 4 | 636G→C | V212V | 0 | [103] |
| 4 | 671G→A | R224G | 1 | [79] |
| 4 | 676C→G | R226G | 1 | [104] |
| 4 | 689A→T | Y230F | 2 | [81] |
| 4 | c.del691–693 | Y231X | 2 | [105] |
| 4 | 696C→T | P232P | 0 | [79] |
| 4 | 697C→T | Q233X | 2 | [106] |
| 4 | c.dup794 | W267fs | 2 | [107] |
| 4 | 724G→A | D242D | 0 | [102] |
| 4 | 747G→A | K249K | 0 | [102] |
| 4 | 814G→T | V272L | 0 | [108] |
| 4 | 829G→A | E277K | 0 | [103] |
| 4 | 845G→A | C282Y | 2 | [13] |
| 4 | 845G→C | C282S | 2 | [109] |
| 4 | 4845G→A + 842C→A | C282Y + T281K | 1 | [110] |
| 4 | 847C→T | G283X | 2 | [111] |
| 4 | 848A→C | Q283P | 2 | [112] |
| 4 | 867C→G | L289L | 0 | [79] |
| 4 | 884T→A | V295E | 1 | [102] |
| 4 | 884T→C | V295A | ? | [89] |
| 4 | 867G→C | L289L | 0 | [79] |
| IVS4(+37)A→G | — | 0 | [85] | |
| IVS4(+48)G→A | — | 0 | [113] | |
| IVS4(+109)A→G | — | 0 | [85] | |
| IVS4(−44)T→C | — | ? | [114] | |
| IVS4(−50)A→G | — | ? | [115] | |
| IVS4(+115)T→C | — | 0 | [85] | |
| 942T→C | A314A | 0 | [98] | |
| 5 | 989G→T | R330M | 2 | [85] |
| IVS5(+1)G→A | — | 1 | [116] | |
| IVS5(−47)G→A | — | ? | [114] | |
| 6 | c.1022–1034del13 | H341X | 2 | [83] |
| — | 5HFEdel | — | 1 or 2 | [117] |
Modified from C.Q. Edwards, J.C. Barton, Hemochromatosis in: J.P. Greer, D.A. Arber, B. Glader, A.F. List, R.T. Means Jr., F. Paraskevas, and G.M. Rodgers (Eds.), Wintrobe’s Clinical Hematology, Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia, 2014, pp. 662–81. Permission to publish requested from publisher.
Most alleles were identified in persons with hemochromatosis phenotypes or their family members. HFE Y231X was identified in a hemochromatosis cell line.
Phenotype: 0 = none known; 1 = probably weak effect on iron homeostasis; 2 = probably strong effect on iron homeostasis.
Complex allele with two mutations in cis.
An Alu-mediated recombination caused loss of the complete HFE gene sequence. Homozygosity for the corresponding chromosome 6p is a common cause of hemochromatosis in Sardinia [118].
4.4. HFE hemochromatosis modifiers
HFE p.C282Y homozygosity is necessary but not sufficient to cause hemochromatosis phenotypes [119]. Several investigators reported that iron phenotypes were more severe in cohorts of hemochromatosis patients who inherited the common hemochromatosis ancestral haplotype [30,120–122]. Other genetic attributes reported to be “modifiers” of iron phenotypes in large cohorts of p.C282Y homozygotes include common alleles of TF [123,124]; BMP2 (rs235756) [125,126]; single-nucleotide polymorphisms (SNPs) at ARNTL and TFR2 [123]; CYBRD1 (rs235756) [127]; GNPAT (rs11558492) [128]; and a microhaplotype on chromosome 6p [129].
4.5. Digenic hemochromatosis
Iron overload has been reported in persons who have digenic inheritance of one or more HFE mutations and a mutation of a non-HFE gene that is also involved in iron metabolism [79,102,130–135]. An example is the development of hemochromatosis in persons who are double heterozygotes for one or more HFE mutations and a mutation of the hepcidin gene (HAMP) [102,132]. Iron loading has occurred in persons with digenic inheritance of a HFE mutation and either a mutation of the hemojuvelin gene (HJV) [102,128,135] or the TFR2 gene (TFR2) [79]. Iron loading is often interpreted to be a synergistic effect of the two mutations because it is unlikely that either mutation alone would cause iron overload. Regardless, evidence is usually inadequate to prove that mutations of the two genes account for additive or multiplicative effects on iron absorption and retention within the same individual.
4.6. HFE alleles and porphyria cutanea tarda
Sporadic porphyria cutanea tarda (S-PCT), the most common of the porphyrias, is characterized by decreased activity of uroporphyrinogen decarboxylase (URO-D) in hepatocytes, accumulation of uroporphyrinogen I, photosensitivity dermatitis, and increased storage iron [136,137]. In persons with decreased URO-D activity, increased storage iron causes oxidation of uroporphyrinogen, resulting in the production of uroporphomethene. Uroporphomethene inhibits the decreased activity of URO-D further [138,139]. Prevalences of p.C282Y and p.H63D are much greater in persons with PCT [136,140–143]. Some persons with S-PCT or familial PCT and severe iron overload are homozygous for HFE mutations [144].
4.7. HFE C282Y frequency and geography
Molecular studies demonstrate that p.C282Y arose ~4000 years ago in the Neolithic Age [145,146] in Europe [147], possibly in Celtic people [148,149]. Although Vikings may have dispersed p.C282Y [146,149–151], especially in the late 8th-11th C, p.C282Y arose much earlier than the Viking era and thus may have also been spread in Europe by earlier seafarers [150]. Today, there are clines of decreasing p.C282Y frequency from Northwestern Europe to more eastern and southern regions of the continent [64,150]. Allele frequencies of p.C282Y in ethnically diverse western European white populations are 5–14% [152,153] and in North American non-Hispanic whites are 6–7% [154]. p.C282Y exists as a polymorphism only in Western European white and derivative populations, although p.C282Y may have arisen independently in non-whites outside Europe [155].
HFE p.C282Y arose on an ancestral chromosome 6p haplotype that included either HLA-A*03, B*07 or -A*03, B*14 and other alleles [5,6]. Part or all of the ancestral haplotypes is detectable in a majority of p.C282Y homozygotes with hemochromatosis phenotypes in Northwestern European and derivative countries [72,156–159]. Haplotypes A*03, B*35 and A*01, B*08, presumed to be linked to p.C282Y by the effects of migration and recombination, are common in subjects with p.C282Y homozygosity in northern Italy [160,161] and Sweden [156,162], respectively. Relative frequencies of haplotypes A*02, B*12 and A*09, B*05 are increased in Portuguese subjects with p.C282Y homozygosity [163].
4.8. HFE H63D frequency and geography
p.H63D is cosmopolitan but its frequency is greatest in whites of European descent [164,165]. Allele frequencies of p.H63D in ethnically diverse western European populations are 10–29% [166] and in North American non-Hispanic whites are 14–15% [154]. European haplotypes bearing p.H63D are typically associated with intronic haplotype TTG [114]. In northern Portuguese subjects, there is linkage disequilibrium between p.H63D and HLA-A*29-containing haplotypes [167]. In 19 populations of Central Eurasia, p.H63D was associated with three intronic haplotypes [168]. p.H63D, common in Indians, is associated with the European haplotype [169]. In Chinese subjects, p.H63D was detected on a variety of HLA haplotypes, indicating that p.H63D may predate the more genetically and geographically restricted p.C282Y mutation [170]. In Australian Aborigines, p.H63D was associated with HLA haplotypes common in Caucasians, suggesting that p.H63D was introduced by admixture. p.H63D (and p.C282Y) is absent in Brazilian Amerindians [171]. These observations suggest that p.H63D originated in Europe although multicentric origin, especially in Asia, cannot be excluded.
4.9. HFE p.S65C frequency and geography
In European whites, p.S65C is typically linked to intronic haplotype CCA [114]. In the Azores, p.S65C occurred in linkage disequilibrium with HLA-A*29 and -B*44 and with haplotype A*29, B*44 [172]. In Alabama whites with iron overload, p.S65C was linked to HLA-A*32 [91].
Allele frequencies of p.S65C in French and Basque cohorts were 2.5% and 2.9%, respectively [70,74]. In Sweden and Lithuania, allele frequencies were 1.6% and 1.9%, respectively [151,173]. In Canadian blood donors, the frequency of p.S65C was 2.0% [174]. In the Hemochromatosis and Iron Overload Screening (HEIRS) Study, the allele frequency of p.S65C in North American whites without high iron phenotypes was 0.7%. p.S65C was very uncommon in populations in Spain and the Mediterranean basin [175,176] and was not detected in Roma-Gypsies or Chinese men [177,178]. In the HEIRS Study, p.S65C was not detected in Hispanics, blacks, or Asians [102]. These observations suggest that p.S65C also arose in Europe.
4.10. Advantages of common HFE mutations
HFE appears to be an example of a non-classical MHC locus that evolved a novel function, but what function? “The mutations of the HFE gene have all of the hallmarks of a balanced polymorphism…one in which the beneficial effect of the heterozygous state balances the deleterious effect of the homozygous state” [179]. It is plausible that protection against deficiency of iron or other trace metals absorbed by the same pathways, especially divalent metal-ion transporter (DMT) [180], and resistance to infectious disease are advantages that may have accrued to p.C282Y heterozygotes and resulted in fixation of p.C282Y in European Caucasian populations.
Women heterozygous for p.C282Y had higher values of hemoglobin, serum iron, and TS than women homozygous for the wild-type HFE allele in a small study [181]. In a larger study of similar design, a protective role against iron deficiency was not detected [179]. Prevalence of iron deficiency without anemia was lower among women heterozygous for p.C282Y than women homozygous for the wild-type HFE allele. There was a small but significant upward shift in the mid-range of the hemoglobin distribution among p.C282Y heterozygotes, consistent with an increased mean hemoglobin level without significant changes in the anemia range [182]. In one study, there was no demonstrable effect of p.C282Y on absorption of either heme or non-heme iron [183]. Taken together, these results suggest that putative evolutionary benefits of p.C282Y heterozygosity with respect to iron absorption, if any, are too small to measure. p.C282Y homozygotes absorb or retain increased amounts of zinc, manganese, and cobalt [184–186]. It has not been reported that p.C282Y heterozygotes absorb or retain greater proportions of non-ferrous metals with physiologic function.
It has been postulated that HFE is a receptor for microorganisms and that p.C282Y would protect against infection, although no specific microorganism(s) was proposed [155]. Although malaria in Europe was more common in areas adjacent to the Mediterranean Sea, malaria was endemic and epidemic in areas adjacent to the North Sea from late Antiquity until the latter half of the 19th C [187–189]. The larger erythrocytes of p.C282Y heterozygotes than those of HFE wild-type subjects [182,190] may have provided relative protection against malaria, although this is unproven. Mice lacking one or both Hfe genes were protected from Salmonella typhimurium septicemia because loss of Hfe induced the iron-capturing peptide LCN2 [191]. In northern Portuguese subjects, there is linkage disequilibrium between p.H63D and all HLA-A*29-containing haplotypes. Persons who have both p.H63D and A*29 have higher blood CD8+ T-lymphocyte counts [167].
5. HFE protein structure, cell and tissue expression, and function
5.1. HFE structure
HFE is a protein of 343 amino acids that includes a signal peptide, an extracellular transferrin receptor-binding region (α1 and α2), an immunoglobulin-like domain (α3), a transmembrane region, and a short cytoplasmic tail [13] (Fig. 1). HFE binds β2M to form a heterodimer expressed at the cell surface [13]. HFE is glycosylated at asparagine residues 110, 130 and 234 during transport to the cell membrane [192]. Glycosylation is important for normal intracellular trafficking and function [192]. HFE interacts with TFRC [193].
Figure 1.
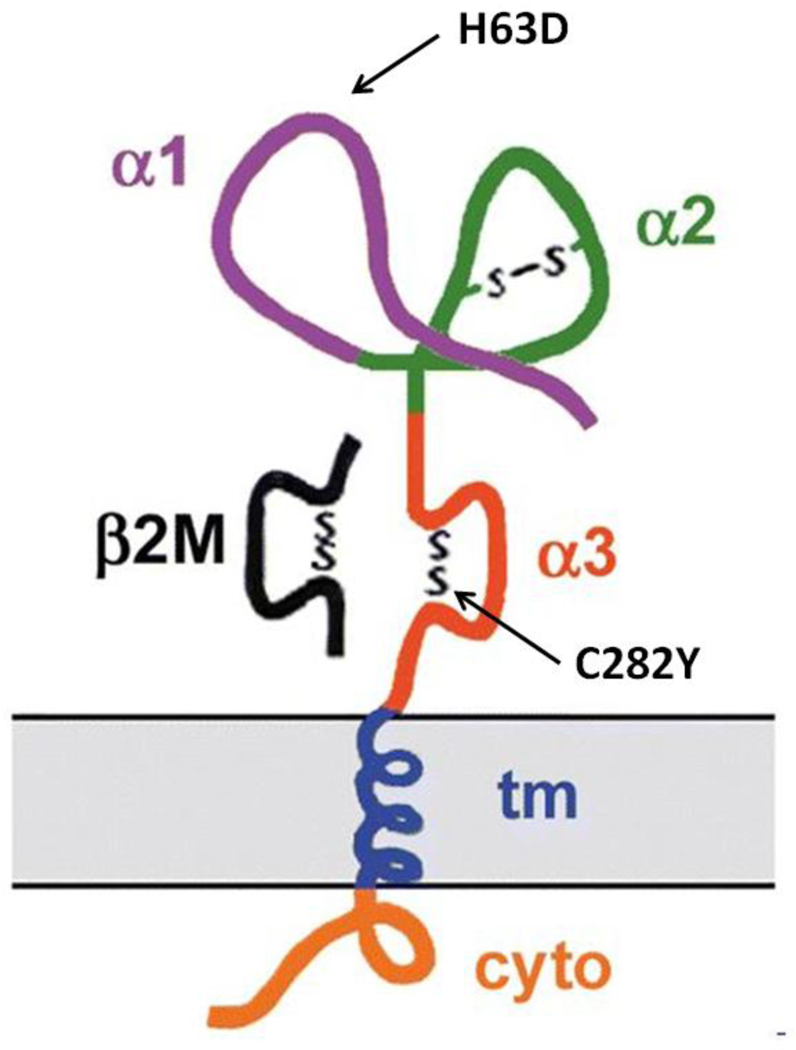
HFE protein in association with beta-2 microglobulin (β2M) at the cell surface. The three extracellular domains of HFE are designated α1, α2, and α3. β2M is shown associated with the α3 domain. Abbreviations: cyto, cytoplasmic tail; tm, transmembrane domain. Adapted from R.E. Fleming, W. S. Sly, Mechanisms of iron accumulation in hereditary hemochromatosis. Annu Rev Physiol 64 (2002) 663–680. Used with permission.
