

Abstract
Exosomes (Exos) are nanoscale natural vehicles for transporting biomolecules to facilitate cell-to-cell communication, indicating a high potential of them for delivering therapeutics/diagnostics. To improve their delivery capacity, a simple, noninvasive, and efficient strategy for functionalizing Exos with effective targeting ligands as well as elucidation of the cellular uptake mechanism of these functionalized Exos was found be to necessary, but remained a challenge. In this work, we used diacyllipid–aptamer conjugates as the targeting ligand to develop an aptamer-functionalized Exos (Apt-Exos) nanoplatform for cell type-specific delivery of molecular therapeutics. The cellular uptake mechanism of Apt-Exos was investigated in details, and distinct behavior was observed in comparison to free Exos. By combining the excellent molecular recognition capability of aptamers and the superiority of Exos as natural vehicles, Apt-Exos can efficiently deliver molecular drugs/fluorophores to target cancer cells, providing a promising delivery platform for cancer theranostics.
Graphical Abstract
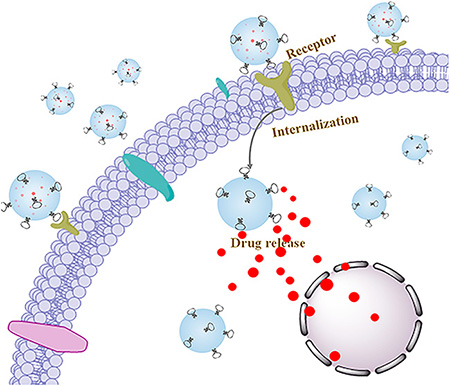
Despite many advances in synthetic delivery vehicles, their transition from bench to bedside was still hindered by low specificity, high biotoxicity, and high immunogenicity.1 Naturally occurring vehicles, for instance, exosomes (Exos), are expected to provide promising alternatives for efficient delivery of therapeutics/diagnostics.2 Exos are nanoscale membrane-enclosed vesicles (30–100 nm) secreted by various eukaryotic cells (e.g., spithelial cells and myeloid cells).3 They play critical roles in cell–cell communication through packaging and transporting specific bioactive molecules.4 In the recent years, pioneered by the Wood group, Exos have been developed as vehicles for delivering various therapeutic/diagnostic agents, such as small-molecule chemotherapeutics, anti-inflammatory agents, RNAs, and proteins.5–7 As demonstrated, Exos have several superiorities over many synthetic vehicles: (1) the narrow nanoscale size, to some extent, facilitated tumor accumulation through enhanced permeability and retention (EPR) effect;8 (2) Exos could be derived from patients’ own cells and body fluids (e.g., blood and urine), and thus can be immunologically inert;9 (3) Exos exhibited intrinsic capability for crossing physiologic barriers.5
While some positive results have been obtained by using Exos as the vehicle, to achieve efficient delivery, equipping them with targeting capability was found to be necessary.10 Several strategies have been developed to functionalize the Exos surface with tumor-targeting ligands. Despite enhanced tumor targetability, these strategies involved either complicated cell-based genetic manipulation,11 specific affinity interaction,12 or toxic covalent cross-linking reaction.13 Meanwhile, the cellular uptake mechanism of these functionalized Exos was rarely studied, limiting their practical applications. Alternative targeting strategies of Exos with merits of convenient operation, high efficiency, high specificity, and clear mechanism are being sought.14
As specfic recognition elements, aptamers have attracted extensive interest in both fundamental and applied research, because of their advantages including easy synthesis, convenient modifications, small size, low immunogenicity, and low toxicity.15,16 Especially, those aptamers selected against living cancer cells have been widely used for designing cancer-targeted delivery systems.17,18 With advanced chemistry of nucleic acids, aptamers could be modified with various functional groups. Recently, our group has successfully conjugated aptamers with a hydrophobic diacyllipid tail. We demonstrated well that such conjugated aptamers can be efficiently anchored onto the cell membrane through the hydrophobic interaction between the diacyllipid tail and the cellular phospholipid layer.19 Since the Exos membrane contains a similar component as that of the cell membrane, this strategy could be extended for functionalization of Exos with cancer-specfic aptamers.
