

Abstract
Two OCD genome-wide association studies (GWAS) have been published by independent OCD consortia, the International Obsessive-Compulsive Disorder Foundation Genetics Collaborative (IOCDF-GC) and the OCD Collaborative Genetics Association Study (OCGAS), but many of the top-ranked signals were supported in only one study. We therefore conducted a meta-analysis from the two consortia, investigating a total of 2,688 individuals of European ancestry with OCD, and 7,037 genomically matched controls. No SNPs reached genome-wide significance. However, in comparison to the two individual GWASs, the distribution of p-values shifted towards significance. The top haplotypic blocks were tagged with rs4733767 (p=7.1×10−7; OR=1.21;(CI: 1.12,1.31); CASC8/CASC11), rs1030757 (p=1.1×10−6; OR=1.18;CI:1.10,1.26, GRID2) and rs12504244 (p=1.6×10−6; OR=1.18;CI: 1.11,1.27, KIT). Variants located in or near the genes ASB13, RSPO4, DLGAP1, PTPRD, GRIK2, FAIM2, and CDH20, identified in linkage peaks and the original GWASs, were amongst the top signals. Polygenic risk scores for each individual study predicted case/control status in the other by explaining 0.9% (p=0.003) and 0.3% (p=0.0009) of the phenotypic variance in OCGAS and the European IOCDF-GC target samples, respectively. The common SNP heritability in the combined OCGAS and IOCDF-GC sample was estimated to be 0.28 (s.e. = 0.04). Strikingly, approximately 65% of the SNP based heritability in the OCGAS sample was accounted for by SNPs with minor allele frequencies equal to or greater than 40%.This joint analysis constituting the largest single OCD genome-wide study to date represents a major integrative step in elucidating the genetic causes of OCD.
INTRODUCTION
Obsessive-compulsive disorder (OCD) is a psychiatric condition characterized by persistent, intrusive thoughts and urges (obsessions) and repetitive, intentional behaviors (compulsions), typically, but not always, performed to reduce anxiety caused by obsessions1. The estimated lifetime prevalence of OCD is 1– 3%, based on national surveys2. Individuals with OCD experience a chronic or episodic course with exacerbations that can substantially impair social and occupational functioning1.
Since the early twentieth century, clinicians have suspected that heredity plays an important role in susceptibility to OCD. Consistent with this, several family studies have found a substantially greater prevalence of OCD (approximately 10-fold increase) in the first-degree relatives of probands, compared to relatives of controls3–6. Family studies of OCD in child and adolescent probands report even greater differences in the risk of OCD in relatives of cases compared to controls7, 8, consistent with previous reports of increased familial loading with an early age-at-onset3, 4.
The few existing studies that have examined twin concordance rates for OCD are insufficient in size to allow for accurate heritability estimates9. However, population-based twin studies estimate the heritability of dimensional measures of obsessive-compulsive symptoms (OCS) to be 40 −50%, with a similar contribution from non-shared environment, and no significant contribution from shared environment10–14. More recently, direct interrogation of the genome using Genome-Wide Complex Trait Analysis (GCTA) on data from the International OCD Foundation-Genetics Consortium (IOCDF-GC) genome wide association study (GWAS) provided heritability estimates of 0.37 (se = 0.07, p = 1.5 × 10−07) for OCD. In the same sample, the estimate of heritability for childhood-onset OCD (symptoms before the age of 1715) was 0.43 (se = 0.10, p = 1 × 10−05). Partitioning by minor allele frequency (MAF) suggested that the vast majority of the heritability was accounted for by SNPs with MAF > 0.30; little heritability was accounted for by SNPs with a MAF of less than 5%15.
To date, eight whole-genome studies of OCD or OCS have been published, including five linkage studies16–22, two genome-wide association studies (GWAS) of OCD23, 24, and one GWAS of obsessive compulsive symptoms (OCS)25. The five linkage studies identified several chromosomal regions with suggestive evidence for linkage16–20, although there was little overlap between them and only one (1p36) met criteria for statistical significance for linkage16. Consistent with sample size expectations for highly polygenic traits, no individual susceptibility variants have yet been identified for OCD using these methods.
The two published GWAS of OCD were conducted by independent OCD consortia: the International OCD Foundation Genetics Collaborative (IOCDF-GC)24 and the OCD Collaborative Genetics Association Study (OCGAS)23. The IOCDF-GC published the first OCD GWAS, comprising 1,465 cases and 5,557 ancestry-matched controls, as well as 400 complete trios, from 22 sites worldwide 26. The top signal from the combined trio-case-control sample was rs297941 on chromosome 19p13.2, near FAIM2 (p =4.99 × 10−7). Although no SNPs were found to be associated with OCD at a genome-wide significance level, a significant enrichment of methylation quantitative trait loci (p<0.001) and frontal lobe expression quantitative trait loci (p=0.001) were observed within the top-ranked SNPs, providing evidence, consistent with other disease reports27, 28, that biologically relevant associations are present within subthreshold GWAS results. The OCGAS reported a second GWAS, conducted by six research centers in the United States23. In this study, 1,065 families (containing 1,406 patients with OCD), combined with population-based control samples (resulting in a total sample of 5,061 individuals), were studied. The smallest p-value (p=4.13 × 10−7) was detected for a SNP on chromosome 9p23, in close proximity to the protein tyrosine phosphate receptor D gene (PTPRD). The second smallest p-value was 1.76 × 10−6 near the cadherin type 9 and 10 (CDH9 and CDH10) genes on chromosome 5p15.
A third GWAS, this one examining quantitative obsessive compulsive symptoms (OCS), was conducted in 6,931 individuals from the Netherlands Twin Registry (NTR)25. This study identified one gene that met criteria for genome-wide significance, the myocyte enhancer factor 2B neighbor (MEF2BNB) (p=2.56 × 10−8), on chromosome 19p13. The total SNP-based heritability for OCS in this sample was 0.14 (se=0.05, p=0.003), and the polygenic risk score (PRS) derived from the IOCDF-GC GWAS was significantly associated with OCS, explaining 0.2% of the variance.
