

Abstract
Lipoprotein lipase (LPL), identified in the 1950s, has been studied intensively by biochemists, physiologists, and clinical investigators. These efforts uncovered a central role for LPL in plasma triglyceride metabolism and identified LPL mutations as a cause of hypertriglyceridemia. By the 1990s, with an outline for plasma triglyceride metabolism established, interest in triglyceride metabolism waned. In recent years, however, interest in plasma triglyceride metabolism has awakened, in part because of the discovery of new molecules governing triglyceride metabolism. One such protein—and the focus of this review—is GP IHBP1, a protein of capillary endothelial cells. GPIHBP1 is LPL’s essential partner: it binds LPL and transports it to the capillary lumen; it is essential for lipoprotein margination along capillaries, allowing lipolysis to proceed; and it preserves LPL’s structure and activity. Recently, GPIHBP1 was the key to solving the structure of LPL. These developments have transformed the models for intravascular triglyceride metabolism.
Keywords: hypertriglyceridemia, chylomicronemia, lipoprotein lipase, lipid transport, endothelial cells
The identification of lipoprotein lipase (LPL) as an intravascular triglyceride hydrolase (Korn, 1955; Korn and Quigley, 1955) was a momentous discovery, one that suggested a mechanism by which triglycerides in the bloodstream are converted to fatty acids for utilization by vital tissues (Havel, 2010). This discovery triggered interest in triglyceride metabolism by scientists around the globe. Soon thereafter, a deficiency of LPL was shown to cause familial chylomicronemia (Havel and Gordon, 1960), the first example of an inborn error in lipoprotein metabolism (Havel, 2010). LPL-mediated processing of triglyceride-rich lipoproteins was shown to be important for utilization of dietary lipids, for generating atherogenic remnant lipoproteins, and for contributing to the biogenesis of high-density lipoproteins (HDL) (Havel, 2010; Havel and Kane, 2001; Rader, 2006; Rye and Barter, 2014). The observation that LPL could be released into the bloodstream with an injection of heparin (Korn, 1955), combined with the fact that LPL binds to heparan sulfate proteoglycans (HSPGs) in vitro (Lookene et al., 1997b; Simionescu and Simionescu, 1986; Vijayagopal et al., 1980), led to the proposal that the LPL inside blood vessels is bound to HSPGs within the glycocalyx lining of blood vessels (Blanchette-Mackie et al., 1989; Olivecrona and Bengtsson-Olivecrona, 1993). LPL was shown to be activated by apolipoprotein (apo) CII (Bengtsson and Olivecrona, 1979; Breckenridge et al., 1978; Havel et al., 1970; LaRosa et al., 1970), and several lines of evidence suggested that LPL is active as a homodimer (Garfinkel et al., 1983; Iverius and Ostlund-Lindqvist, 1976; Kobayashi et al., 2002; Lookene and Bengtsson-Olivecrona, 1993; Lookene et al., 1994; Olivecrona and Bengtsson-Olivecrona, 1990; Olivecrona et al., 1985; Osborne et al., 1985; Peterson et al., 2002; Vannier and Ailhaud, 1989; Wong et al., 1994; Wong et al., 1997). The cloning of the LPL cDNA in the 1980s (Enerbäck et al., 1987; Kirchgessner et al., 1987; Wion et al., 1987) triggered studies on LPL sequences required for substrate binding, catalytic activity, and heparin binding (Emmerich et al., 1992; Lookene et al., 1996; Lookene et al., 1997a; Lookene et al., 1997b; Ma et al., 1994; Sendak et al., 1998), and also opened the door to understanding LPL mutations underlying the familial chylomicronemia syndrome (Mailly et al., 1997; Peterson et al., 2002). Together, these efforts created an outline for intravascular lipoprotein processing—one that was taught to a generation of students and practicing physicians. The outline changed little over several decades, contributing to a perception, occasionally voiced at conferences during the 1990s, that the field of plasma triglyceride metabolism was approaching maturity.
For the scientists who were actively working on plasma triglyceride metabolism, any suggestion that the field was close to maturity would have made little sense. After all, no one understood how LPL, an enzyme that is secreted by adipocytes and myocytes, reached its site of action in the capillary lumen; the regulation of LPL activity was not understood; the structure of LPL remained elusive; and many cases of chylomicronemia, both genetic and acquired, were unexplained. Also, some features within the outline of triglyceride metabolism needed more investigation. For example, it was unclear why the LPL secreted by parenchymal cells would end up being attached to HSPGs in the lumen of blood vessels, given that HSPGs are abundant within the interstitial spaces around parenchymal cells. Also, the idea that LPL is only active as a homodimer needed more study. Finally, the mechanisms by which specific apolipoproteins affected the efficiency of triglyceride hydrolysis were incompletely understood.
Over the past decade, there has been considerable progress in understanding plasma triglyceride metabolism. Much of that progress has centered around GPIHBP1, a GPI-anchored protein of capillary endothelial cells (Beigneux et al., 2007). GPIHBP1 is a dedicated partner for LPL: it captures LPL from within the subendothelial spaces and shuttles it to the capillary lumen (Davies et al., 2012; Davies et al., 2010); it is essential for the margination of lipoproteins along capillaries, allowing LPL-mediated triglyceride processing to proceed (Goulbourne et al., 2014); and it stabilizes the structure and catalytic activity of LPL (Mysling et al., 2016a; Mysling et al., 2016b). Recently, GPIHBP1 proved to be crucial for solving the structure of LPL (Birrane et al., 2019; Horton, 2019; Arora et al., 2019). Here, we review recent insights into the functions of GPIHBP1 and LPL in intravascular lipolysis, including insights derived from the structures of those proteins.
GPIHBP1 and plasma triglyceride metabolism
GPIHBP1 was initially identified during a screen of a mouse liver cDNA library for clones that render CHO cells capable of binding HDL (Ioka et al., 2003). The screen uncovered SR-B1, long recognized as an HDL-binding protein (Acton et al., 1996), and GPIHBP1, a GPI-anchored Ly6/uPAR (LU) protein with a long stretch of negatively charged amino acids at its amino terminus (Ioka et al., 2003). To this day, it is unclear why GPIHBP1 appeared in that screen, given that Gin and colleagues found little evidence of HDL, apo-AI, or apo-E binding to GPIHBP1-expressing cells (Gin et al., 2011). Our best guess is that the HDL used in the initial screen contained small amounts of LPL (which binds to GPIHBP1 avidly) or was enriched in apo-AV (a “heparin-binding” apolipoprotein that has been reported to bind to the negatively charged domain at the amino terminus of GPIHBP1) (Gin et al., 2011; Gin et al., 2008).
The first clue that GPIHBP1 was important for plasma triglyceride metabolism was finding severe hypertriglyceridemia in Gpihbp1 knockout mice (Gpihbp1–/–) (Beigneux et al., 2007; Weinstein et al., 2010a; Weinstein et al., 2010b). That discovery, combined with the finding that GPIHBP1 binds LPL and is expressed by endothelial cells, suggested that GPIHBP1 could be the binding site for LPL on endothelial cells (i.e., the “platform” for LPL-mediated lipoprotein processing) (Beigneux et al., 2007).
Subsequent studies showed that GPIHBP1’s function is more substantial than simply serving as a binding site for LPL within capillaries. GPIHBP1 is a crucial partner for LPL in four ways (Figure 1). First, the GPIHBP1 on the abluminal surface of capillary endothelial cells is important for capturing LPL from within the subendothelial spaces (Davies et al., 2010; Kristensen et al., 2018). Second, GPIHBP1 binding stabilizes the structure and activity of LPL (Kristensen et al., 2018; Mysling et al., 2016a; Mysling et al., 2016b). Third, GPIHBP1 shuttles LPL across capillary endothelial cells to its site of action in the capillary lumen (Davies et al., 2010). Fourth, GPIHBP1-bound LPL is critical for the margination of lipoproteins in the bloodstream (Goulbourne et al., 2014), allowing LPL-mediated lipoprotein processing to proceed.
Figure 1. A cartoon representation of GPIHBP1’s functions in lipoprotein lipase (LPL)-mediated lipoprotein processing.

GPIHBP1 serves four important functions in plasma triglyceride processing. The first two are illustrated in the left panel. First, GPIHBP1 on the abluminal plasma membrane of capillary endothelial cells captures LPL (yellow) from heparin-sulfate proteoglycan (HSPG) binding sites (green tree-like structures) within the subendothelial spaces (Davies et al., 2010; Kristensen et al., 2018). GPIHBP1’s intrinsically disordered acidic domain (green), which projects from GPIHBP1’s three-fingered LU domain (blue), is important for capturing LPL and bringing it into high-affinity interactions with GPIHBP1’s LU domain (Kristensen et al., 2018). Second, GPIHBP1 stabilizes the structure of LPL and protects it from unfolding, even in the face of physiologic inhibitor proteins such as ANGPTL4 (Kristensen et al., 2018; Mysling et al., 2016a; Mysling et al., 2016b). The preservation of LPL structure is illustrated by changing the color of LPL from yellow to purple. The middle panel depicts the third function of GPIHBP1 in LPL biology, which is to transport LPL to its site of action in the capillary lumen (Davies et al., 2010). The movement of GPIHBP1 across endothelial cells is bidirectional (Davies et al., 2012); thus, GPIHBP1 is able to return to the abluminal surface of endothelial cells, pick up more LPL, and then replenish LPL stores along the capillary lumen. The right panel depicts the fourth function of GPIHBP1, which is to mediate the margination of triglyceride-rich lipoproteins (TRLs) along the luminal surface of capillaries. GPIHBP1-bound LPL is required for TRL margination (Goulbourne et al., 2014), allowing triglyceride hydrolysis to proceed.
GPIHBP1’s function in transporting LPL across capillaries was evident from the distribution of LPL in the tissues of wild-type and Gpihbp1–/– mice (Davies et al., 2010). In wild-type mice, most of the LPL in tissues is bound to GPIHBP1 on capillary endothelial cells (Figure 2A), where it is distributed evenly between the luminal and abluminal plasma membranes (Davies et al., 2012; Davies et al., 2010). In Gpihbp1–/– mice, LPL never reaches the capillary lumen (Davies et al., 2010). Instead, the LPL in Gpihbp1–/– mice remains stranded within the interstitial spaces, presumably bound to HSPGs in close proximity to the surface of cells (both parenchymal cells and capillary endothelial cells) (Davies et al., 2010) (Figure 2A).
Figure 2. Localization of GPIHBP1 and lipoprotein lipase (LPL) in brown adipose tissue (BAT).
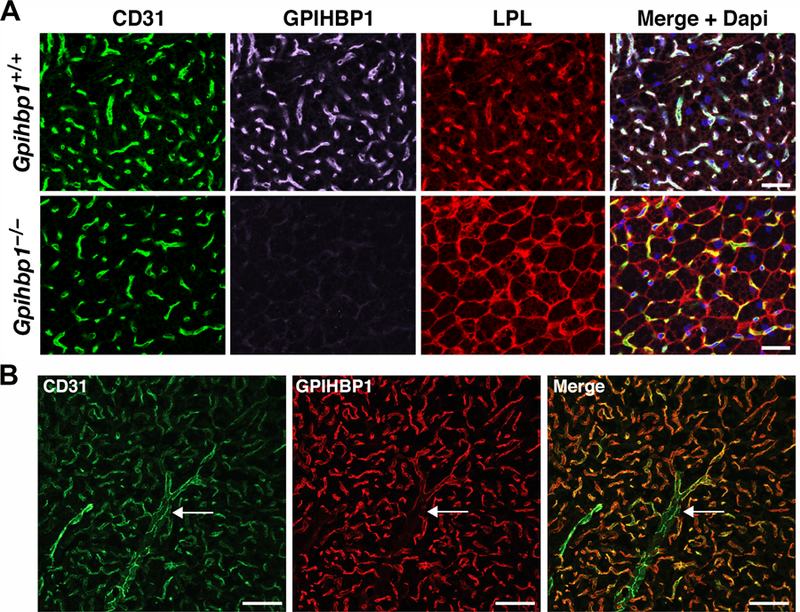
(A) Mislocalization of LPL in the BAT of Gpihbp1–/– mice. Sections of BAT from a Gpihbp1+/+ and Gpihbp1–/– mouse were stained with antibodies against CD31 (green), GPIHBP1 (magenta), and LPL (red), and imaged by confocal fluorescence microscopy. DNA was stained with DAPI (blue). Scale bar, 50 µm. (B) GPIHBP1 is expressed in endothelial cells of capillaries, but not in endothelial cells of larger blood vessels. Sections of BAT were stained with antibodies against CD31 (green) and GPIHBP1 (red). GPIHBP1 expression in endothelial cells decreases as capillaries merge into a venule (arrow). Scale bar, 50 µm.