In this work, by using diacyllipid–aptamer conjugates as the functional ligand, we developed an aptamer-equipped Exos platform for efficient cancer-targeted delivery of chemotherapeutics. As illustrated in Scheme 1, the diacyllipid–aptamer conjugate consists of three segments: a diacyllipid tail, an aptamer sequence, and a PEG linker in between. Exos were first loaded with chemotherapeutic drugs by electroporation, and then functionalized with cancer-specific aptamer. The resultant aptamer-modified Exos (Apt-Exos), with the guiding function of the aptamer, can specifically deliver chemotherapeutics to target cancer cells. With the natural delivery advantages of Exos, the current Apt-Exos is expected to provide an efficient delivery platform for targeted cancer theranostics.
Scheme 1. Illustration of Construction and Targeted Delivery of Aptamer-Functionalized Drug-Loaded Exos (Termed Apt-Exos-D)a.

aExos were loaded with chemotherapeutic drugs via electroporation. Then, the drug-loaded Exos (Exos-D) were functionalized with aptamer. With the guidance system of the aptamer, Apt-Exos-D can efficiently deliver molecular drug to target cancer cells for enhanced cancer chemotherapy.
EXPERIMENTAL SECTION
Solid-Phase Synthesis of Oligonucleotides.
All DNA synthesis reagents were purchased from Glen Research. DNA synthesis, including conjugation of DMT-hexaethyloxy glycol (PEG) phosphoramidite and diacyllipid phosphoramidite, was conducted via solid-phase phosphoramidite chemistry on a 12-column DNA/RNA synthesizer (Polygen) at 1.0 mM scale. Two PEG phosphoramidite molecules were incorporated between the diacyllipid tail and the DNA sequence. The diacyllipid phosphoramidite was synthesized following a previously reported procedure.15 All the detailed sequences were listed in Table S1. The obtained oligonucleotides were cleaved and deprotected from the controlled pore glass (CPG), and then were precipitated in cold salted ethanol solution at −20 °C overnight. After centrifugation to remove supernatant, the DNAs were dissolved with triethylamineacetic acid (TEAA, 100 mM, pH 7.5). Then, DNAs without diacyllipid tail were purified by reversed-phase HPLC using a C18 reversed-phase column and diacyllipid DNAs were purified using a Bio Basic-4 column. Acetonitrile plus TEAA was used as a mobile phase. The resulting oligonucleotides products were dried with a rotary vacuum pump and subsequently quantified by measuring absorbance at 260 nm.
Collection and Purification of Exos.
Immature dendritic cells (imDCs) were cultured with 18 mL of FBS-free 1640 culture medium in 175 cm2 culture flask at 37 °C for 48 h. Exos were collected and purified from the harvested supernatant according to the previous literatures.12 First, the supernatant was centrifuged at 300 g for 10 min to eliminate cells. To further remove cell debris, the supernatant was centrifuged at 1200 g for 10 min, and then 10 000 gfor 30 min at 4 °C. The resulting supernatant was filtered using a 0.22 μm filter, and the filtrate was centrifuged at 120 000 gfor 70 min at 4 °C using an L-100XP ultracentrifuge (Beckman Coulter, Brea, CA, U.S.A.) with a type Ti70 rotor. The pellet was washed with 0.01 M phosphate-buffered solution (PBS, pH7.4) and ultracentrifuged again at 120 000 gfor another 70 min. The final Exos pellet was resuspended in PBS and stored at −80 °C prior to use.
Characterization of Exos.