The 2.6 Å crystal structure of HFE revealed that its ligand TFRC binds in a 2:1 TFRC:HFE molar ratio [61]. Most class I MHC molecules have a peptide-binding groove. Because the α1 and α2 helices are closer in HFE, the analogous site in HFE is too narrow for peptide binding [61]. TFRC and HFE bind tightly at the basic pH of cell surfaces, but not at the acidic pH of intracellular vesicles [61]. The 2.8 Å crystal structure of a complex between the extracellular portions of HFE and TFRC reveals that binding alters configurations of both HFE and its ligand [194]. The structures of TFRC alone and TFRC complexed with HFE differ in their domain arrangements and dimer interfaces [194].
Studies of cultured 293 cells overexpressing HFE wild-type proteins revealed that HFE forms stable complexes with TFR. In 293 cells overexpressing HFE C282Y, the association of HFE protein with TFR was markedly decreased [195]. Normal HFE protein decreased the affinity of TFRC for TF by inhibiting TFRC:TF-Fe interaction in an assay using purified proteins and a biosensor chip [61]. When HFE binds to TFRC in vitro, HFE changes the conformation of the Tf-Fe binding site as detected by biosensor assays, decreasing iron entry into Chinese hamster ovary cells [14]. In co-immunoprecipitation or surface plasmon resonance-based assay experiments using soluble HFE and TFR2, no evidence of binding of HFE and TFR2 was detected [196]. The pertinence of these in vitro results to iron homeostasis in vivo, if any, is unclear.
5.2. HFE function
Numerous factors, including HFE, act as upstream regulators of hepcidin transcription [197] (Fig. 2). The expression of HAMP was significantly lower in untreated patients with hemochromatosis, C282Y homozygosity, and iron overload than controls [198]. There was a significant correlation between hepatic iron concentration and expression of HAMP and SLC40A1 in untreated hemochromatosis patients [198]. In iron-loaded Hfe knockout mice, liver hepcidin expression is relatively decreased [199]. These observations indicate that HFE plays an important part in the regulation of hepcidin expression in response to iron overload and that the liver is important in the pathophysiology of HFE-associated hemochromatosis [198]. These results also suggest that ferroportin could facilitate removal of excess iron from the liver [198]. Thus, HFE C282Y homozygosity results in decreased hepcidin responsiveness to iron and relative or absolute hepcidin deficiency [197].
Figure 2.
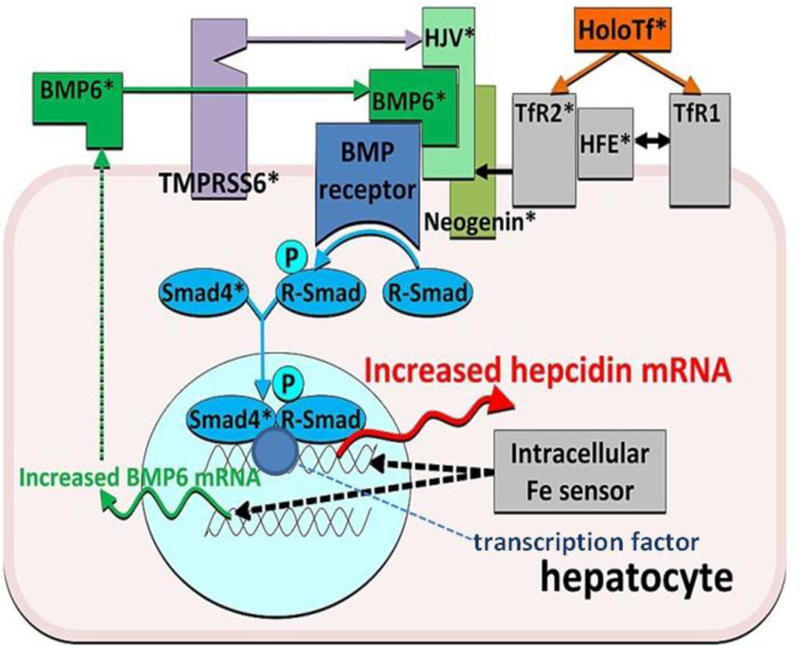
A model of regulation of hepcidin transcription by iron. Iron as holotransferrin is shown in orange, iron sensors and associated molecule in gray, bone morphogenic protein (BMP) receptor and its transduction pathway in shades of blue, the ligands and co-receptors of the BMP receptor in shades of green, and the negative regulator protease in purple. *Molecules the ablation of which caused iron dysregulation. Adapted from T. Ganz, Hepcidin and iron regulation, 10 years later, Blood 117 (2011) 4425–4433. Used with permission.
5.3. HFE in cells and tissues
Small amounts of HFE are expressed in almost all normal cells and tissues [200]. Antibodies used to localize HFE in cells and tissues in several studies were raised against protein-specific peptides, not intact HFE [201–204]. Accordingly, uncertainty remains about the utility of these antibodies in localizing intact HFE protein in cells and tissues. HFE in human enterocytes appears in a decreasing gradient from villous crypts to villous tips and from duodenum to ileum [201,202], although the physiologic significance of these observations is unclear. Knockout of duodenal Hfe in mice does not lead to iron overload [205]. Expression is prominent in gastric epithelial cells, tissue macrophages, and blood monocytes and granulocytes [202,203]. Using immunohistochemical technique, staining for HFE in human liver was positive in the basolateral plasma membranes of bile ductular epithelium and sinusoidal lining cells [206]. In another study, liver immunostaining for HFE was most prominent in human Kupffer cells [207]. In rat liver, there was high expression of HFE mRNA, predominantly in hepatocytes, using quantitative real-time polymerase chain reaction [208]. These results differ from those of two previous studies [206,207]. A possible cause of the discrepancy could be non-specific immunostaining of non-HFE MHC class 1 molecules. HFE is expressed on apical plasma membranes of the syncytiotrophoblast, an iron transport tissue in the placenta [204].
Duodenal expression of HFE and TFR2 (but not TFRC) in wild-type mice and humans was restricted to small intestinal crypt cells where the respective proteins are co-localized [209]. HFE and TFRC are also co-localized in 293 and HeLa cells [195]. In human Caco-2 cells, HFE and TFR2 co-localized to a vesicular compartment that had marked signal enhancement on exposure to iron-saturated transferrin ligand, indicating that HFE preferentially interacts with TFR2 in an early endosomal transport pathway for TF:Fe [209]. In HuH7 hepatoma-derived cells, normal HFE, TFR2, and hemojuvelin form a membrane-associated complex that functions to regulate hepcidin [210].
In cells from HFE p.C282Y homozygotes with hemochromatosis, p.C282Y is not present at cell surfaces, has a diffuse cytoplasmic localization, does not co-localize with β2M and TFRC, and is retained in the endoplasmic reticulum [50]. Cell surface-associated HFE signal is reduced in gastric epithelial cells, monocytes, and macrophages [203]. The cellular distribution of TFR2 in small intestinal crypt cells from p.C282Y homozygotes is also altered [209]. p.C282Y is retained in the endoplasmic reticulum and middle Golgi compartment, fails to undergo late Golgi processing, and is subject to accelerated degradation [201].
6. Mice with Hfe knockouts
6.1. Discovery of murine Hfe
Hashimoto and colleagues isolated the mouse ortholog of human HFE and designated the mouse gene as “MR2” [211], now widely known as Hfe. Compared with human HFE, mouse Hfe has a predicted amino acid sequence similarity of ~66% and is analogously expressed in various tissues. Eight amino acid residues between mouse Hfe α1 and α2 domains that are not present in human HFE are due to a coding sequence from the intron [211]. Whereas human HFE is telomeric to the MHC on chromosome 6p, mouse Hfe has been translocated from a site telomeric to the MHC on chromosome 17 to chromosome 13, along with other genes [211]. Soon after human HFE was reported [13], structural details of mouse Hfe were described [212].
6.2. Hfe knockouts
Zhu and colleagues produced a targeted knockout of all six Hfe exons in the mouse [213]. The mRNA transcript of 1.9 kb, present in multiple tissues from Hfe+/+ mice, was present in reduced amounts in Hfe+/− mice, and was not detectable in the livers, kidneys, and spleens of Hfe−/− mice. Thus, the knockout produced a null allele. On a standard diet, Hfe−/− mice had elevated TS and increased liver iron, predominantly in hepatocytes. Iron measures in heterozygous Hfe+/− mice were normal at age 10 weeks. Iron-related traits of Hfe−/− mice were inherited in an autosomal recessive pattern. This mouse Hfe knockout model simulates genetic and biochemical abnormalities of HFE hemochromatosis [213]. Iron-related characteristics of different Hfe−/− mouse strains vary [214]. Hepatic gene expression profiles differ according to strain and age [215]. The pattern of hepatic iron loading inheritance in Hfe−/− mice is polygenic [216].
HFE interacts with TFRC in the α1–α2 groove [14]. Bahram and colleagues created a knockout mouse by deleting the second and third Hfe exons (corresponding to α1 and α2 domains of Hfe) [217]. Mice homozygous for this deletion had increased duodenal iron absorption, elevated plasma iron and TS levels, and iron overload, predominantly in hepatocytes [217].
Levy and colleagues used targeted mutagenesis to produce two mutations in Hfe. The first deleted a large portion of the coding sequence, generating a null allele. The second introduced a missense mutation (C282Y) [218]. Homozygosity for both mutations caused iron loading, although effects of the null mutation were more severe. Mice heterozygous for either mutation accumulated more iron than normal controls. Thus, the murine Hfe C282Y mutation does not result in a null allele [218].
6.3. Tissue-specific Hfe knockouts
Mice with deletion of Hfe in crypt and villous enterocytes had normal plasma iron and TS values, normal unbound iron-binding capacity, normal liver and spleen iron concentrations, and normal hepcidin mRNA expression [205]. These observations demonstrate that small intestinal Hfe is not necessary for the physiologic control of systemic iron homeostasis [205]. Mice with tissue-specific Hfe knockout in macrophages had normal plasma iron measures and normal iron concentrations in liver and spleen [219]. This is consistent with observations in wild-type mice subjected to macrophage depletion which have normal hepatic iron levels and hepcidin responses to iron challenges [220,221]. Mice with tissue-specific Hfe knockout in hepatocytes developed iron phenotypes similar to those of Hfe−/− mice, including elevated serum iron and TS values, severe hepatic iron accumulation, and reduced splenic iron content [219]. These findings indicate that Hfe must be expressed in hepatocytes to prevent iron overload because it is important for appropriate hepcidin mRNA expression [219].
6.4. Hfe knockins
Levy et al. used a targeting vector to introduce HFE C282Y into murine Hfe codon 282 and a second vector to construct a Hfe null allele [218]. Mice heterozygous for either mutant allele developed more iron loading than wild-type control mice. Mice homozygous for the null allele developed massive iron overload. Mice homozygous for the C282Y knockin had an iron phenotype intermediate between that of null homozygotes and wild-type mice. This indicates that C282Y caused hepatic iron accumulation without total loss of function. Mice homozygous for each Hfe mutation had less non-heme splenic iron than wild-type mice [218]. Relative resistance of the spleen to iron loading also occurs in HFE hemochromatosis [4,222].
Ajioka et al. performed additional studies of Hfe mutant (both C282Y knockins and Hfe knockouts) and wild-type mice [223]. In Hfe mutant mice and wild-type mice, down-regulation of iron absorption occurred with dietary iron loading and with age, although the Hfe mutant mice continued to absorb more iron than wild-type mice. Iron absorption increased in response to reduced iron stores and accelerated erythropoiesis to a similar degree in Hfe mutant and wild-type mice. These results suggest that mouse Hfe plays a minor role in down-regulation of iron absorption but does not influence its up-regulation [223].
