

Abstract
Understanding transporter‐mediated drug–drug interactions is an integral part of risk assessment in drug development. Recent studies support the use of hexadecanedioate (HDA), tetradecanedioate (TDA), coproporphyrin (CP)‐I, and CP‐III as clinical biomarkers for evaluating organic anion‐transporting polypeptide (OATP)1B1 (SLCO1B1) inhibition. The current study investigated the effect of OATP1B1 genotype c.521T>C (OATP1B1‐Val174Ala) on the extent of interaction between cyclosporin A (CsA) and pravastatin, and associated endogenous biomarkers of the transporter (HDA, TDA, CP‐I, and CP‐III), in 20 healthy volunteers. The results show that the levels of each clinical biomarker and pravastatin were significantly increased in plasma samples of the volunteers following administration of pravastatin plus CsA compared with pravastatin plus placebo. The overall fold change in the area under the concentration–time curve (AUC) and maximum plasma concentration (Cmax) was similar among the four biomarkers (1.8–2.5‐fold, paired t‐test P value < 0.05) in individuals who were homozygotes or heterozygotes of the major allele, c.521T. However, the fold change in AUC and Cmax for HDA and TDA was significantly abolished in the subjects who were c.521‐CC, whereas the respective fold change in AUC and Cmax for pravastatin and CP‐I and CP‐III were slightly weaker in individuals who were c.521‐CC compared with c.521‐TT/TC genotypes. In addition, this study provides the first evidence that SLCO1B1 c.521T>C genotype is significantly associated with CP‐I but not CP‐III levels. Overall, these results suggest that OATP1B1 genotype can modulate the effects of CsA on biomarker levels; the extent of modulation differs among the biomarkers.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ There is emerging interest in the use of biomarkers as new tools to predict transporter‐mediated DDIs. Various endogenous metabolites have been investigated as potential OATP1B1 biomarkers in clinical studies involving different OATP1B1/1B3 inhibitors. In addition, OATP1B1‐Val174Ala is known to significantly affect the levels of these OATP1B1 metabolites. However, little is known about the effects of OATP1B1 genotype in modulating the levels of CP‐I and CP‐III and in modulating CsA effects on pravastatin drug levels and levels of various biomarkers.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This study addresses three main questions, which were not previously addressed. (i) What is the impact of OATP1B1‐Val174Ala on CP‐I and CP‐III levels? (ii) What is the effect of CsA, a nonselective OATP1B1 inhibitor, on the levels of the OATP1B1 biomarkers, HDA, TDA, CP‐I, and CP‐III? (iii) How does the genotype of OATP1B1 and OATP1B1‐Val174Ala affect the extent of the OATP1B1‐mediated DDI between CsA and pravastatin and the effect of CsA on OATP1B1 biomarkers?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This study provides further information about metabolomic biomarkers of the transporter OATP1B1. In brief, we now know that the nonsynonymous variant in OATP1B1, OATP1B1‐Val174, associates with plasma levels of HDA, TDA, and CP‐I but not CP‐III. We also know that the OATP1B1 inhibitor, CsA, significantly increases plasma levels of the biomarkers HDA, TDA, CP‐I, and CP‐III and that the extent of increase is modulated by the OATP1B1 genotype.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This study provides critical information about the effects of an important OATP1B1 genetic polymorphism on the levels of biomarkers of OATP1B1 and on the effects of CsA inhibition of OATP1B1 on the pharmacokinetics of pravastatin and biomarkers of OATP1B1. Validated biomarkers may change our ability to predict and understand the mechanisms of transporter‐mediated clinical DDIs.
SLCO1B1 encodes for the organic anion transporter polypeptide (OATP)1B1 (also known as OATP‐C, OATP2, and LST‐1). Mainly expressed on the basolateral (sinusoidal) membrane of hepatocytes, the sodium‐independent transporter mediates the influx of various endogenous substrates, such as bilirubin, bile acids, conjugated steroids, and thyroid hormones (see review1), as well as clinically used drugs, such as angiotensin‐converting enzyme inhibitors, angiotensin II receptor antagonists, and 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase inhibitors (statins).2
Pravastatin is a widely used statin that is prescribed primarily for the treatment of dyslipidemia and the prevention of cardiovascular disease. The drug is a well‐established substrate of OATP1B1 and OATP1B3.3 The OATP1B1‐Val174Ala (rs4149056, c.521T>C) polymorphism is known to have a reduced transporter activity, due to lower protein levels on the cell surface membrane.4, 5 As a result, individuals with the variants have increased plasma levels of OATP1B1 substrates, for example, atorvastatin, rosuvastatin, and repaglinide, by ~ 1.5–3‐fold.6, 7, 8, 9 In addition, individuals who are carriers of the variant also have higher levels of endogenous metabolites, which are OATP1B1 substrates, for example, bile acids, bilirubin, and fatty acid dicarboxylic acids (Table S1).10, 11, 12
Drug–drug interactions (DDIs) are common in clinical practice and frequently involve membrane transporters, such as OATP1B1. As a result, various regulatory agencies, such as the US Food and Drug Administration, the European Medicines Agency, and the Pharmaceutical and Medical Devices Agency in Japan have provided guidances for drug developers for testing new chemical entities (NCEs) in in vitro studies to determine their potential to cause clinically relevant transporter‐mediated or enzyme‐mediated DDIs.13, 14, 15 These guidances have helped drug developers enormously in making decisions to conduct clinical DDI studies and in the design of such studies. Nevertheless, there remain a number of false‐positive and false‐negative predictions resulting in the conduct of unnecessary clinical studies (false‐positives) or in failure to predict important clinical DDIs.16 In the past several years, multiple groups, including ours, have evaluated the OATP1B1 endogenous metabolites, coproporphyrin I (CP‐I), coproporphyrin III (CP‐III), tetradecanedioic acid (TDA), hexadecanedioic acid (HDA), and bile acids for their sensitivity and selectivity as DDI predictive OATP1B1 biomarkers (Table S1).10, 17, 18, 19, 20 Importantly, the levels of many of these clinical biomarkers, TDA, HDA, and bile acids conjugates, have been associated with SLCO1B1 polymorphisms at genomewide significance (P < 5 × 10−8) in multiple studies and in several ethnic groups.21, 22, 23, 24 In brief, significantly higher levels of the biomarkers are observed in individuals who harbor reduced function variants of SLCO1B1, the gene encoding OATP1B1. These biomarkers have the potential for use in phase I clinical studies to determine whether the NCE is an in vivo OATP1B1 inhibitor. That is, inhibitors of OATP1B1 would phenocopy or mimic reduced function genetic polymorphisms, resulting in higher plasma levels of the biomarkers. Thus, measuring these metabolites along with the NCE in early clinical studies may provide important in vivo information on the potential of the NCE to inhibit OATP1B1 and cause a DDI.
The extent of pharmacokinetic DDIs may be modulated by genetic variants, as has been observed in several DDI studies involving cytochrome P450 (CYP) enzymes. For example paroxetine, a CYP2D6 inhibitor, increased the plasma concentrations of the CYP2D6 substrate, desipramine, and this effect was greater in CYP2D6 extensive metabolizers than in poor metabolizers.25 Although fewer, similar observations have been made about the extent of DDIs with polymorphisms in transporters. For example, the extent of modulation of OATP1B1 genotype on interactions between gemfibrozil and repaglinide, rifampicin and atorvastatin, cyclosporin A (CsA) and repaglinide, and darunavir/ritonavir and pravastatin has been characterized for the OATP1B1‐Val174Ala (SLCO1B1 c.521T>C) variant.8, 26, 27, 28 These studies are consistent with studies of CYPs and, in general, show that reduced function polymorphisms attenuate the extent of the interaction. To date, only one study has evaluated the effect of homozygotes for SLCO1B1 c.521T>C (phenotype with poor transporter activity) on both a clinical DDI and the levels of an OATP1B1 biomarker in the presence of a perpetrator of a DDI.
The goal of this study was to determine the influence of the common OATP1B1 functional variant c.521T>C (rs4149056) on OATP1B1/1B3‐mediated CsA inhibition of pravastatin and four previously documented OATP1B1 biomarkers: HDA, TDA, CP‐I, and CP‐III (Table S1). Using a cohort of healthy volunteers with known SLCO1B1 genotype c.521T>C (rs4149056), we determined the extent of CsA interaction on pravastatin disposition and also its effect on the four clinical biomarkers. Consistent with previous studies,29, 30 pravastatin levels as well as levels of biomarker were increased in the presence of CsA. Notably, the effect of c.521T>C in modulating the CsA‐dependent increases in biomarker levels was dependent on the individual biomarker.