Exos (20 μL, 0.5 μg/μL) were fixed with 2% paraformaldehyde for 30 min at room temperature, and then dropped onto copper grids. After being drying, Exos were stained with 1% phosphotungstic acid. Then, the dried grids were imaged on an H-7000 NAR transmission electron microscope (Hitachi) with a working voltage of 200 kV. Dynamic light scattering (DLS) measurements were performed on a Malvern Zetasizer Nano ZS90 (Malvern Instruments, Ltd., Worcestershire, U.K.).
Modification of Exos with Diacyllipid–DNA.
Exos (100 μL 1.5 ug/μL) were incubated with 5-carboxyfluorescein (FAM)-labeled diacyllipid–DNA probes (1 μM) in PBS buffer at 37 °C for 30 min. To remove free probes, Exos were then washed two times with PBS by ultracentrifuging at 120 000 g for 70 min at 4 °C. The pellet was resuspended in PBS buffer. The decoration of diacyllipid–DNA on the surface of Exos was measured with a Fluoromax-4 spectrofluorometer (HORIBA JobinYvon, Edison, NJ, λex = 488 nm, λem = 520 nm).
Dox Loading into Exos.
For Dox loading, Exos of different concentrations were mixed with Dox (60 μg/mL) in a0.4-cm cuvette (Bio-Rad) containing 250 μL of electroporation buffer at 4 °C. Then, electroporation was carried out at 400 V and 150 μF on a Bio-Rad Gene Pulser Xcell electroporation system. After electroporation, the mixture was incubated at 37 °C for 30 min to guarantee the complete recovery of the Exos plasma membrane. To remove free Dox, the final products termed as Exos-D were washed with cold PBS twice by ultracentrifugation at 120 000 gfor 70 min. The payload of Dox in Exos was calculated on the basis of Dox fluorescence (at 594 nm) measured by a Fluoromax-4 spectrofluorometer (HORIBA JobinYvon, Edison, NJ, λex = 488 nm, λem = 593 nm).
Drug Release Kinetics.
In the Dox release study, Exos-D (100 μL, 1 μM) or equivalent free Dox were transferred into a dialysis tube with a molecular weight cut off of 6–10 kDa. The tube was placed into a 15 mL centrifuge tube and immersed in PBS buffer (1 mL, pH 7.4) at 37 °C. The Dox fluorescence of the dialysis buffer was measured with a Fluoromax-4 spectrofluorometer (HORIBA JobinYvon, Edison, NJ, λex =488 nm, λem = 595 nm).
Cell Culture.
ImDC, CEM cells, and Ramos cells were all cultured in RPMI 1640 medium containing 10% fetal bovine serum (Invitrogen, Carlsbad, CA, U.S.A.) and 0.5 mg/mL penicillin–streptomycin (KeyGEN Biotech, Nanjing, China) at 37 °C with 5% CO2.
Selective Binding Assay.
To evaluate the cellular binding affinity of different Exos, the 3′ terminus of the aptamer sgc8 was labeled with a FAM fluorophore. CEM cells or Ramos cells (2 × 105) were incubated with fluorescent sgc8-Exos or lib-Exos in binding buffer [Dulbecco’s phosphate-buffered saline (DPBS, Gibco) supplemented with 4.5 g/L of glucose, 5 mM of MgCl2, 0.1 mg/mL of yeast tRNA, and 1 mg/mL of BSA] on ice for 30 min, followed by washing with washing buffer (DPBS supplemented with 4.5 g/L of glucose and 5 mM of MgCl2) three times and resuspension in PBS (400 μL). Then, the fluorescence signal of the cells was analyzed with flow cytometry (BD FACSVerse) and confocal laser scanning microscopy (FV1000-X81, Olympus).
Investigating the Uptake Mechanism.
To inhibit specific endocytosis pathways, CEM cells were pretreated with 10 mM NaN3 for 30 min, 2.5 mM amiloride for 1 h, 10 mM methyl-β-cyclodextrin (M-β-CD) for 30 min, 10 μg mL−1 chlorpromazine for 30 min, or 80 μM dynasore for 30 min. The cells were then washed with DPBS, followed by incubation with Cy5-labeled sgc8, Dil-labeled Exos, or sgc8-Exos for 2 h. Subsequently, all samples were treated with tyrosine and washed extensively to remove the signal bound on the cell surface. After that, the cells were imaged with confocal laser scanning microscopy (CLSM).