Tomatsu et al. generated knockin mice homozygous for Hfe H67D (corresponding to human H63D), homozygous for Hfe C294Y (corresponding to human C282Y), and Hfe C294Y/H67D compound heterozygotes. Hepatic iron loading was significantly greater in all three groups of mice with Hfe mutations than in control wild-type mice. Iron loading was most severe in C294Y homozygotes, less severe in C294Y/H67D compound heterozygotes, and even less in H67D homozygotes. TS was increased only in C294Y homozygotes. Tomatsu et al. concluded that Hfe H67D in a homozygous configuration or in compound heterozygosity with C294Y results in partial loss of Hfe function and increased hepatic iron loading [224].
Schmidt et al. introduced mutations into a ubiquitously expressed Tfr1 transgene or the endogenous Tfr1 locus to promote or prevent Hfe/Tfr1 interaction [225]. In one mouse model, Hfe constitutively interacted with Tfr1. In two other models, most or all Hfe was free of Tfr1. Under conditions favoring constitutive Hfe/Tfr1 interaction, mice developed iron overload attributed to inappropriately low expression of hepcidin. Mice with a mutation that interferes with Hfe/Tfr1 interaction developed iron deficiency associated with inappropriately high hepcidin expression. High-level expression of a liver-specific Hfe transgene in Hfe−/− mice was also associated with increased hepcidin production and iron deficiency. These results suggest that Hfe induces hepcidin expression when it is not complexed with Tfr1 [225].
6.5. Iron absorption
In a study of iron uptake by duodenal enterocytes of Hfe−/− mice, Herrmann et al. examined ferric reductase duodenal cytochrome b (Dcytb) mRNA and speculated that Dcytb may be important in iron uptake[226]. In persons with HFE hemochromatosis, CYBRD1 (DYCTB) expression was increased in some reports [227,228] and not in others [229–231]. In Hfe−/− mice, DMT mRNA transcripts containing an iron-responsive element are greatly increased, despite the existence of iron overload [232]. Immunoreactive DMT is up-regulated in Hfe−/− mice [233]. Absorption of both ferrous and ferric iron by Hfe−/− mice is greater than that of Hfe+/+ mice [233]. In HFE hemochromatosis, DMT mRNA levels are also increased [234].
Down-regulation of iron absorption occurred in Hfe−/− mice with dietary iron loading and with age, although to a lesser extent than in wild-type mice [223]. Up-regulation of iron absorption consequent to phlebotomy-induced iron deficiency or phenylhydrazine-induced hemolysis was similar in Hfe−/− and wild-type mice [223]. Levels of liver hepcidin mRNA are higher in Hfe+/+ than Hfe−/− mice [235]. In Hfe−/− mice, an iron challenge down-regulated hepcidin production and decreased hepatic Tfr2 levels [235]. Iron overload was abrogated in Hfe−/− mice with constitutive overexpression of the gene that encodes hepcidin (Hamp) [236]. The normal relationship between body iron stores and liver hepcidin mRNA levels is altered in Hfe−/− mice, such that liver hepcidin expression is relatively decreased despite iron overload [199]. These observations substantiate that hepcidin produced in the liver is a central controller of iron absorption [16].
6.6. Iron uptake by hepatocytes
Chua et al. studied hepatocyte iron uptake in Hfe−/− mice and in iron-loaded and non-iron-loaded wild-type mice [237]. Tfr1-mediated iron and Tf uptake of hepatocytes were significantly greater in Hfe knockout mice than in wild-type mice with similar iron levels and Tfr1 expression. This indicates that Tfr1-mediated hepatocyte iron uptake is regulated by Hfe. Hepatocyte iron uptake was much greater via the Tfr1-independent pathway than the Tfr1 pathway but the former was not regulated by Hfe [237]. Tfr2 levels are higher in livers of Hfe−/− than Hfe+/+ mice, indicating that loss of Hfe function does not interfere with iron-responsive regulation of Tfr2 [238]. Diferric transferrin up-regulated hepatocyte Tfr2 protein expression but not iron uptake, suggesting that Tfr2 has a limited role in the Tfr1-independent pathway [237]. Mice with hepatocyte-specific knockout of Hfe also develop hepatic iron overload [219]. In Hfe−/− mice, hepatocyte export of iron via ferroportin is decreased [239].
6.7. Iron and erythropoiesis
Hfe is expressed by splenic erythroid cells and in vitro splenic erythroid colonies of phlebotomized wild-type mice [240]. These results suggest but do not prove that Hfe is not a sensor for hepcidin in erythroid cells [240]. Hfe−/− mice down-regulate hepcidin expression to the same extent as wild-type mice in response to both phlebotomy and erythropoietin injections but recover more rapidly from phlebotomy- or phenylhydrazine-induced anemia than wild-type mice with iron overload of similar severity [240]. Erythroid cell uptake of iron per Tfr1 is greater in Hfe−/− than wild-type mice, suggesting that Hfe interferes with erythroid Tf-Fe uptake [240]. Iron absorption increased similarly in response to hypoxia in Hfe−/− and Hfe+/+ mice [241]. Thus, at least two independent mechanisms regulate iron absorption, only one of which requires Hfe [241].
7. HFE mutations and iron overload in other mammals
Black rhinoceroses (Diceros bicornis) develop iron overload [91,242,243]. To determine whether the HFE gene of black rhinoceroses has undergone mutation as an adaptive mechanism to improve iron absorption from iron-poor diets, Beutler et al. sequenced the entire HFE coding region of four species of rhinoceros (two browsing and two grazing species). Although HFE was well conserved across the species, numerous nucleotide differences were found between rhinoceros and human or mouse, some of which changed deduced amino acids. Only one allele, p.S88T in the black rhinoceros, was a candidate that might adversely affect HFE function. p.S88T occurs in a highly conserved region involved in the interaction of HFE and TFRC [244]. Bottle-nosed dolphins (Tursiops truncatus) also develop iron overload [245,246] but sequencing dolphin hfe did not reveal deleterious mutations [247]. Red deer with iron storage disease (Cervus elaphus elaphus) did not have pathogenic HFE mutations [248].
Highlights.
HFE, the hemochromatosis gene, is linked to the major histocompatibility complex on chromosome 6p
HFE encodes HFE, an extracellular protein that binds beta-2 microglobulin
HFE is a positive upstream regulator of hepcidin.
Common HFE mutations account for most hemochromatosis cases
Iron phenotypes of mice homozygous for Hfe knockouts are similar to those of HFE hemochromatosis
Acknowledgments
This review and the corresponding Gene Wiki article were written as part of the Cardiac Gene Wiki Review series - a series resulting from a collaboration between the journal GENE, the Gene Wiki Initiative, and the BD2K initiative. The Cardiac Gene Wiki Initiative is supported by National Institutes of Health (GM089820). Additional support for Gene Wiki Reviews is provided by Elsevier, the publisher of GENE. This work was supported in part by Southern Iron Disorders Center. The corresponding Gene Wiki entry for this review can be found here:<<https://en.wikipedia.org/wiki/HFE_%28gene%29>>.
Abbreviations
- BLAST
Basic Local Alignment Search Tool
- β2M
beta-2 microglobulin
- chromosome 6p
short arm of chromosome 6
- DMT
divalent metal-ion transporter
- HEIRS Study
Hemochromatosis and Iron Overload Screening Study
- HFE
hemochromatosis gene (human)
- Hfe
hemochromatosis gene (mouse)
- HLA
human leukocyte antigen
- MHC
major histocompatibility complex
- OMIM
Online Mendelian Inheritance in Man
- PARP1
poly (ADP-ribose) polymerase 1
- PCT
porphyria cutanea tarda
- sHFE
soluble HFE
- TF
transferrin (human)
- Tf
transferrin (mouse)
- TF-Fe
iron-loaded transferrin (human)
- Tf-Fe
iron-loaded transferrin (mouse)
- TFRC
transferrin receptor (human)
- Tfr1
transferrin receptor (mouse)
- TFR2
transferrin receptor-2 (human)
- Tfr2
transferrin receptor-2 (mouse)
- TS
transferrin saturation
- SNP
single nucleotide polymorphism
- URO-D
uroporphyrinogen decarboxylase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Concluding Comments
Studies of HFE and HFE protein in humans and mice have greatly advanced knowledge about non-classical MHC class I genes and proteins, iron homeostasis in mammals, and HFE hemochromatosis and other iron overload disorders in humans.
References
- [1].Trousseau A, Glycosurie, diabète sucre, Clinique médicale de l’Hôtel-Dieu de Paris 2 (1865) 663–698. [Google Scholar]
- [2].Troisier M, Diabète sucre, Bull Soc Anat Paris 44 (1871) 231–235. [Google Scholar]
- [3].von Recklinghausen FD, Über hämochromatose, Tagebl Versamml Natur Ärtze Heidelberg 62 (1889) 324–325. [Google Scholar]
- [4].J.H. Sheldon Haemochromatosis, Oxford University Press, Oxford, 1935. [Google Scholar]
- [5].Simon M, Pawlotsky Y, Bourel M, Fauchet R, Genetet B, [Letter: Idiopathic hemochromatosis associated with HL-A 3 tissular antigen], Nouv Presse Med 4 (1975) 1432–1435. [PubMed] [Google Scholar]
- [6].Simon M, Bourel M, Fauchet R, Genetet B, Association of HLA-A3 and HLA-B14 antigens with idiopathic haemochromatosis, Gut 17 (1976) 332–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Edwards CQ, Cartwright GE, Skolnick MH, Amos DB, Genetic mapping of the hemochromatosis locus on chromosome six, Hum Immunol 1 (1980) 19–22. [DOI] [PubMed] [Google Scholar]
- [8].Simon M, Le ML, Fauchet R, Yaouanq J, David V, Edan G, Bourel M, A study of 609 HLA haplotypes marking for the hemochromatosis gene: (1) mapping of the gene near the HLA-A locus and characters required to define a heterozygous population and (2) hypothesis concerning the underlying cause of hemochromatosis-HLA association, Am J Hum Genet 41 (1987) 89–105. [PMC free article] [PubMed] [Google Scholar]
- [9].Rothenberg BE, Voland JR, beta2 knockout mice develop parenchymal iron overload: A putative role for class I genes of the major histocompatibility complex in iron metabolism, Proc Natl Acad Sci U S A 93 (1996) 1529–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Santos M, Schilham MW, Rademakers LH, Marx JJ, de SM, Clevers H, Defective iron homeostasis in beta 2-microglobulin knockout mice recapitulates hereditary hemochromatosis in man, J Exp Med 184 (1996) 1975–1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Muckenthaler MU, Rodrigues P, Macedo MG, Minana B, Brennan K, Cardoso EM, Hentze MW, de SM, Molecular analysis of iron overload in beta2-microglobulin-deficient mice, Blood Cells Mol Dis 33 (2004) 125–131. [DOI] [PubMed] [Google Scholar]
- [12].Walker AP, Wallace DF, Partridge J, Bomford AB, Dooley JS, Atypical haemochromatosis: phenotypic spectrum and beta2-microglobulin candidate gene analysis, J Med Genet 36 (1999) 537–541. [PMC free article] [PubMed] [Google Scholar]
- [13].Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R Jr., Ellis MC, Fullan A, Hinton LM, Jones NL, Kimmel BE, Kronmal GS, Lauer P, Lee VK, Loeb DB, Mapa FA, McClelland E, Meyer NC, Mintier GA, Moeller N, Moore T, Morikang E, Prass CE, Quintana L, Starnes SM, Schatzman RC, Brunke KJ, Drayna DT, Risch NJ, Bacon BR, Wolff RK, A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis, Nat Genet 13 (1996) 399–408. [DOI] [PubMed] [Google Scholar]
- [14].Lebron JA, Bjorkman PJ, The transferrin receptor binding site on HFE, the class I MHC-related protein mutated in hereditary hemochromatosis, J Mol Biol 289 (1999) 1109–1118. [DOI] [PubMed] [Google Scholar]