As is evident from the data above, although multiple regions of interest have been reported, there is currently little convergence of results to identify OCD susceptibility variants. This is likely due to genetic and phenotypic heterogeneity, and insufficient sample sizes. Thus, a logical next step is to use comparable datasets in combined analyses to increase power. Here, we report findings from combined analyses of the IOCDF-GC and OCGAS GWAS data aimed at further exploring the genetic underpinnings of OCD. We first used the genotypes of these two studies, after imputation to a common reference, to conduct a joint GWAS. We then used each individual study as a discovery sample for polygenic risk score (PRS) analysis and predicted case/control status in the alternate dataset to investigate the amount of phenotypic variation explained by the respective polygenic risk scores. To replicate the finding that SNPs with high MAF account for the majority of the heritability in OCD, we next computed the common variation heritability of the OCGAS sample using GCTA and performed the same partitioning, as previously reported15. Finally, we used LD score regression29 to estimate the heritability of OCD based on the combined meta-analysis cohort.
METHODS
Samples
For these analyses we used only individuals of European ancestry from the original GWAS samples, yielding 1,429 cases, 5,089 controls and 285 trios from IOCDF-GC and 344 cases and 1,033 controls and 630 trios from OCGAS (Table S1), after the addition of screened controls from the Genomic Psychiatry Cohort (GPC) 30, matched to the OCGAS cases. All cases met DSM-IV criteria for OCD1. Controls from the IOCDF-GC GWAS were unscreened. Additional information on the IOCDF-GC and OCGAS samples and methods have been previously published23, 26. This work was approved by the relevant institutional review boards at all participating sites, and all participants provided written informed consent.
GWAS Analyses
We imputed genotype level data from the IOCDF-GC (except the Dutch samples, which were imputed separately, see below), OCGAS, and GPC samples using IMPUTE231 and reference haplotypes from the 1000 Genomes Project (Phase I integrated variant set release); NCBI build 37 (hg19) were constructed with SHAPEIT232. We assessed genetic relatedness between samples through IBD estimation between all sample pairs using PLINK33 and retained only one member of each pair of samples with pi_hat >0.2. Samples were excluded if they had a call rate <0.98, an absolute value of F_HET >0.20, or absence or unambiguous correct genotypic sex. SNPs were excluded from pre-imputation data set if the call rate was <0.98, MAF<0.01, case-control differential missingness was >0.02, or the p-value of Hardy–Weinberg equilibrium (HWE) was <1.0×10−6 for controls and <1.0×10−10 for cases. After imputation, SNPs were excluded if IMPUTE2 info was <0.6, IMPUTE2 certainty was <0.9, or MAF<0.01. We assessed population structure using Multidimensional scaling (MDS), and as previously observed24, samples of Ashkenazi Jewish or Afrikaans (South African) ethnicity clustered as separate groups (Supplemental Figures S1–S5). We conducted separate association analyses for each case-control subpopulation (IOCDF-GC European (IOEU), IOCDF-GC Ashkenazi Jewish (IOAJ), IOCDF-GC South African (IOSA), OCGAS case-control (OCCC)) and trio sample (IOCDF-GC trios (IOTR) and OCGAS trios (OCTR); as probands versus pseudo-controls). We defined “pseudo-controls” as the non-transmitted haplotype pairs from parents to affected offspring in the trio samples.
Due to more stringent data sharing restrictions for Dutch cases, imputation and summary statistics for the Dutch cases and population-matched controls (IODU) were calculated separately by the site investigators following the same imputation and quality control procedures. We then performed meta-analysis using the summary statistics of all case-control subpopulations (including IODU) and trio samples using METAL34 with the inverse variance method on SNPs that passed QC in at least 500 cases and 500 controls. Results were visualized with Assocplots35. The top loci of meta-analysis were defined by the regions of linkage disequilibrium (LD) pruned independent top SNPs passing predefined P-value threshold (r2<0.2, 500kb window, clump function in PLINK) and their tagged SNPs (r2>0.2, 1000kb window, show-tags function in PLINK) using 1KG samples of European independent founders (EUR, TSI, and GBR, phase 1) as the reference panel.
Polygenic Risk Score Analysis
We conducted risk polygenic score (PRS) analyses using PLINK, as previously described36, to test whether multiple variants of small effect jointly contribute to OCD. PRSs for subsets of the IOCDF-GC sample (IOEU) and the OCGAS sample (OCTR) (target samples) were calculated based on the SNP effect size estimated from the discovery samples, the OCGAS and European ancestry IOCDF-GC samples (excluding IOAJ), respectively. Imputed SNPs, with high quality (IMPUTE2 info>0.95, MAF>0.05), and GWAS p-values passing predetermined significance thresholds (p<0.0001, 0.001, 0.01, 0.1, 0.2, 0.3, 0.4, and 0.5) in the discovery samples, were extracted along with their risk alleles and odds ratios, and then linkage disequilibrium (LD) pruned within 500kb window at r2>0.2 (clump function in PLINK) using 1KG samples of European independent founders (EUR, TSI, and GBR, phase 1) as the reference panel.
For each significance threshold, a quantitative aggregate risk score was calculated for each individual in the target sample IOEU and OCTR, defined as the sum of the number of risk alleles present at each locus weighted by the log of the odds ratio for that locus estimated from the discovery sample. We examined the relationship between aggregate risk score and case-control status in the target samples, IOEU and OCTR, at each significance threshold, using logistic regression controlling for population stratification. We then estimated the percentage of phenotypic variance explained by the aggregate risk score (Nagelkerke’s pseudo-R2).
Heritability and genetic correlations
Genetic Complex Trait Analysis
We used GCTA v1.24 (http://cnsgenomics.com/software/gcta/) to estimate the proportion of phenotypic variance explained by directly genotyped and imputed SNPs in the OCGAS sample, as has previously been done in the IOCDF-GC sample15. Due to the sensitivity of GCTA to low quality SNPs and remotely related samples, we conducted more stringent QC for these analyses by removing SNPs with HWE p<0.05 and platform effects with p<0.01 (detected by GWAS comparison of platforms among population-matched controls) and removing one member of each sample pair with pi_hat>0.05. For directly genotyped and imputed SNPs respectively, this resulted in 487,459 directly genotyped and 5,843,119 imputed SNPs on 999 cases and 1,064 controls. After quality control, we used the GCTA software to generate a genetic relationship matrix (GRM) file, which included IBD relationships calculated from genotype data. Genomic restricted maximum likelihood (GREML) analysis was conducted using the respective GRM estimated from all the SNPs and 20 principal component quantitative covariates. In order to account for the oversampling of cases, we used the OCD population prevalence (2.5%) to transform the estimate of variance explained to the liability scale. Finally, we estimated the chromosome specific heritability and heritability partitioned by minor allele frequency (MAF) for five MAF bins (0.01~0.1, 0.1~0.2, 0.2~0.3, 0.3~0.4, and 0.4~0.5).