For colocalization assay, after incubation with Dil-labeled sgc8-Exos or lib-Exos, the CEM cells were treated with nucleic indicator Hoechst 33258 (0.2 μg/mL) and lysosomal indicator LysoTracker green (100 nM) for 30 min. After being washed, the cells were imaged with CLSM. Hoechst 33258 was excited with a violet 405 nm laser diode, and the emission was collected from 450 to 500 nm. LysoTracker green was excited at 488 nm, and the emissions were collected from 505 to 535 nm. Dil was excited at 543 nm, and the emissions were collected from 560 to 620 nm.
Cytotoxicity Assay.
Cytotoxicity was determined using 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay. Briefly, CEM cells or Ramos cells (3 × 104 cells per well) were treated with free Dox or sgc8-Exos-D of different concentrations in binding buffer at 37 °C with 5% CO2 for 1 h. Subsequently, 90% of the supernatant medium free therapeutic agents was removed and replaced with 200 μL of fresh medium plus 10% FBS. The cells were cultured for another 48 h. After removal of cell medium, Cell-Titer reagent (20 μL) diluted in fresh medium (100 μL) was added to each well, and the mixture was incubated for 1–2h. The absorbance at 490 nm was recorded using a Synergy H4 hybrid reader (BioTek).
RESULTS AND DISCUSSION
Preparation and Characterization of Apt-Exos.
As a proof of concept, immature dendritic cells (imDCs) with high production yield of Exos, were chosen as the donor cell line.20 The biosafety of Exos derived from imDCs was proved in several clinical trials.21 Exos were collected and purified from the culture medium of imDCs through differential ultra-centrifugation. By quantifying the protein content with a BCA protein assay, about 70 μg of Exos was obtained from each 107 cells. In the transmission electron microscope (TEM) images, Exos displayed a cuplike shape of nanoscale size (dm = 55.0 ±12.4 nm) (Figure 1A). The hydrodynamic size, as measured with DLS, was 91.2 nm with a particle dispersion index (PDI) of 0.128 (Figure 1B). In addition, as illustrated by Western blot (Figure S1), the Exos showed a positive staining of CD63 which was a marker protein of Exos. All of these characterization results were consistent with the typical features of Exos, demonstrating their successful harvest.22
Figure 1.

(A) TEM images of Exos. (B) DLS profiles of Exos and sgc8-Exos. (C) Zeta-Potential of Exos and sgc8-Exos.
To endow the Exos delivery platform with cancer-targeting capability, a diacyllipid–aptamer conjugate was synthesized. Aptamer sgc8, which can specifically recognize membrane-expressed protein tyrosine kinase 7 (PTK7), was used as the model targeting ligand.15,23 The aptamer was decorated onto the Exos surface through hydrophobic interaction between the diacyllipid tail and the phospholipid bilayer of Exos. To establish a fluorescence signal for characterizing the successful modification of Exos as well as the cancer targeting events, the 3′ terminus of the aptamer sequence was labeled with a FAM fluorophore. For aptamer functionalization, Exos were simply mixed with the diacyllipid–sgc8 conjugate (1 μM) at 37 °C for 30 min, and then washed with ultracentrifugation. The resultant sgc8-Exos was resuspended in PBS, and a strong FAM fluorescence was detected (Figure S2). In contrast, little fluorescence was observed in the control samples of both diacyllipid–sgc8 conjugates only or Exos-incubated FAM-labeled sgc8 without the diacyllipid tail, revealing that the diacyllipid conjugation was essential for modifying aptamer onto the Exos surface. The successful fabrication of sgc8-Exos was further confirmed with the increased hydrodynamic size (111.4 nm, PDI = 0.183) and the reduced ζ-potential (−2.06 mV), in comparison to the hydrodynamic size (91.2 nm, PDI =0.128) and the ζ-potential (3.2 mV) of Exos only, respectively (Figure 1C).