By immunohistochemistry, GPIHBP1 is expressed solely in capillary endothelial cells and is absent in endothelial cells of venules, arterioles, and larger blood vessels (Beigneux et al., 2007; Davies et al., 2010) (Figure 2B). GPIHBP1 is found in capillaries of all peripheral tissues, with particularly high levels in the heart and adipose tissue (where LPL-mediated processing of lipoproteins is robust), but it is absent from capillaries of the brain (Beigneux, 2010; Beigneux et al., 2007; Davies et al., 2010; Olafsen et al., 2010), which uses glucose for fuel (Mergenthaler et al., 2013). Consistent with immunohistochemistry studies, Gpihbp1 transcripts are absent from capillary endothelial cells of the brain, as judged by single-cell RNA-seq studies (He et al., 2018b; Vanlandewijck et al., 2018). Interestingly, high levels of GPIHBP1 expression are observed in capillaries of the lung, a tissue where LPL expression is low (Olafsen et al., 2010). GPIHBP1 in lung capillaries appears to play a role in scavenging LPL that escapes into the plasma compartment (Goulbourne et al., 2014), but the physiologic importance of that role is unclear. GPIHBP1 deficiency does not appear to affect pulmonary function.
Why GPIHBP1 expression is restricted to the endothelial cells of the capillaries of peripheral tissues and why GPIHBP1 is expressed at higher levels in some tissues than others are poorly understood. An enhancer element was recently identified ~3.6 kb upstream of exon 1 of Gpihbp1 (Allan et al., 2018). Deleting that enhancer reduced Gpihbp1 transcripts in heart and brown adipose tissue by ~50% but nearly eliminated transcripts in liver and kidney (Allan et al., 2018).
By immunohistochemistry, the tissue distribution of LPL in peripheral tissues mirrors that of GPIHBP1. Even though LPL is synthesized and secreted by parenchymal cells, the vast majority of the LPL in tissues is bound to GPIHBP1 on capillary endothelial cells (Beigneux et al., 2007; Davies et al., 2010).
GPIHBP1’s transport function was established by studies showing that GPIHBP1 transports a GPIHBP1-specific monoclonal antibody from one side of an endothelial cell to the other (Davies et al., 2010). The movement of GPIHBP1 across endothelial cells is bidirectional (Davies et al., 2012). When wild-type mice are injected intravenously with a fluorescently labeled GPIHBP1-specific monoclonal antibody, the antibody initially binds to GPIHBP1 within the capillary lumen but then quickly appears on the abluminal surface of capillaries. Conversely, when the antibody is injected into tissues, it first associates with GPIHBP1 on the outside of capillaries but then quickly appears in the capillary lumen. Antibody transport did not occur in Gpihbp1–/– mice. GPIHBP1 appears to be a long-lived protein (Olafsen et al., 2010), implying that GPIHBP1 could make multiple trips across endothelial cells, with each trip providing an opportunity to capture LPL and bring it to the capillary lumen.
The intravascular processing of triglyceride-rich lipoproteins by LPL requires that the lipoproteins in the bloodstream stop (marginate) along capillaries. For years, lipoprotein margination was assumed to involve electrostatic interactions between positively charged apolipoproteins on lipoproteins and the negatively charged HSPGs that line the luminal surface of endothelial cells (de Beer et al., 1999; Lookene et al., 1997b; Wang and Eckel, 2009). More recent studies revealed that lipoprotein margination is utterly dependent on GPIHBP1—and more specifically on the GPIHBP1–LPL complex (Goulbourne et al., 2014). GPIHBP1 expression alone is not sufficient for lipoprotein margination along capillaries; lipoprotein margination is negligible in lung capillaries, where GPIHBP1 is abundant but LPL expression is low. However, when the GPIHBP1 in lung capillaries was artificially loaded with exogenous bovine LPL, lipoprotein margination along lung capillaries was robust (Goulbourne et al., 2014).
Fluorescently labeled lipoproteins, when injected intravenously into wild-type mice, rapidly marginate along capillaries of the heart, co-localizing with LPL and GPIHBP1. In contrast, lipoprotein margination is negligible along capillaries in hearts of Gpihbp1–/– mice, which are devoid of GPIHBP1–LPL complexes (Goulbourne et al., 2014). In vitro studies indicated that lipoprotein binding to the GPIHBP1–LPL complex depends on a tryptophan-rich loop within the carboxyl-terminal domain of LPL (Goulbourne et al., 2014).
Because the GPIHBP1–LPL complex is anchored to the plasma membrane of endothelial cells and because the luminal surface of endothelial cells is covered by an HSPG-rich glycocalyx, it was initially unclear how the GPIHBP1–LPL complex would be able to participate in lipoprotein margination. However, electron microscopy (EM) studies revealed that the glycocalyx on the surface of capillaries is patchy, with tufts of glycocalyx interrupted by “meadows” where the plasma membrane is exposed (Goulbourne et al., 2014). EM tomography suggested that lipoproteins bind to capillaries within the gaps in the glycocalyx (Goulbourne et al., 2014).
Relevance of GPIHBP1 expression to uptake of lipoprotein-derived nutrients by tissues
The turnover of triglyceride-rich lipoproteins in the bloodstream is quite rapid (Havel, 2010). Recent NanoSIMS imaging studies revealed that the movement of the fatty acid products of lipolysis across endothelial cells and into surrounding parenchymal cells is also rapid (He et al., 2018a). After administering [2H]triglyceride-enriched lipoproteins (2H-TRLs) to wild-type mice by an intravenous injection, it was possible to visualize the margination of 2H-TRLs in heart capillaries as early as 30 sec after the injection (He et al., 2018a). As early as 2 min after the injection, substantial amounts of deuterium had already entered the mitochondria and cytosolic lipid droplets of cardiomyocytes (He et al., 2018a) (Figure 3A). Interestingly, deuterium enrichment in capillary endothelial cells and cardiomyocytes was similar (i.e., there was little evidence that endothelial cells play a gatekeeper role in the movement of fatty acids into cardiomyocytes). When isolated hearts from wild-type mice were perfused with 2H-TRLs, similar findings were observed—rapid margination of 2H-TRLs along capillaries and rapid uptake of fatty acids into cytosolic lipid droplets (He et al., 2018a). When isolated hearts from Gpihbp1–/– mice were perfused with 2H-TRLs, the margination of 2H-TRLs was absent and 2H enrichment in the cytosolic triglyceride droplets of cardiomyocytes was markedly reduced (He et al., 2018a).
Figure 3. NanoSIMS imaging to examine intravascular lipolysis.
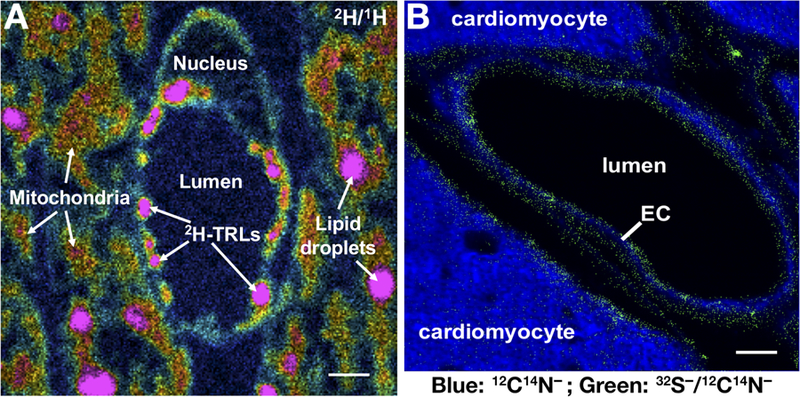
(A) 2H/1H ratio image of a capillary in a mouse heart 2 min after an intravenous injection of 2H-TRLs (triglyceride-rich lipoproteins enriched in 2H-triglycerides). The 2H/1H image shows 2H-TRLs that have marginated along the luminal surface of capillary endothelial cells and entry of 2H-enriched lipids into endothelial cells as well as the mitochondria and lipid droplets of cardiomyocytes. The 2H/1H ratio is multiplied by 10,000; the range spans from slightly higher than 2H natural abundance to approximately six times natural abundance. Scale bar, 1 µm. (B) Composite 12C14N− and 32S− /12C14N− images of mouse heart capillaries. The signal from 12C14N− secondary ions (blue), reflecting 14N distribution, was robust in both capillary endothelial cells (EC) and cardiomyocytes. The 32S− /12C14N− ratio (green) was set from 0.05 to 0.06. Pixels with a 32S− /12C14N− ratio above 0.05 are shown, revealing features rich in 32S, relative to 14N, on the surface of endothelial cells and cardiomyocytes. Scale bar, 1 µm.
Properties of GPIHBP1 required for LPL binding
Mature GPIHBP1 contains two key domains, a disordered amino-terminal acidic domain and an LU domain containing 10 cysteines, all arranged in a characteristic spacing pattern and all disulfide bonded (Beigneux et al., 2011; Mysling et al., 2016a). The five disulfide bonds stabilize the assembly of three loops and form the hallmark three-fingered scaffold of the LU domain (Leth et al., 2019). The acidic domain of human GPIHBP1 is impressive, with 21 acidic amino acids (glutamates or aspartates) in 26 consecutive residues. A sulfated tyrosine, located in the middle of the acidic domain (Kristensen et al., 2018), adds to the negative charge. The GPIHBP1 in every mammalian species contains an acidic domain, but the precise sequence and length of the domain is variable. For example, the acidic domain in opossum GPIHBP1 contains 32 aspartates or glutamates in 39 consecutive residues, including a stretch of 23 consecutive aspartates (Fong et al., 2016). Like all GPI-anchored proteins, GPIHBP1 contains a carboxyl-terminal hydrophobic signal peptide that triggers the addition of a GPI anchor within the endoplasmic reticulum (Ferguson et al., 2009).
GPIHBP1’s LU domain is required for the stability of LPL binding, contributing to a long half-life for the GPIHBP1–LPL complex (t½ ~ 35 s). When GPIHBP1’s LU domain is replaced with an LU domain from another member of the LU family (Gin et al., 2008) or when any of the 10 cysteines in GPIHBP1’s LU domain is mutated (Beigneux et al., 2009b), stable interactions with LPL are abolished. Aside from the 10 cysteines, alanine-scanning mutagenesis of GPIHBP1’s LU domain uncovered 12 additional residues required for LPL binding, with the majority located in conserved segments within the second finger of the LU domain (amino acids 89–110) (Beigneux et al., 2011). Many mutations in GPIHBP1’s LU domain interfered with disulfide bonding and promoted the formation of disulfide-linked GPIHBP1 dimers and multimers (Beigneux et al., 2015; Plengpanich et al., 2014), but there was one noteworthy exception. Mutations in GPIHBP1 Trp-109 abolished LPL binding without interfering with disulfide bonding or promoting protein multimerization, suggesting that Trp-109 plays a direct role in the GPIHBP1–LPL binding interface (Beigneux et al., 2015). Consistent with that idea, hydrogen–deuterium exchange mass spectrometry (HDX-MS) studies showed that the binding of LPL to GPIHBP1 protects GPIHBP1 residues 104–128 from solvent exposure and reduces deuterium uptake into GPIHBP1 (Mysling et al., 2016a).
Studies by Gin and coworkers (Gin et al., 2012) revealed that GPIHBP1 binds to the carboxyl-terminal domain of LPL (residues 325–475). Consistent with those findings, an LPL-specific monoclonal antibody with an epitope within the carboxyl-terminal domain of LPL, 88B8, blocks LPL binding to GPIHBP1 (Allan et al., 2016). Also, two different missense mutations within the carboxyl-terminal domain of LPL, C445Y and E448K, first identified in patients with severe hypertriglyceridemia (Henderson et al., 1998; Henderson et al., 1996), abolish LPL binding to GPIHBP1 (Voss et al., 2011). Finally, HDX-MS studies revealed that when GPIHBP1 is bound to LPL, residues 429–446 in LPL’s carboxyl-terminal domain are protected from solvent exposure and deuterium uptake, implying that those residues are involved in GPIHBP1 binding (Mysling et al., 2016a).
Deciphering functions for GPIHBP1’s intrinsically disordered acidic domain
To explore the contributions of GPIHBP1’s LU and acidic domains to the formation of the GPIHBP1–LPL complex, the kinetics of LPL binding to full-length GPIHBP1 and “Δacidic-GPIHBP1” (a GPIHBP1 mutant lacking the amino-termin al acidic domain) were analyzed by surface plasmon resonance (SPR). In these studies, the two different GPIHBP1 proteins were passed over a sensor chip coated with LPL (which had been tethered to the sensor chip with the LPL-specific monoclonal antibody 5D2) (Kristensen et al., 2018; Mysling et al., 2016a). Both full-length GPIHBP1 and Δacidic-GPIHBP1 bound LPL with nanomolar affinities, but the binding of full-length GPIHBP1 was stronger. Interestingly, the stability of the GPIHBP1–LPL complex, as judged by the dissociation rate constant (koff), was nearly identical for full-length GPIHBP1 and Δacidic-GPIHBP1 (2 × 10−2 s−1) (Mysling et al., 2016a). However, the association rate constants (kon) for Δacidic-GPIHBP1 (0.1 × 106 M−1 s−1) and full-length GPIHBP1 (2.6 × 107 M−1 s−1) were very different, highlighting a key role for GPIHBP1’s acidic domain in accelerating the encounter rate and promoting the formation of the GPIHBP1–LPL complex (Kristensen et al., 2018). Furthermore, the kon for full-length GPIHBP1–LPL binding was sensitive to the ionic strength of the buffer and was as high as 3 × 108 M−1 s−1 under physiological conditions, underscoring the importance of electrostatic steering in the initial interaction between LPL and full-length GPIHBP1 (Kristensen et al., 2018). Under physiologic conditions, GPIHBP1’s disordered acidic domain increases the velocity of LPL binding by >2500-fold. These observations are likely relevant to the ability of GPIHBP1 to capture LPL from within the interstitial spaces along the basolateral surface of capillaries.