Methods
Clinical study
A blinded, randomized, two‐treatment crossover study was conducted on 20 healthy, white volunteer subjects selected for the SLCO1B1 c.521T>C genotype (rs4149056; Val174Ala) at the University of California, San Francisco Clinical and Translational Science Institute Clinical Resources Services (NCT01497483). To be eligible for the study, subjects had to provide written informed consent, be between the ages of 18 and 45 years, be in good health (as evidenced by medical histories, physical examination, and routine clinical laboratory evaluations), not be on any medications other than oral contraceptives, and not have any known allergies to pravastatin or CsA. Subjects were admitted to the clinical facility the evening before and remained onsite for the duration of the study (24 hours). The evening before the study, each study participant received either placebo or CsA (100 mg). Study participants then fasted for at least 10 hours before receiving another dose of placebo or CsA (100 mg) the next morning. One hour after the second dose of placebo or CsA, participants were given a single 40 mg dose of pravastatin. Plasma samples were obtained at baseline and at various times for 12 hours. Each study participant received both treatments that included a washout period of at least 2 weeks in between treatments. Plasma pravastatin concentrations were determined by high‐performance liquid chromatography–tandem accurate mass spectrometry. The liquid chromatography–tandem mass spectrometry analysis of pravastatin, TDA, HDA, CP‐I, and CP‐III are available in the Supplemental Material section.
Genotyping of SLCO1B1 c.521T>C (rs4149056)
Genomic DNA samples from 186 unrelated European‐Americans of the San Francisco Bay Area (part of the Studies of Pharmacogenetics in Ethnically Diverse Populations cohort) were obtained and genotyped for the single nucleotide polymorphism rs4149056 (SLCO1B1 c.521T>C). A TaqMan single nucleotide polymorphism genotyping assay (assay ID C__30633906_10) was used for genotyping on the ABI 7900 Fast HT Sequence Detection Systems (Applied Biosystems, Foster City, CA). Genomic DNA (10 ng) was amplified using a TaqMan Genotyping Master Mix (Applied Biosystems). The polymerase chain reaction conditions were as follows: 1 cycle of 95°C for 10 minutes followed by 40 cycles of two‐step polymerase chain reaction with denaturation at 95°C for 15 seconds and annealing and extension at 60°C for 1.5 minutes. Data were analyzed by allele discrimination.
Statistical analysis
The results are expressed as mean values ± SD in the text and tables and as mean values ± SEM in the figures. The maximum plasma concentration (Cmax) and area under the concentration–time curve (AUC) data were obtained using Phoenix WinNonlin 6.4. (Certara USA, Princeton, NJ) before the statistical analysis. Statistical comparisons of pharmacokinetic parameters between subjects with the c.521‐TT, TC, and CC genotypes during the three treatment phases were carried out using repeated‐measures analysis of variance (ANOVA) with treatment phase as a within‐subjects factor and genotype group as a between‐subjects factor. A priori testing between genotypes during each treatment phase was carried out using the Fisher's least significant difference method and between phases within each genotype group using the paired t‐test. The pharmacokinetic variables of pravastatin and the four endogenous metabolites were compared between the genotype groups using ANOVA or Student's t‐test if comparing two genotype groups (c.521‐TT/TC vs. c.521‐CC). The statistical analysis was performed using Prism 7 (GraphPad, San Diego, CA). Differences were considered statistically significant when P value was < 0.05.
Results
Clinical cohort
Eleven men (SLCO1B1 c.521T>C, N = 5 TT, 5 TC, 1 CC) and nine women (SLCO1B1 c.521T>C, N = 4 TT, 3 TC, 2 CC) participated in the clinical studies (Table S2). For the clinical biomarker studies, only 16 individuals (8 with c.521TT, 6 with c.521TC, and 2 with c.521CC) who had sufficient samples to measure the four metabolites (TDA, HDA, CP‐I, and CP‐III) were included in the analyses for metabolites–CsA interaction (Table S2).
Plasma pravastatin levels were increased following CsA administration in noncarriers and carriers of the SLCO1B1 c.521T>C allele
To determine the effects of CsA on pravastatin disposition, we examined the plasma pravastatin levels after placebo or CsA administration from 0−12 hours in 20 healthy, white volunteers (Figure 1 a). The mean AUC0→12 (AUClast) of pravastatin was significantly greater following administration of CsA (554 ± 380 ng·hour/mL) compared with placebo (162 ± 129 ng·hour/mL; Figure 1 b, Table 1). The mean Cmax of pravastatin was also significantly higher following administration of CsA (339 ± 266 ng/mL) compared with placebo (106 ± 100 ng/mL; Figure 1 c, Table 1). Furthermore, the volume of distribution and clearance for orally administered pravastatin were significantly different in the placebo and CsA treatment groups (Table 1). Similar effect was also observed when we examined the Cmax and AUClast of the pravastatin isomer 3′α‐hydroxy‐pravastatin peak only or pravastatin peak only without the isomer (Figure S1 , Table S3). The pravastatin concentration–time profile for each genotype group following pravastatin and placebo or CsA administration is shown in Figure 2 a. Although the AUC0→12 and Cmax of pravastatin in individuals who were homozygous carriers of the c.521‐CC were greater than the corresponding values for the other genotype groups, they were not statistically significant using ANOVA or Kruskal–Wallis tests (AUC0→12: c.521‐TT = 154 ± 153 ng/mL, TC = 124 ± 58 ng/mL, CC = 290 ± 148 ng/mL; Cmax: c.521‐TT = 98.8 ± 114 ng/mL, TC = 79.1 ± 50 ng/mL, CC = 201 ± 136 ng/mL; see Table 1). On the other hand, when we examined the pravastatin peak only, there were statistically significant results using ANOVA test among the three genotype groups (AUC0→12: c.521‐TT = 43 ± 15 ng/mL, TC = 48 ± 23 ng/mL, CC = 154 ± 147 ng/mL; Cmax: c.521‐TT = 22 ± 9.3 ng/mL, TC = 31 ± 19 ng/mL, CC = 111 ± 126 ng/mL; see Table S3). Notably, the 3′α‐hydroxy‐pravastatin peak only was not statistically significant among the three genotype groups (Figure S1 , Table S3). Figure 2 b shows the AUC0→12 and Cmax of individuals upon administration of placebo or CsA separated by genotype group. CsA administration resulted in increased AUC0→12 and Cmax in all c.521T>C groups (Figure 2 b, Table 1). Although the mean fold change of both AUC0→12 and Cmax was reduced in carriers of the SLCO1B1 c.521T>C allele compared with those with the reference allele, the paired t‐tests were significant except AUC0→12 in carriers of the homozygous SLCO1B1 c.521T>C allele (Table 1 , Figure 2 d). Similar trend is also observed when we examined the 3α‐hydroxy‐pravastatin peak only or pravastatin peak only, where the mean fold change of both AUC0→12 and Cmax was reduced in carriers of the SLCO1B1 c.521T>C allele compared with those with the reference allele (Figure S1 , Table S3). Overall, the ratio of mean AUC0→12 in SLCO1B1 c.521CC and SLCO1B1 c.521TT were 1.9, 3.6, and 1.2 for combined pravastatin and 3α‐hydroxy‐pravastatin, pravastatin only, and 3α‐hydroxy‐pravastatin only, respectively (Table S3). Pravastatin is extensively metabolized to the 3α‐hydroxy isomer, which has one‐tenth to one‐fortieth of the inhibitory activity of the parent compound on 3‐hydroxy‐3‐methylglutaryl coenzyme A reductase.31
Figure 1.
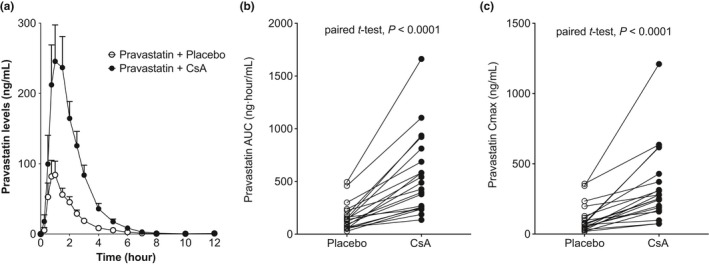
Plasma concentration–time profiles and pharmacokinetic parameters of pravastatin in healthy volunteers following administration of pravastatin plus placebo and pravastatin plus cyclosporin A (CsA). (a) Pravastatin plasma concentration–time profile in each treatment group (mean ± SEM); (b) pravastatin area under the concentration–time curve from time of administration up to the time of the last quantifiable concentration (AUC 0–last(0–12 hour)); and (c) peak plasma concentration (Cmax) of each individual in the pravastatin plus placebo and pravastatin plus CsA group.
Table 1.