Selective Cellular Recognition of Apt-Exos.
In order to test whether sgc8-Exos can specifically recognize target cells. CEM, a T-leukemia cell line that overexpressed PTK7, was used as the target cell model, while Ramos cells with low expression level of PTK7 were used as a control. After incubation with FAM-labeled sgc8-Exos at 4 °C for 30 min, the cells were analyzed with flow cytometry. As illustrated in Figure 2A, an obvious shift of the FAM fluorescence profile was observed in CEM cells treated with sgc8-Exos, while little fluorescence increment could be detected in control Ramos samples. The sequence specificity of sgc8 was also verified by negative fluorescence signal in both CEM and Ramos samples with treatment of lib-Exos (lib represents a random library sequence). The cell-type-specific binding of sgc8-Exos was further visualized with CLSM (Figure 2B). Of note, the direct incubation of equivalent diacyllipid–sgc8 conjugates induced a remarkable and unbiased fluorescence signal on both CEM and Ramos cells (Figure S3), which was attributed to the strong nonspecific hydrophobic interaction. This, from another angle, verified that the selective fluorescence signal in the samples of sgc8-Exos was not due to the diacyllipid–sgc8 conjugates detached from Exos.
Figure 2.

(A) Flow cytometry assays of CEM cells or Ramos cells treated with fluorescent sgc8-Exos or lib-Exos. (B) CLSM images of CEM cells treated with fluorescent sgc8-Exos (i) or lib-Exos (ii), or Ramos cells treated with fluorescent sgc8-Exos (iii) or lib-Exos (iv). The cell binding tests were all performed at 4 °C for 30 min. The scale bar represents 50 μm.
Selective Cellular Uptake Mechanism of Apt-Exos.
To study the interaction of Apt-Exos with cells, a plasma membrane staining dye Dil was used to fluorescently label Exos. The dye leakage can be ignored over the study period (Figure S4), indicating a reliable fluorescence signal for Exos tracking. After incubation with Dil-labeled sgc8-Exos or lib-Exos at 37 °C with 5% CO2, where the cellular activity was relatively high, the CEM cells were washed, and then imaged with CLSM. A dotted cellular distribution pattern was observed in CEM cells treated with sgc8-Exos, but not in that treated with lib-Exos (Figure 3A). The fluorescence signal of sgc8-Exos was mainly colocalized with that of the LysoTracker, a fluorescence indicator of lysosome, revealing the lysosomal location of sgc8-Exos. Of note, the cell membrane was lighted up rapidly and uniformly after incubation with nonfunctionalized Exos, indicating that the aptamer functionalization could change the cellular uptake process of Exos (Figure S5).
Figure 3.

(A) CLSM images of CEM cells incubated with Dil-labeled sgc8-Exos (i) or lib-Exos (ii) at 37 °C for 2 h, and then stained with LysoTracker green and Hoechst 33258. The scale bar represents 30 μm. (B) Histogram analysis of the intracellular fluorescence signal of CEM cells treated with Dil-labeled sgc8-Exos or Exos at different conditions, in which 4 °C and NaN3 were used to inhibit energy-dependent endocytosis, and amiloride, chlorpromazine, M-β-CD, and dynasore were used to inhibit specific endocytosis pathways.