Kristensen and coworkers recently investigated GPIHBP1–LPL interactions in more detail (Kristensen et al., 2018). They established that the stoichiometry of GPIHBP1 binding to LPL is 1:1 (Kristensen et al., 2018). Remarkably, a peptide corresponding to GPIHBP1’s acidic domain also bound to LPL with 1:1 stoichiometry. They also found, by SPR, that the sulfated tyrosine (Tyr-38) in GPIHBP1’s acidic domain contributes to the strength of LPL binding. When Tyr-38 was replaced with Phe, the KD for GPIHBP1–LPL binding increased threefold (Kristensen et al., 2018). Finally, they documented, by SPR, large differences in the abilities of full-length GPIHBP1 and Δacidic-GPIHBP1 to dislodge LPL when it was bound to a sensor chip containing immobilized short-chain heparin moieties. Full-length GPIHBP1 dislodged heparin-bound LPL efficiently, whereas Δacidic-GPIHBP1 did not (despite the fact that Δacidic-GPIHBP1 was able to bind to the LPL) (Kristensen et al., 2018).
The fact that full-length GPIHBP1 effectively captures LPL from heparin is probably relevant to LPL trafficking to capillaries. As noted earlier, the LPL in Gpihbp1–/– mice is stranded within the interstitial spaces, bound to HSPGs near the surface of cells (Davies et al., 2010). The interactions between LPL and HSPGs within the interstitium are transient (i.e., with a high koff), but the abundance of HSPG binding sites means that LPL in Gpihbp1–/– tissues is retained within the interstitium rather than escaping into the lymphatic circulation. The same interstitial HSPG binding sites exist in wild-type mice; however, the dynamic nature of LPL–HSPG interactions allows the LPL to move to more stable GPIHBP1 binding sites on capillaries. The ability of GPIHBP1 to recruit LPL away from interstitial HSPG binding sites has been documented experimentally. When GPIHBP1-coated agarose beads were implanted into the brown adipose tissue of Gpihbp1–/– mice, HSPG-bound LPL within the interstitial spaces moved to the GPIHBP1-coated beads, depleting LPL stores within the interstitium (Allan et al., 2017b).
Recent immunohistochemical studies on LPL localization in tissues of Gpihbp1–/– mice yielded further insights into the trafficking of LPL within the interstitium (Kristensen et al., 2018). As expected, the LPL in the heart and skeletal muscle of Gpihbp1–/– mice was mislocalized to the interstitium. Interestingly, however, the amount of LPL adjacent to endothelial cells was greater than the amount adjacent to myocytes (Kristensen et al., 2018). Thus, even in the absence of GPIHBP1, the LPL within the interstitium appears to associate preferentially with HSPGs on the outside of capillary endothelial cells. We propose that “directed diffusion” (Duchesne et al., 2012) along an HSPG electrostatic gradient is responsible for the flow of LPL from the HSPGs near myocytes to HSPGs on the outer surface of endothelial cells. The same directional LPL movement presumably exists in tissues of wild-type mice, generating a pool of LPL for capture by GPIHBP1 on endothelial cells. Small-angle X-ray scattering (SAXS) studies showed that GPIHBP1’s disordered acidic domain, as it projects from the LU domain, occupies a large mushroom-shaped space with a diameter of 112 Å (Kristensen et al., 2018). In tissues, GPIHBP1’s acidic domain likely projects at least 100 Å from the basolateral plasma membrane of endothelial cells and probably functions in a fly-casting–like mechanism (Shoemaker et al., 2000) to engage the LPL that is bound in a dynamic fashion to HSPGs and drive it into a stable complex with GPIHBP1’s LU domain.
The explanation for the preferential association of interstitial LPL with endothelial cells in Gpihbp1–/– mice is unknown, but the existence of HSPGs on endothelial cells is well documented. Collagen XVIII, a basement membrane HSPG, is found on capillary endothelial cells, as judged by immunohistochemistry (Bishop et al., 2010). Also, NanoSIMS analyses of 32S distribution in tissues appear consistent with the existence of HSPGs adjacent to cardiomyocytes and endothelial cells (Figure 3B). 32S is found in methionines and cysteines of all cellular proteins, but the amount of 32S (relative to 14N) is greater within the interstitial spaces near the surface of cardiomyocytes and endothelial cells than in the 14N-rich cytoplasm of those cells. Larger amounts of 32S, relative to 14N, are also observed in the capillary lumen, likely due to HSPGs in the glycocalyx of endothelial cells (Figure 3B).
GPIHBP1 stabilizes the structure and activity of LPL, even in the presence of endogenous LPL inhibitors
For years, biochemists and physiologists have noted that catalytic activity disappears rapidly when purified preparations of LPL are incubated at room temperature. HDX-MS studies by Mysling and coworkers (Mysling et al., 2016a) provided a likely explanation. When LPL is incubated at 25°C with deuterated water (2H2O), deuterium uptake into specific regions of LPL results in bimodal isotope envelopes, reflecting progressive unfolding of those regions (Figure 4A). Bimodal isotope envelopes were observed in peptides corresponding to LPL’s amino-terminal hydrolase domain, including in a peptide spanning LPL’s catalytic triad (Figure 4B), but they were nearly absent in peptides from LPL’s carboxyl-terminal domain, which has a more stable conformation. The appearance of bimodal deuterium signatures in the amino-terminal domain was accompanied by a parallel fall in LPL catalytic activity (Mysling et al., 2016a). The binding of GPIHBP1 to LPL markedly reduced both the unfolding of LPL’s amino-terminal domain and the loss of catalytic activity, demonstrating that GPIHBP1 preserves the structural integrity and catalytic activity of LPL. Interestingly, the protective effect of GPIHBP1 was largely due to GPIHBP1’s intrinsically disordered acidic domain (Mysling et al., 2016a). First, the preservation of LPL structure and catalytic activity was much greater with full-length GPIHBP1 than with Δacidic-GPIHBP1 (Mysling et al., 2016a). Second, a synthetic peptide corresponding to GPIHBP1’s acidic domain reduced both LPL unfolding and the loss of triglyceride hydrolase activity.
Figure 4. Inherent instability of LPL, as judged by hydrogen–deuterium exchange–mass spectrometry (HDX-MS) studies.
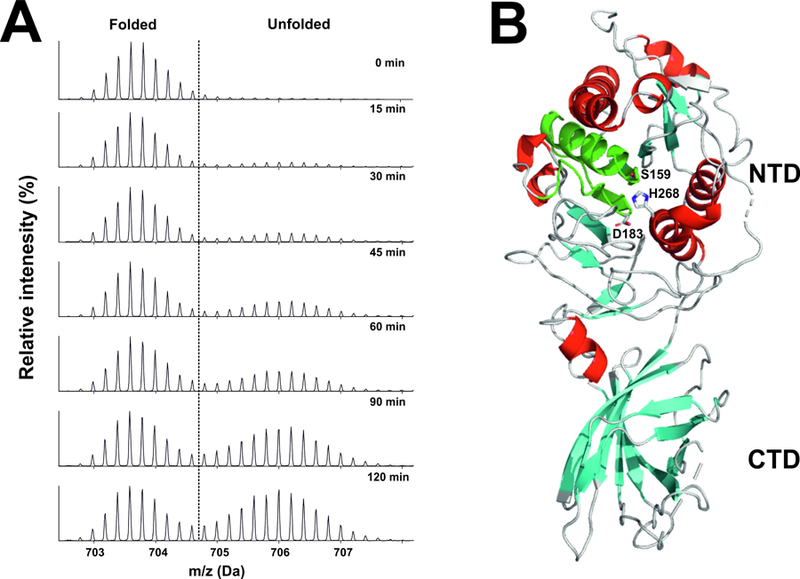
Bovine LPL was incubated at 25°C in deuterium oxide . Panel A shows the emergence of a bimodal isotope envelope of deuterium uptake in a peptic peptide covering the catalytic triad of LPL [from (Mysling et al., 2016a)]. Bimodality of the isotope envelope is a hallmark of protein unfolding (Mysling et al., 2016a; Mysling et al., 2016b). Substantial portions of the amino-terminal catalytic domain of LPL (NTD) exhibited the bimodal isotope envelope, while peptides from the carboxyl-terminal lipid-binding domain (CTD) exhibit a unimodal exchange pattern, reflecting greater stability. Panel B illustrates the position of the catalytic triad peptic peptide (green) in the crystal structure of human LPL [modified from the crystal structure by Birrane and coworkers (Birrane et al., 2019)]. The structure of human LPL is shown as a cartoon representation, with α-helices in red and β-strands in cyan. The location of the three catalytic triad residues (S159, D183, H268) are noted. The structure was visualized with PyMol (Schrödinger) u sing PDB coordinates 6E7K.
The spontaneous unfolding of LPL, as judged by HDX-MS studies, is more than an in vitro biochemical curiosity. In follow-up studies, Mysling and coworkers (Mysling et al., 2016b) showed that ANGPTL4, a physiologic inhibitor of LPL, inactivates LPL by catalyzing LPL unfolding. Spontaneous unfolding of LPL, as judged by HDX-MS, is relatively slow, with ~7% unfolding after 5 min at 25°C and 29% unfolding after 30 min. In contrast, incubating LPL with sub-stoichiometric amounts of ANGPTL4 (a 10:1 LPL to ANGPTL4 molar ratio) resulted in ~70% unfolding of LPL in 5 min. Mysling and coworkers also tested the capacity of an ANGPTL4 polymorphic variant, ANGPTL4E40K, to catalyze LPL unfolding (Mysling et al., 2016b). The ANGPTL4E40K variant, present in 3% of Caucasians, is associated with lower plasma triglyceride levels and reduced risk of coronary disease (Dewey et al., 2016; Helgadottir et al., 2016; Investigators et al., 2016). The capacity of ANGPTL4E40K to catalyze LPL unfolding was ~60% lower than that of wild-type ANGPTL4. Consistent with those findings, LPL incubated with ANGPTL4E40K retained 40–50% more catalytic activity than LPL incubated with wild-type ANGPTL4. The reduced capacity of ANGPTL4E40K to inactivate LPL [which is almost certainly due to instability of a functionally important α-helix in ANGPTL4 (Mysling et al., 2016b)], presumably explains the reduced plasma triglyceride levels in ANGPTL4E40K carriers.
The fact that LPL activity is regulated by an inhibitory protein (i.e., ANGPTL4) that promotes unfolding is, to the best of our knowledge, a novel mechanism for controlling the activity of an enzyme. Mysling and colleagues (Mysling et al., 2016b) proposed that ANGPTL4 interacts transiently with LPL and promotes an unstable conformation that leads inexorably to enzyme inactivation. Another group reported that inactivation of LPL by ANGPTL4 is reversible when the inactivation of LPL is performed in the presence of deoxycholate (Gutgsell et al., 2019; Lafferty et al., 2013), a detergent that stabilizes LPL activity (Bengtsson and Olivecrona, 1979). The significance of this finding is unclear, in part because the relevance of deoxycholate-stabilized LPL to the normal physiology of intravascular lipolysis is open to question. Also, the LPL activity in those studies was assessed with a soluble substrate rather than with an emulsified triglyceride substrate. Finally, the ANGPTL4 proved to be a relatively inefficient inhibitor under the conditions that were studied, with a Ki of 0.86–1.7 µM (Gutgsell et al., 2019; Lafferty et al., 2013).
In the studies by Mysling and coworkers, full-length GPIHBP1 protected LPL from ANGPTL4-mediated unfolding, but there was little protection by Δacidic-GPIHBP1 or an acidic domain peptide (Mysling et al., 2016b). Of note, the inactivation of LPL by ANGPTL4 was not reversible; the loss of LPL activity with ANGPTL4 could not be reversed by adding GPIHBP1 to the incubation mixture (Mysling et al., 2016b). Mysling and coworkers proposed that the anchoring of GPIHBP1’s LU domain to LPL results in optimal positioning of GPIHBP1’s acidic domain, allowing it to stabilize LPL structure and activity (Mysling et al., 2016b). From the standpoint of plasma triglyceride physiology, the ability of GPIHBP1 to preserve LPL structure and activity (even in the presence of inhibitory proteins) probably serves to focus catalytically active LPL where it needs to be for lipoprotein processing (i.e., in the capillary lumen). It seems likely that at least some of the regulation of LPL by ANGPTL4 occurs before LPL reaches the capillary lumen. Kersten and collaborators showed that ANGPTL4 can inactivate LPL in the secretory pathway of adipocytes (Dijk et al., 2016). Also, the LPL in the interstitial spaces, as it moves towards endothelial cells, could be susceptible to inhibition by ANGPTL4. Nilsson and coworkers suggested that the interstitial spaces may be a site for ANGPTL4-mediated regulation of LPL (Nilsson et al., 2012).