- [15].Lebron JA, West AP Jr., Bjorkman PJ, The hemochromatosis protein HFE competes with transferrin for binding to the transferrin receptor, J Mol Biol 294 (1999) 239–245. [DOI] [PubMed] [Google Scholar]
- [16].Nemeth E, Ganz T, The role of hepcidin in iron metabolism, Acta Haematol 122 (2009) 78–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Feder JN, Thomas W, The cloning of the gene for hereditary hemochromatosis, Clin Chem 58 (2012) 1485–1486. [DOI] [PubMed] [Google Scholar]
- [18].Lloyd DA, Adams P, Sinclair NR, Stiller CR, Valberg LS, Histocompatibility antigens as markers of abnormal iron metabolism in idiopathic hemochromatosis, Can Med Assoc J 119 (1978) 1051–1056. [PMC free article] [PubMed] [Google Scholar]
- [19].Edwards CQ, Griffen LM, Dadone MM, Skolnick MH, Kushner JP, Mapping the locus for hereditary hemochromatosis: localization between HLA-B and HLA-A, Am J Hum Genet 38 (1986) 805–811. [PMC free article] [PubMed] [Google Scholar]
- [20].Radisky ES, Ajioka RS, Edwards CQ, Griffen LM, Kushner JP, Mapping recombinant events with molecular markers in hemochromatosis pedigrees, Cytogenet Cell Genet 67 (1994) 126–128. [DOI] [PubMed] [Google Scholar]
- [21].Gasparini P, Borgato L, Piperno A, Girelli D, Olivieri O, Gottardi E, Roetto A, Dianzani I, Fargion S, Schinaia G, Linkage analysis of 6p21 polymorphic markers and the hereditary hemochromatosis: localization of the gene centromeric to HLA-F, Hum Mol Genet 2 (1993) 571–576. [DOI] [PubMed] [Google Scholar]
- [22].Calandro LM, Baer DM, Sensabaugh GF, Characterization of a recombinant that locates the hereditary hemochromatosis gene telomeric to HLA-F, Hum Genet 96 (1995) 339–342. [DOI] [PubMed] [Google Scholar]
- [23].Litt M, Luty JA, Dinucleotide repeat polymorphism at the D6S89 locus, Nucleic Acids Res 18 (1990) 4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Polymeropoulos MH, Rath DS, Xiao H, Merril CR, Tetranucleotide repeat polymorphism at the human coagulation factor XIII A subunit gene (F13A1), Nucleic Acids Res 19 (1991) 4306. [PMC free article] [PubMed] [Google Scholar]
- [25].Ranum LP, Chung MY, Duvick LA, Zoghbi HY, Orr HT, Dinucleotide repeat polymorphism at the D6S109 locus, Nucleic Acids Res 19 (1991) 1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Weber JL, Kwitek AE, May PE, Zoghbi HY, Dinucleotide repeat polymorphism at the D6S105 locus, Nucleic Acids Res 19 (1991) 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Jazwinska EC, Lee SC, Webb SI, Halliday JW, Powell LW, Localization of the hemochromatosis gene close to D6S105, Am J Hum Genet 53 (1993) 347–352. [PMC free article] [PubMed] [Google Scholar]
- [28].Worwood M, Raha-Chowdhury R, Dorak MT, Darke C, Bowen DJ, Burnett AK, Alleles at D6S265 and D6S105 define a haemochromatosis-specific genotype, Br J Haematol 86 (1994) 863–866. [DOI] [PubMed] [Google Scholar]
- [29].Raha-Chowdhury R, Bowen DJ, Burnett AK, Worwood M, Allelic associations and homozygosity at loci from HLA-B to D6S299 in genetic haemochromatosis, J Med Genet 32 (1995) 446–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Barton JC, Harmon L, Rivers C, Acton RT, Hemochromatosis: association of severity of iron overload with genetic markers, Blood Cells Mol Dis 22 (1996) 195–204. [DOI] [PubMed] [Google Scholar]
- [31].Gandon G, Jouanolle AM, Chauvel B, Mauvieux V, Le TA, Feingold J, Le Gall JY, David V, Yaouanq J, Linkage disequilibrium and extended haplotypes in the HLA-A to D6S105 region: implications for mapping the hemochromatosis gene (HFE), Hum Genet 97 (1996) 103–113. [DOI] [PubMed] [Google Scholar]
- [32].Raha-Chowdhury R, Bowen DJ, Stone C, Pointon JJ, Terwilliger JD, Shearman JD, Robson KJ, Bomford A, Worwood M, New polymorphic microsatellite markers place the haemochromatosis gene telomeric to D6S105, Hum Mol Genet 4 (1995) 1869–1874. [DOI] [PubMed] [Google Scholar]
- [33].Camaschella C, Roetto A, Gasparini P, Piperno A, Fortina P, Surrey S, Rappaport E, Allelic association of microsatellites of 6p in Italian hemochromatosis patients, Hum Genet 97 (1996) 476–481. [DOI] [PubMed] [Google Scholar]
- [34].Tay GK, Cattley SK, Chorney MJ, Hollingsworth PN, Roth MP, Dawkins RL, Witt CS, Conservation of ancestral haplotypes telomeric of HLA-A, Eur J Immunogenet 24 (1997) 275–285. [DOI] [PubMed] [Google Scholar]
- [35].Beutler E, Gelbart T, West C, Kuhl W, Lee P, A strategy for cloning the hereditary hemochromatosis gene, Blood Cells Mol Dis 21 (1995) 207–216. [DOI] [PubMed] [Google Scholar]
- [36].Venditti CP, Harris JM, Geraghty DE, Chorney MJ, Mapping and characterization of non-HLA multigene assemblages in the human MHC class I region, Genomics 22 (1994) 257–266. [DOI] [PubMed] [Google Scholar]
- [37].Bodmer JG, Parham P, Albert ED, Marsh SG, Putting a hold on “HLA-H’. The WHO Nomenclature Committee for Factors of the HLA System, Nat Genet 15 (1997) 234–235. [DOI] [PubMed] [Google Scholar]
- [38].Mercier B, Mura C, Ferec C, Putting a hold on ‘HLA-H’, Nat Genet 15 (1997) 234. [DOI] [PubMed] [Google Scholar]
- [39].Dorak MT, HFE (hemochromatosis), Atlas Genet Cytogenet Oncol Haematol 13 (2009) 11–14. [Google Scholar]
- [40].Sanchez M, Queralt R, Bruguera M, Rodes J, Oliva R, Cloning, sequencing and characterization of the rat hereditary hemochromatosis promoter: comparison of the human, mouse and rat HFE promoter regions, Gene 225 (1998) 77–87. [DOI] [PubMed] [Google Scholar]
- [41].Mura C, le GG, Jacolot S, Ferec C, Transcriptional regulation of the human HFE gene indicates high liver expression and erythropoiesis coregulation, FASEB J 18 (2004) 1922–1924. [DOI] [PubMed] [Google Scholar]
- [42].Pelham C, Jimenez T, Rodova M, Rudolph A, Chipps E, Islam MR, Regulation of HFE expression by poly(ADP-ribose) polymerase-1 (PARP1) through an inverted repeat DNA sequence in the distal promoter, Biochim Biophys Acta 1829 (2013) 1257–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Thénié AC, Gicquel IM, Hardy S, Ferran H, Fergelot P, Le Gall JY, Mosser J, Identification of an endogenous RNA transcribed from the antisense strand of the HFE gene, Hum Mol Genet 10 (2001) 1859–1866. [DOI] [PubMed] [Google Scholar]
- [44].Jeffrey GP, Basclain K, Hajek J, Chakrabarti S, Adams PC, Alternate splicing produces a soluble form of the hereditary hemochromatosis protein HFE, Blood Cells Mol Dis 25 (1999) 61–67. [DOI] [PubMed] [Google Scholar]
- [45].Rhodes DA, Trowsdale J, Alternate splice variants of the hemochromatosis gene Hfe, Immunogenetics 49 (1999) 357–359. [DOI] [PubMed] [Google Scholar]
- [46].Thénié A, Orhant M, Gicquel I, Fergelot P, Le Gall JY, David V, Mosser J, The HFE gene undergoes alternate splicing processes, Blood Cells Mol Dis 26 (2000) 155–162. [DOI] [PubMed] [Google Scholar]
- [47].Sanchez M, Bruguera M, Rodes J, Oliva R, Complete characterization of the 3’ region of the human and mouse hereditary hemochromatosis HFE gene and detection of novel splicing forms, Blood Cells Mol Dis 27 (2001) 35–43. [DOI] [PubMed] [Google Scholar]
- [48].Hviid TV, HLA-G in human reproduction: aspects of genetics, function and pregnancy complications, Hum Reprod Update 12 (2006) 209–232. [DOI] [PubMed] [Google Scholar]
- [49].Sangrouber D, Marcou C, Le DM, Chang CC, Carosella ED, Moreau P, Cellular co-localization of intron-4 containing mRNA and HLA-G soluble protein in melanoma analyzed by fluorescence in situ hybridization, J Immunol Methods 326 (2007) 54–62. [DOI] [PubMed] [Google Scholar]
- [50].Martins R, Silva B, Proenca D, Faustino P, Differential HFE gene expression is regulated by alternative splicing in human tissues, PLoS One 6 (2011) e17542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Skop A, Protein homology (Hereditary Hemochromatosis; http://reesegen564s14.weebly.com/homology.html), Hereditary Hemochromatosis. Department of Genetics, University of Wisconsin-Madison; 2015. [Google Scholar]
- [52].Castro LF, Furlong RF, Holland PW, An antecedent of the MHC-linked genomic region in amphioxus, Immunogenetics 55 (2004) 782–784. [DOI] [PubMed] [Google Scholar]
- [53].Hashimoto K, Nakanishi T, Kurosawa Y, Isolation of carp genes encoding major histocompatibility complex antigens, Proc Natl Acad Sci U S A 87 (1990) 6863–6867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hashimoto K, Nakanishi T, Kurosawa Y, Identification of a shark sequence resembling the major histocompatibility complex class I alpha 3 domain, Proc Natl Acad Sci U S A 89 (1992) 2209–2212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Okamura K, Nakanishi T, Kurosawa Y, Hashimoto K, Expansion of genes that encode MHC class I molecules in cyprinid fishes, J Immunol 151 (1993) 188–200. [PubMed] [Google Scholar]
- [56].Hashimoto K, Okamura K, Yamaguchi H, Ototake M, Nakanishi T, Kurosawa Y, Conservation and diversification of MHC class I and its related molecules in vertebrates, Immunol Rev 167 (1999) 81–100. [DOI] [PubMed] [Google Scholar]
- [57].Neves JV, Caldas C, Wilson JM, Rodrigues PN, Molecular mechanisms of hepcidin regulation in sea bass (Dicentrarchus labrax), Fish Shellfish Immunol 31 (2011) 1154–1161. [DOI] [PubMed] [Google Scholar]
- [58].Sambrook JG, Figueroa F, Beck S, A genome-wide survey of Major Histocompatibility Complex (MHC) genes and their paralogues in zebrafish, BMC Genomics 6 (2005) 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Fraenkel PG, Gibert Y, Holzheimer JL, Lattanzi VJ, Burnett SF, Dooley KA, Wingert RA, Zon LI, Transferrin-a modulates hepcidin expression in zebrafish embryos, Blood 113 (2009) 2843–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Fleming RE, Sly WS, Mechanisms of iron accumulation in hereditary hemochromatosis, Annu Rev Physiol 64 (2002) 663–680. [DOI] [PubMed] [Google Scholar]
- [61].Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, Bjorkman PJ, Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor, Cell 93 (1998) 111–123. [DOI] [PubMed] [Google Scholar]
- [62].Garboczi DN, Biddison WE, Shapes of MHC restriction, Immunity 10 (1999) 1–7. [DOI] [PubMed] [Google Scholar]
- [63].Madden DR, Gorga JC, Strominger JL, Wiley DC, The three-dimensional structure of HLA-B27 at 2.1 A resolution suggests a general mechanism for tight peptide binding to MHC, Cell 70 (1992) 1035–1048. [DOI] [PubMed] [Google Scholar]
- [64].Edwards CQ, Barton JC, Hemochromatosis, in: Greer JP, Arber DA, Glader B, List AF, Means RT Jr., Paraskevas F, and Rodgers GM(Eds.), Wintrobe’s Clinical Hematology, Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia, 2014, pp. 662–681. [Google Scholar]
- [65].Feder JN, Tsuchihashi Z, Irrinki A, Lee VK, Mapa FA, Morikang E, Prass CE, Starnes SM, Wolff RK, Parkkila S, Sly WS, Schatzman RC, The hemochromatosis founder mutation in HLA-H disrupts beta2-microglobulin interaction and cell surface expression, J Biol Chem 272 (1997) 14025–14028. [DOI] [PubMed] [Google Scholar]
- [66].Waheed A, Parkkila S, Zhou XY, Tomatsu S, Tsuchihashi Z, Feder JN, Schatzman RC, Britton RS, Bacon BR, Sly WS, Hereditary hemochromatosis: effects of C282Y and H63D mutations on association with beta2-microglobulin, intracellular processing, and cell surface expression of the HFE protein in COS-7 cells, Proc Natl Acad Sci U S A 94 (1997) 12384–12389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk VR, Leiendecker-Foster C, Speechley M, Snively BM, Holup JL, Thomson E, Sholinsky P, Hemochromatosis and iron-overload screening in a racially diverse population, N Engl J Med 352 (2005) 1769–1778. [DOI] [PubMed] [Google Scholar]
- [68].Puckett T, The National Geographic Style Manual, National Geographic Society 12–7-2010. 7–5-2015.