We then combined the two largest European IOCDF-GC and OCGAS datasets (not including trios, IOAJ, IOSA, or Dutch), and performed a second round of QC to remove any samples which fell outside the European genetic cluster, and one sample of any pair with a pi-hat > 0.05, resulting in 1,323 cases and 4,938 controls. At the SNP level, we filtered the imputed SNPs based on the imputation info quality metric (info>0.6), certainty (<80%), and MAF (<0.05), which resulted in 5,235,858 SNPs. A prevalence of 2.5% and 20 principal components were used for the GREML analysis.
LD score regression analysis
We applied the LD score regression (LDSC) method29 to 1,159,580 imputed and directly genotyped SNPs (which overlapped with a panel of high confidence HapMap3 SNPs) measured on all 9,725 (2,688 cases and 7,037 controls) individuals included in the OCD meta-analysis. Regression weights were calculated using the HapMap European reference sample provided by Bulik-Sullivan and colleagues. To transform from the observed heritability scale to the liability scale we used a population prevalence of 2.5%. Using LDSC, we calculated heritability, checked for residual population stratification (based on the LDSC intercept), and calculated genetic correlation between the two consortium sample collections.
RESULTS
GWAS
After data cleaning to remove samples falling outside the European genetic cluster, we had a total sample size of 2,688 OCD cases and 7,037 gnomically matched controls, comprised of seven subsamples (Table S1) that were analyzed individually and then combined by meta-analysis to provide overall p-values on 8,693,187 autosomal SNPs. We generated quantile-quantile plots of the observed versus expected log(P) values under the null hypothesis and calculated the genomic control lambda for the final sample. We observed no evidence for significant residual stratification effects (Supplementary Figure S6; λ=1.028; λ1000=1.007). In addition, the LDSC analysis demonstrated no evidence of residual population stratification (LDSC Intercept = 1.0005; s.e. = 0.0068).
No SNP exceeded the genome-wide threshold for significance (Figure 1, Table 1). 130 SNPs (Supplementary Table S2) from 29 LD independent loci (Table 1) were observed with p<1.0×10-5. The SNP with the lowest p-value was rs4733767 (p=7.1×10−7; OR=1.21;CI:1.12,1.31), which tagged a haplotypic block of 53.7 kb (chr8:128,568,359–128,622,083) on chromosome 8q24.21 and is 87.2 kb 5’ to CASC8 (Cancer Susceptibility Candidate 8, also known as LOC727677) (Table 1, Supplementary Table S2, Supplementary Figure S7). The SNP with the second lowest p-value, rs1030757 (p=1.1×10−6; OR=1.18;CI:1.10,1.26), on chromosome 4q22.1, tagged the second best haplotype block (chr4:93,479,275–94,230,511; 751.2 kb; Supplementary Figure S8) that lies wholly within GRID2 (Glutamate Ionotropic Receptor Delta Type Subunit 2). Within this haplotype there were SNPs with heterogeneity p-values < 0.05, we conducted random-effects meta-analysis using PLINK; the findings were different. We identified that the heterogeneity came from the isolate subpopulation South African. Excluding this sample (post hoc) from the meta analysis, the association with OCD for the SNPs with heterogeneity became more significant (e.g. rs7683744: p = 8.0×10−7; OR=0.84). The SNP with the third lowest p-value, rs12504244 (p=1.6×10−6; OR=1.18;CI:1.11,1.27), on chromosome 4q12, tagged the third best haplotypic block (chr4:55,476,381–55,580,596; 104.2 kb; Supplementary Figure S9) which overlies the promoter and much of the gene body of KIT (KIT Proto-Oncogene Receptor Tyrosine Kinase).
Figure 1.
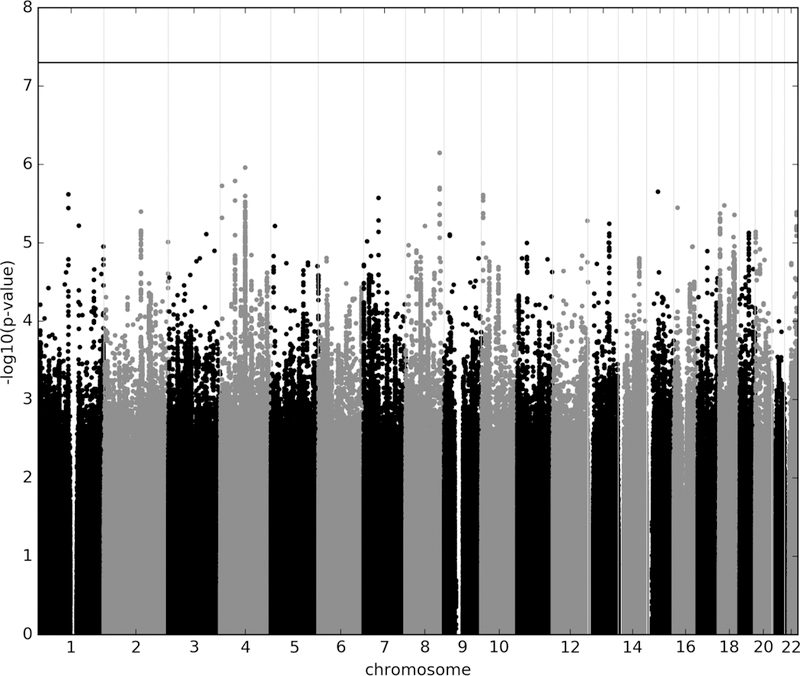
Manhattan plot of genotyped and imputed single-nucleotide polymorphisms (SNPs) for 2,688 OCD cases and 7,031 controls from the IOCDF-GC, OCGAS, and GPC consortia. Red line indicates the genome-wide significance threshold of p=5×10−8.
Table 1.