ANGPTL3, another physiologic inhibitor of LPL, also catalyzes LPL unfolding, but it is less potent than ANGPTL4—at least under the experimental conditions that were tested (Mysling et al., 2016b). ANGPTL3-catalyzed LPL unfolding is also inhibited by the binding of GPIHBP1. In the case of ANGPTL3, several studies have demonstrated that the functional inhibitory unit is actually a complex of ANGPTL3 and ANGPTL8 (Chi et al., 2015; Haller et al., 2017; Quagliarini et al., 2012) and that this complex is as efficient as ANGPTL4 in inactivating LPL (Kovrov et al., 2019).
Familial chylomicronemia from GPIHBP1 mutations
Soon after the discovery of chylomicronemia in Gpihbp1–/– mice, GPIHBP1 mutations were identified in patients with familial chylomicronemia syndrome (Ariza et al., 2016; Beigneux et al., 2009a; Charriere et al., 2011; Coca-Prieto et al., 2011; Franssen et al., 2010; Gonzaga-Jauregui et al., 2014; Olivecrona et al., 2010; Plengpanich et al., 2014; Rabacchi et al., 2016; Rios et al., 2012; Yamamoto et al., 2013). The chylomicronemia that results from GPIHBP1 mutations presents in infancy (Rios et al., 2012) and persists lifelong, with plasma triglycerides often exceeding 2000 mg/dl. Two mutant GPIHBP1 alleles are required for disease; the plasma triglyceride levels in heterozygotes are normal. A variety of different GPIHBP1 mutations have been uncovered but the majority have involved one of the cysteines in the LU domain (e.g., C65Y, C65S, C68Y, C68G, C68R, C83R, C89F) or a residue in close proximity to a cysteine (e.g., Q115P, T111P). In one kindred, chylomicronemia was caused by the introduction of an unpaired cysteine into the LU domain (S107C) (Plengpanich et al., 2014). Chylomicronemia was also observed in the setting of a T80K mutation, which interferes with N-linked glycosylation and trafficking of GPIHBP1 to the plasma membrane (Ariza et al., 2016). A G175R substitution has been observed in several patients with hypertriglyceridemia (Beigneux et al., 2017; Charriere et al., 2011; Chyzhyk et al., 2019) but is unlikely to be causative. G175 is located in the signal peptide that is normally replaced by the GPI anchor (i.e., it is not present in mature GPIHBP1). The G175R substitution has no significant effect on the addition of the GPI anchor (Beigneux et al., 2017). No one has yet uncovered a GPIHBP1 mutation that results in the deletion of all or part of GPIHBP1’s acidic domain.
Low levels of LPL in the pre- and post-heparin plasma are a hallmark of GPIHBP1 deficiency and presumably reflect reduced amounts of LPL within capillaries (Beigneux et al., 2009a; Olivecrona et al., 2010; Plengpanich et al., 2014). Low levels of GPIHBP1 in the plasma are another hallmark (Beigneux et al., 2017). Heterozygous carriers of GPIHBP1 mutations have ~half-normal plasma levels of GPIHBP1 (Beigneux et al., 2017; Miyashita et al., 2018b).
GPIHBP1 missense mutations causing chylomicronemia abolish the capacity of GPIHBP1 to bind LPL (Franssen et al., 2010; Olivecrona et al., 2010; Plengpanich et al., 2014). In cell transfection studies, missense mutations involving cysteine residues (e.g., C65Y, C65S, C68G) interfere with disulfide bonding and result in the appearance of disulfide-linked GPIHBP1 dimers and multimers on the surface of cells (Beigneux et al., 2015). The GPIHBP1 dimers and multimers have no capacity to bind LPL (Beigneux et al., 2015; Plengpanich et al., 2014). The fact that dimeric and multimeric GPIHBP1 proteins were able to reach the surface of the transfected cells was somewhat surprising, given that a cysteine mutation in CD59 (another LU protein) abolished CD59 trafficking to the plasma membrane (Nevo et al., 2013). Given that observation, it seemed possible that the appearance of GPIHBP1 dimers and multimers on the surface of transfected cells was simply an artifact of protein overexpression. To explore that possibility, Allan and coworkers (Allan et al., 2017a) created knock-in mice harboring a cysteine substitution in the LU domain [C63Y, corresponding to a mutation observed in humans (Franssen et al., 2010)]. Homozygotes exhibited severe chylomicronemia, even on a low-fat chow diet. GPIHBP1-C63Y reached the surface of endothelial cells and could be easily detectable by immunohistochemistry, but the amount was reduced by 70% (Allan et al., 2017a). No LPL was bound to GPIHBP1-C63Y in the capillary lumen, demonstrating that the mutant protein had no capacity to bind LPL. Interestingly, most of the GPIHBP1-C63Y on the surface of endothelial cells in vivo was monomeric; only small amounts of disulfide-linked dimers were detected. Thus, the capillary endothelial cells in vivo appear to be more efficient than transfected CHO cells in removing misfolded GPIHBP1 proteins (including dimers and multimers), thereby limiting the amounts of GPIHBP1 that reach the surface of cells.
Chylomicronemia from GPIHBP1 autoantibodies
Chylomicronemia occasionally appears spontaneously, and some of these “acquired” cases are caused by GPIHBP1 autoantibodies (“GPIHBP1 autoanti body syndrome”) (Beigneux et al., 2017; Eguchi et al., 2019; Hu et al., 2017a; Kersten, 2017). This disorder was discovered fortuitously while characterizing a monoclonal antibody–based ELISA designed to measure plasma levels of GPIHBP1 (Beigneux et al., 2017; Miyashita et al., 2018a). In those studies, two plasma samples, both from patients with chylomicronemia, had abnormally low GPIHBP1 levels, and the GPIHBP1 levels remained low even after “spiking” the plasma samples with recombinant GPIHBP1. The latter finding strongly suggested “immunoassay interference,” which can be caused by autoantibodies. Additional studies revealed that the plasma from the two chylomicronemia patients did indeed contain GPIHBP1 autoantibodies (Beigneux et al., 2017). Of note, the autoantibodies abolished the capacity of GPIHBP1 to bind LPL. Patients with the GPIHBP1 autoantibody syndrome invariably have low plasma levels of LPL, consistent with the inability of GPIHBP1 to bind LPL and transport it to the capillary lumen (Beigneux et al., 2017). Subsequent studies revealed that GPIHBP1 autoantibodies are directed against GPIHBP1’s LU domain, not the acidic domain (Kristensen et al., 2018).
The plasma triglyceride levels in patients with the GPIHBP1 autoantibody syndrome are extremely high, typically exceeding 1500 mg/dl, and many of the patients had experienced an episode of acute pancreatitis. In some cases, GPIHBP1 autoantibodies were the sole manifestation of autoimmune disease, but the majority of patients had clinical or serological evidence of an autoimmune disease (e.g., systemic lupus erythematosus; SLE) (Beigneux et al., 2017). One patient with SLE and GPIHBP1 autoantibodies became pregnant and delivered a baby girl. The newborn infant manifested severe chylomicronemia that resolved after several weeks, presumably coinciding with disappearance of maternal GPIHBP1 autoantibodies (Beigneux et al., 2017). In another patient with autoimmune thyroid disease and multiple sclerosis (Kawahara et al., 2017), GPIHBP1 autoantibodies appeared after initiating β-interferon treatment (Eguchi et al., 2019). [β-interferon can, in some patients, cause worsening of autoimmune diseases (Banchereau and Pascual, 2006; Di Domizio and Cao, 2013; Silva, 2012).] In that patient, the GPIHBP1 autoantibodies disappeared and the plasma triglycerides normalized after β-interferon therapy was discontinued (Eguchi et al., 2019).
In the patient treated with β-interferon, the GPIHBP1 autoantibodies were largely subclass IgG4 (Eguchi et al., 2019). IgG4 autoantibodies have been reported to cause a variety of autoimmune diseases, typically by disrupting protein–protein interactions rather than by fixing complement and inducing tissue injury (Huijbers et al., 2018; Koneczny, 2018). Whether most patients with the GPIHBP1 autoantibody syndrome have IgG4 autoantibodies remains to be determined. It will also be important to determine if immune complexes are present in the plasma of affected patients. The presence of immune complexes could have been overlooked by the ELISAs used to detect GPIHBP1 autoantibodies (Beigneux et al., 2017; Miyashita et al., 2018a).
The frequency of the GPIHBP1 autoantibody syndrome among patients with acquired forms of chylomicronemia is unclear, but our experience, while limited, suggests that the syndrome may not be particularly rare (Beigneux et al., 2017). In our experience, if any GPIHBP1 autoantibodies can be detected, the patient invariably has severe chylomicronemia. We have no evidence that GPIHBP1 autoantibodies explain cases of mild-to-moderate hypertriglyceridemia. In one patient, the chylomicronemia resulting from GPIHBP1 autoantibodies resolved spontaneously (Hu et al., 2017a). In two cases, the chylomicronemia resolved after initiating immunosuppressive treatment, and in one of those cases, the normalization of triglyceride levels was accompanied by the disappearance of GPIHBP1 autoantibodies (Beigneux et al., 2017). More experience is needed to establish the natural history and treatment strategies for the GPIHBP1 autoantibody syndrome.
A crystal structure for the GPIHBP1–LPL complex
The structure of LPL remained elusive for decades, forcing investigators to rely on a homology model (Kobayashi et al., 2002) derived from the structure of pancreatic lipase, a distantly related lipase family member (Winkler et al., 1990). The absence of a structure for LPL was not for lack of trying; several laboratories attempted to generate LPL crystals but none succeeded, likely because of the propensity of LPL to unfold (Mysling et al., 2016a). Inspired by the discovery that GPIHBP1 stabilizes LPL structure and activity, Birrane and coworkers solved the crystal structure of a GPIHBP1–LPL complex at 2.8 Å resolut ion (Birrane et al., 2019; Horton, 2019).
The crystallographic unit contained two LPL–GPIHBP1 complexes, with the two LPL molecules arranged in a head-to-tail orientation, with the Trp-rich loop in LPL’s carboxyl-terminal domain interacting with the catalytic pocket in the LPL’s amino-terminal domain. The same conformation was observed for the complex in solution by small-angle X-ray scattering (SAXS) studies (Birrane et al., 2019). The structure of LPL contained an amino-terminal hydrolase domain with six α-helices and ten β-strands and a carboxyl-terminal flattened β-barrel domain containing twelve β-strands (Birrane et al., 2019). LPL contained two N-linked glycans and one calcium ion in the amino-terminal catalytic domain.
Portions of the Trp-rich loop and the lid covering the catalytic pocket were not well defined in the structure by Birrane and coworkers (Birrane et al., 2019). However, very recently, Arora and coworkers solved the crystal structure for a GPIHBP1–LPL complex in the presence of a weak competitive inhibitor of LPL (Arora et al., 2019). That LPL structure was similar to the one reported earlier (Birrane et al., 2019); however, the presence of the inhibitor in LPL’s active site permitted a higher definition of the Trp-rich loop and the lid domain.
The GPIHBP1 was bound to the carboxyl-terminal domain of LPL and, as expected, contained a classic LU fold with three loops stabilized by five disulfide bonds (Leth et al., 2019). Electron densities corresponding to GPIHBP1’s acidic domain could not be identified in the GPIHBP1–LPL crystal structures, which was not surprising, given that the acidic domain is disordered and binds LPL in a transient and dynamic fashion (Kristensen et al., 2018).
The interface between GPIHBP1 and LPL is large (~940 Å2) and mediated largely by hydrophobic contacts (Birrane et al., 2019). Consistent with results from mutagenesis and HDX-MS studies (Beigneux et al., 2011; Beigneux et al., 2015; Mysling et al., 2016a), Trp-109 and adjacent residues in the second finger of GPIHBP1 were directly involved in the binding interface. Extensive sequences within the carboxyl-terminal domain of LPL, including sequences that had been implicated by mutagenesis and HDX-MS studies (Hu et al., 2017b; Mysling et al., 2016a; Voss et al., 2011), participated in the GPIHBP1–LPL binding interface.
With the GPIHBP1–LPL crystal structure in hand, one of our early questions was whether the binding site for GPIHBP1 on LPL might resemble the binding site for colipase on pancreatic lipase (PL). Like GPIHBP1, colipase is a small, multi-fingered cysteine-knot protein stabilized by five disulfide bonds (van Tilbeurgh et al., 1992). A comparison of crystal structures revealed that GPIHBP1 and colipase bind to opposite sides of the carboxyl-terminal domains of LPL and PL, respectively (Birrane et al., 2019). Also, colipase–PL interactions were discrete and mediated by electrostatic interactions (van Tilbeurgh et al., 1992), whereas GPIHBP1–LPL interactions were extensive and driven primarily by hydrophobic contacts (Birrane et al., 2019). In hindsight, the existence of distinct binding sites for GPIHBP1 and colipase was not surprising. While the two proteins have superficial similarities, they are evolutionarily distinct and play very different functions, with colipase serving to activate triglyceride hydrolysis and GPIHBP1 serving to transport LPL and anchor it to capillary endothelial cells.