Influence of cyclosporin A and SLCO1B1 c.521T>C genotype on the pharmacokinetics of pravastatin in volunteers
| Treatment | Pravastatin + placebo | Pravastatin + cyclosporine | ||||||
|---|---|---|---|---|---|---|---|---|
| Genotype | All genotype | TT (N = 9) | TC (N = 8) | CC (N = 3) | All genotype | TT (N = 9) | TC (N = 8) | CC (N = 3) |
| t1/2 (hour) | 1.12 ± 0.65 | 1.16 ± 0.82 | 1.21 ± 0.56 | 0.76 ± 0.25 | 0.87 ± 0.27 | 0.90 ± 0.26 | 0.84 ± 0.34 | 0.88 ± 0.08 |
| Tmax (hour) | 1.05 ± 0.36 | 1.00 ± 0.40 | 1.09 ± 0.35 | 1.08 ± 0.38 | 1.25 ± 0.56 | 1.25 ± 0.57 | 1.16 ± 0.60 | 1.50 ± 0.5 |
| Cmax (ng/mL) | 106 ± 100 | 98.8 ± 114 | 79.1 ± 50.0 | 201 ± 136 |
339 ± 266****
(4.3) |
364 ± 360*
(5.3) |
266 ± 153*
(3.9) |
458 ± 166*
(2.6) |
| AUClast (ng·hour/mL) | 162 ± 129 | 154 ± 153 | 124 ± 58.0 | 290 ± 148 |
554 ± 380****
(4.2) |
572 ± 488*
(5.0) |
436 ± 198**
(3.8) |
818 ± 351 (3.1) |
| V/F (L/kg) | 8.39 ± 8.39 | 9.56 ± 8.74 | 9.49 ± 9.08 | 1.94 ± 1.37 | 1.60 ± 1.02** | 1.92 ± 1.34* | 1.52 ± 0.54* | 0.85 ± 0.70* |
| CL/F (L/hour/kg) | 5.17 ± 4.44 | 6.12 ± 5.34 | 5.42 ± 3.77 | 1.65 ± 0.68 | 1.34 ± 0.85*** | 1.47 ± 0.98* | 1.45 ± 0.74* | 0.64 ± 0.48 |
Pharmacokinetic parameters of pravastatin when administered with placebo and cyclosporin A (CsA) are shown. Data are presented as mean ± SD for each genotype group and all subjects combined. Paired t‐test was used to determine the significance of each pharmacokinetic parameter between the treatment groups. The numbers in the parentheses represent the fold increase of pravastatin levels in pravastatin and CsA group over pravastatin + placebo.
AUClast, area under the curve from time zero to the last quantifiable concentration (12 hour); CL/F, apparent oral clearance (F is oral bioavailability) based on dose divided by AUClast; Cmax, peak plasma concentration; t1/2, half‐life; Tmax, time to reach peak plasma concentration; V/F, apparent volume of distribution.
The significant P values are: *P < 0.05; **P < 0.005; ***P < 0.0005; and ****P < 0.0001.
Figure 2.
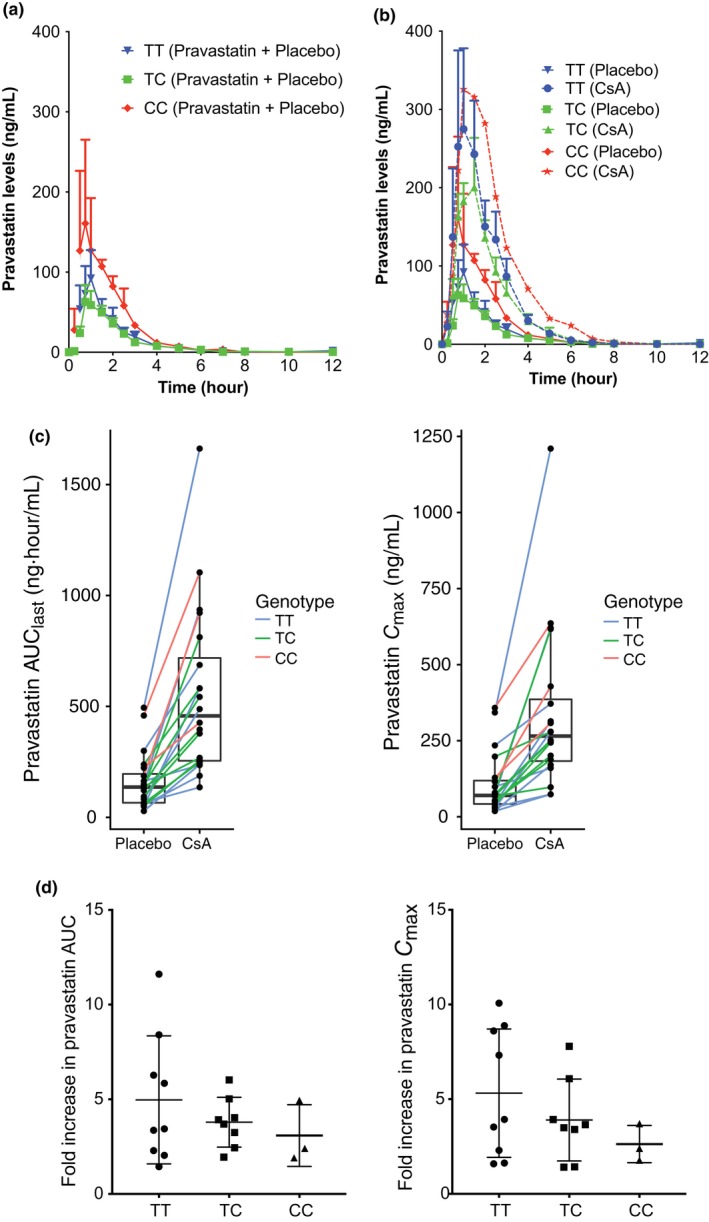
The effect of cyclosporin A (CsA) administration on plasma levels of pravastatin in volunteers from three different genotype groups: SLCO1B1 c.521TT,SLCO1B1 c.521TC, and SLCO1B1 c.521CC (TT, TC, and CC). (a) Plasma concentration–time profiles of pravastatin following placebo administration in the three different SLCO1B1 c.521T>C genotype groups. (b) Plasma concentration–time profiles of pravastatin following placebo or CsA administration in the three different SLCO1B1 c.521T>C genotype groups. Data in a and b are presented as mean ± SEM. (c) Pravastatin area under the concentration–time curve from time of administration up to the time of the last quantifiable concentration (AUC 0–last(0–12 hour)) and peak plasma concentration (Cmax) for each individual following placebo or CsA administration in the three different SLCO1B1 c.521T>C genotype groups. Each line connects data in the same individual who participated in the crossover study. (d) Fold increase in pravastatin AUC 0–last(0–12 hour) and Cmax in the three different SLCO1B1 c.521T>C genotype groups. Fold increase is calculated by dividing the parameters from the pravastatin plus CsA group with pravastatin plus placebo group.
Plasma HDA, TDA, CP‐I, and CP‐III levels were increased following CsA administration
Plasma concentration–time profile of the four biomarkers, HDA, TDA, CP‐I, and CP‐III, showed increased levels after pravastatin plus CsA administration compared with pravastatin plus placebo administration (Figure 3 a, Table 2). After administration of CsA plus pravastatin, average plasma concentration (Cavg), Cmax, and AUClast(0.25–12 hour)) of the four biomarkers, HDA, TDA, CP‐I, and CP‐III, increased 1.8–2.1‐fold, 1.9–2.2‐fold, 1.7–2.2‐fold, and 1.5–1.9‐fold in comparison to the placebo plus pravastatin period, respectively. All increases were statistically significant (P < 0.05, paired t‐test) (Table 2). The mean C0 (plasma levels 12 hours after the first dose of CsA or placebo and right before the second dose of CsA or placebo) of HDA was significantly higher (Table 2). For TDA, the mean C0 also increased significantly (P < 0.05), but only after data from the two subjects who were homozygous for the minor allele c.521‐CC were removed from the analysis (Table 3). In contrast, CP‐I and CP‐III at baseline (C0) were below the levels of quantification for 12 of 16 samples and only reached measurable levels at 15 minutes (or later) after CsA administration. In addition, correlation analysis showed that the pravastatin AUC significantly correlates (P < 0.005) with each of the biomarkers (Figure 4 , Table S3). Overall, the magnitude of the fold increase in Cavg and Cmax for HDA, TDA, and CP‐I were similar and greater than that of CP‐III, whereas the magnitude of AUClast was similar across the four metabolites (Table 2).
Figure 3.