- [69].Altes A, Ruiz A, Barcelo MJ, Remacha AF, Puig T, Maya AJ, Castell C, Amate JM, Saz Z, Baiget M, Prevalence of the C282Y, H63D, and S65C mutations of the HFE gene in 1,146 newborns from a region of Northern Spain, Genet Test 8 (2004) 407–410. [DOI] [PubMed] [Google Scholar]
- [70].De Juan D, Reta A, Castiella A, Pozueta J, Prada A, Cuadrado E, HFE gene mutations analysis in Basque hereditary haemochromatosis patients and controls, Eur J Hum Genet 9 (2001) 961–964. [DOI] [PubMed] [Google Scholar]
- [71].Floreani A, Rosa RE, Basso D, Navaglia F, Zaninotto M, Petridis I, A.O. DI, Testa R, Marra M, Baldo V, Chiaramonte M, An open population screening study for HFE gene major mutations proves the low prevalence of C282Y mutation in Central Italy, Aliment Pharmacol Ther 26 (2007) 577–586. [DOI] [PubMed] [Google Scholar]
- [72].Murphy S, Curran MD, McDougall N, Callender ME, O’Brien CJ, Middleton D, High incidence of the Cys 282 Tyr mutation in the HFE gene in the Irish population--implications for haemochromatosis, Tissue Antigens 52 (1998) 484–488. [DOI] [PubMed] [Google Scholar]
- [73].Kirk L, Bird J, Ramadan S, Samad A, Adebayo G, Lourens W, Williams J, Haemochromatosis gene frequency in a control and diabetic Irish population, Ir J Med Sci 178 (2009) 39–42. [DOI] [PubMed] [Google Scholar]
- [74].Mura C, Raguenes O, Ferec C, HFE mutations analysis in 711 hemochromatosis probands: evidence for S65C implication in mild form of hemochromatosis, Blood 93 (1999) 2502–2505. [PubMed] [Google Scholar]
- [75].Lim EM, Rossi E, De Boer WB, Reed WD, Jeffrey GP, Hepatic iron loading in patients with compound heterozygous HFE mutations, Liver Int 24 (2004) 631–636. [DOI] [PubMed] [Google Scholar]
- [76].Walsh A, Dixon JL, Ramm GA, Hewett DG, Lincoln DJ, Anderson GJ, Subramaniam VN, Dodemaide J, Cavanaugh JA, Bassett ML, Powell LW, The clinical relevance of compound heterozygosity for the C282Y and H63D substitutions in hemochromatosis, Clin Gastroenterol Hepatol 4 (2006) 1403–1410. [DOI] [PubMed] [Google Scholar]
- [77].Gurrin LC, Bertalli NA, Dalton GW, Osborne NJ, Constantine CC, McLaren CE, English DR, Gertig DM, Delatycki MB, Nicoll AJ, Southey MC, Hopper JL, Giles GG, Anderson GJ, Olynyk JK, Powell LW, Allen KJ, HFE C282Y/H63D compound heterozygotes are at low risk of hemochromatosis-related morbidity, Hepatology 50 (2009) 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].del-Castillo-Rueda A, Moreno-Carralero MI, Alvarez-Sala-Walther LA, Cuadrado-Grande N, Enriquez-de-Salamanca R, Mendez M, Moran-Jimenez MJ, Two novel mutations in the SLC40A1 and HFE genes implicated in iron overload in a Spanish man, Eur J Haematol 86 (2011) 260–264. [DOI] [PubMed] [Google Scholar]
- [79].Biasiotto G, Belloli S, Ruggeri G, Zanella I, Gerardi G, Corrado M, Gobbi E, Albertini A, Arosio P, Identification of new mutations of the HFE, hepcidin, and transferrin receptor 2 genes by denaturing HPLC analysis of individuals with biochemical indications of iron overload, Clin Chem 49 (2003) 1981–1988. [DOI] [PubMed] [Google Scholar]
- [80].Dupradeau FY, Pissard S, Coulhon MP, Cadet E, Foulon K, Fourcade C, Goossens M, Case DA, Rochette J, An unusual case of hemochromatosis due to a new compound heterozygosity in HFE (p.[Gly43Asp;His63Asp]+[Cys282Tyr]): structural implications with respect to binding with transferrin receptor 1, Hum Mutat 29 (2008) 206. [DOI] [PubMed] [Google Scholar]
- [81].Mendes AI, Ferro A, Martins R, Picanco I, Gomes S, Cerqueira R, Correia M, Nunes AR, Esteves J, Fleming R, Faustino P, Non-classical hereditary hemochromatosis in Portugal: novel mutations identified in iron metabolism-related genes, Ann Hematol 88 (2009) 229–234. [DOI] [PubMed] [Google Scholar]
- [82].Kinkely SM, Brown BD, Lyng AT, Harrison WK, Schep GN, Goddard-Hill AC, Aubrey ME, Lillicrap D, Taylor SA, Absence of overt iron overload in two individuals compound heterozygotes for a 22 base pair deletion of exon 2 and the C282Y missense mutation of the HFE gene, Clin Genet 63 (2003) 163–165. [DOI] [PubMed] [Google Scholar]
- [83].Aguilar-Martinez P, Grandchamp B, Cunat S, Cadet E, Blanc F, Nourrit M, Lassoued K, Schved JF, Rochette J, Iron overload in HFE C282Y heterozygotes at first genetic testing: a strategy for identifying rare HFE variants, Haematologica 96 (2011) 507–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Beutler L, Beutler E, Hematologically important mutations: iron storage diseases, Blood Cells Mol Dis 33 (2004) 40–44. [DOI] [PubMed] [Google Scholar]
- [85].de Villiers JN, Hillermann R, Loubser L, Kotze MJ, Spectrum of mutations in the HFE gene implicated in haemochromatosis and porphyria, Hum Mol Genet 8 (1999) 1517–1522. [DOI] [PubMed] [Google Scholar]
- [86].Spriggs EL, Harris PE, Best LG, Hemochromatosis mutations C282Y and H63D in “cis” phase [Abstract], Am J Hum Genet 65 (1999) A492. [DOI] [PubMed] [Google Scholar]
- [87].Thorstensen K, Asberg A, Kvitland M, Svaasand E, Hveem K, Bjerve KS, Detection of an unusual combination of mutations in the HFE gene for hemochromatosis, Genet Test 4 (2000) 371–376. [DOI] [PubMed] [Google Scholar]
- [88].Henz S, Reichen J, Liechti-Gallati S, HLA-H mutations and haemochromatosis: the likely association of H63D with mild phenotype and the detection of S65C, a novel variant in exon 2, J Hepatol 26 (1997) 57A. [Google Scholar]
- [89].Jones DC, Young NT, Pigott C, Fuggle SV, Barnardo MC, Marshall SE, Bunce M, Comprehensive hereditary hemochromatosis genotyping, Tissue Antigens 60 (2002) 481–488. [DOI] [PubMed] [Google Scholar]
- [90].Beutler E, West C, New diallelic markers in the HLA region of chromosome 6, Blood Cells Mol Dis 23 (1997) 219–229. [DOI] [PubMed] [Google Scholar]
- [91].Barton JC, Sawada-Hirai R, Rothenberg BE, Acton RT, Two novel missense mutations of the HFE gene (I105T and G93R) and identification of the S65C mutation in Alabama hemochromatosis probands, Blood Cells Mol Dis 25 (1999) 147–155. [DOI] [PubMed] [Google Scholar]
- [92].Barton JC, West C, Lee PL, Beutler E, A previously undescribed frameshift deletion mutation of HFE (c.del277; G93fs) associated with hemochromatosis and iron overload in a C282Y heterozygote, Clin Genet 66 (2004) 214–216. [DOI] [PubMed] [Google Scholar]
- [93].Cukjati M, Koren S, Curin S, V, B. Vidan-Jeras, R. Rupreht, A novel homozygous frameshift deletion c.471del of HFE associated with hemochromatosis, Clin Genet 71 (2007) 350–353. [DOI] [PubMed] [Google Scholar]
- [94].Pointon JJ, Lok CY, Shearman JD, Suckling RJ, Rochette J, Merryweather-Clarke AT, Robson KJ, A novel HFE mutation (c.del478) results in nonsense-mediated decay of the mutant transcript in a hemochromatosis patient, Blood Cells Mol Dis 43 (2009) 194–198. [DOI] [PubMed] [Google Scholar]
- [95].Oberkanins C, Moritz A, de Villiers JN, Kotze MJ, Kury F, A reverse-hybridization assay for the rapid and simultaneous detection of nine HFE gene mutations, Genet Test 4 (2000) 121–124. [DOI] [PubMed] [Google Scholar]
- [96].Piperno A, Arosio C, Fossati L, Vigano M, Trombini P, Vergani A, Mancia G, Two novel nonsense mutations of HFE gene in five unrelated italian patients with hemochromatosis, Gastroenterology 119 (2000) 441–445. [DOI] [PubMed] [Google Scholar]
- [97].Menardi G, Perotti L, Prucca M, Martini S, Prandi G, Peano G, A very rare association of three mutations of the HFE gene for hemochromatosis, Genet Test 6 (2002) 331–334. [DOI] [PubMed] [Google Scholar]
- [98].Imanishi H, Liu W, Cheng J, Ikeda N, Amuro Y, Hada T, Idiopathic hemochromatosis with the mutation of Ala176Val heterozygous for HFE gene, Intern Med 40 (2001) 479–483. [DOI] [PubMed] [Google Scholar]
- [99].Swinkels DW, Venselaar H, Wiegerinck ET, Bakker E, Joosten I, Jaspers CA, Vasmel WL, Breuning MH, A novel (Leu183Pro-)mutation in the HFE-gene co-inherited with the Cys282Tyr mutation in two unrelated Dutch hemochromatosis patients, Blood Cells Mol Dis 40 (2008) 334–338. [DOI] [PubMed] [Google Scholar]
- [100].Wallace DF, Dooley JS, Walker AP, A novel mutation of HFE explains the classical phenotype of genetic hemochromatosis in a C282Y heterozygote, Gastroenterology 116 (1999) 1409–1412. [DOI] [PubMed] [Google Scholar]
- [101].Beutler E, Gelbart T, A common intron 3 mutation (IVS3 −48c→g) leads to misdiagnosis of the c.845G→A (C282Y) HFE gene mutation, Blood Cells Mol Dis 26 (2000) 229–233. [DOI] [PubMed] [Google Scholar]
- [102].Barton JC, Lafreniere SA, Leiendecker-Foster C, Li H, Acton RT, Press RD, Eckfeldt JH, HFE, SLC40A1, HAMP, HJV, TFR2, and FTL mutations detected by denaturing high-performance liquid chromatography after iron phenotyping and HFE C282Y and H63D genotyping in 785 HEIRS Study participants, Am J Hematol 84 (2009) 710–714. [DOI] [PubMed] [Google Scholar]
- [103].Bradbury R, Fagan E, Payne SJ, Two novel polymorphisms (E277K and V212V) in the haemochromatosis gene HFE, Hum Mutat 15 (2000) 120. [DOI] [PubMed] [Google Scholar]
- [104].Cézard C, Rabbind SA, le GG, Gourlaouen I, Ferec C, Rochette J, Phenotypic expression of a novel C282Y/R226G compound heterozygous state in HFE hemochromatosis: molecular dynamics and biochemical studies, Blood Cells Mol Dis 52 (2014) 27–34. [DOI] [PubMed] [Google Scholar]
- [105].Takano A, Niimi H, Atarashi Y, Sawasaki T, Terasaki T, Nakabayashi T, Kitajima I, Tobe K, Takahara T, A novel Y231del mutation of HFE in hereditary haemochromatosis provides in vivo evidence that the Huh-7 is a human haemochromatotic cell line, Liver Int 31 (2011) 1593–1597. [DOI] [PubMed] [Google Scholar]
- [106].Mariani R, Pelucchi S, Arosio C, Coletti S, Pozzi M, Paolini V, Trombini P, Piperno A, Genetic and metabolic factors are associated with increased hepatic iron stores in a selected population of p.Cys282Tyr heterozygotes, Blood Cells Mol Dis 44 (2010) 159–163. [DOI] [PubMed] [Google Scholar]
- [107].Cunat S, Giansily-Blaizot M, Bismuth M, Blanc F, Dereure O, Larrey D, Quellec AL, Pouderoux P, Rose C, Raingeard I, Renard E, Schved JF, Aguilar-Martinez P, Global sequencing approach for characterizing the molecular background of hereditary iron disorders, Clin Chem 53 (2007) 2060–2069. [DOI] [PubMed] [Google Scholar]
- [108].Worwood M, Jackson HA, Feeney GP, Edwards C, Bowen DJ, A single tube heteroduplex PCR for the common HFE genotypes, Blood 94 (1999) A405. [Google Scholar]
- [109].Rosmorduc O, Poupon R, Nion I, Wendum D, Feder J, Bereziat G, Hermelin B, Differential HFE allele expression in hemochromatosis heterozygotes, Gastroenterology 119 (2000) 1075–1086. [DOI] [PubMed] [Google Scholar]
- [110].Phillips M, Meadows CA, Huang MY, Millson A, Lyon E, Simultaneous detection of C282Y and H63D hemochromatosis mutations by dual-color probes, Mol Diagn 5 (2000) 107–116. [DOI] [PubMed] [Google Scholar]
- [111].Padula MC, Martelli G, Larocca M, Rossano R, Olivieri A, A novel homozygous stop-codon mutation in human HFE responsible for nonsense-mediated mRNA decay, Blood Cells Mol Dis 53 (2014) 138–143. [DOI] [PubMed] [Google Scholar]
- [112].le Gac G, Dupradeau FY, Mura C, Jacolot S, Scotet V, Esnault G, Mercier AY, Rochette J, Ferec C, Phenotypic expression of the C282Y/Q283P compound heterozygosity in HFE and molecular modeling of the Q283P mutation effect, Blood Cells Mol Dis 30 (2003) 231–237. [DOI] [PubMed] [Google Scholar]