LPL contains a single large basic patch
LPL contains several positively-charged heparin-binding motifs, scattered within the primary sequence, and their functional importance has been investigated by several groups (Hata et al., 1993; Hill et al., 1998; Ma et al., 1994; Sendak et al., 1998). Interestingly, an electrostatic surface potential map, based on the GPIHBP1–LPL crystal structure, revealed that the various heparin-binding motifs converge to create a single large contiguous basic patch (covering ~2400 Å2) (Birrane et al., 2019) (Figure 5A–B ). This patch extends into both the amino- and carboxyl-terminal domains of LPL and spans the narrow hinge region connecting the two domains. Of note, two lysines (K472 and K473) located within the last five residues of LPL were not visualized in the electron density map (Birrane et al., 2019). Had they been visualized, the size of LPL’s basic patch would have appeared even larger.
Figure 5. Three-dimensional structure of the human LPL–GPIHBP1 complex.
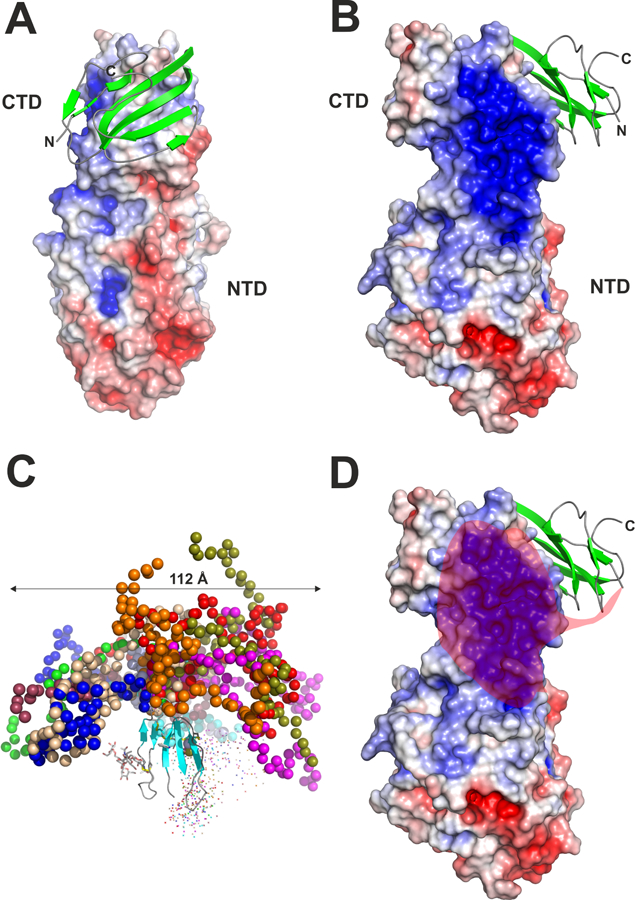
Panels A, B and D show the crystal structure of LPL as a surface representation colored by its electrostatic potential [acidic (red), neutral (white), basic (blue)] and human GPIHBP1 in a cartoon representation with β-strands colored green [modified from the crystal structure by Birrane and coworkers (Birrane et al., 2019)]. For clarity, only one of the two GPIHBP1–LPL complexes in the crystal structure is shown. The amino- and carboxyl-terminal regions of GPIHBP1 are marked by N and C, respectively. The positions of the amino- and carboxyl-terminal domains of LPL are marked with NTD and CTD, respectively. Panel C shows the model of GPIHBP1 in solution, as defined by SAXS analyses, illustrating the large space occupied by GPIHBP1’s intrinsically disordered acidic domain [modified from (Kristensen et al., 2018)]. Different ensembles from the modeling of the disordered acidic domain are illustrated by colored spheres, while the folded LU domain is represented by a cartoon representation. The acidic domain of GPIHBP1 is absent in panels A and B, as it was not defined by electron densities. The semi-transparent red “cloud” in Panel D illustrates the region of LPL that is potentially engaged by GPIHBP1’s intrinsically disordered acidic domain in a “fuzzy complex.”
As noted earlier, GPIHBP1’s intrinsically disordered acidic domain increases the initial encounter rate with LPL and stabilizes LPL’s structure. These effects are mediated by transient interactions (Kristensen et al., 2018; Mysling et al., 2016a; Mysling et al., 2016b), resulting in the formation of a “fuzzy complex” (Borgia et al., 2018; Ivic et al., 2019). Given the transient nature of the interactions, it was not surprising that the acidic domain could not be visualized in the crystal structure. However, it is noteworthy that GPIHBP1 was positioned such that its acidic domain, >60 Å in length (Kristensen et al., 2018), would be expected to project towards—and potentially interact with—the entirety of LPL’s bas ic patch (Figure 5C–D). It seems likely that these interactions stabilize LPL structure and activity (Birrane et al., 2019). GPIHBP1’s acidic domain could provide a “backbone” spanning LPL’s amino- and carboxyl-terminal domains—one that provides sufficient flexibility for enzyme function but also the rigidity required to prevent the unfolding that might otherwise be initiated by torsion of LPL’s hinge region (or collapse of LPL’s two domains upon each other).
Structural insights into LPL mutations causing chylomicronemia
Because the interface between GPIHBP1’s LU domain and the carboxyl-terminal domain of LPL is large, Birrane and coworkers predicted that it would be possible to identify mutations that would disrupt GPIHBP1–LPL interactions (Birrane et al., 2019). They were intrigued by a missense mutation in LPL, M404R, identified in a Middle Eastern patient with chylomicronemia. That mutation was reported to abolish LPL secretion from cells (Pingitore et al., 2016), but the crystal structure revealed that M404 is located within the GPIHBP1–LPL binding interface—implying that the mutation might actually cause disease by interfering with LPL’s ability to bind to GPIHBP1. Indeed, GPIHBP1–LPL binding assays reve aled that the M404R mutation abolishes LPL binding to GPIHBP1 but does not affect LPL secretion from cells (Birrane et al., 2019).
The GPIHBP1–LPL crystal structure also yielded insi ghts into a D201V mutation in LPL, identified in two Lebanese families with chylomicronemia (Abifadel et al., 2004). The crystal structure revealed that D201 participates in the coordination of LPL’s sole calcium ion. A valine cannot fulfill that function. Birrane and coworkers suspected that the inability of LPL-D201V to coordinate calcium would interfere with LPL folding and abolish LPL secretion from cells. Indeed, when LPL-D201V was introduced into CHO cells, the mutant LPL was produced in normal amounts but none was secreted from cells, providing a clear explanation for the chylomicronemia phenotype. A mutation in D202, another residue involved in coordinating the calcium ion, also abolished LPL secretion from cells (Birrane et al., 2019). Insights from the GPIHBP1–LPL crystal structure, along with straightf orward biochemical assays, should make it possible to understand and classify most LPL mutations associated with chylomicronemia.
Relevance of the head-to-tail LPL homodimer conformation in the crystal structure
The fact that the crystal structure revealed two LPL molecules in a head-to-tail orientation appeared, at least at first glance, to support long-held assumptions about the structure of LPL. For decades, the functional unit of active LPL has been assumed to be a homodimer (Berryman and Bensadoun, 1995; Garfinkel et al., 1983; Iverius and Ostlund-Lindqvist, 1976; Krapp et al., 1995; Lookene and Bengtsson-Olivecrona, 1993; Lookene et al., 1994; Olivecrona and Bengtsson-Olivecrona, 1990; Olivecrona et al., 1985; Osborne et al., 1985; Peterson et al., 2002; Vannier and Ailhaud, 1989; Wong et al., 1994; Zhang et al., 2005), and homodimer assembly has been considered essential for LPL secretion from cells (Doolittle et al., 2010; Doolittle and Peterfy, 2010). Several studies concluded that active LPL homodimers have a head-to-tail orientation (Kobayashi et al., 2002; Wong et al., 1997), with the Trp-rich loop in the carboxyl-terminal domain of one monomer supplying substrates to the catalytic pocket in the amino-terminal domain of the partner monomer. However, in the crystal structures for the GPIHBP1–LPL complex (Birrane et al., 2019; Horton, 2019; Arora et al., 2019), the Trp-rich loop in the carboxyl-terminal domain of one monomer interacted with the catalytic pocket of the partner monomer, raising doubts about whether that particular conformation would be compatible with either lipoprotein binding or the hydrolysis of triglyceride substrates. One possibility is that the reciprocal interactions between the two LPL monomers do not represent physiologic interactions and instead reflect a stable conformation that LPL adopts at high protein concentrations—one in which the catalytic pocket shields the hydrophobic Trp-rich loop from the aqueous environment. It is important to emphasize, however, that all of the GPIHBP1–LPL crystal structures were generated in the absence of triglyceride substrates or lipoproteins. The conformation of the GPIHBP1–LPL complex in the presence of lipoproteins remains to be determined.
In recent studies, Beigneux and coworkers revisited accepted dogma holding that LPL is only active as a homodimer (Beigneux et al., 2019). When they subjected freshly secreted human LPL to density gradient ultracentrifugation, they found that the LPL mass and activity peaks corresponded to the size of monomers (~55 kDa)—over lapping with or slightly smaller than the 66-kDa albumin standard and with minimal overlap with a 97-kDa phosphorylase b standard. Incubating the LPL with guanidine hydrochloride inactivated catalytic activity but did not alter the apparent size of LPL, as judged by density gradient ultracentrifugation. Also, the GPIHBP1–LPL complex, expected to have a size of ~75 kDa, was slightly larger than the albumin standard by density gradient ultracentrifugation, with minimal overlap with the phosphorylase b standard (Beigneux et al., 2019).
When LPL is loaded onto a heparin–Sepharose column and then eluted with a linear gradient of sodium chloride, there are two distinct LPL peaks—a low-salt peak with little catalytic activity and a high-salt peak containing large amounts of catalytic activity. For years, the low-salt peak was presumed to contain inactive LPL monomers, whereas the high-salt peak was assumed to contain active homodimers. However, when Beigneux and coworkers subjected the active LPL in the high-salt peak to density gradient ultracentrifugation, they found that it exhibited the size of a monomer (i.e., slightly smaller than the albumin standard) (Beigneux et al., 2019). Most of the LPL in the low salt peak was inactive and in the form of aggregates at the bottom of the density gradient tube. Beigneux and coworkers also examined the LPL in the low-and high-salt peaks with a sandwich ELISA in which a single LPL–specific monoclonal antibody (either 5D2 or 88B8) was used to both capture and detect the LPL. This single-antibody sandwich ELISA format had been touted as being specific for catalytically active LPL homodimers (Peterson et al., 2002). However, Beigneux and coworkers found that the catalytically active LPL in the high-salt peak exhibited little reactivity in single-antibody sandwich ELISAs, whereas the aggregate-containing LPL species in the low-salt peak yielded a robust signal (Beigneux et al., 2019). Importantly, the LPL in both the low- and high-salt fractions yielded robust signals in sandwich ELISAs employing two different monoclonal antibodies (one to capture the LPL and the other to detect the LPL). The two-antibody sandwich ELISAs are sensitive in detecting LPL monomers as well as aggregated LPL species. The findings by Beigneux and coworkers implied that the catalytically active LPL in the high-salt peak, long presumed to be in the form of homodimers, is largely in the form of monomers (Beigneux et al., 2019).
Beigneux and coworkers also tested whether CHO cells, when co-transfected with two LPL constructs harboring different epitope tags, would secrete catalytically active “mixed homodimers” ( i.e., LPL species containing both epitope tags) (Beigneux et al., 2019). The medium of the co-transfected cells contained large amounts of both differentially tagged LPL proteins, and both LPL proteins were catalytically active. However, the amount of mixed LPL species in the medium was very low, as judged by specific ELISAs, but still detectable. Interestingly, the mixed species in the medium were primarily located in the inactive low-salt peak, as judged by heparin-Sepharose chromatography, not in the catalytically active high-salt peak (Beigneux et al., 2019). These studies provided further support for the notion that freshly secreted, catalytically active LPL is primarily in the form of monomers. The concept that LPL can be active as a monomer (Beigneux et al., 2019) was endorsed by Arora and coworkers (Arora et al., 2019), based in part on their suspicion that the head-to-tail LPL homodimers in their crystal structure were unlikely to be physiologically active in binding lipoproteins or hydrolyzing triglycerides.
The view that LPL can be active as a monomer is not inconsistent with the existence of LPL homodimers in specific settings. LPL homodimers can exist, as documented by the crystallography and SAXS studies (Arora et al., 2019; Birrane et al., 2019), but the relevance of those conformations to the physiologic function of LPL is questionable. It is important to emphasize that the LPL homodimers in the crystallization and SAXS studies were observed when the protein concentrations were high, whereas the density gradient and immunochemical studies supporting the existence of active LPL monomers were performed at far lower concentrations of LPL (Beigneux et al., 2019). It is possible that LPL homodimers that appear at high protein concentrations dissociate into active monomers at lower protein concentrations. However, at this point, the existence of stable and catalytically active LPL homodimers seems to be questionable. Moreover, dogma that has accompanied the notion that LPL is active only when it is in the form of homodimers may need additional scrutiny. For example, the notion that ANGPTL4 functions by converting active LPL homodimers into inactive LPL monomers (Sukonina et al., 2006) may need to be revisited. Also, the concept that lipase maturation factor 1 (LMF-1) (an integral membrane protein of the ER) functions to convert newly synthesized LPL monomers into catalytically active and secretion-competent LPL homodimers (Doolittle et al., 2010; Doolittle and Peterfy, 2010) may also require additional investigation.