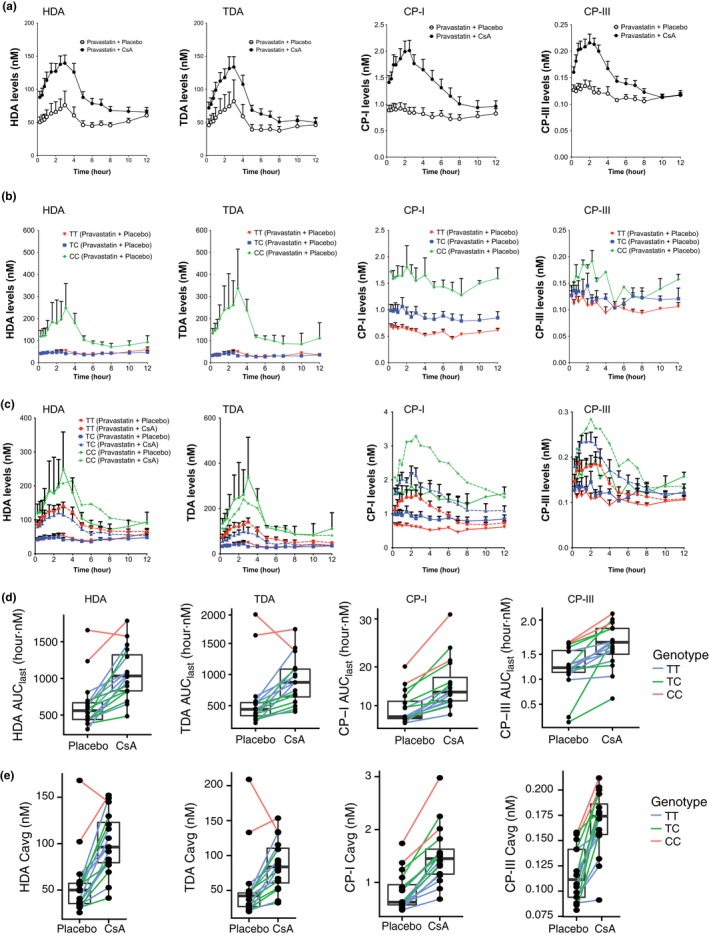
Plasma levels of organic anion‐transporting polypeptide (OATP)1B1 biomarker in carriers of the SLCO1B1 c.521T>C allele and the influence of SLCO1B1 c.521T>C genotype on OATP1B1 biomarker levels and on the effects of cyclosporin A (CsA). Plasma concentration–time profiles of the four OATP1B1 biomarkers in (a) pravastatin plus placebo and pravastatin plus CsA administration; in (b) pravastatin plus placebo administration separated by SLCO1B1 c.521T>C genotype; in (c) pravastatin plus placebo and pravastatin plus CsA administration separated by SLCO1B1 c.521T>C genotype. OATP1B1 biomarkers (d) area under the concentration–time curve from time of administration up to the time of the last quantifiable concentration (AUC 0–last (0–12 hour)) and (e) Cmax of each individual in the pravastatin plus placebo and pravastatin plus CsA group separated by SLCO1B1 c.521T>C genotype (TT, TC, and CC). Each line connects the same individual who did the crossover study. Data in a–c are presented as mean ± SEM. Solid lines in c represent levels from pravastatin plus placebo administration and dashed lines represent levels from pravastatin plus CsA administration. Cavg, average plasma concentration; Cmax, peak plasma concentration; CP, coproporphyrin; HDA, hexadecanedioate; TDA, tetradecanedioate.
Table 2.
Levels of the four OATP1B1 biomarkers, HDA, TDA, CP‐I, and CP‐III, in each treatment group
| Treatment | Pravastatin + placebo (N = 16) | Pravastatin + cyclosporine (N = 16) | ||||||
|---|---|---|---|---|---|---|---|---|
| Biomarker | HDA | TDA | CP‐I | CP‐III | HDA (fold change) | TDA (fold change) | CP‐I (fold change) | CP‐III (fold change) |
| C0 (nM) | 60.1 ± 37 | 55 ± 44 [N = 11] | BLOQ in most samples | BLOQ in most samples | 84 ± 33 ** [N = 11] (1.5) | 68 ± 26 [N = 11] (1.6) | BLOQ in most samples | BLOQ in most samples |
| Cmax (nM) | 90.6 ± 77.3 | 94.7 ± 118.5 | 1.04 ± 0.45 | 0.15 ± 0.039 |
155 ± 55.1** (2.1) |
149 ± 71.0* (2.2) |
2.16 ± 0.71**** (2.2) |
0.24 ± 0.05**** (1.7) |
| Cavg (nM) | 56.4 ± 35.1 | 53.3 ± 49.3 | 0.83 ± 0.36 | a0.12 ± 0.03 |
100 ± 33.3**** (2.0) |
86.0 ± 35.8** (2.0) |
1.51 ± 0.56**** (1.9) |
a0.17 ± 0.03**** (1.5) |
| AUClast (nM·hour) | 649 ± 346 | 593 ± 489 | 9.4 ± 4.2 | a1.21 ± 0.46 |
1,070 ± 357**** (1.8) |
902 ± 378** (1.9) |
15.5 ± 6.7**** (1.7) |
a1.68 ± 0.44*** (1.9) |
Arithmetic mean ± SD for each biomarker's parameters in the two treatment groups are reported in this table. Paired t‐test was used to determine the significance of each parameter between the treatment groups. Number in parentheses is the fold increase between pravastatin plus cyclosporin A group over pravastatin plus placebo group. A total of 16 subjects were available for biomarker analysis; however, for some biomarkers, the levels are below the level of quantification; the number in square brackets is number of subjects available for the analysis.
AUClast, area under the curve from time 15 minutes after second dose of cyclosporin A administration or placebo to 12 hours; BLOQ, below the limit of quantification; C0, concentrations at time 0, baseline (this level is taken after 12 hours from cyclosporin A administration from the night before and right before the second dose of cyclosporin A administration); Cavg, average concentrations from the collected samples, 15 minutes after second dose of placebo or cyclosporin A administration; Cmax, maximum concentrations from the collected samples, 15 minutes after second dose of placebo or cyclosporin A administration; CP, coproporphyrin; HDA, hexadecanedioate; OATP, organic anion‐transporting polypeptide; TDA, tetradecanedioate.
Overall, CP‐III are very low and many timepoints have below quantification levels.
The significant P values are: *P < 0.05; **P < 0.005; ***P < 0.0005; ****P < 0.0001.
Table 3.