LD-Independent Genomic Regions With p <1×10−5 in the Combined OCD Genome-Wide Association Study (GWAS)
| SNP | CHR | BP | A1/A2 | A1 FRQ | INFO | Odd Ratio (95%CI) |
P | LD block (hg19) |
Genes |
|---|---|---|---|---|---|---|---|---|---|
| rs4733767 | 8 | 128,581,578 | A/G | 0.274 | 0.867 | 1.21 (1.12, 1.31) | 7.1E-07 | 128568359-128622083 | |
| rs1030757 | 4 | 93,697,153 | C/A | 0.488 | 0.979 | 1.18 (1.10, 1.26) | 1.1E-06 | 93479275-94230511 | GRID2 |
| rs12504244 | 4 | 55,485,188 | G/C | 0.393 | 0.974 | 1.18 (1.11, 1.27) | 1.6E-06 | 55476381-55580596 | KIT |
| rs13141765 | 4 | 6,243,646 | C/T | 0.393 | 0.713 | 1.31 (1.17, 1.46) | 1.9E-06 | 6239015-6249840 | LOC285484 |
| rs116347760 | 1 | 114,201,251 | A/T | 0.020 | 0.793 | 1.88 (1.44, 2.44) | 2.4E-06 | 113751726-114621340 | AP4B1,AP4B1-AS1, BCL2L15, DCLRE1B,HIPK1, LOC643441,LOC101928846, MAGI3,OLFML3,PHTF1,PTPN22,RSBN1,SYT6 |
| rs72781967 | 10 | 5,622,426 | C/T | 0.344 | 0.995 | 1.18 (1.10, 1.27) | 2.4E-06 | 5607539-5659916 | |
| rs55687617 | 7 | 56,775,429 | A/G | 0.119 | 0.881 | 0.76 (0.68, 0.85) | 2.7E-06 | 56314381-57203788 | DKFZp434L192,LOC650226,LOC100130849, LOC100240728,LOC101928401,MIR4283-1, MIR4283-2,ZNF479 |
| rs117310268 | 18 | 19,675,267 | T/C | 0.038 | 0.780 | 1.57 (1.30, 1.89) | 3.3E-06 | 19505213-19683750 | |
| rs5019028 | 4 | 94,222,825 | G/T | 0.279 | 0.995 | 1.19 (1.11, 1.28) | 3.4E-06 | 93665778-94232270 | GRID2 |
| rs72783425 | 16 | 14,148,431 | A/C | 0.053 | 0.960 | 1.40 (1.22, 1.62) | 3.5E-06 | 13982844-14318912 | ERCC4,LOC101927311,LOC101927348,MKL2 |
| rs56343802 | 2 | 139,823,241 | T/A | 0.281 | 0.999 | 1.19 (1.10, 1.27) | 4.0E-06 | 139821308-139882668 | |
| rs909701 | 22 | 44,973,368 | G/C | 0.500 | 0.999 | 1.17 (1.09, 1.25) | 4.1E-06 | 44971548-44988209 | LINC00207,LINC00229 |
| rs9952159 | 18 | 3,660,801 | T/C | 0.220 | 1.026 | 1.20 (1.11, 1.30) | 4.2E-06 | 3638103-3710355 | DLGAP1 |
| rs77885126 | 18 | 58,420,429 | C/T | 0.015 | 0.932 | 1.83 (1.41, 2.36) | 4.4E-06 | 58250265-59372219 | CDH20 |
| rs10773765 | 12 | 130,767,334 | T/C | 0.252 | 0.907 | 1.20 (1.11, 1.30) | 5.2E-06 | 130739141-130857497 | PIWIL1 |
| rs9544927 | 13 | 79,505,864 | G/A | 0.208 | 0.996 | 0.82 (0.76, 0.90) | 5.7E-06 | 79496764-79551012 | |
| rs28599745 | 1 | 153,396,665 | A/G | 0.160 | 0.650 | 0.70 (0.59, 0.81) | 6.0E-06 | 153349194-153396665 | S100A7A,S100A7L2,S100A8,S100A9,S100A12 |
| rs1652783 | 8 | 73,279,728 | G/A | 0.225 | 0.764 | 1.31 (1.16, 1.47) | 6.1E-06 | 73279414-73312929 | |
| rs190543171 | 5 | 15,840,912 | T/C | 0.012 | 0.787 | 2.32 (1.61, 3.35) | 6.1E-06 | 15726922-16139424 | FBXL7,MARCH11 |
| rs56025909 | 20 | 955,893 | T/C | 0.032 | 0.960 | 1.51 (1.26, 1.81) | 7.2E-06 | 931170-991579 | RSPO4 |
| rs3097331 | 19 | 34,648,956 | C/T | 0.368 | 1.000 | 0.85 (0.80, 0.92) | 7.5E-06 | 34632485-34727202 | KIAA0355,LSM14A |
| rs138445568 | 3 | 143,922,936 | T/A | 0.013 | 0.630 | 2.53 (1.68, 3.80) | 7.7E-06 | 142971745-144868438 | C3orf58,SLC9A9,SLC9A9-AS1 |
| rs139286049 | 9 | 20,688,387 | G/A | 0.019 | 0.760 | 1.82 (1.40, 2.37) | 7.8E-06 | 20688387-21100533 | FOCAD,IFNB1,MIR491,PTPLAD2 |
| rs146238482 | 9 | 20,856,226 | C/G | 0.010 | 0.823 | 2.68 (1.74, 4.13) | 8.1E-06 | 20836624-21016372 | FOCAD,PTPLAD2 |
| rs35894340 | 18 | 54,307,062 | G/A | 0.252 | 0.948 | 1.19 (1.10, 1.29) | 8.4E-06 | 54246712-54518866 | TXNL1,WDR7 |
| rs149952789 | 7 | 12,716,170 | T/A | 0.010 | 0.603 | 3.33 (1.96, 5.68) | 9.6E-06 | 12716170-12716170 | ARL4A |
| rs4444795 | 4 | 93,355,172 | T/C | 0.183 | 0.912 | 1.22 (1.12, 1.33) | 9.7E-06 | 93195208-94077524 | GRID2 |
| rs75740353 | 2 | 242,741,686 | A/G | 0.103 | 0.869 | 0.65 (0.54, 0.79) | 9.7E-06 | 242685298-242764651 | D2HGDH,GAL3ST2,ING5,NEU4 |
| rs116969557 | 13 | 77,337,736 | A/G | 0.016 | 0.957 | 1.77 (1.38, 2.29) | 9.9E-06 | 77082597-77566923 | BTF3P11,CLN5,FBXL3,IRG1,KCTD12 |
Chr=chromosome; SNP=single-nucleotide polymorphism; A1=minor allele; A2=major allele; A1 FRQ=frequency of A1 allele; LD block= tagged region by the index SNP at r2>0.2; Genes=Genes and their 20kb flanking regions on each side overlapped with the LD block
Polygenic Risk Score Analysis
We used SNP effect sizes derived from the individual OCGAS and IOCDF-GC meta-analyses to calculate PRS and predict OCD status in individuals of European ancestry from the IOCDF-GC sample (IOEU) and in trios of European ancestry from the OCGAS sample (OCTR), respectively. As shown in Figure 2 and Table S3, the PRS derived from meta-analysis of the European IOCDF-GC samples (excluding the IOAJ samples to avoid heterogeneity in the discovery sample) reasonably predicted case-control status in the OCGAS trio target sample (p=0.003), explaining approximately 0.9% of the phenotypic variance. Conversely, risk scores derived using the OCGAS as a discovery sample explained 0.3% of the phenotypic variance in the IOEU samples (p=0.0009).