Concluding remarks
Studies over the past decade have established that GPIHBP1 is LPL’s partner—required for transporting LPL into capillaries, lipoprotein margination, and for preserving LPL structure and activity. Impaired GPIHBP1–LPL interactions, whether from genetic defects or autoantibodies, cause chylomicronemia. Biophysical studies have demonstrated that GPIHBP1’s acidic domain stabilizes LPL structure and activity, likely by creating a fuzzy complex with LPL’s large basic patch. For years, the properties of LPL have been analyzed with purified preparations of LPL, but future research will need to focus on the properties of the GPIHBP1–LPL complex. We need a better understanding of lipoprotein interactions with the GPIHBP1–LPL complex and how regulators of lipolysis (e.g., apo-CII, apo-AV) influence the activity of the GPIHBP1–LPL complex. We also need a clear understanding of how the “ANGPTL proteins” initiate unfolding of LPL and a more precise understanding of the interactions between GPIHBP1’s acidic domain and LPL’s basic patch. Finally, commonly occurring LPL polymorphisms are known to influence plasma triglyceride levels (Almeida et al., 2007; Jensen et al., 2009; Rip et al., 2006); the impact of those polymorphisms needs to be investigated in the context of the GPIHBP1–LPL complex.
Young et al. review mechanisms for intravascular triglyceride metabolism, focusing on the role of GPIHBP1 in capturing lipoprotein lipase (LPL) within the subendothelial spaces, transporting LPL across endothelial cells to its site of action in the capillary lumen, stabilizing the structural integrity of LPL, and mediating lipoprotein margination along capillaries.
Acknowledgments
Supported by grants (HL090553, HL087228, and HL125335) from the National Heart, Lung, and Blood Institute, a Transatlantic Network Grant (12CVD04) from the Leducq Foundation, and NOVO Nordisk Foundation Grants (NNF17OC0026868 and NNF18OC0033864).
Declaration of Interests
Author M.M. is an employee of Shire/Takeda and holds stock and stock options in the company. Author K.N. holds stock in Immunobiologic Laboratories and serves as a consultant for Skylight and Sysmex.
Stephen Young, Anne Beigneux, Loren Fong, and Katsuyuki Nakashima and their respective institutions have applied for a patent on an assay for a diagnostic test for the GPIHBP1 autoantibody syndrome.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Abifadel M, Jambart S, Allard D, Rabes JP, Varret M, Derre A, Chouery E, Salem N, Junien C, Aydenian H, et al. (2004). Identification of the first Lebanese mutation in the LPL gene and description of a rapid detection method. Clin. Genet 65, 158–161. [DOI] [PubMed] [Google Scholar]
- Acton S, Rigotti A, Landschulz KT, Xu S, Hobbs HH, and Krieger M (1996). Identification of scavenger receptor SR-BI as a high density lipoprotein receptor. Science 271, 518–520. [DOI] [PubMed] [Google Scholar]
- Allan CM, Heizer PJ, Tu Y, Sandoval NP, Jung RS, Morales JE, Sajti E, Troutman TD, Saunders TL, Cusanovich DA, et al. (2019). An upstream enhancer regulates Gpihbp1 expression in a tissue-specific manner. J. Lipid Res 60, 869–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan CM, Jung CJ, Larsson M, Heizer PJ, Tu Y, Sandoval NP, Dang TLP, Jung RS, Beigneux AP, de Jong PJ, et al. (2017a). Mutating a conserved cysteine in GPIHBP1 reduces amounts of GPIHBP1 in capillaries and abolishes LPL binding. J. Lipid Res 58, 1453–1461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan CM, Larsson M, Hu X, He C, Jung RS, Mapar A, Voss C, Miyashita K, Machida T, Murakami M, et al. (2016). An LPL-specific monoclonal antibody, 88B8, that abolishes the binding of LPL to GPIHBP1. J. Lipid Res 57, 1889–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan CM, Larsson M, Jung RS, Ploug M, Bensadoun A, Beigneux AP, Fong LG, and Young SG (2017b). Mobility of “HSPG-bound” LPL explains how LPL is able to reach GPIHBP1 on capillaries. J. Lipid Res 58, 216–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida KA, Strunz CM, Maranhao RC, and Mansur AP (2007). The S447X polymorphism of lipoprotein lipase: effect on the incidence of premature coronary disease and on plasma lipids. Arq. Bras. Cardiol 88, 297–303. [DOI] [PubMed] [Google Scholar]
- Ariza MJ, Martinez-Hernandez PL, Ibarretxe D, Rabacchi C, Rioja J, Grande-Aragon C, Plana N, Tarugi P, Olivecrona G, Calandra S, et al. (2016). Novel mutations in the GPIHBP1 gene identified in 2 patients with recurrent acute pancreatitis. J. Clin. Lipidol 10, 92–100 e1. [DOI] [PubMed] [Google Scholar]
- Arora R, Nimonkar AV, Baird D, Wang C, Chiu CH, Horton PA, Hanrahan S, Cubbon R, Weldon S, Tschantz WR, et al. (2019). Structure of lipoprotein lipase in complex with GPIHBP1. Proc Natl Acad Sci U S A (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, and Pascual V (2006). Type I interferon in systemic lupus erythematosus and other autoimmune diseases. Immunity 25, 383–392. [DOI] [PubMed] [Google Scholar]
- Beigneux AP (2010). GPIHBP1 and the processing of triglyceride-rich lipoproteins. Clin. Lipidol 5, 575–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Allan CM, Sandoval NP, Cho GW, Heizer PJ, Jung RS, Stanhope KL, Havel PJ, Birrane G, Meiyappan M, et al. (2019). Lipoprotein lipase is active as a monomer. Proc. Natl. Acad. Sci. USA 116, 6319–6328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Davies B, Gin P, Weinstein MM, Farber E, Qiao X, Peale P, Bunting S, Walzem RL, Wong JS, et al. (2007). Glycosylphosphatidylinositol-anchored high density lipoprotein–binding protein 1 plays a critical role in the lipolytic processing of chylomicrons. Cell Metab 5, 279–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Davies BS, Tat S, Chen J, Gin P, Voss CV, Weinstein MM, Bensadoun A, Pullinger CR, Fong LG, et al. (2011). Assessing the role of the glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) three-finger domain in binding lipoprotein lipase. J. Biol. Chem 286, 19735–19743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Fong LG, Bensadoun A, Davies BS, Oberer M, Gardsvoll H, Ploug M, and Young SG (2015). GPIHBP1 missense mutations often cause multimerization of GPIHBP1 and thereby prevent lipoprotein lipase binding. Circ. Res 116, 624–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Franssen R, Bensadoun A, Gin P, Melford K, Peter J, Walzem RL, Weinstein MM, Davies BS, Kuivenhoven JA, et al. (2009a). Chylomicronemia with a mutant GPIHBP1 (Q115P) that cannot bind lipoprotein lipase. Arterioscler. Thromb. Vasc. Biol 29, 956–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Gin P, Davies BSJ, Weinstein MM, Bensadoun A, Fong LG, and Young SG (2009b). Highly conserved cysteines within the Ly6 domain of GPIHBP1 are crucial for the binding of lipoprotein lipase. J. Biol. Chem 284, 30240–30247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beigneux AP, Miyashita K, Ploug M, Blom DJ, Ai M, Linton MF, Khovidhunkit W, Dufour R, Garg A, McMahon MA, et al. (2017). Autoantibodies against GPIHBP1 as a cause of hypertriglyceridemia. N. Engl. J. Med 376, 1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bengtsson G, and Olivecrona T (1979). Apolipoprotein CII enhances hydrolysis of monoglycerides by lipoprotein lipase, but the effect is abolished by fatty acids. FEBS Lett 106, 345–348. [DOI] [PubMed] [Google Scholar]
- Berryman DE, and Bensadoun A (1995). Heparan sulfate proteoglycans are primarily responsible for the maintenance of enzyme activity, binding, and degradation of lipoprotein lipase in Chinese hamster ovary cells. J. Biol. Chem 270, 24525–24531. [DOI] [PubMed] [Google Scholar]
- Birrane G, Beigneux AP, Dwyer B, Strack-Logue B, Kristensen KK, Francone OL, Fong LG, Mertens HDT, Pan CQ, Ploug M, et al. (2019). Structure of the lipoprotein lipase-GPIHBP1 complex that mediates plasma triglyceride hydrolysis. Proc. Natl. Acad. Sci. USA 116, 1723–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop JR, Passos-Bueno MR, Fong L, Stanford KI, Gonzales JC, Yeh E, Young SG, Bensadoun A, Witztum JL, Esko JD, et al. (2010). Deletion of the basement membrane heparan sulfate proteoglycan type XVIII collagen causes hypertriglyceridemia in mice and humans. PLoS One 5, e13919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette-Mackie EJ, Masuno H, Dwyer NK, Olivecrona T, and Scow RO (1989). Lipoprotein lipase in myocytes and capillary endothelium of heart: immunocytochemical study. Am. J. Physiol 256, E818–E828. [DOI] [PubMed] [Google Scholar]
- Borgia A, Borgia MB, Bugge K, Kissling VM, Heidarsson PO, Fernandes CB, Sottini A, Soranno A, Buholzer KJ, Nettels D, et al. (2018). Extreme disorder in an ultrahigh-affinity protein complex. Nature 555, 61–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breckenridge WC, Little JA, Steiner G, Chow A, and Poapst M (1978). Hypertriglyceridemia associated with deficiency of apolipoprotein C-II. N. Engl. J. Med 298, 1265–1273. [DOI] [PubMed] [Google Scholar]
- Charriere S, Peretti N, Bernard S, Di Filippo M, Sassolas A, Merlin M, Delay M, Debard C, Lefai E, Lachaux A, et al. (2011). GPIHBP1 C89F neomutation and hydrophobic C-terminal domain G175R mutation in two pedigrees with severe hyperchylomicronemia. J. Clin. Endocrinol. Metab 96, E1675–1679. [DOI] [PubMed] [Google Scholar]
- Chi X, Shetty SK, Shows HW, Hjelmaas AJ, Malcolm EK, and Davies BS (2015). Angiopoietin-like 4 modifies the interactions between lipoprotein lipase and its endothelial cell transporter GPIHBP1. J. Biol. Chem 290, 11865–11877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chyzhyk V, Kozmic S, Brown AS, Hudgins LC, Starc TJ, Davila AD, Blevins TC, Diffenderfer MR, He L, Geller AS, et al. (2019). Extreme hypertriglyceridemia: genetic diversity, pancreatitis, pregnancy, and prevalence. J. Clin. Lipidol 13, 89–99. [DOI] [PubMed] [Google Scholar]
- Coca-Prieto I, Kroupa O, Gonzalez-Santos P, Magne J, Olivecrona G, Ehrenborg E, and Valdivielso P (2011). Childhood-onset chylomicronaemia with reduced plasma lipoprotein lipase activity and mass: identification of a novel GPIHBP1 mutation. J. Intern. Med 270, 224–228. [DOI] [PubMed] [Google Scholar]
- Davies BS, Goulbourne CN, Barnes RH 2nd, Turlo KA, Gin P, Vaughan S, Vaux DJ, Bensadoun A, Beigneux AP, Fong LG, et al. (2012). Assessing mechanisms of GPIHBP1 and lipoprotein lipase movement across endothelial cells. J. Lipid. Res 53, 2690–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies BSJ, Beigneux AP, Barnes RH II, Tu Y, Gin P, Weinstein MM, Nobumori C, Nyrén R, Goldberg IJ, Olivecrona G, et al. (2010). GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab 12, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Beer F, Hendriks WL, van Vark LC, Kamerling SW, van Dijk KW, Hofker MH, Smelt AH, and Havekes LM (1999). Binding of beta-VLDL to heparan sulfate proteoglycans requires lipoprotein lipase, whereas ApoE only modulates binding affinity. Arterioscler. Thromb. Vasc. Biol 19, 633–637. [DOI] [PubMed] [Google Scholar]
- Dewey FE, Gusarova V, O’Dushlaine C, Gottesman O, Trejos J, Hunt C, Van Hout CV, Habegger L, Buckler D, Lai KM, et al. (2016). Inactivating variants in ANGPTL4 and risk of coronary artery disease. N. Engl. J. Med 374, 1123–1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Domizio J, and Cao W (2013). Fueling autoimmunity: type I interferon in autoimmune diseases. Expert Rev. Clin. Immunol 9, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijk W, Beigneux AP, Larsson M, Bensadoun A, Young SG, and Kersten S (2016). Angiopoietin-like 4 promotes intracellular degradation of lipoprotein lipase in adipocytes. J. Lipid. Res 57, 1670–1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle MH, Ehrhardt N, and Peterfy M (2010). Lipase maturation factor 1: structure and role in lipase folding and assembly. Curr. Opin. Lipidol 21, 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doolittle MH, and Peterfy M (2010). Mechanisms of lipase maturation. Clin. Lipidol 5, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchesne L, Octeau V, Bearon RN, Beckett A, Prior IA, Lounis B, and Fernig DG (2012). Transport of fibroblast growth factor 2 in the pericellular matrix is controlled by the spatial distribution of its binding sites in heparan sulfate. PLoS Biol 10, e1001361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi J, Miyashita K, Fukamachi I, Nakajima K, Murakami M, Kawahara Y, Yamashita T, Ohta Y, Abe K, Nakatsuka A, et al. (2019). GPIHBP1 autoantibody syndrome during interferon beta1a treatment. J. Clin. Lipidol 13, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich J, Beg OU, Peterson J, Previato L, Brunzell JD, Brewer HB Jr., and Santamarina-Fojo S (1992). Human lipoprotein lipase. Analysis of the catalytic triad by site-directed mutagenesis of Ser-132, Asp-156, and His-241. J. Biol. Chem 267, 4161–4165. [PubMed] [Google Scholar]