Levels of the OATP1B1 biomarkers, HDA, TDA, CP‐I, and CP‐III, in each genotype and treatment group
| Treatment | Pravastatin + placebo [N = 16] | Pravastatin + cyclosporine [N = 16] | bPaired t‐test, P value (only TT/TC) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| HDA | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | |
| C0 (nM) | 60.1 ± 37.0*, b [N = 11] | 52.0 ± 29.6 [N = 5] | 43.1 ± 13.4 [N = 4] | 114 ± 47.5 [N = 2] | 84 ± 33 [N = 11] (1.5) | 84.6 ± 39.1**, b [N = 5] (1.7) | 70.3 ± 23.4*, b [N = 4] (1.6) | 111 ± 34.6 [N = 2] (1.0) | 0.0009 (2.8E‐5) |
| Cavg (nM) | 56.4 ± 35.1****, b | 46.1 ± 14.3 | 44.0 ± 11.5 | 135 ± 46.6 | 100 ± 33.3 (2.0) | 98.5 ± 27.8***, b (2.2) | 86.9 ± 33.0*, b (2.0) | 149 ± 5.0 (1.2) | 2.1E‐5 (5.2E‐6) |
| AUClast (nM·hour) | 649 ± 346****, b | 557 ± 161 | 506 ± 116 | 1,450 ± 299 | 1,070 ± 357*, b (1.8) | 1,070 ± 239***, b (2.0) | 853 ± 303*, b (1.7) | 1,680 ± 148 (1.2) | 1.9E‐5 (1.8E‐5) |
| TDA | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | bPaired t‐test P‐value (only TT/TC) |
|---|---|---|---|---|---|---|---|---|---|
| C0 (nM) | 55 ± 44**, b [N = 11] | 44 ± 24.5 [N = 5] | 31.3 ± 5.0 [N = 4] | 131 ± 48.9 [N = 2] |
68 ± 26*,
b [N = 11] (1.6) |
77.7 ± 22.0**,
b [N = 5] (2.1) |
44.2 ± 14.8 [N = 4] (1.4) |
91.3 ± 23.9 [N = 2] (0.7) |
0.20 (0.002) |
| Cavg (nM) | 53.3 ± 49.3****, b | 38 ± 13.7 | 34.6 ± 10.4 | 171 ± 53.7 |
86.0 ± 35.8*,
b
(2.0) |
87.6 ± 29.5**,
b
(2.5) |
64.5 ± 26.4*,
b
(1.8) |
144 ± 13.9 (0.9) |
0.003 (2.5E‐5) |
| AUClast (nM·hour) | 593 ± 489****, b | 448 ± 153 | 386 ± 84 | 1,800 ± 237 |
902 ± 378**,
b
(1.9) |
927 ± 282**,
b
(2.2) |
652 ± 246*,
b
(1.7) |
1,550 ± 249 (0.9) |
0.0025 (6.0E‐5) |
| CP‐I | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | bPaired t‐test P‐value (only TT/TC) |
|---|---|---|---|---|---|---|---|---|---|
| Cavg (nM) | 0.83 ± 0.36****, b | 0.6 ± 0.1 | 0.88 ± 0.26 | 2.19 ± 0.64 |
1.51 ± 0.56**,
b
(1.9) |
1.15 ± 0.28***,
b
(1.9) |
1.67 ± 0.33***,
b
(2.0) |
2.50 ± 0.68 (1.6) |
5.0E‐8 (2.2E‐7) |
| AUClast (nM·hour) | 9.4 ± 4.2***, b | 6.9 ± 1.2 | 9.8 ± 3.3 | 17.8 ± 3.2 |
15.5 ± 6.7**,
b
(1.7) |
11.8 ± 2.3***,
b
(1.7) |
16.5 ± 5.7***,
b
(1.7) |
27.6 ± 8.4 (1.5) |
2.7E‐6 (8.3E‐6) |
| CP‐III | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | All genotype | TT (N = 8) | TC (N = 6) | CC (N = 2) | bPaired t‐test P‐value (only TT/TC) |
|---|---|---|---|---|---|---|---|---|---|
| cCavg (nM) | 0.12 ± 0.03 | 0.11 ± 0.02 | 0.11 ± 0.03 | 0.15 ± 0.004 |
0.17 ± 0.03*,
b
(1.5) |
0.15 ± 0.03**,
b
(1.4) |
0.18 ± 0.02**,
b
(1.7) |
0.21 ± 0.008*,
b
(1.3) |
1.2E‐6 (1.3E‐5) |
| cAUClast (nM·hour) | 1.21 ± 0.46 | 1.2 ± 0.17 | 1.0 ± 0.68 | 1.7 ± 0.013 |
1.68 ± 0.44 (1.9) |
1.56 ± 0.24**,
b
(1.3) |
1.65 ± 0.60*,
b
(3.0) |
2.22 ± 0.13 (1.2) |
0.0001 (0.0006) |
Arithmetic mean ± SD for each biomarker's parameters in the two treatment groups are reported in the table. Paired t‐test was used to determine the significance of each parameter between the treatment groups. Possible associations of SLCO1B1 c.521T>C with biomarkers in the pravastatin plus placebo group were investigated by use of analysis of variance. Differences were considered statistically significant at P < 0.05. The fold increase between pravastatin + cyclosporin A (CsA) group over pravastatin + placebo group is shown in the table. The number in parentheses is the fold increased between pravastatin plus CsA group over pravastatin plus placebo group for each genotype group. A total of 16 subjects were included in the biomarker analysis; however, for some biomarkers, the levels were below the level of quantification; the number in square brackets is the number of subjects available for the analysis. See footnote below for the different statistical analyses performed.
AUClast, area under the curve from time 15 minutes after second dose of CsA administration or placebo to 12 hours; C0, concentrations at time 0, baseline (this level is taken 12 hours after the first CsA dose and right before the second dose of CsA); Cavg, average concentrations from the collected samples, 15 minutes after second dose of placebo or CsA administration; CP, coproporphyrin; HDA, hexadecanedioate; OATP, organic anion‐transporting polypeptide; TDA, tetradecanedioate.
Paired t‐test to test whether there were significant differences between the individuals in the two treatment groups. The P value for significance including all genotype groups and the P value for significance including the SLCO1B1 c.521T>C, TT, and TC groups are shown in the parentheses.
Analysis of variance (ANOVA) was used to test whether the three genotype groups’ means are all equal. Possible associations of SLCO1B1 c.521T>C with biomarkers in the pravastatin plus placebo group were investigated by use of ANOVA. The P values for significance are *P < 0.05; **P < 0.005; ***P < 0.0005; and ****P < 0.0001.
Paired t‐test to test whether there were significant differences between the individuals in the two treatment groups with the same genotype group. The P values for significance are *P < 0.05; **P < 0.005; *** P < 0.0005; and **** P < 0.0001.
Overall, CP‐III are very low and many time points have below quantification levels.
Figure 4.
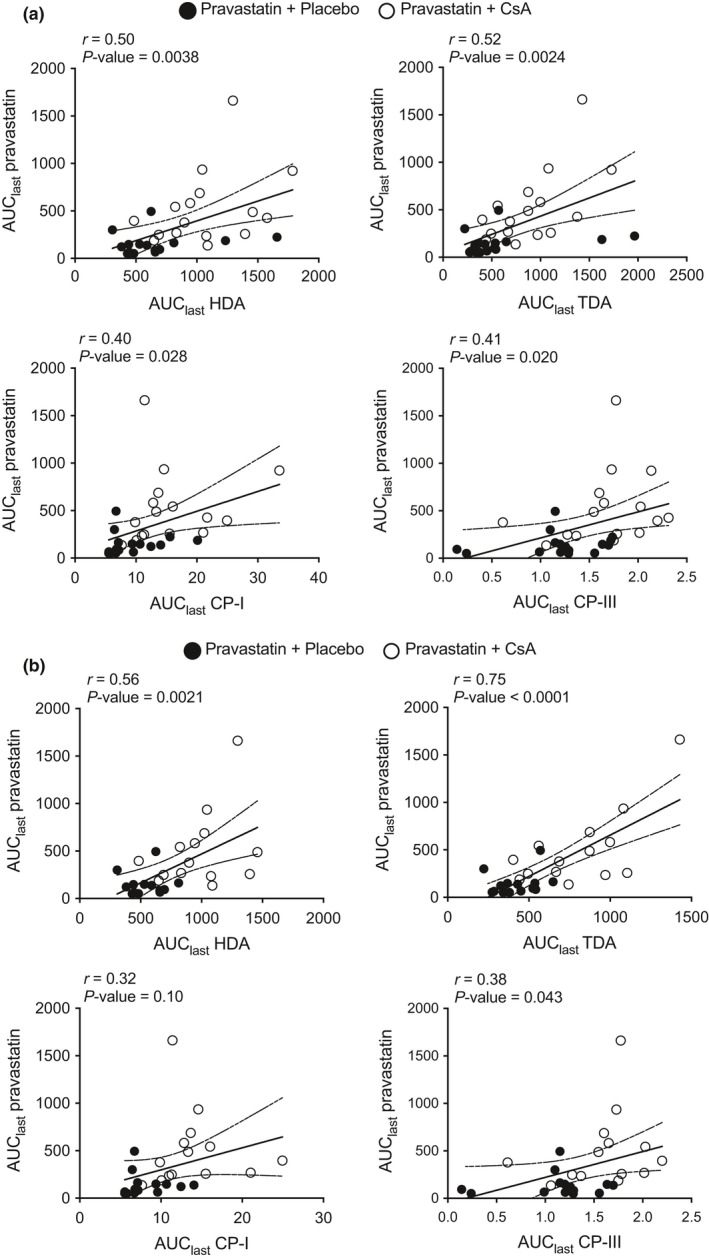
Correlation between pravastatin area under the concentration–time curve from time of administration up to the time of the last quantifiable concentration (AUC last) and each of the biomarkers in pravastatin plus placebo group (solid circles) and pravastatin plus cyclosporin A (CsA) group (open circles). Correlation in (a) individual irrespective of SLCO1B1 c.521T>C genotype and (b) individual with SLCO1B1 c.521‐TT or c.521‐TC genotype. The correlation, r, and P value for the Pearson's correlation is shown in the figure and in Table S4. CP, coproporphyrin; HDA, hexadecanedioate; TDA, tetradecanedioate.
Plasma HDA, TDA, and CP‐I levels were increased in carriers of the SLCO1B1 c.521T>C allele
Results comparing the plasma concentration–time curves of the four biomarkers and the Cavg and AUClast(0.25–12 hour) for each of the biomarkers are shown in Figure 3 b and Table 3. After administration of placebo plus pravastatin, the Cavg and AUClast of HDA, TDA, and CP‐I were significantly different among the three genotype groups, c.521‐TT, TC, and CC. In contrast, CP‐III Cavg and AUClast were similar among the genotype groups (Table 3 , Figure 3 b, Figure S2). One unexpected observation is noted in Figure 3 a,b. That is, levels of CP‐I and CP‐III did not change following administration of pravastatin plus placebo; however, the plasma concentrations of HDA and TDA in individuals who were homozygous for the c.521‐CC increased by at least twofold (Cmax over baseline) and peaked at around 3 hours (Figure 3 a,b, Figure S2).