- [113].Jeffrey GP, Chakrabarti S, Hegele RA, Adams PC, Polymorphism in intron 4 of HFE may cause overestimation of C282Y homozygote prevalence in haemochromatosis, Nat Genet 22 (1999) 325–326. [DOI] [PubMed] [Google Scholar]
- [114].Mikhailova SV, Babenko VN, Voevoda MI, Romashchenko AG, The ethnospecific distribution of the HFE haplotypes for IVS2(+4)t/c, IVS4(−44)t/c, and IVS5(−47)g/a in populations of Russia and possible effects of these single-nucleotide polymorphisms in splicing, Genet Test Mol Biomarkers 14 (2010) 461–469. [DOI] [PubMed] [Google Scholar]
- [115].Zaahl MG, Merryweather-Clarke AT, Kotze MJ, van der Merwe S, Warnich L, Robson KJ, Analysis of genes implicated in iron regulation in individuals presenting with primary iron overload, Hum Genet 115 (2004) 409–417. [DOI] [PubMed] [Google Scholar]
- [116].Steiner M, Ocran K, Genschel J, Meier P, Gerl H, Ventz M, Schneider ML, Buttner C, Wadowska K, Kerner W, Schuff-Werner P, Lochs H, Schmidt H, A homozygous HFE gene splice site mutation (IVS5+1 G/A) in a hereditary hemochromatosis patient of Vietnamese origin, Gastroenterology 122 (2002) 789–795. [DOI] [PubMed] [Google Scholar]
- [117].le Gac G, Gourlaouen I, Ronsin C, Geromel V, Bourgarit A, Parquet N, Quemener S, Le MC, Chen JM, Ferec C, Homozygous deletion of HFE produces a phenotype similar to the HFE p.C282Y/p.C282Y genotype, Blood 112 (2008) 5238–5240. [DOI] [PubMed] [Google Scholar]
- [118].le Gac G, Congiu R, Gourlaouen I, Cau M, Ferec C, Melis MA, Homozygous deletion of HFE is the common cause of hemochromatosis in Sardinia, Haematologica 95 (2010) 685–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Beutler E, The HFE Cys282Tyr mutation as a necessary but not sufficient cause of clinical hereditary hemochromatosis, Blood 101 (2003) 3347–3350. [DOI] [PubMed] [Google Scholar]
- [120].Piperno A, Arosio C, Fargion S, Roetto A, Nicoli C, Girelli D, Sbaiz L, Gasparini P, Boari G, Sampietro M, Camaschella C, The ancestral hemochromatosis haplotype is associated with a severe phenotype expression in Italian patients, Hepatology 24 (1996) 43–46. [DOI] [PubMed] [Google Scholar]
- [121].Barton JC, Shih WW, Sawada-Hirai R, Acton RT, Harmon L, Rivers C, Rothenberg BE, Genetic and clinical description of hemochromatosis probands and heterozygotes: evidence that multiple genes linked to the major histocompatibility complex are responsible for hemochromatosis, Blood Cells Mol Dis 23 (1997) 135–145. [DOI] [PubMed] [Google Scholar]
- [122].Pratiwi R, Fletcher LM, Pyper WR, Do KA, Crawford DH, Powell LW, Jazwinska EC, Linkage disequilibrium analysis in Australian haemochromatosis patients indicates bipartite association with clinical expression, J Hepatol 31 (1999) 39–46. [DOI] [PubMed] [Google Scholar]
- [123].Benyamin B, Esko T, Ried JS, Radhakrishnan A, Vermeulen SH, Traglia M, Gogele M, Anderson D, Broer L, Podmore C, Luan J, Kutalik Z, Sanna S, van der Meer P, Tanaka T, Wang F, Westra HJ, Franke L, Mihailov E, Milani L, Haldin J, Winkelmann J, Meitinger T, Thiery J, Peters A, Waldenberger M, Rendon A, Jolley J, Sambrook J, Kiemeney LA, Sweep FC, Sala CF, Schwienbacher C, Pichler I, Hui J, Demirkan A, Isaacs A, Amin N, Steri M, Waeber G, Verweij N, Powell JE, Nyholt DR, Heath AC, Madden PA, Visscher PM, Wright MJ, Montgomery GW, Martin NG, Hernandez D, Bandinelli S, van der Harst P, Uda M, Vollenweider P, Scott RA, Langenberg C, Wareham NJ, van DC, Beilby J, Pramstaller PP, Hicks AA, Ouwehand WH, Oexle K, Gieger C, Metspalu A, Camaschella C, Toniolo D, Swinkels DW, Whitfield JB, Novel loci affecting iron homeostasis and their effects in individuals at risk for hemochromatosis, Nat Commun 5 (2014) 4926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].deTayrac M, Roth MP, Jouanolle AM, Coppin H, le GG, Piperno A, Ferec C, Pelucchi S, Scotet V, Bardou-Jacquet E, Ropert M, Bouvet R, Genin E, Mosser J, Deugnier Y, Genome-wide association study identifies TF as a significant modifier gene of iron metabolism in HFE hemochromatosis, J Hepatol 62 (2015) 664–672. [DOI] [PubMed] [Google Scholar]
- [125].Milet J, Dehais V, Bourgain C, Jouanolle AM, Mosser A, Perrin M, Morcet J, Brissot P, David V, Deugnier Y, Mosser J, Common variants in the BMP2, BMP4, and HJV genes of the hepcidin regulation pathway modulate HFE hemochromatosis penetrance, Am J Hum Genet 81 (2007) 799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Milet J, le GG, Scotet V, Gourlaouen I, Theze C, Mosser J, Bourgain C, Deugnier Y, Ferec C, A common SNP near BMP2 is associated with severity of the iron burden in HFE p.C282Y homozygous patients: a follow-up study, Blood Cells Mol Dis 44 (2010) 34–37. [DOI] [PubMed] [Google Scholar]
- [127].Constantine CC, Anderson GJ, Vulpe CD, McLaren CE, Bahlo M, Yeap HL, Gertig DM, Osborne NJ, Bertalli NA, Beckman KB, Chen V, Matak P, McKie AT, Delatycki MB, Olynyk JK, English DR, Southey MC, Giles GG, Hopper JL, Allen KJ, Gurrin LC, A novel association between a SNP in CYBRD1 and serum ferritin levels in a cohort study of HFE hereditary haemochromatosis, Br J Haematol 147 (2009) 140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].McLaren CE, Emond MJ, Subramaniam VN, Phatak PD, Barton JC, Adams PC, Goh JB, McDonald CJ, Powell LW, Gurrin LC, Allen KJ, Nickerson DA, Louie T, Ramm GA, Anderson GJ, McLaren GD, Exome sequencing in HFE C282Y homozygous men with extreme phenotypes identifies a GNPAT variant associated with severe iron overload, Hepatology 62 (2015) 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Costa M, Cruz E, Barton JC, Thorstensen K, Morais S, da Silva BM, Pinto JP, Vieira CP, Vieira J, Acton RT, Porto G, Effects of highly conserved major histocompatibility complex (MHC) extended haplotypes on iron and low CD8+ T lymphocyte phenotypes in HFE C282Y homozygous hemochromatosis patients from three geographically distant areas, PLoS One 8 (2013) e79990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Lee PL, Gelbart T, West C, Halloran C, Felitti V, Beutler E, A study of genes that may modulate the expression of hereditary hemochromatosis: transferrin receptor-1, ferroportin, ceruloplasmin, ferritin light and heavy chains, iron regulatory proteins (IRP)-1 and −2, and hepcidin, Blood Cells Mol Dis 27 (2001) 783–802. [DOI] [PubMed] [Google Scholar]
- [131].Lee P, Gelbart T, West C, Halloran C, Beutler E, Seeking candidate mutations that affect iron homeostasis, Blood Cells Mol Dis 29 (2002) 471–487. [DOI] [PubMed] [Google Scholar]
- [132].Merryweather-Clarke AT, Cadet E, Bomford A, Capron D, Viprakasit V, Miller A, McHugh PJ, Chapman RW, Pointon JJ, Wimhurst VL, Livesey KJ, Tanphaichitr V, Rochette J, Robson KJ, Digenic inheritance of mutations in HAMP and HFE results in different types of haemochromatosis, Hum Mol Genet 12 (2003) 2241–2247. [DOI] [PubMed] [Google Scholar]
- [133].Biasiotto G, Roetto A, Daraio F, Polotti A, Gerardi GM, Girelli D, Cremonesi L, Arosio P, Camaschella C, Identification of new mutations of hepcidin and hemojuvelin in patients with HFE C282Y allele, Blood Cells Mol Dis 33 (2004) 338–343. [DOI] [PubMed] [Google Scholar]
- [134].Jacolot S, le GG, Scotet V, Quere I, Mura C, Ferec C, HAMP as a modifier gene that increases the phenotypic expression of the HFE pC282Y homozygous genotype, Blood 103 (2004) 2835–2840. [DOI] [PubMed] [Google Scholar]
- [135].Pietrangelo A, Caleffi A, Henrion J, Ferrara F, Corradini E, Kulaksiz H, Stremmel W, Andreone P, Garuti C, Juvenile hemochromatosis associated with pathogenic mutations of adult hemochromatosis genes, Gastroenterology 128 (2005) 470–479. [DOI] [PubMed] [Google Scholar]
- [136].Bulaj ZJ, Phillips JD, Ajioka RS, Franklin MR, Griffen LM, Guinee DJ, Edwards CQ, Kushner JP, Hemochromatosis genes and other factors contributing to the pathogenesis of porphyria cutanea tarda, Blood 95 (2000) 1565–1571. [PubMed] [Google Scholar]
- [137].Ajioka RS, Phillips JD, Weiss RB, Dunn DM, Smit MW, Proll SC, Katze MG, Kushner JP, Down-regulation of hepcidin in porphyria cutanea tarda, Blood 112 (2008) 4723–4728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Phillips JD, Kushner JP, Bergonia HA, Franklin MR, Uroporphyria in the Cyp1a2−/−mouse, Blood Cells Mol Dis 47 (2011) 249–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Phillips JD, Bergonia HA, Reilly CA, Franklin MR, Kushner JP, A porphomethene inhibitor of uroporphyrinogen decarboxylase causes porphyria cutanea tarda, Proc Natl Acad Sci U S A 104 (2007) 5079–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Roberts AG, Whatley SD, Morgan RR, Worwood M, Elder GH, Increased frequency of the haemochromatosis Cys282Tyr mutation in sporadic porphyria cutanea tarda, Lancet 349 (1997) 321–323. [DOI] [PubMed] [Google Scholar]
- [141].Sampietro M, Piperno A, Lupica L, Arosio C, Vergani A, Corbetta N, Malosio I, Mattioli M, Fracanzani AL, Cappellini MD, Fiorelli G, Fargion S, High prevalence of the His63Asp HFE mutation in Italian patients with porphyria cutanea tarda, Hepatology 27 (1998) 181–184. [DOI] [PubMed] [Google Scholar]
- [142].Stuart KA, Busfield F, Jazwinska EC, Gibson P, Butterworth LA, Cooksley WG, Powell LW, Crawford DH, The C282Y mutation in the haemochromatosis gene (HFE) and hepatitis C virus infection are independent cofactors for porphyria cutanea tarda in Australian patients, J Hepatol 28 (1998) 404–409. [DOI] [PubMed] [Google Scholar]
- [143].Tannapfel A, Stolzel U, Kostler E, Melz S, Richter M, Keim V, Schuppan D, Wittekind C, C282Y and H63D mutation of the hemochromatosis gene in German porphyria cutanea tarda patients, Virchows Arch 439 (2001) 1–5. [DOI] [PubMed] [Google Scholar]
- [144].Bottomley SS, Porphyrias, in: Greer JP, Arber DA, Glader B, List AF, Means RT Jr., Paraskevas F, and Rodgers GM (Eds.), Wintrobe’s Clinical Hematology, Wolters Kluwer/Lippincott Williams & Wilkins, Philadelphia, 2014, pp. 682–706. [Google Scholar]
- [145].Toomajian C, Ajioka RS, Jorde LB, Kushner JP, Kreitman M, A method for detecting recent selection in the human genome from allele age estimates, Genetics 165 (2003) 287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Distante S, Robson KJ, Graham-Campbell J, Arnaiz-Villena A, Brissot P, Worwood M, The origin and spread of the HFE-C282Y haemochromatosis mutation, Hum Genet 115 (2004) 269–279. [DOI] [PubMed] [Google Scholar]
- [147].Symonette CJ, Adams PC, Do all hemochromatosis patients have the same origin? An analysis of mitochondrial DNA and Y-DNA, Can J Gastroenterol 25 (2011) 324–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Simon M, Alexandre JL, Fauchet R, Genetet B, Bourel M, The genetics of hemochromatosis, Prog Med Genet 4 (1980) 135–168. [PubMed] [Google Scholar]
- [149].Lucotte G, Dieterlen F, A European allele map of the C282Y mutation of hemochromatosis: Celtic versus Viking origin of the mutation?, Blood Cells Mol Dis 31 (2003) 262–267. [DOI] [PubMed] [Google Scholar]
- [150].Olsson KS, Konar J, Dufva IH, Ricksten A, Raha-Chowdhury R, Was the C282Y mutation an Irish Gaelic mutation that the Vikings helped disseminate? HLA haplotype observations of hemochromatosis from the west coast of Sweden, Eur J Haematol 86 (2011) 75–82. [DOI] [PubMed] [Google Scholar]
- [151].Kucinskas L, Juzenas S, Sventoraityte J, Cedaviciute R, Vitkauskiene A, Kalibatas V, Kondrackiene J, Kupcinskas L, Prevalence of C282Y, H63D, and S65C mutations in hereditary HFE-hemochromatosis gene in Lithuanian population, Ann Hematol 91 (2012) 491–495. [DOI] [PubMed] [Google Scholar]