- Enerbäck S, Semb H, Bengtsson-Olivecrona G, Carlsson P, Hermansson M-L, Olivecrona T, and Bjursell G (1987). Molecular cloning and sequence analysis of cDNA encoding lipoprotein lipase of guinea pig. Gene 58, 1–12. [DOI] [PubMed] [Google Scholar]
- Ferguson MAJ, Kinoshita T, and Hart GW (2009). Glycosylphosphatidylinositol Anchors. In Essentials of Glycobiology, Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, and Etzler ME, eds. (Cold Spring Harbor; (NY: )). [PubMed] [Google Scholar]
- Fong LG, Young SG, Beigneux AP, Bensadoun A, Oberer M, Jiang H, and Ploug M (2016). GPIHBP1 and plasma triglyceride metabolism. Trends Endocrinol. Metab 27, 455–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franssen R, Young SG, Peelman F, Hertecant J, Sierts JA, Schimmel AWM, Bensadoun A, Kastelein JJP, Fong LG, Dallinga-Thie GM, et al. (2010). Chylomicronemia with low postheparin lipoprotein lipase levels in the setting of GPIHBP1 defects. Circ. Cardiovasc. Genet 3, 169–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garfinkel AS, Kempner ES, Ben-Zeev O, Nikazy J, James SJ, and Schotz MC (1983). Lipoprotein lipase: size of the functional unit determined by radiation inactivation. J. Lipid Res 24, 775–780. [PubMed] [Google Scholar]
- Gin P, Beigneux AP, Voss C, Davies BS, Beckstead JA, Ryan RO, Bensadoun A, Fong LG, and Young SG (2011). Binding preferences for GPIHBP1, a glycosylphosphatidylinositol-anchored protein of capillary endothelial cells. Arterioscler. Thromb. Vasc. Biol 31, 176–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gin P, Goulbourne CN, Adeyo O, Beigneux AP, Davies BS, Tat S, Voss CV, Bensadoun A, Fong LG, and Young SG (2012). Chylomicronemia mutations yield new insights into interactions between lipoprotein lipase and GPIHBP1. Hum. Mol. Genet 21, 2961–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gin P, Yin L, Davies BS, Weinstein MM, Ryan RO, Bensadoun A, Fong LG, Young SG, and Beigneux AP (2008). The acidic domain of GPIHBP1 is important for the binding of lipoprotein lipase and chylomicrons. J. Biol. Chem 284, 29554–29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzaga-Jauregui C, Mir S, Penney S, Jhangiani S, Midgen C, Finegold M, Muzny DM, Wang M, Bacino CA, Gibbs RA, et al. (2014). Whole-exome sequencing reveals GPIHBP1 mutations in infantile colitis with severe hypertriglyceridemia. J.Pediatr. Gastroenterol. Nutr 59, 17–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goulbourne CN, Gin P, Tatar A, Nobumori C, Hoenger A, Jiang H, Grovenor CR, Adeyo O, Esko JD, Goldberg IJ, et al. (2014). The GPIHBP1-LPL complex is responsible for the margination of triglyceride-rich lipoproteins in capillaries. Cell Metab 19, 849–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutgsell AR, Ghodge SV, Bowers AA, and Neher SB (2019). Mapping the sites of the lipoprotein lipase (LPL)-angiopoietin-like protein 4 (ANGPTL4) interaction provides mechanistic insight into LPL inhibition. J. Biol. Chem 294, 2678–2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haller JF, Mintah IJ, Shihanian LM, Stevis P, Buckler D, Alexa-Braun CA, Kleiner S, Banfi S, Cohen JC, Hobbs HH, et al. (2017). ANGPTL8 requires ANGPTL3 to inhibit lipoprotein lipase and plasma triglyceride clearance. J. Lipid. Res 58, 1166–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hata A, Ridinger DN, Sutherland S, Emi M, Shuhua Z, Myers RL, Ren K, Cheng T, Inoue I, Wilson DE, et al. (1993). Binding of lipoprotein lipase to heparin. Identification of five critical residues in two distinct segments of the amino-terminal domain. J. Biol. Chem 268, 8447–8457. [PubMed] [Google Scholar]
- Havel RJ (2010). Triglyceride-rich lipoproteins and plasma lipid transport. Arterioscler. Thromb. Vasc. Biol 30, 9–19. [DOI] [PubMed] [Google Scholar]
- Havel RJ, and Gordon RS Jr. (1960). Idiopathic hyperlipemia: metabolic studies in an affected family. J. Clin. Invest 39, 1777–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Havel RJ, and Kane JP (2001). Introduction: Structure and metabolism of plasma lipoproteins. In The Metabolic and Molecular Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, and Vogelstein B, eds. (New York: McGraw-Hill: ), pp. 2705–2716. [Google Scholar]
- Havel RJ, Shore VG, Shore B, and Bier DM (1970). Role of specific glycopeptides of human serum lipoproteins in the activation of lipoprotein lipase. Circ. Res 27, 595–600. [DOI] [PubMed] [Google Scholar]
- He C, Weston TA, Jung RS, Heizer P, Larsson M, Hu X, Allan CM, Tontonoz P, Reue K, Beigneux AP, et al. (2018a). NanoSIMS analysis of intravascular lipolysis and lipid movement across capillaries and into cardiomyocytes. Cell Metab 27, 1055–1066.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He L, Vanlandewijck M, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. (2018b). Single-cell RNA sequencing of mouse brain and lung vascular and vessel-associated cell types. Sci. Data 5, 180160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helgadottir A, Gretarsdottir S, Thorleifsson G, Hjartarson E, Sigurdsson A, Magnusdottir A, Jonasdottir A, Kristjansson H, Sulem P, Oddsson A, et al. (2016). Variants with large effects on blood lipids and the role of cholesterol and triglycerides in coronary disease. Nat. Genet 48, 634–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henderson H, Leisegang F, Hassan F, Hayden M, and Marais D (1998). A novel Glu421Lys substitution in the lipoprotein lipase gene in pregnancy-induced hypertriglyceridemic pancreatitis. Clin. Chim. Acta 269, 1–12. [DOI] [PubMed] [Google Scholar]
- Henderson HE, Hassan F, Marais D, and Hayden MR (1996). A new mutation destroying disulphide bridging in the C-terminal domain of lipoprotein lipase. Biochem. Biophys. Res. Commun 227, 189–194. [DOI] [PubMed] [Google Scholar]
- Hill JS, Yang D, Nikazy J, Curtiss LK, Sparrow JT, and Wong H (1998). Subdomain chimeras of hepatic lipase and lipoprotein lipase. Localization of heparin and cofactor binding. J. Biol. Chem 273, 30979–30984. [DOI] [PubMed] [Google Scholar]
- Horton JD (2019). Intravascular triglyceride lipolysis becomes crystal clear. Proc Natl Acad Sci U S A 116, 1480–1482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Dallinga-Thie GM, Hovingh GK, Chang SY, Sandoval NP, Dang TLP, Fukamachi I, Miyashita K, Nakajima K, Murakami M, et al. (2017a). GPIHBP1 autoantibodies in a patient with unexplained chylomicronemia. J. Clin. Lipidol 11, 964–971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu X, Sleeman MW, Miyashita K, Linton MF, Allan CM, He C, Larsson M, Tu Y, Sandoval NP, Jung RS, et al. (2017b). Monoclonal antibodies that bind to the Ly6 domain of GPIHBP1 abolish the binding of LPL. J. Lipid Res 58, 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huijbers MG, Plomp JJ, van der Maarel SM, and Verschuuren JJ (2018). IgG4-mediated autoimmune diseases: a niche of antibody-mediated disorders. Ann. NY Acad. Sci 1413, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Investigators MIG a.C.E.C., Stitziel NO, Stirrups KE, Masca NG, Erdmann J, Ferrario PG, Konig IR, Weeke PE, Webb TR, Auer PL, et al. (2016). Coding variation in ANGPTL4, LPL, and SVEP1 and the risk of coronary disease. N. Engl. J. Med 374, 1134–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioka RX, Kang M-J, Kamiyama S, Kim D-H, Magoori K, Kamataki A, Ito Y, Takei YA, Sasaki M, Suzuki T, et al. (2003). Expression cloning and characterization of a novel glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein, GPI-HBP1. J. Biol. Chem 278, 7344–7349. [DOI] [PubMed] [Google Scholar]
- Iverius PH, and Ostlund-Lindqvist AM (1976). Lipoprotein lipase from bovine milk. Isolation procedure, chemical characterization, and molecular weight analysis. J. Biol. Chem 251, 7791–7795. [PubMed] [Google Scholar]
- Ivic N, Potocnjak M, Solis-Mezarino V, Herzog F, Bilokapic S, and Halic M (2019). Fuzzy interactions form and shape the histone transport complex. Mol. Cell 73, 1191–1203.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen MK, Rimm EB, Rader D, Schmidt EB, Sorensen TI, Vogel U, Overvad K, and Mukamal KJ (2009). S447X variant of the lipoprotein lipase gene, lipids, and risk of coronary heart disease in 3 prospective cohort studies. Am. Heart J 157, 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara Y, Yamashita T, Ohta Y, Sato K, Tsunoda K, Takemoto M, Hishikawa N, Eguchi J, and Abe K (2017). Marked hypertriglyceridemia induced by interferon-beta1a therapy in a clinically isolated syndrome patient. J. Neurol. Sci 373, 144–146. [DOI] [PubMed] [Google Scholar]
- Kersten S (2017). Triglyceride metabolism under attack. Cell Metab 25, 1209–1210. [DOI] [PubMed] [Google Scholar]
- Kirchgessner TG, Svenson KL, Lusis AJ, and Schotz MC (1987). The sequence of cDNA encoding lipoprotein lipase. A member of a lipase gene family. J. Biol. Chem 262, 8463–8466. [PubMed] [Google Scholar]
- Kobayashi Y, Nakajima T, and Inoue I (2002). Molecular modeling of the dimeric structure of human lipoprotein lipase and functional studies of the carboxyl-terminal domain. Eur. J. Biochem 269, 4701–4710. [DOI] [PubMed] [Google Scholar]
- Koneczny I (2018). A new classification system for IgG4 autoantibodies. Front. Immunol 9, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn ED (1955). Clearing factor, a heparin-activated lipoprotein lipase. I. Isolation and characterization of the enzyme from normal rat heart. J. Biol. Chem 215, 1–14. [PubMed] [Google Scholar]
- Korn ED, and Quigley TW Jr. (1955). Studies on lipoprotein lipase of rat heart and adipose tissue. Biochim. Biophys. Acta 18, 143–145. [DOI] [PubMed] [Google Scholar]
- Kovrov O, Kristensen KK, Larsson E, Ploug M, and Olivecrona G (2019). On the mechanism of angiopoietin-like protein 8 for control of lipoprotein lipase activity. J. Lipid Res 60, 783–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapp A, Zhang H, Ginzinger D, Liu MS, Lindberg A, Olivecrona G, Hayden MR, and Beisiegel U (1995). Structural features in lipoprotein lipase necessary for the mediation of lipoprotein uptake into cells. J. Lipid Res 36, 2362–2373. [PubMed] [Google Scholar]
- Kristensen KK, Midtgaard SR, S., M., Kovrov O, H. L.B., Skar-Gislinge N, Beigneux AP, Kragelund BB, G., O., S.G., Y., et al. (2018). A disordered acidic domain in GPIHBP1 harboring a sulfated tyrosine regulates lipoprotein lipase. Proc. Natl. Acad. Sci. USA 115, E6020–E6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafferty MJ, Bradford KC, Erie DA, and Neher SB (2013). Angiopoietin-like protein 4 inhibition of lipoprotein lipase: evidence for reversible complex formation. J. Biol. Chem 288, 28524–28534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaRosa JC, Levy RI, Herbert P, Lux SE, and Fredrickson DS (1970). A specific apoprotein activator for lipoprotein lipase. Biochem. Biophys. Res. Commun 41, 57–62. [DOI] [PubMed] [Google Scholar]