Figure 2. Polygenic Risk Score Analysis in OCD.
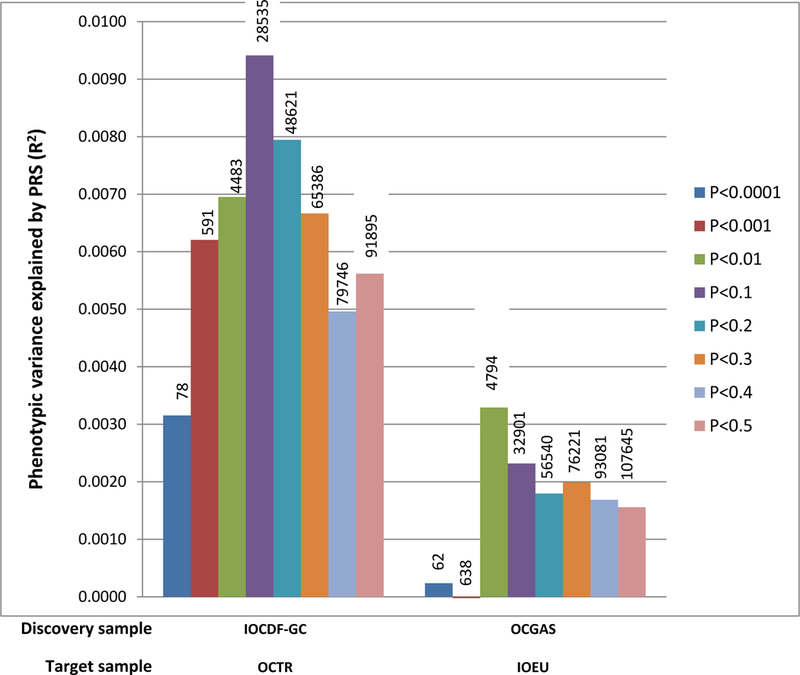
The variance explained in two target samples (OCTR, consisting of 630cases and 630 pseudo-controls; IOEU, consisting of 1032 cases and 4100 controls) is based on risk scores derived from an aggregated sum of weighted single-nucleotide polymorphism risk allele effect sizes estimated from the discovery samples (IOCDF-GC without IOAJ, consisting of 1623 cases and 5113 controls; OCGAS, consisting of 974 cases and 663 controls) at eight significance thresholds. The numbers of SNPs used at each significance thresholds for PRS were listed on the top of the corresponding bars. The y-axis indicates Nagelkerke’s pseudo R2.
Heritability Analyses
GCTA-based heritability in the OCGAS sample alone (999 cases and 1,064 controls (Table 2)) was 0.25 (se=0.11, p=0.0096). The GCTA heritability estimate of OCD in the combined OCGAS and IOCDF-GC European sample was also 0.25 (se=0.05; p=0.0096). LD score regression (LDSC) analysis yielded a heritability estimate of 0.28 (se=0.04) for the combined OCD sample (Table 2). When the sample was then split into its constituent parts (OCGAS and IOCDF-GC), we observed a significant genetic correlation between the two (rg = 0.83; s.e. = 0.28; p = 0.003).
Table 2.
Heritability estimates for OCD in the IOCDF-GC and OCGAS samples.
| Sample characteristics | Method | Cases | Controls | Number of SNPs |
Reference | V(g)/V(p)_Liability (SE) |
|---|---|---|---|---|---|---|
| IOCDF-GC case/control | GCTA | 1,061 | 4,236 | 7,657,106 | Davis et al., 2013 | 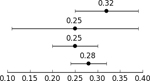 |
| OCGAS case/control and trio controls | GCTA | 999 | 1,064 | 5,843,119 | Current Manuscript | |
| OCGAS and IOCDF-GC cases/control EU only | GCTA | 1,323 | 4,398 | 5,235,858 | Current Manuscript | |
| OCGAS and IOCDF-GC case/control and pseudo-control | LDSC | 2,936 | 7,279 | 1,159,580 | Current Manuscript |
In parallel with the univariate and bivariate analyses, we partitioned heritability by allele frequency in the OCGAS sample using five minor allele frequency (MAF) bins as follows, 0.01~0.1, 0.1~0.2, 0.2~0.3, 0.3~0.4, and 0.4~0.5 in the GRMEL joint analysis; results are presented in Table S4. As has been previously reported for the IOCDF-GC sample15, the largest proportion of heritability was observed in the highest frequency allele bins (MAF>0.4).
DISCUSSION
We report the results of a meta-analysis of GWAS from the two published genome-wide association studies of OCD, with a sample of 2,688 individuals with OCD that include both family-based and singleton cases, and 7,037 controls. Polygenic Risk Score (PRS) and LD score regression (LDSC) analyses confirm that the two samples share genetic risk factors for OCD, and are thus appropriate for combined GWAS analyses. With LDSC we observed a strong genetic correlation between the IOCDF-GC and OCGAS samples (rg = 0.83, s.e. = 0.28; p = 0.003). PRS derived from each sample significantly predicted case-control status in the other sample. Although the phenotypic variance explained was relatively small (R2 = 0.9% for the OCGAS trios and 0.3% for the IOCDF-GC European cases and controls), they are comparable to those found for schizophrenia, using similar discovery sample sizes37 (PGC2 SCZ GWAS, 2014).
Using GCTA, the heritability tagged by the SNPs in the OCGAS sample was slightly lower (25%) than previously observed for the IOCDF-GC sample (32%)15. The ascertainment strategies differed in IOCDF-GC and OCGAS studies, with the former recruiting individuals, and the latter, primarily families (trios), which may underestimate the heritability tagged by SNPs, as the polygenic load in family members of affected individuals is elevated in comparison to controls38. Joint heritability analyses of the two samples, using GCTA and LDSC, resulted in similar estimates (0.25 and 0.28, respectively), suggesting that the common variation heritability of OCD is between 25–30% (i.e., 50% or more of the total heritability than estimated by twin studies.
We also examined the allele frequency distribution of the common variation heritability of OCD. Although the confidence intervals of each allele frequency bin are large, due to the limited sample sizes, the majority of the heritability (~65%) was accounted for by SNPs with high MAF (e.g., above 40%) in both the OCGAS sample alone and combined sample (Table S4).