- [152].Ryan E, O’Keane C, Crowe J, Hemochromatosis in Ireland and HFE, Blood Cells Mol Dis 24 (1998) 428–432. [DOI] [PubMed] [Google Scholar]
- [153].Porto G, de Sousa M, Variation of hemochromatosis prevalence and genotype in national groups, in: Barton JC and Edwards CQ (Eds.), Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment, Cambridge University Press, Cambridge, 2000, pp. 51–62. [Google Scholar]
- [154].Acton RT, Barton JC, Snively BM, McLaren CE, Adams PC, Harris EL, Speechley MR, McLaren GD, Dawkins FW, Leiendecker-Foster C, Holup JL, Balasubramanyam A, Geographic and racial/ethnic differences in HFE mutation frequencies in the Hemochromatosis and Iron Overload Screening (HEIRS) Study, Ethn Dis 16 (2006) 815–821. [PubMed] [Google Scholar]
- [155].Rochette J, Pointon JJ, Fisher CA, Perera G, Arambepola M, Arichchi DS, De SS, Vandwalle JL, Monti JP, Old JM, Merryweather-Clarke AT, Weatherall DJ, Robson KJ, Multicentric origin of hemochromatosis gene (HFE) mutations, Am J Hum Genet 64 (1999) 1056–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Ritter B, Safwenberg J, Olsson KS, HLA as a marker of the hemochromatosis gene in Sweden, Hum Genet 68 (1984) 62–66. [DOI] [PubMed] [Google Scholar]
- [157].Milman N, Graudal N, Nielsen LS, Fenger K, An HLA study in 74 Danish haemochromatosis patients and in 21 of their families, Clin Genet 41 (1992) 6–11. [DOI] [PubMed] [Google Scholar]
- [158].Jazwinska EC, Pyper WR, Burt MJ, Francis JL, Goldwurm S, Webb SI, Lee SC, Halliday JW, Powell LW, Haplotype analysis in Australian hemochromatosis patients: evidence for a predominant ancestral haplotype exclusively associated with hemochromatosis, Am J Hum Genet 56 (1995) 428–433. [PMC free article] [PubMed] [Google Scholar]
- [159].Barton JC, Acton RT, HLA-A and -B alleles and haplotypes in hemochromatosis probands with HFE C282Y homozygosity in central Alabama, BMC Med Genet 3 (2002) 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Piperno A, Fargion S, Panaiotopoulos N, Del NE, Taddei MT, Fiorelli G, Idiopathic haemochromatosis and HLA antigens in Italy: is A3 Bw35 HLA haplotype a marker for idiopathic haemochromatosis gene in north east regions?, J Clin Pathol 39 (1986) 125–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Panajotopoulos N, Piperno A, Conte D, Mandelli C, Cesana M, Mercuriali F, Fiorelli G, Bianchi PA, Fargion S, HLA typing in 67 Italian patients with idiopathic hemochromatosis and their relatives, Tissue Antigens 33 (1989) 431–436. [DOI] [PubMed] [Google Scholar]
- [162].Olsson KS, Ritter B, Hansson N, Chowdhury RR, HLA haplotype map of river valley populations with hemochromatosis traced through five centuries in Central Sweden, Eur J Haematol 81 (2008) 36–46. [DOI] [PubMed] [Google Scholar]
- [163].Porto G, da Silva BM, Vincente C, de SM, Idiopathic haemochromatosis in north Portugal: association with haplotype A3B7, J Clin Pathol 42 (1989) 667–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Merryweather-Clarke AT, Pointon JJ, Shearman JD, Robson KJ, Global prevalence of putative haemochromatosis mutations, J Med Genet 34 (1997) 275–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Merryweather-Clarke AT, Pointon JJ, Jouanolle AM, Rochette J, Robson KJ, Geography of HFE C282Y and H63D mutations, Genet Test 4 (2000) 183–198. [DOI] [PubMed] [Google Scholar]
- [166].Fairbanks VF, Hemochromatosis: population genetics, in: Barton JC and Edwards CQ (Eds.), Hemochromatosis: Genetics, Pathophysiology, Diagnosis and Treatment, Cambridge University Press, Cambridge, 2000, pp. 42–50. [Google Scholar]
- [167].Cardoso CS, Alves H, Mascarenhas M, Goncalves R, Oliveira P, Rodrigues P, Cruz E, de SM, Porto G, Co-selection of the H63D mutation and the HLA-A29 allele: a new paradigm of linkage disequilibrium?, Immunogenetics 53 (2002) 1002–1008. [DOI] [PubMed] [Google Scholar]
- [168].Khusainova RI, Khusnutdinova NN, Litvinov SS, Khusnutdinova EK, [Analysis of H63D mutation in hemochromatosis (HFE) gene in populations of central Eurasia], Genetika 49 (2013) 269–278. [DOI] [PubMed] [Google Scholar]
- [169].Dhillon BK, Prakash S, Chandak GR, Chawla YK, Das R, H63D mutation in HFE gene is common in Indians and is associated with the European haplotype, J Genet 91 (2012) 229–232. [DOI] [PubMed] [Google Scholar]
- [170].Cullen LM, Gao X, Easteal S, Jazwinska EC, The hemochromatosis 845 G>A and 187 C->G mutations: prevalence in non-Caucasian populations, Am J Hum Genet 62 (1998) 1403–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Agostinho MF, Arruda VR, Basseres DS, Bordin S, Soares MC, Menezes RC, Costa FF, Saad ST, Mutation analysis of the HFE gene in Brazilian populations, Blood Cells Mol Dis 25 (1999) 324–327. [DOI] [PubMed] [Google Scholar]
- [172].Couto AR, Peixoto MJ, Garrett F, Laranjeira F, Cipriano T, Armas JB, Linkage disequilibrium between S65C HFE mutation and HLA A29-B44 haplotype in Terceira Island, Azores, Hum Immunol 64 (2003) 625–628. [DOI] [PubMed] [Google Scholar]
- [173].Holmstrom P, Marmur J, Eggertsen G, Gafvels M, Stal P, Mild iron overload in patients carrying the HFE S65C gene mutation: a retrospective study in patients with suspected iron overload and healthy controls, Gut 51 (2002) 723–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Arya N, Chakrabrati S, Hegele RA, Adams PC, HFE S65C variant is not associated with increased transferrin saturation in voluntary blood donors, Blood Cells Mol Dis 25 (1999) 354–357. [DOI] [PubMed] [Google Scholar]
- [175].Remacha AF, Barcelo MJ, Sarda MP, Blesa I, Altes A, Baiget M, The S65C mutation in Spain. Implications for iron overload screening, Haematologica 85 (2000) 1324–1325. [PubMed] [Google Scholar]
- [176].Campo S, Restuccia T, Villari D, Raffa G, Cucinotta D, Squadrito G, Pollicino T, Raimondo G, Analysis of haemochromatosis gene mutations in a population from the Mediterranean Basin, Liver 21 (2001) 233–236. [DOI] [PubMed] [Google Scholar]
- [177].Gabrikova D, Bernasovska J, Macekova S, Bozikova A, Bernasovsky I, Balisinova A, Sovicova A, Behulova R, Petrejcikova E, Sotak M, Boronova I, Unique frequencies of HFE gene variants in Roma/Gypsies, J Appl Genet 53 (2012) 183–187. [DOI] [PubMed] [Google Scholar]
- [178].He X, Lu X, Hu J, Xi J, Zhou D, Shang H, Liu L, Zhou H, Yan B, Yu L, Hu F, Liu Z, He L, Yao X, Xu Y, H63D polymorphism in the hemochromatosis gene is associated with sporadic amyotrophic lateral sclerosis in China, Eur J Neurol 18 (2011) 359–361. [DOI] [PubMed] [Google Scholar]
- [179].Beutler E, Iron absorption in carriers of the C282Y hemochromatosis mutation, Am J Clin Nutr 80 (2004) 799–800. [DOI] [PubMed] [Google Scholar]
- [180].Andrews NC, The iron transporter DMT1, Int J Biochem Cell Biol 31 (1999) 991–994. [DOI] [PubMed] [Google Scholar]
- [181].Datz C, Haas T, Rinner H, Sandhofer F, Patsch W, Paulweber B, Heterozygosity for the C282Y mutation in the hemochromatosis gene is associated with increased serum iron, transferrin saturation, and hemoglobin in young women: a protective role against iron deficiency?, Clin Chem 44 (1998) 2429–2432. [PubMed] [Google Scholar]
- [182].Beutler E, Felitti V, Gelbart T, Waalen J, Haematological effects of the C282Y HFE mutation in homozygous and heterozygous states among subjects of northern and southern European ancestry, Br J Haematol 120 (2003) 887–893. [DOI] [PubMed] [Google Scholar]
- [183].Hunt JR, Zeng H, Iron absorption by heterozygous carriers of the HFE C282Y mutation associated with hemochromatosis, Am J Clin Nutr 80 (2004) 924–931. [DOI] [PubMed] [Google Scholar]
- [184].Adams PC, Bradley C, Frei JV, Hepatic zinc in hemochromatosis, Clin Invest Med 14 (1991) 16–20. [PubMed] [Google Scholar]
- [185].Altstatt LB, Pollack S, Feldman MH, Reba RC, Crosby WH, Liver manganese in hemochromatosis, Proc Soc Exp Biol Med 124 (1967) 353–355. [DOI] [PubMed] [Google Scholar]
- [186].Olatunbosun D, Corbett WE, Ludwig J, Valberg LS, Alteration of cobalt absorption in portal cirrhosis and idiopathic hemochromatosis, J Lab Clin Med 75 (1970) 754–762. [PubMed] [Google Scholar]
- [187].Dobson MJ, Malaria in England: a geographical and historical perspective, Parassitologia 36 (1994) 35–60. [PubMed] [Google Scholar]
- [188].Reiter P, From Shakespeare to Defoe: malaria in England in the Little Ice Age, Emerg Infect Dis 6 (2000) 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Knotterus OS, Malaria Around the North Sea: A Survey, in: Wefer G, Berger WH, Behre K-E, and Jansen E (Eds.), Climatic Development and History of the North Atlantic Realm: Hanse Conference Report, Springer-Verlag, Berlin, 2002, pp. 339–353. [Google Scholar]
- [190].Barton JC, Bertoli LF, Rothenberg BE, Peripheral blood erythrocyte parameters in hemochromatosis: evidence for increased erythrocyte hemoglobin content, J Lab Clin Med 135 (2000) 96–104. [DOI] [PubMed] [Google Scholar]
- [191].Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, Seifert M, Crouch ML, Hantke K, Akira S, Fang FC, Weiss G, Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2, Blood 114 (2009) 3642–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [192].Bhatt L, Murphy C, O’Driscoll LS, Carmo-Fonseca M, McCaffrey MW, Fleming JV, N-glycosylation is important for the correct intracellular localization of HFE and its ability to decrease cell surface transferrin binding, FEBS J 277 (2010) 3219–3234. [DOI] [PubMed] [Google Scholar]
- [193].Salter-Cid L, Brunmark A, Li Y, Leturcq D, Peterson PA, Jackson MR, Yang Y, Transferrin receptor is negatively modulated by the hemochromatosis protein HFE: implications for cellular iron homeostasis, Proc Natl Acad Sci U S A 96 (1999) 5434–5439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Bennett MJ, Lebron JA, Bjorkman PJ, Crystal structure of the hereditary haemochromatosis protein HFE complexed with transferrin receptor, Nature 403 (2000) 46–53. [DOI] [PubMed] [Google Scholar]
- [195].Feder JN, Penny DM, Irrinki A, Lee VK, Lebron JA, Watson N, Tsuchihashi Z, Sigal E, Bjorkman PJ, Schatzman RC, The hemochromatosis gene product complexes with the transferrin receptor and lowers its affinity for ligand binding, Proc Natl Acad Sci U S A 95 (1998) 1472–1477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [196].West AP Jr., Bennett MJ, Sellers VM, Andrews NC, Enns CA, Bjorkman PJ, Comparison of the interactions of transferrin receptor and transferrin receptor 2 with transferrin and the hereditary hemochromatosis protein HFE, J Biol Chem 275 (2000) 38135–38138. [DOI] [PubMed] [Google Scholar]
- [197].Ganz T, Hepcidin and iron regulation, 10 years later, Blood 117 (2011) 4425–4433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Bridle KR, Frazer DM, Wilkins SJ, Dixon JL, Purdie DM, Crawford DH, Subramaniam VN, Powell LW, Anderson GJ, Ramm GA, Disrupted hepcidin regulation in HFE-associated haemochromatosis and the liver as a regulator of body iron homoeostasis, Lancet 361 (2003) 669–673. [DOI] [PubMed] [Google Scholar]
- [199].Ahmad KA, Ahmann JR, Migas MC, Waheed A, Britton RS, Bacon BR, Sly WS, Fleming RE, Decreased liver hepcidin expression in the Hfe knockout mouse, Blood Cells Mol Dis 29 (2002) 361–366. [DOI] [PubMed] [Google Scholar]
- [200].GeneCards® Human Gene Database: Expression for HFE gene, Weizmann Institute of Science 2015.