Next, we systematically studied the cellular uptake mechanism of sgc8-Exos under different conditions. The energy-dependent endocytosis, which could be suppressed by temperature reduction and ATP depletion, is a popular way for cells to internalize nanoscale cargos.24 By cooling the cells to 4 °C or pretreating them with sodium azide (NaN3) to reduce the ATP level, the cellular uptake of sgc8-Exos was apparently inhibited, indicating an energy-dependent endocytosis process (Figure 3B, Figure S6). To further investigate the possible endocytosis pathways of sgc8-Exos, different endocytosis inhibitors, including dynasore (dynamin), chlorpromazine (clathrin), M-β-CD (caveolae), and amiloride (micropinocytosis), were applied. When CEM cells were pretreated with dynasore, a specific inhibitor of dynamin that could affect the endocytosis mediated by either caveolae or clathrin,25 their uptake capability for sgc8-Exos was significantly reduced. In addition, a remarkable signal decline was observed in the CEM cells pretreated with chlorpromazine,26 indicating a noticeable contribution of the clathrin-mediated endocytosis pathway in cellular uptake of sgc8-Exos. On the other hand, with pretreatment of M-β-CD,27 a strong signal was retained inside the CEM cells, suggesting that caveolae-mediated endocytosis was involved weakly in the sgc8-Exos internalization. The contribution of the micropinocytosis pathway was also demonstrated by the reduced sgc8-Exos signal in the amiloride-pretreated28 CEM cells. Collectively, these results showed that sgc8-Exos was internalized through multiple endocytosis pathways, while clathrin-mediated endocytosis was a major pathway, whereas micropinocytosis and caveolae-mediated endocytosis play a less important role. The cellular uptake mechanism of free sgc8 aptamer was also studied and showed a similar pattern as that of sgc8-Exos (Figure S7).
Of note, the cellular internalization of free Exos was also regulated through multiple endocytosis pathways. Inhibition of either energy-mediated endocytosis, micropinocytosis, or caveolae-mediated endocytosis could significantly reduce the uptake of Exos, whereas much less impact was observed by inhibition of clathrin-mediated endocytosis (Figure 3B and Figure S8). These results demonstrated that the functionalization of Exos with aptamer could change their cellular uptake pathways, which could be potentially useful for artificially manipulating the Exos-related biological processes.29
Selective Cytotoxicity of Therapeutics Delivered by Apt-Exos.
To engineer the Exos-carried drug delivery system, doxorubicin (Dox), a commonly used anticancer drug,30 was used as the drug model and loaded into Exos through electroporation. After removal of free Dox with ultra-centrifugation, the resultant Dox-loaded Exos (Exos-D) were resuspended in PBS. The Dox loading capacity of Exos was tested by fixing the initial Dox concentration at 60 μg/mL while changing the Exos concentration from 0 to 560 μg/mL. As demonstrated with the fluorescence intensity of Dox (at 593 nm) in the Exos pellet, the loading of Dox showed a dose-dependent pattern (Figure S9A). Meanwhile, little Dox fluorescence was observed by replacing Exos with equivalent bovine serum albumin (BSA), indicating the successful loading of Dox into Exos. By interpolating with a standard calibration curve, the payload of Dox was 14.157 ng per 1 μg of Exos. The leakage of Dox from Exos was studied with a dialysis experiment. In comparison to the rapid release of free Dox, the Dox release from Exos was relatively slow, and no more than 45% of Dox leakage was observed after a 5 h incubation (Figure S9B), indicating that Exos can, to some extent, prevent the leakage of Dox before reaching the target site. The cell-type-specific accumulation of Dox via sgc8-Exos was well-demonstrated with CLSM (Figure S10). Only the target CEM cells treated with sgc8-Exos-D displayed a strong Dox fluorescence signal, while relatively weaker signal was observed in those control samples of CEM cells treated with lib-Exos-D and Ramos cells treated with either sgc8-Exos-D or lib-Exos-D.
Having demonstrated the cell-type-specific uptake of sgc8-Exos-D, we next investigated their selective cytotoxicity with an MTS assay. As shown in Figure 4, both sgc8-Exos-D and free Dox induced a dose-dependent cytotoxicity on target CEM cells, while the therapeutic efficacy of sgc8-Exos-D tended to be stronger than that of free Dox, revealing enhanced cellular accumulation of Dox via the sgc8-Exos-D delivery system, whereas, for CEM cells treated with control lib-Exos-D, a relatively lower cytotoxicity was observed (Figure S11). In addition, for nontarget Ramos cells, a lower cytotoxicity was induced by sgc8-Exos-D in comparison to that induced by free Dox. All these results proved that the sgc8-Exos-D was able to induce selective and potent therapeutic efficacy on target cancer cells.