The extent of fatty acid dicarboxylic acid–CsA interaction, but not CP‐I interaction, was reduced in carriers of the SLCO1B1 c.521T>C allele
Figure 3 c–e and Table 3 show the results comparing the plasma concentration–time curves and the Cavg and AUClast of the four biomarkers. We determined whether SLCO1B1 c.521T>C genotype modulated the effect of CsA on the levels of the four metabolites by comparing change in baseline, Cavg, and AUClast of the metabolites among the three genotype groups following CsA (plus pravastatin) administration. Figure 3 c–e shows that the fold change in HDA and TDA Cavg and AUClast following CsA administration was small in the two individuals who were c.521‐CC. That is, the change in Cavg was 1.2 and 0.9 for HDA and TDA, respectively, and for AUClast it was identical, 1.2 and 0.9 for HDA and TDA, respectively. In contrast, greater changes were observed in both Cavg and AUClast following CsA administration in the individuals who were c.521‐TT and c.521‐TC (Table 3). Importantly, the paired t‐test P value for the effect of CsA on Cavg and AUClast were more significant when individuals who were c.521‐CC were excluded from the analysis (Table 3). In contrast, Cavg and AUClast of CP‐I and CP‐III were increased upon CsA administration in all three genotype groups. Although the fold change is reduced in individuals with c.521‐CC, the paired t‐test P values remained similar whether or not the c.521‐CC was excluded (Table 3).
Pravastatin AUC correlates significantly with AUC of all four biomarkers
Pearson's correlation analysis showed that pravastatin AUC positively correlates (P < 0.05) with all biomarkers (Figure 4 a, Table S4). When individual with SLCO1B1 c.521‐CC were removed, the AUC correlation remained significant, except with CP‐I (P = 0.10; Figure 4 b). Notably, when one individual who had the highest pravastatin AUClast (in both placebo and CsA group) was removed from the correlation analysis, the correlation of pravastatin AUC with each of the four biomarkers became significant (P < 0.05).
Discussion
Clinical DDIs contribute to drug toxicities by increasing systemic or tissue‐specific levels of drugs. Thus, in order to ensure patient safety, regulatory agencies require that all new drug applications include information on DDI liabilities. For new drugs that are shown in in vitro transporter studies to be substrates or inhibitors of major transporters in the liver and/or kidneys, clinical DDI studies may be required to determine the effect of the drug candidates on co administered drugs or vice versa. To enhance the ability to predict clinical DDIs and reduce false‐positive and false‐negative results, other tools are needed in addition to in vitro transporter assays. A number of groups have explored the use of endogenous metabolites as biomarkers of transporter‐mediated DDIs in early clinical studies (Table S1). In this study, we further characterized four endogenous metabolites that have been previously identified as potential biomarkers of OATP1B1 inhibition. The goal of our study was to assess the effect of a reduced function genetic variant in SLCO1B1 (c.521T>C) in association with baseline levels and modulating the effects of CsA on the levels of the four metabolites as well as on pravastatin pharmacokinetics. Our study revealed four major findings. First, the effect of CsA on pravastatin pharmacokinetics was reduced in individuals who were homozygous for c.521C in comparison with heterozygotes and individuals homozygous for the reference allele, c.521T. Second, baseline levels of CP‐I, but not CP‐III, were associated with the SLCO1B1 c.521T>C genotype. Third, CsA plus pravastatin did not significantly increase the levels of HDA and TDA over pravastatin plus placebo in the individuals who were homozygous for SLCO1B1 c.521C; however, levels of CP‐I and CP‐III increased following CsA administration in all genotype groups. Finally, the pravastatin AUClast correlated significantly with all four biomarkers. Below, we discuss the four major findings in the context of the literature.
Our first finding, that CsA increased pravastatin plasma concentrations in accordance with SLCO1B1 c.521T>C genotype, was not unexpected. First, CsA has been found to increase pravastatin levels in previous studies in transplant patients.29, 30 Second, pharmacogenetic studies have revealed that pravastatin levels are significantly higher in patients who are homozygous for SLCO1B1 c.521T>C.32, 33 A few studies have determined the effect of SLCO1B1 c.521T>C genotype on OATP1B1‐mediated DDIs (see Table S5). To our knowledge, this is the first report to show the effect of genotype in modulating an OATP1B1‐mediated DDI with pravastatin. We rationalized that if the transporter activity is already substantially reduced by the genetic polymorphism, CsA will not be able to further reduce the activity. Our study with pravastatin and CsA demonstrated similar SLCO1B1 genotype effects with a published study of atorvastatin and rifampicin.8 That is, the effects of perpetrator (OATP1B1 inhibitor, CsA, or rifampicin) on victim drug (pravastatin or atorvastatin) plasma levels are much lower in individuals who are homozygous for the reduced function variant, c.521T>C, in comparison with heterozygotes or individuals who were homozygous for the reference allele. As CsA (or rifampicin) is not a selective inhibitor for OATP1B1 and also inhibits OATP1B3 and other hepatic transporters, such as OATP2B1,34 the data suggest that OATP1B1 is a major CsA sensitive influx transporter for pravastatin in the liver. If other CsA sensitive transporters were significantly involved in pravastatin hepatic uptake, then CsA would have provided additional inhibitory effects in individuals homozygous for the reduced function OATP1B1 variant. For example, following administration of gemfibrozil, the AUC of repaglinide increased more in subjects who were SLCO1B1 c.521‐CC than in subjects who were SLCO1B1 c.521‐TT.28
Our second finding, that baseline levels of CP‐I but not CP‐III were associated with SLCO1B1 c.521T>C genotype, was somewhat surprising, and suggests that there may be quantitative differences in the influx pathways of the two metabolites. Recent study from Mori et al.35 also showed that CP‐I level was significantly associated with SLCO1B1 c.521T>C. Previously, we have shown that HDA and TDA are substrates of OATP1B1 and their levels are significantly higher in individuals who are carriers of c.521T>C.10 Accordingly, because our previous studies demonstrated that CP‐I and CP‐III are substrates of OATP1B136 (Table S1), we would have expected that levels of both of these metabolites would be elevated in individuals who carried the reduced function allele of SLCO1B1 (Figure 3). It was previously shown that patients with Rotor's syndrome, who have deletions in SLCO1B1 and SLCO1B3, have greater urinary excretion rates of CP‐I.37, 38 Our finding that CP‐III levels were not sensitive to SLCO1B1 genotype suggests that other transporters (or pathways) may be involved in its hepatic uptake. Indeed, in vitro studies showed the CP‐III, but not CP‐I, is a substrate of OATP2B1.36 The observation that there was a trend toward elevated CP‐III levels in the two individuals with a c.521‐CC genotype (Figure 3 , Table 3) is consistent with the notion that OATP1B1 plays at least a minor role in the hepatic uptake of CP‐III. Our observations are also supported by studies in patients with Rotor's syndrome (i.e., CP‐III excretion rates are either mildly increased or not increased in patients with Rotor's syndrome, whereas CP‐I excretion rates are increased significantly; Wolkoff et al.38 and Shimidzu et al.37). Overall, the ratio of mean AUC in SLCO1B1 c.521‐CC to c.521TT were 2.6, 4.0, and 2.6 for HDA, TDA, and CP‐I, respectively, similar to the corresponding values of pravastatin plus 3α‐hydroxy pravastatin peaks (1.9‐fold) and of pravastatin peak only (3.6‐fold).
Our third finding was that CsA plus pravastatin did not significantly increase the levels of HDA and TDA in the individuals who were homozygous for SLCO1B1 c.521C in comparison to pravastatin plus placebo; however, levels of CP‐I and CP‐III increased following CsA administration in all genotype groups. These differences in the effects of OATP1B1 genotype in modulating the inhibitory effects of CsA and even pravastatin itself on the levels of the biomarkers suggest differences in the importance of OATP1B1 in the kinetics of the individual biomarkers. Interestingly, HDA and TDA levels increased after pravastatin plus placebo administration, but only in the two individuals who had the SLCO1B1 c.521‐CC genotype. In these same two individuals, HDA and TDA levels did not increase further following CsA plus pravastatin administration (Figure 3 b–e, Figure S2). These data are consistent with the idea that pravastatin may have maximally inhibited the residual transport activity of OATP1B1 in the individuals who are homozygous for the reduced function allele. Thus, CsA plus pravastatin was not able to further affect the levels of the two metabolites over pravastatin alone. Notably, Figures 2 a,b and 3 a,b showed that HDA and TDA levels in the two individuals who are c.521‐CC peaked after levels of pravastatin increased (time to maximum concentration, 1.5 hours with pravastatin and 3.0 hours with HDA and TDA) consistent with a causal relationship. That is, pravastatin levels are affecting HDA and TDA levels. Pravastatin may not have been able to affect the levels of CP‐I and CP‐III in the homozygotes for the reduced function allele because CP‐I and CP‐III may have other important routes of elimination. Thus, inhibiting residual OATP1B1 activity in individuals who are homozygous for reduced activity of OATP1B1 may have little effect on the overall levels of the two CP metabolites. Notably, CP‐I and CP‐III are substrates of both OATP1B1 and OATP1B3, whereas HDA and TDA are selective substrates of OATP1B1 and also renal transporters, organic anion transporter (OAT)1 and OAT3. Thus, CsA, as a nonselective inhibitor, would be expected to increase the levels of CP‐I and CP‐III in all patients irrespective of genotype.