- Leth J, Leth-Espensen K, Kristensen K, Kumari A, Winther A, Young S, and Ploug M (2019). Evolution and medical significance of LU domain−containing proteins. Int J Molecular Sciences. (In revision) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lookene A, and Bengtsson-Olivecrona G (1993). Chymotryptic cleavage of lipoprotein lipase. Identification of cleavage sites and functional studies of the truncated molecule. Eur. J. Biochem 213, 185–194. [DOI] [PubMed] [Google Scholar]
- Lookene A, Chevreuil O, Ostergaard P, and Olivecrona G (1996). Interaction of lipoprotein lipase with heparin fragments and with heparan sulfate: stoichiometry, stabilization, and kinetics. Biochemistry 35, 12155–12163. [DOI] [PubMed] [Google Scholar]
- Lookene A, Groot NB, Kastelein JJ, Olivecrona G, and Bruin T (1997a). Mutation of tryptophan residues in lipoprotein lipase. Effects on stability, immunoreactivity, and catalytic properties. J. Biol. Chem 272, 766–772. [DOI] [PubMed] [Google Scholar]
- Lookene A, Savonen R, and Olivecrona G (1997b). Interaction of lipoproteins with heparan sulfate proteoglycans and with lipoprotein lipase. Studies by surface plasmon resonance technique. Biochemistry 36, 5267–5275. [DOI] [PubMed] [Google Scholar]
- Lookene A, Skottova N, and Olivecrona G (1994). Interactions of lipoprotein lipase with the active-site inhibitor tetrahydrolipstatin (Orlistat). Eur. J. Biochem 222, 395–403. [DOI] [PubMed] [Google Scholar]
- Ma Y, Henderson HE, Liu MS, Zhang H, Forsythe IJ, Clarke-Lewis I, Hayden MR, and Brunzell JD (1994). Mutagenesis in four candidate heparin binding regions (residues 279–282, 291–304, 390–393, and 439–448) and identification of residues affecting heparin binding of human lipoprotein lipase. J. Lipid Res 35, 2049–2059. [PubMed] [Google Scholar]
- Mailly F, Palmen J, Muller DP, Gibbs T, Lloyd J, Brunzell J, Durrington P, Mitropoulos K, Betteridge J, Watts G, et al. (1997). Familial lipoprotein lipase (LPL) deficiency: a catalogue of LPL gene mutations identified in 20 patients from the UK, Sweden, and Italy. Hum. Mutat 10, 465–473. [DOI] [PubMed] [Google Scholar]
- Mergenthaler P, Lindauer U, Dienel GA, and Meisel A (2013). Sugar for the brain: the role of glucose in physiological and pathological brain function. Trends Neurosci 36, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita K, Fukamachi I, Machida T, Nakajima K, Young SG, Murakami M, Beigneux AP, and Nakajima K (2018a). An ELISA for quantifying GPIHBP1 autoantibodies and making a diagnosis of the GPIHBP1 autoantibody syndrome. Clin. Chim. Acta 487, 174–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyashita K, Fukamachi I, Nagao M, Ishida T, Kobayashi J, Machida T, Nakajima K, Murakami M, Ploug M, Beigneux AP, et al. (2018b). An enzyme-linked immunosorbent assay for measuring GPIHBP1 levels in human plasma or serum. J. Clin. Lipidol 12, 203–210.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysling S, Kristensen KK, Larsson M, Beigneux AP, Gardsvoll H, Fong LG, Bensadouen A, Jorgensen TJ, Young SG, and Ploug M (2016a). The acidic domain of the endothelial membrane protein GPIHBP1 stabilizes lipoprotein lipase activity by preventing unfolding of its catalytic domain. eLife 5, e12095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysling S, Kristensen KK, Larsson M, Kovrov O, Bensadouen A, Jorgensen TJ, Olivecrona G, Young SG, and Ploug M (2016b). The angiopoietin-like protein ANGPTL4 catalyzes unfolding of the hydrolase domain in lipoprotein lipase and the endothelial membrane protein GPIHBP1 counteracts this unfolding. eLife 5, e20958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nevo Y, Ben-Zeev B, Tabib A, Straussberg R, Anikster Y, Shorer Z, Fattal-Valevski A, Ta-Shma A, Aharoni S, Rabie M, et al. (2013). CD59 deficiency is associated with chronic hemolysis and childhood relapsing immune-mediated polyneuropathy. Blood 121, 129–135. [DOI] [PubMed] [Google Scholar]
- Nilsson SK, Anderson F, Ericsson M, Larsson M, Makoveichuk E, Lookene A, Heeren J, and Olivecrona G (2012). Triacylglycerol-rich lipoproteins protect lipoprotein lipase from inactivation by ANGPTL3 and ANGPTL4. Biochim. Biophys. Acta 1821, 1370–1378. [DOI] [PubMed] [Google Scholar]
- Olafsen T, Young SG, Davies BS, Beigneux AP, Kenanova VE, Voss C, Young G, Wong KP, Barnes RH 2nd, Tu Y, et al. (2010). Unexpected expression pattern for glycosylphosphatidylinositol-anchored HDL-binding protein 1 (GPIHBP1) in mouse tissues revealed by positron emission tomography scanning. J. Biol. Chem 285, 39239–39248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivecrona G, Ehrenborg E, Semb H, Makoveichuk E, Lindberg A, Hayden MR, Gin P, Davies BS, Weinstein MM, Fong LG, et al. (2010). Mutation of conserved cysteines in the Ly6 domain of GPIHBP1 in familial chylomicronemia. J. Lipid Res 51, 1535–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivecrona T, and Bengtsson-Olivecrona G (1990). Lipoprotein lipase and hepatic lipase. Curr. Opin. Lipidol 1, 222–230. [Google Scholar]
- Olivecrona T, and Bengtsson-Olivecrona G (1993). Lipoprotein lipase and hepatic lipase. Curr. Opin. Lipidol 4, 187–196. [Google Scholar]
- Olivecrona T, Bengtsson-Olivecrona G, Osborne JC Jr., and Kempner ES (1985). Molecular size of bovine lipoprotein lipase as determined by radiation inactivation. J. Biol. Chem 260, 6888–6891. [PubMed] [Google Scholar]
- Osborne JC Jr., Bengtsson-Olivecrona G, Lee NS, and Olivecrona T (1985). Studies on inactivation of lipoprotein lipase: role of the dimer to monomer dissociation. Biochemistry 24, 5606–5611. [DOI] [PubMed] [Google Scholar]
- Peterson J, Ayyobi AF, Ma Y, Henderson H, Reina M, Deeb SS, Santamarina-Fojo S, Hayden MR, and Brunzell JD (2002). Structural and functional consequences of missense mutations in exon 5 of the lipoprotein lipase gene. J. Lipid Res 43, 398–406. [PubMed] [Google Scholar]
- Pingitore P, Lepore SM, Pirazzi C, Mancina RM, Motta BM, Valenti L, Berge KE, Retterstol K, Leren TP, Wiklund O, et al. (2016). Identification and characterization of two novel mutations in the LPL gene causing type I hyperlipoproteinemia. J. Clin. Lipidol 10, 816–823. [DOI] [PubMed] [Google Scholar]
- Plengpanich W, Young SG, Khovidhunkit W, Bensadoun A, Karnman H, Ploug M, Gardsvoll H, Leung CS, Adeyo O, Larsson M, et al. (2014). Multimerization of glycosylphosphatidylinositol-anchored high density lipoprotein-binding protein 1 (GPIHBP1) and familial chylomicronemia from a serine-to-cysteine Substitution in GPIHBP1 Ly6 domain. J. Biol. Chem 289, 19491–19499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quagliarini F, Wang Y, Kozlitina J, Grishin NV, Hyde R, Boerwinkle E, Valenzuela DM, Murphy AJ, Cohen JC, and Hobbs HH (2012). Atypical angiopoietin-like protein that regulates ANGPTL3. Proc. Natl. Acad. Sci. USA 109, 19751–19756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabacchi C, D’Addato S, S, P., T, L., S, B., S, C., and P, T. (2016). Clinical and genetic features of 3 patients with familial chylomicronemia due to mutations in GPIHBP1 gene. J. Clin. Lipidol 10, 915–921.e4. [DOI] [PubMed] [Google Scholar]
- Rader DJ (2006). Molecular regulation of HDL metabolism and function: implications for novel therapies. J. Clin. Invest 116, 3090–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios JJ, Shastry S, Jasso J, Hauser N, Garg A, Bensadoun A, Cohen JC, and Hobbs HH (2012). Deletion of GPIHBP1 causing severe chylomicronemia. J. Inherit. Metab. Dis 35, 531–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rip J, Nierman MC, Ross CJ, Jukema JW, Hayden MR, Kastelein JJ, Stroes ES, and Kuivenhoven JA (2006). Lipoprotein lipase S447X: a naturally occurring gain-of-function mutation. Arterioscler. Thromb. Vasc. Biol 26, 1236–1245. [DOI] [PubMed] [Google Scholar]
- Rye KA, and Barter PJ (2014). Regulation of high-density lipoprotein metabolism. Circ. Res 114, 143–156. [DOI] [PubMed] [Google Scholar]
- Sendak RA, Melford K, Kao A, and Bensadoun A (1998). Identification of the epitope of a monoclonal antibody that inhibits heparin binding of lipoprotein lipase: new evidence for a carboxyl-terminal heparin-binding domain. J. Lipid Res 39, 633–646. [PubMed] [Google Scholar]
- Shoemaker BA, Portman JJ, and Wolynes PG (2000). Speeding molecular recognition by using the folding funnel: the fly-casting mechanism. Proc. Natl. Acad. Sci. USA 97, 8868–8873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva MO (2012). Risk of autoimmune complications associated with interferon therapy. Gastroenterol. Hepatol. (NY) 8, 540–542. [PMC free article] [PubMed] [Google Scholar]
- Simionescu M, and Simionescu N (1986). Functions of the endothelial cell surface. Annu. Rev. Physiol 48, 279–293. [DOI] [PubMed] [Google Scholar]
- Sukonina V, Lookene A, Olivecrona T, and Olivecrona G (2006). Angiopoietin-like protein 4 converts lipoprotein lipase to inactive monomers and modulates lipase activity in adipose tissue. Proc. Natl. Acad. Sci. USA 103, 17450–17455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Tilbeurgh H, Sarda L, Verger R, and Cambillau C (1992). Structure of the pancreatic lipase–procolipase complex. Nature 359, 159–162. [DOI] [PubMed] [Google Scholar]
- Vanlandewijck M, He L, Mae MA, Andrae J, Ando K, Del Gaudio F, Nahar K, Lebouvier T, Lavina B, Gouveia L, et al. (2018). A molecular atlas of cell types and zonation in the brain vasculature. Nature 554, 475–480. [DOI] [PubMed] [Google Scholar]
- Vannier C, and Ailhaud G (1989). Biosynthesis of lipoprotein lipase in cultured mouse adipocytes. II. Processing, subunit assembly, and intracellular transport. J. Biol. Chem 264, 13206–13216. [PubMed] [Google Scholar]
- Vijayagopal P, Radhakrishnamurthy B, Srinivasan SR, and Berenson GS (1980). Studies of biologic properties of proteoglycans from bovine aorta. Lab. Invest 42, 190–196. [PubMed] [Google Scholar]
- Voss CV, Davies BS, Tat S, Gin P, Fong LG, Pelletier C, Mottler CD, Bensadoun A, Beigneux AP, and Young SG (2011). Mutations in lipoprotein lipase that block binding to the endothelial cell transporter GPIHBP1. Proc. Natl. Acad. Sci. USA 108, 7980–7984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, and Eckel RH (2009). Lipoprotein lipase: from gene to obesity. Am. J. Physiol. Endocrinol. Metab 297, E271–288. [DOI] [PubMed] [Google Scholar]
- Weinstein MM, Tu Y, Beigneux AP, Davies BS, Gin P, Voss C, Walzem RL, Reue K, Tontonoz P, Bensadoun A, et al. (2010a). Cholesterol intake modulates plasma triglyceride levels in glycosylphosphatidylinositol HDL-binding protein 1-deficient mice. Arterioscler. Thromb. Vasc. Biol 30, 2106–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinstein MM, Yin L, Tu Y, Wang X, Wu X, Castellani LW, Walzem RL, Lusis AJ, Fong LG, Beigneux AP, et al. (2010b). Chylomicronemia elicits atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol 30, 20–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler FK, D’Arcy A, and Hunziker W (1990). Structure of human pancreatic lipase. Nature 343, 771–774. [DOI] [PubMed] [Google Scholar]
- Wion KL, Kirchgessner TG, Lusis AJ, Schotz MC, and Lawn RM (1987). Human lipoprotein lipase complementary DNA sequence. Science 235, 1638–1641. [DOI] [PubMed] [Google Scholar]
- Wong H, Davis RC, Thuren T, Goers JW, Nikazy J, Waite M, and Schotz MC (1994). Lipoprotein lipase domain function. J. Biol. Chem 269, 10319–10323. [PubMed] [Google Scholar]
- Wong H, Yang D, Hill JS, Davis RC, Nikazy J, and Schotz MC (1997). A molecular biology-based approach to resolve the subunit orientation of lipoprotein lipase. Proc. Natl. Acad. Sci. USA 94, 5594–5598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto H, Onishi M, Miyamoto N, Oki R, Ueda H, Ishigami M, Hiraoka H, Matsuzawa Y, and Kihara S (2013). Novel combined GPIHBP1 mutations in a patient with hypertriglyceridemia associated with CAD. J. Atheroscler. Thromb 20, 777–784. [DOI] [PubMed] [Google Scholar]
- Zhang L, Lookene A, Wu G, and Olivecrona G (2005). Calcium triggers folding of lipoprotein lipase into active dimers. J. Biol. Chem 280, 42580–42591. [DOI] [PubMed] [Google Scholar]