Figure 4.

Selective cytotoxicity of sgc8-Exos-D. CEM cells or Ramos cells were incubated with different concentrations of free Dox and sgc8-Exos-D at 37 °C for 48 h.
CONCLUSIONS
In summary, we have developed an aptamer (sgc8)-functionalized Exos platform for efficient cancer-targeted delivery of chemotherapeutics. The cellular uptake mechanism of the resultant sgc8-Exos has been studied. We found that the specific cellular uptake of sgc8-Exos was facilitated through multiple endocytosis pathways, and the clathrin-mediated endocytosis was a major one, which was different from that of free Exos, indicating that the functionalization of Exos with targeting ligands could change their interaction with cells. In addition, by combining the advantages of Exos as endogenous nanoscale vehicles and the superiorities of aptamers as the cell-type-specific recognition ligand, these Apt-Exos were proved to offer a promising nanoplatform for efficient cancer-targeted delivery of therapeutics/diagnostics.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NSFC Grants (91753109, 21505039, and 2016QNRC001), the HPNSFC 2018JJ3034, the CPSF 2017M622567, the National Institutes of Health GMR35 127130, and the NSF1645215.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: (10.1021/acs.anal-chem.8b05204).
DNA sequences designed in this work, Western blot analysis of specific Exos marker proteins, fluorescence spectra of the precipitant solution in different samples, relative fluorescence intensity of Dil that remained in the Exos, CLSM images of CEM cells or Ramos cells treated with different samples, fluorescence spectra of Dox after incubation with different concentrations of Exos or BSA, and release profiles of free Dox and Dox from Exos-D in PBS (PDF)
The authors declare no competing financial interest.
REFERENCES
- (1).Murphy EA; Majeti BK; Barnes LA; Makale M; Weis SM; Lutu-Fuga K; Wrasidlo W; Cheresh DA Proc. Natl. Acad. Sci. U. S. A 2008, 105, 9343–9348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Akers JC; Gonda D; Kim R; Carter BS; Chen CC J. Neuro-Oncol 2013, 113, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Théry C; Zitvogel L; Amigorena S Nat. Rev. Immunol 2002, 2, 569–579. [DOI] [PubMed] [Google Scholar]
- (4).Théry C; Ostrowski M; Segura E Nat. Rev. Immunol 2009, 9, 581–593. [DOI] [PubMed] [Google Scholar]
- (5).Alvarez-Erviti L; Seow Y; Yin H; Betts C; Lakhal S; Wood MJ Nat. Biotechnol 2011, 29, 341–345 [DOI] [PubMed] [Google Scholar]
- (6).Kooijmans SA; Vader P; Van Dommelen SM; Van Solinge WW; Schiffelers RM Int. J. Nanomed 2012, 7, 1525–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Johnsen KB; Gudbergsson JM; Skov MN; Pilgaard L; Moos T; Duroux M Biochim. Biophys. Acta, Rev. Cancer 2014, 1846, 75–87. [DOI] [PubMed] [Google Scholar]
- (8).Jang SC; Kim OY; Yoon CM; Choi DS; Roh TY; Park J; Nilsson J; Lötvall J; Kim YK; Gho YS ACS Nano 2013, 7, 7698–7710. [DOI] [PubMed] [Google Scholar]
- (9).Van Dommelen SM; Vader P; Lakhal S; Kooijmans S; Van Solinge WW; Wood MJ; Schiffelers RM J. Controlled Release 2012, 161, 635–644. [DOI] [PubMed] [Google Scholar]
- (10).Lakhal S; Wood MJ BioEssays 2011, 33, 737–741. [DOI] [PubMed] [Google Scholar]