Although there were no genome-wide significant findings, the 53.7 kb (chr8:128,568,359–128,622,083) haplotype block encompassing the top SNP, rs4733767 (Figure S7), contains 25 H3K27Ac peaks in the ENCODE/ROADMAP data, suggesting it has regulatory potential39, although the current release of the Genotype-Tissue Expression project (GTEx Release V6 (dbGaP Accession phs000424.v6.p1))40 has no eQTLs in the block. The closest genes on either side of rs4733767 (CASC8 and CASC11), are long non-coding RNAs (lincRNAs), which are thought as a class to have regulatory functions 41. Both are only expressed at low levels in the brain in the GTEx database. The potential transcriptional consequences of genomic risk for OCD in this region are unclear, at present.
The second best haplotypic block (chr4:93,479,275–94,230,511) lies entirely within GRID2, a gene expressing a subunit of an ionotropic glutamate receptor, and contains about 300 H3K27Ac peaks. The region between ~94,120,000–94,230,000 kb contains multiple SNPs that regulate GRID2, in both brain (www.BRAINEAC.org, intralobular white matter (WHMT, n=131))42 and testis, (GTEx Release V6). In the latter, two of these SNPs overlap with those observed in this study (rs7684707 and rs5019028), and the OCD risk allele is predicted to increase expression. These eQTLs were not detected in brain in the GTEx study, most likely as a consequence of small sample size. GRID2 is highly expressed in the cerebellum, but is also expressed in other regions of the brain throughout the lifespan (www.BrainSpan.org), with detectable expression in the caudate, putamen, nucleus accumbens and the anterior cingulate cortex, all regions that have been implicated in OCD43, and is part of the glutamatergic signaling system, which is thought to be important in OCD44. Deletions of portions of GRID2 in humans are responsible for spinocerebellar ataxia, autosomal recessive 18 (SCAR18; http://omim.org/entry/602368), which are severe when homozygous and milder when heterozygous44. These observations suggest that lower GRID2, particularly in the cerebellum causes ataxia, while higher GRID2, especially in the non-cerebellar brain, may increase risk for OCD.
The third best haplotypic block observed contains the promotor and much of the gene body of KIT (KIT Proto-Oncogene Receptor Tyrosine Kinase). No eQTLs for KIT are found in GTEx (v6), but the haplotypic block is likely to regulate the gene, as it contains 47.7 kb 5’ to the transcription start site, and has 76 H3K27Ac peaks. KIT is expressed in multiple brain regions (BrainSpan and GTEx) and across the human lifespan, with highest expression during fetal development (BrainSpan). Allelic variants of KIT in humans have been observed in individuals with piebaldism, various leukemias, and gastrointestinal stromal tumors (http://omim.org/entry/164920?search=kit&highlight=kit). Of note, both KIT and GRID2 are regulated by transforming growth factor beta1 in rodents46,47,48.
Comparison of findings in prior linkage or GWAS studies of OCD
Of the signals with p values <1 × 10−5 in this meta-analysis, two were in genomic regions that have been previously identified in genome-wide linkage studies. These include 7 SNPs on chromosome 10p1518, all of which are eQTLS of ankyrin repeat and SOCS box containing 13 (ASB13), in EBV-transformed lymphocytes, and predict high expression (GTEx Release V6). A 60.4 kb haplotypic block was seen in a linkage peak on 20p1316 that encompasses the gene for RSPO4 and part of its promotor. The block contains SNPs that are eQTLs for RSPO4 in about a dozen tissues and an eQTL for SRXN1 in thyroid. As mentioned above, the number of brain samples in the V6 release is ≤100, limiting the power to detect brain eQTLs. Overall, most of the eQTLs being detected at the present sample sizes in the GTEx project affect multiple tissues, so it is plausible these SNPs may also be regulating the same genes in brain. Final determination of this will require more data.
Of the signals identified in the three prior OCD GWASs23,24,25 ,SNPs within DLGAP1, which was identified in the IOCDF-GC GWAS, represented the signal most strongly supported in this meta-analysis (best was rs9952159, p=4.2×10−6, OR = 1.20). Among the other genes of interest, signals in or near PTPRD, which was previously identified in the OCGAS GWAS (p=2.4×10−4, OR=1.45), GRIK2 (rs116966225, p=5.4×10−4, −158.5 kb and rs78014006, p= 7.2×10−3, intronic) and FAIM2 (rs297941, p=6.1×10−5, 21.3 kb) were also identified in this meta-analysis, although not among the top hits. Although no signal was identified in this meta-analysis for either CDH9 or CDH10, we did identify a strong signal for a related cadherin gene CDH20 (rs77885126, p=4.4×10–6, OR=1.83). It should be noted that according to a power analysis, the sample had low power to detect genome-wide significant association with a common SNP conferring an OR of 1.2 or less. The OCS GWAS was omitted from this study because it employed a different phenotype; i.e. it used a self-report assessment tool that measured the presence of symptoms and not the diagnosis of OCD, as opposed to a clinical assessment that was based on OCD diagnostic criteria. We omitted it because of these differences in phenotypes, and the desire to not introduce additional heterogeneity into the study. The OCS GWAS identified RFXANK as a top signal; it was also identified among the top SNPs of this meta-analysis (rs11666960, p = 6.3 × 10–4, OR = 0.87).
Overall, the results from this meta-analysis support some of the pre-existing findings generated from two previous GWASs of OCD. Among the best findings in this study are several glutamatergic system genes (i.e., GRID2, DLGAP1). Evidence has implicated abnormalities in this system as part of the etiology of OCD and the most robust candidate gene study results have consistently identified genes involved in this neurotransmitter system (GRIN2B 48 & SLC1A1 50). Therefore, further dissection of glutamatergic system genes along with increasing sample sizes will improve our understanding of the underlying mechanism of OCD.
As sample sizes grow and sequencing costs reduce further, we anticipate that genetic associations with OCD will become increasingly robust, and that a proportion of these currently suggestive findings will reach genome-wide significance.
Supplementary Material
ACKNOWLEDGEMENTS
The OCD Collaborative Genetics Association Study (OCGAS) is a collaborative research study and was funded by the following NIMH Grant Numbers: MH071507 (G N), MH079489 (DAG), MH079487 (JM), MH079488 (AF), and MH079494 (JK). Yao Shugart and Wei Guo were also supported by the Intramural Research Program of the NIMH (MH002930-06)
The International Obsessive Compulsive Foundation Genetics Collaborative (IOCDF-GC) was supported by a grant from the David Judah Foundation (a private, non-industry related foundation established by a family affected by OCD), MH079489 (DLP), MH073250 (DLP), S40024 (JMS), MH 085057 (JMS), and MH087748 (CAM).