Our final finding showed that pravastatin AUC significantly correlated with all four biomarkers in all individuals irrespectively of SLCO1B1 c.521T>C genotype (Figure 4 a). The correlation between the endogenous metabolites and the exogenous pravastatin gives further strong evidence that the endogenous metabolites are useful biomarkers of OATP1B1. Future studies are needed to see whether these correlations remained significant with larger sample size and with other OATP1B1 inhibitors and probe substrates.
There are some limitations in this study. There were only two individuals with the SLCO1B1 c.521‐CC genotype who had plasma samples of sufficient quantity for quantification of biomarkers. Future studies with more samples from individuals homozygous for SLCO1B1 c.521T>C are needed to confirm the influence of the polymorphisms on OATP1B1‐mediated drug–drug or drug–metabolite interactions and also to confirm the genetic associations with CP‐I and CP‐III. Another limitation is that other OATP1B1 biomarkers, such as bile acids conjugates (sulfates and glucuronide), were not measured in this study.39 Thus, extensive comparisons of other OATP1B1 biomarkers could not be made. Notably, many of these biomarkers, like other drugs, are not entirely specific to a single transporter (Table S1). Further information and comparisons of other OATP1B1 inhibitors and inhibitors of other OATs will provide valuable information to guide the use of various types of these biomarkers.
In conclusion, we identified significant associations of the major reduced function OATP1B1 polymorphism with CP‐I levels. Our studies suggest that SLCO1B1 genotype modulates the effect of CsA on pravastatin pharmacokinetics and on the pharmacokinetics of the OATP1B1 biomarkers tested, although the effects were slightly different with each biomarker. Although there will be a low prevalence of individuals who are homozygous for c.521T>C in most populations, genotyping should be done for clarity in the interpretation of the data. Our results suggest that all three metabolites, CP‐I, HDA, and TDA, have similar sensitivity to the OATP1B1/1B3 inhibitor, CsA, and that OATP1B1‐Ala174Val variant is associated with the levels of these metabolites. However, none of these metabolites are substrates of a specific transporter (Table S1). Further studies are needed to evaluate the effect of the OATP1B1 polymorphism on other clinical biomarkers and in the presence of other OATP1B1 inhibitors. Overall, these results along with other literature reports strongly support the use of HDA/TDA and CP‐I as useful biomarkers for clinical monitoring of OATP function. A single OATP1B1 biomarker may not be sufficient for interpretation of the mechanisms responsible for clinical DDIs, and the use of multiple biomarkers is warranted.
Funding
This study was supported partly by the National Institutes of Health (NIH) grant (R01 GM117163, U19 GM061390, 1R01DK108722, T32 GM07546) and the Bristol‐Myers Squibb Company.
Conflict of Interest
As Deputy Editor‐in‐Chief for Clinical and Translational Science, D.L.K. was not involved in the review or decision process for this paper. The other authors declared no conflict of interest.
Author Contributions
S.W.Y., M.M.G., D.L.K., and K.M.G. wrote the manuscript. S.W.Y., M.M.G., H.S., W.G.H., W.B., Y.L, D.L.K., and K.M.G. designed the research. M.M.G. and H.H. performed the research. S.W.Y., M.M.G., H.S., W.G.H., W.B., Y.L, D.L.K., and K.M.G. analyzed the data.
Supporting information
Figure S1. Concentration–time curves of pravastatin and 3α‐hydroxy pravastatin after oral administration of pravastatin and placebo or pravastatin plus cyclosporin A (CsA).
Figure S2. Concentration–time curves of the four OATP1B1 biomarkers, HDA, TDA, CP‐I, and CP‐III, after administration of pravastatin plus placebo and pravastatin plus CsA.
Table S1. Summary of endogenous metabolites, which are substrates of OATP1B1 and have been evaluated as OATP1B1‐mediated drug–drug interaction biomarkers.
Table S2. Characteristics of healthy volunteers who participated in the pravastatin‐cyclosporin A (CsA) interaction study.
Table S3. Influence of cyclosporin A and SLCO1B1 c.521T>C genotype on the Cmax and AUC of pravastatin alone or pravastatin isomer (3′α‐hydroxy pravastatin) alone in volunteers.
Table S4. Pearson correlation, r, between pravastatin AUC with each of the biomarkers after administration with CsA.
Table S5. Overall summary of previous and current studies comparing the effect of SLCO1B1 genotype on drug levels and its effect by OATP1B1 inhibition.
Supplemental Material: Liquid chromatography–tandem mass spectrometry (LC‐MS/MS) analysis of pravastatin, TDA, HDA, CP‐I, and CP‐III.
Acknowledgments
We would like to acknowledge staff at the University of California, San Francisco Clinical Translational Science Institute (CTSI), and Sophie Stocker, Richard A. Castro, and Anabelle Coelho for their guidance in clinical protocol and subject recruitment.
Contributor Information
Deanna L. Kroetz, Email: deanna.kroetz@ucsf.edu
Kathleen M. Giacomini, Email: kathy.giacomini@ucsf.edu
References
- 1. Niemi, M. , Pasanen, M.K. & Neuvonen, P.J. Organic anion transporting polypeptide 1B1: a genetically polymorphic transporter of major importance for hepatic drug uptake. Pharmacol. Rev. 63, 157–181 (2011). [DOI] [PubMed] [Google Scholar]
- 2. Maeda, K. Organic anion transporting polypeptide (OATP)1B1 and OATP1B3 as important regulators of the pharmacokinetics of substrate drugs. Biol. Pharm. Bull. 38, 155–168 (2015). [DOI] [PubMed] [Google Scholar]
- 3. Seithel, A. et al The influence of macrolide antibiotics on the uptake of organic anions and drugs mediated by OATP1B1 and OATP1B3. Drug Metab. Dispos. 35, 779–786 (2007). [DOI] [PubMed] [Google Scholar]
- 4. Kameyama, Y. , Yamashita, K. , Kobayashi, K. , Hosokawa, M. & Chiba, K. Functional characterization of SLCO1B1 (OATP‐C) variants, SLCO1B1*5, SLCO1B1*15 and SLCO1B1*15+C1007G, by using transient expression systems of HeLa and HEK293 cells. Pharmacogenet. Genomics 15, 513–522 (2005). [DOI] [PubMed] [Google Scholar]
- 5. Tirona, R.G. , Leake, B.F. , Merino, G. & Kim, R.B. Polymorphisms in OATP‐C: identification of multiple allelic variants associated with altered transport activity among European‐ and African‐Americans. J. Biol. Chem. 276, 35669–35675 (2001). [DOI] [PubMed] [Google Scholar]
- 6. Kalliokoski, A. , Neuvonen, P.J. & Niemi, M. SLCO1B1 polymorphism and oral antidiabetic drugs. Basic Clin. Pharmacol. Toxicol. 107, 775–781 (2010). [DOI] [PubMed] [Google Scholar]
- 7. Niemi, M. et al Polymorphic organic anion transporting polypeptide 1B1 is a major determinant of repaglinide pharmacokinetics. Clin. Pharmacol. Ther. 77, 468–478 (2005). [DOI] [PubMed] [Google Scholar]
- 8. He, Y.J. et al Rifampicin alters atorvastatin plasma concentration on the basis of SLCO1B1 521T>C polymorphism. Clin. Chim. Acta 405, 49–52 (2009). [DOI] [PubMed] [Google Scholar]
- 9. Pasanen, M.K. , Fredrikson, H. , Neuvonen, P.J. & Niemi, M. Different effects of SLCO1B1 polymorphism on the pharmacokinetics of atorvastatin and rosuvastatin. Clin. Pharmacol. Ther. 82, 726–733 (2007). [DOI] [PubMed] [Google Scholar]
- 10. Yee, S.W. et al Metabolomic and genome‐wide association studies reveal potential endogenous biomarkers for OATP1B1. Clin. Pharmacol. Ther. 100, 524–536 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Xiang, X. et al Effect of SLCO1B1 polymorphism on the plasma concentrations of bile acids and bile acid synthesis marker in humans. Pharmacogenet. Genomics 19, 447–457 (2009). [DOI] [PubMed] [Google Scholar]
- 12. Zhang, W. et al OATP1B1 polymorphism is a major determinant of serum bilirubin level but not associated with rifampicin‐mediated bilirubin elevation. Clin. Exp. Pharmacol. Physiol. 34, 1240–1244 (2007). [DOI] [PubMed] [Google Scholar]
- 13. European Medicines Agency . Guideline on the investigation of drug interactions. <http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2012/07/WC500129606.pdf> (2012). Accessed November 1, 2018.