- (11).Kamerkar S; LeBleu VS; Sugimoto H; Yang S; Ruivo CF; Melo SA; Lee JJ; Kalluri R Nature 2017, 546, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Qi H; Liu C; Long L; Ren Y; Zhang S; Chang X; Qian X; Jia H; Zhao J; Sun J; Hou X; Yuan X; Kang C ACS Nano 2016, 10, 3323–3333. [DOI] [PubMed] [Google Scholar]
- (13).Smyth T; Petrova K; Payton NM; Persaud I; Redzic JS; Graner MW; Smith-Jones P; Anchordoquy TJ Bioconjugate Chem. 2014, 25, 1777–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Ha D; Yang N; Nadithe V Acta Pharm. Sin. B 2016, 6, 287–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Shangguan D; Li Y; Tang Z; Cao ZC; Chen HW; Mallikaratchy P; Sefah K; Yang CJ; Tan W Proc. Natl. Acad. Sci. U. S. A 2006, 103, 11838–11843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Fang X; Tan W Acc. Chem. Res 2010, 43, 48–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tan W; Fang X Aptamers Selected by Cell-SELEX for Theranostics; Springer: Berlin, Germany, 2015; pp 301–337. [Google Scholar]
- (18).Qiu L; Wu C; You M; Han D; Chen T; Zhu G; Jiang J; Yu R; Tan WJ Am. Chem. Soc 2013, 135, 12952–12955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Xiong X; Liu H; Zhao Z; Altman MB; Lopez Colon D.; Yang CJ; Chang LJ; Liu C; Tan W Angew. Chem., Int. Ed 2013, 52, 1472–1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).El-Andaloussi S; Lee Y; Lakhal-Littleton S; Li J; Seow Y; Gardiner C; Alvarez-Erviti L; Sargent IL; Wood MJ Nat. Protoc 2012, 7, 2112–2126. [DOI] [PubMed] [Google Scholar]
- (21).Escudier B; Dorval T; Chaput N; Andre F; Caby MP; Novault S; Flament C; Leboulaire C; Borg C; Amigorena S; Boccaccio C; Bonnerot C; Dhellin O; Movassagh M; Piperno S; Robert C; Serra V; Valente N; Le Pecq JB; Spatz A; Lantz O; Tursz T; Angevin E; Zitvogel LJ Transl. Med 2005, 3, 10–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Jiang Y; Shi M; Liu Y; Wan S; Cui C; Zhang L; Tan W Angew. Chem., Int. Ed 2017, 56, 11916–11920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Shangguan D; Cao Z; Meng L; Mallikaratchy P; Sefah K; Wang H; Li Y; Tan WJ Proteome Res. 2008, 7, 2133–2139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kim JS; Yoon TJ; Yu KN; Noh MS; Woo M; Kim BG; Lee KH; Sohn BH; Park SB; Lee JK; Cho MH J. J. Vet. Sci 2006, 7, 321–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Macia E; Ehrlich M; Massol R; Boucrot E; Brunner C; Kirchhausen T Dev. Cell 2006, 10, 839–850. [DOI] [PubMed] [Google Scholar]
- (26).Rejman J; Oberle V; Zuhorn IS; Hoekstra D Biochem. J 2004, 377, 159–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Park TE; Kang B; Kim YK; Zhang Q; Lee WS; Islam MA; Kang SK; Cho MH; Choi YJ; Cho CS Biomaterials 2012, 33, 7272–7281. [DOI] [PubMed] [Google Scholar]
- (28).Mao D; Liang Y; Liu Y; Zhou X; Ma J; Jiang B; Liu J; Ma D Angew. Chem 2017, 129, 12788–12792. [Google Scholar]
- (29).Ludwig AK; Giebel B Int. J. Biochem. Cell Biol 2012, 44, 11–15. [DOI] [PubMed] [Google Scholar]
- (30).Tian Y; Li S; Song J; Ji T; Zhu M; Anderson GJ; Wei J; Nie G Biomaterials 2014, 35, 2383–2390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.