- [201].Waheed A, Parkkila S, Saarnio J, Fleming RE, Zhou XY, Tomatsu S, Britton RS, Bacon BR, Sly WS, Association of HFE protein with transferrin receptor in crypt enterocytes of human duodenum, Proc Natl Acad Sci U S A 96 (1999) 1579–1584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Griffiths WJ, Kelly AL, Smith SJ, Cox TM, Localization of iron transport and regulatory proteins in human cells, QJM 93 (2000) 575–587. [DOI] [PubMed] [Google Scholar]
- [203].Parkkila S, Parkkila AK, Waheed A, Britton RS, Zhou XY, Fleming RE, Tomatsu S, Bacon BR, Sly WS, Cell surface expression of HFE protein in epithelial cells, macrophages, and monocytes, Haematologica 85 (2000) 340–345. [PubMed] [Google Scholar]
- [204].Parkkila S, Waheed A, Britton RS, Bacon BR, Zhou XY, Tomatsu S, Fleming RE, Sly WS, Association of the transferrin receptor in human placenta with HFE, the protein defective in hereditary hemochromatosis, Proc Natl Acad Sci U S A 94 (1997) 13198–13202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Vujic SM, Kiss J, Herrmann T, Kessler R, Stolte J, Galy B, Rathkolb B, Wolf E, Stremmel W, Hentze MW, Muckenthaler MU, Physiologic systemic iron metabolism in mice deficient for duodenal Hfe, Blood 109 (2007) 4511–4517. [DOI] [PubMed] [Google Scholar]
- [206].Parkkila S, Waheed A, Britton RS, Feder JN, Tsuchihashi Z, Schatzman RC, Bacon BR, Sly WS, Immunohistochemistry of HLA-H, the protein defective in patients with hereditary hemochromatosis, reveals unique pattern of expression in gastrointestinal tract, Proc Natl Acad Sci U S A 94 (1997) 2534–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Bastin J, Drakesmith H, Rees M, Sargent I, Townsend A, Localisation of proteins of iron metabolism in the human placenta and liver, Br J Haematol 134 (2006) 532–543. [DOI] [PubMed] [Google Scholar]
- [208].Zhang AS, Xiong S, Tsukamoto H, Enns CA, Localization of iron metabolism-related mRNAs in rat liver indicate that HFE is expressed predominantly in hepatocytes, Blood 103 (2004) 1509–1514. [DOI] [PubMed] [Google Scholar]
- [209].Griffiths WJ, Cox TM, Co-localization of the mammalian hemochromatosis gene product (HFE) and a newly identified transferrin receptor (TfR2) in intestinal tissue and cells, J Histochem Cytochem 51 (2003) 613–624. [DOI] [PubMed] [Google Scholar]
- [210].d’Alessio F, Hentze MW, Muckenthaler MU, The hemochromatosis proteins HFE, TfR2, and HJV form a membrane-associated protein complex for hepcidin regulation, J Hepatol 57 (2012) 1052–1060. [DOI] [PubMed] [Google Scholar]
- [211].Hashimoto K, Hirai M, Kurosawa Y, Identification of a mouse homolog for the human hereditary haemochromatosis candidate gene, Biochem Biophys Res Commun 230 (1997) 35–39. [DOI] [PubMed] [Google Scholar]
- [212].Riegert P, Gilfillan S, Nanda I, Schmid M, Bahram S, The mouse HFE gene, Immunogenetics 47 (1998) 174–177. [DOI] [PubMed] [Google Scholar]
- [213].Zhou XY, Tomatsu S, Fleming RE, Parkkila S, Waheed A, Jiang J, Fei Y, Brunt EM, Ruddy DA, Prass CE, Schatzman RC, O’Neill R, Britton RS, Bacon BR, Sly WS, HFE gene knockout produces mouse model of hereditary hemochromatosis, Proc Natl Acad Sci U S A 95 (1998) 2492–2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Fleming RE, Holden CC, Tomatsu S, Waheed A, Brunt EM, Britton RS, Bacon BR, Roopenian DC, Sly WS, Mouse strain differences determine severity of iron accumulation in Hfe knockout model of hereditary hemochromatosis, Proc Natl Acad Sci U S A 98 (2001) 2707–2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Lee SM, Loguinov A, Fleming RE, Vulpe CD, Effects of strain and age on hepatic gene expression profiles in murine models of HFE-associated hereditary hemochromatosis, Genes Nutr 10 (2015) 443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Bensaid M, Fruchon S, Mazeres C, Bahram S, Roth MP, Coppin H, Multigenic control of hepatic iron loading in a murine model of hemochromatosis, Gastroenterology 126 (2004) 1400–1408. [DOI] [PubMed] [Google Scholar]
- [217].Bahram S, Gilfillan S, Kuhn LC, Moret R, Schulze JB, Lebeau A, Schumann K, Experimental hemochromatosis due to MHC class I HFE deficiency: immune status and iron metabolism, Proc Natl Acad Sci U S A 96 (1999) 13312–13317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Levy JE, Montross LK, Cohen DE, Fleming MD, Andrews NC, The C282Y mutation causing hereditary hemochromatosis does not produce a null allele, Blood 94 (1999) 9–11. [PubMed] [Google Scholar]
- [219].Vujic SM, Kiss J, Herrmann T, Galy B, Martinache S, Stolte J, Grone HJ, Stremmel W, Hentze MW, Muckenthaler MU, Hfe acts in hepatocytes to prevent hemochromatosis, Cell Metab 7 (2008) 173–178. [DOI] [PubMed] [Google Scholar]
- [220].Lou DQ, Lesbordes JC, Nicolas G, Viatte L, Bennoun M, Van RN, Kahn A, Renia L, Vaulont S, Iron-and inflammation-induced hepcidin gene expression in mice is not mediated by Kupffer cells in vivo, Hepatology 41 (2005) 1056–1064. [DOI] [PubMed] [Google Scholar]
- [221].Montosi G, Corradini E, Garuti C, Barelli S, Recalcati S, Cairo G, Valli L, Pignatti E, Vecchi C, Ferrara F, Pietrangelo A, Kupffer cells and macrophages are not required for hepatic hepcidin activation during iron overload, Hepatology 41 (2005) 545–552. [DOI] [PubMed] [Google Scholar]
- [222].Anderson GJ, Halliday JW, Powell LW, Transferrin receptors in hemochromatosis, Hepatology 7 (1987) 967–969. [DOI] [PubMed] [Google Scholar]
- [223].Ajioka RS, Levy JE, Andrews NC, Kushner JP, Regulation of iron absorption in Hfe mutant mice, Blood 100 (2002) 1465–1469. [DOI] [PubMed] [Google Scholar]
- [224].Tomatsu S, Orii KO, Fleming RE, Holden CC, Waheed A, Britton RS, Gutierrez MA, Velez-Castrillon S, Bacon BR, Sly WS, Contribution of the H63D mutation in HFE to murine hereditary hemochromatosis, Proc Natl Acad Sci U S A 100 (2003) 15788–15793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Schmidt PJ, Toran PT, Giannetti AM, Bjorkman PJ, Andrews NC, The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression, Cell Metab 7 (2008) 205–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Herrmann T, Muckenthaler M, van der Hoeven F, Brennan K, Gehrke SG, Hubert N, Sergi C, Grone HJ, Kaiser I, Gosch I, Volkmann M, Riedel HD, Hentze MW, Stewart AF, Stremmel W, Iron overload in adult Hfe-deficient mice independent of changes in the steady-state expression of the duodenal iron transporters DMT1 and Ireg1/ferroportin, J Mol Med (Berl) 82 (2004) 39–48. [DOI] [PubMed] [Google Scholar]
- [227].Dostalikova-Cimburova M, Kratka K, Balusikova K, Chmelikova J, Hejda V, Hnanicek J, Neubauerova J, Vranova J, Kovar J, Horak J, Duodenal expression of iron transport molecules in patients with hereditary hemochromatosis or iron deficiency, J Cell Mol Med 16 (2012) 1816–1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Nelson JE, Mugford VR, Kilcourse E, Wang RS, Kowdley KV, Relationship between gene expression of duodenal iron transporters and iron stores in hemochromatosis subjects, Am J Physiol Gastrointest Liver Physiol 298 (2010) G57–G62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Stuart KA, Anderson GJ, Frazer DM, Powell LW, McCullen M, Fletcher LM, Crawford DH, Duodenal expression of iron transport molecules in untreated haemochromatosis subjects, Gut 52 (2003) 953–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [230].Zoller H, Theurl I, Koch RO, McKie AT, Vogel W, Weiss G, Duodenal cytochrome b and hephaestin expression in patients with iron deficiency and hemochromatosis, Gastroenterology 125 (2003) 746–754. [DOI] [PubMed] [Google Scholar]
- [231].Gleeson F, Ryan E, Barrett S, Russell J, Kelleher B, Crowe J, Duodenal Dcytb and hephaestin mRNA expression are not significantly modulated by variations in body iron homeostasis, Blood Cells Mol Dis 35 (2005) 303–308. [DOI] [PubMed] [Google Scholar]
- [232].Fleming RE, Migas MC, Zhou X, Jiang J, Britton RS, Brunt EM, Tomatsu S, Waheed A, Bacon BR, Sly WS, Mechanism of increased iron absorption in murine model of hereditary hemochromatosis: increased duodenal expression of the iron transporter DMT1, Proc Natl Acad Sci U S A 96 (1999) 3143–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Griffiths WJ, Sly WS, Cox TM, Intestinal iron uptake determined by divalent metal transporter is enhanced in HFE-deficient mice with hemochromatosis, Gastroenterology 120 (2001) 1420–1429. [DOI] [PubMed] [Google Scholar]
- [234].Zoller H, Koch RO, Theurl I, Obrist P, Pietrangelo A, Montosi G, Haile DJ, Vogel W, Weiss G, Expression of the duodenal iron transporters divalent-metal transporter 1 and ferroportin 1 in iron deficiency and iron overload, Gastroenterology 120 (2001) 1412–1419. [DOI] [PubMed] [Google Scholar]
- [235].Ludwiczek S, Theurl I, Bahram S, Schumann K, Weiss G, Regulatory networks for the control of body iron homeostasis and their dysregulation in HFE mediated hemochromatosis, J Cell Physiol 204 (2005) 489–499. [DOI] [PubMed] [Google Scholar]
- [236].Nicolas G, Viatte L, Lou DQ, Bennoun M, Beaumont C, Kahn A, Andrews NC, Vaulont S, Constitutive hepcidin expression prevents iron overload in a mouse model of hemochromatosis, Nat Genet 34 (2003) 97–101. [DOI] [PubMed] [Google Scholar]
- [237].Chua AC, Herbison CE, Drake SF, Graham RM, Olynyk JK, Trinder D, The role of Hfe in transferrin-bound iron uptake by hepatocytes, Hepatology 47 (2008) 1737–1744. [DOI] [PubMed] [Google Scholar]
- [238].Robb A, Wessling-Resnick M, Regulation of transferrin receptor 2 protein levels by transferrin, Blood 104 (2004) 4294–4299. [DOI] [PubMed] [Google Scholar]
- [239].Chua AC, Drake SF, Herbison CE, Olynyk JK, Leedman PJ, Trinder D, Limited iron export by hepatocytes contributes to hepatic iron-loading in the Hfe knockout mouse, J Hepatol 44 (2006) 176–182. [DOI] [PubMed] [Google Scholar]
- [240].Ramos P, Guy E, Chen N, Proenca CC, Gardenghi S, Casu C, Follenzi A, Van RN, Grady RW, de SM, Rivella S, Enhanced erythropoiesis in Hfe-KO mice indicates a role for Hfe in the modulation of erythroid iron homeostasis, Blood 117 (2011) 1379–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Laftah AH, Simpson RJ, Beaumont N, Bahram S, Schumann K, Srai SK, Hypoxic response of iron absorption is not affected by the Hfe gene knock-out in mice, Br J Haematol 123 (2003) 170–172. [DOI] [PubMed] [Google Scholar]
- [242].Paglia DE, Radcliffe RW, Anthracycline cardiotoxicity in a black rhinoceros (Diceros bicornis): evidence for impaired antioxidant capacity compounded by iron overload, Vet Pathol 37 (2000) 86–88. [DOI] [PubMed] [Google Scholar]
- [243].Olias P, Mundhenk L, Bothe M, Ochs A, Gruber AD, Klopfleisch R, Iron overload syndrome in the black rhinoceros (Diceros bicornis): microscopical lesions and comparison with other rhinoceros species, J Comp Pathol 147 (2012) 542–549. [DOI] [PubMed] [Google Scholar]
- [244].Beutler E, West C, Speir JA, Wilson IA, Worley M, The hHFE gene of browsing and grazing rhinoceroses: a possible site of adaptation to a low-iron diet, Blood Cells Mol Dis 27 (2001) 342–350. [DOI] [PubMed] [Google Scholar]
- [245].Johnson SP, Venn-Watson SK, Cassle SE, Smith CR, Jensen ED, Ridgway SH, Use of phlebotomy treatment in Atlantic bottlenose dolphins with iron overload, J Am Vet Med Assoc 235 (2009) 194–200. [DOI] [PubMed] [Google Scholar]
- [246].Mazzaro LM, Johnson SP, Fair PA, Bossart G, Carlin KP, Jensen ED, Smith CR, Andrews GA, Chavey PS, Venn-Watson S, Iron indices in bottlenose dolphins (Tursiops truncatus), Comp Med 62 (2012) 508–515. [PMC free article] [PubMed] [Google Scholar]
- [247].Phillips BE, Venn-Watson S, Archer LL, Nollens HH, Wellehan JF Jr., Preliminary investigation of bottlenose dolphins (Tursiops truncatus) for hfe gene-related hemochromatosis, J Wildl Dis 50 (2014) 891–895. [DOI] [PubMed] [Google Scholar]
- [248].Olias P, Weiss AT, Gruber AD, Klopfleisch R, Iron storage disease in red deer (Cervus elaphus elaphus) is not associated with mutations in the HFE gene, J Comp Pathol 145 (2011) 207–213. [DOI] [PubMed] [Google Scholar]