The authors thank the Psychiatric Genomics Consortium (PGC) for the use of their servers for data integration and analysis, the many families who have participated in the study, as well as the clinicians, study managers, and clinical interviewers at the respective study sites for their efforts in participant recruitment and clinical assessments.
The views expressed here do not reflect the view of the National Institutes of Health, the Department of Health and Human Services, or the United States government.
Footnotes
Supplementary information is available at Molecular Psychiatry’s website
Conflict of Interest: None of the authors report a conflict related to this article.
References
- 1.American Psychiatric Association A. Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition American Psychiatric Association: Arlington,VA, 2013. [Google Scholar]
- 2.Karno M, Golding JM. Obsessive compulsive disorder. In: Robins LN, Regier DA (eds). Psychiatric disorders in America : the epidemiologic catchment area study. Free Press ;Collier Macmillan Canada ; Maxwell Macmillan International: New York Toronto, 1991, pp 204–219. [Google Scholar]
- 3.Pauls DL, Alsobrook JP 2nd, Goodman W, Rasmussen S, Leckman JF. A family study of obsessive-compulsive disorder. Am J Psychiatry 1995; 152(1): 76–84. [DOI] [PubMed] [Google Scholar]
- 4.Nestadt G, Samuels J, Riddle M, Bienvenu OJ 3rd, Liang KY, LaBuda M et al. A family study of obsessive-compulsive disorder. Arch Gen Psychiatry 2000; 57(4): 358–363. [DOI] [PubMed] [Google Scholar]
- 5.Grabe HJ, Ruhrmann S, Ettelt S, Buhtz F, Hochrein A, Schulze-Rauschenbach S et al. Familiality of obsessive-compulsive disorder in nonclinical and clinical subjects. The American journal of psychiatry 2006; 163(11): 1986–1992. [DOI] [PubMed] [Google Scholar]
- 6.Fyer AJ, Lipsitz JD, Mannuzza S, Aronowitz B, Chapman TF. A direct interview family study of obsessive-compulsive disorder. I. Psychological medicine 2005; 35(11): 1611–1621. [DOI] [PubMed] [Google Scholar]
- 7.Hanna GL, Himle JA, Curtis GC, Gillespie BW. A family study of obsessive-compulsive disorder with pediatric probands. Am J Med Genet B Neuropsychiatr Genet 2005; 134(1): 13–19. [DOI] [PubMed] [Google Scholar]
- 8.do Rosario-Campos MC, Leckman JF, Curi M, Quatrano S, Katsovitch L, Miguel EC et al. A family study of early-onset obsessive-compulsive disorder. Am J Med Genet B Neuropsychiatr Genet 2005; 136(1): 92–97. [DOI] [PubMed] [Google Scholar]
- 9.van Grootheest DS, Cath DC, Beekman AT, Boomsma DI. Twin studies on obsessive-compulsive disorder: a review. Twin Res Hum Genet 2005; 8(5): 450–458. [DOI] [PubMed] [Google Scholar]
- 10.Clifford CA, Murray RM, Fulker DW. Genetic and environmental influences on obsessional traits and symptoms. Psychol Med 1984; 14(4): 791–800. [DOI] [PubMed] [Google Scholar]
- 11.Jonnal AH, Gardner CO, Prescott CA, Kendler KS. Obsessive and compulsive symptoms in a general population sample of female twins. Am J Med Genet 2000; 96(6): 791–796. [DOI] [PubMed] [Google Scholar]
- 12.Eley TC, Bolton D, O’Connor TG, Perrin S, Smith P, Plomin R. A twin study of anxiety-related behaviours in pre-school children. J Child Psychol Psychiatry 2003; 44(7): 945–960. [DOI] [PubMed] [Google Scholar]
- 13.Hudziak JJ, Van Beijsterveldt CE, Althoff RR, Stanger C, Rettew DC, Nelson EC et al. Genetic and environmental contributions to the Child Behavior Checklist Obsessive-Compulsive Scale: a cross-cultural twin study. Arch Gen Psychiatry 2004; 61(6): 608–616. [DOI] [PubMed] [Google Scholar]
- 14.Taylor S Etiology of obsessions and compulsions: a meta-analysis and narrative review of twin studies. Clin Psychol Rev 2011; 31(8): 1361–1372. [DOI] [PubMed] [Google Scholar]
- 15.Davis LK, Yu D, Keenan CL, Gamazon ER, Konkashbaev AI, Derks EM et al. Partitioning the heritability of tourette syndrome and obsessive compulsive disorder reveals differences in genetic architecture. PLoS Genet 2013; 9(10): e1003864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mathews CA, Badner JA, Andresen JM, Sheppard B, Himle JA, Grant JE et al. Genome-wide Linkage Analysis of Obsessive-Compulsive Disorder Implicates Chromosome 1p36. Biol Psychiatry 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ross J, Badner J, Garrido H, Sheppard B, Chavira DA, Grados M et al. Genomewide linkage analysis in Costa Rican families implicates chromosome 15q14 as a candidate region for OCD. Hum Genet 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanna GL, Veenstra-Vanderweele J, Cox NJ, Van Etten M, Fischer DJ, Himle JA et al. Evidence for a susceptibility locus on chromosome 10p15 in early-onset obsessive-compulsive disorder. Biol Psychiatry 2007; 62(8): 856–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shugart YY, Samuels J, Willour VL, Grados MA, Greenberg BD, Knowles JA et al. Genomewide linkage scan for obsessive-compulsive disorder: evidence for susceptibility loci on chromosomes 3q, 7p, 1q, 15q, and 6q. Mol Psychiatry 2006; 11(8): 763–770. [DOI] [PubMed] [Google Scholar]
- 20.Hanna GL, Veenstra-VanderWeele J, Cox NJ, Boehnke M, Himle JA, Curtis GC et al. Genome-wide linkage analysis of families with obsessive-compulsive disorder ascertained through pediatric probands. Am J Med Genet 2002; 114(5): 541–552. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y, Samuels JF, Chang YC, Grados MA, Greenberg BD, Knowles JA et al. Gender differences in genetic linkage and association on 11p15 in obsessive-compulsive disorder families. Am J Med Genet B Neuropsychiatr Genet 2009; 150B(1): 33–40. [DOI] [PubMed] [Google Scholar]
- 22.Samuels J, Shugart YY, Grados MA, Willour VL, Bienvenu OJ, Greenberg BD et al. Significant linkage to compulsive hoarding on chromosome 14 in families with obsessive-compulsive disorder: results from the OCD Collaborative Genetics Study. Am J Psychiatry 2007; 164(3): 493–499. [DOI] [PubMed] [Google Scholar]