- 14. Food and Drug Administration . Draft Guidance for Clinical Drug Interaction Studies. <http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm292362.pdf> (2017) Accessed November 1, 2018.
- 15. Maeda, K. , Hisaka, A. , Saito, Y. , Nagai, N. & Kume, T. Brief guide for “Drug interaction guideline for drug development and labeling recommendations (final draft)”. J. Pharm. Sci. Technol. Jpn. 74, 406–413 (2014). [Google Scholar]
- 16. Lee, S.C. , Arya, V. , Yang, X. , Volpe, D.A. & Zhang, L. Evaluation of transporters in drug development: current status and contemporary issues. Adv. Drug Deliv. Rev. 116, 100–118 (2017). [DOI] [PubMed] [Google Scholar]
- 17. Lai, Y. et al Coproporphyrins in plasma and urine can be appropriate clinical biomarkers to recapitulate drug‐drug interactions mediated by organic anion transporting polypeptide inhibition. J. Pharmacol. Exp. Ther. 358, 397–404 (2016). [DOI] [PubMed] [Google Scholar]
- 18. Shen, H. et al Comparative evaluation of plasma bile acids, dehydroepiandrosterone sulfate, hexadecanedioate, and tetradecanedioate with coproporphyrins I and III as markers of OATP inhibition in healthy subjects. Drug Metab. Dispos. 45, 908–919 (2017). [DOI] [PubMed] [Google Scholar]
- 19. Shen, H. et al Coproporphyrins I and III as functional markers of OATP1B activity: in vitro and in vivo evaluation in preclinical species. J. Pharmacol. Exp. Ther. 357, 382–393 (2016). [DOI] [PubMed] [Google Scholar]
- 20. Shen, H. et al Cynomolgus monkey as a clinically relevant model to study transport involving renal organic cation transporters: in vitro and in vivo evaluation. Drug Metab. Dispos. 44, 238–249 (2016). [DOI] [PubMed] [Google Scholar]
- 21. Long, T. et al Whole‐genome sequencing identifies common‐to‐rare variants associated with human blood metabolites. Nat. Genet. 49, 568–578 (2017). [DOI] [PubMed] [Google Scholar]
- 22. Shin, S.Y. et al An atlas of genetic influences on human blood metabolites. Nat. Genet. 46, 543–550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yousri, N.A. et al Whole‐exome sequencing identifies common and rare variant metabolic QTLs in a Middle Eastern population. Nat. Commun. 9, 333 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yu, B. et al Loss‐of‐function variants influence the human serum metabolome. Sci. Adv. 2, e1600800 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brosen, K. , Hansen, J.G. , Nielsen, K.K. , Sindrup, S.H. & Gram, L.F. Inhibition by paroxetine of desipramine metabolism in extensive but not in poor metabolizers of sparteine. Eur. J. Clin. Pharmacol. 44, 349–355 (1993). [DOI] [PubMed] [Google Scholar]
- 26. Aquilante, C.L. et al Influence of SLCO1B1 polymorphisms on the drug‐drug interaction between darunavir/ritonavir and pravastatin. J. Clin. Pharmacol. 52, 1725–1738 (2012). [DOI] [PubMed] [Google Scholar]
- 27. Kajosaari, L.I. et al Cyclosporine markedly raises the plasma concentrations of repaglinide. Clin. Pharmacol. Ther. 78, 388–399 (2005). [DOI] [PubMed] [Google Scholar]
- 28. Kalliokoski, A. , Backman, J.T. , Kurkinen, K.J. , Neuvonen, P.J. & Niemi, M. Effects of gemfibrozil and atorvastatin on the pharmacokinetics of repaglinide in relation to SLCO1B1 polymorphism. Clin. Pharmacol. Ther. 84, 488–496 (2008). [DOI] [PubMed] [Google Scholar]
- 29. Hedman, M. , Neuvonen, P.J. , Neuvonen, M. , Holmberg, C. & Antikainen, M. Pharmacokinetics and pharmacodynamics of pravastatin in pediatric and adolescent cardiac transplant recipients on a regimen of triple immunosuppression. Clin. Pharmacol. Ther. 75, 101–109 (2004). [DOI] [PubMed] [Google Scholar]
- 30. Regazzi, M.B. et al Altered disposition of pravastatin following concomitant drug therapy with cyclosporin A in transplant recipients. Transplant. Proc. 25, 2732–2734 (1993). [PubMed] [Google Scholar]
- 31. Bristol‐Myers Squibb . Pravachol (pravastatin) prescribing information. <https://http://www.bms.com/assets/bms/ca/documents/productmonograph/PRAVACHOL_EN_pm.pdf> (1990). Accessed October 6, 2018.
- 32. Deng, J.W. et al The effect of SLCO1B1*15 on the disposition of pravastatin and pitavastatin is substrate dependent: the contribution of transporting activity changes by SLCO1B1*15. Pharmacogenet. Genomics 18, 424–433 (2008). [DOI] [PubMed] [Google Scholar]
- 33. Niemi, M. , Pasanen, M.K. & Neuvonen, P.J. SLCO1B1 polymorphism and sex affect the pharmacokinetics of pravastatin but not fluvastatin. Clin. Pharmacol. Ther. 80, 356–366 (2006). [DOI] [PubMed] [Google Scholar]
- 34. Gertz, M. et al Cyclosporine inhibition of hepatic and intestinal CYP3A4, uptake and efflux transporters: application of PBPK modeling in the assessment of drug‐drug interaction potential. Pharm. Res. 30, 761–780 (2013). [DOI] [PubMed] [Google Scholar]
- 35. Mori, D. et al Effect of OATP1B1 genotypes on plasma concentrations of endogenous OATP1B1 substrates and drugs, and their association in healthy volunteers. Drug Metab. Pharmacokinet. 34, 78–86 (2019). [DOI] [PubMed] [Google Scholar]
- 36. Bednarczyk, D. & Boiselle, C. Organic anion transporting polypeptide (OATP)‐mediated transport of coproporphyrins I and III. Xenobiotica 46, 457–466 (2016). [DOI] [PubMed] [Google Scholar]
- 37. Shimizu, Y. , Naruto, H. , Ida, S. & Kohakura, M. Urinary coproporphyrin isomers in Rotor's syndrome: a study in eight families. Hepatology 1, 173–178 (1981). [DOI] [PubMed] [Google Scholar]
- 38. Wolkoff, A.W. , Wolpert, E. , Pascasio, F.N. & Arias, I.M. Rotor's syndrome. A distinct inheritable pathophysiologic entity. Am. J. Med. 60, 173–179 (1976). [DOI] [PubMed] [Google Scholar]
- 39. Chu, X. , Chan, G.H. & Evers, R. Identification of endogenous biomarkers to predict the propensity of drug candidates to cause hepatic or renal transporter‐mediated drug‐drug interactions. J. Pharm. Sci. 106, 2357–2367 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Concentration–time curves of pravastatin and 3α‐hydroxy pravastatin after oral administration of pravastatin and placebo or pravastatin plus cyclosporin A (CsA).
Figure S2. Concentration–time curves of the four OATP1B1 biomarkers, HDA, TDA, CP‐I, and CP‐III, after administration of pravastatin plus placebo and pravastatin plus CsA.
Table S1. Summary of endogenous metabolites, which are substrates of OATP1B1 and have been evaluated as OATP1B1‐mediated drug–drug interaction biomarkers.
Table S2. Characteristics of healthy volunteers who participated in the pravastatin‐cyclosporin A (CsA) interaction study.
Table S3. Influence of cyclosporin A and SLCO1B1 c.521T>C genotype on the Cmax and AUC of pravastatin alone or pravastatin isomer (3′α‐hydroxy pravastatin) alone in volunteers.
Table S4. Pearson correlation, r, between pravastatin AUC with each of the biomarkers after administration with CsA.
Table S5. Overall summary of previous and current studies comparing the effect of SLCO1B1 genotype on drug levels and its effect by OATP1B1 inhibition.
Supplemental Material: Liquid chromatography–tandem mass spectrometry (LC‐MS/MS) analysis of pravastatin, TDA, HDA, CP‐I, and CP‐III.