- 23.Mattheisen M, Samuels JF, Wang Y, Greenberg BD, Fyer AJ, McCracken JT et al. Genome-wide association study in obsessive-compulsive disorder: results from the OCGAS. Mol Psychiatry 2015; 20(3): 337–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA et al. Genome-wide association study of obsessive-compulsive disorder. Mol Psychiatry 2013; 18(7): 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.den Braber A, Zilhao NR, Fedko IO, Hottenga JJ, Pool R, Smit DJ et al. Obsessive-compulsive symptoms in a large population-based twin-family sample are predicted by clinically based polygenic scores and by genome-wide SNPs. Translational psychiatry 2016; 6: e731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart SE, Yu D, Scharf JM, Neale BM, Fagerness JA, Mathews CA et al. Genome-wide association study of obsessive-compulsive disorder. Molecular psychiatry 2013; 18(7): 788–798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davis LK, Gamazon ER, Kistner-Griffin E, Badner JA, Liu C, Cook EH et al. Loci nominally associated with autism from genome-wide analysis show enrichment of brain expression quantitative trait loci but not lymphoblastoid cell line expression quantitative trait loci. Mol Autism 2012; 3(1): 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Below JE, Parra EJ, Gamazon ER, Torres J, Krithika S, Candille S et al. Meta-analysis of lipid-traits in Hispanics identifies novel loci, population-specific effects, and tissue-specific enrichment of eQTLs. Sci Rep 2016; 6: 19429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bulik-Sullivan BK, Loh PR, Finucane HK, Ripke S, Yang J, Schizophrenia Working Group of the Psychiatric Genomics C et al. LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat Genet 2015; 47(3): 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pato MT, Sobell JL, Medeiros H, Abbott C, Sklar BM, Buckley PF et al. The genomic psychiatry cohort: partners in discovery. American journal of medical genetics Part B, Neuropsychiatric genetics : 2013; 162B(4): 306–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 (Bethesda) 2011; 1(6): 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Delaneau O, Howie B, Cox AJ, Zagury JF, Marchini J. Haplotype estimation using sequencing reads. American journal of human genetics 2013; 93(4): 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007; 81(3): 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010; 26(17): 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khramtsova Ekaterina A., and Stranger Barbara E.. 2016 “Assocplots: A Python Package for Static and Interactive Visualization of Multiple-Group GWAS Results.” Bioinformatics (2017) 33 (3): 432–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu D, Mathews CA, Scharf JM, Neale BM, Davis LK, Gamazon ER et al. Cross-Disorder Genome-Wide Analyses Suggest a Complex Genetic Relationship Between Tourette’s Syndrome and OCD. Am J Psychiatry 2015; 172(1): 82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014; 511(7510): 421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Klei L, Sanders SJ, Murtha MT, Hus V, Lowe JK, Willsey AJ et al. Common genetic variants, acting additively, are a major source of risk for autism. Mol Autism 2012; 3(1): 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malik AN, Vierbuchen T, Hemberg M, Rubin AA2, Ling E, et al. Genome-wide identification and characterization of functional neuronal activity-dependent enhancers. Nat Neurosci 2014; 17(10):1330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Consortium G Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015; 348(6235): 648–660.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ng SY, Lin L, Soh BS, Stanton LW.Long noncoding RNAs in development and disease of the central nervous system. Trends in Genetics 2013; 29(8): 461–468. [DOI] [PubMed] [Google Scholar]
- 42.Ramasamy A, Trabzuni D, Guelfi S, Varghese V, Smith C, Walker R, De T; UK Brain ExpressionConsortium.; North American Brain Expression Consortium., Coin L, de Silva R, Cookson MR, Singleton AB, Hardy J, Ryten M, Weale ME. Genetic variability in the regulation of gene expression in ten regions of the human brain. Nat Neurosci. 2014. ;17(10):1418–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graybiel AM, Rauch SL. Toward a neurobiology of obsessive-compulsive disorder. Neuron. 2000. ; 28(2):343–7. [DOI] [PubMed] [Google Scholar]
- 44.Pittenger C1, Bloch MH, Williams K. Glutamate abnormalities in obsessive compulsive disorder: neurobiology, pathophysiology, and treatment. Pharmacol Ther. 2011;132(3):314–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Coutelier M, Burglen L, Mundwiller E, Abada-Bendib M, Rodriguez D, et al. GRID2 mutations span from congenital to mild adult-onset cerebellar ataxia. Neurology. 2015. 28; (17):1751–9. [DOI] [PubMed] [Google Scholar]
- 46.Fernando J, Faber TW, Pullen NA, Falanga YT, Kolawole EM, Oskeritzian CA, et al. Genotype-dependent effects of TGF-β1 on mast cell function: targeting the Stat5 pathway. J Immunol. 2013. 1;191(9):4505–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kawakami T, Soma Y, Kawa Y, Ito M, Yamasaki E, Watabe H, et al. Transforming growth factor beta1 regulates melanocyte proliferation and differentiation in mouse neural crest cells via stem cell factor/KIT signaling. J Invest Dermatol. 2002; 118(3):471–8. [DOI] [PubMed] [Google Scholar]
- 48.Cacheaux LP, Ivens S, David Y, Lakhter AJ, Bar-Klein G, Shapira M, et al. Transcriptome profiling reveals TGF-beta signaling involvement in epileptogenesis. J Neurosci. 2009. 15; 29(28):8927–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Arnold PD, Rosenberg DR, Mundo E, Tharmalingam S, Kennedy JL, Richter MA. Association of a glutamate (NMDA) subunit receptor gene (GRIN2B) with obsessive-compulsive disorder: a preliminary study. Psychopharmacology (Berl). 2004;174(4):530–8. [DOI] [PubMed] [Google Scholar]
- 50.Stewart SE, Mayerfeld C, Arnold PD, Crane JR, O’Dushlaine C, Fagerness JA, et al. Meta-analysis of association between obsessive-compulsive disorder and the 3’ region of neuronal glutamate transporter gene SLC1A1. Am J Med Genet B Neuropsychiatr Genet. 2013;162B(4):367–79 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.